[level-membership-for-critical-care-medicine-category]21
Circulatory Shock
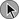 PATHOGENESIS AND PATHOPHYSIOLOGY OF SHOCK
PATHOGENESIS AND PATHOPHYSIOLOGY OF SHOCK
Introduction
The syndrome of shock in humans is often the final pathway through which a variety of pathologic processes lead to cardiovascular failure and death. As such, it is perhaps the most common and important problem with which critical care physicians contend. The importance of shock as a medical problem can be appreciated by the prominence of its three dominant forms. Cardiogenic shock related to pump failure is a major component of the mortality associated with cardiovascular disease, the leading cause of death in the United States with almost 800,000 deaths annually.1 Similarly, hypovolemic shock remains a major contributor to early mortality from trauma, the most common cause of death in those between the ages of 1 and 45 (approximately 200,000 cases annually.1,2 Finally, despite improving medical and surgical therapy, overall mortality coded as septicemia has increased from the 13th to the 11th most frequent cause of death in the United States.1,3 Most current estimates suggest that there are more than 100,000 cases of septic shock annually in the United States alone.4,5 In addition, all forms of shock increase the probability of other major comorbidities such as serious infection, acute respiratory distress syndrome (ARDS), and multiple organ dysfunction syndrome (MODS).
History
Despite recognition of a posttraumatic syndrome by Greek physicians such as Hippocrates and Galen, the origin of the term shock is generally credited to the French surgeon Henri Francois Le Dran who, in his 1737 “A Treatise of Reflections Drawn from Experience with Gunshot Wounds,” coined the term choc to indicate a severe impact or jolt.6 An inappropriate translation by the English physician Clarke, in 1743, led to the introduction of the word shock to the English language to indicate the sudden deterioration of a patient’s condition with major trauma.6 It was Edwin A. Moses,7 however, who began to popularize the term, using it in his 1867 “A Practical Treatise on Shock after Operations and Injuries.” He defined it as “a peculiar effect on the animal system, produced by violent injuries from any cause, or from violent mental emotions.” Prior to this definition, the rarely used term shock referred in a nonspecific sense to the immediate and devastating effects of trauma, not a specific posttrauma syndrome. Although not entirely accurate by today’s standards, his definition was one of the first to separate the syndrome involving the body’s response to massive trauma from the immediate, direct manifestations of trauma itself.
By the late 1800s, two theories of traumatic shock physiology dominated. The first, based on observations by Bernard, Charcot, Goltz, and others, was proposed by Fischer in 1870.8–10 He suggested that traumatic shock was caused by generalized “vasomotor paralysis” resulting in splanchnic blood pooling. The corollary was that total circulating blood volume is preserved in shock. The second dominant theory, articulated by Mapother in 1879, suggested that decreased cardiac output in traumatic shock is caused by intravascular volume loss due to extrusion of plasma through the vessel wall from the intravascular space to the interstitium.11 He proposed that this was a consequence of the failure of “vasodilator nerves” in traumatic shock and subsequent generalized arteriolar vasoconstriction. With the 1899 publication of “An Experimental Research into Surgical Shock” (perhaps the first experimental studies of shock), George W. Crile provided scientific data supporting a variation of the vasomotor paralysis theory.12 After documenting the importance of decreased central venous pressure and venous return in experimental shock due to hemorrhage and demonstrating the potential for intravascular volume replacement as therapy, he proposed that traumatic shock was caused by exhaustion of the overstimulated “vasomotor center” and subsequent generalized relaxation of large vessels (veins) leading to decreased ventricular filling and cardiac output.
Further advances in shock research were substantially driven by military concerns. During World War I, Walter B. Cannon and other physiologists/physicians studied the early clinical response to battlefield trauma. Their work eventually led to the publication of the classic monograph “Traumatic Shock” in 1923.13 Cannon and his colleagues were the first to relate trauma-associated hypotension in a large group of patients to a fall in blood volume, loss of bicarbonate, and accumulation of organic acids. Others, using dye dilution techniques, demonstrated that severity of shock was directly related to the decrease in intravascular volume.14 Clinical data from war casualties also suggested the importance of reduced blood flow (independent of blood pressure) in shock.15 The observation that blood in the capillaries of victims of massive trauma was hemoconcentrated compared to venous blood would lead to the practice of resuscitating trauma patients with dried pooled plasma rather than whole blood in the early part of World War II.16
Although work originating from the battlefields of World War I clearly linked traumatic shock associated with substantial, obvious bleeding to a loss of circulating blood volume, the origin of traumatic shock in the absence of defined hemorrhage was unclear. The accepted explanation for this phenomenon remained a variation of the vasomotor paralysis theory of shock. It was postulated that nonhemorrhagic, posttraumatic shock (“wound shock”) was caused by the liberation of “wound toxins” (histamine or other substances), which resulted in neurogenic vasodilation and peripheral blood pooling. However, after the war, Blalock and others demonstrated in animal models that nonhemorrhagic traumatic shock was due to the loss of blood and fluids into injured tissue rather than circulating toxins resulting in stasis of blood within the circulation.17
Additional advances occurred during World War II. Using injured subjects from the European front, Henry Beecher confirmed that hemorrhage and fluid loss leading to metabolic acidosis was a major cause of shock.18 In the first use of indicator dye techniques in humans for studying blood flow, Cournand and Richard, in 1943, demonstrated that cardiac output was typically reduced in shock.19 They also reinforced Blalock’s findings regarding nonhemorrhagic “wound shock” in trauma patients by demonstrating that circulating blood volume was reduced in such patients through loss of fluid into damaged tissues. The importance of maintaining intravascular volume in traumatic and hemorrhagic shock was supported by the well-known cardiovascular physiologist, Carl J. Wiggers, who published a landmark series of studies20 in the 1940s using a standardized animal model, which showed that prolonged hypovolemic shock resulted in a resuscitation-resistant state that he termed irreversible shock. He defined it as a condition resulting from “a depression of many functions but in which reduction of the effective circulating blood volume is of basic importance and in which impairment of the circulation steadily progresses until it eventuates in a state of irreversible circulatory failure.” Aggressive fluid support became the standard of resuscitation for trauma and shock.
Subsequently, the Korean War fueled the research that demonstrated the relationship of acute tubular necrosis (ATN) and acute renal failure (ARF) to circulatory shock.21 In addition, studies of battlefield casualties clearly demonstrated the relationship between early resuscitation and survival.21 During the Vietnamese conflict, with the widespread use of ventilator technology, the dominant research concern became postshock infection and “shock lung” (ARDS), a concern that has evolved to the present interest in shock-related MODS.
Definitions and Categorization of Shock
The definition of shock has evolved in parallel with our understanding of the phenomenon. As noted, until the late 1800s, the term shock was used to indicate the immediate response to massive trauma, without regard to a specific posttrauma syndrome. The definition consisted of descriptions of its obvious clinical signs. In 1895, John Collins Warren22 referred to shock as “a momentary pause in the act of death,” which was characterized by an “imperceptible” or “weak, threadlike” peripheral pulse and a “cold, clammy sweat.”
Subsequently, with the introduction of noninvasive blood pressure monitoring devices, most clinical definitions of shock added the requirement for arterial hypotension. In 1930, Blalock8 included arterial hypotension as one of the required manifestations of shock when he defined it as “peripheral circulatory failure resulting from a discrepancy in the size of the vascular bed and the volume of the intravascular fluid.” Simeone,23 as recently as 1964, suggested that shock exists when “the cardiac output is insufficient to fill the arterial tree with blood under sufficient pressure to provide organs and tissues with adequate blood flow.”
Current technology, which allows for the assessment of perfusion independent of arterial pressure, has shown that hypotension does not define shock. The emphasis in defining shock is now on tissue perfusion in relation to cellular function. According to Fink,24 shock is “a syndrome precipitated by a systemic derangement of perfusion leading to widespread cellular hypoxia and vital organ dysfunction.” Cerra25 has emphasized supply/demand mismatch in his definition: “a disordered response of organisms to an inappropriate balance of substrate supply and demand at a cellular level.”
Effective tissue perfusion, as opposed to tissue perfusion per se, is an important issue. Effective tissue perfusion may be reduced by either a global reduction of systemic perfusion (cardiac output) or by increased ineffective tissue perfusion due to a maldistribution of blood flow or a defect of substrate utilization at the subcellular level (Box 21.1).
Classification
Although hypovolemic shock associated with trauma was the first form of shock to be recognized and studied, by the early 1900s it became broadly recognized that other clinical conditions could result in a similar constellation of signs and symptoms. Sepsis as a distinct cause of shock was initially proposed by Laennec (1831) and subsequently supported by Boise (1897).26,27 In 1934, Fishberg and colleagues introduced the concept of primary cardiogenic shock due to myocardial infarction.28 Later the same year, Blalock developed the precursor of the most commonly used classification systems of the present.29 He subdivided shock into four etiologic categories: hematogenic or oligemic (hypovolemic), cardiogenic, neurogenic (e.g., shock after spinal injury), and vasogenic (primarily septic shock). Shubin and Weil, in 1967, proposed the additional etiologic categories of hypersensitivity (i.e., anaphylactic), bacteremic (i.e., septic), obstructive, and endocrinologic shock.30 However, as the hemodynamic profiles of the different forms of shock were uncovered, a classification based on cardiovascular characteristics, initially proposed in 1972 by Hinshaw and Cox,31 came to be accepted by most clinicians. The categories include (1) hypovolemic shock, due to a decreased circulating blood volume in relation to the total vascular capacity and characterized by a reduction of diastolic filling pressures and volumes; (2) cardiogenic shock related to cardiac pump failure due to loss of myocardial contractility/functional myocardium or structural/mechanical failure of the cardiac anatomy and characterized by elevations of diastolic filling pressures and volumes; (3) extracardiac obstructive shock involving obstruction to flow in the cardiovascular circuit and characterized by either impairment of diastolic filling or excessive afterload; and (4) distributive shock caused by loss of vasomotor control resulting in arteriolar and venular dilation and (after resuscitation with fluids) characterized by increased cardiac output with decreased systemic vascular resistance. We have adapted these categories into an etiologic/physiologic classification of shock that is summarized in Figure 21.1 and Box 21.2. This figure and box represent our current understanding of the causes and typical hemodynamic features of different forms of shock.
Despite this hemodynamic-based categorization system, it is important to note the mixed nature of most forms of clinical shock. Septic shock is nominally considered a form of distributive shock. However, prior to resuscitation with fluids, a substantial hypovolemic component may exist due to venodilatation and third-spacing. In addition, depression of the myocardium in human septic shock is well documented (see Fig. 21.1).32–34 Similarly, hemorrhagic shock in experimental models has been linked to both myocardial depression35,36 and vascular dysfunction (see Fig. 21.1).37,38 Cardiogenic shock typically presents with increased ventricular filling pressures. However, many patients have been aggressively diuresed prior to the onset of shock and may have a relative hypovolemic component. In addition, systemic vascular resistance (SVR) is only inconsistently increased in cardiogenic shock, suggesting that an inflammatory element may exist under some circumstances. Finally, shock from any cause may cause a deterioration of the coronary perfusion pressure, the difference between mean arterial pressure (MAP) and the higher of left ventricular diastolic pressure or the right atrial pressure, resulting in some degree of myocardial ischemia and myocardial dysfunction.39 Thus, although four categories of shock exist based on hemodynamic profile, clinical shock states tend to combine components of each.
Hypovolemic Shock
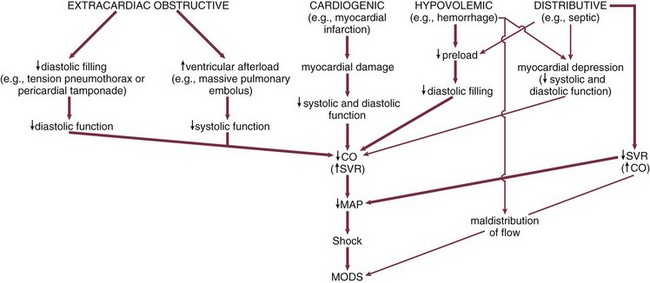
Hypovolemic shock may be related to dehydration, internal or external hemorrhage, gastrointestinal fluid losses (diarrhea or vomiting), urinary losses due to either diuretics or kidney dysfunction, or loss of intravascular volume to the interstitium due to decrease of vascular permeability (in response to sepsis or trauma). In addition, venodilatation due to a number of causes (sepsis, spinal injury, various drugs and toxins) may result in a relative hypovolemic state (see Box. 21.2, Fig. 21.1). Hemodynamically, hypovolemic shock is characterized by a fall in ventricular preload resulting in decreased ventricular diastolic pressures and volumes (Table 21.1). Cardiac index (CI) and stroke volume index (SVI) are typically reduced. In addition to hypotension, a decreased pulse pressure may be noted. Due to a decreased output and unchanged or increased metabolic demand, mixed venous oxygen saturation (MVO2) may be decreased and the arteriovenous oxygen content difference widened. Clinical characteristics include pale, cool, clammy skin (often mottled); tachycardia (or if severe shock, bradycardia)7,40; tachypnea; flat, nondistended peripheral veins; decreased jugular venous pulse; decreased urine output; and altered mental status.
Table 21.1
Hemodynamic Profiles of Shock*
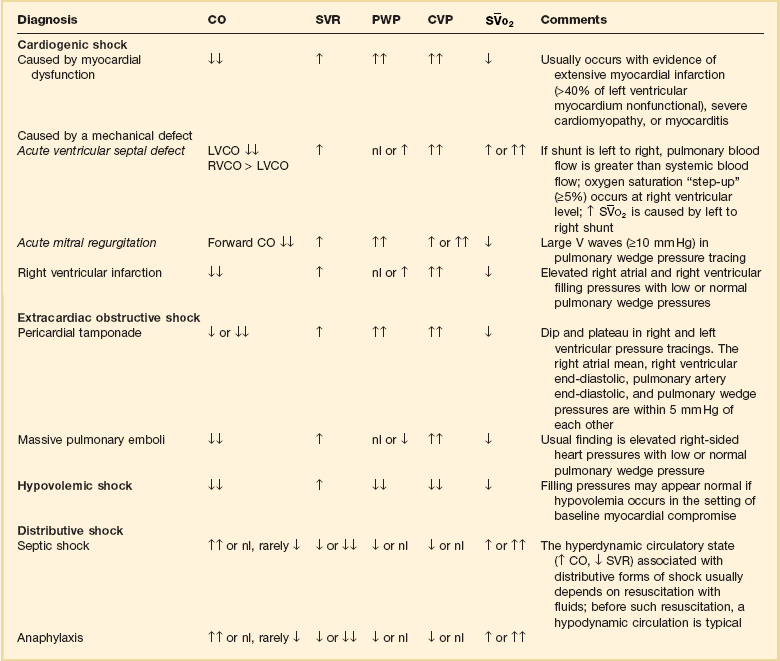
 , mixed venous oxygen saturation; ↑↑ or ↓, mild to moderate increase or decrease; ↑↑ or ↓ ↓, moderate to severe increase or decrease.
, mixed venous oxygen saturation; ↑↑ or ↓, mild to moderate increase or decrease; ↑↑ or ↓ ↓, moderate to severe increase or decrease.*The hemodynamic profiles summarized in this table refer to patients with the diagnosis listed in the left column who are also in shock (mean arterial blood pressure < 60-65 mm Hg).
Modified from Parrillo JE: Septic shock: Clinical manifestations, pathogenesis, hemodynamics, and management in a critical care unit. In Parrillo JE, Ayers SM (eds): Major Issues in Critical Care Medicine. Baltimore, Williams & Wilkins, 1984.
A number of factors may influence the development and hemodynamic characteristics of hypovolemic shock in humans. Studies in animals and humans have demonstrated a clear relationship between the degree of circulating blood volume loss and clinical response.41–44 Acute loss of 10% of the circulating blood volume is well tolerated, with tachycardia the only obvious sign. CI may be minimally decreased despite a compensatory increase in myocardial contractility. SVR typically increases slightly, particularly if sympathetic stimulation augments mean arterial pressure (MAP). Compensatory mechanisms begin to fail with a 20% to 25% volume loss. Mild to moderate hypotension and decreased CI may be present. Orthostasis (with a blood pressure decrease of 10 mm Hg and increased heart rate of 20 to 30 beats/minute) may become apparent. There is a marked increase in SVR and serum lactate may begin to rise. With decreases of the circulating volume of 40% or more, marked hypotension with clinical signs of shock is noted. CI and tissue perfusion may fall to less than half normal. Lactic acidosis is usually present at this stage and predicts a poor outcome.45,46 The case fatality rate can exceed 50% in hemorrhagic shock associated with trauma.47
The rate of loss of intravascular volume and the preexisting cardiac reserve is of substantial importance in the development of hypovolemic shock. As an example, whereas an acute blood loss of 1 L in a healthy adult may result in mild to moderate hypotension with a reduced pulmonary artery occlusion pressure (PAOP) and central venous pressure (CVP),42 the same loss over a longer period of time may be well tolerated due to compensatory responses such as tachycardia, increased myocardial contractility, increased red blood cell 2,3-diphosphoglycerate (2,3 DPG), and increased fluid retention. On the other hand, a similar slow loss may lead to substantial hemodynamic compromise in a person with a limited cardiac reserve, even while the person’s PAOP and CVP remain elevated.
Hypovolemic shock represents more than a simple mechanical response to loss of circulating volume. It is a dynamic process involving competing adaptive (compensatory) and maladaptive responses at each stage of development. Thus, although intravascular volume replacement is always a necessary component of resuscitation from hypovolemia or hypovolemic shock, the biologic responses to the insult may progress to the point where such resuscitation is insufficient to reverse the progression of the shock syndrome. Patients who have sustained a greater than 40% loss of blood volume for 2 hours or more may be unable to be effectively resuscitated.37,41,44 A series of inflammatory mediator, cardiovascular, and organ responses to shock are initiated, which supersede the importance of the initial insult in driving further injury.
Cardiogenic Shock
Cardiogenic shock results from the failure of the heart as a pump (see Box 21.2, Fig. 21.1). It is the most common cause of in-hospital mortality in patients with Q-wave myocardial infarction.48,49 Hemodynamically, cardiogenic shock is characterized by increased ventricular preload (increased ventricular volumes, pulmonary wedge pressure [PWP] and CVP) (see Table 21.1). Otherwise hemodynamic characteristics are similar to those for hypovolemic shock (see Table 21.1). In particular, both involve reduced CI, SVI, and ventricular stroke work indices with increased SVR. Due to inadequate tissue perfusion, the MVO2 is substantially reduced and the arteriovenous oxygen content difference increased. The degree of lactic acidosis may predict mortality.50 Clinically, the specific signs of shock are similar. However, signs of congestive heart failure (volume overload) are typically present in cardiogenic shock. The jugular and peripheral veins may be distended. An S3 and evidence of pulmonary edema are usually found.
Cardiogenic shock is most commonly due to ischemic myocardial injury with a total of 40% of the myocardium nonfunctional.49,51–53 Such damage may involve a single large myocardial infarction or may involve accumulation of damage from multiple infarctions. In addition, viable but dysfunctional “stunned” myocardium may temporarily contribute to cardiogenic shock postinfarction. Cardiogenic shock usually involves an anterior myocardial infarction with left main or proximal left anterior descending artery occlusion. Historically, the incidence of cardiogenic shock due to Q-wave infarction has ranged from 8% to 20%.48,54–56 Although several large studies demonstrate lower incidence rates (4% to 7%) when patients receive thrombolytic interventions,55,57–60 retrospective community studies suggest no overall decrease in the incidence of postinfarction cardiogenic shock or cardiogenic shock mortality (70% to 90%) in the first decades following the introduction of this therapy.48 Further, no trials have demonstrated that thrombolytic therapy reduces mortality rates in patients with established cardiogenic shock.60,61 In contrast, several major studies suggest that mortality of infarction-related cardiogenic shock may be improved by emergent angioplasty.56,62–64 Accordingly, data suggest a reduction in the incidence of acute infarction-related cardiogenic shock to <2% in 2003 in association with widespread use of emergent percutaneous coronary intervention.62 This intervention has also been associated with a reduction of cardiogenic shock mortality risk from 60% to 84% to 43% to 47% in two large analyses.56,62
Mortality is better for cardiogenic shock due to surgically remediable cardiac lesions. Mitral valve failure may be associated with rupture or dysfunction of chordae or papillary muscles due to myocardial ischemia or infarction, endocarditis, blunt chest trauma, or prosthetic valve deterioration and is characterized by “v” waves of greater than 10 mm Hg on a PAOP tracing. Ischemic papillary muscle rupture frequently occurs 3 to 7 days after an infarct in left anterior descending coronary artery territory and may be preceded by the onset of a mitral regurgitant murmur.65 Mortality is high in the absence of surgical therapy.65 Acute aortic valve failure is most commonly due to endocarditis but may involve mechanical failure of prosthetic valves, or aortic dissection. Ventricular septal defects caused by myocardial infarction may also result in the abrupt onset of cardiogenic shock and can be diagnosed by a 5% step up in hemoglobin oxygen saturation between the right atrium and the pulmonary artery (due to left-to-right shunting of blood through the septum).66 As with ischemic papillary muscle rupture, rupture of the intraventricular septum is most frequently seen with occlusions of the left anterior descending artery, a few days after infarction.66
The pathophysiology of cardiogenic shock due to a right ventricular infarction and failure is different from other forms of cardiogenic shock. Although some degree of right ventricular involvement is seen in half of inferior myocardial infarctions, only the largest 10% to 20% result in right ventricular failure and cardiogenic shock.67 These infarctions usually involve part of the left ventricular wall as well. Isolated infarctions of the right ventricle are rare.67,68
Because therapy of this form of shock requires fluid resuscitation and inotropes (rather than vasopressors), differentiation from other causes of cardiogenic shock is crucial. Conditions compromising right ventricular function such as cardiac tamponade, restrictive cardiomyopathy, constrictive pericarditis, and pulmonary embolus are also included in the differential diagnosis. Each of these conditions may present with some of the typical clinical and hemodynamic findings of right ventricular infarction including Kussmaul’s sign, and pulsus paradoxus with elevation and equalization of CVP, right ventricular systolic pressure, pulmonary artery diastolic pressure, and PAOP. Prognosis in this form of cardiogenic shock is distinctly better than that of cardiogenic shock due to left ventricular infarction69,70; however, an inferior infarction with right ventricular injury has a substantially worse prognosis than such an infarction without significant right-sided involvement.71
As with hypovolemic shock, a number of interactions may complicate the development of cardiogenic shock. Optimal cardiac performance in patients with impaired myocardial contractility may occur at substantially higher than normal PAOP (i.e., 20 to 24 mm Hg). Yet patients who develop cardiogenic shock are frequently initially treated with diuretics and may have a degree of hypovolemia (relative to their optimal requirements). Thus, patients should not be diagnosed with cardiogenic shock unless hypotension (MAP < 65 mm Hg) and reduced cardiac output (CI < 2.2 L/min/m2) coexist with an elevated ventricular filling pressure.72 Cautious fluid challenge may be required (in the absence of overt pulmonary edema) to increase the filling pressures to an optimal range. Other interactions include increased right ventricular ischemia due to decreased right coronary perfusion pressure (MAP decreased while right ventricular end-diastolic pressure is increased) and increased right ventricular afterload due to pulmonary hypertension. Right ventricular ischemia may also lead to right ventricular dilatation, septal shift, and impairment of left ventricular function.
Other causes of cardiogenic shock include acute myocarditis, end-stage cardiomyopathy, brady- or tachyarrhythmias, hypertrophic cardiomyopathy with obstruction, and traumatic myocardial contusion (see Box 21.2).
Obstructive Shock
Extracardiac obstructive shock results from an obstruction to flow in the cardiovascular circuit (see Box 21.2, Fig. 21.1). Pericardial tamponade and constrictive pericarditis directly impair diastolic filling of the right ventricle. Tension pneumothorax and intrathoracic tumors indirectly impair right ventricular filling by obstructing venous return. Massive pulmonary emboli (two or more lobar arteries with >50% of the vascular bed occluded), nonembolic acute pulmonary hypertension, large systemic emboli (e.g., saddle embolus), and aortic dissection may result in shock due to increased ventricular afterload.
The characteristic hemodynamic/metabolic patterns are, in most ways, similar to other low output shock states (see Table 21.1). CI, SVI, and stroke work indices are usually decreased. Because tissue perfusion is decreased, the MVO2 is low, the arteriovenous oxygen content difference increased, and serum lactate frequently elevated. Other hemodynamic parameters are dependent on the site of the obstruction. Tension pneumothorax and mediastinal tumors may obstruct the great thoracic veins, resulting in a hemodynamic pattern (decreased CI and elevated SVR) similar to hypovolemia (although distended jugular and peripheral veins may be seen). Cardiac tamponade typically causes increased and equalized right and left heart ventricular diastolic pressures, pulmonary artery diastolic pressure, CVP, and PAOP. In constrictive pericarditis, right and left ventricular diastolic pressures are elevated and within 5 mm Hg of each other. Mean right and left atrial pressures may or may not be equal as well. Massive pulmonary embolus will result in right ventricular failure with elevated pulmonary artery and right heart pressures whereas PAOP remains normal. A systemic saddle embolus or aortic occlusion due to dissection causes peripheral hypotension and signs of left ventricular failure including an elevated PAOP. Clinical signs are similarly dependent on the site of the obstruction.
As with other forms of shock, the time course of development of the insult has a substantial impact on the clinical response. Ischemic rupture of the left ventricular free wall (usually 3 to 7 days after myocardial infarction) leads to immediate cardiac tamponade and shock with as little as 150 mL blood in the pericardium.73–75 Survival requires emergency surgery.74,75 Similar situations may develop with bleeding into the pericardium after blunt chest trauma or thrombolytic therapy. Pericardial tamponade due to malignant or inflammatory pericardial effusions usually develop much more slowly. Although shock may still develop, it usually requires substantially more pericardial fluid (1 to 2 L) to cause critical failure of right ventricular diastolic filling.73 No large reliable studies examining mortality rates with and without therapy in these conditions are available due to the small numbers of cases.
A similar time course–dependent risk is seen with major pulmonary emboli. In those without preexisting cardiopulmonary disease, a massive embolus involving two or more lobar arteries and 50% to 60% of the vascular bed76,77 may result in obstructive shock. However, if recurrent smaller pulmonary emboli result in right ventricular hypertrophy, a substantially larger total occlusion of the pulmonary vascular bed may be required to cause right ventricular decompensation. Analyses have suggested that the presence of shock due to pulmonary embolus (regardless of underlying chronic cardiopulmonary dysfunction) indicates a three- to sevenfold increase in mortality risk with the majority of deaths occurring within an hour of presentation.78,79 An analysis of more than 70,000 unstable (hemodynamic instability or ventilator-requiring) patients with pulmonary embolus in the national inpatient sample shows that mortality in untreated patients is approximately 47%.80 Systemic thrombolysis is associated with a substantial reduction in mortality to 15%. Where available, catheter-directed therapy may be even more efficacious with a lower risk of serious hemorrhage.81 Shock due to pulmonary embolism is an indication for urgent thrombolytic or catheter-directed intervention.
Distributive Shock
Hemodynamically, distributive shock is characterized by an overall decrease in SVR (see Table 21.1). However, resistance in any specific organ bed or tissue may be decreased, increased, or unchanged. Initially, CI may be depressed and ventricular filling pressures decreased. After fluid resuscitation, when filling pressures are normalized or increased, CI is usually elevated. Due to hypotension, left and right ventricular stroke work indices are normally decreased. MVO2 is increased above normal. Concomitantly, arteriovenous oxygen content difference is narrowed despite the fact that oxygen demand is usually increased (particularly in sepsis). The basis of this phenomenon may be that because total body perfusion (CI) is increased, perfusion is not effective in that either it does not reach the necessary tissues or the tissues cannot utilize the substrates presented. As a reflection of this inadequate “effective” tissue perfusion, lactic acidosis may ensue. Clinical characteristics of resuscitated distributive shock include, in contrast to the other forms of shock, warm, well-perfused extremities, a decreased diastolic blood pressure, and an increased pulse pressure. Nonspecific signs of shock include tachycardia, tachypnea, decreased urine output, and altered mentation. In addition, evidence of the primary insult may exist (urticaria for anaphylaxis, spinal injury for neurogenic shock, and evidence of infection in septic shock).
Septic shock (shock due to infection) and sepsis-associated multiple organ failure are the most common causes of death in ICUs of the industrialized world. As many as 800,000 cases of sepsis are admitted every year to American hospitals (comparable to the incidence of first myocardial infarctions) with half of those developing septic shock and about half of those (200,000) dying.82 Since the 1970s there has been a progressive increase in the incidence of and total deaths from sepsis and septic shock.5 The total toll of septic deaths is comparable to deaths from myocardial infarction and far exceeds the impact of illnesses such as AIDS or breast cancer.82,83
Septic shock is caused by the systemic activation of the inflammatory cascade. Numerous mediators including cytokines, kinins, complement, coagulation factors, and eicosanoids are activated or systemically released, resulting in profound disturbances of cardiovascular and organ system function84 (Table 21.2). These mediators, particularly tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), platelet activating factor (PAF), and prostaglandins are thought to mediate reduced peripheral vascular resistance seen in septic shock.
Table 21.2
Inflammatory Mediators in Sepsis and Septic Shock
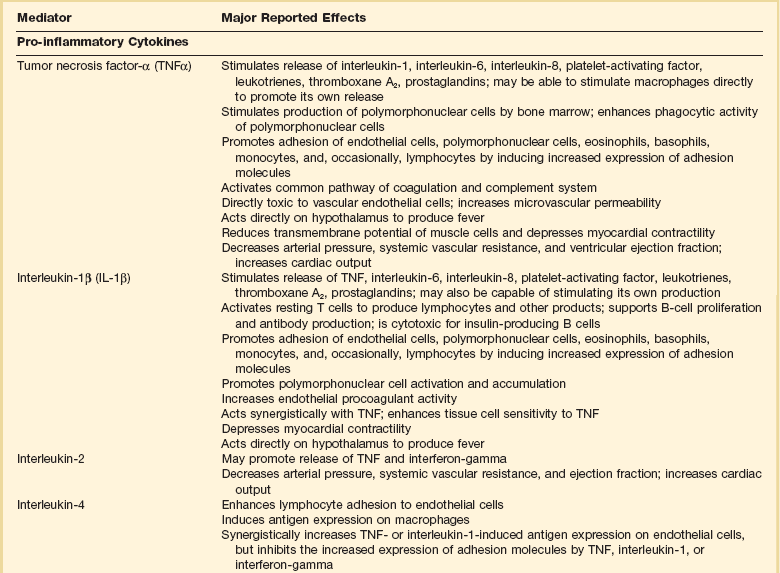
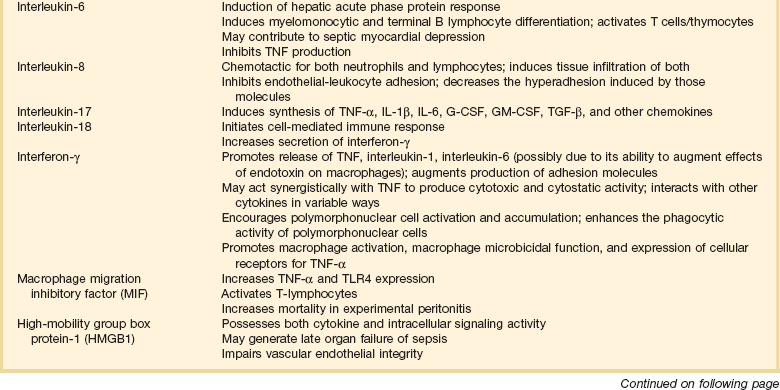
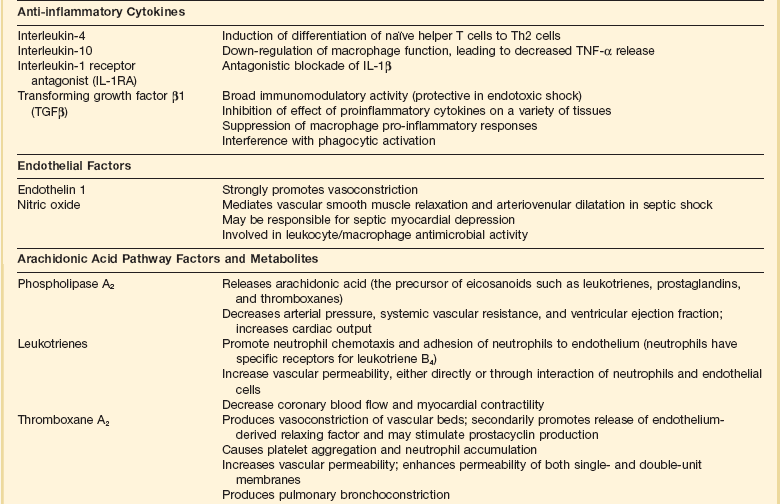

Adapted from Bone RC: The pathogenesis of sepsis. Ann Intern Med 1991;115:457.
Loss of vascular autoregulatory control may explain some of the typical metabolic findings of sepsis and septic shock. An early theory postulated the existence of microanatomic shunts between the arterial and venous circulations. During sepsis, these shunts were said to result in decreased SVR and increased MVO2.85 However, although microanatomic shunting has been noted in localized areas of inflammation, systemic evidence of this phenomenon in sepsis and septic shock is lacking.85–89 “Functional” shunting due to defects of microcirculatory regulation in sepsis has also been suggested.90,91 Overperfusion of tissues with low metabolic requirements would increase MVO2 and narrow the arteriovenous oxygen content difference. Relative vasoconstriction of vessels supplying more metabolically active tissues would result in tissue hypoxia and lactate production due to anaerobic metabolism. Observations that some capillary beds may be occluded by platelet microaggregates, leukocytes, fibrin deposits, and endothelial damage support this theory.86,90,92 Additional support comes from studies that demonstrate evidence of oxygen supply–dependent oxygen consumption in sepsis.93–97 A third theory suggests that circulating mediators cause an intracellular metabolic defect involving substrate utilization, which results in bioenergetic failure (decreased high-energy phosphate production) and lactate production.98,99 Increased mixed venous oxygen saturation could then be explained by perfusion, which is increased in excess of tissue oxygen utilization capability. However, animal studies using nuclear magnetic resonance (NMR) spectroscopy demonstrate that high-energy phosphates are not depleted in septic animals as is expected in all of these theories.100–102 According to these and other studies, cellular ischemia is not the dominant factor in metabolic dysfunction in sepsis.100–106 Rather, circulating mediators may result in cellular dysfunction, aerobic glycolysis, and lactate production in the absence of global ischemia.101 This position is weakened by data suggesting that increased lactate in septic shock is also associated with decreased pH (which would not be expected in aerobic glycolysis)101 and, to some extent, by studies that support the existence of oxygen supply–dependent oxygen consumption in sepsis.94–97
The trigger for systemic activation of the inflammatory cascade is the presence of gram-negative bacilli in 50% to 75% of cases of septic shock. Gram-positive bacteria account for most of the remainder, but infection with fungi, protozoa, and viruses can also result in septic shock.107–109 Investigations suggest a surprising commonality of signaling mechanisms in septic shock via Toll-like receptors from a broad range of etiologic agents.110–114 Despite aggressive supportive care and antibiotic treatment, mortality is 50% overall and may exceed 70% for gram-negative septic shock.107 Of those succumbing to septic shock, approximately 75% are early deaths (within 1 week of shock), primarily due to hyperdynamic circulatory failure.115 Late mortality is usually due to MODS.115
More than any other form of shock, distributive and, particularly, septic shock involves substantial elements of the hemodynamic characteristics of other shock categories (see Fig. 21.1, Table 21.1). As noted, all forms of distributive shock involve decreased mean peripheral vascular resistance. Prior to fluid resuscitation, distributive shock also involves a relative hypovolemic component. The first element of this relative hypovolemia is an increase of the vascular capacitance due to venodilatation. This phenomenon has been directly supported in animal models of sepsis116–120 and is reinforced by the fact that clinical hypodynamic septic shock (low cardiac output) can usually be converted to hyperdynamic shock (high cardiac output) with adequate fluid resuscitation.115,121,122 Relaxation of vascular smooth muscle is attributed to a number of the mediators known to circulate during sepsis. These same mediators also contribute to the second cause of hypovolemia in sepsis, third-spacing of fluid to the interstitium due to a loss of endothelial integrity. In addition, a number of studies have demonstrated that human septic shock is characterized by myocardial depression (biventricular dilatation and decreased ejection fraction).32–34 Circulating substances such as TNFα, IL-1β, platelet activating factor (PAF), leukotrienes, and, most recently, interleukin-6 (IL-6) have been implicated in this process.123–130
Anaphylactic shock is a form of distributive shock caused by the release of mediators from tissue mast cells and circulating basophils. Anaphylaxis, an immediate hypersensitivity reaction, is mediated by the interaction of IgE antibodies on the surface of mast cells and basophils with the appropriate antigen. Antigen binding results in the release of the primary mediators of anaphylaxis contained in the basophilic granules of mast cells and basophils. These include histamine, serotonin, eosinophil chemotactic factor, and various proteolytic enzymes.131 Subsequently, a number of secondary lipid mediators are synthesized and released including PAF, bradykinin prostaglandins, and leukotrienes (slow-reacting substance of anaphylaxis).131 An anaphylactoid reaction (clinically indistinguishable from anaphylaxis) results from the direct, nonimmunologic release of mediators from mast cells and basophils and can also result in shock.
Anaphylaxis is triggered by insect envenomations (Hymenoptera bees, hornets, and wasps) and certain drugs, especially antibiotics (beta-lactams, cephalosporins, sulfonamides, vancomycin).131 In addition, less frequently, heterologous serum (e.g., tetanus antitoxin, snake antitoxin, antilymphocyte antisera), blood transfusion, immunoglobulin (particularly in IgA-deficient patients), and egg-based vaccine products have been implicated.131 Anaphylactoid reactions can be caused by a wide range of medical agents including ionic contrast media, protamine, opiates, polysaccharide volume expanders such as dextran and hydroxyethyl starch, muscle relaxants, and anesthetics.131
The hemodynamic features of anaphylactic shock are very similar to those for septic shock and include elements of hypovolemia (due to interstitial edema and venodilatation) and myocardial depression.132–136 Cardiac output and ventricular filling pressures may be reduced until patients are fluid resuscitated.136,137 In addition to typical findings of shock, patients may demonstrate urticaria, angioedema, laryngeal edema, and severe bronchospasm.
Adrenal crisis (see also Chapter 59) is an uncommon cause of shock, which can be difficult to diagnose as it occurs in patients with other active disease processes and the clinical features may mimic infection. It is a life-threatening emergency that requires prompt diagnosis and management.
Adrenal crisis is caused by a deficiency of adrenal production of mineralocorticoids and glucocorticoids. It may occur de novo in patients with critical illness or may occur against a background of occult adrenal insufficiency. In the critical care setting, the most common cause of de novo acute adrenal insufficiency is bilateral adrenal hemorrhage in association with overwhelming infections (classically meningococcal, but frequently gram-negative bacteria), human immunodeficiency virus infection, or anticoagulation.138,139 In addition, fungal infections such as histoplasmosis, blastomycosis, and coccidioidomycosis and malignant infiltration of the adrenals may cause acute adrenal insufficiency in ICU patients.139 In some patients, steroid production remains adequate for the baseline state despite adrenal disease. Once stressed, however, the adrenal response is inadequate, leading to decompensation and adrenal crisis. Stressors may be relatively innocuous or may be severe. A febrile illness, infection, trauma, surgery, dehydration, or any other intercurrent illness may trigger the crisis. Abrupt cessation of glucocorticoid therapy or replacement may also result in adrenal crisis.
Symptoms are generally nonspecific and may include anorexia, nausea, vomiting, diarrhea, abdominal pain, myalgia, joint pains, headache, weakness, confusion, and agitation or delirium.139,140 Fever (often out of proportion to any minor infection) is almost always present, and hypotension, initially due to hypovolemia, is frequent.139 The initial hemodynamic pattern may resemble hypovolemic shock (if shock is due only to adrenal crisis). With volume resuscitation, a high output, vasopressor-refractory shock may become apparent.141,142
An unrecognized “relative” adrenal insufficiency has been implicated in the pathogenesis of human septic shock.143–147 In this circumstance, sepsis is associated with a suboptimal adrenal response with an improvement in cardiovascular parameters or outcome with “stress” dose corticosteroid administration.143,145,146,148,149 One randomized controlled trial has suggested that prospective “stress dose” therapy with a combination of hydrocortisone (50 mg intravenous every 6 hours) and fludrocortisone (50 µg oral/nasogastric daily) for 7 days improved outcome in nonresponders to corticotropin challenge.150 Unfortunately, confirmatory randomized trials have failed to reproduce this finding.151,152
Compensatory Responses to Shock
Various sensing mechanisms involved in physiologic compensatory responses exist to recognize hemodynamic and metabolic dyshomeostasis (Fig. 21.2). Low-pressure right atrial and pulmonary artery stretch receptors sense volume changes. A decrease in circulating volume (or an increase of venous capacitance) results in an increase in sympathetic discharge from the medullary vasomotor center.153,279,280 Aortic arch, carotid, and splanchnic high pressure baroreceptors sense early blood pressure changes close to the physiologic range.153,279,280 An increase of sympathetic discharge from the medullary vasomotor center results from a small to moderate decrease in blood pressure associated with early shock. However, once mean arterial pressure falls below about 80 to 90 mm Hg, aortic baroreceptor activity is absent. Subsequently, carotid baroreceptor response is eliminated as mean pressure falls below 60 mm Hg. As blood pressure falls further, carotid and aortic chemoreceptors, sensitive to decreased PO2, increased PCO2, and increased hydrogen ion concentrations (decreased pH), dominate the response. These receptor complexes, active only when mean blood pressure is less than approximately 80 mm Hg, are of minimal relevance during physiologic states.153 During shock, they make a substantial contribution to increases of sympathetic tone.
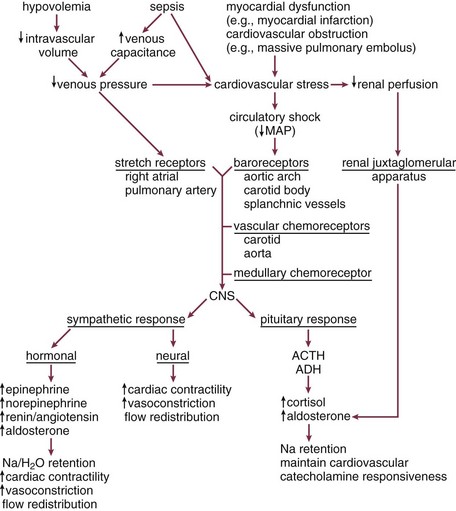
During severe shock, the most powerful stimulus to sympathetic tone is the central nervous system ischemic response.153 The lower medullary chemoreceptors for this response (thought to be sensitive to increased CO2 associated with decreased cerebral perfusion) become active when mean blood pressure falls below 60 mm Hg. Sympathetic stimulation provided by these receptors peaks at mean pressures of 15 to 20 mm Hg and results in maximal stimulation of the cardiovascular system.153 The Cushing response to increased intracranial pressure is an example of activation of this reflex under different circumstances.
Other mechanisms also play a role in the compensatory response to shock. Vasopressin release is regulated by alterations of serum osmolality. During effective hypovolemia due to intravascular volume loss or increased vascular capacitance, low-pressure, right atrial stretch receptors can override osmolar control of vasopressin response to result in the retention of body water.153,281 Similarly, during hypovolemia and shock, the juxtaglomerular apparatus in the kidneys responds to decreased perfusion pressure by renin release.153
All compensatory responses to shock, whether hemodynamic, metabolic, or biochemical, support oxygen delivery to vital organs. These responses are similar (to varying extents) for different classes of shock and can be broken down into four components: (1) preserving mean circulatory pressure (a measure of venous pressure) by either maintaining total intravascular volume or increasing stressed volume (i.e., increasing venous tone), (2) optimizing cardiac performance, (3) redistributing perfusion to vital organs, and (4) optimizing the unloading of oxygen at the tissues (Box 21.3, Fig. 21.E3).
Mean circulatory pressure and venous return are sustained in early shock by a number of mechanisms. Acutely, total intravascular volume is supported by alterations of capillary hydrostatic pressure as described by Starling.282 Sympathetic activation results in precapillary vasoconstriction. In combination with initial hypotension, this results in decreased capillary hydrostatic pressure.282 A decrease in capillary hydrostatic pressure enhances intravascular fluid shift due to maintained plasma oncotic pressures. Transcapillary fluid influx following the removal of 500- to 1000-mL blood volumes in humans can be as high as 2 mL/minute with full correction of intravascular volume by 24 to 48 hours.283 The intravascular volume may also be supported by the osmotic activity of glucose generated by glycogenolysis. Increased extracellular osmolarity results in fluid redistribution from the intracellular to the extracellular space.
Pathogenesis and Pathophysiology of Shock
Hemodynamic Basis of Shock
From a hemodynamic perspective, shock is the failure of cardiovascular adaptation to systemic dyshomeostasis induced by trauma, infection, or other insult such that cardiac output or blood pressure are compromised. This failure is manifested by inadequate organ and tissue perfusion. Although effective perfusion also depends on microcirculatory and intracellular factors (see Box 21.1), the hemodynamic aspects of shock can be described, in part, by the contributions of cardiac and arterial vascular function to blood pressure and cardiac output.
Arterial Pressure
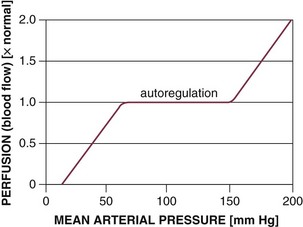
The ability of all organ vascular beds to support normal blood flow depends on the maintenance of blood pressure within the defined range for that organ (Fig. 21.E1).153 Vital organs such as the brain and heart, in particular, are able to autoregulate blood flow over a wide range of blood pressure. Failure to maintain the minimal MAP and perfusion pressure required for autoregulation during hypodynamic circulatory shock indicates a severe reduction in cardiac output. Pharmacologic support of blood pressure in such situations (with alpha-adrenergic agonists) usually results in decreased total systemic perfusion as sensitive vascular beds constrict and overall vascular resistance increases. However, due to their strong autoregulatory capacity, vital organs maintain increased perfusion under these conditions.
In addition to sufficient cardiac output, effective perfusion requires appropriate distribution of blood flow. Failure to maintain blood pressure within the autoregulatory range results in a distribution of blood flow that strictly depends on the passive mechanical properties of the vasculature.154 This may result in inappropriate distribution of perfusion between and within tissues and organs. Late hemorrhagic shock has been shown to be characterized by abnormal microvascular flow with dilatation of precapillary sphincters.37
Cardiac Output
Preload represents the extent of precontraction myocardial fiber (or sarcomere) stretch. In vivo, preload is the end-diastolic ventricular volume. Because measurement of such volumes in the clinical context is difficult, intracardiac pressures, which can be determined more easily, are frequently substituted. There are difficulties with this approach. The relationship of ventricular end-diastolic volume (preload) to end-diastolic pressure is nonlinear. Further, alterations of myocardial compliance render CVP and PAOP unreliable as estimates of preload in critically ill patients.155
Preload is dependent on circulating volume, venous tone, atrial contraction, and intrathoracic pressure among other factors.153,156 Atrial contraction is particularly important in those with impaired ventricular function. Although it accounts for only 5% to 10% of cardiac output in healthy humans, synchronized atrial contraction contributes as much as 40% to 50% of the cardiac output in patients with severe left ventricular dysfunction.156 Increased intrathoracic pressure or increased venous capacitance affects preload by reducing venous return.153,157 Nitrovasodilators such as nitroglycerin may decrease cardiac output despite arteriolar vasodilation due to their venodilatory (decreased preload) effects. Conversely, the earliest increases in cardiac output seen with sympathetic stimulation and exogenous catecholamine infusion are related to venoconstriction-induced increases of venous return and preload.158 Cardiogenic and some forms of obstructive shock are typically characterized by increased preload. Preresuscitation distributive shock and hypovolemic shock are uniformly associated with decreased preload.
Afterload has been suggested to be equivalent to systolic myocardial wall stress. This definition suggests that afterload is substantially dependent on intrinsic cardiac mechanical and functional properties.159 An alternative approach equates left ventricular afterload with the mechanical properties of the arterial side of the circulatory system. Aortic input impedance, which represents the total resistance to flow from outside the left ventricle, is determined by the inertial and viscous properties of blood and the resistive and viscoelastic properties of the arterial system. The term covers SVR, heart rate effects, and pulse wave reflections in the arterial tree.159 Although it is an accurate measure of afterload in pulsatile systems, assessment of impedance is technically difficult, requiring continuous harmonic analysis of rhythmic variations of aortic pressure and flow. Systemic vascular resistance is a limited approximation of aortic input impedance based on a model that assumes nonpulsatile flow. At a heart rate of 0, SVR and aortic input impedance are equivalent. From a clinical point of view, SVR is the most practical way of assessing afterload.
Contractility refers to the intrinsic ability of myocardial fibers to shorten under given loading conditions. Under normal conditions, determinants of contractility include myocardial mass and sympathoadrenal activation state. In pathologic states (e.g., shock), hypoperfusion/ischemia, myocardial cell injury (e.g., reperfusion injury, myocarditis), acidosis, and circulating myocardial depressant substances (such as seen in sepsis) depress cardiac contractility (Fig. 21.E2).
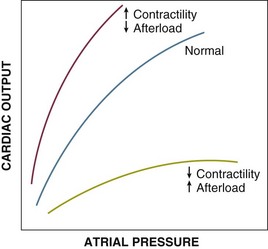
As with preload and afterload, the ex vivo/in vitro assessment of contractility is straightforward. Assessment of in vivo contractility (even in experimental animals) is substantially more difficult due to the intrinsic lability of preload and afterload. Relatively load-independent variables such as peak systolic pressure/end-systolic volume ratio may be the most clinically useful measures of contractility.160 Many of these variables can be obtained echocardiographically.
Venous Function in Shock
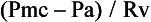
where Pmc is the mean circulatory pressure (the upstream driving pressure of the systemic venous circulation, i.e., the intravascular pressure measured when the heart is stopped), Pa is the right atrial pressure (the downstream pressure that opposes flow to the right ventricle), and Rv is the venous resistance (resistance of the conduit to flow). Pmc equals the stressed volume or Vs portion of the vascular volume, which contributes to venous pressure, divided by the mean vascular compliance (C):
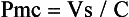
Stressed volume is dependent on total vascular volume (Vt) and the state of venous tone (i.e., venoconstriction). It is defined as the difference between Vt and the unstressed vascular volume (Vo), the intravascular volume that remains when the vascular circuit is equalized to atmospheric pressure (i.e., the volume remaining after passive exsanguination). Stressed volume is approximately 30% of total blood volume in both humans and experimental animals.161–163 Compliance refers to the total elastic properties of the entire cardiovascular circuit inclusive of the heart and vasculature.
As Equation 1 shows, the only direct role the heart plays on venous return is to alter right atrial pressure (Pa). Mean arterial pressure has no direct effect at all despite the fact that it is closely related to cardiac output in the systemic circulation (MAP − CVP = CO × SVR).
The venous return relationship is shown in Figure 21.E3.153,164,165 Because the systemic venous bed comprises the bulk of venous capacitance, the systemic venous vasculature dominates the physiology of venous return. Venous return is linearly related to Pa (right atrial pressure) down to 0 cm H2O (= atmospheric pressure), at which point intermittent collapse of the great veins results in limitation of return producing the plateau.153,166 The slope of the line representing venous return is the inverse of the resistance (1/Rv). The ordinate intercept denotes the right atrial pressure (Pa) at which venous return is zero. But according to Equation 1, venous return is zero only when right atrial pressure (Pa) equals the Pmc. Thus, the intercept of the atrial pressure axis represents Pmc. Changes of Pmc shift the curve to the left or right without changing the slope of the line (see Fig. 21.E3, line a to line b and c). Changes of Rv change the slope of the line without changing the Pmc (see Fig. 21.E3, line a to line d and e).
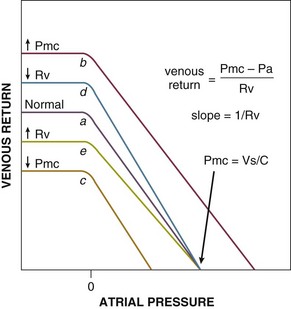
Graphic Analysis of Venous-Cardiac Interactions during Shock
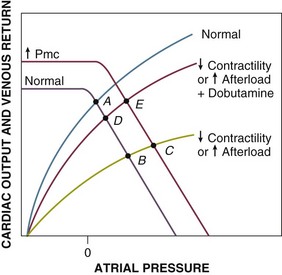
Cardiogenic shock and obstructive shock due to increased afterload of the right or left ventricle (e.g., massive pulmonary embolus) result in a common change of the right ventricular Starling function curves. In the case of primary left ventricular loading or damage, this occurs because increased left ventricular filling pressures are passively transmitted to the right ventricle. The Starling curves are shifted downward and to the right (flatter) (Fig. 21.E4, point A to B), resulting in decreased cardiac output at increased atrial pressures. Therapy can consist of fluid resuscitation (increased Vt and Pmc), which may result in only modest augmentation of cardiac output despite significant increases of atrial pressures and ventricular filling pressures (point C); dobutamine, a beta-1 and beta-2 agonist that increases cardiac output by increasing contractility (point D); and both fluids and dobutamine (point E). Other catecholamines such as dopamine and norepinephrine, which both increase myocardial contractility and reduce venous capacitance, also increase afterload and have variable effects on cardiac output and venous return depending on which effect is dominant. Resistance to therapy may be noted if myocardial damage is sufficiently severe to flatten the Starling function curve to the point that increasing Pmc has little effect on increasing venous return/cardiac output and insufficient functional myocardium remains to respond to inotropes with increased contractility (a steeper Starling relationship).
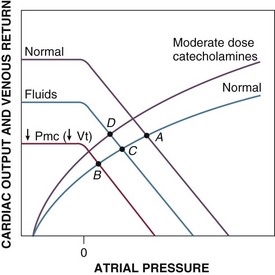
Hypovolemic shock results from decreased Vt, Vs, and Pmc (Fig. 21.E5, point A to B). The venous return curve is shifted downward and to the left, resulting in a reduced venous return and cardiac output at lower right atrial pressures. Although late depression of myocardial contractility with shift of the Starling function curve downward and to the right (analogous to myocardial depression during cardiogenic shock) has been noted in experimental hemorrhagic shock,35,36 this phenomenon is be considered here. Volume therapy, whether with crystalloid or colloid, tends to correct Pmc and venous return toward the original value (point C). Although optimal therapy of hypovolemic shock involves volume resuscitation, low-dose catecholamines exert similar hemodynamic effects; Pmc (and venous return) are augmented by an increase of the stressed volume (Vs), whereas the total (reduced from baseline) circulating volume (Vt) is unchanged (also point C).165,167,168 These changes outweigh any deleterious effect on increasing venous resistance (Rv). Cardiac contractility and vascular resistance are minimally affected at these doses. At moderate infusion rates (and with sympathetic stimulation), cardiac contractility is also augmented (point D). With higher catecholamine infusion rates, venous resistance and afterload may increase to the point of decreasing cardiac output and venous return (not shown). For that reason, vasopressors may be used only with great caution in hypovolemic shock.
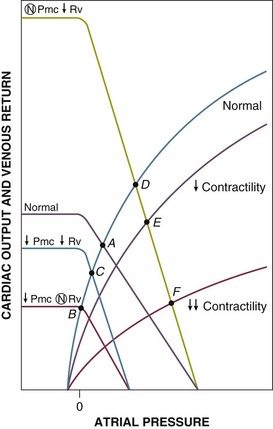
Septic shock is especially complicated. Sepsis may involve elements of hypovolemia, myocardial depression, and altered distribution of cardiac output. Total circulating volume (Vt) and stressed volume (Vs) are decreased due to loss of fluids to the interstitium (third-spacing) and due to insensible losses. Stressed circulating volume (Vs) is further decreased due to active dilation of small venules/veins resulting in increased venous capacitance. This increase in unstressed volume (Vt) and decrease in stressed volume (Vs) have been confirmed in experimental animal models of canine and porcine endotoxemia.120,169 Thus, in unresuscitated septic shock, Pmc is almost universally decreased, resulting in reduced venous return and cardiac output (Fig. 21.E6, point A to B). Sepsis is also associated with dilatation of large veins and shunting of arterial blood flow to low-resistance (fast time constant) vascular beds, both of which decrease venous resistance and tend to augment venous return. Decreased venous resistance, however, does not fully compensate for decreased Pmc in unresuscitated septic shock. Cardiac output remains depressed (point B to C). With fluid resuscitation, Pmc may be corrected back toward normal, allowing the decreased Rv to be manifested by supernormal cardiac output and venous return (point D).120 Patients with septic shock also develop myocardial depression, which is typically masked by the overall increase in cardiac output (point E). In about a fifth of patients, however, myocardial depression is sufficiently severe that venous return and, therefore, cardiac output remain depressed even after resuscitation (point F).
Human data suggest that sepsis is associated with a decrease of total vascular compliance.170,171 However, it is unclear whether this represents a primary septic phenomenon or a neurohumorally mediated compensatory response.170,172,173 Following fluid resuscitation, therapy of septic shock primarily involves catecholamines such as norepinephrine and dopamine. These affect the venous and cardiac function curves as specified earlier, although there are some data to suggest that both vascular and myocardial responsiveness to sympathomimetics may be reduced. In addition, they may also affect vascular compliance similar to the potential compensatory neurohumoral effects described earlier.
Obstructive forms of shock such as those due to pericardial tamponade and tension pneumothorax can also be analyzed in the context of venous-cardiac interactions. For a detailed review of this subject matter, the reader is referred to several excellent reviews.158,174
Ventricular function of each distinct form of shock can also be examined by using end-systolic and end-diastolic pressure-volume analysis. This analysis can be demonstrated graphically using ventricular pressure-volume loops. Changes in stroke volume and ventricular contractility can be examined with respect to ventricular volume and pressure alterations in circulatory shock states. Although this represents a useful approach to the study of circulatory shock physiology, a review of the subject is beyond the scope of this chapter. The interested reader is referred to a number of cogent reviews.175–177
Microvascular Function in Shock
Intrinsic control (autoregulation) of blood flow is thought to occur through two mechanisms. Rapid alterations of microvascular tone are mediated though endothelial stretch receptors so that sudden changes in perfusion pressure can be compensated by opposing changes in vascular resistance in order to maintain perfusion.178 In addition, increases in metabolic activity within tissues and organs are thought to cause local elevation of various metabolites (CO2, H+, etc.), resulting in vasodilation and increased perfusion to match substrate demand.178
Extrinsic control of vascular tone is primarily exerted through the autonomic nervous system. Parasympathetic release of acetylcholine to blood vessels results in nitric oxide and cyclic guanosine monophosphate (cGMP) generation in endothelial cells and vascular smooth muscle leading to vascular relaxation. Increases of sympathetic tone cause local norepinephrine release, activation of vascular alpha-adrenoreceptors, and increased vascular tone (Fig. 21.E7). Under stress, epinephrine and norepinephrine can be systemically released by sympathetic stimulation of the adrenal medulla. Basal control of blood pressure and flow resides in the activity of the renin-angiotensin system.
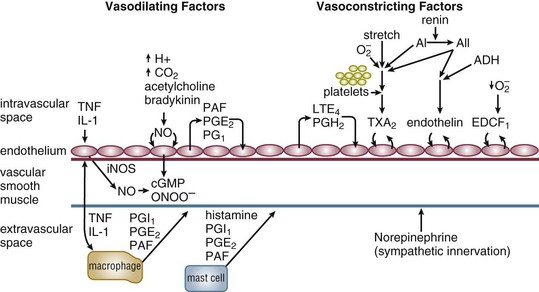
Alterations in microvascular function are effected through pre- and postcapillary sphincters that are sensitive to both intrinsic and extrinsic control mechanisms. Because the exchange of carbon dioxide, oxygen, and other substrates/metabolites as well as the compartmental regulation of fluids occurs at the capillary level, alteration of tone of either sphincter may have varying effects. Opening of either non-nutrient capillary sphincters (microanatomic shunts)85 or increased flow to hypometabolic tissues (functional shunts)86 will result in a suboptimal distribution of substrate supply with increased MVO2. Failure to dilate sphincters supplying metabolically active tissues may result in ischemia and anaerobic metabolism with lactate production. Increased precapillary tone as seen with sympathetic stimulation results in increased blood pressure systemically and decreased hydrostatic pressure locally. This decreased hydrostatic pressure favors redistribution of volume from the interstitium to the circulation. Increased postcapillary tone (relative to precapillary) results in vascular pooling of blood and loss of fluid to the interstitium (due to increased hydrostatic pressure).
Organ blood flow changes are well characterized in shock states. Autoregulation of blood flow is dependent on maintenance of blood pressure within a defined range that varies among organs. The autoregulatory capacity of various organs can be determined by mechanically altering blood pressure in the organ vascular bed. With isolated local hypotension, the brain exhibits dominant autoregulatory capability with the ability to maintain blood flow over a wide range of pressures (30 to 200 mm Hg in dogs).154 Coronary perfusion is also substantially autoregulated between 40 and 100 mm Hg. In contrast, mesenteric and renal blood flow becomes pressure dependent below about 60 mm Hg, whereas the vascular bed of skeletal muscle behaves in a passive manner at pressures outside 50 and 100 mm Hg. Human data suggest that overall, good autoregulation of blood flow exists in humans between pressures of 60 and 100 mm Hg.154 In the context of normal physiology, blood flow is not effectively autoregulated outside this range. Without local adaptation, this would result in a mismatching of blood flow and metabolic demands producing organ failure and the metabolic correlates of shock. However, extrinsic adaptive mechanisms to protect the most vital organs come into play.
During hypovolemia and other hypodynamic forms of shock, extrinsic blood flow regulatory mechanisms overwhelm the autoregulatory response of most vascular beds. Blood flow to the heart and brain are well preserved due to dominant local autoregulation of flow. Blood flow to other organs is reduced relative to the decrease in total cardiac output as organ vascular resistance increases to maintain blood pressure.179 This effect is mediated in part by both sympathetic neural activity and adrenal release of catecholamines.154,179 This adaptive mechanism maintains perfusion to vital organs at mild to moderate levels of reduced cardiac output. If the insult is sufficiently severe or prolonged, organ ischemia and subsequent organ failure may develop. Even if resuscitation restores systemic circulatory hemodynamics, microvascular perfusion abnormalities persist for days.180 Experimental data suggest that perfusion of brain, kidneys, liver, and other splanchnic organs remains impaired following resuscitation from hemorrhagic shock.180 Persistence of inadequate matching of tissue substrate demand and delivery after resuscitation of shock can lead to continued ischemia/hypoxia of some tissues. This may explain why hemorrhagic shock–related tissue injury can be irreversible if its duration and severity are excessive. Animal models suggest this irreversible phase of severe hemorrhagic shock is characterized by vasodilatation of precapillary sphincters.37
During sepsis and septic shock, organ blood flow is disturbed at higher mean arterial pressures, suggesting a primary defect of microvascular function. Cerebral blood flow to the brain in humans has been shown to be depressed even before the onset of septic shock in patients with systemic inflammatory response syndrome.181 This pathologic vasoconstriction (apparently a unique response of the cerebral circulation to sepsis) does not appear to be the cause of septic encephalopathy. Cerebral autoregulation remains intact during sepsis.182 The greater decrease in coronary than systemic vascular resistance during human septic shock may suggest that myocardial autoregulation also remains intact despite the fact that, in contrast to the brain, myocardial perfusion is often increased during septic shock.183,184
Animal models demonstrate that all other vascular beds (splanchnic, renal, skeletal, cutaneous) exhibit decreased vascular resistance, with flow in these beds becoming increasingly dependent on cardiac output. This suggests both an active vasodilatory process and failure of extrinsic control of blood flow.154 Inappropriate levels of splanchnic and skeletal muscle perfusion are also observed in humans during sepsis.88 Other experimental data suggest that sepsis and septic shock are also associated with aberrant distribution of flow within organs.86 In sepsis, vasodilatation and autoregulatory failure of the microvasculature may be responsible for mismatches of oxygen delivery and demand, resulting in anaerobic glycolysis with lactate production despite increased mixed venous oxygen saturation.
During both irreversible hemorrhagic shock and septic shock, peripheral vascular failure results in worsened matching of tissue demand and substrate supply, leading to failure of all organs and death. Among the potential responsible mechanisms are (1) tissue acidosis,185 (2) catecholamine depletion and mediator-related vascular resistance to catecholamines,186 (3) release of vasodilating and vasoconstricting arachidonic acid metabolites,187 (4) decreased sympathetic tone due to altered central nervous system perfusion,188 and (5) pathologic generation of nitric oxide by vascular smooth muscle cells.38,189
In addition to vasomotor dysfunction, shock is associated with other microvascular pathology. Prime among these is disruption of endothelial cell barrier integrity. The endothelial layer is responsible for maintaining oncotic proteins (mostly albumin) within the circulatory space. During shock, capillary permeability increases, resulting in a loss of plasma proteins into the interstitium. Endothelial injury, through the action of neutrophil-generated free radicals190 and nitric oxide/peroxynitrite generation,191,192 may account for this phenomenon. The release of vasoactive intermediaries such as histamine, bradykinin, PAF, leukotrienes, and TNFα appear to drive this pathologic process. Injury is initiated by leukocyte-endothelial cell interactions via adhesion molecules (integrins, selectins) that allow emigration of neutrophils to the tissues. Blockade of such activity or depletion of neutrophils attenuates tissue injury in animal models of shock.193 With the loss of plasma proteins, the plasma oncotic pressure drops, interstitial edema develops, and circulating volume falls.
There is also evidence of intravascular hemagglutination of red cells, white cells, and platelets in almost all shock syndromes.86,194 This may be due to primary microvascular clotting leading to microthrombi. Alternately, clotting may occur as a consequence of primary endothelial damage due to circulating cytokines, free radicals produced by reperfusion and neutrophils, or complement activation. In any case, the result may be further endothelial cell injury, microvascular abnormalities, and inadequate distribution of perfusion within tissues. Decreased deformability of erythrocytes due to membrane free radical injury may also play a role in microcirculatory alterations in hemorrhagic and septic shock.195
Mechanisms of Cellular Injury in Shock
Although different forms of shock have their different precipitants, they do share common mechanisms of cellular dysfunction and injury. Eventually, events at the cellular level result in organ dysfunction and death. The pathogenesis of the cellular dysfunction is a combination of the interrelated precipitants and consequences of shock, including (1) ischemia, (2) inflammation, and (3) free radical injury (Fig. 21.E8). Genetic factors are also felt to play a role in an individual’s susceptibility and response to shock.
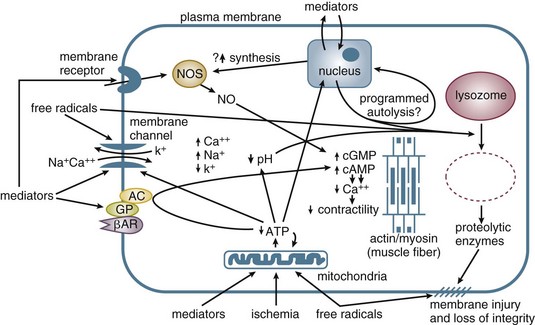
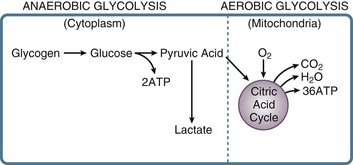
In the stressed preshock phase, physiologic adaptive mechanisms attempt to compensate, and perfusion to vital organs is maintained. With the onset of shock, these mechanisms fail, and thus oxygen delivery to tissues is not maintained. At the cellular level, a lack of oxygen prevents the mitochondrial citric acid cycle from functioning, and pyruvate accumulates (Fig. 21.E9). The failure of aerobic respiration requires shunting of pyruvate into the lactic acid pathway in order to recycle NAD and allow glycolysis to continue. However, the loss of the aerobic citric acid cycle results in a large decrease in adenosine triphosphate (ATP) production (net 2 molecules of ATP per molecule of glucose, versus a theoretical net of 38 molecules of ATP per glucose molecule in the entire aerobic pathway). In addition, shunting to the lactic acid pathway results in the accumulation of hydrogen ions. Thus, loss of perfusion results in a rapid decrease in cellular energy stores, with inadequate ATP regeneration rates. As ATP is the primary source of energy in the cell, energy-dependent cellular systems cease functioning. This includes energy-dependent enzymes, maintenance of transmembrane gradients including electrical potential,196 mitochondrial function,197 carbohydrate metabolism,198 and even the glycolysis pathway itself, which requires two ATP molecules to prime each glucose molecule at the entry point into the pathway. Some cell types are more sensitive than others to the depletion of energy stores, such as in the liver and kidney, but eventually all organs are affected.196,199–202 With the loss of energy-dependent cell maintenance functions, ultrastructural breakdown, including mitochondria, occurs.203 Systemically, the combination of worsening acidosis and loss of energy stores results in a positive feedback loop of worsening shock, organ failure, and eventual death.
Ischemia is felt to play a minor role in early sepsis, with high cardiac outputs maintaining perfusion to more sensitive, higher energy-consuming tissues, and that ATP levels remain normal in these tissues, with mitochondrial function preserved. Instead, early septic organ dysfunction results from other causes.101,102,105 However, localized areas of ischemia are not ruled out. Potentially, microthrombi may result in small areas of decreased microcirculatory flow, resulting in localized ischemia. Evidence to support a role for ischemia in cellular dysfunction in sepsis includes oxygen-supply–dependent oxygen consumption, washout of organic acids from ischemic areas in patients in septic shock on vasodilators, and elevated ATP degradation products. In any case, the role of ischemia in septic shock remains a question. For example, alterations of liver and skeletal muscle transmembrane potential occur early in shock prior to the decrease in levels of high-energy phosphates and onset of hypotension. Further, this membrane defect is not prevented by the administration of membrane-permeable forms of high-energy phosphates such as ATP-MgCl2.204
Inflammation, on the other hand, is a major contributor to the development of septic shock and its effects on cellular and organ dysfunction.205,206 Other forms of shock also activate the inflammatory cascade—for example, hemorrhagic shock with tissue trauma.207 Indeed, shock is associated with systemic activation of the inflammatory cascade. The resulting cellular and tissue dysfunction, and thus hypermetabolic state, contributes to organ dysfunction and failure. The inflammatory cascade in sepsis can be roughly broken down into initiation, transduction, and release of inflammatory mediators.
Initiation of the inflammatory cascade in sepsis is thought to rely largely on the innate immune response.206,208,209 This ancient immune pathway evolved to recognize non-self molecular signatures, produced by pathogens including bacteria, yeast, and viruses, which have remained invariant over time. These signatures are called pathogen-associated molecular patterns (PAMPs), and they are recognized by a series of receptors known as pattern recognition receptors (PRRs). In addition, signature host proteins can also be released extracellularly in response to cellular damage, and these proteins, also called alarmins, are known as damage-associated molecular patterns (DAMPs), and these too may be recognized by PRRs. Thus, both non-self PAMPs and self-expressed DAMPs may lead to initiation of the inflammatory cascade via the PRRs.
The TLRs and CLRs, being transmembrane, are presented on the cell surface or within the lumens of endosomes or lysosomes.206,210,211 Ten families of TLRs have been identified in humans. Different TLR families on the cell surface recognize various bacterial lipoproteins, lipopolysaccharides, or flagellins, with sources that may be bacterial, viral, or protozoan. In contrast, TLR families located in endosomal lumens recognize ssRNA, dsRNA, and modified CpG-DNA, which may be of viral or bacterial origin. Interestingly, different individual families of TLRs can form both homodimers and heterodimers on the cell surface, increasing their range of ligand recognition. Meanwhile, the CLRs are presented on the cell surface, and being lectins, they recognize polysaccharides present in yeast and fungi, mycobacteria, and viruses.
In contrast, the NLRs and RLRs are intracellular and require that their ligands be present in the cytoplasm of the cell.211 NLRs recognize bacterial peptidoglycans; the RLR ligands consist primarily of dsRNA, cytoplasmically produced either by dsRNA viruses or as an intermediate step in the replication of ssRNA viruses.
The TLRs have been shown to use two primary pathways.211 All the TLRs, with the exception of TLR3, use a pathway dependent on the adaptor protein MyD88, which binds to the intracellular sections of the TLRs; for some of the TLRs, another adaptor protein is required to bind MyD88. Upon activation of the TLR, MyD88 recruits a serine/threonine kinase, IRAK-4, in turn recruiting further kinases such as IRAK-1 and IRAK-2. Ultimately, the cytoplasmic cascade results in phosphorylation and degradation of IκB, an inhibitor of the transcription factor NF-κB. This allows NF-κB to enter the nucleus, resulting in the activation of transcription of a suite of inflammatory mediators and modulators. In contrast, TLR3 uses a pathway dependent on the TRIF adaptor protein, which also ultimately results in NF-κB activation. In both pathways, other adaptor proteins may be required for individual TLRs to activate these cascades; for example, TLR4 can also utilize the TRIF-dependent pathway but is dependent on the TRAM adaptor protein to do this. Different cell types may express different adaptor proteins, resulting in intertissue variation in the cascades. In addition, protein inhibitors of the cascades can be expressed variably in different cell types.
In contrast, the NLRs act via formation of a multiprotein complex, termed the inflammasome.206,211,212 Central to this cascade are the caspases,213 a family of cysteine proteases. Upon activation, the NLR will recruit a pro-caspase, along with an adaptor protein ASC, which recruits a second pro-caspase. This complex allows the pro-caspase pair to autoactivate, resulting in active caspase. The best studied of these is caspase 1, although other caspases may be involved. Once activated, caspase 1 in turn activates the inflammatory cytokine IL-1β, allowing its release from the cell. Similarly, caspase1 is also responsible for the activation and release of other cytokines, including IL-18. In this way, the NLR’s prime function is to regulate inflammatory mediator release through a posttranslation system. Crosstalk from the TLR cascades may also activate some NLRs and result in inflammasome formation.
At the center of these cascades is transcriptional regulation of inflammatory cytokines and mediators. The transcription factor NF-κB sits at the end of many of these cascades and plays a crucial role in the regulation of expression of many of these factors.206,211,214 NF-κB is present in an inactive form in the cytoplasm, in complex with repressor proteins.215 When the repressors, known as IκB proteins, are phosphorylated and subsequently degraded, NF-κB is released and transported into the nucleus where it can act on its target sequences. By being stored in inactive form in the cytoplasm, no new NF-κB need be expressed, allowing for rapid response to the signals received by the PRRs. Inflammatory mediators under the control of NF-κB include cytokines such as TNFα, IL-1β and IL-6, enzymes involved in inflammation such as iNOS, and others.206
Inflammatory mediator effects on cellular metabolism are of prime importance in organ dysfunction due to sepsis and septic shock. Circulating inflammatory mediators may also play a substantial role in other forms of shock, including hemorrhagic shock associated with extensive tissue trauma.216,217 Both sepsis and trauma are associated with generalized, systemic activation of the inflammatory response. Resulting cell injury and hypermetabolism may culminate in organ failure. A number of triggers can result in activation of the inflammatory cascade. The best studied is endotoxin from gram-negative bacteria, but other bacterial antigens and cell injury itself can also initiate the cascade. Macrophage production of cytokines such as TNFα, IL-1β, and IL-6 appears to be central.
Tumor necrosis factor-alpha (TNFα) is a 51-kD trimeric peptide produced by macrophages in response to a variety of inflammatory stimuli including bacterial antigens and other cytokines. Circulating levels of TNFα are transiently elevated soon after the onset of shock (particularly septic shock).218 Administration of TNFα to animals or humans results in a hyperdynamic circulatory state (± dose-dependent hypotension) similar to untreated sepsis and septic shock.126 Although clinical trials to date have yielded disappointing results, anti-TNFα strategies protect animals from experimental endotoxic and septic shock.219 Among the many effects of TNFα are the release of IL-1β, IL-6, IL-8, PAF, leukotrienes, thromboxanes, and prostaglandins; stimulation of production and activity of polymorphonuclear leukocytes; promotion of immune cell adhesion to endothelium; activation of coagulation and complement systems; direct endothelial cell cytotoxicity; depression of myocardial contractility; and fever production by the hypothalamus.126,220 Notably, TNFα causes alterations of skeletal transmembrane electrical potential similar to those described in hemorrhagic and septic shock.221 These membrane effects precede hemodynamic alterations, suggesting that TNFα exerts a primary effect on cell metabolism independent of perfusion alterations. Although TNFα appears to be of central importance in the pathogenesis of septic shock, it is also known to be elevated in congestive heart failure222 and hemorrhagic shock.216
Other substances involved in the inflammatory process include IL-1β, which can potentiate the in vivo effects of TNFα; IL-2, which can cause hemodynamic abnormalities in humans; IL-6, which is involved in the acute phase response and has been implicated in septic myocardial depression; interferon-gamma, which promotes the release of other cytokines, enhances adhesion of immune cells, and promotes macrophage activation; IL-10, which is an anti-inflammatory cytokine that limits macrophage generation of pro-inflammatory cytokines; TGFβ, which is another anti-inflammatory cytokine that, in addition to limiting macrophage pro-inflammatory responses, also blocks the effects of proinflammatory cytokines on target cells; endothelin-1, a cytokine that strongly promotes vasoconstriction, particularly in the renal vascular bed, possibly resulting in renal hypoperfusion and decreased glomerular filtration rate; PAF, which stimulates TNFα, thromboxane, and leukotriene release, stimulates free radical formation, and alters microvascular permeability; leukotrienes, which release other arachidonic acid metabolites, alter vascular endothelial permeability, and may mediate vascular and myocardial depression in shock; thromboxanes, which may contribute to altered microvascular vasomotor and permeability function; prostaglandins, which produce fever, induce vasodilatation, and inhibit thrombus formation; and complement fragments C3a and C5a, which constrict vascular smooth muscle, release histamine, and promote chemotaxis.130,220,223
Several newly recognized mediators/mediator groups have been shown to have important roles in shock, particularly septic shock. These include high-mobility group 1 protein (HMG-1), myocardial depressant substances and nitric oxide/peroxynitrites. HMG-1 appears to have a key role in the late pathogenesis of sepsis224,225 and may also have a role in traumatic/hemorrhagic shock.226–228 HMG-1 is a late mediator of inflammation. Mice show increased levels of HMG-1 in serum 8 to 32 hours after endotoxin administration. Patients succumbing to septic shock also demonstrate increased serum HMG-1 levels.224 Administration of HMG-1 to normal and endotoxin-resistant mice induces dose-dependent mortality with signs consistent with endotoxic shock.224,225 Several anti-HMG-1 therapies are in development.228
A circulating myocardial depressant substance is present in the blood of patients with septic shock who exhibit myocardial depression with biventricular dilatation and reduced ventricular ejection fractions.229 Similar substances have been shown to be present in animal models of hemorrhagic shock.230 Other data suggest canine myocardial infarction231 and human cardiogenic shock232 may also be associated with circulating myocardial depressant substances. Serum from appropriate septic patients or animal models depresses myocardial tissue in vitro.32,229 Myocardial depressant substances from both septic and hemorrhagic shock appear to be dependent on calcium.233 The substance implicated in human sepsis may represent a synergistic combination of TNFα and IL-1β that produces depression by inducing myocardial nitric oxide production.229,234,235 TNFα and IL-1β are both elevated in shock and cause similar depression of myocardial tissue.234,236 Other data suggest that IL-6 may have a central role.130,223,237
Another important mediator, nitric oxide (NO), has a vital role in normal intracellular signal transduction.238 Of particular importance to shock, NO is the mediator through which endothelial cells normally cause relaxation of adjacent smooth muscle.238 Endothelial cells, through a constitutive nitric oxide synthetase, produce picomolar quantities of nitric oxide in response to a number of vasodilatory mediators such as acetylcholine and bradykinin. This NO diffuses to adjacent smooth muscle and activates guanylate cyclase to produce cyclic GMP, which affects vascular relaxation. Nitrovasodilators bypass nitric oxide synthetase to relax smooth muscle directly though the guanylate cyclase pathway. During septic shock, an inducible NO synthetase capable of producing nanomolar quantities of NO is generated in vascular smooth muscle.189,238 Studies have also implicated NO in late vascular dysfunction seen in hemorrhagic shock.38 Nitric oxide–mediated generation of cyclic GMP may explain the profound loss of arterial vascular tone and venodilatation seen in septic shock189,239 and may, in part, explain the irreversible vascular collapse seen late in hemorrhagic shock.38 A potential role for NO in inflammation-associated edema and third-spacing during shock has also been suggested.191 The in vitro myocardial depressant effects of TNFα, IL-1β, and serum from septic humans may be mediated by a similar NO– and cyclic GMP–dependent pathway.126,234
An alternative pathway by which NO may play a role in the cardiovascular pathophysiology of shock and sepsis was described by Beckman and colleagues in 1990.240 Peroxynitrite (ONOO-), a highly reactive oxidant, is produced from the interaction of superoxide (OH-) and nitric oxide (NO-). It is known to react rapidly with proteins, lipids, and DNA during sepsis and shock states.241–244 Lipids may be peroxidized, and although proteins may be oxidized, nitrated, or nitrosated, the latter result in nitrotyrosine residues.240,245,246 Peroxynitrite inactivates mitochondrial aconitase disrupting the Krebs cycle and otherwise interferes with ATP production and utilization,247–252 an activity similar to that described for NO.253–255 It also generates DNA strand breaks leading to poly-ADP ribose synthetase (PARS) activation that may itself have significant pathophysiologic effects.256,257 Peroxynitrite, like NO, also activates guanylate cyclase in vascular tissues.258,259 In the periphery, the result may be cellular energetic failure, vascular contractile dysfunction (vasodilation), and reperfusion injury.256,260–262 Many other molecular targets of NO relevant to the cardiovascular system exist and are well reviewed elsewhere.245,263
Free radical injury induced by reperfusion or neutrophil activity is another mechanism of organ injury during hemorrhagic and septic shock as well as burns and myocardial infarction.264 During tissue ischemia, oxygen deficiency leads to accumulation of ATP degradation products including adenosine, inosine, and hypoxanthine (Fig. 21.E10).265 With resuscitation and reperfusion of ischemic areas, oxygen drives the generation of superoxide (O2−), the most common precursor of reactive oxidants, by xanthine oxidase, in endothelial cells. Most of the superoxide is converted, either spontaneously or through superoxide dismutase, to hydrogen peroxide (H2O2−). This further reacts to produce tissue-damaging hydroxyl radicals (or other highly reactive free radicals).264 These radicals interact with critical cell targets such as the plasma membrane, lipid membranes of organelles, and various enzymes, resulting in cell lysis and tissue injury. Oxidant activity, directly and through endothelial damage, attracts and activates neutrophils, resulting in amplification of superoxide generation by a neutrophil NADPH-oxidase and in further tissue damage due to neutrophil protease release.264 Injured tissue may release xanthine oxidase into the circulation, resulting in systemic microvascular injury.266
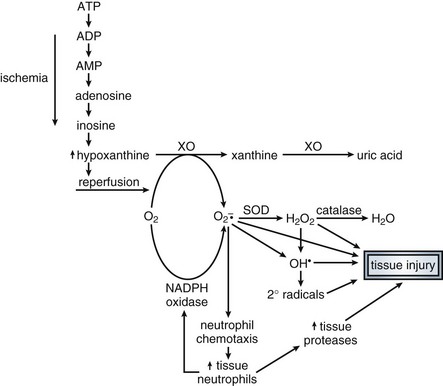
A parallel process is found during reperfusion of ischemic myocardium following myocardial infarction.267 Thrombolytic therapy or balloon angioplasty results in sudden delivery of oxygen to ischemic myocardium. Although substantial salvage of myocardial function results, free oxygen radical–mediated reperfusion injury can contribute to myocardial “stunning.”268 Cardiogenic shock during this phase may resolve as the reperfusion injury settles. Free radical damage likely also plays a role in tissue damage during sepsis and septic shock. Following activation by inflammatory mediators and during phagocytosis, polymorphonuclear leukocytes undergo a respiratory burst during which they consume oxygen and generate both superoxide and hydrogen peroxide through a membrane-associated NADPH-oxidase.264 Macrophages similarly produce oxygen radicals upon activation. Activation also enhances adhesion and tissue migration of leukocytes so that both vascular endothelial and parenchymal tissue damage may result. Free radical injury may play an important role in the development of organ failure following shock.269
The clinical presentation of shock, progression of the syndrome, and final outcome may be substantially controlled by genetic factors.270 Genetic factors have been best studied in septic shock. Studies have demonstrated that the human TNFα promoter polymorphism, TNF2, imparts an increased susceptibility to and mortality from septic shock.271 Other studies suggest increased TNFα generation, severity of sepsis, and mortality with another human TNFα gene polymorphism.272 A specific locus on chromosome 12 in mice has been shown to be associated with resistance to mortality due to TNFα-induced shock.273 A human IL-1β receptor antagonist gene polymorphism has been linked to increased susceptibility to sepsis.274 Several additional linked polymorphisms have been described.275 It appears likely that gene polymorphisms may play similar roles in other forms of shock.
Beyond the role of gene alleles in the development and clinical response to shock, the progression of irreversible circulatory shock and MODS may have its basis in genetically driven vascular or parenchymal responses. Production of cytokines by macrophages during shock requires acute expression of the genes coding for TNFα, IL-1β, and other proinflammatory cytokines. The production of adhesion molecules by endothelial cells and inducible nitric oxide synthetase by vascular smooth muscle during shock requires active up-regulation of gene expression. Both events are thought to be key to the development of MODS following shock in humans. In addition, human and animal research indicates that apoptosis, a genetically programmed process of cell autolysis, occurs in a variety of organs during shock and subsequent organ failure.276,277 Data suggest that a variety of transcription factors may be activated in models of sepsis in association with the process.278 Further research should elucidate the important link between irreversible/refractory shock/shock-associated MODS and genetically programmed cell responses to inflammatory stimulation or injury.
constriction results in a fall in glomerular filtration rate and urine output. In addition, decreased renal perfusion pressure, sympathetic stimulation, and compositional changes in tubular fluid153 result in renin release from the juxtaglomerular apparatus. Renin release leads to the adrenal cortical release of aldosterone (via angiotensin II), which increases sodium reabsorption in the distal tubules of the kidney in exchange for potassium or hydrogen ion.153 Angiotensin II also exerts a powerful direct vasoconstricting effect (particularly on mesenteric vessels) while increasing sympathetic outflow and adrenal epinephrine release. As noted, vasopressin (antidiuretic hormone) release occurs through activation of right atrial low pressure. Angiotensin II augments this release by increasing sympathetic outflow. The release of vasopressin from the posterior pituitary results in water retention at the expense of osmolarity. Hyponatremia can result. Vasopressin, like angiotensin II, also results in vasoconstriction, particularly of the splanchnic circulation.
Redistribution of blood flow during shock has already been discussed. Increased sympathetic vasoconstrictor tone, systemic release of epinephrine from the adrenals, vasopressin, endothelin, and angiotensin II cause vasoconstriction in all sensitive vascular beds including the skin, skeletal muscle, kidneys, and splanchnic organs.154 Dominant autoregulatory control of blood flow spares brain and heart blood work from these effects. Redistribution of flow to these vital organs is the effective result.
The effects of decreased delivery of oxygen to the tissues during shock can be attenuated by local adaptive responses. Hypoperfusion and tissue ischemia will result in local acidosis due to decreased clearance of CO2 and anaerobic metabolism. Local acidosis decreases the affinity between oxygen and hemoglobin at the capillary level.153 The resultant rightward shift of the oxyhemoglobin dissociation curve allows greater unloading of oxygen from hemoglobin for a given PO2. Tissue ischemia is also accompanied by decreased tissue PO2 (relative to normal), which further augments the unloading of oxygen. Pyrexia associated with sepsis may also contribute to a rightward shift of the oxyhemoglobin dissociation curve, whereas hypothermia is associated with a leftward shift. For that reason, maintenance of normothermia during resuscitation from shock helps to optimize oxygen unloading.
Organ System Dysfunction Due to Shock (Table 21.3)
Central Nervous System
Central nervous system neurons are extremely sensitive to ischemia. Fortunately, the central nervous system vascular supply is highly resistant to extrinsic regulatory mechanisms. Although cerebral perfusion is clearly impaired in shock, flow remains relatively well preserved until the later stages.284,285 Absent primary cerebrovascular impairment, cerebral function is well supported until mean arterial pressure falls below approximately 50 to 60 mm Hg.286 Eventually, irreversible ischemic injury may occur to the most sensitive areas of the brain (cerebral cortex). Before this fixed injury, an altered level of consciousness, varying from confusion to unconsciousness, may be seen depending on the degree of perfusion deficit. Disturbances of acid/base/ electrolytes may also contribute. Electroencephalographic (EEG) recordings demonstrate nonspecific changes compatible with encephalopathy. Sepsis-related encephalopathy may occur at higher blood pressures (due in part to the effects of circulating inflammatory mediators) and is associated with increased mortality.287
Heart
The major clinically apparent manifestations of shock on the heart are due to sympathoadrenal stimulation. Increased heart rate, in the absence of disturbances of cardiac conduction, is almost universally present. Vagally mediated paradoxical bradycardia may seen on occasion in severe hemorrhage.40 In patients predisposed to myocardial ischemia or irritability, catecholamine-driven supraventricular tachycardias and ventricular ectopy with ischemic electrocardiogram (ECG) changes are not common. Like the brain, the blood supply to the heart is autoregulated. This, in combination with the resilient nature of myocardial tissue, renders it resistant to sympathetically driven vasoconstriction and shock-related hypoperfusion injury. Overt necrosis does not typically occur, although evidence of cellular injury may be present.
Most forms of shock are associated with increased contractility of healthy myocardium. Regardless, shock can have a substantial impact on myocardial contractility and compliance. Hypotension during cardiogenic (and other forms of shock) is associated with decreased coronary artery perfusion pressure. In patients with coronary artery disease or increased filling pressures, decreased coronary artery perfusion pressure may lead to overt ischemia. Further, circulating myocardial depressant substances contribute to myocardial depression in septic229 and hemorrhagic230 shock. This has been linked to decreased beta adrenoreceptor affinity and density as well as potential defects of intracellular signal transduction involving nitric oxide, G proteins, cAMP, and cGMP.126 Circulating depressant substances may also be present during cardiogenic shock.232
Respiratory System
Early alterations of pulmonary function seen during acute circulatory shock are primarily related to changes in central drive or muscle fatigue. Increased minute volume occurs as a result of augmented respiratory drive due to peripheral stimulation of pulmonary J receptors and carotid body chemoreceptors as well as hypoperfusion of the medullary respiratory center. This results in hypocapnia and primary respiratory alkalosis.153,288 With increased minute volume and decreased cardiac output, the V/Q ratio increases. Unless arterial hypoxemia complicates shock, pulmonary resistance is initially unchanged or minimally increased. Coupled with an increased workload, respiratory and diaphragmatic muscle impairment due to hypoperfusion (manifested by decreased transmembrane electrical potential) may lead to early respiratory failure.289 Adult respiratory distress syndrome (ARDS) due to inflammatory or free radical injury to the alveolar capillary cell layers following established shock may develop as a late cause of respiratory failure.
Kidney
Acute renal failure is a major complication of circulatory shock with associated mortality rates between 35% and 80%.290 Although initial injury manifested by decreased urine output occurs, other clinical manifestations of renal dysfunction (increased creatinine, urea, and potassium) may not be noted for 1 to 3 days. Once hemodynamic stabilization has been achieved, it becomes apparent that urine output does not immediately improve and both serum creatinine and urea continue to rise. The single most common cause of acute renal failure is renal hypoperfusion resulting in acute tubular necrosis (ATN). The most frequent cause of renal hypoperfusion is hemodynamic compromise from septic shock, hemorrhage, hypovolemia, trauma, and major operative procedures. ATN that occurs in the setting of circulatory shock is associated with a higher mortality than in other situations.
Part of the reason for the kidney’s sensitivity to hypoperfusion has to do with the nature of its vascular supply. The renal vascular bed is moderately autoregulated. Increases of efferent arteriolar tone can initially maintain glomerular perfusion despite compromise of renal flow.291 Renal hypoperfusion does not become critical until relatively late in shock when maximal vasoconstriction of renal preglomerular arterioles291 results in cortical, then medullary, ischemic injury.
Decreased urine output in shock can pose a diagnostic dilemma, as it can be associated with both oliguric ATN and hypoperfusion-related prerenal failure without ATN. Indices suggestive of the latter include a benign urine sediment, a urine sodium concentration <20 mEq/L, fractional urine sodium excretion <1%, urine osmolality >450 mOsm/L, and a urine/plasma creatinine ratio >40. Useful markers of acute renal failure due to ATN include hematuria and heme granular casts, a urine sodium concentration >40 mEq/mL, fractional excretion of urine sodium to >2%, urine osmolarity <350 mOsm/L, and a urine:plasma creatinine ratio <20.292 Of note, ATN caused by circulatory shock may be associated with urine sodium <20 mEq/L and fractional excretion < 1% if the acute renal injury is superimposed upon chronic effective volume depletion as may be seen with cirrhosis and congestive heart failure.293
Gastrointestinal System
The gut is relatively sensitive to circulatory failure. The splanchnic vasculature is highly responsive to sympathetic vasoconstriction. Typical clinical gut manifestations of hypoperfusion, sympathetic stimulation, and inflammatory injury associated with shock include ileus, erosive gastritis, pancreatitis, acalculous cholecystitis, and colonic submucosal hemorrhage. Enteric ischemia produced by circulatory shock and free radical injury with resuscitation may breach gut barrier integrity with translocation of enteric bacteria and antigens (notably endotoxin) from the gut lumen to the systemic circulation, resulting in the propagation and amplification of shock and MODS.294,295
Liver
Like the gut, the liver is highly sensitive to hypotension and hypoperfusion injury. “Shock liver,” associated with massive ischemic necrosis and major elevations of transaminases, is atypical in the absence of extensive hepatocellular disease on very severe insult.296 Centrilobular injury with mild increases of transaminases and lactate dehydrogenase is more typical. Transaminases usually peak within 1 to 3 days of the insult and resolve over 3 to 10 days. In either case, early increases in bilirubin and alkaline phosphatase are modest. Despite the production of acute phase reactants in early circulatory shock, synthetic functions may be impaired with decreased generation of prealbumin, albumin, and hepatic coagulation factors. After hemodynamic resolution of shock, evidence of biliary stasis with increased bilirubin and alkaline phosphatase can develop, even though the patient is otherwise improving. Postshock MODS involves similar hepatic pathology.
Hematologic System
Hematologic manifestations of circulatory shock tend to depend on the nature of shock. Disseminated intravascular coagulation (DIC), characterized by microangiopathic hemolysis, consumptive thrombocytopenia, consumptive coagulopathy, and microthrombi with tissue injury, is most commonly seen in association with septic shock. Because it is due to simultaneous systemic activation of coagulation and fibrinolysis cascades, it can be differentiated from the coagulopathy of liver failure by determination of endothelial cell–produced factor 8 (normal or increased with hepatic dysfunction). In the absence of extensive tissue injury/trauma, hemorrhagic shock is rarely associated with DIC.297 Dilutional thrombocytopenia is the most common cause of coagulation deficits after resuscitation for hemorrhage.298
Metabolic Alterations
Metabolic alterations associated with shock occur in a predictable pattern. Early in shock, when hemodynamic instability triggers compensatory responses, sympathoadrenal activity is enhanced. Increased release of ACTH, glucocorticoids, and glucagon and a decreased release of insulin results in glycogenolysis, gluconeogenesis, and hyperglycemia.281,299 An increased release of epinephrine results in skeletal muscle insulin resistance sparing glucose for use by glucose-dependent organs (heart and brain). Late in shock, hypoglycemia may develop, possibly due to glycogen depletion or failure of hepatic glucose synthesis. Fatty acids are increased early in shock but fall later as hypoperfusion of adipose containing peripheral tissue progresses. Hypertriglyceridemia is often seen during shock as a consequence of catecholamine stimulation and reduced lipoprotein lipase expression induced by circulating TNFα.299 Increased catecholamines, glucocorticoids, and glucagon also increase protein catabolism, resulting in a negative nitrogen balance.299
Immune System
Immune dysfunction, frequent during and after circulatory shock and trauma, rarely has immediate adverse effects but likely contributes to late mortality. Underlying mechanisms of immune dysfunction include ischemic injury to barrier mucosa (particularly of the gut), leading to anatomic breaches (colonic ulceration) and potential mucosal translocation of bacteria and bacterial products; parenchymal tissue injury due to associated trauma, inflammation, ischemia or free radical injury; and direct ischemic or mediator (immunosuppressant cytokines, corticosteroids, prostaglandins, catecholamines, endorphins)-induced dysfunction of the cellular and humoral immune systems.300,301 In particular, macrophage function is adversely affected during trauma and circulatory shock. A decrease in antigen presenting ability impairs the activation of T and B lymphocytes. Associated with this defect are a decrease in Ia antigen expression, a decrease in membrane IL-1β receptors, and the presence of suppressor T-lymphocytes. Phagocytic activity of the reticuloendothelial system is also compromised, partially due to an acute decrease in fibronectin levels. Suppression of T lymphocyte immune function is manifested by decreased responsiveness to antigenic stimulation and a decreased helper:suppressor ratio. Decreased production of IgG and IgM suggests B cell suppression. Nonspecific immune suppression is expressed as decreased neutrophil bactericidal function, chemotaxis, opsonization, and phagocytosis.
Resuscitation agents used in shock may also substantially depress immune function. For example, red blood cell transfusion used in traumatic hemorrhagic shock and in support of oxygen transport in septic shock302 has been shown to suppress immune function and lead to increased infections (and improved allograft survival).303,304 Similarly, dopamine, used for hemodynamic support in shock, has been shown to suppress pituitary production of prolactin (required for optimal immune function), thereby suppressing T-cell proliferative responses.305 Thus, dopamine may contribute, along with stress-induced increases in immunosuppressive glucocorticoids, to T-cell anergy seen in critically ill patients.
Diagnostic Approach and Evaluation
Because shock is the common end point of a variety of insults, evaluation and management for all forms of shock involve a common approach (Box 21.4).
Clinical Evaluation
Impending shock is characterized by the typical compensatory response to cardiovascular stress. Tachycardia, tachypnea, and oliguria (<0.5 mL/kg/h) are usually present. Cool extremities are seen in hypodynamic shock. The blood pressure may be elevated or normal with maximal sympathetic stimulation. With progression, however, blood pressure falls, whereas pulse pressure narrows (except in the case of distributive shock). Frank hypotension (mean arterial pressure <60 to 65 mm Hg in adults) may ensue. The chronic level of blood pressure must be considered. Normotension in a normally hypertensive patient may denote a critical degree of hypoperfusion. With further progression anuria may develop, extremities may become mottled and dusky (except in distributive shock), and the sensorium may become clouded. It is important to note that clinical parameters can underestimate initial resuscitative requirements in critically ill subjects including those with septic shock.306–309
Laboratory Studies
Lactate levels (particularly serial determinations) are of use in assessment of prognosis. Substantial data suggest that lactate levels, as a marker of tissue oxygen debt, can predict outcome in shock.45,46,310–314 The utility of lactate assessment is limited by the fact that it is a relatively late marker of tissue hypoperfusion.315,316 Significant tissue ischemia and injury are present by the time it is elevated. In addition, the liver clears lactate. Liver failure may markedly increase elevated lactate levels during hypoperfusion. Conversely, normal hepatic clearance may obscure limited lactate production by ischemic tissues. Glycolysis and alkalosis will also nonspecifically increase lactate levels.317,318 However, since in the appropriate setting, arterial lactate levels beyond 2 mEq/L are associated with increased mortality,46,50,312,313,319 such levels should be considered to represent tissue ischemia in the absence of another clearly defined etiology. The adequacy of resuscitation of shock can also be assessed using serial changes of systemic lactate.314,319,320 Resolution of elevated lactate, however, may lag following the implementation of effective resuscitation.321
Central venous oxygen saturation can also be used to assess the prognosis and efficacy of resuscitation in shock states including septic shock.302,309,322,323 However, central venous saturation, which has been suggested to represent a reliable marker of resuscitation efficacy in sepsis,302 does not appear to correlate well with lactate clearance in some sepsis studies, perhaps reflecting elements other than oxygen debt in those conditions.320 Poor correlation with mixed venous oxygen saturation has also been noted in septic shock.324
Invasive Hemodynamic Monitoring
All patients suspected of having circulatory shock should have an indwelling arterial pressure catheter placed. Blood pressure assessment by manual sphygmomanometry or automated noninvasive oscillometric techniques may be inaccurate during shock due to marked peripheral vasoconstriction.325 In addition, neither technique supplies continuous monitoring of the rapidly changing hemodynamic status of unstable patients. Further, an arterial catheter allows ready access for arterial blood gas samples and other laboratory tests. In most cases, a peripheral site, such as the radial artery, is utilized. However, given the potential for disparity of pressures between central and peripheral sites,326 if marked peripheral vasoconstriction due to either sympathetic stimulation or exogenous catecholamines obscures the peripheral pulses, a central site such as the femoral artery may be preferred.
Central venous pressure monitoring is frequently used during the perioperative period to assess the intravascular volume status in patients without critical illness. Because of the relatively stable hemodynamic status of these patients and the questionable benefit of pulmonary artery catheterization in these patients, such an approach is adequate. Similarly, CVP monitoring for otherwise healthy patients being resuscitated for hypovolemic shock may provide useful data. In the appropriate clinical context, CVP monitoring may also occasionally be useful in differentiating among different forms of shock (e.g., low CVP in hypovolemic shock, high CVP in cardiac tamponade). As a rule, though, CVP monitoring is inadequate for the hemodynamic assessment of critically ill patients, particularly those with shock. A number of studies have conclusively shown that central venous pressure does not accurately estimate left ventricular preload in critically ill patients.327,328
The use of the flow-directed balloon-tipped pulmonary artery catheters with thermodilution cardiac output determination capability has been the standard of practice for the hemodynamic assessment of circulatory shock. Their use is supported by a number of studies that demonstrate that experienced physicians cannot accurately determine cardiac filling pressures or cardiac output based on clinical evaluation alone.329,330 In addition to cardiac output determination, pulmonary artery catheters provide continuous monitoring of central venous and pulmonary artery waveforms and pressures. Pulmonary artery occlusion pressure (as an estimate of left ventricular end-diastolic pressure and volume) and waveform can be obtained intermittently. Waveform analysis may be useful in cardiovascular diagnoses of cardiac tamponade, restrictive cardiomyopathy, congestive heart failure, ventricular hypertrophy, and mitral or tricuspid regurgitation. These devices also allow withdrawal of blood from the pulmonary artery, enabling the determination of MVO2 in order to verify sufficient oxygen delivery during hypodynamic shock. In addition, they can demonstrate evidence of right heart and pulmonary artery oxygen saturation “stepups” for the diagnosis of left-to-right shunts associated with cardiac anatomic abnormalities such as ventricular septal defect. In shock, the flow-directed balloon-tipped pulmonary artery catheter is useful for etiologic classification, determination of optimal management, and to follow the response to therapy. Typical hemodynamic profiles of different forms of shock have been described in Table 21.1.
The utility of pulmonary artery catheterization has been questioned. In a case-matched study, researchers suggested that increased resource utilization and mortality were associated with the use of pulmonary artery catheters in the ICU.331 A series of randomized studies performed since then have reported on the role of the pulmonary artery catheter (PAC) in the setting of major noncardiac surgery,332 congestive heart failure (CHF),333 sepsis and adult respiratory distress syndrome (ARDS),334 acute lung injury,335 and in the general ICU setting.336,337 In the perioperative management of patients undergoing major noncardiac surgery, the use of a PAC did not impact mortality and was associated with a greater incidence of pulmonary embolism.332 In critically ill patients diagnosed with CHF, management directed with the use of a PAC did not influence mortality but resulted in more in-hospital adverse events.333 Another study comparing the use of a PAC versus central venous monitoring in acute lung injury found no difference in organ failure or mortality.335 A large trial evaluating the role of the PAC in the management of patients with ARDS secondary to sepsis also found no significant differences in mortality if a PAC was utilized or not.334 Two randomized studies of general ICU patients similarly concluded that the use of PAC among critically ill patients neither increased nor decreased mortality.336,337 Most, although not all, meta-analyses have shown no consistent benefit with the PAC.338–341 These studies failing to show a lack of clinical benefit have been paralleled by other studies questioning some of the basic premises of PAC utility. Several studies have shown a lack of correlation between PAOP/CVP and ventricular volumes in the critically ill.342,343 One study has even demonstrated that PAOP and CVP fail to predict ventricular filling volume, cardiac performance, or the response to volume infusion in normal volunteer subjects.344 Despite these data, no studies have examined the use of pulmonary artery catheters in cases of shock specifically. Nonetheless, the use of the PAC in the United States has decreased dramatically since the 1990s.345
One parameter that a pulmonary artery catheter uniquely provides is mixed venous oxygen saturation. This measure may assess the adequacy of resuscitation of low output states prior to the presence of anaerobic metabolism (as signified by increased lactate). MVO2 rises with increases of perfusion above requirements and falls, with increasing oxygen extraction ratio, as perfusion becomes inadequate (Fig. 21.3). Normal MVO2 falls within the 65% to 75% range. During myocardial infarction, saturations of less than 60% are found with congestive heart failure, and less than 40% with cardiogenic shock.323 Lactate accumulation and supply-dependent oxygen consumption begin to appear as saturation levels fall below 30% to 40%.346,347 MVO2 is especially useful in determining whether low cardiac outputs indicate supply-dependent oxygen consumption (MVO2 low) or normally depressed metabolic demands (normal MVO2). Due to the maldistribution of perfusion in distributive shock (or substrate utilization defect in septic shock) and left-to-right shunting in cardiogenic shock associated with ventricular septal defects, MVO2 is not useful in assessment in those conditions. Central venous oxygen saturation (cSVO2) has been proposed as an alternate way to examine the adequacy of resuscitation.302,309
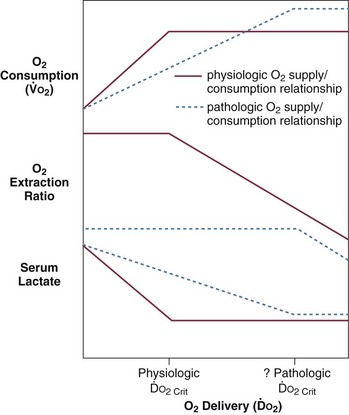
Ancillary Monitoring Techniques
Oximetry
Because oxygen delivery is dependent on arterial oxygen saturation, pulse oximetry should, in theory, provide useful data during circulatory shock. However, limitations of the technique include the fact that ambient light sources, dyshemoglobinemias (methemoglobin, carboxyhemoglobin), lipemia, and hypothermia can affect results.348 Motion artifact may generate a false signal.349 Shock-associated vasoconstriction also impairs signal aquisition. One study has shown that a cardiac index less than 2.4 L/min/m2 is associated with signal loss.350 Although these problems limit the utility of pulse oximetry in the acute management of circulatory shock, the technique may be more helpful during postresuscitation monitoring. At this time, sequential arterial blood gases provide more reliable data during acute shock.
Transcutaneous and transconjunctival oxygen tension measurement are newer noninvasive techniques used for determining tissue oxygen tension, which show some promise in the management of patients at risk for or following the resuscitation of circulatory shock. In the absence of shock, transcutaneous probes reflect arterial oxygenation.351 A number of studies, however, have suggested that if arterial oxygen tension is stable, the devices may be of use in assessing global or regional changes in perfusion and oxygen metabolism.352–355 Global hypoperfusion due to low output shock (as well as local vasoconstriction) results in decreased transcutaneous oxygen tension and a decreased ratio of transcutaneous to arterial oxygen tension.353 Decreased transcutaneous oxygen tension, seen early in hemorrhagic shock, may precede hypotension.354,355 Decreases in transconjunctival oxygen tension predict hemodynamic collapse in perioperative patients352 and death in patients with cardiogenic shock.355 Transcutaneous and transconjunctival measurement of oxygen tension may also be useful in assessing the adequacy of tissue perfusion during resuscitation from hypodynamic shock.356,357
As anaerobic glycolysis with lactate generation is paralleled by the production of hydrogen ions during hypodynamic shock, noninvasive measurement of tissue pH may provide an attractive, metabolism-based assessment of adequacy of tissue oxygenation/perfusion. Because the stomach is easily accessible, may reflect overall splanchnic perfusion during shock,358 and splanchnic perfusion is known to be altered early in shock,359 most clinical work has focused on gastric mucosal pH. Studies suggest that gastric intramucosal pH correlates closely with systemic and organ oxygen consumption, organ failure, and outcome in critically ill humans.360,361 Normalization of gastric mucosal pH has been suggested as one appropriate target during resuscitation of circulatory shock.362 Limited evidence suggests such an approach may be associated with improved survival.363 However, further supportive studies are required before this can be accepted as an appropriate therapeutic target.
Normal tissue oxygenation is the ultimate expression of normal cardiopulmonary function. Any major perturbation of cardiopulmonary status including incipient shock should ultimately express decreased tissue oxygenation. Because peripheral tissue perfusion is, as part of normal compensatory mechanisms, among the first to become restricted during cardiovascular stress, peripheral tissue oxygenation may serve as an excellent marker of cardiovascular stress. Near-infrared light is known to pass through biologic tissues such as skin and muscle. By illuminating a tissue with a known amount of incident light, the amount recovered depends on the degree of absorption by chromophores within the tissue and the amount of scattering. Only three chromophores are known to absorb light in the near-infrared wavelength spectrum: hemoglobin, myoglobin, and cytochrome aa3. All three are known to vary their absorption of near-infrared light depending on whether they are in an oxygenated or deoxygenated/reduced state. By monitoring these parameters, NIR spectroscopy is a unique tool that allows for real-time assessment of the adequacy of tissue perfusion during both the resuscitation and ongoing management of patients with shock. Studies in both animals and humans have suggested the potential utility of this technique in assessment of shock including hypovolemic/traumatic364–369 and septic.370,371
The application of advanced echocardiography techniques to the ICU environment as intermittent monitoring tools may represent one of the most exciting developments in critical care management. A number of factors have hastened this development. These include questions regarding the safety and efficacy of routine pulmonary artery catheter utilization; the availability of relatively inexpensive, portable echocardiography systems; the application of advanced software algorithms to analyze data; and the development of new, high-resolution echocardiographic techniques including transesophageal and contrast echocardiography. The confluence of these factors has resulted in a substantial increase in the use of echocardiography in the ICU assessing hemodynamic instability and shock. In addition to its long-recognized ability to detect anatomic lesions (pericardial tamponade, pericardial effusion, septal defects, valvular disease, aortic dissection), new techniques allow for the assessment of cardiac output, stroke volume, preload (ventricular volumes), intravascular volume status (inspiratory inferior caval collapse), pulmonary artery pressures, systolic contractility (ejection fraction), diastolic function, and regional motion abnormalities at baseline and under stressed conditions.372 The utility for diagnosis of hemodynamically significant pulmonary embolism has also been established.373 The widespread dissemination of echocardiographic skills among the next generation of intensivists has allowed for a substantial reduction in the use of invasive monitoring techniques including pulmonary artery catheterization.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]21
Circulatory Shock
Introduction
The syndrome of shock in humans is often the final pathway through which a variety of pathologic processes lead to cardiovascular failure and death. As such, it is perhaps the most common and important problem with which critical care physicians contend. The importance of shock as a medical problem can be appreciated by the prominence of its three dominant forms. Cardiogenic shock related to pump failure is a major component of the mortality associated with cardiovascular disease, the leading cause of death in the United States with almost 800,000 deaths annually.1 Similarly, hypovolemic shock remains a major contributor to early mortality from trauma, the most common cause of death in those between the ages of 1 and 45 (approximately 200,000 cases annually.1,2 Finally, despite improving medical and surgical therapy, overall mortality coded as septicemia has increased from the 13th to the 11th most frequent cause of death in the United States.1,3 Most current estimates suggest that there are more than 100,000 cases of septic shock annually in the United States alone.4,5 In addition, all forms of shock increase the probability of other major comorbidities such as serious infection, acute respiratory distress syndrome (ARDS), and multiple organ dysfunction syndrome (MODS).
History
Despite recognition of a posttraumatic syndrome by Greek physicians such as Hippocrates and Galen, the origin of the term shock is generally credited to the French surgeon Henri Francois Le Dran who, in his 1737 “A Treatise of Reflections Drawn from Experience with Gunshot Wounds,” coined the term choc to indicate a severe impact or jolt.6 An inappropriate translation by the English physician Clarke, in 1743, led to the introduction of the word shock to the English language to indicate the sudden deterioration of a patient’s condition with major trauma.6 It was Edwin A. Moses,7 however, who began to popularize the term, using it in his 1867 “A Practical Treatise on Shock after Operations and Injuries.” He defined it as “a peculiar effect on the animal system, produced by violent injuries from any cause, or from violent mental emotions.” Prior to this definition, the rarely used term shock referred in a nonspecific sense to the immediate and devastating effects of trauma, not a specific posttrauma syndrome. Although not entirely accurate by today’s standards, his definition was one of the first to separate the syndrome involving the body’s response to massive trauma from the immediate, direct manifestations of trauma itself.
By the late 1800s, two theories of traumatic shock physiology dominated. The first, based on observations by Bernard, Charcot, Goltz, and others, was proposed by Fischer in 1870.8–10 He suggested that traumatic shock was caused by generalized “vasomotor paralysis” resulting in splanchnic blood pooling. The corollary was that total circulating blood volume is preserved in shock. The second dominant theory, articulated by Mapother in 1879, suggested that decreased cardiac output in traumatic shock is caused by intravascular volume loss due to extrusion of plasma through the vessel wall from the intravascular space to the interstitium.11 He proposed that this was a consequence of the failure of “vasodilator nerves” in traumatic shock and subsequent generalized arteriolar vasoconstriction. With the 1899 publication of “An Experimental Research into Surgical Shock” (perhaps the first experimental studies of shock), George W. Crile provided scientific data supporting a variation of the vasomotor paralysis theory.12 After documenting the importance of decreased central venous pressure and venous return in experimental shock due to hemorrhage and demonstrating the potential for intravascular volume replacement as therapy, he proposed that traumatic shock was caused by exhaustion of the overstimulated “vasomotor center” and subsequent generalized relaxation of large vessels (veins) leading to decreased ventricular filling and cardiac output.
Further advances in shock research were substantially driven by military concerns. During World War I, Walter B. Cannon and other physiologists/physicians studied the early clinical response to battlefield trauma. Their work eventually led to the publication of the classic monograph “Traumatic Shock” in 1923.13 Cannon and his colleagues were the first to relate trauma-associated hypotension in a large group of patients to a fall in blood volume, loss of bicarbonate, and accumulation of organic acids. Others, using dye dilution techniques, demonstrated that severity of shock was directly related to the decrease in intravascular volume.14 Clinical data from war casualties also suggested the importance of reduced blood flow (independent of blood pressure) in shock.15 The observation that blood in the capillaries of victims of massive trauma was hemoconcentrated compared to venous blood would lead to the practice of resuscitating trauma patients with dried pooled plasma rather than whole blood in the early part of World War II.16
Although work originating from the battlefields of World War I clearly linked traumatic shock associated with substantial, obvious bleeding to a loss of circulating blood volume, the origin of traumatic shock in the absence of defined hemorrhage was unclear. The accepted explanation for this phenomenon remained a variation of the vasomotor paralysis theory of shock. It was postulated that nonhemorrhagic, posttraumatic shock (“wound shock”) was caused by the liberation of “wound toxins” (histamine or other substances), which resulted in neurogenic vasodilation and peripheral blood pooling. However, after the war, Blalock and others demonstrated in animal models that nonhemorrhagic traumatic shock was due to the loss of blood and fluids into injured tissue rather than circulating toxins resulting in stasis of blood within the circulation.17
Additional advances occurred during World War II. Using injured subjects from the European front, Henry Beecher confirmed that hemorrhage and fluid loss leading to metabolic acidosis was a major cause of shock.18 In the first use of indicator dye techniques in humans for studying blood flow, Cournand and Richard, in 1943, demonstrated that cardiac output was typically reduced in shock.19 They also reinforced Blalock’s findings regarding nonhemorrhagic “wound shock” in trauma patients by demonstrating that circulating blood volume was reduced in such patients through loss of fluid into damaged tissues. The importance of maintaining intravascular volume in traumatic and hemorrhagic shock was supported by the well-known cardiovascular physiologist, Carl J. Wiggers, who published a landmark series of studies20 in the 1940s using a standardized animal model, which showed that prolonged hypovolemic shock resulted in a resuscitation-resistant state that he termed irreversible shock. He defined it as a condition resulting from “a depression of many functions but in which reduction of the effective circulating blood volume is of basic importance and in which impairment of the circulation steadily progresses until it eventuates in a state of irreversible circulatory failure.” Aggressive fluid support became the standard of resuscitation for trauma and shock.
Subsequently, the Korean War fueled the research that demonstrated the relationship of acute tubular necrosis (ATN) and acute renal failure (ARF) to circulatory shock.21 In addition, studies of battlefield casualties clearly demonstrated the relationship between early resuscitation and survival.21 During the Vietnamese conflict, with the widespread use of ventilator technology, the dominant research concern became postshock infection and “shock lung” (ARDS), a concern that has evolved to the present interest in shock-related MODS.
Definitions and Categorization of Shock
The definition of shock has evolved in parallel with our understanding of the phenomenon. As noted, until the late 1800s, the term shock was used to indicate the immediate response to massive trauma, without regard to a specific posttrauma syndrome. The definition consisted of descriptions of its obvious clinical signs. In 1895, John Collins Warren22 referred to shock as “a momentary pause in the act of death,” which was characterized by an “imperceptible” or “weak, threadlike” peripheral pulse and a “cold, clammy sweat.”
Subsequently, with the introduction of noninvasive blood pressure monitoring devices, most clinical definitions of shock added the requirement for arterial hypotension. In 1930, Blalock8 included arterial hypotension as one of the required manifestations of shock when he defined it as “peripheral circulatory failure resulting from a discrepancy in the size of the vascular bed and the volume of the intravascular fluid.” Simeone,23 as recently as 1964, suggested that shock exists when “the cardiac output is insufficient to fill the arterial tree with blood under sufficient pressure to provide organs and tissues with adequate blood flow.”
Current technology, which allows for the assessment of perfusion independent of arterial pressure, has shown that hypotension does not define shock. The emphasis in defining shock is now on tissue perfusion in relation to cellular function. According to Fink,24 shock is “a syndrome precipitated by a systemic derangement of perfusion leading to widespread cellular hypoxia and vital organ dysfunction.” Cerra25 has emphasized supply/demand mismatch in his definition: “a disordered response of organisms to an inappropriate balance of substrate supply and demand at a cellular level.”
Effective tissue perfusion, as opposed to tissue perfusion per se, is an important issue. Effective tissue perfusion may be reduced by either a global reduction of systemic perfusion (cardiac output) or by increased ineffective tissue perfusion due to a maldistribution of blood flow or a defect of substrate utilization at the subcellular level (Box 21.1).
Classification
Although hypovolemic shock associated with trauma was the first form of shock to be recognized and studied, by the early 1900s it became broadly recognized that other clinical conditions could result in a similar constellation of signs and symptoms. Sepsis as a distinct cause of shock was initially proposed by Laennec (1831) and subsequently supported by Boise (1897).26,27 In 1934, Fishberg and colleagues introduced the concept of primary cardiogenic shock due to myocardial infarction.28 Later the same year, Blalock developed the precursor of the most commonly used classification systems of the present.29 He subdivided shock into four etiologic categories: hematogenic or oligemic (hypovolemic), cardiogenic, neurogenic (e.g., shock after spinal injury), and vasogenic (primarily septic shock). Shubin and Weil, in 1967, proposed the additional etiologic categories of hypersensitivity (i.e., anaphylactic), bacteremic (i.e., septic), obstructive, and endocrinologic shock.30 However, as the hemodynamic profiles of the different forms of shock were uncovered, a classification based on cardiovascular characteristics, initially proposed in 1972 by Hinshaw and Cox,31 came to be accepted by most clinicians. The categories include (1) hypovolemic shock, due to a decreased circulating blood volume in relation to the total vascular capacity and characterized by a reduction of diastolic filling pressures and volumes; (2) cardiogenic shock related to cardiac pump failure due to loss of myocardial contractility/functional myocardium or structural/mechanical failure of the cardiac anatomy and characterized by elevations of diastolic filling pressures and volumes; (3) extracardiac obstructive shock involving obstruction to flow in the cardiovascular circuit and characterized by either impairment of diastolic filling or excessive afterload; and (4) distributive shock caused by loss of vasomotor control resulting in arteriolar and venular dilation and (after resuscitation with fluids) characterized by increased cardiac output with decreased systemic vascular resistance. We have adapted these categories into an etiologic/physiologic classification of shock that is summarized in Figure 21.1 and Box 21.2. This figure and box represent our current understanding of the causes and typical hemodynamic features of different forms of shock.
Despite this hemodynamic-based categorization system, it is important to note the mixed nature of most forms of clinical shock. Septic shock is nominally considered a form of distributive shock. However, prior to resuscitation with fluids, a substantial hypovolemic component may exist due to venodilatation and third-spacing. In addition, depression of the myocardium in human septic shock is well documented (see Fig. 21.1).32–34 Similarly, hemorrhagic shock in experimental models has been linked to both myocardial depression35,36 and vascular dysfunction (see Fig. 21.1).37,38 Cardiogenic shock typically presents with increased ventricular filling pressures. However, many patients have been aggressively diuresed prior to the onset of shock and may have a relative hypovolemic component. In addition, systemic vascular resistance (SVR) is only inconsistently increased in cardiogenic shock, suggesting that an inflammatory element may exist under some circumstances. Finally, shock from any cause may cause a deterioration of the coronary perfusion pressure, the difference between mean arterial pressure (MAP) and the higher of left ventricular diastolic pressure or the right atrial pressure, resulting in some degree of myocardial ischemia and myocardial dysfunction.39 Thus, although four categories of shock exist based on hemodynamic profile, clinical shock states tend to combine components of each.
Hypovolemic Shock
Hypovolemic shock may be related to dehydration, internal or external hemorrhage, gastrointestinal fluid losses (diarrhea or vomiting), urinary losses due to either diuretics or kidney dysfunction, or loss of intravascular volume to the interstitium due to decrease of vascular permeability (in response to sepsis or trauma). In addition, venodilatation due to a number of causes (sepsis, spinal injury, various drugs and toxins) may result in a relative hypovolemic state (see Box. 21.2, Fig. 21.1). Hemodynamically, hypovolemic shock is characterized by a fall in ventricular preload resulting in decreased ventricular diastolic pressures and volumes (Table 21.1). Cardiac index (CI) and stroke volume index (SVI) are typically reduced. In addition to hypotension, a decreased pulse pressure may be noted. Due to a decreased output and unchanged or increased metabolic demand, mixed venous oxygen saturation (MVO2) may be decreased and the arteriovenous oxygen content difference widened. Clinical characteristics include pale, cool, clammy skin (often mottled); tachycardia (or if severe shock, bradycardia)7,40; tachypnea; flat, nondistended peripheral veins; decreased jugular venous pulse; decreased urine output; and altered mental status.
Table 21.1
Hemodynamic Profiles of Shock*
*The hemodynamic profiles summarized in this table refer to patients with the diagnosis listed in the left column who are also in shock (mean arterial blood pressure < 60-65 mm Hg).
Modified from Parrillo JE: Septic shock: Clinical manifestations, pathogenesis, hemodynamics, and management in a critical care unit. In Parrillo JE, Ayers SM (eds): Major Issues in Critical Care Medicine. Baltimore, Williams & Wilkins, 1984.
A number of factors may influence the development and hemodynamic characteristics of hypovolemic shock in humans. Studies in animals and humans have demonstrated a clear relationship between the degree of circulating blood volume loss and clinical response.41–44 Acute loss of 10% of the circulating blood volume is well tolerated, with tachycardia the only obvious sign. CI may be minimally decreased despite a compensatory increase in myocardial contractility. SVR typically increases slightly, particularly if sympathetic stimulation augments mean arterial pressure (MAP). Compensatory mechanisms begin to fail with a 20% to 25% volume loss. Mild to moderate hypotension and decreased CI may be present. Orthostasis (with a blood pressure decrease of 10 mm Hg and increased heart rate of 20 to 30 beats/minute) may become apparent. There is a marked increase in SVR and serum lactate may begin to rise. With decreases of the circulating volume of 40% or more, marked hypotension with clinical signs of shock is noted. CI and tissue perfusion may fall to less than half normal. Lactic acidosis is usually present at this stage and predicts a poor outcome.45,46 The case fatality rate can exceed 50% in hemorrhagic shock associated with trauma.47
The rate of loss of intravascular volume and the preexisting cardiac reserve is of substantial importance in the development of hypovolemic shock. As an example, whereas an acute blood loss of 1 L in a healthy adult may result in mild to moderate hypotension with a reduced pulmonary artery occlusion pressure (PAOP) and central venous pressure (CVP),42 the same loss over a longer period of time may be well tolerated due to compensatory responses such as tachycardia, increased myocardial contractility, increased red blood cell 2,3-diphosphoglycerate (2,3 DPG), and increased fluid retention. On the other hand, a similar slow loss may lead to substantial hemodynamic compromise in a person with a limited cardiac reserve, even while the person’s PAOP and CVP remain elevated.
Hypovolemic shock represents more than a simple mechanical response to loss of circulating volume. It is a dynamic process involving competing adaptive (compensatory) and maladaptive responses at each stage of development. Thus, although intravascular volume replacement is always a necessary component of resuscitation from hypovolemia or hypovolemic shock, the biologic responses to the insult may progress to the point where such resuscitation is insufficient to reverse the progression of the shock syndrome. Patients who have sustained a greater than 40% loss of blood volume for 2 hours or more may be unable to be effectively resuscitated.37,41,44 A series of inflammatory mediator, cardiovascular, and organ responses to shock are initiated, which supersede the importance of the initial insult in driving further injury.
Cardiogenic Shock
Cardiogenic shock results from the failure of the heart as a pump (see Box 21.2, Fig. 21.1). It is the most common cause of in-hospital mortality in patients with Q-wave myocardial infarction.48,49 Hemodynamically, cardiogenic shock is characterized by increased ventricular preload (increased ventricular volumes, pulmonary wedge pressure [PWP] and CVP) (see Table 21.1). Otherwise hemodynamic characteristics are similar to those for hypovolemic shock (see Table 21.1). In particular, both involve reduced CI, SVI, and ventricular stroke work indices with increased SVR. Due to inadequate tissue perfusion, the MVO2 is substantially reduced and the arteriovenous oxygen content difference increased. The degree of lactic acidosis may predict mortality.50 Clinically, the specific signs of shock are similar. However, signs of congestive heart failure (volume overload) are typically present in cardiogenic shock. The jugular and peripheral veins may be distended. An S3 and evidence of pulmonary edema are usually found.
Cardiogenic shock is most commonly due to ischemic myocardial injury with a total of 40% of the myocardium nonfunctional.49,51–53 Such damage may involve a single large myocardial infarction or may involve accumulation of damage from multiple infarctions. In addition, viable but dysfunctional “stunned” myocardium may temporarily contribute to cardiogenic shock postinfarction. Cardiogenic shock usually involves an anterior myocardial infarction with left main or proximal left anterior descending artery occlusion. Historically, the incidence of cardiogenic shock due to Q-wave infarction has ranged from 8% to 20%.48,54–56 Although several large studies demonstrate lower incidence rates (4% to 7%) when patients receive thrombolytic interventions,55,57–60 retrospective community studies suggest no overall decrease in the incidence of postinfarction cardiogenic shock or cardiogenic shock mortality (70% to 90%) in the first decades following the introduction of this therapy.48 Further, no trials have demonstrated that thrombolytic therapy reduces mortality rates in patients with established cardiogenic shock.60,61 In contrast, several major studies suggest that mortality of infarction-related cardiogenic shock may be improved by emergent angioplasty.56,62–64 Accordingly, data suggest a reduction in the incidence of acute infarction-related cardiogenic shock to <2% in 2003 in association with widespread use of emergent percutaneous coronary intervention.62 This intervention has also been associated with a reduction of cardiogenic shock mortality risk from 60% to 84% to 43% to 47% in two large analyses.56,62
Mortality is better for cardiogenic shock due to surgically remediable cardiac lesions. Mitral valve failure may be associated with rupture or dysfunction of chordae or papillary muscles due to myocardial ischemia or infarction, endocarditis, blunt chest trauma, or prosthetic valve deterioration and is characterized by “v” waves of greater than 10 mm Hg on a PAOP tracing. Ischemic papillary muscle rupture frequently occurs 3 to 7 days after an infarct in left anterior descending coronary artery territory and may be preceded by the onset of a mitral regurgitant murmur.65 Mortality is high in the absence of surgical therapy.65 Acute aortic valve failure is most commonly due to endocarditis but may involve mechanical failure of prosthetic valves, or aortic dissection. Ventricular septal defects caused by myocardial infarction may also result in the abrupt onset of cardiogenic shock and can be diagnosed by a 5% step up in hemoglobin oxygen saturation between the right atrium and the pulmonary artery (due to left-to-right shunting of blood through the septum).66 As with ischemic papillary muscle rupture, rupture of the intraventricular septum is most frequently seen with occlusions of the left anterior descending artery, a few days after infarction.66
The pathophysiology of cardiogenic shock due to a right ventricular infarction and failure is different from other forms of cardiogenic shock. Although some degree of right ventricular involvement is seen in half of inferior myocardial infarctions, only the largest 10% to 20% result in right ventricular failure and cardiogenic shock.67 These infarctions usually involve part of the left ventricular wall as well. Isolated infarctions of the right ventricle are rare.67,68
Because therapy of this form of shock requires fluid resuscitation and inotropes (rather than vasopressors), differentiation from other causes of cardiogenic shock is crucial. Conditions compromising right ventricular function such as cardiac tamponade, restrictive cardiomyopathy, constrictive pericarditis, and pulmonary embolus are also included in the differential diagnosis. Each of these conditions may present with some of the typical clinical and hemodynamic findings of right ventricular infarction including Kussmaul’s sign, and pulsus paradoxus with elevation and equalization of CVP, right ventricular systolic pressure, pulmonary artery diastolic pressure, and PAOP. Prognosis in this form of cardiogenic shock is distinctly better than that of cardiogenic shock due to left ventricular infarction69,70; however, an inferior infarction with right ventricular injury has a substantially worse prognosis than such an infarction without significant right-sided involvement.71
As with hypovolemic shock, a number of interactions may complicate the development of cardiogenic shock. Optimal cardiac performance in patients with impaired myocardial contractility may occur at substantially higher than normal PAOP (i.e., 20 to 24 mm Hg). Yet patients who develop cardiogenic shock are frequently initially treated with diuretics and may have a degree of hypovolemia (relative to their optimal requirements). Thus, patients should not be diagnosed with cardiogenic shock unless hypotension (MAP < 65 mm Hg) and reduced cardiac output (CI < 2.2 L/min/m2) coexist with an elevated ventricular filling pressure.72 Cautious fluid challenge may be required (in the absence of overt pulmonary edema) to increase the filling pressures to an optimal range. Other interactions include increased right ventricular ischemia due to decreased right coronary perfusion pressure (MAP decreased while right ventricular end-diastolic pressure is increased) and increased right ventricular afterload due to pulmonary hypertension. Right ventricular ischemia may also lead to right ventricular dilatation, septal shift, and impairment of left ventricular function.
Other causes of cardiogenic shock include acute myocarditis, end-stage cardiomyopathy, brady- or tachyarrhythmias, hypertrophic cardiomyopathy with obstruction, and traumatic myocardial contusion (see Box 21.2).
Obstructive Shock
Extracardiac obstructive shock results from an obstruction to flow in the cardiovascular circuit (see Box 21.2, Fig. 21.1). Pericardial tamponade and constrictive pericarditis directly impair diastolic filling of the right ventricle. Tension pneumothorax and intrathoracic tumors indirectly impair right ventricular filling by obstructing venous return. Massive pulmonary emboli (two or more lobar arteries with >50% of the vascular bed occluded), nonembolic acute pulmonary hypertension, large systemic emboli (e.g., saddle embolus), and aortic dissection may result in shock due to increased ventricular afterload.
The characteristic hemodynamic/metabolic patterns are, in most ways, similar to other low output shock states (see Table 21.1). CI, SVI, and stroke work indices are usually decreased. Because tissue perfusion is decreased, the MVO2 is low, the arteriovenous oxygen content difference increased, and serum lactate frequently elevated. Other hemodynamic parameters are dependent on the site of the obstruction. Tension pneumothorax and mediastinal tumors may obstruct the great thoracic veins, resulting in a hemodynamic pattern (decreased CI and elevated SVR) similar to hypovolemia (although distended jugular and peripheral veins may be seen). Cardiac tamponade typically causes increased and equalized right and left heart ventricular diastolic pressures, pulmonary artery diastolic pressure, CVP, and PAOP. In constrictive pericarditis, right and left ventricular diastolic pressures are elevated and within 5 mm Hg of each other. Mean right and left atrial pressures may or may not be equal as well. Massive pulmonary embolus will result in right ventricular failure with elevated pulmonary artery and right heart pressures whereas PAOP remains normal. A systemic saddle embolus or aortic occlusion due to dissection causes peripheral hypotension and signs of left ventricular failure including an elevated PAOP. Clinical signs are similarly dependent on the site of the obstruction.
As with other forms of shock, the time course of development of the insult has a substantial impact on the clinical response. Ischemic rupture of the left ventricular free wall (usually 3 to 7 days after myocardial infarction) leads to immediate cardiac tamponade and shock with as little as 150 mL blood in the pericardium.73–75 Survival requires emergency surgery.74,75 Similar situations may develop with bleeding into the pericardium after blunt chest trauma or thrombolytic therapy. Pericardial tamponade due to malignant or inflammatory pericardial effusions usually develop much more slowly. Although shock may still develop, it usually requires substantially more pericardial fluid (1 to 2 L) to cause critical failure of right ventricular diastolic filling.73 No large reliable studies examining mortality rates with and without therapy in these conditions are available due to the small numbers of cases.
A similar time course–dependent risk is seen with major pulmonary emboli. In those without preexisting cardiopulmonary disease, a massive embolus involving two or more lobar arteries and 50% to 60% of the vascular bed76,77 may result in obstructive shock. However, if recurrent smaller pulmonary emboli result in right ventricular hypertrophy, a substantially larger total occlusion of the pulmonary vascular bed may be required to cause right ventricular decompensation. Analyses have suggested that the presence of shock due to pulmonary embolus (regardless of underlying chronic cardiopulmonary dysfunction) indicates a three- to sevenfold increase in mortality risk with the majority of deaths occurring within an hour of presentation.78,79 An analysis of more than 70,000 unstable (hemodynamic instability or ventilator-requiring) patients with pulmonary embolus in the national inpatient sample shows that mortality in untreated patients is approximately 47%.80 Systemic thrombolysis is associated with a substantial reduction in mortality to 15%. Where available, catheter-directed therapy may be even more efficacious with a lower risk of serious hemorrhage.81 Shock due to pulmonary embolism is an indication for urgent thrombolytic or catheter-directed intervention.
Distributive Shock
Hemodynamically, distributive shock is characterized by an overall decrease in SVR (see Table 21.1). However, resistance in any specific organ bed or tissue may be decreased, increased, or unchanged. Initially, CI may be depressed and ventricular filling pressures decreased. After fluid resuscitation, when filling pressures are normalized or increased, CI is usually elevated. Due to hypotension, left and right ventricular stroke work indices are normally decreased. MVO2 is increased above normal. Concomitantly, arteriovenous oxygen content difference is narrowed despite the fact that oxygen demand is usually increased (particularly in sepsis). The basis of this phenomenon may be that because total body perfusion (CI) is increased, perfusion is not effective in that either it does not reach the necessary tissues or the tissues cannot utilize the substrates presented. As a reflection of this inadequate “effective” tissue perfusion, lactic acidosis may ensue. Clinical characteristics of resuscitated distributive shock include, in contrast to the other forms of shock, warm, well-perfused extremities, a decreased diastolic blood pressure, and an increased pulse pressure. Nonspecific signs of shock include tachycardia, tachypnea, decreased urine output, and altered mentation. In addition, evidence of the primary insult may exist (urticaria for anaphylaxis, spinal injury for neurogenic shock, and evidence of infection in septic shock).
Septic shock (shock due to infection) and sepsis-associated multiple organ failure are the most common causes of death in ICUs of the industrialized world. As many as 800,000 cases of sepsis are admitted every year to American hospitals (comparable to the incidence of first myocardial infarctions) with half of those developing septic shock and about half of those (200,000) dying.82 Since the 1970s there has been a progressive increase in the incidence of and total deaths from sepsis and septic shock.5 The total toll of septic deaths is comparable to deaths from myocardial infarction and far exceeds the impact of illnesses such as AIDS or breast cancer.82,83
Septic shock is caused by the systemic activation of the inflammatory cascade. Numerous mediators including cytokines, kinins, complement, coagulation factors, and eicosanoids are activated or systemically released, resulting in profound disturbances of cardiovascular and organ system function84 (Table 21.2). These mediators, particularly tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), platelet activating factor (PAF), and prostaglandins are thought to mediate reduced peripheral vascular resistance seen in septic shock.
Table 21.2
Inflammatory Mediators in Sepsis and Septic Shock
Adapted from Bone RC: The pathogenesis of sepsis. Ann Intern Med 1991;115:457.
Loss of vascular autoregulatory control may explain some of the typical metabolic findings of sepsis and septic shock. An early theory postulated the existence of microanatomic shunts between the arterial and venous circulations. During sepsis, these shunts were said to result in decreased SVR and increased MVO2.85 However, although microanatomic shunting has been noted in localized areas of inflammation, systemic evidence of this phenomenon in sepsis and septic shock is lacking.85–89 “Functional” shunting due to defects of microcirculatory regulation in sepsis has also been suggested.90,91 Overperfusion of tissues with low metabolic requirements would increase MVO2 and narrow the arteriovenous oxygen content difference. Relative vasoconstriction of vessels supplying more metabolically active tissues would result in tissue hypoxia and lactate production due to anaerobic metabolism. Observations that some capillary beds may be occluded by platelet microaggregates, leukocytes, fibrin deposits, and endothelial damage support this theory.86,90,92 Additional support comes from studies that demonstrate evidence of oxygen supply–dependent oxygen consumption in sepsis.93–97 A third theory suggests that circulating mediators cause an intracellular metabolic defect involving substrate utilization, which results in bioenergetic failure (decreased high-energy phosphate production) and lactate production.98,99 Increased mixed venous oxygen saturation could then be explained by perfusion, which is increased in excess of tissue oxygen utilization capability. However, animal studies using nuclear magnetic resonance (NMR) spectroscopy demonstrate that high-energy phosphates are not depleted in septic animals as is expected in all of these theories.100–102 According to these and other studies, cellular ischemia is not the dominant factor in metabolic dysfunction in sepsis.100–106 Rather, circulating mediators may result in cellular dysfunction, aerobic glycolysis, and lactate production in the absence of global ischemia.101 This position is weakened by data suggesting that increased lactate in septic shock is also associated with decreased pH (which would not be expected in aerobic glycolysis)101 and, to some extent, by studies that support the existence of oxygen supply–dependent oxygen consumption in sepsis.94–97
The trigger for systemic activation of the inflammatory cascade is the presence of gram-negative bacilli in 50% to 75% of cases of septic shock. Gram-positive bacteria account for most of the remainder, but infection with fungi, protozoa, and viruses can also result in septic shock.107–109 Investigations suggest a surprising commonality of signaling mechanisms in septic shock via Toll-like receptors from a broad range of etiologic agents.110–114 Despite aggressive supportive care and antibiotic treatment, mortality is 50% overall and may exceed 70% for gram-negative septic shock.107 Of those succumbing to septic shock, approximately 75% are early deaths (within 1 week of shock), primarily due to hyperdynamic circulatory failure.115 Late mortality is usually due to MODS.115
More than any other form of shock, distributive and, particularly, septic shock involves substantial elements of the hemodynamic characteristics of other shock categories (see Fig. 21.1, Table 21.1). As noted, all forms of distributive shock involve decreased mean peripheral vascular resistance. Prior to fluid resuscitation, distributive shock also involves a relative hypovolemic component. The first element of this relative hypovolemia is an increase of the vascular capacitance due to venodilatation. This phenomenon has been directly supported in animal models of sepsis116–120 and is reinforced by the fact that clinical hypodynamic septic shock (low cardiac output) can usually be converted to hyperdynamic shock (high cardiac output) with adequate fluid resuscitation.115,121,122 Relaxation of vascular smooth muscle is attributed to a number of the mediators known to circulate during sepsis. These same mediators also contribute to the second cause of hypovolemia in sepsis, third-spacing of fluid to the interstitium due to a loss of endothelial integrity. In addition, a number of studies have demonstrated that human septic shock is characterized by myocardial depression (biventricular dilatation and decreased ejection fraction).32–34 Circulating substances such as TNFα, IL-1β, platelet activating factor (PAF), leukotrienes, and, most recently, interleukin-6 (IL-6) have been implicated in this process.123–130
Anaphylactic shock is a form of distributive shock caused by the release of mediators from tissue mast cells and circulating basophils. Anaphylaxis, an immediate hypersensitivity reaction, is mediated by the interaction of IgE antibodies on the surface of mast cells and basophils with the appropriate antigen. Antigen binding results in the release of the primary mediators of anaphylaxis contained in the basophilic granules of mast cells and basophils. These include histamine, serotonin, eosinophil chemotactic factor, and various proteolytic enzymes.131 Subsequently, a number of secondary lipid mediators are synthesized and released including PAF, bradykinin prostaglandins, and leukotrienes (slow-reacting substance of anaphylaxis).131 An anaphylactoid reaction (clinically indistinguishable from anaphylaxis) results from the direct, nonimmunologic release of mediators from mast cells and basophils and can also result in shock.
Anaphylaxis is triggered by insect envenomations (Hymenoptera bees, hornets, and wasps) and certain drugs, especially antibiotics (beta-lactams, cephalosporins, sulfonamides, vancomycin).131 In addition, less frequently, heterologous serum (e.g., tetanus antitoxin, snake antitoxin, antilymphocyte antisera), blood transfusion, immunoglobulin (particularly in IgA-deficient patients), and egg-based vaccine products have been implicated.131 Anaphylactoid reactions can be caused by a wide range of medical agents including ionic contrast media, protamine, opiates, polysaccharide volume expanders such as dextran and hydroxyethyl starch, muscle relaxants, and anesthetics.131
The hemodynamic features of anaphylactic shock are very similar to those for septic shock and include elements of hypovolemia (due to interstitial edema and venodilatation) and myocardial depression.132–136 Cardiac output and ventricular filling pressures may be reduced until patients are fluid resuscitated.136,137 In addition to typical findings of shock, patients may demonstrate urticaria, angioedema, laryngeal edema, and severe bronchospasm.
Adrenal crisis (see also Chapter 59) is an uncommon cause of shock, which can be difficult to diagnose as it occurs in patients with other active disease processes and the clinical features may mimic infection. It is a life-threatening emergency that requires prompt diagnosis and management.
Adrenal crisis is caused by a deficiency of adrenal production of mineralocorticoids and glucocorticoids. It may occur de novo in patients with critical illness or may occur against a background of occult adrenal insufficiency. In the critical care setting, the most common cause of de novo acute adrenal insufficiency is bilateral adrenal hemorrhage in association with overwhelming infections (classically meningococcal, but frequently gram-negative bacteria), human immunodeficiency virus infection, or anticoagulation.138,139 In addition, fungal infections such as histoplasmosis, blastomycosis, and coccidioidomycosis and malignant infiltration of the adrenals may cause acute adrenal insufficiency in ICU patients.139 In some patients, steroid production remains adequate for the baseline state despite adrenal disease. Once stressed, however, the adrenal response is inadequate, leading to decompensation and adrenal crisis. Stressors may be relatively innocuous or may be severe. A febrile illness, infection, trauma, surgery, dehydration, or any other intercurrent illness may trigger the crisis. Abrupt cessation of glucocorticoid therapy or replacement may also result in adrenal crisis.
Symptoms are generally nonspecific and may include anorexia, nausea, vomiting, diarrhea, abdominal pain, myalgia, joint pains, headache, weakness, confusion, and agitation or delirium.139,140 Fever (often out of proportion to any minor infection) is almost always present, and hypotension, initially due to hypovolemia, is frequent.139 The initial hemodynamic pattern may resemble hypovolemic shock (if shock is due only to adrenal crisis). With volume resuscitation, a high output, vasopressor-refractory shock may become apparent.141,142
An unrecognized “relative” adrenal insufficiency has been implicated in the pathogenesis of human septic shock.143–147
[/not-level-membership-for-critical-care-medicine-category]
