Chapter 2 Clinical Evaluation of the Nervous System
• Step back and observe the patient walking, reading, or moving in bed before beginning the clinical examination. If you focus on an obvious deficit, you may miss many important details. The examiner must master the skill of observing and listening to the patient. A thorough and artfully elicited history and examination are still essential and constitute the cornerstone of what we do and should be used in conjunction with the imaging studies to help direct therapy.
• Signs of pyramidal tract dysfunction include spasticity, weakness, slowing of rapid alternating movements, hyperreflexia, and a Babinski sign. Pyramidal lesions often cause rapid alternating movements to become slowed, but accuracy is preserved, in contrast with cerebellar lesions, which can result in fast but inaccurate, sloppy movements.
• A basal ganglion tremor often is present at rest but disappears with movement, in contrast with a cerebellar tremor, which is minimal at rest and exaggerated with movement (intention tremor).
• Use caution investigating the cause of the dilated pupil on one side, because the larger pupil is always the more impressive, even though the patient actually has a constricted pupil on the opposite side because of Horner syndrome.
• A compressive lesion, such as an aneurysm, may produce a dilated pupil with ptosis and painful ophthalmoplegia, in contrast with a pupil-sparing, painless ophthalmoplegia due to diabetes.
• The presence of optokinetic nystagmus can be used to confirm cortical vision and rule out hysterical blindness; its absence, however, is inconclusive.
• A fourth cranial nerve lesion causes weakness of the superior oblique muscle and results in a compensatory head tilt away from the side of the affected eye to compensate for the diplopia. Patients with a fourth nerve paresis have difficulty walking down steps or looking down when they walk.
• Note any asymmetry or marked preference for one hand or the other in a young child; the presence of definite hand preference before 24 months may raise the suspicion of central nervous system or peripheral nerve impairment.
• Asymmetry of the Babinski response is abnormal at any age and may reflect an upper motor neuron lesion.
• The open fontanelle in a child under 15 months of age provides good access for checking intracranial pressure. If it is bulging in a quiet child in an upright posture, you can assume that the intracranial pressure is high.
The history and neurological examination is still the centerpiece in the evaluation of a patient with a surgically correctable neurological disease. The neurosurgeon’s job requires basic investigative work, a thorough knowledge of neuroanatomy, appropriate utilization of the currently available diagnostic tools, and last, substantial interpersonal skills. Correctly identifying the neurological problem is one of the most satisfying parts of a neurosurgeon’s job, for it is a mandatory skill that must precede a successful surgical outcome for the patient. It is what everything we do is built upon.
Focal Cortical Signs
We will begin this tour at the top with the cerebral cortex and then continue down the line. In general, conversation with the patient during the course of the examination will elicit the cortical deficits that are obvious. The ability to talk and respond to questions in a sensible and coherent fashion reveals a great deal about the cerebral cortices. Asking a patient to perform a simple task such as reading a newspaper to the examiner requires activation of an incredibly complex set of neural circuits. In so doing, the examiner is able to test the visual system, cranial nerves, and the motor and sensory systems as well as higher cortical function. This seemingly straightforward, everyday task helps the examiner quickly close down on a wide spectrum of neurological functions that may be affected by the patient’s disease. More subtle cortical deficits require meticulous testing, often by neuropsychological examinations, the interpretation of which requires specific training. Neuropsychological examinations are performed more commonly in the pre- and postoperative stages of modern neurosurgical intervention.1 It is simply not sufficient to know if the patient did “OK” after complex intracranial surgery. It is important to understand what subtle deficits existed preoperatively and how well the deficits improved postoperatively, or which new deficits will require active rehabilitative intervention to improve after surgery.
In broad strokes, the examiner must understand two major types of pathognomonic cortical signs: focal and bihemispheric. Focal cortical signs direct the examiner to a specific area of cortex in one hemisphere, or if bihemispheric, in both hemispheres. Certain portions of the cerebral hemispheres are also termed “silent” areas, because the localizing evidence for lesions here may be absent.2,3
Left occipital lobe dysfunction produces a right homonomous hemianopia (loss of the right half of a visual field), although loss of this field can theoretically result from a lesion of the left optic tract or left thalamic lateral geniculate body. A right or left hemianopia can therefore result from any retrochiasmal lesion (behind the chiasm). Color dysnomia (inability to name colors) is the result of an interruption of fibers streaming from the occipital lobe to Wernicke’s area, the comprehension center in the left temporal lobe. In 98% of right-handed people, Wernicke’s area is located in the left temporal lobe. In most left-handed people, Wernicke’s area is still located in either the left temporal lobe alone or in both temporal lobes.4,5 In only a minority of left-handed people is Wernicke’s area confined to the right temporal lobe.6 A lesion in Wernicke’s area results in a sensory or receptive aphasia characterized by fluent speech filled with gibberish words. Written words come from the occipital cortex, while spoken words may come from both temporal lobes. A mistake in naming results in a paraphasia and is often the result of a lesion in the posterosuperior temporal lobe, but can have quite variable localization. Adjacent to Wernicke’s area in the temporal lobe is another area called the “dysnomia center,” which shows variable localization from person to person. Another pathognomonic sign of temporal lobe dysfunction is a focal, temporal lobe seizure, described as fits consisting of a sense of fear, smell, pleasure, or déjà vu. Another common manifestation of temporal lobe seizures is the automatism, a brief episode of automatic behavior during which the patient is unaware of his or her surroundings and is unable to communicate with others. Patients with complex partial seizures may experience sudden unpleasant smells (e.g., burning rubber) of brief duration which constitute olfactory auras. Temporal lobe dysfunction may also cause a superior quadrantopia (loss of a quarter of the visual field), described as a “pie in the sky,” as a result of a disruption of the optic radiations, called Meyer’s loop, which dip into the temporal lobe.
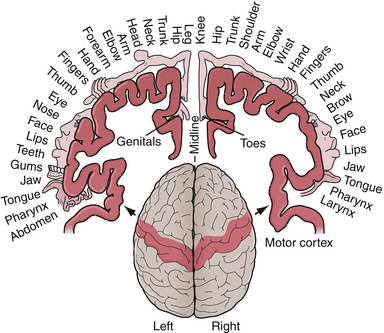
Pathognomonic signs of left parietal dysfunction include right-sided cortical sensory loss, right-sided sensory-motor seizures, or a Gerstmann syndrome, characterized by finger agnosia (inability to recognize one’s fingers), acalculia (inability to calculate numbers), right/left confusion, and agraphia without alexia (an ability to read but not write). Another sign of left parietal cortical dysfunction is cortical sensory loss and results in agraphesthesia (inability to identify numbers written on his/her skin). Sensory seizures may spread up or down the sensory strip and have been described as the jacksonian march. The movement, usually clonic, begins in one portion of the body, for example, the thumb or fingers, and spreads to involve the wrist, arm, face, and leg on the same side along the stereotypical pattern of cortical organization termed the homunculus (Fig. 2.1). A Todd’s paralysis may then occur following the attack, with the same distribution.
Left frontal lobe dysfunction can result in Broca’s aphasia, also known as motor or expressive aphasia, and is characterized by halting, slow, and nonfluent speech.7 Speech lesions in the arcuate fasciculus, a dense bundle of fibers connecting Wernicke’s area to Broca’s, prevent patients from repeating phrases but does not impair comprehension (Table 2.1).
| Lesion | Deficit | Aphasia Type |
|---|---|---|
| Temporal | Retained repetition and fluency, no comprehension, no naming | Transcortical sensory |
| Wernicke’s | Retained fluency, no comprehension, repetition, or naming | Wernicke’s |
| Parietal | Retained comprehension and fluency, no repetition | Conduction |
| Broca’s | Retained comprehension, no fluency, repetition, or naming | Broca’s |
| Frontal | Retained comprehension and repetition, no fluency or naming | Transcortical motor |
Signs such as lethargy, stupor, coma, disorientation, confusion, amnesia, dementia, and delirium often result from bihemispheric dysfunction and are not derived from a simple focal cortical lesion.2
Pyramidal Tract
The pyramidal tract begins in the motor strip of the cortex and courses downward through the brain and into the spinal cord. In the hemispheres it is called the coronal radiata and then becomes the internal capsule, cerebral peduncle, and pyramidal tract, which crosses at the medulla–spinal cord junction, and finally in the spinal cord becomes the corticospinal tract. Functionally, a lesion anywhere along this tract can produce the same long tract signs. Signs of pyramidal tract dysfunction include spasticity, weakness, slowing of rapid alternating movements, hyperreflexia, and a Babinski sign.8 Muscle tone is examined by manipulating the major joints and determining the degree of resistance. Spasticity is one type of increased tone (resistance of a relaxed limb to flexion and extension). Muscle strength is commonly graded from 0 to 5 using the grading system shown in Table 2.2.
| Grade | Strength |
|---|---|
| 0 | No muscle contraction |
| 1 | Flicker or trace of contraction |
| 2 | Active movement with gravity eliminated |
| 3 | Active movement against gravity |
| 4 | Active movement against gravity and resistance |
| 5 | Normal power |
Acute lesions anywhere along the pyramidal tract may also produce flaccid hemiparesis, at least initially, with spasticity developing later. If the whole area of cortex supplying a limb is damaged, the extrapyramidal pathways may be unable to take over and an acute global flaccid weakness of the limb can occur. Intraoperative monitoring has been used to mitigate injury to the corticospinal tract.3 Pyramidal tract lesions typically produce weakness of an arm and leg, or face and arm, or all three together.9 Facial weakness may manifest with a slight flattening of the nasolabial fold; however, the forehead will not be weak (frontalis muscle) because the muscles on each side of the forehead have dual innervation by both cerebral hemispheres (corticopontine fibers). The less affected muscles are the antigravity muscles (wrist flexors, biceps, gluteus maximus, quadriceps, and gastrocnemius). Specific tests of grouped muscle strength can also be quite useful (Table 2.3): pronator drift (arms outstretched with the palms up), standing on each foot, hopping on one foot, walking on toes (gastrocnemius), walking on heels (tibialis anterior), and deep knee bend (proximal hip muscles). Typically, pyramidal lesions often cause rapid alternating movements to become slowed but accuracy is preserved. This is in contrast to cerebellar lesions (see later discussion), which can result in fast but inaccurate, sloppy movements.
TABLE 2.3 Deep Tendon Reflexes
| Reflex | Segmental Level∗ | Peripheral Nerve |
|---|---|---|
| Biceps | C5-C6 | Musculocutaneous |
| Triceps | C6, C7, C8 | Radial |
| Brachioradialis | C5-C7 | Radial |
| Quadriceps | L2, L3, L4 | Femoral |
| Achilles | L4, L5, S1, S2 | Sciatic |
∗ Roots in bold type indicate spinal segment with greatest contribution.
Reflexes can also be quite important in detecting subtle pyramidal tract lesions, especially if asymmetrical. Reflexes are graded by a numerical system: 0 indicates an absent reflex, trace describes a reflex that is palpable but not visible, 1+ is hypoactive but present, 2+ is normal, 3+ is hyperactive, 4+ implies unsustained clonus, and 5+ is sustained clonus. Clonus is a series of rhythmic involuntary muscle contractions induced by sudden stretching of a spastic muscle such as at the ankle. The cutaneous reflex (abdominal twitch obtained when you gently stroke someone’s abdomen) and the cremasteric reflex (L1, L2 innervation; retraction of the testicle upward with a brush along the inner thigh) may also be lost in pyramidal tract lesions. The abdominal cutaneous reflexes in the upper quadrant of the abdomen are mediated by segments T8 and T9; the lower by T10 to T12. If, for example, the lower abdominal reflexes are absent but the upper are preserved, the lesion may be between T9 and L1. The Hoffmann reflex is reflective of hyperreflexia and spasticity on that side and suggests pyramidal tract involvement. It is elicited by snapping the distal phalanx of the middle finger; a pathological response consists of thumb flexion. The Babinski reflex is the best-known sign of disturbed pyramidal tract function. The Babinski reflex is an important sign of upper motor neuron disease, but should not be confused with a more delayed voluntary knee and toe withdrawal due to oversensitive soles of the feet.10 The Babinski reflex is sought by stroking the lateral border of the sole of the foot, beginning at the heel and moving toward the toes. The stimulus should be firm but not painful. The abnormal response, referred to as the Babinski sign, consists of immediate dorsiflexion of the big toe and subsequent separation (fanning) of the other toes. The Babinski sign is present in infancy but usually disappears at about 10 months of age (range 6-12 months). When planar responses produce equivocal results, a related reflex may be tested by stroking the lateral aspect of the dorsum of the foot, and is known as the Chaddock sign.
In general, the more spasticity is present, the more likely the pyramidal tract lesion is in the spinal cord, especially if the spasticity is bilateral.11 Conversely, it is unusual for a pyramidal tract lesion in the spinal cord to produce a hemiparesis or monoparesis. A hemiparesis that involves the face places the lesion somewhere above the facial nucleus, although if the hemiparesis spares the face, the lesion need not be below the facial nucleus. Mild or more chronic hydrocephalus may also cause impressive pyramidal tract dysfunction in the legs more than in the arm fibers. Bladder axons also become stretched by the dilated ventricles associated with hydrocephalus and cause urinary urgency and incontinence. Finally, it should be remembered that the spinal cord terminates normally at the level of the L1-L2 vertebral body, and therefore, neurologically L5 is anatomically in the lower thoracic region.
The Extrapyramidal System
Unlike the pyramidal tracts, which govern strength and fine dexterity, the basal ganglia govern the speed and spontaneity of movements. Two basic patterns emerge with basal ganglia dysfunction: either too much or not enough movement. The number one characteristic of a basal ganglia tremor is its presence at rest and disappearance with movement, in contrast to a cerebellar tremor which is minimal at rest and exaggerated with movement (intention tremor). The strength and deep tendon reflexes are normal in extrapyramidal diseases and there is no Babinski sign. However, the tone is either hypotonic, as occurs in choreiform disorders, or increased (rigid), as in the bradykinetic (slowness of movements) varieties with rachety rigidity appropriately called cogwheeling. Choreiform movements are involuntary random jerky movements of small muscles of the hands, feet, or face and may be proximal enough to cause the whole arm to jerk gently. If instead of the small distal muscles, the larger more proximal muscles involuntarily flinch, the patient may have ballismus. Ballismus can be unilateral, but chorea is almost always bilateral. Athetoid movements are slower, more continuous, and sustained, and may involve the head, neck, limb girdles, and distal extremities. Dystonic movements resemble a fixation of athetoid movements involving larger portions of the body. Torticollis, or torsion of the neck, is an example of a neck dystonia that is the result of the continuous contraction of the sternocleidomastoid muscle on one side. Postural and gait abnormalities of extrapyramidal disease are most diagnostic in patients with Parkinson’s disease (tremor, bradykinesia, and rigidity).12 A blank expression and infrequent blinking, walking with a leaning forward posture, and a festinating gait (running, shuffling feet) are typical findings of a Parkinson’s patient. Once in gear, the initially bradykinetic patient may have difficulty stopping. At the same time, the patient’s hand is coarsely shaking at three times a second and the patient’s speech is also devoid of normal changes in pitch and cadence.
Cranial Nerves
Cranial Nerve I
Cranial nerve I, the olfactory nerve, begins at the cribriform plate and travels back underneath the frontal lobe to the temporal lobe without relaying in the thalamus. To test olfaction, test each nostril independently and avoid using a caustic substance such as ammonia, which tests the trigeminal nerve (V) in addition to the olfactory nerve due to irritation of the nasal mucosa. An olfactory groove tumor may present with unilateral anosmia (loss of smell), although the most likely explanation is local nasal obstruction. Foster-Kennedy syndrome is characterized by ipsilateral anosmia, ipsilateral scotoma with optic atrophy (direct pressure on the optic nerve), and contralateral papilledema (elevated intracranial pressure) and is classically due to an olfactory groove or medial sphenoid wing meningioma. Loss of smell can also complicate up to 30% of head injuries as a result of shearing of the nerves as they pass through the cribriform plate.
Cranial Nerve II
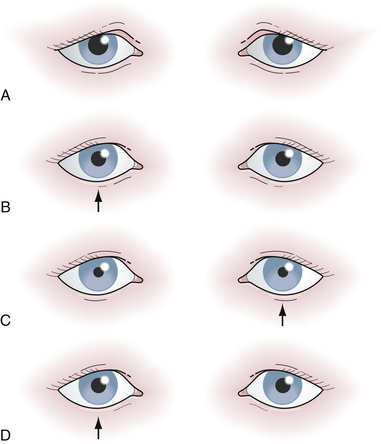
The second cranial nerve, the optic nerve, is the most complex. Visual acuity, color vision, Marcus Gunn pupil, visual fields, and direct ophthalmoscopic observation must all be assessed. Visual acuity is affected early in optic neuropathies, because 20% to 25% of all optic fibers come from the macula and travel in the center of the nerve. If the patient’s visual acuity is not 20/20 and cannot be improved by refraction (looking through a pinhole in a piece of cardboard is a good bedside test), then the visual impairment is most likely neurological. The size, shape, and symmetry of the pupils in moderate lighting conditions should be noted. If the pupils are unequal it is important to decide which pupil is the abnormal one. One frequent mistake is to investigate for the cause of the dilated pupil on one side, because the larger pupil is always the more impressive, even though the patient actually has a constricted pupil on the opposite side because of Horner syndrome. If there is ptosis of the eyelid on the side of the small pupil, the patient may have Horner syndrome, although if the ptosis is on the side of the large pupil, the patient may have an ipsilateral partial third cranial nerve lesion. Furthermore, the light and accommodation reflexes will be normal in a Horner syndrome and impaired in a partial third nerve lesion. Whenever a patient is found to have a widely dilated pupil that is fixed to light and accommodation without accompanying ptosis, there is a possibility of a pharmacological pupil (e.g., atropine drops instilled into the eye). A Marcus Gunn pupil (afferent pupillary defect), a form of optic nerve dysfunction, is elicited by the swinging flashlight test: shine a dim light into the right eye, and note how small the right pupil constricts (left pupil also constricts). Swing the light over to the left eye and carefully note the left pupil. If the very first reaction of that pupil is dilation instead of maintaining its previous small size, then there may be left optic nerve dysfunction, i.e., an afferent papillary defect (Fig. 2.2). The examiner must ignore “hippus,” which is a normal phasic instability of the pupil with waves of alternating constriction and dilatation. An optic nerve lesion can be corroborated with visual field testing and direct funduscopy, both of which will be discussed later in this chapter in the neuro-ophthalmology section. The approach to patients with diplopia also requires a systematic approach because double vision may arise from ocular, neurological, or extraocular muscle disorders (i.e., thyrotoxicosis). The Cover test can be useful in the evaluation of a patient with binocular diplopia. The test is based on the fact that the separation of two images becomes greatest as the eyes attempt to look in the direction of the action of the weak muscle. By determining which eye must be covered to obliterate the outer image, the affected eye is identified, because the false image is always projected as the outer image.
Cranial Nerves III, IV, and VI
The third cranial nerve, or oculomotor nerve, is one of the three nerves that move the eye, the others being the fourth (trochlear) and the sixth (abducens) cranial nerves. Defective adduction and elevation with outward and downward displacement of the eye suggests a third cranial nerve palsy. The third cranial nerve also innervates the levator palpebrae superioris, the muscle that opens the eyelid. Parasympathetic fibers travel within the superior and medial perimeter of the third cranial nerve to constrict the iris and stimulate the ciliary body to round up the lens. As a general rule, if the pupil is affected, the cause is more likely to be surgical (compressive) and if spared, the cause is more likely to be medical (diabetes, cranial arteritis, arteriosclerosis, syphilis, migraine). A compressive lesion, such as an aneurysm, selectively injures these superficially situated parasympathetic fibers, producing a dilated pupil with ptosis and painful ophthalmoplegia. In contrast, diabetes more often causes a pupil-sparing, painless ophthalmoplegia by damaging the interior motor axons through arterial thrombosis.13 The sympathetic nerves supply Müller’s muscle, which also slightly elevates the eyelid and when injured causes the upper eyelid to droop and results in ptosis and miosis (eyelid droop and a dilated pupil), or Horner syndrome. If the sympathetic nerves to the eye are interrupted prior to the carotid bifurcation, ipsilateral facial anhidrosis (no sweating) may also result. Some of the sympathetic nerves also ascend the common carotid and follow the external carotid onto the face to stimulate the facial sweat glands.
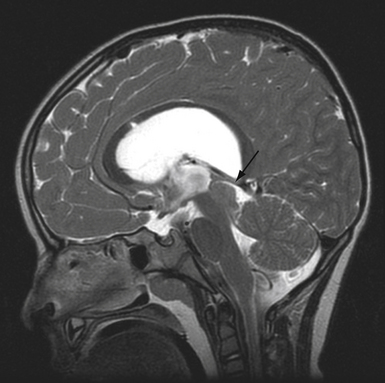
If the pupils do not react to light, the anatomical differential diagnosis includes the afferent limb (retina, optic nerves, optic tracts) and the efferent limb (pretectum, Edinger-Westphal nucleus, parasympathetic fibers in the oculomotor nerves, and the pupillary constrictor muscle in the iris). A pupil able to accommodate to near vision but not react to light is referred to as an Argyll Robertson pupil and has been classically seen in patients with tertiary syphilis. This, of course, is a rare finding because of the decrease in this disease over the past century. Light-near dissociation is also seen in Adie’s pupil, which is usually unilateral and is caused by parasympathetic dysfunction. When parasympathetic innervation is first lost in Adie syndrome, the pupil is relatively large, but with time and reinnervation the pupil constricts. This is a curious but benign disorder of unknown cause, usually affecting one eye, and results from injury or illness to the ciliary ganglion, usually inflammatory in nature. Pineal region tumors can also damage the midbrain pretectum and cause light-near dissociation. Pineal region tumors more classically damage the midbrain upgaze center and cause a constellation of dorsal midbrain signs called Parinaud syndrome: (1) impaired upward or downward gaze; (2) bilateral light-near dissociation; (3) pupillary dilatation; and (4) retraction of the eyelids.
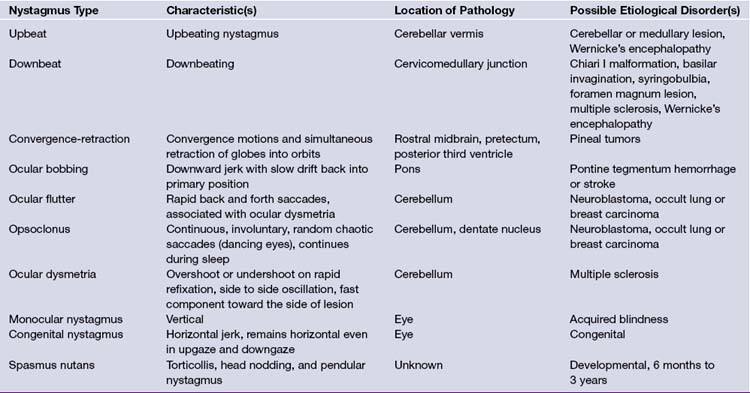
In general, nystagmus can be due to labyrinthine or brainstem/cerebellar pathology, may be central or peripheral, and is defined in the direction of the fast movement (Table 2.4). Upbeat or downbeat nystagmus is almost always of central origin, and represents disrupted connections between the cerebellum and brainstem (Chiari malformations, basilar invagination, platybasia, or a midline cerebellar lesion such as medulloblastoma in children). Horizontal nystagmus is more commonly peripheral in origin, especially if the patient can stop the nystagmus by fixating on a target. Two axes of nystagmus, as seen in rotary nystagmus, suggest a disturbance of two semicircular canals. Opsoclonus is another form of nystagmus and is characterized by chaotic, repetitive, saccadic movements in all directions, preventing fixation, and has also been termed dancing eyes.14 In an adult, opsoclonus is associated with postinfectious encephalopathy as well as with carcinomas of the lung or breast, although in younger children it has been described in association with neuroblastoma. The presence of optokinetic nystagmus can be used to confirm cortical vision; its absence, however, is inconclusive.15 When the optokinetic tape (a series of vertical black lines on a white background) is pulled from the patient’s left to his or her right, the right parieto-occipital lobe tracks the target to the right (smooth pursuit, slow phase). The eyes saccade left to track each newly arriving target (fast phase). In right parieto-occipital lesions, a smooth pursuit (slow phase) to the right is lost. However, occipital stroke due to a posterior cerebral artery infarct do not usually impair optokinetic nystagmus. A tumor, in contrast, may cross vascular boundaries and interrupt a smooth pursuit generators.
Looking left involves two cranial nerves: the left sixth (left lateral rectus) and the right third (right medial rectus) (Fig. 2.3). There are three classic signs of a pontine medial longitudinal fasciculus (MLF) lesion, or internuclear ophthalmoplegia (INO): (1) weakness of the contralateral medial rectus muscle causing paralysis of adduction on lateral gaze, because the MLF cannot transmit its message to the third cranial nerve to pull the eye medially; (2) nystagmus in the abducting eye; and (3) the retained ability to converge, demonstrating that the reason for medial rectus weakness on adduction is not in the third cranial nerve or muscle itself. In the setting of a third nerve lesion, the eye will be deviated downward (secondary depressant action of superior oblique) and outward (lateral rectus action) and the diplopia would improve when testing lateral gaze in the affected eye.
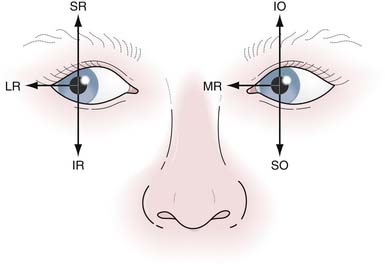
A fourth or trochlear nerve lesion causes weakness of the superior oblique muscle and diplopia. This weakness results in a compensatory head tilt away from the side of the affected eye to compensate and is called the Bielschowsky sign. Patients with a fourth cranial nerve palsy can lessen or extinguish their double vision by tilting their head toward the unaffected side. Tilting their head toward the shoulder on the affected side makes the diplopia worse. The diplopia is particularly troublesome on looking downward and thus especially problematic when the patient is attempting to walk down a set of stairs. In some patients, this head tilt may be misdiagnosed as torticollis. A sixth or abducens nerve palsy is the most disabling eye movement abnormality because the diplopia persists in nearly all directions of gaze. At rest, the affected eye is pulled medially by the unopposed action of the medial rectus muscle. Multiple sclerosis is the most common cause of an isolated sixth cranial nerve palsy due to a plaque in the brainstem. The sixth nerve takes a ventral course from the pontine tegmentum over the petrous ridge to the dorsum sellae and into the cavernous sinus lateral to the carotid nerve and medial to cranial nerves III, IV, V1, and V2. A posterior fossa tumor can cause hydrocephalus, which can also stretch the sixth nerve over the petrous tip and cause diplopia. As the sixth nerve has a rather long course, it is the most vulnerable cranial nerve to closed head injury. Benign transient sixth nerve palsies can also occur in children following mild infections. More severe cases of mastoiditis (Gradenigo syndrome) can cause ear pain and a combination of sixth, seventh, eighth, and occasionally fifth cranial nerve lesions. These symptoms must be differentiated from Ramsay Hunt syndrome (geniculate herpes zoster), in which there is vesicular eruption in the ear and a seventh cranial nerve palsy.
Cranial Nerve V
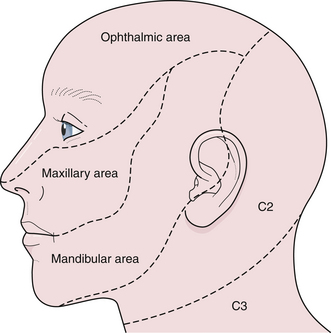
The fifth cranial nerve, the trigeminal, controls both sensory and motor function: sensation of the forehead and face (including inside the mouth) and strength of chewing muscles (temporalis, masseter, pterygoids). The three branches of the trigeminal nerve are denoted as V1, V2, and V3. It is important to recognize that there is a large area over the angle of the jaw supplied by nerve roots C2 and C3 and that patients with nonorganic sensory loss over the face usually claim anesthesia extending to the line of the jaw and the hairline (Fig. 2.4). When testing for a corneal reflex, a wisp of cotton wool should not be allowed to cross in front of the pupil or the patient will see it and blink (false positive). In addition, both eyes should shut simultaneously if the corneal reflex is present. A depressed or absent corneal reflex can be an early physical sign of an acoustic neuroma in the cerebellopontine angle. Intracavernous lesions (i.e., aneurysms, meningiomas, carotid-cavernous fistulas, and pituitary tumors) can all cause facial numbness. However, jaw numbness does not typically occur with cavernous sinus lesions because nerve V3 does not enter the cavernous sinus like nerves III, IV, VI, V1, and V2. Patients suffering from lightning jabs of terrible facial and jaw pain, often precipitated by a trigger point along the gums or lips, may have trigeminal neuralgia.16 The cardinal feature of trigeminal neuralgia is pain without any objective neurological abnormality (i.e., sensory or motor dysfunction). The cause is thought to be an arterial or venous loop that pulsates against the trigeminal nerve at the pontine root entry zone or sometimes a small plaque of brainstem demyelination as in multiple sclerosis, although attacks of trigeminal pain may occur with any tumor of the cerebellopontine angle or petrous apex. Although herpes zoster can affect any nerve in the body, the thoracic roots are usually affected in younger age groups, although in the elderly, the virus has a predilection for nerve V1.
Cranial Nerve VII
The seventh cranial nerve, the facial, controls all the facial and forehead muscles. The seventh nerve does not contribute to normal eye opening but instead contributes to forced eye opening. Paralysis of facial movement including both the cheek and forehead on one side of the face, both volitional and emotional, indicates a lesion in the seventh cranial nerve (peripheral) somewhere between the pontine facial nerve nucleus and the facial muscles. In general, if the forehead is spared, then the facial paralysis is “central” and is the result of a lesion in the descending corticopontine upper motor neuron. Eye closure and forehead movement will remain relatively intact because the intact hemisphere pathways provide adequate cross-innervation. Recent evidence also suggests that upper facial motor neurons receive little direct cortical input, whereas lower facial neurons do and are therefore more affected.17 As the facial nerve leaves its nucleus in the brainstem, other nerves piggyback it on their way to the lacrimal gland, stapedius muscle (dampens loud noises in the ear), and the taste buds along the anterior two thirds of the tongue. Ipsilateral loss of taste and tear production, and the presence of hyperacusis (noises sound too loud) confirm that the patient’s facial weakness is the result of lower motor neuron dysfunction. Most often, an acute peripheral facial weakness without associated sensory loss is the result of Bell’s palsy, a poorly understood acute inflammatory attack on the facial nerve within the facial canal. This disorder has an excellent prognosis for recovery within weeks or months. Blepharospasm is a recurrent involuntary spasm of forceful eye closure (both eyes) with some spread into other facial muscles. Hemifacial spasm is characterized by recurrent spasms of one side of the face and is most likely the result of an irritation of the facial nerve as it leaves the brainstem by a pulsating arterial loop.17,18
Cerebellum
The smooth and efficient performance of volitional movements depends on the coordination of agonist and antagonist muscles, acting in synergy. A failure of a group of muscles to act harmoniously is a sign of cerebellar dysfunction. Dysdiadochokinesis is characterized by difficulty in performing rapid alternating movements. Dysmetria is the difficulty in reaching a target accurately or past-pointing. The rate, rhythm, amplitude, and smoothness of movement may all be affected in cerebellar disease. A relatively common cerebellar tremor, called titubation, affects elderly people with a rapid, fine, bobbing motion of the head. The side-to-side imbalance of cerebellar ataxia is in contrast to the front-to-back imbalance of parkinsonian patients. However, unlike the crossed pyramidal and extrapyramidal systems, the right cerebellar hemisphere controls the right arm and right leg and vice versa. Often a cerebellar tremor is present with the arms outstretched (postural tremor), but the tremor almost always worsens with intention (intention tremor). Speech is also affected in cerebellar disorders, causing ataxia of speech called scanning speech. In addition, the inability to perform finger-to-nose movements or to tandem walk is characteristic of cerebellar dysfunction. Postural instability can be best evaluated by the Romberg test, which is a nonspecific test of vestibular function and often used to demonstrate loss of joint position sense. A positive test results when the patient falls with his or her eyes closed when standing with the feet together. In unilateral vestibular or cerebellar disease, the patient sways toward the damaged side.
Spinal Cord, Nerve Roots, and Muscles
The last neural circuits to consider are the spinal cord, nerve roots, and muscles. However, before distinguishing between a root and peripheral nerve lesion it is important to discriminate an upper motor neuron from a lower motor neuron lesion. As discussed earlier, upper motor neuron signs include spasticity, weakness, slowing of rapid alternating movements, hyperreflexia, and a Babinski sign. Lower motor neuron lesions (root or peripheral nerve) can cause muscular atrophy, fasciculations, hypotonia, or weakness in a particular root or peripheral nerve distribution, and diminished reflexes. To diagnose a myelopathy, the long tract signs need to be combined with root or segmental signs (Fig. 2.5). Fasciculations are spontaneous, random contractions of muscle, usually too small to move a joint but visible when the skin over the affected muscle is inspected. However, in order to call a spontaneous muscular twitch a fasciculation, the muscle must be fully at rest. The presence of fasciculations implies a lower motor neuron dysfunction; however, the abnormality may be in the spinal cord (ventral horn) or anywhere along the peripheral nerve up to the point of muscle insertion. Fibrillations are the smallest potentials obtainable from individual muscle fibers and occur in denervated muscle fibers after 3 weeks when the motor neurons supplying a muscle are damaged, either in their cell bodies, the ventral roots, or the peripheral nerve itself.
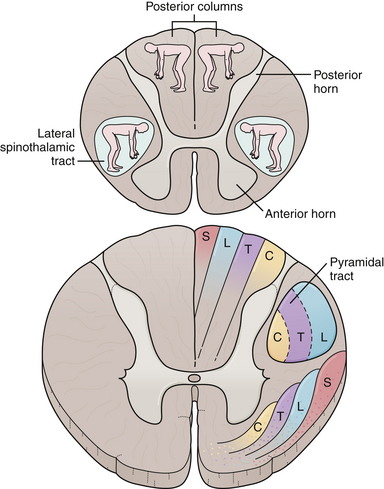
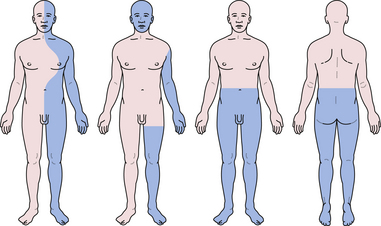
A Brown-Sequard syndrome affects the left or right half of the spinal cord and is characterized by ipsilateral weakness, contralateral pain and temperature loss, and ipsilateral vibration and proprioception loss below the lesion (Fig. 2.6). Anterior spinal artery syndrome is characterized by flaccidity followed by spasticity, weakness, slowing of rapid alternating movements, hyperreflexia, and a Babinski sign, as well as bilateral pain and temperature loss below the lesion but no vibratory or proprioceptive loss (dissociated sensory loss).19 Syringomyelia (slowly expanding cyst of the spinal cord) or a centrally located spinal cord tumor can also cause dissociated sensory loss. A constellation of lower motor neuron signs and upper extremity dissociated sensory loss is virtually pathognomonic of syringomyelia in the cervical spinal cord. A syrinx can be congenital, developmental, or even post-traumatic and can present in a delayed fashion following a spinal cord injury. Occasionally, the syrinx extends up into the medulla (called syringobulbia) and causes atrophy, fasciculations, and weakness of the tongue and pharynx.20,21 Vitamin B12 deficiency causes another type of dissociated sensory loss called combined systems disease. In this disease, vibration and proprioception are lost but pain and temperature sensation are spared. Lower motor neuron signs may also be present from the peripheral neuropathy due to vitamin B12 deficiency. Central cord syndrome is another spinal cord syndrome characterized by post-traumatic quadriparesis (worse in the arms) without sensory loss following a hyperextension cervical injury and usually occurs in the elderly with preexisting cervical canal stenosis. Injury to the ventral horns can cause the lower motor signs in the arms and hands, and injury to the corticospinal tracts results in a spastic quadriparesis.22 The center of the spinal cord is a vascular watershed zone, which renders it more vulnerable to injury from edema, and furthermore, the cervical fibers are located more medially than lumbar fibers for the lower extremity. A cruciate paralysis due to a foramen magnum lesion may also result in hand weakness that will start in one hand and then go to the ipsilateral leg and the contralateral side.
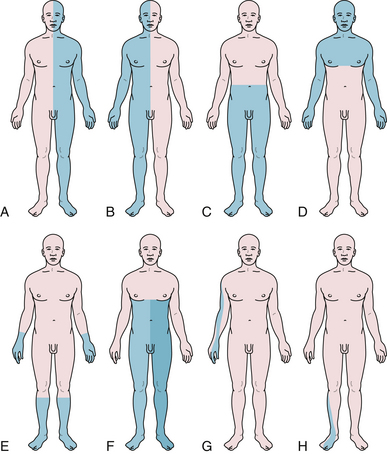
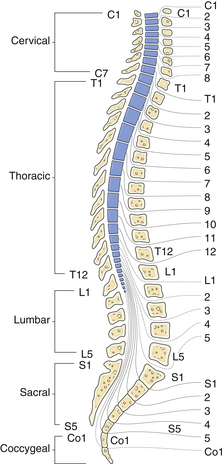
There are a few pitfalls to consider when examining a patient with a potential myelopathy.3 First, remember that the pyramidal tracts to the legs terminate neurologically at about L4 (Babinski is extensor hallucis longus: L5) and anatomically at around the T12 vertebral body (Fig. 2.7). Therefore, a spastic paraparesis warrants a cervical or thoracic MRI and not a lumbar MRI. A spastic paraparesis does not automatically place the lesion between the thoracic and lumbar regions, because compressive lesions of the upper cervical cord can damage the cord’s blood supply, and in addition, the descending leg fibers in the corticospinal tracts are more vulnerable to ischemia than the arm fibers. Second, a hemiparesis sparing the face is not necessarily the result of a cervical myelopathy because a pyramidal tract lesion in the internal capsule can also spare the face. Myelopathies, however, rarely result in a hemiparesis. Third, atrophy of the hands and arms may be the result of a high cervical extramedullary mass at the foramen magnum. An extramedullary mass is one that lies outside the spinal cord, either intradurally or extradurally. It is difficult by history and examination to distinguish an intramedullary from an extramedullary spinal cord lesion. In general, extramedullary lesions stretch nerve roots and can be more painful than intramedullary lesions and can cause compression of the spinal cord and nerve roots at the affected segment. Also, extramedullary lesions cause more pain in the supine position, which is the opposite of a herniated disk, in which lying flat can relieve the pain. Palpation of the spinous processes and straight leg raising will often elicit pain from an extramedullary lesion but not an intramedullary lesion. Intramedullary lesions, in contrast, are more likely to produce atrophy, dissociated sensory loss, and early bowel and bladder problems. Sacral sparing can also be helpful, since the sacral sensory fibers are lateral in the spinothalamic tracts and may not be affected in a patient suffering from an intramedullary lesion. The cauda equina and conus medullaris syndromes are also important to distinguish from peripheral root symptoms. Lesions at either location interrupt multiple motor and sensory roots to the legs, producing bilateral lower extremity atrophy and weakness, depressed reflexes, down-pointing toes, and often a sensory level.
Proximal weakness alone is the most common sign of a myopathy. Patients with proximal weakness waddle, because the weak gluteus medius muscles allow the pelvis to tilt from side to side. A patient may also have to lean forward and push off with both hands to get up from a chair, signifying pelvic girdle weakness. When trying to get up off the floor, children also adapt to pelvic girdle weakness and from the all-fours position, lock both their knees and push their trunk back over their legs by bracing their hands on their thighs (Gower’s sign). Myotonia is a myopathic sign resulting from delayed relaxation after the muscle contracts and occurs in myotonia congenita, myotonic dystrophy, and paramyotonia congenita. As a rule, a myotonic patient shakes your hand and does not let go. In patients with widespread symmetrical weakness, pay attention to any sensory loss so that myopathic weakness is not confused with Guillain-Barré syndrome, an acute peripheral neuropathy with some distal vibratory loss and areflexia. Muscles above the shoulders are particularly susceptible to myasthenia gravis and botulism, two illnesses that attack the neuromuscular junction. Patients may present with a pure motor syndrome dominated by ophthalmoplegia, ptosis, weakness of chewing, difficulty sucking through a straw, dysphagia, and tongue weakness, but without pyramidal signs in the arms and legs. Almost any external ophthalmoplegia can be mimicked by myasthenia gravis23: internuclear ophthalmoplegia, up- or down-gaze palsy, sixth cranial nerve palsy, and a pupil-sparing third cranial nerve palsy. Neuromuscular blockade produces “fatigable” weakness that worsens with each contraction.
The Pediatric Patient
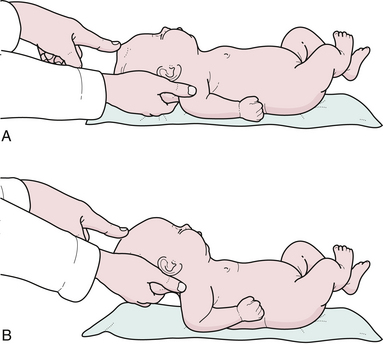
The neurological evaluation of the infant or child begins with the birth history, social history, developmental history, family history, and physical examination. The general appearance of the child should be noted, particularly the presence of any dysmorphic features or neurocutaneous abnormalities such as café au lait spots, neurofibromas,24 facial port-wine stain in Sturge-Weber disease, depigmented lesion nevi in tuberous sclerosis, as well as a craniofacial dysmorphism seen with craniosynostosis. It is important to inspect the midline of the neck, back, and pilonidal area for any defects, particularly for small dimples above the level of the gluteal fold in the midline that might indicate the presence of occult spinal dysraphism or a dermal sinus tract. The head should be examined by inspection, palpation, and auscultation. The shape, size, and asymmetry may point to microcephaly, hydrocephalus, craniosynostosis (premature cranial suture fusion), or cerebral atrophy. Maximum head circumference should be recorded on a standard chart according to the patient’s age and sex. The charting of the head circumference by the primary care provider and the neurosurgeon examining that plotted curve are essential parts of the examination and may indicate an intracranial pathology before it becomes symptomatic. The general appearance of the skull, prominence of venous pattern, and palpation of the anterior fontanelle may suggest increased intracranial pressure. The palpation of the anterior fontanelle is another essential part of the neurological examination. In the sitting position the fontanelle should be concave or sunken; in the supine position it may be more full (Fig. 2.8). Intracranial pressure can be estimated within several millimeters of water by palpating the anterior fontanelle. The baby should be laid flat and the head should be gently raised off the examining table. At the point the fontanelle becomes flat, the intracranial pressure equals the extracranial pressure. If one measures the height the head has been raised in millimeters above a horizontal line drawn through the child’s heart (the physiological zero point), then one has a fairly good estimate of the child’s intracranial pressure. If the patent’s anterior fontanelle consistently remains bulging and full when a quiet child is fully erect or sitting, then that denotes increased intracranial pressure. An imaging study such as a computed tomography (CT) or MRI of the head is a reasonable option. The anterior fontanelle is often closed by 18 to 24 months of age, although the posterior fontanelle closes after 2 to 3 months. Transillumination of the head with a flashlight in absolute darkness up to the age of approximately 9 months is an old-fashioned but useful way to detect severe hydrocephalus, arachnoid cysts, or subdural effusions at the bedside. However, it has become a lost art and cranial ultrasound has replaced this once important historical diagnostic modality. Percussion of the head, also of historical note, may produce a hollow or “cracked pot” resonance in patients with severe hydrocephalus (Macewen’s sign). Cranial nerve examination can be more reliably tested beyond 30 weeks’ gestation since prior to that time, the pupillary response to light is not predictably present, and the gag reflex is also not easily elicited.4 The “blink reflex” is often used to determine the presence of functional vision in small infants but is absent in the newborn.25 A slight degree of anisocoria is not unusual, particularly in infants and small children. The funduscopic examination is an essential part of the neurological examination (see discussion under neuro-ophthalmology). True papilledema with early obliteration of the disk margins and absent pulsations of the central veins is rare in patients under the age of 2 years because of the ability of the expansile skull to dissipate a rise in intracranial pressure. Medulloblastoma, a midline cerebellar tumor that can infiltrate the superior medullary velum, may produce bilateral fourth nerve palsies. A setting sun sign is the forced downward deviation of the eyes at rest with associated upward gaze palsy. Parinaud syndrome is an upward gaze palsy that can also be seen in any patient as a result of pressure on the upward gaze eye center in the region of the suprapineal recess and quadrigeminal plate due to hydrocephalus or a pineal region mass lesion.
Neuro-Ophthalmology
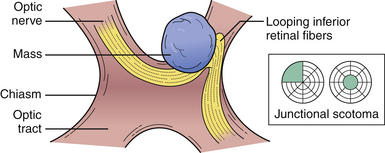
No neurological examination is complete without a detailed study of the visual system. Because of the extent of the visual system and its intimate relations with other areas of the brain, much valuable information can be obtained. Color vision is especially important in neuro-ophthalmology in the detection of pregeniculate pathway lesions. The visual field to a red object is interestingly more affected by damage in these areas. Similarly an optic tract lesion may produce an incongruous hemianopic defect of color vision. The results of confrontation testing are conventionally recorded as seen by the patient, which means reversing the defect as seen by the examiner during confrontation testing. The nature of the field defect should be carefully documented: left central scotoma (optic nerve lesion), bitemporal hemianopia (chiasmatic lesion), right upper quadrantic hemianopia (left temporal), macula-sparing hemianopia (lesions of the optic tract), and right homonomous hemianopia’s scotoma lesion (tip of occipital pole) (Fig. 2.9). The areas of calcarine cortex subserving the peripheral fields lie anteriorly and those subserving macular vision are concentrated at the extreme tip: the upper fields are represented in the lower half below the calcarine sulcus and the lower fields in the upper half of the cortex. Special attention should be paid to whether the defect crosses the horizontal meridian, because retinal lesions due to vascular occlusion cannot do so. The defect may extend to the blind spot, and defects due to vitamin B12 deficiency, toxins, or glaucoma usually extend into it. Lastly, the defect may cross the vertical meridian because organic visual field defects have a sharp vertical edge at the midline. The macula of the retina responsible for central vision is situated to the temporal side of the optic nerve head, which then moves centrally into the optic nerve as it joins the chiasm. This papillomacular bundle conveying central vision in the optic nerve is very vulnerable to extrinsic compression by mass lesions. It is equally important to check for an early temporal field cut (contralateral junctional scotoma) in the opposite eye due to damage to the decussating nasal fibers (anterior chiasmatic syndrome of Traquair) (Fig. 2.10).
Ancillary Diagnostic Tests
MRI is noninvasive and has a number of diverse clinical uses in the evaluation of neurological disorders without exposure to ionizing radiation. However, as a result of the longer acquisition time and less access to the patient during the study, it is not routinely used for acute trauma or unstable patients. MRI offers superb anatomical detail in the detection of structural causes for neurological dysfunction such as tumors, arteriovenous malformations, demyelinating disease, or stroke. Generally, T1-weighted images provide a better view of structural anatomy, whereas T2-weighted images are exquisitely sensitive to water (hydrocephalus) and cerebral edema and are preferred for the detection of pathology. MRI has been shown to be superior to CT for the detection and characterization of posterior fossa lesions. MRI has been used to define the anatomy in epilepsy patients with mesial temporal sclerosis or with anomalies of cortical architecture, depict the compression of the trigeminal nerve by a vascular loop, and evaluate CSF flow in patients with Chiari malformation and syringomyelia or normal-pressure hydrocephalus. Diffusion-weighted MRI is extremely sensitive to the brownian motion of water protons and is used in the early evaluation of stroke in evolution. MRA (arteriography or venography) is an excellent way to evaluate the vascular structures and avoids invasive cerebral angiography. MR perfusion techniques have evolved to quantify blood flow to areas of ischemia or hyperemia and have been used in patients with brain tumors, stroke, and subarachnoid hemorrhage. MRI can be combined with high-resolution MR spectroscopy to evaluate the spectral peaks obtained that reflect the concentrations of the metabolites and some neurotransmitters in the voxel area under investigation. Spinal MRI is the most efficient way to screen for spinal disease and can be combined with a gadolinium contrast agent in the setting of neoplasia or infection. Functional MRI is useful in the preoperative localization of the motor and somatosensory cortex based on the identification of cortical activation by detecting changes in venous oxygen.
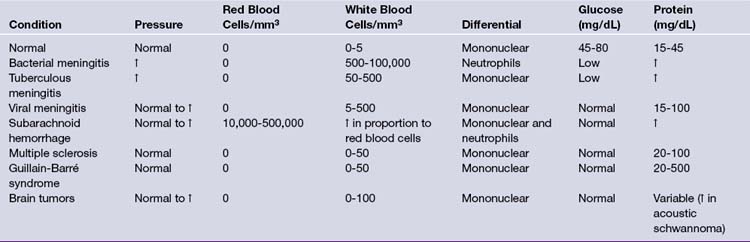
Following funduscopic and CT or MRI examinations, CSF analysis is indicated in patients suspected of having central nervous system bacterial, fungal, or viral infection as well as subarachnoid hemorrhage. Lumbar CSF pressure recordings are also useful in the diagnosis of pseudotumor cerebri and normal-pressure hydrocephalus, although it is important to recognize that falsely high pressures result when the knees are pressed against the abdomen and when the patient holds his breath. The chief danger of a lumbar puncture (spinal tap) is uncal herniation in patients with raised intracranial pressure because of focal disease. The spinal fluid is normally clear and colorless. Turbidity can result from the presence of leukocytes or bacteria, and hemorrhage can result from a “bloody tap” or a subarachnoid hemorrhage. In normal adult CSF, there are 0 to 4 lymphocytes or mononuclear cells per mm3, and no polymorphonuclear lymphocytes or red blood cells. Polymorphonuclear lymphocytes can be present in the newborn, but they are not normally found in CSF taken from healthy children older than 1 year of age. In general, 1 white blood cell can be subtracted for every 700 red blood cells in the CSF. The CSF/plasma ratio for glucose is normally 0.60 to 0.80. Low protein content suggests its relative exclusion by the blood-brain or blood-CSF barriers, although high protein levels are found in patients with blood (1000 red blood cells raise the total protein level by 1.5 mg/dL) or intraspinal tumors. Among spinal cord tumors, intradural extramedullary tumors such as meningiomas or neurofibromas often have elevated CSF protein values greater than 100 mg/dL (Table 2.5).
Electromyography and nerve conduction studies can often aid in the evaluation of neuromuscular disorders or spinal disease, such as herniated disks or spondylosis. A needle electrode is inserted into the muscle and action potentials are generated by muscle activity. Normal resting muscle is electrically silent except for the insertion potential produced by needle insertion. After denervation of the muscle, fibrillation potentials appear. Nerve conduction velocities can be used to differentiate demyelination and axonal degeneration from muscular disorders. Conduction rates of motor nerves can be measured by stimulating the nerve at two points and recording the latency between each stimulus and the muscle contraction. Somatosensory evoked potentials are recorded after stimulation of peripheral nerves and are sensitive to compare side to side.
Diagnosis and Investigation of Cerebral Tumors
History and physical examination remain the gold standard for the initial assessment of any patient suspected of suffering from a primary or secondary cerebral neoplasm. However, the advent of CT and MRI has transformed the investigation.26 The cardinal symptoms and signs of a cerebral tumor are headache, vomiting, malaise, cognitive decline, and papilledema. These are most commonly seen with posterior fossa tumors or those which have blocked the flow of CSF. However, in general, less than 0.1% of patients referred to the hospital for headaches have a cerebral tumor. Thus lies the diagnostic dilemma for the primary care physician. A first (nonfebrile, nonmetabolic-induced, nontraumatic) epileptic seizure occurring in an adult patient warrants an electroencephalogram (EEG) or an imaging study such as an MRI or CT scan. An EEG is of value in the assessment of patients who have presented with an epileptic fit, although it may be misleadingly normal. The EEG does not exclude the presence of epilepsy or organic disease and a single normal EEG is of little value. The basic rhythm observed in an adult is called the alpha rhythm (frequency of 8-13 Hz) and is present when the patient is relaxed with his or her eyes closed and suppressed when the patient opens his or her eyes or concentrates.
Headache
Luckily not all bad headaches are the result of brain tumors or aneurysms and the seriousness of a headache does not uniformly correlate with its pathological severity. However, a careful headache history is essential in deciding on a further workup, especially is acute situations. We often suggest a headache log for a patient whose headaches are problematic. All the extracranial structures, including the arteries and muscles, are pain sensitive. Intracranially the dura and dura-based vessels are pain sensitive, although the brain itself, cortical vessels, and pia-arachnoid are pain insensitive. Pain can also be referred to the head from other structures sharing its innervation such as the eye, ear, sinuses, and teeth.6,7,27 The temporal pattern of the headache; its site and radiation; precipitating, aggravating, and mitigating factors; accompanying symptoms; and family history should all be considered.28
A typical tension headache is characterized by a tight feeling in the suboccipital muscles which spreads over the top of the head and is exacerbated by stress. A typical pressure headache is one that occurs on waking or at the end of the day, is aggravated by bending or movement, and may be responsive to analgesics.29 A patient with a Chiari I malformation–related headache often complains of “tussive” symptoms that are felt in the back of the head or neck and are worse with coughing, laughing, bending, or any Valsalva-related action. They are better at rest. Analgesics often do not work for these headaches. A headache starting after the age of 60 with pain and tenderness over the temporal region may represent temporal arteritis and is associated with a high erythrocyte sedimentation rate, visual impairment, and generalized malaise.
Vascular Diseases
To make the clinical differentiation between a neoplastic or other space-occupying lesion and a stroke one must substantially rely on the temporal history. Following a stroke, it is unusual to find a vessel actually occluded because most occlusions are the result of temporary embolic blockage with rapid subsequent recanalization. Stroke or “brain attack” emphasizes the abrupt onset of symptoms, the single characteristic feature of a vascular accident. It is important to recognize a stuttering onset of symptoms, characterized by repeated identical brief episodes of hemiparesis with full recovery, known as transient ischemic attacks. There are three main types of hemorrhagic strokes: (1) classic hypertensive intracerebral hemorrhage as a result of rupture of one of the peripheral lenticulostriate arteries; (2) hemorrhage associated with a cerebral arteriovenous malformation; and (3) subarachnoid hemorrhage as a result of aneurysms arising from the vessels traversing the subarachnoid space. Thunderclap headache, acute nausea, vomiting, and neck stiffness are the hallmarks of both subarachnoid hemorrhage and meningitis. Three considerations should be applied to working up a patient with cerebrovascular disease. (1) Is the extent of the lesion typical of occlusion of an identifiable vessel? (2) Is there any hematological disorder that could have predisposed to or mimicked a cerebrovascular accident? (3) Are there any causative factors or comorbidity such as hypertension, atrial fibrillation, vessel stenosis, or myocardial infarction? If the clinical suspicion is high for a subarachnoid hemorrhage and the CT scan is normal, a lumbar puncture should be performed. Normally the CSF is clear and colorless, and therefore, if the CSF is pink or bloody, differentiation between a traumatic lumbar puncture and a subarachnoid hemorrhage must be made by performing cell counts on three sequential samples of CSF. Classically, a small amount of CSF is centrifuged and the supernatant is inspected. If the supernatant is xanthochromic, there is a high likelihood that a subarachnoid hemorrhage has occurred. However, an ultra-early lumbar puncture (<2 hours) may precede the window to establish whether or not a subarachnoid hemorrhage has occurred. On occasion, an ophthalmological examination may reveal retinal or vitreous hemorrhage thought secondary to the subarachnoid hemorrhage.8,30 This syndrome of retinal hemorrhage in association with subarachnoid hemorrhage is known as Terson’s syndrome.
Hysteria and Malingering
A single disease can often explain all the symptoms and signs; however, a patient may have a variety of diseases, old and new, organic and functional.3 Although the accuracy of neurodiagnostic tests is superb, it is the additional duty of the neurosurgeon to identify the malingering patient, for hysterical signs present more often in the nervous system than in any other organ system.3
Reactive pupils do not necessarily indicate hysterical blindness, because there may still be a lesion behind the midbrain, damaging the optic radiations or occipital cortex. Normal optokinetic nystagmus in the face of blindness does indicate hysteria or malingering, because optokinetic nystagmus requires intact connections from the retina to the occipital cortex. A constricted visual field should also be cone-shaped. Each time the examiner doubles the distance between him and the patient, the intact field should double in diameter and not leave only a central core of retained tunnel vision. Some patients can mimic a sixth cranial nerve palsy by converging the eyes while looking to the side. The tip-off is that convergence also constricts the pupil. Diplopia should often disappear when one covers one eye; however, monocular diplopia does occur in rare cases such as a retinal detachment or lens dislocation.31
Eliciting collapsing or ratchety weakness versus true weakness when the muscle gradually gives way can be a challenge. While vigorously testing the strength of an individual muscle, the examiner suddenly lets go. If the muscle fails to spring back to its contracted position, hysterical weakness may be present. When testing grip strength, watch the thumb; if the flexor pollicis longus does not flex the distal interphalangeal joint, the patient was not really giving maximal effort. The Hoover maneuver is another method of detecting insufficient effort by the patient. After placing one palm under the patient’s heel, the examiner asks the patient to lift the other leg against the examiner’s other hand. If the examiner does not feel the heel digging into his palm, the patient is not really trying to lift his leg. Hysterically hemiparetic patients also forget that pyramidal lesions selectively weaken the tibialis anterior. Although they drag the leg, there’s no circumduction; in fact, they purposely elevate the toe to keep it from scraping the floor. Withdrawal of a limb to pain also belies another common hysterical complaint: marked sensory loss. Hysterical hemihypesthesia may be uncovered by demonstrating nonorganic splitting of vibration at the midline (Fig. 2.11). Several nonorganic physical signs have also been described by Waddel, such as pain with gently tapping the lower back or with toe dorsiflexion,32 and are collectively referred as Waddel signs.
Damasio A.R. Aphasia. N Engl J Med. 1992;326:531-539.
Ojemann G.A. Cortical organization of language. J Neurosci. 1991;11:2281-2287.
Strub R.L., Black F.W. The Mental Status Examination in Neurology. Philadelphia: FA Davis; 1985.
Schneider R.C., Crosby E.C., Russo R.H., Gosch H.H. Traumatic spinal cord syndromes and their management. Clin Neurosurg. 1973;20:424-492.
Shareef A.H., Dafer R.M., Jay W.M. Neuro-ophthalmologic manifestations of primary headache disorders. Semin Ophthalmol. 2008;23(3):169-177.
Waddel G. Nonorganic physical signs in low-back pain. Spine. 1980;5:117.
Please go to expertconsult.com to view complete list of references.
1. Tysnes O.B. Neurological examination of cortical function deficits. Acta Neurol Scand Suppl. 2009;189:58-62.
2. Strub R.L., Black F.W. The Mental Status Examination in Neurology. Philadelphia: FA Davis; 1985.
3. Patton J. Neurological Differential Diagnosis. London: Springer-Verlag; 1998.
4. Ojemann G.A. Cortical organization of language. J Neurosci. 1991;11:2281-2287.
5. Ojemann G.A. Individual variability in cortical localization of language. J Neurosurg. 1979;50:164-169.
6. Billingsley-Marshall R.L., Simos P.G., Papanicolaou A.C. Reliability and validity of functional neuroimaging techniques for identifying language-critical areas in children and adults. Dev Neuropsychol. 2004;26(2):541-563.
7. Damasio A.R. Aphasia. N Engl J Med. 1992;326:531-539.
8. Lance J.W. The control of muscle tone, reflexes, and movement. Robert Wartenberg lecture. Neurology. 1980;30:1303-1313.
9. Sala F., Manganotti P., Tramontano V., et al. Monitoring of motor pathways during brainstem surgery: what we have achieved and what we still miss.[?]. Neurophysiol Clin. 2007;37(6):399-406. Epub 2007 Oct 29
10. Van Gijn J. The Babinski sign and the pyramidal syndrome. J Neurol Neurosurg Psychiatr. 1978;41:865-873.
11. Burke D., Knowles L., Andrews C., Ashby P. Spasticity, decerebrate rigidity and the clasp-knife phenomenon: an experimental study in the cat. Brain. 1972;95:31-48.
12. Rao G., Fisch L., Srinivasan S., et al. Does this patient have Parkinson disease? JAMA. 2003;289:347-353.
13. Trobe J.D. Isolated pupil-sparing third nerve palsy. Ophthalmology. 1985;92:58-61.
14. Bellur S.N. Opsoclonus: its clinical value. Neurology. 1975;25:502-507.
15. Baloh R.W., Yee R.D., Honrubia V. Optokinetic nystagmus and parietal lobe lesions. Ann Neurol. 1980;7:269-276.
16. Fromm G.H., Terrence C.F., Maroon J.C. Trigeminal neuralgia. Current concepts regarding etiology and pathogenesis. Arch Neurol. 1984;41:1204-1207.
17. Loeser J.D., Chen J. Hemifacial spasm: treatment by microsurgical facial nerve decompression. Neurosurgery. 1983;13:141-146.
18. Janetta P. Etiology and definitive microsurgical treatment of hemifacial spasm: operative techniques and results in 47 patients. J Neurosurg. 1977;47:321.
19. Schneider R.C., Crosby E.C., Russo R.H., Gosch H.H. Traumatic spinal cord syndromes and their management. Clin Neurosurg. 1973;20:424-492.
20. Bertrand G. Dynamic factors in the evolution of syringomyelia and syringobulbia. Clin Neurosurg. 1973;20:322-333.
21. Williams B. On the pathogenesis of syringomyelia: a review. J R Soc Med. 1980;73:798-806.
22. Schneider R.C. The syndrome of acute central cervical cord injury. With special reference to the mechanism involved in hyperextension injuries of cervical spine. J Neurosurg. 1954;11:546.
23. Engel A.G. Myasthenia gravis and myasthenic syndromes. Ann Neurol. 1984;16:519-534.
24. Friedman J.M. Neurofibromatosis 1: clinical manifestations and diagnostic criteria. J Child Neurol. 2002;17:548-554. discussion 571-572, 646-651
25. Volpe J.J. Neonatal neurologic evaluation by the neurosurgeon. Neurosurg Clin North Am. 1998;9(1):1-16.
26. Jamieson D.G., Hargreaves R. The role of neuroimaging in headache. J Neuroimaging. 2002;12:42-51.
27. Bogduk N., Govind J. Cervicogenic headache: an assessment of the evidence on clinical diagnosis, invasive tests, and treatment. Lancet Neurol. 2009;8(10):959-968.
28. Shareef A.H., Dafer R.M., Jay W.M. Neuro-ophthalmologic manifestations of primary headache disorders. Semin Ophthalmol. 2008;23(3):169-177.
29. Rapoport A.M. New acute treatments for headache. Neurol Sci. 2010;31(Suppl 1):S129-S132.
30. McCarron M.O., Alberts M.J., McCarron P. A systematic review of Terson’s syndrome: frequency and prognosis after subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2004;75(3):491-493.
31. Keane J.R. Neuro-ophthalmic signs and symptoms of hysteria. Neurology. 1982;32:757-762.
32. Waddel G. Nonorganic physical signs in low-back pain. Spine. 1980;5:117.