[level-membership-for-anesthesiology-category]
Chapter 3 Clinical Cardiac and Pulmonary Physiology
Hemodynamics
Preload
14. How can “preload” be measured clinically?
15. When will central venous pressure (CVP) poorly reflect filling pressures in the left heart?
16. What is the Frank-Starling mechanism?
17. What are common causes of low preload?
18. What is systolic pressure variation and how might it be useful in analyzing hypotension?
Pulmonary gas exchange
Oxygen
38. How is blood oxygen measured?
39. What is the “P50”? What is a normal value?
40. What are common clinical factors that shift the oxyhemoglobin dissociation curve left and right?
41. What are the benefits of a rightward shift in the oxyhemoglobin dissociation curve?
42. What is the equation describing the effect of ventilation on oxygenation?
43. How does increased FIO2 improve oxygenation during hypercapnia?
44. Is it possible to deliver hypoxic gas mixtures with a modern anesthesia machine?
45. What does an A-a gradient mean with respect to a problem in oxygenation?
46. What is intrapulmonary shunt?
47. What is the shunt equation?
48. Is diffusion limitation a significant clinical cause of hypoxemia?
49. Which causes of hypoxemia are very responsive to supplemental oxygen and therefore easily treated with higher FIO2?
50. How does low mixed venous oxygen saturation affect arterial oxygenation?
Carbon dioxide
51. What are the three forms in which carbon dioxide is carried in the blood?
52. Why is hypercapnia a problem clinically?
53. What are the four physiologic causes of hypercapnia?
54. What are significant causes of increased CO2 production under anesthesia?
55. What are the various types of dead space?
56. What pathologic conditions may increase dead space?
57. What is a normal value for physiologic dead space?
Control of breathing
Integration of the heart and lungs
Oxygen Extraction
89. Why is examining oxygen extraction clinically useful?
90. What is normal mixed venous oxygen saturation?
91. How would the arterial to venous oxygen content difference change with higher FIO2?
92. Why is the oxygen extraction ratio useful?
93. How can the body respond physiologically to anemia or increased metabolic demand (oxygen consumption)?
Answers*
Hemodynamics
Arterial Blood Pressure
1. Mean arterial pressure (MAP) is the average blood pressure. On modern monitors, MAP is calculated from integrating the arterial waveform over time. MAP can often be estimated by adding one third of the pulse pressure to the diastolic blood pressure. (50)
2. MAP is the product of cardiac output (CO) and SVR, or MAP = CO × SVR. This is similar to electricity where voltage = current × resistance. (If we were to be exactly correct, we would use the pressure drop across the systemic vascular system, or MAP – CVP.) (50)
3. Pulse pressure is the difference between systolic and diastolic blood pressure. (50)
4. Pulse pressure is produced from the stroke volume being pushed into the aorta. The compliance features of the aorta therefore have a very significant effect on pulse pressure so that a stiff aorta results in a higher pulse pressure, a common feature of aging. A lower diastolic pressure can reduce pulse pressure by moving to a more compliant part of the aortic compliance curve. A higher stroke volume generally increases pulse pressure. Lower SVR can decrease pulse pressure because part of the stroke volume “runs off” rapidly during ejection. Aortic insufficiency can increase pulse pressure as the diastolic pressure drops significantly during backward flow into the left ventricle. (50-51)
Systemic vascular resistance
5. Classic pathologic causes of low SVR include sepsis, anaphylactic and anaphylactoid reactions, and reperfusion of ischemic organs. Many anesthetic drugs and neuraxial anesthesia also lower SVR.
6. SVR = 80 × 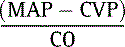 , where MAP is mean arterial pressure, SVR is systemic vascular resistant, CVP is central venous pressure, and CO is cardiac output. The factor “80” converts the SVR to the proper units. (51)
, where MAP is mean arterial pressure, SVR is systemic vascular resistant, CVP is central venous pressure, and CO is cardiac output. The factor “80” converts the SVR to the proper units. (51)
7. Most of the resistance in the vascular system is in the arterioles. Despite having smaller diameters, there are large numbers of capillaries in parallel, resulting in overall lower resistance at this level of the vascular tree. (51)
8. Resistance is inversely proportional to the fourth power of the radius of the vessel. (51)
Cardiac output
9. Cardiac output can be determined by thermodilution with a PA catheter. In addition, transesophageal echocardiography (TEE) may be used to estimate cardiac output. A variety of other noninvasive monitors are available and being developed that estimate cardiac output, including Doppler of the ascending aorta and arterial pressure waveform analysis. Thermodilution is still the dominant technique. The Fick equation can also be used to calculate cardiac output from the oxygen consumption, and arterial and mixed venous oxygen content. (51)
10. Stroke volume is the cardiac output divided by heart rate. It is important to calculate stroke volume, because a high heart rate may make cardiac output appear normal despite inadequate stroke volume. (51)
11. Because the appropriate cardiac output changes with body size, the cardiac “index” is used to normalize for body size by dividing cardiac output by body surface area. (51)
12. An excessively rapid heart rate might not leave sufficient time to fill the ventricle. Loss of a “p” wave with certain rhythms will also lead to inadequate ventricular filling from loss of atrial contraction. (51)
13. Ejection fraction is the percentage of ventricular blood volume that is pumped during a single contraction or SV/end-diastolic volume. Unlike stroke volume, ejection fraction does not change with body size. A normal ejection fraction is 60% to 70%. (51, Figure 6-3)
Preload
14. The volume of the heart at end-diastole can be directly measured by transesophageal echocardiography (TEE). Ventricular filling pressures can be measured on the right side of the heart with central venous pressure and on the left side of the heart by pulmonary capillary wedge pressure. A complete picture of preload would still require both pressure and volume information to more fully understand the compliance of the heart. Systolic pressure variation may also be an important indicator of low preload. (51-52, Figure 6-1)
15. CVP will poorly reflect filling of the left ventricle in a number of pathologic conditions. With pulmonary disease and elevated PVR, right heart failure may develop with elevated CVP despite poor filling of the left ventricle. With left ventricular failure, CVP may be normal despite elevated left heart filling pressures as long as right ventricular function is preserved. (51)
16. The Frank-Starling mechanism describes how the heart responds to increased filling by increasing contraction and stroke volume. This can be described by the cardiac function curves in Figure 6-2. (51)
17. “Hypovolemia” or low circulating blood volume is a key cause of low preload. Blood loss and fluid loss from other sources are commonly faced during surgery. Low preload can also occur with venodilation from an anesthetic agent and neuraxial anesthesia. Pathologic problems such as pericardial tamponade and tension pneumothorax may result in low preload (inadequate filling of the heart) despite normal blood volume and high CVP. (52)
18. Systolic pressure variation describes the regular changes in systolic pressure that occur with ventilation. During mechanical ventilation, significant systolic pressure variation reflects low preload. Systolic pressure variation may be more useful than other monitors in determining which patients will appropriately respond to fluid administration. In cases of hypotension, SPV may indicate low preload. Extreme SPV may indicate other important causes of hypotension, such as pericardial tamponade or tension pneumothorax. Pulse pressure variation, which is closely related, requires a computer to evaluate; systolic pressure variation can be measured with a standard arterial line and monitor. (52)
Contractility
19. Contractility, or inotropic state, describes the force of contraction independent of preload and afterload. It is reflected in the rate of rise of pressure over time. Graphically, it is reflected in the systolic pressure volume relationship. (52, Figure 6-3)
20. Important causes of poor contractility that may be associated with hypotension include myocardial ischemia, previous myocardial infarction, cardiomyopathy, and myocardial depression from a number of different drugs. In addition, when considering a differential diagnosis of hypotension, valvular heart disease would be considered as low contractility. (52, Table 6-1)
Afterload
21. Low SVR or afterload increases ejection fraction, which can approach 75% or even 80% in low SVR states. This is a classic feature of low SVR conditions such as liver failure. (52-53, Figure 6-4)
22. Low SVR or afterload lowers cardiac filling pressure (central venous pressure or pulmonary capillary wedge pressure) via the Frank-Starling mechanism. Vasodilation can therefore cause relative hypovolemia and a volume-responsive condition. Likewise high SVR or afterload increases cardiac filling pressure. (52-53, Figure 6-4)
23. Low SVR or afterload leads to low end-systolic left ventricular volume. This is a pathognomonic sign of low SVR on TEE. (52-53, Figure 6-4)
Cardiac reflexes
Autonomic Nervous System
24. The parasympathetic nervous system primarily affects the cardiovascular system by decreasing heart rate through vagal innervation of the sinoatrial node. Mild negative effects on contractility are probably less important. The sympathetic nervous system can increase heart rate and contractility, but it also causes peripheral vasoconstriction. (54)
Receptor Systems
25. Baroreceptors are present in the carotid sinus and aortic arch. Increased blood pressure will stimulate baroreceptors, leading to parasympathetic stimulation and a decrease in sympathetic stimulation. (54)
26. The Bainbridge reflex describes the increase in heart rate from atrial stretch. This helps increase cardiac output in response to increased venous return. (54)
27. Anesthetic agents decrease cardiac reflex responsiveness. This increases the likelihood of hypotension under anesthesia. (54)
Coronary blood flow
28. The myocardium extracts a higher percentage of oxygen than other tissues in the body, up to 60% to 70%. Normal whole body oxygen extraction is approximately 25%. (54)
29. Intramural pressure of the myocardium during systole stops blood flow to the subendocardium. Therefore, blood flow to the subendocardium occurs predominantly during diastole. (54)
Pulmonary circulation
Pulmonary artery pressure
30. The pulmonary circulation has much lower pressures than the system circulation. This is due to lower PVR compared to the systemic vascular resistance, since both systems accept the entire cardiac output. Since these pressures can be measured clinically with a PA catheter, the anesthesiologist should be familiar with normal and pathologic values, which are shown in Table 6-2. (54-55, Table 6-2)
Pulmonary Vascular Resistance
31. Pulmonary artery pressure stays remarkably constant over a wide range of cardiac output. PVR accommodates to increased cardiac output by distention and recruitment of capillaries, so that resistance decreases as cardiac output increases. (55)
32. Both high and low lung volumes increase PVR. At high lung volumes, intraalveolar vessels are compressed. At low lung volumes, extraalveolar vessels are compressed. Increased PVR at low lung volumes may be physiologically helpful in diverting blood flow from a collapsed lung. (55)
33. Elevated PVR can be very difficult to treat. Inhaled nitric oxide, prostaglandins, and phosphodiesterase inhibitors may lower PVR, but cannot always completely reverse elevated PA pressure. (55)
34. Hypoxia increases PVR through “hypoxic pulmonary vasoconstriction” (HPV). This process may significantly improve gas exchange by lowering blood flow to areas of poor ventilation. However, global hypoxia, such as occurs at high altitude, can result in increased PA pressure through HPV. (55)
35. Pathologic elevation in PVR may occur with pulmonary emboli. In addition, arteriolar hyperplasia may occur with certain congenital cardiac diseases (Eisenmenger syndrome), idiopathically (primary pulmonary hypertension), and associated with cirrhosis (portopulmonary hypertension). Intrinsic lung disease from a variety of causes can also increase PVR. (55)
Zones of the lung
36. Because the hydrostatic changes due to gravity are of a similar order of magnitude as PA pressure, gravity can have significant effects on pulmonary blood flow. Notable effects are in zone 1, where airway pressure is higher than pulmonary artery pressure, leading to no perfusion and therefore dead space. If areas of poor gas exchange are in an elevated position, lower perfusion can result, improving gas exchange. In lung surgery, the lower PA pressure in the nondependent collapsed lung helps gas exchange. (55)
Pulmonary Edema
37. Hydrostatic leak can occur in the lung when pulmonary capillary pressure is elevated. Pulmonary edema (hydrostatic) results when lymphatic system removal of fluid is overwhelmed. The risk of pulmonary edema increases as pulmonary capillary wedge pressure exceeds 20 mm Hg. Capillary leak can also occur with pulmonary injury from a variety of causes, such as aspiration or sepsis. The adult respiratory distress syndrome (ARDS) represents very significant lung injury with a high risk of mortality. (56)
Pulmonary gas exchange
Oxygen
38. Three measurements of blood oxygen are used clinically: partial pressure (in mm Hg), oxygen saturation (in %), and oxygen content (in mL O2/dL). The oxyhemoglobin dissociation curve (Figure 6-5) relates oxygen partial pressure and saturation. “Content,” really a concentration, is the sum of the amount of oxygen in hemoglobin (1.39 mL O2/dL/g hemoglobin) and in the dissolved (0.003 mL O2/mm Hg). (56)
39. The “P50” is the partial pressure at which hemoglobin is 50% saturated, normally 26.8 mm Hg. Sigmoidal curves are usually defined by such midpoints. This is shown graphically in Figure 6-5. (56, Figure 6-5)
40. The most important factors shifting the oxyhemoglobin dissociation curve to the right are metabolic acidosis and hypercapnia. Metabolic alkalosis and hypocapnia shift the curve to the left. Lower 2-3 DPG in stored blood leads to a significant left shift. (56, Table 6-3)
41. Right shifts of the oxyhemoglobin dissociation curve improve unloading of oxygen in the tissues. For the same tissue PO2 more oxygen will be unloaded because of a right shift. Because of the sigmoidal shape of the curve, little change in loading of oxygen in the lungs will occur because of the rightward shift. (56)
42. The “alveolar gas equation” is used most to determine the effect of ventilation on oxygenation. The equation describes the transfer of oxygen from the environment to the alveoli, and therefore contains all the determinants of alveolar oxygen: barometric pressure, FIO2, and ventilation. (57)
43. FIO2 is another determinant of alveolar oxygen, and it can overcome the effect of higher CO2 on alveolar oxygen. The effect of hypoventilation with and without supplemental oxygen is shown in Figure 6-8. (57-58)
44. Modern anesthesia machines can effectively prevent delivery of hypoxic gas mixtures. Multiple features are necessary, including pin indexing of tanks and gas hoses, shut-off valves for nitrous oxide, and use of oxygen to drive the bellows. These safety mechanisms might be overcome if a gas other than oxygen were delivered through the oxygen piping, which has occurred because of construction mishaps. A monitor measuring FIO2 is therefore still critical. Hypoxemia still occurs because of unintentional delivery of room air in patients requiring supplemental oxygen. (58)
45. Calculation of an A-a gradient divides the potential causes of hypoxemia into two groups of causes. Figure 6-7 illustrates this division. The first group of causes includes all the factors that determine alveolar oxygen: FIO2, barometric pressure (altitude), and ventilation. A normal A-a gradient would indicate that this first group is the problem. An abnormal A-a gradient indicates a gas exchange issue, usually 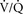 mismatch or shunt. (58)
mismatch or shunt. (58)
46. Shunt describes the passage of mixed venous blood through the lung, unexposed to alveolar gas. This commonly occurs because alveoli are collapsed, or filled with fluid such as in pneumonia or pulmonary edema. Mixed venous blood combines with blood passing through normal lung, lowering the PaO2, which is the end result of the mixture. (58-59, Figure 6-9)
47. The shunt equation quantitatively describes the physiologic effect of shunt on oxygenation. Since 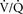 mismatch may also be present, the shunt equation really describes a simple two-compartment model analyzing oxygenation as if it were all pure shunt. (59)
mismatch may also be present, the shunt equation really describes a simple two-compartment model analyzing oxygenation as if it were all pure shunt. (59)
48. Diffusion impairment or limitation is not a major clinical cause of hypoxemia. However, diffusion limitation is often misunderstood. If an alveolus is filled with fluid, such that no diffusion of oxygen occurs, this is shunt, not diffusion limitation. Diffusion limitation occurs when a partial pressure gradient still exists between the alveolus and the capillary blood after the blood has passed through. Sufficient time for diffusion usually occurs, such that equilibration occurs early in the process. Even alveolar thickening, which may slow diffusion, does not usually result in diffusion limitation because equilibration of PO2 between the alveolus and capillary blood does occur. Diffusion limitation may be a clinically significant physiologic problem at extreme altitude during exercise. (59)
49. Hypoventilation, diffusion impairment, and 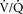 mismatch are all very responsive to supplemental oxygen. High FIO2 can effectively eliminate hypoxemia from these causes. Shunt is much more resistant to supplemental oxygen. At shunt fractions over 30%, hypoxemia may remain despite administration of 100% oxygen. Higher FIO2 does improve oxygenation with pure shunt, although there is an incorrect impression that this impact is minimal. The effect of FIO2 is difficult to calculate and is not linear, so it is best graphically illustrated as in Figure 6-9. (59)
mismatch are all very responsive to supplemental oxygen. High FIO2 can effectively eliminate hypoxemia from these causes. Shunt is much more resistant to supplemental oxygen. At shunt fractions over 30%, hypoxemia may remain despite administration of 100% oxygen. Higher FIO2 does improve oxygenation with pure shunt, although there is an incorrect impression that this impact is minimal. The effect of FIO2 is difficult to calculate and is not linear, so it is best graphically illustrated as in Figure 6-9. (59)
50. Low mixed venous oxygen levels may affect PaO2, but only in the presence of intrapulmonary shunt. For the same shunt, lower mixed venous oxygen results in a lower PaO2. (59)
Carbon dioxide
51. In the blood, CO2 is carried as dissolved gas, as bicarbonate, and bound to hemoglobin as carbaminohemoglobin. The greatest total quantity of CO2 is as bicarbonate, which is in fairly rapid equilibrium with CO2 through carbonic acid. Despite being the smallest total, the CO2 from carbaminohemoglobin represents about one third of the arterial to venous CO2 movement. (59)
52. Hypercapnia can be well tolerated, although at higher levels, probably approaching 80 mm Hg or greater, hypercapnia can cause CO2 narcosis. The most significant problem is what hypercapnia represents. A major cause of hypercapnia is oversedation or narcotization. This could progress to apnea and anoxia. Hypercapnia may also represent impending respiratory failure from a variety of causes. (59-60)
53. Physiologically, hypercapnia can be caused by (1) rebreathing (elevated inspired CO2), (2) hypoventilation, (3) elevated CO2 production, and (4) elevated dead space. (60-61)
54. The most concerning cause of significant CO2 production under general anesthesia is malignant hyperthermia (MH). While fever alone will increase CO2 production, the increase is not dramatic. MH may increase CO2 production several fold. Thyroid storm may increase CO2 production. Absorption of CO2 introduced during laparoscopy may be quite significant for certain procedures, particularly if subcutaneous CO2 emphysema develops. The CO2 removed through the lungs appears as if it is CO2 production. (60, Table 6-4)
55. Dead space is described as anatomic, alveolar, or physiologic (total). Anatomic dead space consists of the conducting airways, which are not involved in gas exchange, plus the larynx and pharynx. Alveolar dead space consists of alveoli that are not involved in gas exchange, usually from lack of blood flow. Physiologic or total dead space consists of all dead space, and is the easiest to measure. “Equipment” dead space may be produced by the addition of tubing beyond the y-connector of the anesthesia circuit. (60-61)
56. Many forms of end-stage lung disease, such as emphysema, are characterized by elevated dead space. Pulmonary emboli of any source increase dead space. Hypovolemic shock increases dead space, since very low PA pressures result in more zone 1 of the lung, where alveoli are not perfused and therefore represent dead space. (60-61)
57. Normal dead space is 25% to 30% and consists almost entirely of anatomic dead space. (60-61)
58. The Bohr equation is used to calculate dead space, Vd/Vt. It requires measuring PaCO2 and mixed-expired CO2 by collecting exhaled gas. The gradient from PaCO2 to end-tidal PaCO2 is a reflection of alveolar dead space and is a simple semiquantitative way of evaluating dead space under general anesthesia. (61)
59. CO2 jumps up fairly rapidly during the first 30 seconds to one minute of apnea. This jump is due to rapid transition to mixed venous CO2 levels, which usually means an increase of about 6 mm Hg. This occurs because the lungs do not continue to store CO2, so once equilibration of CO2 occurs across the alveoli, PaCO2 will jump to mixed venous levels. Thereafter, CO2 increases due to metabolism at a slower rate of about 2 to 3 mm Hg/min. (61)
Pulmonary mechanics
60. Pulmonary mechanics describes the pressure, volume, and flow relationships of gas within the lungs and the tracheobronchial tree. (61)
Static Properties
61. The lung itself has elastic properties. It requires pressure to expand. The chest wall and abdominal cavity produce a pressure effect on the lung. Surface tension, which exists at any air-fluid interface, also contributes. (61)
62. Without surfactant, surface tension would make the lungs much stiffer. Additionally, alveoli would be less stable and would tend to collapse. (61-62)
63. Static compliance is the change in volume divided by the change in pressure. By static, this means that the pressure and volume measurements are made at a point of no gas flow, which would contribute a resistive pressure component. Low or poor compliance would indicate that more pressure is needed to inflate the lungs. (62-63, Figures 6-12 and 6-13)
64. The FRC is simply the balance point between the lungs collapsing and the chest wall expanding. Stiffer lungs will produce a lower FRC, because this balance point will occur at a lower lung volume. On the other hand a disease such as emphysema, with loss of elastic recoil, results in a higher FRC. (62)
Dynamic Properties and Airway Resistance
65. Similar to the vascular system, resistance is largely determined by airway diameter. However, turbulent gas flow can add a significant resistance component, which can happen at airway narrowing. (62)
66. Pressure from resistance only occurs during gas flow. By ceasing gas flow with an inspiratory pause, one can determine the static or plateau pressure. (62, Figure 6-12)
67. High airway resistance can be caused by a number of common clinical conditions. A useful differential might trace the potential resistance anatomically, starting with airway equipment, including the endotracheal tube. Cause of resistance in the upper airways can include compression, foreign bodies, and secretions. In the lower airway, bronchoconstriction becomes the dominant cause. (62-63, Table 6-5)
Control of breathing
Central Chemoreceptors
68. The central chemoreceptors are located on the ventral surface of the brainstem. (62)
69. Carbon dioxide is the main stimulus for the central chemoreceptors. While the signal transduction may be through protons, the mechanisms are not completely understood. Because CO2 crosses the blood-brain barrier, for clinical purposes, we consider that CO2 is the primary stimulus. (62-63)
70. The central chemoreceptors are protected from metabolic acid by the blood-brain barrier. Cerebrospinal fluid pH will change in response to peripheral blood pH changes, but this may take days. An acute lactic acidosis will therefore have no effect on central chemoreceptors, except due to decreases in PaCO2 that may occur from the ventilatory response to the peripheral acidosis. (63)
Peripheral Chemoreceptors
71. The carotid bodies are the primary peripheral chemoreceptors in humans. Aortic bodies do not appear to have a significant clinical effect (which was studied in humans who had aortic body denervation). (63)
72. The peripheral chemoreceptors are stimulated by low pH, high PaCO2, and low PaO2. Unlike the central chemoreceptors, the peripheral chemoreceptors are not protected from an acute metabolic acidosis, which will cause stimulation and hyperventilation (the lower PaCO2 from this hyperventilation will affect the central chemoreceptors). (63)
73. High blood flow relative to metabolic rate creates a tissue with hardly any arterial to venous PO2 difference. This allows the carotid bodies to effectively sense arterial values. (63)
Hypercapnic Ventilatory Response
74. While a variety of techniques are used to obtain ventilatory data, the slope of CO2 versus minute ventilation is the primary measure of hypercapnic ventilatory responsiveness. The slope is the change in minute ventilation divided by the change in CO2 (usually end-tidal PCO2 since a noninvasive measurement can be preferable). (63-64, Figure 6-14)
75. The central chemoreceptors are the major receptor system responsible for hypercapnic drive. However, in room air, approximately one third of the CO2 response is from peripheral chemoreceptor drive. Usually hypercapnic drive is measured at higher FIO2 where the majority of the response will then be from central chemoreceptors. (63-64)
76. Below a certain value of PaCO2, ventilation usually ceases. In an awake person, this can be difficult to measure due to an awake drive to breath. Under general anesthesia, this phenomenon is easy to observe. With mechanical ventilation, if a patient is hyperventilating, spontaneous ventilatory efforts cease at a PCO2 about approximately 5 mm Hg lower than the set point. As CO2 is allowed to build up again, ventilation begins slowly and will stabilize again at the set point. (63-64)
77. CO2 ventilatory drive is a slow response, with a time constant of approximately 2 minutes. This is rarely appreciated, although it is easy to observe that ventilation takes noticeable time to stabilize as CO2 rises to a patient’s set point. (64)
Hypoxic Ventilatory Response
78. Hypoxic ventilatory drive can be measured from a plot of PO2 versus minute ventilation or SaO2 versus minute ventilation. Because the relationship of PO2 to minute ventilation is nonlinear, more complex parameters would be needed to describe the relationship, which then are not very clinically useful. A plot of SaO2 (SpO2 is conveniently and noninvasively measured by pulse oximetry) versus minute ventilation is quite linear. Hypoxic responsiveness can then be measured by a simple slope (which will be negative), the change in minute ventilation divided by the change in SpO2. (64, Figure 6-15)
79. Hypoxic ventilatory stimulation is from the carotid bodies. (64)
80. Central nervous effects of hypoxia lead to a slower development of ventilatory depression known as hypoxic ventilatory decline. The carotid bodies initially lead to increased minute ventilation, but if hypoxia is prolonged, ventilation drops to a level lower than peak ventilation, but still above baseline. This central response is a regulated response probably involving several inhibitory neurotransmitters. (64)
81. Hypoxic drive from the peripheral chemoreceptors develops extremely rapidly. The time constant is 10 to 20 seconds. Peak ventilation will therefore usually occur within 1 minute. The response is rapid enough that carotid body output will actually vary in response to the small oscillations of PO2 and PCO2 that occur with tidal breathing. (64)
82. The hypoxic drive is significantly higher with a higher PCO2. This synergistic response between PO2 and PCO2 will be most noticeable during apnea. (64)
Effects of anesthesia
83. Opioids and most ventilatory depressants work on neurons in the integratory area of the brainstem. They do not affect detection of hypoxia or hypercapnia per se. The clinically observed respiratory depression therefore affects both hypercapnic and hypoxic ventilatory drive equally. (64)
Disorders of Ventilatory Control
84. Premature infants less than 60 weeks of postconceptual age can be at risk of apnea following general anesthesia. (64)
85. Originally described following surgery near the high cervical spinal cord, Ondine curse describes patients with a nearly absent drive to breath. While awake, they may breathe fairly normally. But asleep, or under general anesthesia, breathing can be significantly depressed. This is due to abnormalities in the central integratory system that seem to blunt the hypoxic and hypercapnic ventilatory responses. Idiopathic forms of Ondine curse, which can be seen in children, are usually referred to as primary central alveolar hypoventilation syndrome. (64)
86. Periodic breathing, most commonly Cheyne-Stokes breathing, occurs frequently when some degree of hypoxia is present. The stimulation of the carotid bodies can lead to overshoots and undershoots of ventilation. Periodic breathing can often be observed on sedated patients with some degree of hypoxia during sleep. This is a major cause of sleep disturbance at high altitudes. Some patients with central sleep apnea have problems primarily with periodic breathing. Periodic breathing will not usually be observed in patients who are awake. (64)
Integration of the heart and lungs
87. The Fick equation describes the relationship between cardiac output, oxygen consumption, and oxygen about (arterial to venous content difference). (64)
Oxygen Extraction
89. Examining oxygen extraction provides a better global indication of whether cardiac output is matched to the body’s oxygen needs. Oxygen extraction may provide clinically and diagnostically useful clues as to disease state. In cardiogenic shock, oxygen extraction is high because cardiac output is insufficient. In sepsis and liver failure, oxygen extraction may be very low. (65)
90. Normal whole body mixed venous oxygen saturation is about 75%. Individual organs and tissues can differ significantly. (65)
91. Arterial to venous oxygen content difference (CaO2 – CvO2) is independent of FIO2, whereas the mixed venous oxygen saturation (SvO2) can increase significantly with higher PaO2. (65)
92. Oxygen extraction ratio is probably the most reliable index. It is the oxygen extraction value most independent of FIO2 and hemoglobin level. (65)
93. The two major compensatory mechanisms for increased demand or less availability of oxygen is (1) increased cardiac output and (2) increased extraction. This is readily apparent by examining the Fick equation. In anemia without general anesthesia, the primary compensation is increased cardiac output. Increased extraction occurs with more severe anemia. Under anesthesia, the cardiac output compensation may be blunted, and oxygen extraction is more important. In exercise, both increased cardiac output and increased extraction are utilized. (65)
* Numbers in parentheses: numbers refer to pages, figures, or tables in Miller RD, Pardo MC: Basics of Anesthesia, 6th ed. Philadelphia, Elsevier Saunders, 2011.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 3 Clinical Cardiac and Pulmonary Physiology
Hemodynamics
Preload
14. How can “preload” be measured clinically?
15. When will central venous pressure (CVP) poorly reflect filling pressures in the left heart?
16. What is the Frank-Starling mechanism?
17. What are common causes of low preload?
18. What is systolic pressure variation and how might it be useful in analyzing hypotension?
Pulmonary gas exchange
Oxygen
38. How is blood oxygen measured?
39. What is the “P50”? What is a normal value?
40. What are common clinical factors that shift the oxyhemoglobin dissociation curve left and right?
41. What are the benefits of a rightward shift in the oxyhemoglobin dissociation curve?
42. What is the equation describing the effect of ventilation on oxygenation?
43. How does increased FIO2 improve oxygenation during hypercapnia?
44. Is it possible to deliver hypoxic gas mixtures with a modern anesthesia machine?
45. What does an A-a gradient mean with respect to a problem in oxygenation?
46. What is intrapulmonary shunt?
47. What is the shunt equation?
48. Is diffusion limitation a significant clinical cause of hypoxemia?
49. Which causes of hypoxemia are very responsive to supplemental oxygen and therefore easily treated with higher FIO2?
50. How does low mixed venous oxygen saturation affect arterial oxygenation?
Carbon dioxide
51. What are the three forms in which carbon dioxide is carried in the blood?
52. Why is hypercapnia a problem clinically?
53. What are the four physiologic causes of hypercapnia?
54. What are significant causes of increased CO2 production under anesthesia?
55. What are the various types of dead space?
56. What pathologic conditions may increase dead space?
57. What is a normal value for physiologic dead space?
Control of breathing
Integration of the heart and lungs
Oxygen Extraction
89. Why is examining oxygen extraction clinically useful?
90. What is normal mixed venous oxygen saturation?
91. How would the arterial to venous oxygen content difference change with higher FIO2?
92. Why is the oxygen extraction ratio useful?
93. How can the body respond physiologically to anemia or increased metabolic demand (oxygen consumption)?
Answers*
Hemodynamics
Arterial Blood Pressure
1. Mean arterial pressure (MAP) is the average blood pressure. On modern monitors, MAP is calculated from integrating the arterial waveform over time. MAP can often be estimated by adding one third of the pulse pressure to the diastolic blood pressure. (50)
2. MAP is the product of cardiac output (CO) and SVR, or MAP = CO × SVR. This is similar to electricity where voltage = current × resistance. (If we were to be exactly correct, we would use the pressure drop across the systemic vascular system, or MAP – CVP.) (50)
3. Pulse pressure is the difference between systolic and diastolic blood pressure. (50)
4. Pulse pressure is produced from the stroke volume being pushed into the aorta. The compliance features of the aorta therefore have a very significant effect on pulse pressure so that a stiff aorta results in a higher pulse pressure, a common feature of aging. A lower diastolic pressure can reduce pulse pressure by moving to a more compliant part of the aortic compliance curve. A higher stroke volume generally increases pulse pressure. Lower SVR can decrease pulse pressure because part of the stroke volume “runs off” rapidly during ejection. Aortic insufficiency can increase pulse pressure as the diastolic pressure drops significantly during backward flow into the left ventricle. (50-51)
Systemic vascular resistance
5. Classic pathologic causes of low SVR include sepsis, anaphylactic and anaphylactoid reactions, and reperfusion of ischemic organs. Many anesthetic drugs and neuraxial anesthesia also lower SVR.
6. SVR = 80 × , where MAP is mean arterial pressure, SVR is systemic vascular resistant, CVP is central venous pressure, and CO is cardiac output. The factor “80” converts the SVR to the proper units. (51)
7. Most of the resistance in the vascular system is in the arterioles. Despite having smaller diameters, there are large numbers of capillaries in parallel, resulting in overall lower resistance at this level of the vascular tree. (51)
8. Resistance is inversely proportional to the fourth power of the radius of the vessel. (51)
