[level-membership-for-pathology-category]
Cirrhosis
James M. Crawford
Introduction
Cirrhosis is an important cause of morbidity and mortality. In the United States, cirrhosis accounts for more than 33,000 deaths per year and is the 12th leading cause of death.1 It is reported on hospital discharge in at least 1% of adult patients2 and is found in 4% to 12% of patients at autopsy in developed countries.3–5 Cirrhosis is the morphologic result of many different types of chronic insult to the liver. It may develop rapidly during a period of months, but most often it is a product of many years of chronic injury. The causes include chronic viral or autoimmune hepatitis, biliary obstruction, alcohol toxicity, and a variety of metabolic abnormalities.
The term cirrhosis derives from the Greek word κίρρoς, meaning “tawny,” and was chosen early in the 19th century to describe the gross appearance (tawny, nodular, and firm), and later the microscopic appearance of the chronically diseased, physiologically burned out, and dysfunctional liver.6,7 In 1977, an international panel sponsored by the World Health Organization (WHO) defined cirrhosis as “a diffuse process characterized by fibrosis and the conversion of normal liver architecture into structurally abnormal nodules.”8 The WHO stated that “cirrhosis is a chronic progressive condition that results in liver cell failure and portal hypertension” and observed that vascular abnormalities were an important feature of cirrhosis. These abnormalities included thrombosis, obliteration and recanalization of veins, formation of arteriovenous shunts, and “capillarization” of sinusoids.9 Etiology was not an important consideration, because there was no cure for cirrhosis. However, reports in the 1980s described the partial “reversal” of cirrhosis,10 and there is now consistent evidence that effective treatment of the underlying cause may mitigate the fibrotic progression of certain liver diseases, particularly in viral hepatitis.11,12
In 2012, an international group of liver pathologists recommended discontinuing use of the term cirrhosis, substituting instead the diagnostic concept of “an advanced stage of chronic liver disease,” with emphasis on etiologic cause, grade of activity, and whether features are present that suggest progression or regression of the fibrotic process, presence of other diseases, or features indicating risk for malignancy.13 Liver morphology should be used to help create an integrated assessment of the patient’s clinicopathologic status. This recommendation is in accord with the increasing awareness in the hepatology community concerning the need for pathophysiologic, rather than morphologic, classification of cirrhosis.14 Indeed, identifying when a liver truly becomes “cirrhotic” is a major diagnostic dilemma, depending on whether one relies on the clinical features of advancing liver disease, imaging modalities, liver biopsy, or an increasing array of noninvasive tests of liver fibrotic status.15,16 All this being said, the term cirrhosis has been used for two centuries and will remain in use for the foreseeable future. This chapter is titled and written with that historical premise in mind.
Cirrhosis is defined by the presence of certain anatomic abnormalities of liver structure. However, a presumptive diagnosis of cirrhosis can often be made when certain clinical consequences of cirrhosis are found. These consequences may be mechanical, functional, or neoplastic. The mechanical effects are related to obstruction of blood flow in the liver, which leads to increased pressure in the splanchnic veins and a high risk for rupture of esophageal varices. Obstruction progresses gradually as cirrhosis develops but may worsen suddenly after thrombosis of the portal vein. Obstruction is associated with the opening of multiple intrahepatic and extrahepatic collateral channels that allow shunting of splanchnic blood past the hepatic parenchyma. Hepatic functional deficits are, in part, related to this portosystemic shunting but also to loss of hepatocellular mass and intracellular retention of bile salts and other toxic substances. Many of the clinical effects of cirrhosis are found in other organs. Renal and pulmonary failure may occur secondary to the systemic hemodynamic response to cirrhosis. Hemorrhage may occur because of platelet dysfunction, platelet sequestration in the spleen, and decreased synthesis of proteins of the coagulation cascade. Neoplasia is a not infrequent late occurrence in patients with cirrhosis. The lifetime risk of hepatocellular carcinoma exceeds 50% in patients with some forms of cirrhosis. Cholangiocarcinoma may be a late complication of intrahepatic or extrahepatic biliary disease. Hence, the development and consequences of cirrhosis are of concern in every form of chronic liver disease.
Definition
Using the 1977 consensus criteria,8 cirrhosis is defined anatomically by the presence throughout the liver of fibrous septa that subdivide the parenchyma into nodules (Fig. 50.1).17–19 Several features in this definition should be emphasized.

The progression of chronic liver disease is highly variable. The point at which a liver becomes cirrhotic is rather subjective and is a frequent source of interobserver disagreement. Establishing a diagnosis of cirrhosis on the basis of percutaneous or transjugular needle biopsy sampling (which samples less than 1/10,000 of the liver mass) can be difficult, particularly if fibrous septa are widely spaced or have regressed. Fortunately, clinical data often provide valuable guidance as to whether any abnormal findings observed in percutaneous liver biopsy tissue are representative of the entire liver. Supporting clinical data include physical examination findings (e.g., ascites, caput medusae, spider angiomas, gynecomastia) and impressions gained from imaging studies or intraoperative visualization of the organ. Laboratory data may not reveal abnormalities, because serum levels of albumin, clotting factors, urea, alkaline phosphatase, aminotransferases, and bilirubin can be normal in a patient who has quiescent cirrhosis with minimal ongoing damage but has not yet experienced hepatic failure, whereas a patient with massive hepatic necrosis and hepatic failure is not cirrhotic despite profound abnormalities in the above serum parameters. Therefore, laboratory data, per se, do not establish a diagnosis of cirrhosis, although various laboratory multiparametric indices have been advanced as capable of providing diagnostic guidance.20,21
Occasionally, a severe focal injury to the liver results in focal histologic changes indistinguishable from cirrhosis on percutaneous needle biopsy; this focal change is not considered true cirrhosis. When this question arises, having definitive information from clinical evaluation and from imaging studies on the general status of the liver, or a biopsy sample from elsewhere in the liver, is critical to determine whether a fibrotic process is focal or diffuse.
Pathogenesis
The 1977 definition of cirrhosis is used by pathologists to recognize cirrhosis, but the definition does not rely on understanding its pathogenesis. Liver cirrhosis is not, strictly, the end stage of hepatic scarring. Rather, it is a dynamic, biphasic process dominated on the one hand by progressive parenchymal fibrosis and on the other by severe disruption of vascular architecture and distortion of the normal lobular architecture. The main anatomic elements (Box 50.1) include deposition of collagen in the parenchyma and portal tracts, arterialization of parenchymal sinusoids, obliteration of small hepatic and portal veins with resultant loss of hepatocytes through a process referred to as parenchymal extinction, abnormal vascular physiology, and regeneration of hepatocytes. Although the importance of these elements is widely appreciated, there is healthy debate concerning the role of each in the pathogenesis of cirrhosis,18,22–24 particularly because the design of effective interventions to delay or reverse the development of cirrhosis depends on understanding its pathogenesis. The concepts thought to be operative in the genesis of cirrhosis of any cause are summarized here.22,25 A discussion of the causes of hepatocellular death is beyond the scope of this chapter.
Collagen in the Liver
Collagen accumulation is a prominent feature of cirrhosis. In the normal liver, collagen types I and III are concentrated in the portal tracts and around terminal hepatic veins, with bundles occasionally located between hepatocytes and endothelial cells in the space of Disse. Strands of type IV collagen (reticulin) are present in the space of Disse, where they form a delicate and uniform network that supports the liver cell plates. In cirrhosis, excessive amounts of types I and III collagen are deposited in the portal tracts, along individual liver cell plates in the space of Disse, and in regions of necroinflammatory collapse. A variety of noncollagenous matrix proteins are also deposited in the space of Disse. In cirrhosis, the amounts of collagen, glycoproteins, and proteoglycans can increase severalfold.26 On a percent area basis, total extracellular matrix components can increase from 5% in normal liver to 25% to 40% in cirrhosis.27 Some of this is only an apparent increase, because condensation of the normal structural collagen and other matrix components occurs during parenchymal collapse and extinction.
The two main cell types that synthesize collagen in the liver are hepatic stellate cells23 and portal fibroblasts.28 Hepatic stellate cells reside in the subendothelial space of Disse in the sinusoidal walls. They are normally distended with lipid droplets containing retinyl esters and other fat-soluble vitamins. During hepatic injury, stellate cells are stimulated by inflammatory mediators to become myofibroblasts: They lose their fat globules, express α-smooth muscle actin in the cytoplasm, and commence proliferation and collagen synthesis. It has recently been discovered that autophagy of the lipid droplets and catabolism of the retinyl esters and triglycerides provide the energy to drive the activation of stellate cells.29 Experimental inhibition of stellate cell autophagy inhibits their activation.30
Stellate cell activation and sinusoidal fibrosis are readily reversible within weeks after cessation of injury.19 When stellate cells are activated in chronic low-grade disease, the liver cell plates are able to maintain their structure while collagen is deposited in the space of Disse, giving an appearance known as pericellular fibrosis or sinusoidal fibrosis. This type of delicate collagen is most easily appreciated in the perivenular regions (Rappaport zone 3). With time, collagen is deposited along the entire length of the sinusoid. Alternatively, widespread injury to hepatocytes, as in alcoholic hepatitis or some forms of drug injury (e.g., amiodarone), may activate stellate cells throughout the liver, leading to extensive deposition of sinusoidal collagen. In either instance, the total matrix in the space of Disse increases and changes from one that contains delicate interspersed strands of fibrillar collagen (types III and IV) to one composed of a dense matrix of basement membrane–type matrix proteins, which closes the space of Disse to protein exchange between hepatocytes and plasma. In general, abnormal matrix deposition within the space of Disse occurs in those parts of the parenchyma where cell injury and inflammation are greatest.
Portal tract fibroblasts differ from stellate cells in location and physiology.31–34 These cells are activated by injury within the portal tracts, particularly in biliary disease, which leads to fibrosis in the region of the ducts and ductules. Peribiliary myofibroblasts are capable of rapid proliferation and deposition of collagen. The epithelial-to-mesenchymal transition of bile duct epithelial cells to a myofibroblast phenotype also appears to contribute to the fibrosis that develops around bile ducts in chronic biliary disease.35 As a result, fibrosis arising from biliary tract disease can run an aggressive course; an example is complete biliary obstruction in infants with extrahepatic biliary atresia, in whom the liver becomes cirrhotic by 9 weeks of age (see Chapter 54). At the opposite end of the spectrum is the exceedingly indolent progression of portal tract fibrosis to cirrhosis in primary biliary cirrhosis, which may extend for 20 or more years (see Chapter 47). In either instance, bridging fibrous septa between portal tracts develop throughout the liver, thereby fulfilling the criteria for cirrhosis. A curious feature of biliary-type fibrosis is that the lobular parenchyma is not induced to regenerate substantially until the liver is extensively fibrotic. Hence, biliary-type fibrosis subdivides the liver into a jigsaw-like pattern during its progression, and cirrhosis may be a very late feature in the course of the disease.
Hepatic Arterialization and Capillarization
Arterialization of the liver in cirrhosis has been known for more than a century. In 1907, Herrick perfused cadaver livers and demonstrated that resistance to flow in the hepatic artery of cirrhotic livers was markedly decreased.36 This fact is documented daily by ultrasonographers when they examine patients with cirrhotic livers and find increased arterial flow in the liver, along with sluggish or even reversed flow in the portal vein. Moschcowitz first used the term capillarization to describe the light microscopic appearance of arterialization in the cirrhotic liver as a granulation tissue response—that is, arterial growth into inflamed tissue.37 Schaffner and Popper used the term capillarization to represent a constellation of ultrastructural changes, including a decrease in the number and size of sinusoidal endothelial fenestrations, loss of hepatocellular microvilli, and an increase in basement membrane material.38,39 By this definition, documentation of capillarization requires electron microscopy. Specifically, in the normal liver, sinusoidal endothelial cells lack a basement membrane and exhibit fenestrations approximately 100 nm in diameter, occupying between 2% to 3% of the area of the endothelial cell. Deposition of extracellular matrix in the space of Disse is accompanied by loss of fenestrations in the sinusoidal endothelial cells.39 With the development of cirrhosis, the diameter of the fenestrations slightly decreases but the area occupancy (porosity) falls to less than 0.5%.
The sinusoidal endothelium in the cirrhotic liver may express CD34. Because this expression is a normal property of arterial endothelium, CD34 positivity is considered a marker of arterialization of the sinusoids. Sinusoidal arterialization is accompanied by α-smooth muscle actin staining in the sinusoidal wall (Fig. 50.2), reflecting the accompanying transformation of hepatic stellate cells into myofibroblasts. It appears that capillarization leads to loss of normal sinusoidal endothelial cell generation of nitric oxide (NO). This loss of NO generation creates a microenvironment that is permissive for stellate cell activation.40 Transformation of the stellate cells then increases sinusoidal vascular resistance by tonic contraction of these “myofibroblasts.” Sinusoidal fibrosis in the perivenular region of the lobule may also partially obstruct vascular outflow, creating postsinusoidal vascular resistance.
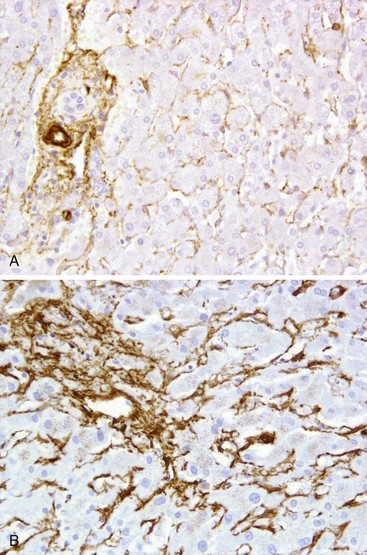
Transformation of sinusoidal vascular channels is widely considered to be an explanation for functional deficits in blood-hepatocyte solute exchange.39,41–43 Rapid flow in sinusoids may represent an effective arteriovenous shunt, resulting in a further decrease in solute exchange.44 Although these effects may decrease solute transport into hepatocytes, capillarization or arterialization may also be viewed as a protective form of adaptation that allows the hepatocytes to survive in a high-pressure, high-flow environment.
Arterialization also occurs at the level of portal tracts, where an increased number (and size) of arterial profiles is seen in a variety of conditions, including cirrhosis and use of oral contraceptives. Arterialization of small portal tracts in cirrhosis is usually accompanied by obliteration of adjacent portal veins. Portal vein loss may result from portal tract inflammation in chronic hepatitis or from congestive changes (congestive portopathy) after hepatic venous outflow is compromised (discussed in the next section). Obliteration of small portal veins increases presinusoidal vascular resistance for blood inflow via the splanchnic system. Resistance to hepatic arterial blood flow decreases, owing to an increased arterial capacity, whereas resistance to portal vein blood inflow increases. Hepatic arterial blood pressure is sufficient to supply blood to the liver, but the low pressure of the splanchnic system is not able to overcome the pressure impedance, leading to portal hypertension.
Parenchymal Extinction
Parenchymal extinction is defined as a focal loss of contiguous hepatocytes (Fig. 50.3). Detailed morphologic studies by Wanless and colleagues demonstrated that parenchymal extinction plays a critical role in the pathogenesis of cirrhosis.45–47
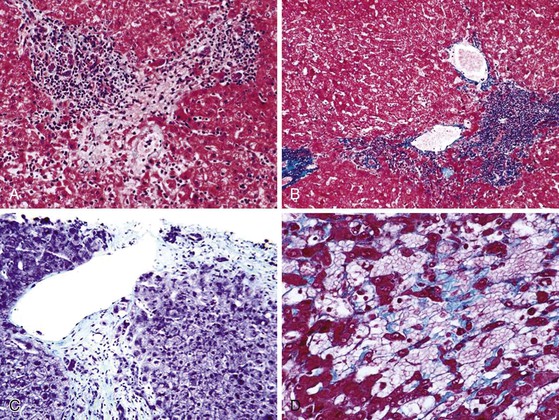
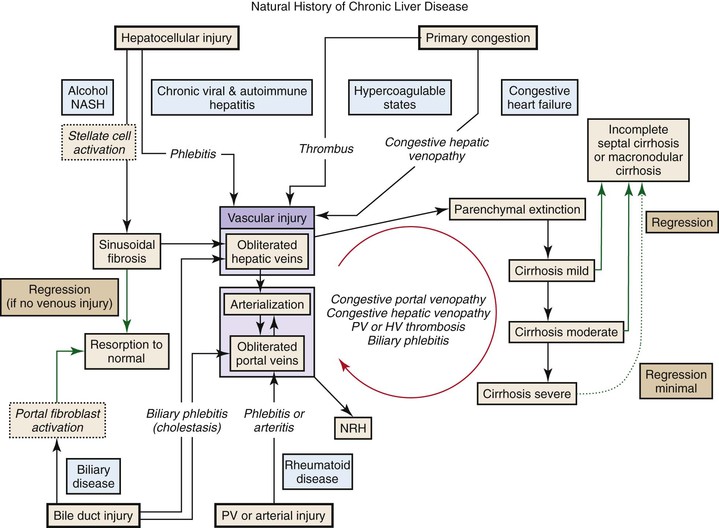
Hepatocyte apoptosis and necrosis occur in all types of liver diseases that progress to cirrhosis. The mechanisms are diverse and include lymphocyte-mediated injury, rupture of triglyceride-laden hepatocytes, bile-salt toxicity, and various metabolic stresses. Most of these injuries, if accompanied by low-grade spotty necrosis or apoptosis, lead to local replacement and complete healing. Progressive disease occurs when these injuries are accompanied by a stromal reaction that includes deposition of extracellular matrix, increased sinusoidal vascular resistance, and obstruction of blood flow. The convergence of these injuries leads to contiguous loss of hepatocytes (see Fig. 50.3).45 These extinction lesions may involve a small portion of an acinus, larger units of one or more adjacent acini, or even a whole lobule. The contiguous cell loss is ultimately the result of focal ischemia resulting from obstruction of veins or sinusoids. Naturally, the size of extinction lesions depends on the size of the obstructed vessels.
The concept of parenchymal extinction is important because it incorporates the following perceptions: (1) parenchymal extinction is not directly caused by the initial hepatocellular injury but is an epiphenomenon caused by innocent bystander injury of the local vessels; (2) each parenchymal extinction lesion (PEL) has its own natural history and may be in an early or late stage of healing; (3) cirrhosis develops simultaneously with the accumulation of numerous independent and discrete PELs throughout the liver; and (4) the form of cirrhosis is largely determined by the distribution of the vascular injury. Importantly, parenchymal extinction may progress long after cirrhosis is already established, leading to slow conversion of a marginally functional liver into a sclerotic organ incapable of sustaining life.
The pathogenesis of vascular obstruction depends on the size of the vessels and is outlined in Figure 50.4 (see also Fig. 50.3). Most small-vessel obliteration is secondary to local inflammation.46,47 Although thrombosis may be important in veins of all sizes, it is especially important for blockage of large veins. Most PELs are produced by blockage of veins larger than 100 µm in diameter, because obstruction at this site cannot be easily circumvented by collateral flow within the sinusoids. Obstruction of several adjacent sinusoids is also difficult to circumvent. This raises the issue of congestion, which occurs whenever blood entering the vasculature exceeds the ability of the outflow tract to carry that blood, a state known as in-out imbalance. Congestion is particularly severe when there is total obstruction of the outflow tract or when there is increased inflow in the presence of partial outflow obstruction. Congestive injury is exacerbated by reactive hyperemia, shunt formation, and angiogenesis. If inflow is marked, congestion occurs even when the tissue has normal outflow capacity.
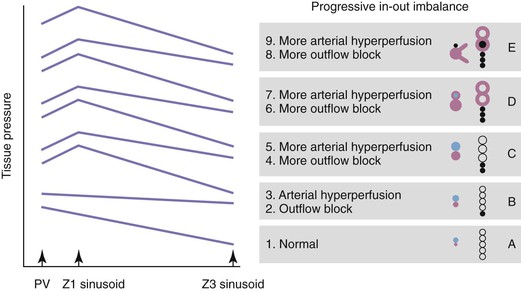
Although the obstructive vascular events leading to parenchymal extinction play out largely in blood vessels larger than 100 µm in diameter, activation of the coagulation cascade within sinusoids releases thrombin, which can interact with the proteinase receptor PAR1 on hepatic stellate cells.48 This is a stimulus for stellate cell activation and transformation to myofibroblasts, which further contributes to the increased vascular resistance and congestive pathophysiology described earlier.
Therefore, PELs are the result of local failure of the microvasculature, usually because of obstruction of hepatic veins or sinusoids. The mechanism for formation of PELs is detailed in Figure 50.5 (see also Fig. 50.4).
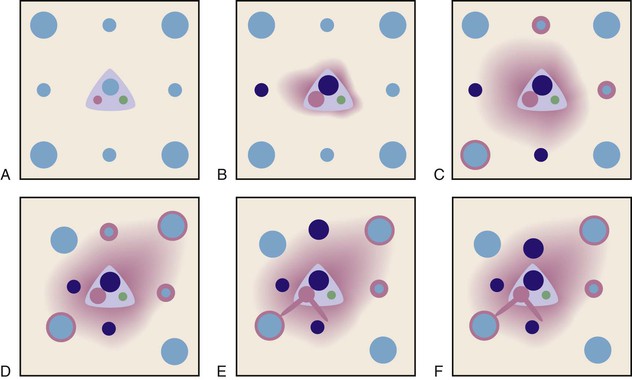
In early chronic liver disease, PELs are recognized by the close approximation of the terminal hepatic vein and the adjacent portal tract (see Figs. 50.3 and 50.5). They may be difficult to recognize, because damaged small hepatic veins only appear as a few collagen bundles lying adjacent to a portal tract. PELs become more evident as they aggregate and involve larger and more easily recognizable hepatic veins. PEL aggregates may be evident as two or more portal tracts that are bound together with a hepatic vein remnant apparent in the intervening space (see Fig. 50.2). With progression of disease, progressive obstruction of hepatic veins and secondary arterial dilatation cause further congestive injury in the tissue located between the lesions. This creates a self-perpetuating pathophysiology that may eventually lead to interconnected portal tracts throughout the whole liver.
Shunt Formation
When a region of parenchyma becomes extinct, it collapses so that a portal tract becomes closely associated with an adjacent terminal hepatic vein. This close approximation offers an opportunity for the artery in the portal tract to drain directly into the collapsed perivenous tissue. Often, these arteries can be seen supplying a pool of blood surrounded by atrophic hepatocytes. In older lesions, there is a well-demarcated blood-filled channel; this suggests a stable, high-flow and high-pressure conduit connecting a small artery to a small hepatic vein. This appearance has been interpreted as an arteriovenous shunt.
After parenchymal extinction, the formation of bona fide bridging fibrous septa between portal tracts and terminal hepatic veins enables portovenous and arteriovenous shunting through de novo vascular channels, effectively bypassing the parenchymal nodules. Shunted blood flow through these “fast” vascular channels leaves the remainder of the hepatic parenchyma almost bereft of meaningful blood flow.44,46 This progression also helps explain the increased blood flow observed in sinusoids of the cirrhotic liver in the midst of relative underperfusion of the liver parenchyma as a whole. A remarkable fraction of nutritive blood flow may pass through these intrahepatic functional shunts, contributing to ongoing hepatocellular necrosis and the further generation of PELs. Unfortunately, compression of the shunt channels by contiguous regenerating nodules maintains an increased transhepatic vascular resistance.
Congestive Hepatopathy
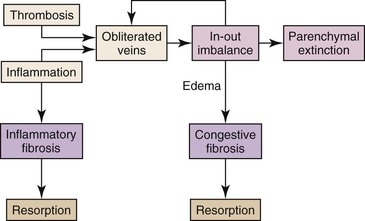
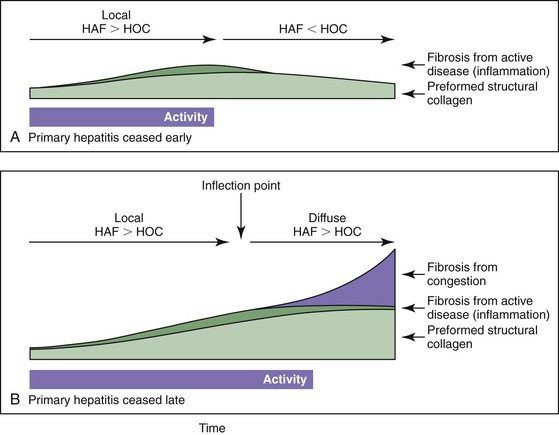
Vascular injury in cirrhosis can be divided into an early primary phase, a later congestive phase, and, ultimately, vascular thrombosis. In the primary phase, venous and sinusoidal obstruction is caused by local inflammation occurring in the course of chronic hepatitis (Fig. 50.6).47 The generation of soluble proinflammatory mediators in this setting is a powerful stimulus of fibrogenesis. However, one of the most powerful stimuli of fibrogenesis in many organs is the organization of exudates, especially those rich in fibrin.49–56 In the chronically congested liver, the development of capsular fibrosis attests to the presence of this fibrogenic mechanism (Fig. 50.7). This is the basis for the schematic diagram in Figure 50.8, which indicates that instances of fibrosis can be divided into those stimulated by conventional inflammation and those stimulated by chronic edema and exudation. These two pathways may converge to generate the pattern of fibrogenesis typically seen in chronically injured livers.
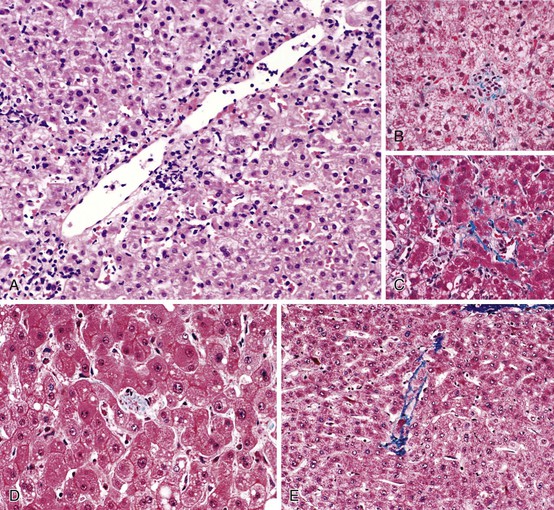
Collagen accumulation is determined by the rates of collagen synthesis and resorption (Fig. 50.9; see also Fig. 50.8). Most fibrosis likely accumulates late, when congestive forces cause interstitial exudate and collagen deposition that exceeds the resorptive capacity of the liver. In this formulation, obstructed hepatic veins and PELs occur before septa and before fibrosis; therefore, these lesions are at the leading edge of the pathogenesis of cirrhosis.19,22,45,47,57 The preexisting structural collagen of the liver condenses during the formation of PELs and is incorporated into septa. Although this is not true fibrosis, the presence of such collagen is a striking feature of these lesions.
In the later congestive phase of liver injury, a self-perpetuating pattern of progressive liver injury is created. Specifically, when there is in-out imbalance of blood flow, the high transmural pressure leads to edema, hemorrhage, and narrowing of venous and sinusoidal lumina, followed by intimal fibrous thickening of these vessels. This injury, known as congestive hepatopathy, causes a decrease in the outflow capacity of the tissue that worsens obstruction and, therefore, congestion. Thus, congestive hepatopathy establishes a positive feedback loop of progressive tissue injury.
Vascular Thrombosis
Thrombosis is an important insult. In angiographic and ultrasonographic studies, portal vein thrombosis has been detected in 0.6% to 16.6% of cirrhotic patients,58 and grossly visible portal vein fibrosis or thrombosis has been found in 39% of cirrhotic livers at autopsy.59 Veno-occlusive lesions of hepatic veins smaller than 0.2 mm in diameter have been found in as many as 74% of cirrhotic livers examined at autopsy.60–62 Obliterative lesions are found in 36% of portal veins and 70% of hepatic veins in livers removed at liver transplantation.45 The distribution of obliterative lesions is more uniform in portal veins than in hepatic veins, consistent with the concept of propagation of multifocal thrombi downstream from their site of origin. Portal vein lesions are associated with prominent regional variation in the size of cirrhotic nodules. Hepatic vein lesions are associated with regions of confluent fibrosis and parenchymal extinction. The compelling conclusion is that thrombosis of medium- and large-sized portal veins and hepatic veins is a common occurrence in cirrhosis and may represent a final common pathway for the propagation of parenchymal extinction to full-blown cirrhosis. Furthermore, cirrhotic livers are susceptible to thrombosis because of sluggish or reversed blood flow and the prothrombotic effects of sepsis and cholestasis, thus creating an opportunity for continued loss of functional residual liver parenchyma.
Regeneration
After childhood, the normal liver becomes a stable organ with slow turnover of hepatocytes. However, on injury or surgical reduction, the liver cells proliferate. Normal human liver can restore approximately three fourths of its own mass within 6 months. Hepatocytes, bile duct epithelial cells, and hepatic progenitor or stem cells maintain the potential to multiply during adult life.63,64 Depending on the severity of the primary injury, liver regeneration may occur by at least two mechanisms.65 In brief, with mild to moderate hepatocellular loss, mature hepatocytes undergo replication. More extensive or massive hepatic necrosis stimulates proliferation of progenitor cells within the periportal region, particularly when necroinflammation occurs at the portal-parenchymal interface. Proliferation of these cells gives rise first to so-called ductular hepatocytes, in which ductular structures containing cuboidal cells and slightly larger cells with mitochondria-rich cytoplasm are present. With time, these cells mature into definitive hepatocytes or cholangiocytes and may repopulate damaged parenchymal regions and bile duct structures, respectively.
Parenchymal regeneration is recognized initially by the twinning of liver cell plates, evident as a double line of hepatocytes with nuclei seemingly running in parallel. Twinning of cell plates may remain for some months after regeneration before new sinusoidal channels develop and the nuclear alignment dissipates.66 If regeneration is recent, the hepatocytes lack lipofuscin, because this pigment accumulates with time in the normal liver. Regeneration also is characterized by increased numbers of binucleate or multinucleate hepatocytes, reflecting replication of nuclear material. Hepatocyte nuclei may be more uniform in size, because anisonucleosis increases with patient age in the normal liver and may not be as evident in regenerating liver. Finally, regeneration imparts a rounded appearance to the expanding contours of residual parenchyma, which is demonstrated with a reticulin stain. To the extent that fibrosis and cell death precede hepatocellular regeneration, a residual viable parenchymal island may stand out in the midst of necroinflammation and developing fibrosis.
It is not until hepatocellular regeneration occurs that the characteristic nodular transformation of cirrhosis becomes manifest. On thickening of the liver cell plates, the parenchyma expands against the constraining fibrous septa and acquires a spherical shape. The hepatocyte plates abutting the fibrous septa become compressed and are bent outward by the less-constrained plates toward the interior of the nodule. Poor regeneration or failure to keep up with the pace of collapse results in variants of cirrhosis in which the feature of rounded contours may not be prominent.
The ultimate size of the nodule is determined, in part, by the anatomic location of the antecedent fibrous septa. If matrix deposition occurs at the acinar level, the resulting nodules will grow out of monoacinar units and will be small. If matrix deposition encompasses many acinar units (multiacinar), the growing nodules may be much larger and will retain components of the preexisting acini, including intact portal tracts.
In some cases of cirrhosis, vast expanses of bile ductules within the fibrous septa coexist with the interspersed hepatocellular nodules. These ductules may occur in cirrhosis of almost any cause and are not necessarily the result of biliary obstruction.67 Hyperplasia of ductules is associated with lengthening and increased tortuosity of existing channels and with extensive sprouting of new channels. This change is reminiscent of the massive proliferation of ductular structures within the hepatic parenchyma, or at the interface between parenchyma and portal tracts, that occurs in massive hepatic necrosis and implicates a proliferation of periportal progenitor cells.63,68,69 With time, these ductular structures may mature into hepatocellular parenchyma or bile ductules. Therefore, the presence of expanses of bile ductules in a cirrhotic liver points toward episodes in the recent past in which there was extensive parenchymal destruction; the ductules represent an intermediate stage of a massive regenerative response.70
Natural History and Reversibility of Cirrhosis
Cirrhosis is traditionally viewed as an end stage in the evolution of many types of chronic liver diseases. However, clinical reports have indicated that on cessation of the injurious process, cirrhosis may reverse, or at least improve, histologically.71–79 Tissue samples from some patients with established cirrhosis have revealed incomplete septal cirrhosis or apparent absence of fibrosis after successful treatment. This evolution has been documented in patients with hemochromatosis, autoimmune hepatitis, Wilson disease, primary biliary cirrhosis, schistosomiasis, extrahepatic biliary obstruction, alcoholic disease, chronic viral hepatitis B or C, or postjejunal bypass steatohepatitis. The concept of reversibility of fibrosis, potentially including patients with a histologic or clinical diagnosis of cirrhosis, is now not only widely accepted but a hoped-for outcome after pharmaceutical treatment of some forms of chronic liver disease, particularly viral hepatitis.11,12
In many different experimental models of cirrhosis reversal, collagen is resorbed within weeks after cessation of injury.27 The mechanism of resorption of fibrous extracellular matrix involves activation of tissue metalloproteinases.80 Likewise, stellate cells and activated portal tract fibroblasts may undergo apoptosis and subside. Fibrosis may even disappear in cases of long-term quiescent cirrhosis. In this situation, the diagnosis of cirrhosis may be made by demonstrating severe paucity of small hepatic and portal veins, even when fibrous septa are highly regressed and not well represented within a biopsy specimen. Indeed, a revised definition of cirrhosis can be made wherein it represents a condition of widespread obliteration of small hepatic veins (i.e., venopenia). This definition is sufficient because if there is severe venopenia, all other histologic features of cirrhosis will ultimately follow.
Therefore, the histologic appearance of cirrhosis depends on the age of accumulated tissue damage and the time of dormancy with an opportunity to resorb fibrous tissue. The presence of PELs is a helpful indicator. If the causative injury is long past, only old PELs will be present. If the causative injury is long past, the liver will contain lesions only in the late stages of repair. New extinction lesions are easily seen in livers with moderate to severe activity. They are recognized as areas of bridging necrosis or of focal intense congestion with atrophy and clusters of apoptotic cells.81 More commonly, especially in low-grade chronic hepatitis, the lesions are recognized as subtle atrophy, sinusoidal dilatation, clusters of apoptotic cells, and approximation of hepatic veins close to portal tracts.
Old extinction lesions predominate in livers in which the primary disease has remitted, either spontaneously or after successful treatment.81 Fibrosis tissue is progressively removed from extinction lesions so that broad septa become delicate and delicate septa become incomplete (“perforated”) or disappear. Thus, micronodular cirrhosis may remodel to macronodular cirrhosis, incomplete septal cirrhosis, or near-normal liver (and be discovered only if there is “noncirrhotic” portal hypertension). The problem of diagnosing regression of cirrhosis is discussed later.
Even with substantial resorption of fibrous septa, restoration of the hepatic architecture to a normal state probably does not occur. Limiting factors are the persistence of vascular abnormalities—that is, outflow obstruction and arterialization (Figs. 50.10 and 50.11). If these vascular factors are sufficient to cause continued hepatocellular injury or interstitial exudation, septa will not resorb, and some degree of cirrhosis or incomplete septal cirrhosis will remain. Arterialization is important because even if hepatic vein outflow is not limiting, elevated sinusoidal pressure prevents the regeneration of obliterated small portal veins so that noncirrhotic portal hypertension may remain.82 Moreover, residual sclerosis in portal tracts may still lead to persistent presinusoidal resistance to splanchnic blood flow, leading to continued clinical evidence of portal hypertension.
The previous description indicates that the equilibrium of injury and repair depends on the local balance of inflow and outflow of blood and on the continued presence (or absence) of the proinflammatory disease environment. This equilibrium is continually affected by activity of the primary disease, congestive hepatopathy, persistence of fibrosis in anatomically strategic locations, and catastrophic events such as portal or hepatic venous obstruction by tumor or thrombosis.
Anatomic Classification and Pathology
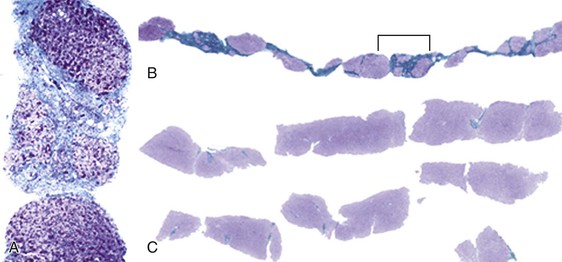
The main macroscopic types of cirrhosis are referred to as micronodular and macronodular. The former consists of nodules mostly smaller than 3 mm in diameter, the latter of nodules mostly larger than 3 mm17,82 (Box 50.2). The term mixed cirrhosis describes livers in which nodules both larger and smaller than 3 mm coexist. The definitions of macronodular and micronodular cirrhosis have been modified over the years, from an original cutoff value of 10 mm (before 1976) to the current WHO cutoff of 3 mm.83,84 This modification was intended to help distinguish alcoholic cirrhosis from other etiologic types. However, since the WHO criteria appeared, hepatitis C cirrhosis has been shown to be mostly micronodular,85 and alcoholic micronodular cirrhosis has been shown to transform to a more macronodular form during inactive disease.86 In addition, the progressive stages of biliary cirrhosis, from incomplete, to macronodular, mixed, and finally micronodular forms of cirrhosis, have been described.87 Therefore, this anatomic classification is not adequate to allow a precise correlation with etiology. Rather, it is clear that the severity of activity and duration of inactivity are important. The evolutionary nature of these anatomic types is shown in Figures 50.12 and 50.13.
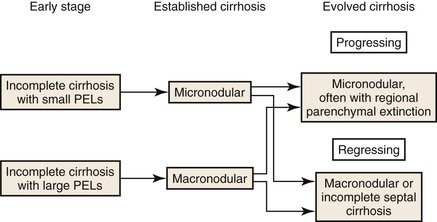
Despite these caveats, certain generalizations can be made. Micronodular cirrhosis is associated with diseases in which there is uniform injury to all acinar units. The result is that most small hepatic veins are damaged, parenchymal extinction extends to involve all portal tracts, and subsequent regenerative nodules are small and devoid of portal tracts. In contrast, livers with macronodular cirrhosis are irregular, with less severe destruction of hepatic veins, so that most regenerative nodules contain some portal tracts and intact hepatic veins. Therefore, based on the single parameter of hepatic vein destruction, micronodular cirrhosis is more severe than macronodular cirrhosis.
Macroscopic Features
Micronodular Cirrhosis
In the early evolution of micronodular cirrhosis, the overall shape and external appearance of the liver may not be greatly altered. The organ weight may be normal or increased, and the left lobe may enlarge disproportionately. Parenchymal nodules may be difficult to discern by observation of the capsule of the liver. With time, organ weight decreases, and the capsular surface may become studded with a myriad of small protruding nodules between the shallow and regular fibrous indentations that define the nodules. On cut section, the nodules are small and uniform but may be difficult to see without slight magnification. As the cirrhotic liver becomes small, the liver becomes progressively firm to the touch, and septa are more wide and prominent. In severe examples, residual small islands of parenchyma appear to float in a sea of fibrous tissue.88
Alcohol abuse is the most frequent cause of micronodular cirrhosis in Europe and North America. However, increasingly common is the cirrhotic outcome of nonalcoholic fatty liver disease. Hepatitis C viral infection, being less prone to bouts of severe necroinflammatory injury, progresses with time from an initial macronodular pattern toward a more micronodular pattern of cirrhosis (unlike hepatitis B viral infection, which remains macronodular). Less common causes for the micronodular pattern of cirrhosis include hemochromatosis, chronic biliary obstruction caused by primary biliary cirrhosis or primary sclerosing cholangitis, drug toxicity, and many types of metabolic diseases of infancy and childhood (see Chapter 54). With chronic extrahepatic venous outflow obstruction (see Chapter 51), the liver also is finely subdivided by fibrous tissue (see Congestive Cirrhosis).
In alcoholic liver disease, the liver may show a diffuse tan or yellow color caused by severe fatty change or, in the setting dominated by alcoholic hepatitis, it may be a characteristic red color. In hemochromatosis, heavy iron deposition imparts a dark reddish-brown color to the liver. Destructive biliary diseases such as primary biliary cirrhosis or primary sclerosing cholangitis, while rendering the liver a deep yellow or green color, impart to the liver capsule a progressive fine septal scarring pattern resembling pigskin. Although any decompensated liver may become yellow or green, it is seldom as deeply pigmented as in patients with a biliary disease. There are rarely any distinguishing macroscopic features in micronodular cirrhosis caused by drug toxicity.
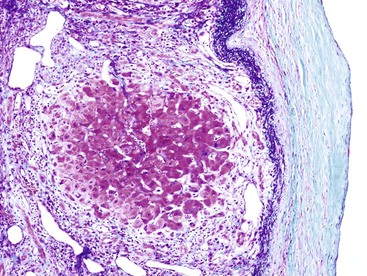
The condition known as cirrhosis with regional parenchymal extinction (also known as confluent hepatic fibrosis) occurs when macroscopic expanses of liver tissue become purely fibrotic (Fig. 50.14).45 This appears to be the natural progression of cirrhosis when there is progressive regional vascular compromise. Regional parenchymal extinction may be 1 to 2 cm in greatest dimension or even much larger. The capsular surface overlying large areas of extinction may be finely granular and wrinkled, rendering a pigskin appearance.
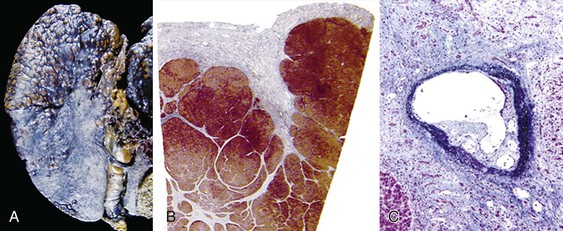
Macronodular Cirrhosis
Liver size and shape in macronodular cirrhosis are highly variable. As with micronodular cirrhosis, the early stages are associated with increased liver weight. The parenchyma exhibits large, bulging nodules separated by fibrous bands that vary considerably in width, but the liver generally retains its overall anatomic shape. Alternatively, the nodules may be more uniform in size (but larger than in micronodular cirrhosis), with intervening marked grooving and retraction that is especially evident on examination of the capsular surface. The more irregular pattern of macronodular cirrhosis reflects the capricious nature of necroinflammatory injury imparted by the causative diseases, which include viral hepatitis (e.g., hepatitis B) and autoimmune hepatitis.
Mixed Cirrhosis
Mixed cirrhosis is not rigorously defined and hence is not a very useful term. It can be interpreted as many nodules with a diameter near the 3-mm cutoff or as the presence of many nodules larger than 3 mm combined with many nodules smaller than 3 mm. The former appearance is frequent in any type of inactive, formerly micronodular cirrhosis that is undergoing evolution to macronodular cirrhosis through gradual enlargement of the nodules while the fibrous septa are resorbed and the mechanical constraint is eased. The latter appearance is often found in primary biliary cirrhosis and in primary sclerosing cholangitis, in which there is a distinction between nodules with duct drainage and those without it. In addition, the progressive parenchymal extinction of evolving macronodular cirrhosis can further subdivide parenchymal nodules, reducing their size. Therefore, a dogmatic distinction between the causation of micronodular and that of macronodular cirrhosis is unwise.
Microscopic Features
The microscopic features of cirrhosis are more easily generalized than the macroscopic features. As expected, fibrous subdivision of the liver parenchyma with isolation of parenchymal islands is the sine qua non for a diagnosis of cirrhosis. However, application of these criteria brings its own challenges, particularly on liver biopsy.
Evolution of Cirrhosis
Before discussing the histology of cirrhosis, further comments must be made on the natural history of this condition. The histologic appearance of cirrhosis depends on the age of the accumulated extinction lesions. If the causative injury continues, new PELs will coexist with the old. Parenchymal extinction, usually accompanied by inflammation, constitutes activity; “active cirrhosis” indicates a cirrhotic liver with continued destruction of residual tissue. Conversely, if the causative injury is remote at the time of examination, the liver will contain only lesions in late stages of repair, and the cirrhosis is considered quiescent, or “inactive.”
New extinction lesions are easily identified as areas of bridging necrosis or focal intense congestion with atrophy and clusters of apoptotic cells. Moreover, careful examination of the vascular channels—both septal and intraparenchymal—may reveal organized thrombi, the result of chaotic and sluggish blood flow, loss of anticoagulant function, and the prothrombotic effects of sepsis and cholestasis. Thrombosis of medium- or large-sized hepatic veins causes large regions of extinction, leading to marked irregularity of the cirrhotic liver.45 Essentially by definition, ongoing parenchymal extinction is seen only in livers with moderate to severe activity.
Old lesions of parenchymal extinction predominate in livers in which the primary disease has remitted, either spontaneously or after successful treatment.71 Fibrosis is progressively removed from extinction lesions so that broad septa become delicate, and delicate septa become incomplete and disappear. Put differently, micronodular cirrhosis may become remodeled to macronodular cirrhosis, thence to incomplete septal cirrhosis, and eventually to a near-normal appearance if given enough time. Portal tracts released from septa are seen as remnants lacking portal veins. Indeed, cirrhotic nodules may eventually be fed entirely by arteries. Arteriovenous shunts can be recognized by focal regions of sinusoidal congestion and hepatocellular atrophy, usually marked by CD34-positive sinusoidal endothelial cells.71
Micronodular Cirrhosis
Normal liver acini measure approximately 1 mm in diameter, and because micronodular cirrhosis arises from subdivision of parenchymal acini, cirrhotic nodules may be less than 1 mm in diameter. The tiny nodules enlarge as they regenerate but are severely confined by the surrounding fibrous tissue. Almost every acinus is affected, and in most cases, no anatomically intact acini are evident. Fibrous septa connect the smallest portal tracts to their adjacent terminal hepatic venules. As a result, zones 1, 2, and 3 of the acinus are transected by fibrovascular septa; regenerative nodules may form concurrently or subsequent to this fibrosing event. Fibrosis may be dominant in one zone—around the portal tracts (zone 1) in hemochromatosis, or around the terminal hepatic venules (zone 3) in alcoholic liver disease—but cirrhosis has not developed if the fibrosis is restricted only to that zone.
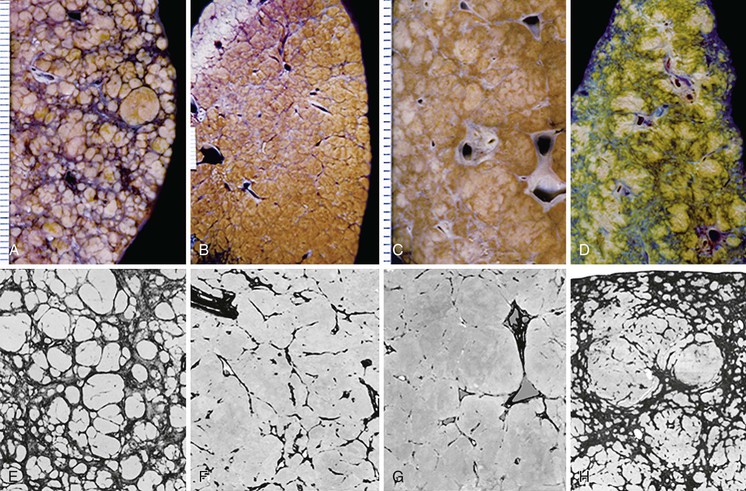
The characteristic feature of micronodular cirrhosis is deposition of fibrous septa along the sinusoidal channels, connecting portal tracts to terminal hepatic veins. Frequently, multiple adjacent sinusoids exhibit fibrosis; this is a characteristic feature of evolving cirrhosis in both alcohol- and drug-induced micronodular cirrhosis. These various manifestations of micronodular cirrhosis are illustrated in Figure 50.15.
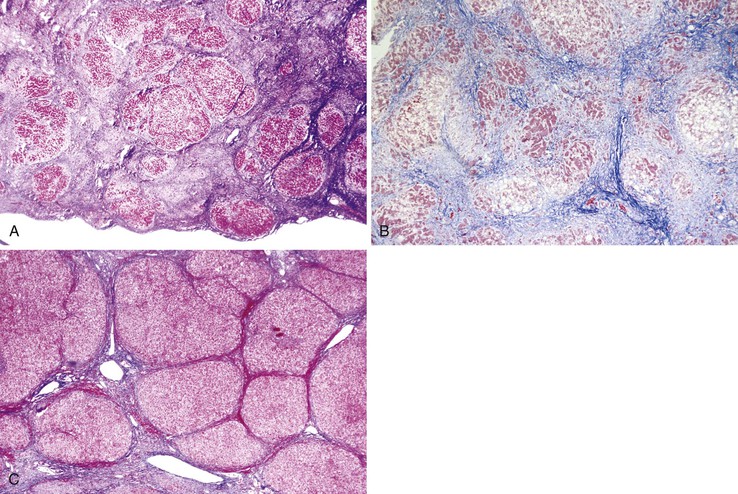
Micronodular cirrhosis seems to be most commonly associated with diseases in which there is a uniform and generalized effect of a hepatotoxic agent or metabolic derangement affecting the smallest parenchymal units in the liver. For example, in alcoholic liver disease every acinus has been regularly exposed to high levels of alcohol. In cirrhosis arising in the setting of severe alcoholic hepatitis, the rate of fibrous tissue deposition can be so rapid (during weeks to months) as to render nodular regeneration almost impossible. In this setting, extensive hepatocellular degeneration with steatosis, ballooning, and Mallory body formation is accompanied by a mixed neutrophilic and mononuclear pattern of inflammation. Nodule formation may be minimal, yet the liver is transformed into a densely fibrotic organ with fibrous septa seeming to traverse almost every sinusoid. This extreme end of micronodular cirrhosis begs the question of whether the definition of cirrhosis is fulfilled, yet few will argue that such a fibrotic liver is not cirrhotic. Alternatively, a smoldering form of alcoholic hepatitis may proceed to a more mixed micronodular-macronodular pattern of cirrhosis, possibly because of a more portal-based pattern of fibrous tissue deposition.
The fibrous septa that connect the smallest portal tracts to their adjacent terminal hepatic venules are devoid of portal tracts. In three dimensions, the fibrous septa are actually sheets of fibrous tissue rich in blood vessels.89 Most small portal and hepatic veins are obliterated. In advanced disease, many medium hepatic veins are also thickened or obstructed,47,60–62 attesting to the role of vascular injury in the pathogenesis of cirrhosis.
In cirrhosis that has long been quiescent, many regressive changes are seen, representing the hepatic repair complex. These changes include thin and incomplete septa, spurs of collagen attached to portal tracts, septa split by ingrowth of hepatocytes, and small buds of hepatocytes and cytokeratin 7 (CK7)-positive ductular cells in broader septa. Sinusoidal endothelium is frequently CD34 positive.71 A feature invoking prior injury is close approximation of portal tracts and terminal hepatic veins, either with or without tethering delicate fibrous septa. This is the residua of the PEL.
Macronodular Cirrhosis
The histology of macronodular cirrhosis is highly variable, particularly when evaluated by percutaneous liver biopsy (see later discussion). This pattern exhibits large nodules delimited by septa in which multiple hepatic acini are incorporated into single nodules. During the early to intermediate stages of evolution, residual portal tracts and portal tract/hepatic vein acinar units may be evident (Fig. 50.16). Cell plates within the multiacinar nodules are often single, with little evidence of twinning, but do not show the regular radial orientation that is present between portal tracts and hepatic veins in normal acini. The abnormal cell plate patterns probably reflect altered blood flow through the parenchyma, as discussed earlier (see Pathogenesis). The hepatic veins are often dilated, giving the impression that they are increased in number. Although this finding cannot be regarded as diagnostic, the possibility of macronodular cirrhosis must be considered when abnormal hepatocellular plate patterns and an apparent excess of veins are present in a needle liver biopsy that does not contain identifiable portal tracts. The presence of identifiable portal tracts correlates inversely with sinusoidal pressure and therefore is a marker of a milder form of cirrhosis.90
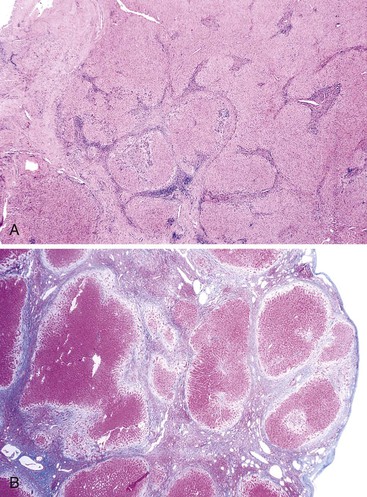
Unusual Variants
Incomplete Septal Cirrhosis
Incomplete septal cirrhosis is a highly regressed form of cirrhosis that is often associated with portal hypertension but exhibits normal hepatocellular function71,91,92 (see Fig. 50.12). Portal vein thrombosis is a complication that leads to portal hypertension in those cases that would otherwise escape clinical attention. Macroscopically, the septa are usually invisible, but variation in the color of the parenchyma on cut section demonstrates the presence of nodules. The liver may be without significant distortion or may exhibit residual bulging nodules. Because healed portal vein thrombosis is often present, gross examination should include consideration of this possibility.
Microscopically, slender fibrovascular septa extend from portal tracts into the parenchyma but often do not connect with other portal tracts or hepatic veins. These septa demarcate large, rather inconspicuous nodules (see Fig. 50.13). Intrasinusoidal collagen away from the septa is not obviously increased, and there is little evidence of hepatocellular damage or inflammation. Portal tracts are variably attenuated, so that venous channels appear relatively increased. This abnormal architecture is present throughout the liver, because there is a variable mixture of thickened hepatocellular plates, dilated sinusoids, and compression of sinusoids between hyperplastic plates.91 The plate pattern is disorganized, with irregular orientation of plates to portal tracts and terminal hepatic veins. Histologic indicators of the original injurious process are usually absent.
In a patient with portal hypertension undergoing liver biopsy, incomplete septal cirrhosis is difficult to distinguish from hepatoportal sclerosis and nodular regenerative hyperplasia. When large expanses of liver are available for examination, incomplete septal cirrhosis exhibits fibrous septa extending out from portal tracts and disorganized plate architecture, both of which are absent in hepatoportal sclerosis. The chief distinctions between incomplete septal cirrhosis and nodular regenerative hyperplasia are subtle parenchymal nodules; in the former condition, these are delineated by incomplete, delicate fibrous septa that lack obvious spherical features, whereas the latter exhibits a complete absence of parenchymal fibrous tissue and the presence of obvious spherical nodules separated by curvilinear compressed hepatocyte plates.93 In every instance, a reticulin stain is very helpful in assessing parenchymal architecture, in addition to the obvious need for a trichrome stain for collagen.
Postnecrotic Cirrhosis
Postnecrotic cirrhosis is the fibrotic stage of severe acute hepatitis; it occurs in association with large contiguous regions of hepatocyte extinction and lesser regions of regenerative tissue.94 The initial massive hepatic necrosis (Fig. 50.17) may subdivide the liver in a very coarse macroscopic fashion, leading to a grossly misshapen liver with large regenerative regions of liver parenchyma separated by very broad regions of parenchymal scar. Because many of these patients suffer subacute hepatic failure, these livers are usually severely cholestatic even though the hepatitis activity may have resolved. The degree of fibrosis depends on the time course. If the patient dies or undergoes transplantation before complete evolution of the scarring process, nodules with expanding contours may be less evident and fibrosis may not be mature, leading to difficulty in classifying this as “cirrhosis” versus late acute hepatitis with regenerative features. The term evolving cirrhosis may suffice for these cases.
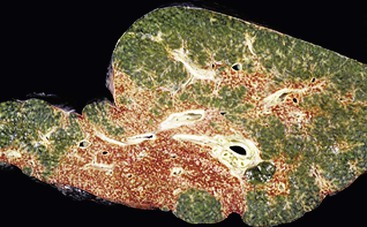
Differential Diagnostic Considerations
Lobar Hypertrophy and Atrophy
In many cirrhotic livers, the left lobe is relatively enlarged compared with the right. In contrast, in patients with Budd-Chiari syndrome (see Chapter 51), the caudate lobe veins are frequently less involved with thrombus, which allows hypertrophy of that lobe.95 This latter situation is not cirrhosis. In addition, regional compromise of the hepatic vessels, as from hilar malignancy, may lead to atrophy of a major hepatic lobe, frequently the left lobe.96 This also is not cirrhosis.
Large Nodules in the Cirrhotic Liver
Occasionally, one encounters hepatocellular nodules substantially larger than those in the background liver. These may be large regenerative nodules, dysplastic nodules, or hepatocellular carcinoma.97,98 Given that malignant transformation can occur in nodules smaller than 0.5 cm in diameter, sectioning at 0.3- to 0.5-cm intervals is requisite for adequate gross examination of the cirrhotic liver. Macroscopic features characteristic of a regenerative nodule are color and texture similar to those of other cirrhotic nodules and minimal bulging of the cut surface. Dysplastic nodules often have a softer texture, paler color, and bulging cut section. Early hepatocellular carcinomas are usually ill-defined but are otherwise similar to dysplastic nodules. Early hepatocellular carcinoma may arise within a dysplastic nodule, giving a “nodule-in-nodule” appearance. Moderately differentiated hepatocellular carcinoma is usually well demarcated, with a pale color, but may be variegated when necrosis is present. Metastatic hepatocellular carcinoma in liver often consists of several small and pale nodules, sometimes visible as a treelike structure within the portal vein. These nodules are usually satellites of the primary lesion in the same lobe.
Nodular Regenerative Hyperplasia
Nodular regenerative hyperplasia is characterized by small nodules separated by regions of atrophy rather than septa.98 It is a response to microvascular vascular derangement, as described in Chapter 51. Cirrhotic livers may show similar hypertrophic/atrophic variation within nodules and might be mistaken for nodular regenerative hyperplasia if a biopsy fails to sample sufficient fibrous tissue or otherwise demonstrate the classic features of cirrhosis.81
Hepar Lobatum
Focal fibrous septation can cause deep linear clefts in the liver capsule such that the liver acquires an irregular shape or hepar lobatum, also known as “potato liver” (Fig. 50.18). This anomaly is caused by obliteration of large hepatic veins and confluent parenchymal necrosis, as occurs in adult-onset syphilis, metastatic breast carcinoma, and Hodgkin disease.99 This process is not true cirrhosis, because the large surviving regions of tissue may be normal. Supernumerary hepatic lobes, a congenital defect, can have a similar appearance.
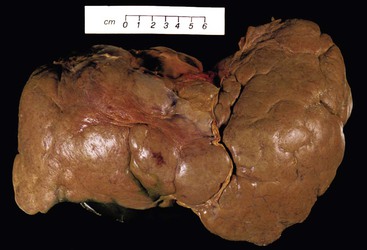
Congenital Hepatic Fibrosis
Focal fibrous septation also is a feature of congenital hepatic fibrosis (see Chapter 54). The liver parenchyma may or may not become nodular. The fibrous septa are broad, with dense collagen bands. The diagnostic feature for congenital hepatic fibrosis is dilated marginal ductal remnants, aligned longitudinally along the septa–parenchyma interface. These do not represent the ductular reaction of an active cirrhosis; rather, they are embryonic remnants of the developing biliary tree (the “ductal plate”). The disease usually manifests with portal hypertension and normal liver function. Portal vein thrombosis occurs in half of the patients. Cholangitis may also occur.
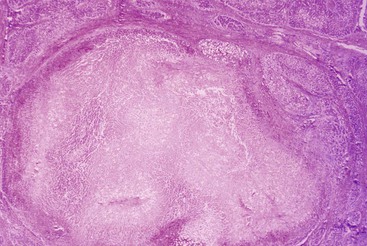
Infarction of Nodules versus Congestive Necrosis of Nodules
Necrosis of cirrhotic nodules may occur in association with systemic hypotension, typically after a variceal hemorrhage. This finding is often a response to radiofrequency ablation or alcohol injection directed at neoplasms. These lesions, characterized by diffuse and uniform coagulative necrosis of established cirrhotic nodules (Fig. 50.19), should be distinguished from congestive necrosis of nodules as part of the development of progressive regional parenchymal extinction and fibrosis that occurs in severe cirrhotic livers. In the latter situation there is patchy congestive sinusoidal injury in various stages of organization within subregions of a cirrhotic nodule.
Portal Vein Thrombosis
Portal vein thrombosis is a frequent complication of cirrhosis; it is found in approximately 10% of livers removed at transplantation.100–102 The healed phase of portal vein thrombosis is easily confused with congestive portal venopathy (see Chapter 51). Portal vein thrombosis is occasionally seen when carcinoma invades the vascular tree.103,104
Patent Paraumbilical Veins
In patients with severe portal hypertension, there is often spontaneous opening of collateral veins within the round ligament.105,106 These paraumbilical veins connect the umbilical portion of the left portal vein to the umbilicus, where caput medusae and a bruit may be detected on physical examination. Histologic inspection of the round ligament will reveal these patent channels when one is evaluating a cirrhotic liver.
Capsular Fibrosis
Capsular fibrous thickening occurs in the liver and spleen when there is chronic exudation or transudation, especially in patients with cirrhosis or severe congestive heart failure.107,108 This condition is also known as sugar-coating (Zukerguss). The capsule may achieve a thickness of as much as several millimeters, with a tough, cartilage-like consistency.
Etiology
Here we give consideration to the diseases that give rise to cirrhosis (Box 50.3), with the goal of highlighting histologic features that may be identified in cirrhotic livers. However, a cautionary note is in order, because most often the histologic features of the originating disease are obliterated or are long past by the time of liver examination. This applies not only to examination of the whole organ (at autopsy or liver transplantation) but also to examination of liver biopsy tissue from a cirrhotic liver. Nevertheless, in many instances, the pathologist’s opinion is sought to determine the cause of the cirrhosis. With that in mind, the following considerations pertain.
Chronic Hepatitis
The most frequent forms of chronic hepatitis are hepatitis B, hepatitis C, and autoimmune hepatitis. These diseases are usually diagnosed with the help of a variety of serologic tests. Histologically, hepatitis B may exhibit “ground glass” cytoplasmic inclusions within hepatocytes; immunohistochemical staining for hepatitis B surface and core antigens is more sensitive. Detection of hepatitis C virions in liver tissue is possible but is not a routine technique. There is no immunohistochemical test for autoimmune hepatitis.
Therefore, the pathologist must still perceive potential histologic features of these chronic hepatitides, despite the difficulty in distinguishing one from another under the best of circumstances. Each may exhibit a portal lymphoplasmacytic infiltrate and diffuse parenchymal lymphocytosis with acidophilic bodies. However, untreated autoimmune hepatitis is usually more active than the other types; along with the requisite plasmacytic features, it also shows extensive necroinflammatory damage of the portal tract–parenchyma interface and regenerative ductular reaction. The pattern of fibrosis also reflects the activity of the autoimmune disease. Both hepatitis B infection and autoimmune hepatitis may produce large regions of extinction that can lead to a pattern of cirrhosis with large regenerative nodules and macronodular cirrhosis. In contrast, as a low-grade necroinflammatory disease, hepatitis C usually exhibits only small regions of extinction, which more often leads to smaller nodules ab initio. These nodules are not so consistently uniform as to necessarily constitute micronodular cirrhosis, but the tendency of hepatitis C cirrhosis is toward smaller nodules, when compared with hepatitis B and autoimmune hepatitis.
Hepatitis C is further suggested by the presence of portal tract lymphoid aggregates and patchy macrovesicular steatosis. However, these findings are not specific. In the former case, portal tract lymphocytes still accumulate with time in hepatitis B and autoimmune hepatitis, even if they do not seem to organize into structured aggregates. In the latter case, obesity and alcoholic disease may be confounding factors generating parenchymal steatosis in this and other diseases. The possibility of coinfection with hepatitis B and C viruses must also be kept in mind. The cirrhosis of autoimmune hepatitis may be active or quiescent, so serologic findings are required in most cases. The presence of numerous plasma cells supports a diagnosis of autoimmune hepatitis, but this finding is not required and is also seen in the more active examples of chronic viral hepatitis. Alcoholic and nonalcoholic steatohepatitis are often associated with a mild portal lymphocytosis. Whenever such lymphocytosis is prominent, it is prudent to suggest that serologic studies for viral and autoimmune disease be performed.
Fatty Liver Disease
Fatty liver disease may be caused by alcohol abuse or states of insulin excess, as in obesity or type 2 diabetes mellitus. Non–alcohol-associated cases are collectively known as nonalcoholic steatohepatitis or nonalcoholic fatty liver disease.109
Fatty liver disease is characterized by large-droplet steatosis, either with or without evidence of activity in the form of steatohepatitis. Steatohepatitis is defined by the presence of ballooning necrosis. Mallory bodies and neutrophils are found in more active cases. The presence of pigmented macrophages scattered in zone 3 is often an indication of recent necrosis and supports a diagnosis of steatohepatitis, even if ballooned hepatocytes are not readily evident.
Alcoholic and nonalcoholic fatty liver disease are often identical in histologic appearance.110 However, severe activity with numerous Mallory bodies and neutrophils is seen more often in alcoholic disease. The diagnosis of steatohepatitis requires large-droplet steatosis and some evidence of liver cell injury, such as ballooning or pigmented macrophages, but Mallory bodies and neutrophils are not required. Minimal steatohepatitis often occurs in patients with chronic hepatitis C, so the cause of elevated aminotransferases may be difficult to determine except by a trial of dietary restraint.
Simple steatosis (without steatohepatitis) on biopsy usually does not progress to cirrhosis.111 However, there are exceptions, probably because features of minimal steatohepatitis may be missed on biopsy, and metabolic derangements in the patient may become more severe with time, leading to steatohepatitis. Fibrosis develops in steatohepatitis with deposition of delicate fibers in the walls of zone 3 sinusoids. When small hepatic veins are obliterated during this process, there is focal parenchymal extinction with approximation of portal tracts and adjacent hepatic veins. Collagen deposition varies from minimal to severe.47
Such sinusoidal (pericellular) fibrosis is often considered suggestive of fatty liver disease. However, this is not entirely reliable. Sinusoidal fibrosis can occur by two mechanisms: as a local response to activation of hepatic stellate cells, and in the repair of quiescent chronic liver disease by the splitting and repopulation of broad fibrous septa. The former condition occurs in early fatty liver disease but also late in cases of severely congested cirrhotic nodules of any cause. The latter mechanism can be seen in any cirrhotic liver undergoing repair and recovery.
As the liver becomes cirrhotic, the presence of steatotic droplets in hepatocytes may substantially decrease. When the cirrhotic liver is examined by microscopy, scattered residual features of macrovesicular steatosis may be the only footprints left of prior steatotic liver disease—alcoholic or nonalcoholic. In end-stage cirrhosis caused by fatty liver disease, steatosis and steatohepatitis may be focal or completely absent, so that biopsies may not be able to confirm the diagnosis.
Mallory bodies are not specific for fatty liver disease, because they are seen in chronic cholestasis and copper-overload states as well.
Chronic Biliary Diseases in Adults
Chronic biliary diseases are characterized by chronic retention of biliary products. Although they usually are caused by duct obstruction, they may also be caused by hepatocellular defects in bile secretion, as seen with cholestatic drug reactions or familial transport defects.112 In adults, chronic duct obstruction is most often caused by primary biliary cirrhosis or primary sclerosing cholangitis.113 Rarer biliary causes of cirrhosis are chronic graft rejection and graft-versus-host disease. Although the clinical settings for the latter are obvious, distinguishing chronic rejection from recurrent biliary disease in liver transplants can be quite difficult (see Chapter 52).
In primary biliary cirrhosis, there is inflammatory destruction of ducts that mostly measure less than 40 µm in diameter. Although these lesions can easily be seen in needle biopsies, it is the exception rather than the rule that a liver biopsy succeeds in sampling diagnostic duct lesions. Indeed, by the time the liver is cirrhotic, these lesions are long gone. In primary sclerosing cholangitis, there is inflammation and concentric periductal fibrosis around ducts larger than 200 µm in diameter, and these lesions are seldom observed in small biopsy specimens. In either disease, when diagnostic duct lesions are not available for examination, chronic biliary disease may be suggested by the presence of a prominent fibrous expansion of the portal tracts accompanied by ductular proliferation, portal edema, and neutrophilic infiltration. A portal mononuclear infiltrate may be prominent, especially in primary biliary cirrhosis. Importantly, with evolution of portal tract fibrosis from any cause, including chronic hepatitis, activation of peribiliary myofibroblasts may impart a periductal “onionskin” pattern of fibrosis. Care must therefore be taken to observe genuine ductal withering before invoking a diagnosis of primary sclerosing cholangitis (see Chapter 47).
In the late cirrhotic stage of any biliary or nonbiliary disease, there may be swelling of periportal hepatocytes (feathery degeneration), often with periseptal Mallory bodies, caused by obstruction to bile outflow. By this time, the phases of inflammatory bile duct destruction in primary biliary cirrhosis and primary sclerosing cholangitis are complete, and therefore diagnostic histologic lesions are not usually present. Instead, only nonspecific features of biliary obstruction may be seen, such as septal edema and neutrophilic inflammation, bile ductular proliferation (although this, too, may be sparse), ductular and hepatocellular cholestasis, degenerative swelling of periseptal hepatocytes (cholate stasis), Mallory body formation and copper retention in periseptal hepatocytes, swelling of periportal hepatocytes, liver cell rosettes, feathery degeneration of parenchymal hepatocytes, and clusters of bile-stained foamy macrophages within the nodules.
Biliary fibrosis is topographically variable, especially when larger ducts are involved, as in primary sclerosing cholangitis and cystic fibrosis. Whole liver segments may undergo extinction or be spared, depending on the distribution of duct obstruction.
The differential diagnosis of chronic biliary disease includes extrahepatic obstruction, hepatolithiasis, choledochal cyst, congenital hepatic fibrosis, oriental cholangiohepatitis (liver fluke disease), cystic fibrosis, and a variety of transport disorders. Onionskin fibrosis with fibrous obliteration of medium-sized ducts is suggestive of primary sclerosing cholangitis, but this may be seen in most diseases with obstruction of medium-sized and large ducts. Peribiliary fibrosis without duct obliteration can occur in any evolving cirrhosis, but a well-delineated fibrous cord in a larger portal tract is a characteristic residuum of primary sclerosing cholangitis (Fig. 50.20). Congenital hepatic fibrosis seldom manifests as cholestasis and is characterized by multiple dilated, ductlike structures in almost all portal tracts. Drug reactions, especially those caused by various antibiotics and angiotensin-converting enzyme inhibitors, can have clinical features similar to those seen in large duct obstruction; observation of necrosis in small ducts in a biopsy specimen is helpful in this differential diagnosis. Cytokeratin 7 (CK-7) stain is also helpful in this assessment. Cirrhosis rarely ensues if drug use is discontinued.
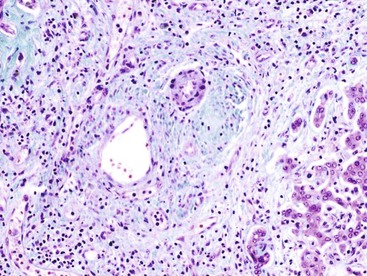
A frequent error is to assume that the presence of visible bile in a biopsy specimen defines biliary disease. Visible bile in the tissue is almost never seen in primary biliary cirrhosis and primary sclerosing cholangitis until there is late cirrhosis with functional decompensation. The finding of bile-stained hepatocytes, bile-stained macrophages, or canalicular bile plugs in a liver specimen without cirrhosis suggests an acute cholestatic disorder, usually a drug reaction or recent complete obstruction of the external biliary tract.
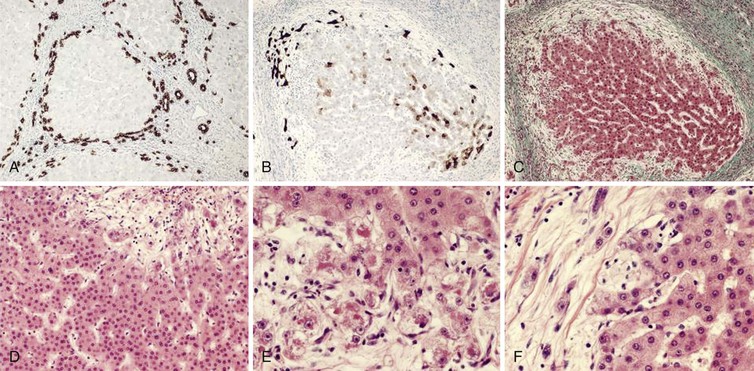
In most cirrhotic livers, there are regenerative bile ductules at the margins of nodules and within septa (Fig. 50.21). This pattern may occur in cirrhosis of almost any cause and is not necessarily indicative of chronic biliary obstruction.70 A similar type of ductular proliferation occurs after massive hepatic necrosis and implies a proliferation of periportal progenitor cells.63,68,69 Proliferating ductular structures are often admixed with small clusters of hepatocytes to form “buds” that appear to enlarge into small cirrhotic nodules.64,71 Therefore, identification of proliferating bile ductules, per se, does not directly implicate chronic biliary disease. Lastly, prominent regenerative changes in cholestatic liver disease may result in thickening of the hepatocellular plates and an increased nucleus-to-cytoplasm ratio of the hepatocytes, features that may be mistaken for dysplasia or malignancy (see Fig. 50.21, D).
Biliary Diseases in Pediatric Patients
Biliary Atresia
The histologic features of biliary atresia in percutaneous liver biopsy specimens, in comparison with neonatal hepatitis, are discussed in Chapter 54. Here we discuss aspects of cirrhosis arising from biliary atresia. Explanted livers demonstrate progressive findings that occur with increasing age in children. These findings include progressive portal and periportal fibrosis and ballooning degeneration of hepatocytes at the portal vein–parenchyma interface, with copper accumulation, Mallory bodies, and bile lakes in severe examples. Although the inciting injury is obliteration of the extrahepatic biliary tree, paucity of intrahepatic bile ducts develops by 4 to 5 months of age, and a biliary pattern of cirrhosis occurs by 8 or 9 months of age. The rapidity of progression to cirrhosis depends on the severity of large duct obstruction remaining after Kasai portoenterostomy.
On occasion, in a liver biopsy specimen from a cholestatic infant, the portal tracts do not contain interlobular bile ducts but rather have residual ductal plate remnants concentrically placed around the periphery of portal tracts, in the absence of ductular proliferation.114–118 Hypertrophic hepatic arterial elements may be present toward the center of portal tracts, embedded in a robust fibrous mesenchyme. Cirrhosis may evolve very rapidly in these young children, within the first months of life. Recognition of these histologic features in a percutaneous liver biopsy specimen, even in the absence of ductular proliferation, may justify consideration of biliary atresia. However, paucity of bile ducts may instead reflect Alagille syndrome, and active bile duct injury or bile duct paucity may be the result of α1-antitrypsin deficiency. Great care must therefore be exercised in interpreting the histology, even in a pediatric liver biopsy exhibiting substantial portal tract fibrosis.
Cystic Fibrosis
In cystic fibrosis, thick, mucus-laden bile focally blocks bile flow, leading to focal biliary fibrosis. Cirrhosis develops in fewer than 10% of patients with cystic fibrosis.119 The pattern of cirrhosis is irregular, showing focal loss of bile ducts accompanied by cholestasis and fibrosis in the obstructed regions.120,121 There may be large duct strictures resembling primary sclerosing cholangitis. Classically, there is inspissated mucinous material in the ducts, but this may be minimal and difficult to demonstrate.
Metal Overload States
Iron and copper are deposited in the liver in hemochromatosis and Wilson disease, respectively (see Chapter 54). However, both iron and copper may accumulate in severe cirrhosis of any etiology, and their presence is not diagnostic for hemochromatosis or Wilson disease. The pattern of fibrosis is not diagnostically useful in these conditions. Although presumptive diagnoses can be made in the appropriate clinical setting, definitive diagnosis rests on molecular analysis of the genome.122,123
Iron
Iron overload in hemochromatosis is genetically determined but also is influenced by gender, oral ingestion, ineffective hematopoiesis (including inherited conditions such as thalassemia), hemolysis, transfusion, and portosystemic shunting.124 Low to moderate levels of iron may accumulate in cirrhosis in the absence of iron overload, resulting from portosystemic shunting.125,126 Iron overload is a well-documented phenomenon in alcoholic cirrhosis127 and is attributable to redistribution of iron stores from other sites in the body.128 In general, a limited degree of hemosiderosis is common in nonbiliary forms of cirrhosis, but it is uncommon in biliary forms of cirrhosis.129
Iron, in the form of hemosiderin, may be detected in the liver by histochemistry. Quantitative iron assay is rarely performed for diagnostic purposes, because there is an acceptable correlation with histochemistry. Genetic testing is less cumbersome than chemical assays and more diagnostically precise.
Iron accumulation may be heterogeneous in cirrhotic livers of hereditary hemochromatosis because regenerating, dysplastic, or malignant hepatocytes will have had less time to accumulate intracellular iron.130 Paradoxically, some dysplastic nodules selectively accumulate stainable iron in otherwise iron-free cirrhotic livers.131
Copper
Wilson Disease
The most common copper overload state is Wilson disease, in which copper deposition occurs in the liver, eyes, and basal ganglia. Copper in the cirrhotic liver is distributed throughout the nodules. In contrast, in biliary cirrhosis, copper accumulates in paraseptal hepatocytes.132 Because not all cirrhotic nodules in Wilson disease are affected, the diagnosis may be missed even with large specimens. Similarly, the copper stain is often negative, so chemical assays on fresh or paraffin-embedded liver are highly recommended if Wilson disease is suspected clinically. Hepatocellular degenerative features include mild steatosis, Mallory bodies, ballooning degeneration, and glycogenated hepatocellular nuclei (Fig. 50.22). These histologic features are often present in obesity and other toxic conditions as well, so care must be taken in interpreting the histology. Hemosiderin deposition may be caused by depressed iron clearance from hepatocytes after therapy that depletes hepatic copper.133
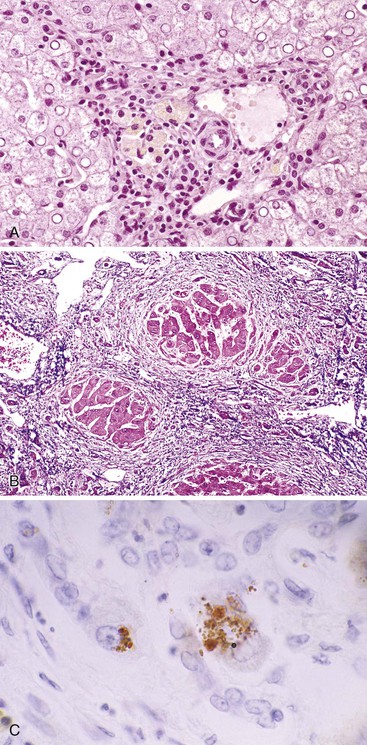
Non-Wilsonian Copper Toxicosis
Cirrhosis may develop in infants with marked hepatic copper overload. Most of these patients live in the Indian subcontinent (Indian childhood cirrhosis) or in Austria (Tyrolean copper toxicosis).134,135 Rare cases have been reported from other countries. The Indian form of the disease has been attributed to the use of brass and copper vessels for storage of milk. In Austria, the disease has been associated with acidic well water delivered to homes by copper pipes and also to the use of unlined copper vessels for food storage. However, this disease has virtually disappeared since these environmental sources have been removed. A genetic predisposition has been suggested but not yet proved.
Liver tissue from this group of patients typically shows hepatocellular ballooning, prominent Mallory bodies, and marked pericellular fibrosis, with progression to micronodular cirrhosis. The appearance is similar to that of alcoholic steatohepatitis but without the steatosis component. A prominent feature is marked accumulation of copper, as detected by copper and orcein stains. Copper chelation therapy has caused remarkable regression of fibrosis and cirrhosis in some cases.136,137
Congestive Cirrhosis
Although there is a congestive element in the genesis of all types of cirrhosis, the term congestive cirrhosis refers only to livers in which the initiating lesion is obstruction of medium- to large-sized hepatic veins, the vena cava, or the heart, as in hepatic vein thrombosis or congestive heart failure (see Chapter 51). In these conditions, congestive features are usually evident at the sinusoidal level, although chronic lesions may remodel and recanalize, making the heritage of large-vessel obstruction difficult to document without access to the whole liver for histologic examination.
Congestive cirrhosis is characterized by venocentric cirrhosis, also known as reversed-nodularity cirrhosis (Fig. 50.23).138 This pattern is caused by dominant hepatic vein outflow obstruction with relatively intact portal veins that serve as outflow tracts. In this situation, hepatic vein to hepatic vein fibrosis results in nodules composed of portal tracts in the center and hepatic veins at the periphery within the fibrous septa. If portal veins also become obstructed because of congestive venopathy or thrombosis, the nodules lose their outflow tract and undergo congestive injury and eventually hepatocyte extinction, leading to fibrous septation that bridges portal tracts and hepatic veins together. This results in a venoportal pattern of septation.
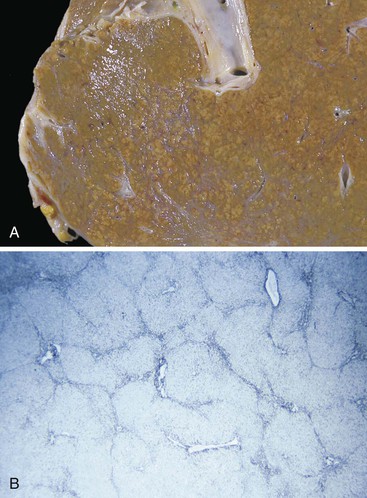
Congestive heart failure, by itself, usually causes only sinusoidal dilatation and mild parenchymal atrophy. Livers in this condition develop parenchymal extinction and fibrous septa only when there is additional obliteration of hepatic and portal veins.139 Venous lesions typically occur in an irregular fashion, making congestive fibrosis quite variable from one region to another. Severe cirrhosis is seldom, if ever, attributed to congestive failure alone. Other causes should always be considered as cofactors, including hepatic vein thrombosis.
In Budd-Chiari syndrome, hepatic vein thrombosis leads to a more severe degree of outflow obstruction than occurs with congestive heart failure alone. This greater degree of obstruction leads to more severe parenchymal extinction. The histology is otherwise similar to that of severe congestive heart failure. Identification of thrombus material within the hepatic veins histologically or on imaging studies is the only certain way to differentiate these two conditions (see Chapter 51).
Sinusoidal obstruction syndrome (SOS) is a distinct cause of congestive hepatic damage that is accompanied by occlusion of the smallest tributaries of the hepatic venous system (see Chapter 51). It is most commonly associated with toxic induction therapy before bone marrow transplantation but may develop from other toxic causes, such as exposure to the alkaloids contained in certain herbal remedies. If the patient survives the initial acute occlusive sequence, there may be brisk and severe deposition of fibrous tissue within the parenchymal sinusoids. Within a few months, the liver may be transformed into a fibrotic organ, and in patients who survive for years, a form of cirrhosis without specific features develops.
Drug-Induced Cirrhosis
Drugs rarely cause cirrhosis. Most drugs cause clinically evident “acute” disease, so that drug exposure is terminated before cirrhosis can develop. However, some drugs such as methotrexate, oxyphenisatin, and amiodarone cause asymptomatic low-grade injury that can lead to cirrhosis. Cofactors such as alcohol and nonalcoholic steatohepatitis are important in many of those patients in whom cirrhosis develops. Severe drug-induced subacute or chronic hepatitis can also lead to cirrhosis, for example with allopurinol, methyldopa, nitrofurantoin, or isoniazid. In addition to the conditions that lead to veno-occlusive disease, thioguanine has been associated with hepatic vein thrombosis as well.
Diagnosis
The diagnosis of cirrhosis has undergone many changes over the past 200 years and continues to evolve. These changes may be divided into three eras, based on the dominant methodology used for diagnosis: (1) clinical, (2) histologic, and (3) posthistologic. Before the advent of needle liver biopsies in approximately 1950, antemortem diagnosis was based on clinical features. The diagnosis of cirrhosis was presumptive, based on clinical criteria such as the presence of varices and ascites. Anatomic or histologic confirmation was occasionally possible at surgery or autopsy. We currently rely on histologic criteria for definitive diagnosis of cirrhosis, to which this chapter is devoted. The posthistologic era is rapidly emerging, with the development of new noninvasive techniques for assessing liver fibrosis, advancing knowledge of the natural history of cirrhosis, and the desire for less invasive tests. This era will use imaging and acoustic techniques and chemical, physiologic, and molecular parameters, usually by analysis of peripheral blood. Determination of liver stiffness, estimated by sound transmission properties of the liver, is also a promising new approach.
Types of Biopsy and Technical Issues
The four main types of liver biopsy procedures are wedge biopsy, cutting liver biopsy, fine-needle aspiration biopsy, and transvenous (transjugular or transfemoral) biopsy. Wedge biopsies of the liver provide the most liver tissue. They are obtained by incising the convexity of the liver surface or by resection of a small portion of the most inferior edge of the right lobe. With the former technique, the Glisson capsule will be present on one of the three faces of the triangular specimen obtained. With the latter approach, the Glisson capsule is present on two of the three faces. The risk in histologic evaluation of such specimens is misinterpretation of normal anatomy. There is a normal array of fibrous septa that penetrate from the Glisson capsule to a depth of approximately 0.5 cm in the liver parenchyma, partially ensheathing subcapsular portal tracts in more than 50% of individuals140 and linking subcapsular portal tracts to terminal hepatic veins in 25% of individuals.141 If this is not understood, normal fibrous tissue may be erroneously diagnosed as cirrhosis. If a wedge biopsy specimen is the only tissue available, then staging of liver fibrosis cannot be performed accurately. Surgeons are encouraged to obtain an open cutting needle biopsy as well, to sample deeper portions of liver tissue. In our experience, a needle biopsy is almost always more informative, despite its smaller size.
Percutaneous cutting needle biopsies are performed with a 16- to 20-gauge needle and can usually provide a diagnosis of cirrhosis on the first attempt; multiple needle passes are needed only if tissue is not obtained on the first pass.142 Unfortunately, biopsy needles, particularly the smaller ones, tend to glance off hard fibrous tissue and selectively sample softer parenchyma.143–146 This yields tissue that is more difficult to interpret. Because a cutting needle may harvest mainly parenchymal nodules, fragmentation of the liver tissue on ejection from the cutting needle is an important clinical observation. This fragmentation carries through into the tissue sections (Fig. 50.24). The degree of fragmentation alone cannot be used as a criterion of cirrhosis, because even normal liver may be fragmented by the time final histologic sections are prepared.147 Rather, a critical feature of a cirrhotic liver is rounded fragments of parenchyma with concentrically oriented, compressed liver cell plates at the periphery and curvilinear rims of connective tissue (see Fig. 50.24, B).
Transjugular biopsies yield useful tissue if a length of 2 to 2.5 cm is obtained. Fine-needle aspiration, which is usually performed with a 21-gauge or smaller needle, yields an array of small fragments of hepatic parenchyma regardless of liver status (see Chapter 42). Applying histologic criteria for diagnosis of cirrhosis to fine-needle biopsy specimens is difficult.
Regardless of the method used to obtain a tissue sample, biopsy specimens from patients with chronic liver disease should be examined for lobular architecture (including the relationship between portal tracts and terminal hepatic veins), degree of hepatocyte damage, degree of fibrosis, inflammatory infiltration, and parenchymal regeneration and nodule formation. When fibrous septa encompass regenerative nodules within tissue samples, a definitive diagnosis of cirrhosis can be established. The pathologist must always be alert to the presence of hepatocellular carcinoma and its antecedent lesions.
Role of Biopsy
The clinical indications for liver biopsy include the following (see Chapters 44 and 55):
• Evaluation of abnormal laboratory or clinical findings
• Staging known chronic liver disease up to and including cirrhosis
• Searching for a cause of portal hypertension of unknown etiology
The determination of whether cirrhosis is histologically present consists of three primary steps148:
If the liver biopsy does not contain sufficient tissue for ready identification of these characteristics, additional features supporting a diagnosis of cirrhosis include the following:
4. Rounding of tissue edges with connective tissue matrix along the edges
5. Loss of terminal hepatic veins between portal tracts or bridging septa
6. Abnormally large spacing of portal tracts and hepatic veins
7. The presence of larger-caliber vascular channels within fibrous areas of extinction or scar
8. Hepatocyte cord thickening and evidence of curvilinear nodule formation
9. Nuclear features of hepatocyte regeneration such as uniformity in size or twinning
10. Hepatocellular cytologic alterations as noted in dysplastic nodules
Of course, connective tissue stains, such as Masson trichrome and reticulin, are essential in making these histologic determinations. As noted earlier, fragmentation of the tissue obtained from percutaneous liver biopsy commonly occurs,147 and transjugular biopsy yields only small fragments of liver tissue. Hence, fragmentation per se is an unreliable indicator.148
Determination of etiology in a cirrhotic liver is a very difficult exercise. If a specific etiology has not been established on clinical grounds before biopsy, there is very limited ability to identify one in end-stage cirrhotic liver tissue. Moreover, accuracy in staging liver disease depends both on the observer and on tissue sampling. Observer error may be minimized if the pathologist has a complete knowledge of the clinical situation, particularly regarding clinical features of portal hypertension and venous pressure measurements. Overinterpretation of the histologic features, in regard to staging fibrosis or establishing an etiology, is often based on the desire to assist the clinician with a specific diagnosis. Limited sampling of liver tissue may preclude assessment of either stage or etiology. Such sampling error is impossible to avoid entirely, because the definition of cirrhosis requires involvement of the entire liver. Rather, sampling error depends on the character of the liver disease and is greatest in diseases that have a low density of diagnostic lesions in the tissue.
In macronodular cirrhosis, septa may be more than 1 cm apart, and a specimen obtained by small-needle biopsy may not contain any septa. In contrast, a biopsy measuring 1 to 2 mm in length (and 0.2 cm in diameter) may be sufficient for the diagnosis of micronodular cirrhosis, because entire nodules may be encompassed within the 0.6- to 1.4-mm diameter of most tissue fragments.
Many authors recommend that needle biopsies should be at least 2.5 cm long, to encompass parenchymal nodules longitudinally.141,142 In one study, agreement of stage and hepatic vein pressure gradient was excellent when specimens longer than 1.5 cm were evaluated.149 Transjugular biopsies are generally smaller than percutaneous biopsies in both length and width. However, the transjugular technique provides an opportunity to measure pressure during the same procedure. Regardless of methodology, biopsy reports should include comments on the size of the specimen and the presence of fragmentation as indicators of possible adequacy of the biopsy.
The goal of liver biopsy is to establish the severity of necroinflammatory and fibrotic liver injury (grade and stage, respectively) and to provide insights into the specific etiology. However, clinically cirrhotic patients are not without comorbidity, and the risks of liver biopsy, especially hematoma and intraabdominal hemorrhage, are potentially increased by the presence of coagulopathy and ascites. If the need for biopsy is high, despite the presence of contraindications, transvenous hepatic biopsy may be required. Although these biopsies typically yield small tissue specimens, in experienced hands diagnostic tissue is obtained in more than 90% of cases.150
While liver biopsy will continue to be important for a variety of diagnostic purposes, it may not remain the dominant modality for the diagnosis of cirrhosis. For example, to estimate prognosis and response to therapy, hepatic vein pressure gradient may be the best measure.151 Importantly, anatomic proof of cirrhosis is not required for clinical management, especially if the cause is known and the clinical syndrome is internally consistent. That said, cirrhosis remains an anatomic diagnosis, and tissue examination may be necessary in difficult cases.
Staging Systems
Many publications describe methods for semiquantitative estimation of fibrosis in chronic hepatitis and steatohepatitis, as reviewed by Brunt152 and Theise153 (see also Chapters 46 and 49). Regardless of the staging system, the stage must be estimated with the use of a connective tissue stain, such as Masson trichrome. The reticulin stain is a useful adjunct to detect delicate, highly resorbed septa. The presence of curved hepatocyte plate contours, well demonstrated by reticulin stain, helps confirm the presence of a nodule and septal rim when the lesion is otherwise indefinite on hematoxylin and eosin (H&E) stain.
Diagnostic Pitfalls
This chapter has given extensive consideration to the manifestations of various liver diseases in cirrhotic liver and how these diseases might manifest in tissue obtained by liver biopsy. Some final considerations pertain.
Pitfalls in Assessing Fibrosis
Assessment of the degree of fibrosis and architectural derangements requires solid knowledge of normal hepatic structure140,147 and the appearance of artifacts and diagnostic pitfalls (Box 50.4).
An overstained trichrome stain may easily be misinterpreted as showing evidence of pericellular fibrosis. When in doubt, notice that the cytoplasm of hepatocytes should not stain the same color as collagen. If fibrosis is truly present, there are usually other clues to confirm the abnormality, such as obliteration of portal or hepatic veins and abnormal distribution of hepatic veins.
Fibrous portal expansion also is frequently overdiagnosed. Normal large portal tracts may be present in biopsy specimens; these contain large vessels and ducts. A normal portal vein should have a thin collagen wall close to the limiting plate. Fibrous mesenchyme may be quite prominent in larger normal portal tracts. The texture of normal portal tract collagen is coarse bundles; acquired fibrosis has a finer pattern of bundles (Fig. 50.25).
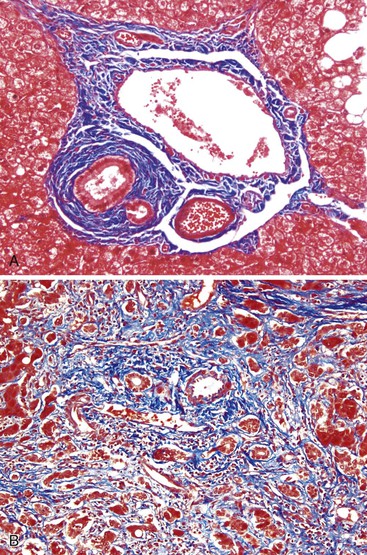
Longitudinally cut normal portal tracts—whether large or small—may traverse the entire width of the biopsy fragment. These can easily be mistaken for fibrous septa. The clue to their normality is that longitudinally arranged blood vessels and ducts are present.
Pitfalls in Diagnosing Cirrhosis
Moderate to severe cirrhosis does not usually present a diagnostic problem, even with tissue obtained by needle biopsy. However, the following considerations apply.
Large regions of parenchymal extinction, commonly present in severe cirrhosis, are often misinterpreted on liver biopsy as severe hepatitis with active bridging necrosis. Misdiagnosis is particularly likely when numerous mononuclear cells within the collapsed area are present. This possibility can be discounted if the margins of the collapsed parenchymal regions show clearcut evidence of a curved fibrous margin, which is an indicator of cirrhosis.
Conversely, massive hepatic necrosis can be misinterpreted as cirrhosis if the collapse of normal parenchymal reticulin fibers are not appreciated as the cause of “increased” connective tissue matrix on either H&E stain or Masson trichrome stain (Fig. 50.26). On Masson trichrome stain, the lighter tinctural qualities of the type III and IV collagen in regions of parenchymal collapse can help distinguish massive hepatic necrosis from cirrhosis, which has strongly staining type I collagen fibers within bridging septa separating parenchymal nodules. Immunostain for CD34 may detect arterialized sinusoids that also indicate advanced disease (Fig. 50.27).
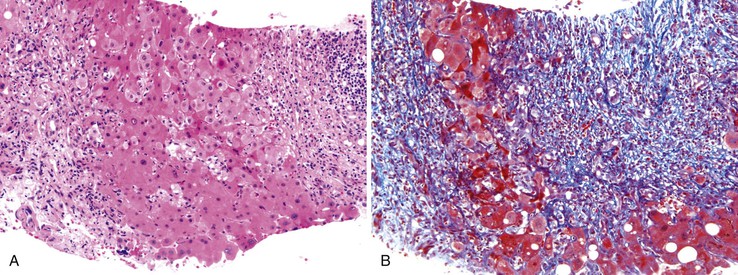
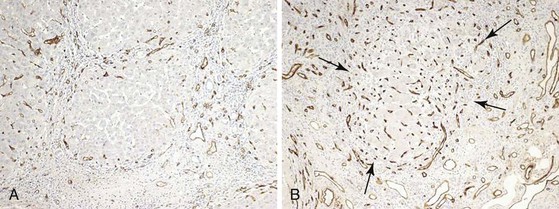
Incomplete septal cirrhosis has delicate fibrous septa within liver tissue that exhibits large expanses of parenchyma devoid of portal tracts. The number of portal tracts, the distance between them, the presence of curved contours within the parenchyma, and the presence of thin and perforated fibrous septa are clues that point toward a regressed form of cirrhosis. Evidence in favor of cirrhosis includes the finding of portal veins or hepatic veins that are obliterated or hepatic veins that are closely approximated to portal tracts. These residual PELs may be all that is left to support incontrovertible clinical evidence of portal hypertension.
Nodular regenerative hyperplasia is suspected when there are narrow curvilinear regions of hepatocellular atrophy that alternate with normal or hypertrophied hepatocytes, in the absence of fibrous septa. As stated earlier, reticulin stain is helpful to accentuate subtle nodular lesions.
Focal nodular hyperplasia, as a lesion with fibrous septation, ductular reaction, and benign-appearing hepatocytes (see Chapter 51), may be mistaken for cirrhosis on percutaneous biopsy. Awareness of the clinical setting of a needle biopsy of a focal lesion is the first step in avoiding an interpretive error. The finding of a large dystrophic artery also points in the correct direction of focal nodular hyperplasia.
Mass lesions often show areas of parenchymal extinction, portal tract inflammation, and parenchymal fibrosis at the margin. These parenchymal changes represent a mass effect and should not be overinterpreted as cirrhosis of the background liver. Peritumoral parenchyma should not be used to stage chronic liver disease.
In the end, interpretation of liver biopsy tissue in a patient with suspected cirrhosis consists of
• Assessment of fibrosis, up to and including a diagnosis of cirrhosis
• Identification of possible etiologic features, as required for clinical management
Cirrhosis is not a disease unto itself; rather, it is a stage of evolution for many forms of chronic liver disease. Likewise, it is not the end stage in all instances, because on occasion cirrhosis may reverse through gradual resorption of connective tissue in the absence of further injury. For the foreseeable future, the pathologist will have a key role to play in the management of cirrhosis.
Acknowledgment
The authors are indebted to Ian R. Wanless, MD, FRCPC, for his extensive work on this chapter in prior editions of this book.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Cirrhosis
James M. Crawford
Introduction
Cirrhosis is an important cause of morbidity and mortality. In the United States, cirrhosis accounts for more than 33,000 deaths per year and is the 12th leading cause of death.1 It is reported on hospital discharge in at least 1% of adult patients2 and is found in 4% to 12% of patients at autopsy in developed countries.3–5 Cirrhosis is the morphologic result of many different types of chronic insult to the liver. It may develop rapidly during a period of months, but most often it is a product of many years of chronic injury. The causes include chronic viral or autoimmune hepatitis, biliary obstruction, alcohol toxicity, and a variety of metabolic abnormalities.
The term cirrhosis derives from the Greek word κίρρoς, meaning “tawny,” and was chosen early in the 19th century to describe the gross appearance (tawny, nodular, and firm), and later the microscopic appearance of the chronically diseased, physiologically burned out, and dysfunctional liver.6,7 In 1977, an international panel sponsored by the World Health Organization (WHO) defined cirrhosis as “a diffuse process characterized by fibrosis and the conversion of normal liver architecture into structurally abnormal nodules.”8 The WHO stated that “cirrhosis is a chronic progressive condition that results in liver cell failure and portal hypertension” and observed that vascular abnormalities were an important feature of cirrhosis. These abnormalities included thrombosis, obliteration and recanalization of veins, formation of arteriovenous shunts, and “capillarization” of sinusoids.9 Etiology was not an important consideration, because there was no cure for cirrhosis. However, reports in the 1980s described the partial “reversal” of cirrhosis,10 and there is now consistent evidence that effective treatment of the underlying cause may mitigate the fibrotic progression of certain liver diseases, particularly in viral hepatitis.11,12
In 2012, an international group of liver pathologists recommended discontinuing use of the term cirrhosis, substituting instead the diagnostic concept of “an advanced stage of chronic liver disease,” with emphasis on etiologic cause, grade of activity, and whether features are present that suggest progression or regression of the fibrotic process, presence of other diseases, or features indicating risk for malignancy.13 Liver morphology should be used to help create an integrated assessment of the patient’s clinicopathologic status. This recommendation is in accord with the increasing awareness in the hepatology community concerning the need for pathophysiologic, rather than morphologic, classification of cirrhosis.14 Indeed, identifying when a liver truly becomes “cirrhotic” is a major diagnostic dilemma, depending on whether one relies on the clinical features of advancing liver disease, imaging modalities, liver biopsy, or an increasing array of noninvasive tests of liver fibrotic status.15,16 All this being said, the term cirrhosis has been used for two centuries and will remain in use for the foreseeable future. This chapter is titled and written with that historical premise in mind.
Cirrhosis is defined by the presence of certain anatomic abnormalities of liver structure. However, a presumptive diagnosis of cirrhosis can often be made when certain clinical consequences of cirrhosis are found. These consequences may be mechanical, functional, or neoplastic. The mechanical effects are related to obstruction of blood flow in the liver, which leads to increased pressure in the splanchnic veins and a high risk for rupture of esophageal varices. Obstruction progresses gradually as cirrhosis develops but may worsen suddenly after thrombosis of the portal vein. Obstruction is associated with the opening of multiple intrahepatic and extrahepatic collateral channels that allow shunting of splanchnic blood past the hepatic parenchyma. Hepatic functional deficits are, in part, related to this portosystemic shunting but also to loss of hepatocellular mass and intracellular retention of bile salts and other toxic substances. Many of the clinical effects of cirrhosis are found in other organs. Renal and pulmonary failure may occur secondary to the systemic hemodynamic response to cirrhosis. Hemorrhage may occur because of platelet dysfunction, platelet sequestration in the spleen, and decreased synthesis of proteins of the coagulation cascade. Neoplasia is a not infrequent late occurrence in patients with cirrhosis. The lifetime risk of hepatocellular carcinoma exceeds 50% in patients with some forms of cirrhosis. Cholangiocarcinoma may be a late complication of intrahepatic or extrahepatic biliary disease. Hence, the development and consequences of cirrhosis are of concern in every form of chronic liver disease.
Definition
Using the 1977 consensus criteria,8 cirrhosis is defined anatomically by the presence throughout the liver of fibrous septa that subdivide the parenchyma into nodules (Fig. 50.1).17–19 Several features in this definition should be emphasized.
The progression of chronic liver disease is highly variable. The point at which a liver becomes cirrhotic is rather subjective and is a frequent source of interobserver disagreement. Establishing a diagnosis of cirrhosis on the basis of percutaneous or transjugular needle biopsy sampling (which samples less than 1/10,000 of the liver mass) can be difficult, particularly if fibrous septa are widely spaced or have regressed. Fortunately, clinical data often provide valuable guidance as to whether any abnormal findings observed in percutaneous liver biopsy tissue are representative of the entire liver. Supporting clinical data include physical examination findings (e.g., ascites, caput medusae, spider angiomas, gynecomastia) and impressions gained from imaging studies or intraoperative visualization of the organ. Laboratory data may not reveal abnormalities, because serum levels of albumin, clotting factors, urea, alkaline phosphatase, aminotransferases, and bilirubin can be normal in a patient who has quiescent cirrhosis with minimal ongoing damage but has not yet experienced hepatic failure, whereas a patient with massive hepatic necrosis and hepatic failure is not cirrhotic despite profound abnormalities in the above serum parameters. Therefore, laboratory data, per se, do not establish a diagnosis of cirrhosis, although various laboratory multiparametric indices have been advanced as capable of providing diagnostic guidance.20,21
Occasionally, a severe focal injury to the liver results in focal histologic changes indistinguishable from cirrhosis on percutaneous needle biopsy; this focal change is not considered true cirrhosis. When this question arises, having definitive information from clinical evaluation and from imaging studies on the general status of the liver, or a biopsy sample from elsewhere in the liver, is critical to determine whether a fibrotic process is focal or diffuse.
Pathogenesis
The 1977 definition of cirrhosis is used by pathologists to recognize cirrhosis, but the definition does not rely on understanding its pathogenesis. Liver cirrhosis is not, strictly, the end stage of hepatic scarring. Rather, it is a dynamic, biphasic process dominated on the one hand by progressive parenchymal fibrosis and on the other by severe disruption of vascular architecture and distortion of the normal lobular architecture. The main anatomic elements (Box 50.1) include deposition of collagen in the parenchyma and portal tracts, arterialization of parenchymal sinusoids, obliteration of small hepatic and portal veins with resultant loss of hepatocytes through a process referred to as parenchymal extinction, abnormal vascular physiology, and regeneration of hepatocytes. Although the importance of these elements is widely appreciated, there is healthy debate concerning the role of each in the pathogenesis of cirrhosis,18,22–24 particularly because the design of effective interventions to delay or reverse the development of cirrhosis depends on understanding its pathogenesis. The concepts thought to be operative in the genesis of cirrhosis of any cause are summarized here.22,25 A discussion of the causes of hepatocellular death is beyond the scope of this chapter.
Collagen in the Liver
Collagen accumulation is a prominent feature of cirrhosis. In the normal liver, collagen types I and III are concentrated in the portal tracts and around terminal hepatic veins, with bundles occasionally located between hepatocytes and endothelial cells in the space of Disse. Strands of type IV collagen (reticulin) are present in the space of Disse, where they form a delicate and uniform network that supports the liver cell plates. In cirrhosis, excessive amounts of types I and III collagen are deposited in the portal tracts, along individual liver cell plates in the space of Disse, and in regions of necroinflammatory collapse. A variety of noncollagenous matrix proteins are also deposited in the space of Disse. In cirrhosis, the amounts of collagen, glycoproteins, and proteoglycans can increase severalfold.26 On a percent area basis, total extracellular matrix components can increase from 5% in normal liver to 25% to 40% in cirrhosis.27 Some of this is only an apparent increase, because condensation of the normal structural collagen and other matrix components occurs during parenchymal collapse and extinction.
The two main cell types that synthesize collagen in the liver are hepatic stellate cells23 and portal fibroblasts.28 Hepatic stellate cells reside in the subendothelial space of Disse in the sinusoidal walls. They are normally distended with lipid droplets containing retinyl esters and other fat-soluble vitamins. During hepatic injury, stellate cells are stimulated by inflammatory mediators to become myofibroblasts: They lose their fat globules, express α-smooth muscle actin in the cytoplasm, and commence proliferation and collagen synthesis. It has recently been discovered that autophagy of the lipid droplets and catabolism of the retinyl esters and triglycerides provide the energy to drive the activation of stellate cells.29 Experimental inhibition of stellate cell autophagy inhibits their activation.30
Stellate cell activation and sinusoidal fibrosis are readily reversible within weeks after cessation of injury.19 When stellate cells are activated in chronic low-grade disease, the liver cell plates are able to maintain their structure while collagen is deposited in the space of Disse, giving an appearance known as pericellular fibrosis or sinusoidal fibrosis. This type of delicate collagen is most easily appreciated in the perivenular regions (Rappaport zone 3). With time, collagen is deposited along the entire length of the sinusoid. Alternatively, widespread injury to hepatocytes, as in alcoholic hepatitis or some forms of drug injury (e.g., amiodarone), may activate stellate cells throughout the liver, leading to extensive deposition of sinusoidal collagen. In either instance, the total matrix in the space of Disse increases and changes from one that contains delicate interspersed strands of fibrillar collagen (types III and IV) to one composed of a dense matrix of basement membrane–type matrix proteins, which closes the space of Disse to protein exchange between hepatocytes and plasma. In general, abnormal matrix deposition within the space of Disse occurs in those parts of the parenchyma where cell injury and inflammation are greatest.
Portal tract fibroblasts differ from stellate cells in location and physiology.31–34 These cells are activated by injury within the portal tracts, particularly in biliary disease, which leads to fibrosis in the region of the ducts and ductules. Peribiliary myofibroblasts are capable of rapid proliferation and deposition of collagen. The epithelial-to-mesenchymal transition of bile duct epithelial cells to a myofibroblast phenotype also appears to contribute to the fibrosis that develops around bile ducts in chronic biliary disease.35 As a result, fibrosis arising from biliary tract disease can run an aggressive course; an example is complete biliary obstruction in infants with extrahepatic biliary atresia, in whom the liver becomes cirrhotic by 9 weeks of age (see Chapter 54). At the opposite end of the spectrum is the exceedingly indolent progression of portal tract fibrosis to cirrhosis in primary biliary cirrhosis, which may extend for 20 or more years (see Chapter 47). In either instance, bridging fibrous septa between portal tracts develop throughout the liver, thereby fulfilling the criteria for cirrhosis. A curious feature of biliary-type fibrosis is that the lobular parenchyma is not induced to regenerate substantially until the liver is extensively fibrotic. Hence, biliary-type fibrosis subdivides the liver into a jigsaw-like pattern during its progression, and cirrhosis may be a very late feature in the course of the disease.
Hepatic Arterialization and Capillarization
Arterialization of the liver in cirrhosis has been known for more than a century. In 1907, Herrick perfused cadaver livers and demonstrated that resistance to flow in the hepatic artery of cirrhotic livers was markedly decreased.36 This fact is documented daily by ultrasonographers when they examine patients with cirrhotic livers and find increased arterial flow in the liver, along with sluggish or even reversed flow in the portal vein. Moschcowitz first used the term capillarization to describe the light microscopic appearance of arterialization in the cirrhotic liver as a granulation tissue response—that is, arterial growth into inflamed tissue.37 Schaffner and Popper used the term capillarization to represent a constellation of ultrastructural changes, including a decrease in the number and size of sinusoidal endothelial fenestrations, loss of hepatocellular microvilli, and an increase in basement membrane material.38,39 By this definition, documentation of capillarization requires electron microscopy. Specifically, in the normal liver, sinusoidal endothelial cells lack a basement membrane and exhibit fenestrations approximately 100 nm in diameter, occupying between 2% to 3% of the area of the endothelial cell. Deposition of extracellular matrix in the space of Disse is accompanied by loss of fenestrations in the sinusoidal endothelial cells.39 With the development of cirrhosis, the diameter of the fenestrations slightly decreases but the area occupancy (porosity) falls to less than 0.5%.
The sinusoidal endothelium in the cirrhotic liver may express CD34. Because this expression is a normal property of arterial endothelium, CD34 positivity is considered a marker of arterialization of the sinusoids. Sinusoidal arterialization is accompanied by α-smooth muscle actin staining in the sinusoidal wall (Fig. 50.2), reflecting the accompanying transformation of hepatic stellate cells into myofibroblasts. It appears that capillarization leads to loss of normal sinusoidal endothelial cell generation of nitric oxide (NO). This loss of NO generation creates a microenvironment that is permissive for stellate cell activation.40 Transformation of the stellate cells then increases sinusoidal vascular resistance by tonic contraction of these “myofibroblasts.” Sinusoidal fibrosis in the perivenular region of the lobule may also partially obstruct vascular outflow, creating postsinusoidal vascular resistance.
Transformation of sinusoidal vascular channels is widely considered to be an explanation for functional deficits in blood-hepatocyte solute exchange.39,41–43 Rapid flow in sinusoids may represent an effective arteriovenous shunt, resulting in a further decrease in solute exchange.44 Although these effects may decrease solute transport into hepatocytes, capillarization or arterialization may also be viewed as a protective form of adaptation that allows the hepatocytes to survive in a high-pressure, high-flow environment.
Arterialization also occurs at the level of portal tracts, where an increased number (and size) of arterial profiles is seen in a variety of conditions, including cirrhosis and use of oral contraceptives. Arterialization of small portal tracts in cirrhosis is usually accompanied by obliteration of adjacent portal veins. Portal vein loss may result from portal tract inflammation in chronic hepatitis or from congestive changes (congestive portopathy) after hepatic venous outflow is compromised (discussed in the next section). Obliteration of small portal veins increases presinusoidal vascular resistance for blood inflow via the splanchnic system. Resistance to hepatic arterial blood flow decreases, owing to an increased arterial capacity, whereas resistance to portal vein blood inflow increases. Hepatic arterial blood pressure is sufficient to supply blood to the liver, but the low pressure of the splanchnic system is not able to overcome the pressure impedance, leading to portal hypertension.
Parenchymal Extinction
Parenchymal extinction is defined as a focal loss of contiguous hepatocytes (Fig. 50.3). Detailed morphologic studies by Wanless and colleagues demonstrated that parenchymal extinction plays a critical role in the pathogenesis of cirrhosis.45–47
Hepatocyte apoptosis and necrosis occur in all types of liver diseases that progress to cirrhosis. The mechanisms are diverse and include lymphocyte-mediated injury, rupture of triglyceride-laden hepatocytes, bile-salt toxicity, and various metabolic stresses. Most of these injuries, if accompanied by low-grade spotty necrosis or apoptosis, lead to local replacement and complete healing. Progressive disease occurs when these injuries are accompanied by a stromal reaction that includes deposition of extracellular matrix, increased sinusoidal vascular resistance, and obstruction of blood flow. The convergence of these injuries leads to contiguous loss of hepatocytes (see Fig. 50.3).45 These extinction lesions may involve a small portion of an acinus, larger units of one or more adjacent acini, or even a whole lobule. The contiguous cell loss is ultimately the result of focal ischemia resulting from obstruction of veins or sinusoids. Naturally, the size of extinction lesions depends on the size of the obstructed vessels.
The concept of parenchymal extinction is important because it incorporates the following perceptions: (1) parenchymal extinction is not directly caused by the initial hepatocellular injury but is an epiphenomenon caused by innocent bystander injury of the local vessels; (2) each parenchymal extinction lesion (PEL) has its own natural history and may be in an early or late stage of healing; (3) cirrhosis develops simultaneously with the accumulation of numerous independent and discrete PELs throughout the liver; and (4) the form of cirrhosis is largely determined by the distribution of the vascular injury. Importantly, parenchymal extinction may progress long after cirrhosis is already established, leading to slow conversion of a marginally functional liver into a sclerotic organ incapable of sustaining life.
The pathogenesis of vascular obstruction depends on the size of the vessels and is outlined in Figure 50.4 (see also Fig. 50.3). Most small-vessel obliteration is secondary to local inflammation.46,47 Although thrombosis may be important in veins of all sizes, it is especially important for blockage of large veins. Most PELs are produced by blockage of veins larger than 100 µm in diameter, because obstruction at this site cannot be easily circumvented by collateral flow within the sinusoids. Obstruction of several adjacent sinusoids is also difficult to circumvent. This raises the issue of congestion, which occurs whenever blood entering the vasculature exceeds the ability of the outflow tract to carry that blood, a state known as in-out imbalance. Congestion is particularly severe when there is total obstruction of the outflow tract or when there is increased inflow in the presence of partial outflow obstruction. Congestive injury is exacerbated by reactive hyperemia, shunt formation, and angiogenesis. If inflow is marked, congestion occurs even when the tissue has normal outflow capacity.
Although the obstructive vascular events leading to parenchymal extinction play out largely in blood vessels larger than 100 µm in diameter, activation of the coagulation cascade within sinusoids releases thrombin, which can interact with the proteinase receptor PAR1 on hepatic stellate cells.48 This is a stimulus for stellate cell activation and transformation to myofibroblasts, which further contributes to the increased vascular resistance and congestive pathophysiology described earlier.
Therefore, PELs are the result of local failure of the microvasculature, usually because of obstruction of hepatic veins or sinusoids. The mechanism for formation of PELs is detailed in Figure 50.5 (see also Fig. 50.4).
In early chronic liver disease, PELs are recognized by the close approximation of the terminal hepatic vein and the adjacent portal tract (see Figs. 50.3 and 50.5). They may be difficult to recognize, because damaged small hepatic veins only appear as a few collagen bundles lying adjacent to a portal tract. PELs become more evident as they aggregate and involve larger and more easily recognizable hepatic veins. PEL aggregates may be evident as two or more portal tracts that are bound together with a hepatic vein remnant apparent in the intervening space (see Fig. 50.2). With progression of disease, progressive obstruction of hepatic veins and secondary arterial dilatation cause further congestive injury in the tissue located between the lesions. This creates a self-perpetuating pathophysiology that may eventually lead to interconnected portal tracts throughout the whole liver.
Shunt Formation
When a region of parenchyma becomes extinct, it collapses so that a portal tract becomes closely associated with an adjacent terminal hepatic vein. This close approximation offers an opportunity for the artery in the portal tract to drain directly into the collapsed perivenous tissue. Often, these arteries can be seen supplying a pool of blood surrounded by atrophic hepatocytes. In older lesions, there is a well-demarcated blood-filled channel; this suggests a stable, high-flow and high-pressure conduit connecting a small artery to a small hepatic vein. This appearance has been interpreted as an arteriovenous shunt.
After parenchymal extinction, the formation of bona fide bridging fibrous septa between portal tracts and terminal hepatic veins enables portovenous and arteriovenous shunting through de novo vascular channels, effectively bypassing the parenchymal nodules. Shunted blood flow through these “fast” vascular channels leaves the remainder of the hepatic parenchyma almost bereft of meaningful blood flow.44,46 This progression also helps explain the increased blood flow observed in sinusoids of the cirrhotic liver in the midst of relative underperfusion of the liver parenchyma as a whole. A remarkable fraction of nutritive blood flow may pass through these intrahepatic functional shunts, contributing to ongoing hepatocellular necrosis and the further generation of PELs. Unfortunately, compression of the shunt channels by contiguous regenerating nodules maintains an increased transhepatic vascular resistance.
Congestive Hepatopathy
Vascular injury in cirrhosis can be divided into an early primary phase, a later congestive phase, and, ultimately, vascular thrombosis. In the primary phase, venous and sinusoidal obstruction is caused by local inflammation occurring in the course of chronic hepatitis (Fig. 50.6).47 The generation of soluble proinflammatory mediators in this setting is a powerful stimulus of fibrogenesis. However, one of the most powerful stimuli of fibrogenesis in many organs is the organization of exudates, especially those rich in fibrin.49–56 In the chronically congested liver, the development of capsular fibrosis attests to the presence of this fibrogenic mechanism (Fig. 50.7). This is the basis for the schematic diagram in Figure 50.8, which indicates that instances of fibrosis can be divided into those stimulated by conventional inflammation and those stimulated by chronic edema and exudation. These two pathways may converge to generate the pattern of fibrogenesis typically seen in chronically injured livers.
Collagen accumulation is determined by the rates of collagen synthesis and resorption (Fig. 50.9; see also Fig. 50.8). Most fibrosis likely accumulates late, when congestive forces cause interstitial exudate and collagen deposition that exceeds the resorptive capacity of the liver. In this formulation, obstructed hepatic veins and PELs occur before septa and before fibrosis; therefore, these lesions are at the leading edge of the pathogenesis of cirrhosis.19,22,45,47,57 The preexisting structural collagen of the liver condenses during the formation of PELs and is incorporated into septa. Although this is not true fibrosis, the presence of such collagen is a striking feature of these lesions.
In the later congestive phase of liver injury, a self-perpetuating pattern of progressive liver injury is created. Specifically, when there is in-out imbalance of blood flow, the high transmural pressure leads to edema, hemorrhage, and narrowing of venous and sinusoidal lumina, followed by intimal fibrous thickening of these vessels. This injury, known as congestive hepatopathy, causes a decrease in the outflow capacity of the tissue that worsens obstruction and, therefore, congestion. Thus, congestive hepatopathy establishes a positive feedback loop of progressive tissue injury.
Vascular Thrombosis
Thrombosis is an important insult. In angiographic and ultrasonographic studies, portal vein thrombosis has been detected in 0.6% to 16.6% of cirrhotic patients,58 and grossly visible portal vein fibrosis or thrombosis has been found in 39% of cirrhotic livers at autopsy.59 Veno-occlusive lesions of hepatic veins smaller than 0.2 mm in diameter have been found in as many as 74% of cirrhotic livers examined at autopsy.60–62 Obliterative lesions are found in 36% of portal veins and 70% of hepatic veins in livers removed at liver transplantation.45 The distribution of obliterative lesions is more uniform in portal veins than in hepatic veins, consistent with the concept of propagation of multifocal thrombi downstream from their site of origin. Portal vein lesions are associated with prominent regional variation in the size of cirrhotic nodules. Hepatic vein lesions are associated with regions of confluent fibrosis and parenchymal extinction. The compelling conclusion is that thrombosis of medium- and large-sized portal veins and hepatic veins is a common occurrence in cirrhosis and may represent a final common pathway for the propagation of parenchymal extinction to full-blown cirrhosis. Furthermore, cirrhotic livers are susceptible to thrombosis because of sluggish or reversed blood flow and the prothrombotic effects of sepsis and cholestasis, thus creating an opportunity for continued loss of functional residual liver parenchyma.
Regeneration
After childhood, the normal liver becomes a stable organ with slow turnover of hepatocytes. However, on injury or surgical reduction, the liver cells proliferate. Normal human liver can restore approximately three fourths of its own mass within 6 months. Hepatocytes, bile duct epithelial cells, and hepatic progenitor or stem cells maintain the potential to multiply during adult life.63,64 Depending on the severity of the primary injury, liver regeneration may occur by at least two mechanisms.65 In brief, with mild to moderate hepatocellular loss, mature hepatocytes undergo replication. More extensive or massive hepatic necrosis stimulates proliferation of progenitor cells within the periportal region, particularly when necroinflammation occurs at the portal-parenchymal interface. Proliferation of these cells gives rise first to so-called ductular hepatocytes, in which ductular structures containing cuboidal cells and slightly larger cells with mitochondria-rich cytoplasm are present. With time, these cells mature into definitive hepatocytes or cholangiocytes and may repopulate damaged parenchymal regions and bile duct structures, respectively.
Parenchymal regeneration is recognized initially by the twinning of liver cell plates, evident as a double line of hepatocytes with nuclei seemingly running in parallel. Twinning of cell plates may remain for some months after regeneration before new sinusoidal channels develop and the nuclear alignment dissipates.66 If regeneration is recent, the hepatocytes lack lipofuscin, because this pigment accumulates with time in the normal liver. Regeneration also is characterized by increased numbers of binucleate or multinucleate hepatocytes, reflecting replication of nuclear material. Hepatocyte nuclei may be more uniform in size, because anisonucleosis increases with patient age in the normal liver and may not be as evident in regenerating liver. Finally, regeneration imparts a rounded appearance to the expanding contours of residual parenchyma, which is demonstrated with a reticulin stain. To the extent that fibrosis and cell death precede hepatocellular regeneration, a residual viable parenchymal island may stand out in the midst of necroinflammation and developing fibrosis.
It is not until hepatocellular regeneration occurs that the characteristic nodular transformation of cirrhosis becomes manifest. On thickening of the liver cell plates, the parenchyma expands against the constraining fibrous septa and acquires a spherical shape. The hepatocyte plates abutting the fibrous septa become compressed and are bent outward by the less-constrained plates toward the interior of the nodule. Poor regeneration or failure to keep up with the pace of collapse results in variants of cirrhosis in which the feature of rounded contours may not be prominent.
The ultimate size of the nodule is determined, in part, by the anatomic location of the antecedent fibrous septa. If matrix deposition occurs at the acinar level, the resulting nodules will grow out of monoacinar units and will be small. If matrix deposition encompasses many acinar units (multiacinar), the growing nodules may be much larger and will retain components of the preexisting acini, including intact portal tracts.
In some cases of cirrhosis, vast expanses of bile ductules within the fibrous septa coexist with the interspersed hepatocellular nodules. These ductules may occur in cirrhosis of almost any cause and are not necessarily the result of biliary obstruction.67 Hyperplasia of ductules is associated with lengthening and increased tortuosity of existing channels and with extensive sprouting of new channels. This change is reminiscent of the massive proliferation of ductular structures within the hepatic parenchyma, or at the interface between parenchyma and portal tracts, that occurs in massive hepatic necrosis and implicates a proliferation of periportal progenitor cells.63,68,69 With time, these ductular structures may mature into hepatocellular parenchyma or bile ductules. Therefore, the presence of expanses of bile ductules in a cirrhotic liver points toward episodes in the recent past in which there was extensive parenchymal destruction; the ductules represent an intermediate stage of a massive regenerative response.70
Natural History and Reversibility of Cirrhosis
Cirrhosis is traditionally viewed as an end stage in the evolution of many types of chronic liver diseases. However, clinical reports have indicated that on cessation of the injurious process, cirrhosis may reverse, or at least improve, histologically.71–79 Tissue samples from some patients with established cirrhosis have revealed incomplete septal cirrhosis or apparent absence of fibrosis after successful treatment. This evolution has been documented in patients with hemochromatosis, autoimmune hepatitis, Wilson disease, primary biliary cirrhosis, schistosomiasis, extrahepatic biliary obstruction, alcoholic disease, chronic viral hepatitis B or C, or postjejunal bypass steatohepatitis. The concept of reversibility of fibrosis, potentially including patients with a histologic or clinical diagnosis of cirrhosis, is now not only widely accepted but a hoped-for outcome after pharmaceutical treatment of some forms of chronic liver disease, particularly viral hepatitis.11,12
In many different experimental models of cirrhosis reversal, collagen is resorbed within weeks after cessation of injury.27 The mechanism of resorption of fibrous extracellular matrix involves activation of tissue metalloproteinases.80 Likewise, stellate cells and activated portal tract fibroblasts may undergo apoptosis and subside. Fibrosis may even disappear in cases of long-term quiescent cirrhosis. In this situation, the diagnosis of cirrhosis may be made by demonstrating severe paucity of small hepatic and portal veins, even when fibrous septa are highly regressed and not well represented within a biopsy specimen. Indeed, a revised definition of cirrhosis can be made wherein it represents a condition of widespread obliteration of small hepatic veins (i.e., venopenia). This definition is sufficient because if there is severe venopenia, all other histologic features of cirrhosis will ultimately follow.
Therefore, the histologic appearance of cirrhosis depends on the age of accumulated tissue damage and the time of dormancy with an opportunity to resorb fibrous tissue. The presence of PELs is a helpful indicator. If the causative injury is long past, only old PELs will be present. If the causative injury is long past, the liver will contain lesions only in the late stages of repair. New extinction lesions are easily seen in livers with moderate to severe activity. They are recognized as areas of bridging necrosis or of focal intense congestion with atrophy and clusters of apoptotic cells.81 More commonly, especially in low-grade chronic hepatitis, the lesions are recognized as subtle atrophy, sinusoidal dilatation, clusters of apoptotic cells, and approximation of hepatic veins close to portal tracts.
Old extinction lesions predominate in livers in which the primary disease has remitted, either spontaneously or after successful treatment.81 Fibrosis tissue is progressively removed from extinction lesions so that broad septa become delicate and delicate septa become incomplete (“perforated”) or disappear. Thus, micronodular cirrhosis may remodel to macronodular cirrhosis, incomplete septal cirrhosis, or near-normal liver (and be discovered only if there is “noncirrhotic” portal hypertension). The problem of diagnosing regression of cirrhosis is discussed later.
Even with substantial resorption of fibrous septa, restoration of the hepatic architecture to a normal state probably does not occur. Limiting factors are the persistence of vascular abnormalities—that is, outflow obstruction and arterialization (Figs. 50.10 and 50.11). If these vascular factors are sufficient to cause continued hepatocellular injury or interstitial exudation, septa will not resorb, and some degree of cirrhosis or incomplete septal cirrhosis will remain. Arterialization is important because even if hepatic vein outflow is not limiting, elevated sinusoidal pressure prevents the regeneration of obliterated small portal veins so that noncirrhotic portal hypertension may remain.82 Moreover, residual sclerosis in portal tracts may still lead to persistent presinusoidal resistance to splanchnic blood flow, leading to continued clinical evidence of portal hypertension.
The previous description indicates that the equilibrium of injury and repair depends on the local balance of inflow and outflow of blood and on the continued presence (or absence) of the proinflammatory disease environment. This equilibrium is continually affected by activity of the primary disease, congestive hepatopathy, persistence of fibrosis in anatomically strategic locations, and catastrophic events such as portal or hepatic venous obstruction by tumor or thrombosis.
Anatomic Classification and Pathology
The main macroscopic types of cirrhosis are referred to as micronodular and macronodular
[/not-level-membership-for-pathology-category]
