Chapter 69
Classification and Natural History of Vascular Anomalies
Carolyn R. Rogers, John B. Mulliken
Based on a chapter in the seventh edition by Byung-Boong Lee and Leonel Villavicencio
Vascular surgery matured as a specialty of general surgery during the twentieth century while the field of vascular anomalies remained obscured by a cloud of confusing nomenclature. This haze of bewildering nosology lifted during the last quarter of that century to reveal a truly interdisciplinary field that overlaps many specialties, including vascular surgery.
Traditionally, vascular surgeons manage disorders of anatomically named arteries and veins. Only a few vascular surgeons, most of them European, have focused part of their practice on malformations distal to the main vascular trunks, particularly those in the extremities. Noteworthy in this regard are the names Malan, Schobinger, Servelle, Azzolini, Belov, Loose, and Matassi. In the mid-1970s, several of these vascular surgeons joined like-minded colleagues from other specialties to initiate a workshop for the study of vascular anomalies—this grew to become the International Society for the Study of Vascular Anomalies (ISSVA). The early gatherings of this organization were punctuated by heated discussions about terminology. Finally, there was a consensus at the 1996 meeting in Rome, and the ISSVA classification was accepted by the membership.1
Nosology
The earliest terminology for vascular anomalies was descriptive. Words such as “strawberry mark,” “port-wine stain,” and, “cherry angioma” referenced food because the mother was blamed for imprinting her unborn child. With the ascent of histopathology in the nineteenth century, all vascular anomalies were called “angiomas” or “lymphangiomas.” With increasing knowledge of cardiovascular embryology in the early twentieth century, vascular anomalies were envisioned as disorders of faulty development. Nevertheless, when put to the test of clinical usefulness, embryologic classifications failed to differentiate between vascular anomalies that regress and those that progress and, thus, offered little help in guiding management.
A prospective study by Mulliken and Glowacki2 defined cellular features of vascular anomalies using histochemistry, radiography, and electron microscopy. This investigation delineated two major categories of vascular anomalies: tumors, which exhibit endothelial hyperplasia; and malformations, which have normal endothelial turnover unless disturbed. This binary “biologic” system became the foundation of the ISSVA classification (Table 69-1).
Table 69-1
International Society for the Study of Vascular Anomalies (ISSVA) Classification of Vascular Anomalies
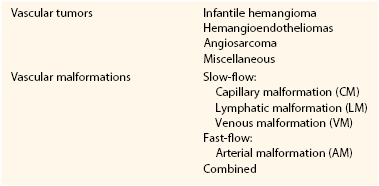
Vascular malformations are structural anomalies, inborn errors of vascular morphogenesis. A single type of channel anomaly may predominate; however, often the malformed channels are combined forms. Vascular malformations are subcategorized on the basis of rheology and channel architecture as slow-flow (capillary, lymphatic, or venous), fast-flow (arterial), or combined. These malformations are more likely to be referred to vascular surgeons than are vascular tumors. The consultant must be wary because vascular tumors can masquerade as vascular malformations in various hues of pink, red, and blue. An estimated 54% of vascular malformations and 30% of vascular tumors in patients referred to one center between 1999 and 2010 were initially diagnosed incorrectly.3
Vascular Tumors
The spectrum of vascular tumors ranges from common infantile hemangiomas to uncommon lesions of borderline malignancy to rare malignant neoplasms (see Chapter 70). Unlike vascular malformations (and other vascular tumors), infantile hemangiomas are immune-positive for glucose transporter protein-1 (GLUT-1).4,5 Because most infantile hemangiomas arise in the head/neck region, vascular surgeons are unlikely to be asked for consultation. Vascular surgeons might be asked to opine on a rare variant of the common infantile tumor that has a predilection for the lower extremities called “reticular hemangioma.” This lesion is associated with intractable ulceration, anogenito-urinary-sacral anomalies, and sometimes cardiac overload. Reticular hemangioma of the lower limb can be confused with vascular malformations, such as capillary malformation, cutis marmorata telangiectatica congenita, and Parkes Weber syndrome.6 Congenital hemangiomas constitute a special category of vascular tumors that are fully grown at birth, behave differently from infantile hemangiomas, and are GLUT-1 negative. There is rare vascular tumors that are categorized as borderline malignant called hemangioendotheliomas and angiosarcomas.
The doubly eponymic “Kasabach-Merritt” thrombocytopenia has been wrongly associated with infantile hemangioma. This disorder of platelet trapping occurs only with kaposiform hemangioendothelioma and tufted angioma, not with common infantile hemangiomas.7–9 Kasabach-Merritt phenomenon has also been used incorrectly to describe the localized intravascular coagulopathy that occurs with extensive venous malformations; this is an entirely different hematologic disorder.10
Vascular Malformations
Vascular malformations are, by definition, congenital, and most are obvious at birth (see Chapters 71 and 72). However, some vascular malformations are undetectable at birth and manifest in childhood, adolescence, or adulthood. Vascular malformations arise because of abnormal signaling processes regulating proliferation, differentiation, maturation, adhesion, and apoptosis of vascular cells, including endothelium, smooth muscle, and pericytes.11 Some are inherited in an autosomal dominant pattern, whereas others occur sporadically and are caused by somatic (postzygotic) mutations.
Slow-Flow Malformations
Capillary Malformation
Capillary malformation (CM) must be differentiated from the common fading macular stain naevus flammeus neonatorum (“angel’s kiss” or “stork bite”) frequently present in a newborn on the face and nuchal region. Capillary malformations (still often called “port-wine stains”) are red macular lesions seen at birth that persist throughout life. They are histopathologically characterized by thin-walled capillary- to venule-sized channels in the papillary and upper reticular dermis with deficient perivascular neural elements. Facial CM often darkens, thickens, and develops a cobblestone appearance in adulthood. In contrast, CM in the trunk and limbs deepens in color with age but does not become nodular, although adjacent veins often become more prominent. Capillary malformations occur in association with several disorders; the best known is Sturge-Weber syndrome.
Sturge-Weber Syndrome.
Sturge-Weber syndrome is characterized by leptomeningeal vascular anomalies and facial CM, commonly manifesting as seizures and glaucoma. The clinical course is highly variable; some children have intractable seizures, behavioral issues, mental retardation, and recurrent stroke-like episodes. Soft tissue overgrowth in the area of facial CM occurs with age. More than half of patients with Sturge-Weber syndrome have noncontiguous, patchy CMs on the torso and extremities.12 Fifteen percent of patients with Sturge-Weber syndrome and extracranial staining have hypertrophy of an extremity.13 Varicose veins are seen in the stained areas in older patients with this disorder.
Cutis Marmorata Telangiectatica Congenita.
The vascular anomaly known as cutis marmorata telangiectatica congenita (CMTC) manifests in a neonate as serpiginous, reticulated bands of deep purple color with localized cutaneous atrophy and often ulceration. The lesions occur in a localized or regional pattern. The legs are most frequently involved; there may be subcutaneous hypoplasia or hypertrophy. The patient with leg involvement may be referred to a vascular surgeon because of concern about vascular occlusion in the limb. There have been rare case reports of hypoplasia of the iliac and femoral veins14 and of iliac arterial stenosis, including stenosis in the unaffected limb.15 Predictably, CMTC shows improvement during the first year of life that continues throughout childhood. Cutaneous atrophy and vascular staining persist, usually with venous ectasias in the involved limb.
Macrocephaly–Capillary Malformation.
Macrocephaly–capillary malformation (M-CM) is characterized by faint CM of the upper lip and scattered staining over much of the body in association with overgrowth, syndactyly or postaxial polydactyly, connective tissue dysplasia of the skin, subcutaneous tissue, and joints, and cortical brain malformations, typically polymicrogyria and megalencephaly/macrocephaly.16 This condition was initially mistakenly called “macrocephaly–cutis marmorata telangiectatica congenita.” Affected patients do not have CMTC; rather, they have diffuse, reticulate capillary staining that fades.17 Patients with M-CM may be referred to a vascular surgeon because their signs and symptoms are often confused with those of combined vascular malformation/overgrowth syndromes.
Venous Malformation
For many years, vascular malformations (VMs) were called “cavernous hemangiomas;” this practice caused confusion with vascular tumors. Although present at birth, VMs may not be obvious and appears later as bluish, compressible swellings. They are histopathologically characterized by thin-walled interconnected vessels or pouches with abnormal investing smooth muscle. VMs often develop thrombi and later phleboliths. Venous anomalies are typically painful, especially in the morning and when an affected limb is dependent. An extensive VM of the limb is known by the old eponym “genuine diffuse phlebectasia of Bockenheimer,”18 in which all soft tissues and long bones are affected. Leg length discrepancy may result, causing pelvic tilt, scoliosis, and gait disturbance.
The blood contained in a large or extensive VM exhibits a localized intravascular coagulopathy—that is, slight decrease in platelets (100,000 to 150,000/mm3), normal PT and aPTT values, low fibrinogen content (150-200 mg/dL), and elevation of fibrin split products (D-dimer).9 Affected patients are at risk for development of a systemic coagulopathy following trauma or surgical intervention; the hematologic findings are similar to those in disseminated intravascular coagulopathy. If a patient presents with a lesion of uncertain vascular nature, elevation of D-dimer can be used as a diagnostic tool to differentiate large VM from another vascular lesion, such as glomuvenous malformation (normal D-dimer), arteriovenous malformation, or lymphatic malformation. D-dimer measurement can also differentiate a slow-flow disorder, such as Klippel-Trenaunay syndrome, from a fast-flow disorder, such as Parkes Weber syndrome.19 Low-molecular-weight heparin may be given to treat the pain caused by localized intravascular coagulopathy and to prevent decompensation to a systemic coagulopathy prior to an operation.10
Glomuvenous Malformation.
A genetic diagnosis should be considered whenever there is a family history of VM, particularly multiple lesions. Glomuvenous malformation (GVM) is the most common heritable VM; it is autosomal dominant. Once known as “glomangiomas,” GVMs are clinically distinguished from common VMs because they have a cobblestone appearance and are painful on palpation.20 Also, unlike VMs, GVMs in limbs do not respond to elastic compressive garments and there is rarely deep extension. The lesions can be small or extensive and plaque-like, particularly on the trunk and extremities. They range from pink in infants to deep blue or purple in children and adults. Seventy-eight percent are located on the extremities.21 Histologically, GVMs have pathognomonic glomus cells within the walls of distended vein-like channels.
Cutaneomucosal Venous Malformation.
Patients with this hereditary disorder known as cutaneomucosal venous malformation (CMVM) typically have small, multifocal, soft, compressible cutaneous lesions appearing in various shades of blue. Unlike GVMs, these lesions are often present on mucosa, typically on the lips and tongue, and may occur in skeletal muscle. There may also be progressive venous ectasia in the neck and upper limb. Fifty percent of CMVMs are located in the cervicofacial area, and 37% on the extremities.21
Blue Rubber Bleb Nevus Syndrome.
Soft, blue, nodular-appearing cutaneous and gastrointestinal VMs characterize blue rubber bleb nevus syndrome. Lesions increase in size and number with age. Gastrointestinal VMs can cause life-threatening bleeding. The genetic cause of this disorder is most likely a postzygotic mutation.
Lymphatic Malformation
Lymphatic malformations (LMs) are clinically and radiologically categorized as microcystic, macrocystic, or combined. These anomalies were previously described with terms such as “lymphangioma” (microcystic LM), “cystic hygroma” (macrocystic LM), and “lymphangiomatosis” (visceral LM). They are composed of anomalous channels or pockets of lymphatic fluid, often with overlying cutaneous lesions. The walls are of variable thickness with smooth and striated muscle components and nodular collections of lymphocytes. The spectrum of LMs is quite variable. Most LMs manifest in the infantile period. Deep LMs appear as generalized swellings to localized areas of overgrowth. Expansion is rapid if infection or intralesional bleeding occurs. Cutaneous LMs most often involve the chest and upper limbs, less often the lower limbs. They manifest as clear or dark red vesicles (due to intravesicular bleeding). Visceral LMs may go undetected until rapid expansion occurs. Congenital lymphedema is a type of LM that typically involves the lower limbs.
Congenital Lymphedema.
Primary (congenital) lymphedema is caused by anomalous lymphatic channels and must be differentiated from secondary (acquired) lymphedema due to injury to the lymph nodes or vessels (from operation, radiation, trauma, etc.) (see Chapter 66). Primary lymphedema is often nonfamilial, but it can be inherited in an autosomal dominant fashion, in which case it is also known as Milroy disease. This condition manifests shortly after birth in 49% of patients, during childhood in 10%, or during adolescence in 41%. Congenital lymphedema manifests as gradually progressive swelling of an extremity, most often the lower extremities (92%). Cellulitis affects 19% of patients and is often recurrent.22 Over time, soft tissue overgrowth develops because of fatty deposition and fibrosis. Compression is the mainstay of initial treatment; suction lipectomy has also been used.22–24 There are reports of success following microvascular lymphaticovenous anastomosis.25,26
Fast-Flow Malformations
Arterial Malformations
Arterial malformations are abnormally formed arteries such as aneurysms, fistulae, ectasias, and stenoses; the combined forms are arteriovenous malformation (AVM) and arteriovenous fistula (AVF).
Arteriovenous Fistula
Congenital AVF occurs in isolation or as part of a complex AVM. It manifests as an enlarging, pulsatile lesion with palpable thrill and possibly signs and symptoms of shunting. The old name “congenital AVF” is often subsumed by the term AVM of which multiple AVFs are a component. Early literature on “AVFs” of the lower extremity documented edema, hypertrophy causing leg length discrepancy, trophic changes, and even cardiovascular sequelae.27–29 Congenital AVFs in the upper limb typically manifest with the same signs and symptoms—pain, distal ischemia, and discoloration of the digits.30 Endovascular and operative techniques, alone or in combination, have been the mainstay of treatment for over 2 decades.31
Arteriovenous Malformations
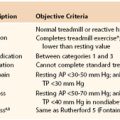
AVMs are present at birth and are usually quiescent during infancy and childhood. They initially are seen as telangiectasias or macular stains that may be mistaken for CMs or infantile hemangiomas. Histopathologically, AVMs are characterized by thick, fibromuscular walls with fragmented elastic lamina and fibrotic stroma. The epicenter of an AVM, called the nidus, is composed of arterial feeding vessels, micro- and macro-AVFs, and ectatic veins. The most common location for AVMs is intracranial, followed by limbs, trunk, and viscera. AVMs enlarge in response to triggers such as puberty and trauma. There is conflicting evidence about whether pregnancy causes expansion of AVMs.32 The clinical course is often slowly progressive and can be documented with the staging system proposed by Schobinger, a Swiss vascular surgeon and first president of ISSVA (Table 69-2).33
Table 69-2
Schobinger Staging System for Arteriovenous Malformations
| Stage I (Quiescence) | Cutaneous blush, warmth (arteriovenous shunting documented by Doppler ultrasonography) |
| Stage II (Expansion) | Bruit, audible pulsations, expanding lesion |
| Stage III (Destruction) | Pain, ulceration, bleeding, infection |
| Stage IV (Decompensation) | Cardiac failure |
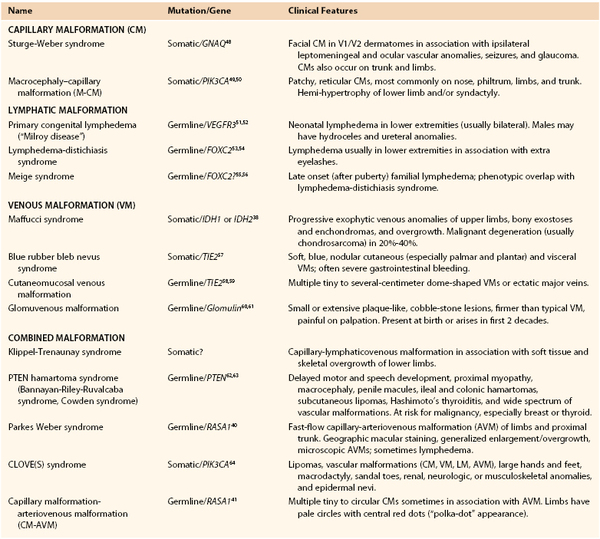
Combined Malformations and Overgrowth Syndromes
Combined vascular malformations can involve any combination of capillary, lymphatic, venous, and arterial channels (Fig. 69-1). Like pure vascular malformations, combined malformations are classified as either slow-flow or fast-flow. Often, these combined malformations constitute an overgrowth syndrome. Some of the well-known eponymous disorders involving combined malformations are described below.
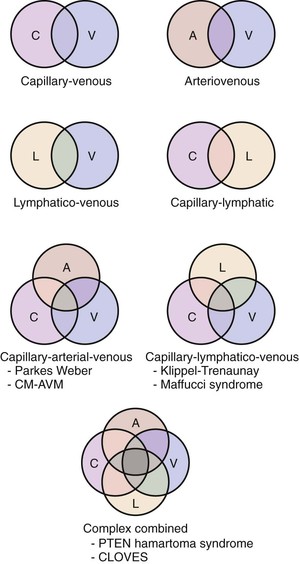
Figure 69-1 Types of combined vascular malformations depicted in Venn diagrams. A, Artery/arterio; AVM, arteriovenous malformation; C, capillary; CLOVES, congenital, lipomatous, overgrowth, vascular malformations, epidermal nevi, and spinal/skeletal anomalies and/or scoliosis; CM, capillary malformation; L, lymphatic; PTEN, tumor suppressor gene; V, vein/venous.
Slow-Flow
Klippel-Trenaunay Syndrome.
Klippel-Trenaunay syndrome, also known as capillary-lymphaticovenous malformation (CLVM), is sporadic. The causative gene is not yet known; it is likely a regional somatic mutation. At birth, there are geographic large or small CMs over the extremity and buttock. Over time, the CMs usually become studded with lymphatic vesicles. Lymphedema and/or microcystic and macrocystic LMs in the limb (often involving the pelvis or retroperitoneum) are present, along with splenic lymphatic cysts. Limb hypertrophy is present at birth and progressively worsens with growth. Complications of Klippel-Trenaunay syndrome include recurrent infections, pulmonary embolism, thrombophlebitis, gastrointestinal bleeding, constipation, bladder outlet obstruction, and hematuria.34–36 Young37 clearly differentiated slow-flow Klippel-Trenaunay syndrome from fast-flow Parkes Weber syndrome. There is ongoing controversy over the diagnostic criteria for Klippel-Trenaunay syndrome. The debate focuses on whether patients with capillary-venous malformation (CVM) or solely CM in a limb should be classified at the minor end of the Klippel-Trenaunay spectrum.
Maffucci Syndrome.
Maffucci syndrome is an extremely rare disorder characterized by cutaneous VMs, long bone enchondromas, and skeletal deformities.38 Enchondromas cause bony distortion and asymmetric growth; it is common for a patient to present with a pathologic fracture. Vascular malformations on the skin usually appear around age 4-to-5 years and are often progressive. These lesions begin as compressible, round, bluish spots; later they become firm, knotty, and warty, and they often contain phleboliths. A benign vascular tumor, spindle cell hemangioma, often arises in these malformed veins. Patients with Maffucci syndrome are at risk of developing chondrosarcoma and various other malignancies.39
Fast-Flow
Parkes Weber Syndrome.
Parkes Weber syndrome is characterized as a fast-flow combined capillary-arteriovenous malformation (CAVM) or capillary-lymphatico-arteriovenous malformation (CLAVM), usually involving the lower limb and proximal trunk. In many patients there is a mutation in the RASA1 gene, but not all patients with phenotypic Parkes Weber syndrome have this mutation.40 All patients with Parkes Weber syndrome and a mutation in RASA1 have the characteristic multiple CMs, as seen in capillary malformation–arteriovenous malformation (CM-AVM). The affected extremity exhibits a cutaneous flush/staining with underlying multiple micro-AVFs and generalized enlargement or overgrowth with or without lymphatic lesions or lymphedema.
Capillary Malformation-Arteriovenous Malformation.
A newly delineated familial disorder, capillary malformation–arteriovenous malformation (CM-AVM) is characterized by single or multiple small (1-2 cm in diameter) pink-to-red, round-to-oval CMs in association with AVM or AVF. Often the small lesions exhibit fast flow on hand-held Doppler ultrasonographic examination. Frequently there is a family history of one or more innocent-appearing capillary stains. There is phenotypic overlap with Parkes Weber syndrome.40,41
Bannayan-Riley-Ruvalcaba Syndrome (PTEN Hamartoma Syndrome).
Bannayan-Riley-Ruvalcaba syndrome, also known as PTEN hamartoma syndrome, is characterized by macrocephaly, multiple lipomas, hamartomatous polyps of distal ileum and colon, Hashimoto thyroiditis, pigmented penile macules, and vascular anomalies including CM, VM, and AVM.42 Affected patients have an increased risk of malignancy, particularly of the thyroid and breast. An estimated 31% of patients have some type of thyroid involvement, in the form of multinodular goiter, thyroid adenoma, or thyroid cancer.43 Bannayan-Riley-Ruvalcaba syndrome is an autosomal dominant condition known to be caused by a mutation in tumor suppressor gene PTEN; it is allelic with Cowden syndrome. The spectrum including both Bannayan-Riley-Ruvalcaba and Cowden syndromes is subsumed by the term PTEN hamartoma syndrome.
CLOVE(S) (Congenital, Lipomatous, Overgrowth, Vascular Malformations, and Epidermal Nevi) Syndrome.
For many years, the disorder now known as CLOVE (congenital, lipomatous, overgrowth, vascular malformations, and epidermal nevi) syndrome was mistakenly designated “thoracic Klippel-Trenaunay syndrome.” CLOVE syndrome is characterized by masses with fatty and lymphatic components in the trunk, retroperitoneum, pelvis, and extremities. Patients exhibit either nonprogressive or slightly progressive overgrowth.44 Capillary malformations are common on the trunk as well as the extremities. Lymphatic vesicles may appear on thorax and axilla, attesting to the underlying LM. Fast-flow lesions (AVMs) may be present in the fatty lesions on the trunk and paraspinal region, the latter of which can cause debilitating spinal myelopathy.45 The acronym was augmented with an “S” to CLOVES to include the frequently present scoliosis, spinal, and/or skeletal anomalies.46 Acral deformities are present, including symmetrically large, wide feet and hands, macrodactyly, and wide first web space (“sandal toe deformity”). Thoracic and central veins may be dilated; patients with this finding are at high risk for thromboembolism.47
Interdisciplinary Care
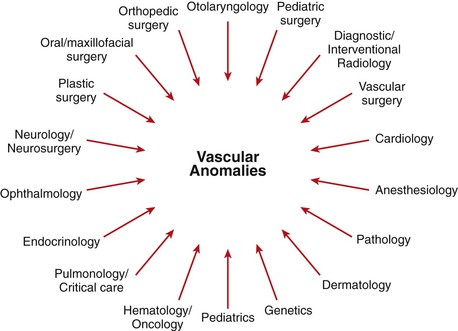
The field of vascular anomalies has emerged at the interface of many medical and surgical specialties. Patients with vascular anomalies were once medical nomads–they wandered from specialist to specialist because no one physician was able to address their many issues. Interdisciplinary teams for vascular anomalies have developed to care for these patients, and vascular surgeons are welcome members of such teams (Fig. 69-2). European vascular surgeons have been in the vanguard of this developing field, contributing endovascular and surgical techniques. The sharing of responsibility among specialties varies considerably from one institution to the next. Thankfully, there are usually no “turf battles,” given the difficulty in managing many patients with these anomalies.
Toward a Molecular Classification
Advances in molecular genetics are illuminating the field of vascular anomalies. Infantile hemangiomas have been shown to be clonal, likely arising by somatic mutation in an endothelial progenitor cell. There are also some families predisposed to development of hemangiomas, suggesting possible germ-line mutations. The causative genes for several vascular malformations have been identified (Table 69-3). The pathogenetics are being elucidated in vitro and in animal models. Once the basic mechanisms are understood, targeted pharmacologic therapies might be possible, and our classification system for vascular anomalies will be a molecular one.
Selected Key References
Dompmartin A, Acher A, Thibon P, Tourbach S, Hermans C, Deneys V, Pocock B, Lequerrec A, Labbé D, Barrellier MT, Vanwijck R, Vikkula M, Boon LM. Association of localized intravascular coagulopathy with venous malformations. Arch Dermatol. 2008;144:873–977.
Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–422.
North PE, Waner M, Mizeracki A, Mihm MC Jr. GLUT1: A newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11–22.
Sarkar M, Mulliken JB, Kozakewich HP, Robertson RL, Burrows PE. Thrombocytopenic coagulopathy (Kasabach-Merritt phenomenon) is associated with kaposiform hemangioendothelioma and not with common infantile hemangioma. Plast Reconstr Surg. 1997;100:1377–1386.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Enjolras O, et al. Vascular tumors and vascular malformations (new issues). Adv Dermatol. 1997;13:375–423.
2. Mulliken JB, et al. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–422.
3. Greene AK, et al. Vascular anomalies in 5,621 patients: guidelines for referral. J Pediatr Surg. 2011;46:1784–1789.
4. North PE, et al. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31:11–22.
5. North PE, et al. A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol. 2001;137:559–570.
6. Mulliken JB, et al. Reticular infantile hemangioma of the limb can be associated with ventral-caudal anomalies, refractory ulceration, and cardiac overload. Pediatr Dermatol. 2007;24:356–362.
7. Sarkar M, et al. Thrombocytopenic coagulopathy (Kasabach-Merritt phenomenon) is associated with Kaposiform hemangioendothelioma and not with common infantile hemangioma. Plast Reconstr Surg. 1997;100:1377–1386.
8. Enjolras O, et al. Infants with Kasabach-Merritt syndrome do not have “true” hemangiomas. J Pediatr. 1997;130:631–640.
9. Mulliken JB, et al. Case records of the Massachusetts General Hospital: weekly clinicopathological exercises: case 13-2004: a newborn girl with a large cutaneous lesion, thrombocytopenia, and anemia. N Engl J Med. 2004;350:1764–1775.
10. Dompmartin A, et al. Association of localized intravascular coagulopathy with venous malformations. Arch Dermatol. 2008;144:873–877.
11. Vikkula M, et al. Molecular genetics of vascular malformations. Matrix Biol. 2001;20:327–335.
12. Sujansky E, et al. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49–58.
13. Greene AK, et al. Sturge-Weber syndrome: soft-tissue and skeletal overgrowth. J Craniofac Surg. 2009;20:617–621.
14. Morgan JM, et al. Cutis marmorata telangiectatica congenita with hypoplasia of the right iliac and femoral veins. Br J Dermatol. 1997;137:119–122.
15. Vogel AM, et al. Iliac artery stenosis in a child with cutis marmorata telangiectatica congenita. J Pediatr Surg. 2005;40:e9–e12.
16. Conway RL, et al. Neuroimaging findings in macrocephaly-capillary malformation: a longitudinal study of 17 patients. Am J Med Genet. 2007;43A:2981–3008.
17. Toriello HV, et al. Accurately renaming macrocephaly-cutis marmorata telangiectatica congenita (M-CMTC) as macrocephaly-capillary malformation (M-CM) [letter]. Am J Med Genet. 2007;143A:3009.
18. Kubiena HF, et al. Genuine diffuse phlebectasia of Bockenheimer: dissection of an eponym. Pediatr Dermatol. 2006;23:294–297.
19. Dompmartin A, et al. Elevated D-dimer level in the differential diagnosis of venous malformations. Arch Dermatol. 2009;145:1239–1244.
20. Brouillard P, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866–887.
21. Boon LM, et al. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971–976.
22. Schook CC, et al. Primary lymphedema: clinical features and management in 138 pediatric patients. Plast Reconstr Surg. 2011;127:2419–2431.
23. Brorson H, et al. Complete reduction of lymphoedema of the arm by liposuction after breast cancer. Scand J Plast Reconstr Surg Hand Surg. 1997;3:1137–1143.
24. Brorson H, et al. Controlled compression and liposuction treatment for lower extremity lymphedema. Lymphology. 2008;41:52–63.
25. Campisi C, et al. Microsurgery for lymphedema: clinical research and long-term results. Microsurgery. 2010;30:256–260.
26. Yamamoto T, et al. Simultaneous multi-site lymphaticovenular anastomoses for primary lower extremity and genital lymphoedema complicated with severe lymphorrhea. J Plast Reconstr Aesthet Surg. 2011;64:812–815.
27. Callander CL. Study of arteriovenous fistula with an analysis of 447 cases. Ann Surg. 1920;71:428–459.
28. Leonard FC, et al. Congenital arteriovenous fistulation of the lower limb: report of a case successfully treated by total excision. N Engl J Med. 1951;245:885–888.
29. Malan E, et al. Congenital angiodysplasias of the extremities II: arterial, arterial and venous, and haemolymphatic dysplasias. J Cardiovasc Surg (Torino). 1965;6:255–345.
30. Upton J, et al. Vascular malformations of the upper limb: a review of 270 patients. J Hand Surg Am. 1999;24:1019–1035.
31. Loose DA. Combined treatment of congenital vascular defects: indications and tactics. Semin Vasc Surg. 1993;6:260–265.
32. Liu AS, et al. Extracranial arteriovenous malformations: natural progression and recurrence after treatment. Plast Reconstr Surg. 2010;125:1185–1194.
33. Kohout MP, et al. Arteriovenous malformations of the head and neck: natural history and management. Plast Reconstr Surg. 1998;102:643–654.
34. Baskerville PA, et al. The Klippel-Trenaunay syndrome: clinical, radiological and haemodynamic features and management. Br J Surg. 1985;72:232–236.
35. Samuel M, et al. Klippel-Trenaunay syndrome: clinical features, complications and management in children. Br J Surg. 1995;82:757–761.
36. Jacob AG, et al. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28–36.
37. Young AE. Combined vascular malformations. Mulliken JB, et al. Vascular birthmarks: hemangiomas and malformations. WB Saunders: Philadelphia; 1988:246–274.
38. Pansuriya TC, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261.
39. Verdegaal SH, et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: an international multicenter study of 161 patients. Oncologist. 2011;16:1771–1779.
40. Revencu N, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–965.
41. Eerola I, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hu Genet. 2003;73:1240–1249.
42. Cohen MM Jr. Bannayan-Riley-Ruvalcaba syndrome: renaming three formerly recognized syndromes as one etiologic entity. Am J Med Genet. 1990;35:291–292.
43. Tan WH, et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. 2007;44:594–602.
44. Sapp JC, et al. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007;143A:2944–2958.
45. Alomari AI, et al. Complex spinal-paraspinal fast-flow lesions in CLOVES syndrome: analysis of clinical and imaging findings in 6 patients. AJNR Am J Neuroradiol. 2011;32:1812–1817.
46. Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009;18:1–7.
47. Alomari AI, et al. CLOVES syndrome with thoracic and central phlebectasia: increased risk of pulmonary embolism. J Thorac Cardiovasc Surg. 2010;140:459–463.
48. Shirley MD, et al. Sturge Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–1979.
50. Lee JH, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nature Genet. 2012;44:941–945.
51. Ferrell RE, et al. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum Molec Genet. 1998;7:2073–2078.
52. Karkkainen MJ, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nature Genet. 2000;25:153–159.
53. Fang J, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388.
54. Erickson RP, et al. Clinical heterogeneity in lymphoedema-distichiasis with FOXC2 truncating mutations. J Med Genet. 2001;38:761–766.
55. Finegold DN, et al. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Molec Genet. 2001;10:1185–1189.
56. Rezaie T, et al. Primary non-syndromic lymphoedema (Meige disease) is not caused by mutations in FOXC2. Eur J Hum Genet. 2008;16:300–304.
57. Soblet J, et al. Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol Syndromol. 2013;4:179.
58. Vikkula M, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190.
59. Calvert JT, et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999;8:1279–1289.
60. Boon LM, et al. A gene for inherited cutaneous venous anomalies (“glomangiomas”) localizes to chromosome 1p21-22. Am J Hum Genet. 1999;65:125–133.
61. Brouillard P, et al. High-resolution physical and transcript map of the locus for venous malformations with glomus cells (VMGLOM) on chromosome 1p21-p22. Genomics. 2000;67:96–101.
62. Marsh DJ, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome [letter]. Nature Genet. 1997;16:333–334.
63. Longy M, et al. Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J Med Genet. 1998;35:886–889.
64. Kurek KC, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115.