CHAPTER 59 Chronic Pancreatitis
The traditional definition of chronic pancreatitis has been permanent and irreversible damage to the pancreas, with histologic evidence of chronic inflammation, fibrosis, and destruction of exocrine (acinar cell) and endocrine (islets of Langerhans) tissue (Fig. 59-1). As a consequence of this histologic damage, varying degrees of permanent and irreversible clinical, morphologic, and functional derangements occur. Chronic pancreatitis was distinguished from acute pancreatitis, characterized by complete recovery of the pancreas after the acute episode. This definition, using histologic criteria, is imperfect for a number of reasons. Most important, pancreatic tissue is rarely available to clinicians. Patients may have histologic evidence of chronic pancreatitis and have no symptoms or complications from the disease. In addition, the histologic features of chronic pancreatitis are often focal, such that a small biopsy, even if available, might miss the disease. Finally, the individual histologic features that are consistent with chronic pancreatitis are not unique, and may be seen in other conditions (such as normal aging). Alternatively, chronic pancreatitis has been defined based on clinical features (pain and exocrine or endocrine insufficiency) or on imaging techniques including ultrasonography (US), computed tomography (CT), endoscopic ultrasonography (EUS), magnetic resonance imaging (MRI), and endoscopic retrograde cholangiopancreatography (ERCP).
Finally, these staging systems make the assumption that acute pancreatitis and chronic pancreatitis are entirely separate entities, when in fact there is now abundant evidence documenting the evolution in many patients from acute to chronic pancreatitis (see Chapter 58). It is probably most accurate to think of the two conditions as separate ends of the same spectrum; acute pancreatitis is an identifiable event, whereas chronic pancreatitis is an ongoing process of variable tempo.
More recent reviews recognize these shortfalls and try to emphasize the importance of etiology, the difficulty of obtaining pancreatic tissue, and the lack of sensitivity of currently available diagnostic tools.1 These proposals have some advantages in that they attempt to define the disease in a more clinically useful manner, classify disease on the basis of etiology, and include advances in technology (e.g., EUS, magnetic resonance cholangiopancreatography [MRCP], genetic analysis) as well as functional and structural damage to the pancreas in the staging criteria.
EPIDEMIOLOGY
Chronic pancreatitis can be demonstrated in up to 5% of autopsies.2,3 Similar, though less pronounced, histologic features are seen even more commonly.4,5 Determining the prevalence of chronic pancreatitis from this type of autopsy data is misleading because the patients from whom the data are taken may not have had clinical symptoms of chronic pancreatitis during life. Long-standing alcohol use, even in moderate amounts, can lead to histologic changes of chronic pancreatitis without symptoms of chronic pancreatitis.6,7 Similarly, aging, chronic kidney disease, and long-standing diabetes mellitus can induce histologic changes within the pancreas that are difficult to distinguish from those of chronic pancreatitis.4,5 Making a diagnosis solely on the basis of autopsy data will therefore overestimate the rate of clinically important (i.e., symptomatic) chronic pancreatitis.
Estimates of annual incidence of chronic pancreatitis in several retrospective studies range from 3 to 9 cases per 100,000 population.8,9 The only prospective study, which was largely limited to alcoholic chronic pancreatitis, noted an annual incidence of 8.2 cases per 100,000 population and an overall prevalence of 27.4 cases per 100,000.10 A nationwide cross-sectional survey in Japan revealed an overall prevalence rate of 35.5 cases per 100,000 population with an estimated incidence of 14.4 per 100,000 and a male-to-female ratio of 3.5 : 1.11 A similar survey in France noted an annual estimated incidence of 7.7 per 100,000 population and a prevalence of 26 per 100,000.12 In most studies, alcohol abuse accounts for two thirds of all cases of chronic pancreatitis. These limited epidemiologic data demonstrate substantial geographic variation.13 The variation may partly be due to differences in alcohol consumption in different populations, but another part of the variation in incidence rates may merely reflect different diagnostic approaches and different diagnostic criteria.
Chronic pancreatitis accounts for substantial morbidity and health care costs. Approximately 26,000 hospital admissions to non-federal hospitals with a first-listed diagnosis of chronic pancreatitis occur yearly; in more than 80,000 yearly admissions, chronic pancreatitis is listed as one of the discharge diagnoses.14,15 The prognosis of chronic pancreatitis is variable and is driven largely by the presence of ongoing alcoholism in persons with chronic alcoholic pancreatitis and equally by concomitant tobacco use. One can estimate prognosis from such features as need for medical care or hospitalization or from the development of complications, reduced quality of life, or mortality.
Data on the quality of life of patients with chronic pancreatitis16–19 document that the presence of abdominal pain and alcohol abuse (in those with alcoholic chronic pancreatitis) are the dominant negative influences on quality of life and that, not surprisingly, quality of life is substantially worse for such patients than for the general population. Mortality in patients with chronic pancreatitis is also substantially influenced by the presence of continued alcoholism. In one large multicenter study, the standardized mortality ratio was 3.6:1 (i.e., those with a diagnosis of any form of chronic pancreatitis died at 3.6 times the rate of age-matched controls), and older subjects, those who smoked, and those with alcoholic chronic pancreatitis had the most significant reduction in survival.20 Continuing alcohol use raised mortality risk by an additional 60%. Overall, 10-year survival in patients with chronic pancreatitis is about 70%, and 20-year survival about 45%. Similar mortality data were noted in an analysis from Japan.21 The cause of death in patients with chronic pancreatitis usually is not the pancreatitis itself but other medical conditions commonly associated with smoking, continued alcohol abuse, pancreatic carcinoma, and postoperative complications.
PATHOLOGY
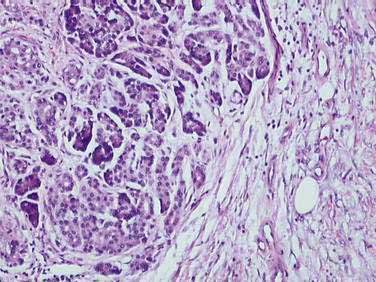
The different etiologies of chronic pancreatitis usually produce similar pathologic findings (see Fig. 59-1), particularly as the disease progresses. In early chronic pancreatitis the damage is varying and uneven. Areas of interlobular fibrosis are seen, with the fibrosis often extending to the ductal structures. Infiltration of the fibrotic area and lobules with lymphocytes, plasma cells, and macrophages is seen.22 The ducts may contain eosinophilic protein plugs. In affected lobules, acinar cells are surrounded and replaced by fibrosis. The islets are usually less severely damaged until very late in the course of the disease. Features of acute pancreatitis also may be seen, such as edema, acute inflammation, and acinar cell or fat necrosis. As the disease progresses, fibrosis within the lobules and between lobules becomes more widespread. The pancreatic ducts become more abnormal with progressive fibrosis, stricture formation, and dilation. The ductal protein plugs may calcify and obstruct major pancreatic ducts. Ductal epithelium may become cuboidal, may develop atrophy or squamous metaplasia, or may be replaced by fibrosis entirely. If special stains are performed, activated pancreatic stellate cells may be identified in close association with fibrosis.
These histologic features are found in most forms of chronic pancreatitis. Many of these changes, in particular perilobular fibrosis and ductal metaplasia, are also commonly seen in patients of advanced age without chronic pancreatitis, and in patients with long-standing diabetes mellitus.4,5 Obstructive chronic pancreatitis (associated with obstruction of the main pancreatic duct by a tumor or stricture) can differ slightly, in that the histologic changes are limited to the gland upstream of the obstruction and protein precipitates and intraductal stones are not seen.23
Autoimmune chronic pancreatitis demonstrates a more robust lymphoplasmacytic infiltrate, including plasma cells, and these are usually positive when stained for immunoglobulin G subtype 4 (IgG4).24 Obstructive phlebitis affecting the major and minor veins and a whorled fibrosis pattern are also characteristic, a pattern termed lymphoplasmacytic sclerosing pancreatitis (LPSP). A variant pattern termed idiopathic duct-centric chronic pancreatitis is characterized by additional neutrophilic infiltration. With time, the pattern may assume a more end-stage chronic pancreatitis appearance and become indistinguishable from other forms of chronic pancreatitis.
PATHOPHYSIOLOGY
The pathophysiology of chronic pancreatitis remains incompletely understood. The pathophysiologic processes must ultimately account for the features of chronic pancreatitis, including loss of parenchymal cells, self-sustaining chronic inflammation, and fibrosis. Any proposed mechanism must therefore include explanations for cellular necrosis or apoptosis, initiation and maintenance of inflammatory cell activation, and fibrogenesis by pancreatic stellate cells. The pancreas, like all other organs, has a limited repertoire of responses to injury and although it is not clear that all of the various etiologies have identical pathophysiology, the end histologic result is similar. The study of mechanisms of disease is hampered by the difficulty of obtaining tissue in humans and the relative lack of animal models of chronic pancreatitis,25,26 as opposed to acute pancreatitis.
Alcoholic chronic pancreatitis, being the most common form, has received the most attention. No single theory explains adequately why only about 10% of heavy alcohol users develop chronic pancreatitis. Alcohol is metabolized by the liver and the pancreas. In the liver the main end product of oxidative alcohol metabolism is acetaldehyde. In the pancreas, an alternative pathway produces fatty acid ethanol esters (FAEEs). Alcohol and its metabolites like FAEE, have direct injurious effects on pancreatic acinar cells. Increased membrane lipid peroxidation, a marker of oxidative stress and free radical production, can be seen in animal models and human alcoholic chronic pancreatitis.27–29 In addition FAEEs are able to induce sustained elevations in cytosolic calcium in acinar cells,29,30 a mechanism shared by other experimental causes of pancreatitis. Alcohol may also lead to pathologic increases in acinar cell sensitivity to physiologic stimuli such as cholecystokinin (CCK)31 and redirect CCK-mediated zymogen exocytosis to the basolateral rather than apical membrane.32 This basolateral secretion would place digestive enzymes in the interstitial space, where they could produce tissue damage. Chronic alcohol ingestion in animal models also alters expression of multiple genes in acinar cells, which could increase the sensitivity to physiologic stress33 and up-regulate the expression and activity of enzymes involved in cell death.34 Alcohol can promote the inflammatory responses involved in pancreatitis.35 These multiple effects of alcohol on the acinar cell are complemented by alcohol injury to ductal cells.36 Finally alcohol and its metabolites appear to stimulate the pancreatic stellate cell.36–38 These cells, as in the liver, appear to be the final common pathway for fibrosis.
Pancreatic stellate cells are found in association with the acinar units. They are typically found in the periacinar space, with long cytoplasmic processes extending to the acini themselves, but are also present in smaller numbers in association with blood vessels and ducts. Quiescent pancreatic stellate cells are recognized by the presence of vitamin A lipid droplets in the cytoplasm. When activated, they assume a stellate or myofibroblastic appearance, express smooth muscle actin, and lose the lipid droplets. This activation is necessary for the cell to begin to secrete extracellular matrix and produce fibrosis within the gland. Activation of pancreatic stellate cells can occur by alcohol or one of its metabolites, but also occurs in response to both inflammatory cytokines that are released following pancreatic acinar cell necrosis and to reactive oxygen species.36–38 In addition, growth factors (platelet-derived growth factor, transforming growth factor-β1), hormones, intracellular signaling molecules, transcription factors, and angiotensin II can activate pancreatic stellate cells.38 Activated pancreatic stellate cells are found in areas of extensive necrosis and inflammation in acute pancreatitis, in human as well as animal tissues. These activated pancreatic stellate cells produce autocrine factors that maintain the activated phenotype. In addition to their role in secretion and modulation of the extracellular matrix, pancreatic stellate cells can proliferate in response to stimulation, migrate to areas of inflammation, and participate in phagocytosis. Activation of pancreatic stellate cells is likely occurring via at least two mechanisms in alcoholic chronic pancreatitis: directly by alcohol and its metabolites and indirectly by cytokines induced by cellular necrosis.36
There have been several hypotheses for the pathophysiology of chronic pancreatitis that attempt to interweave these concepts into a coherent paradigm. One hypothesis focuses on the concept that ductal obstruction (from strictures or stones) is the cause rather than the effect of chronic pancreatitis. This hypothesis, the ductal obstruction hypothesis, is not consistent with most clinical and experimental evidence and with few exceptions (such as the rare condition of obstructive chronic pancreatitis) is not applicable to human chronic pancreatitis. A second paradigm, the toxic-metabolic hypothesis, focuses primarily on the role of alcohol and its metabolites (or other toxins) and their ability to damage the pancreas and activate pancreatic stellate cells. A third model that has been proposed is the necrosis-fibrosis hypothesis, which holds that the occurrence of repeated episodes of acute pancreatitis with cellular necrosis or apoptosis eventually leads to the development of chronic pancreatitis as the healing process replaces necrotic tissue with fibrosis. This last hypothesis has significant supporting evidence from some natural history studies that document the more common development of chronic pancreatitis in patients with more severe and more frequent acute attacks of alcoholic pancreatitis.39,40 The concept that multiple clinical or subclinical attacks of acute pancreatitis lead to chronic pancreatitis is certainly being reinforced by observations in both animal models and in humans.25,29,35,36,41
It is not clear why only a small subset of chronic alcoholics develop chronic pancreatitis. Some of the many possible explanations might include the presence of important co-toxins, differences in the genetic or epigenetic background, or differences in the microenvironment in the pancreas. For example, tobacco use is one very important cofactor for the development of alcoholic (and other types) chronic pancreatitis.42,43 There are also unexplained racial differences in the rates of alcoholic chronic pancreatitis.44 Multiple mutations have been identified in several forms of chronic pancreatitis, suggesting a complex genetic background that provides the relative predisposition to develop chronic pancreatitis (see Chapter 57). Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), cationic trypsinogen gene (PRSS1 gene), serine protease inhibitor Kazal type 1 (SPINK1, a trypsin inhibitor, formerly called PSTI or pancreatic secretory trypsin inhibitor), and many others have been identified.36,41,45–48 These mutations and the many more yet to be identified may be adequate in and of themselves to produce pancreatitis (at this point only PRSS1 satisfies that requirement) or may only predispose to disease. In the case of PRSS1, a number of families with hereditary pancreatitis (see Chapter 57) have been identified with mutations in the PRSS1 gene. These mutations appear to lead to a gain of function mutation in which trypsinogen, once activated to trypsin, is resistant to inactivation. The mutant trypsin might therefore be able to activate other proenzymes and to produce clinical or subclinical episodes of acute pancreatitis, ultimately leading to chronic pancreatitis in affected patients. This presumed pathophysiology provides support for the necrosis-fibrosis theory of chronic pancreatitis, in which repeated episodes of acute pancreatitis ultimately lead to chronic pancreatitis. Cystic fibrosis is associated with abnormalities of bicarbonate secretion by ductal cells and the eventual development of dilated pancreatic ducts, intraductal precipitates, and pancreatic atrophy (see Chapter 57). Several studies have documented an increase in rate of CFTR mutations in patients with presumed “idiopathic” pancreatitis.49–52 SPINK1 mutations have been found in idiopathic chronic pancreatitis, hereditary pancreatitis, tropical pancreatitis, and alcoholic pancreatitis.45,46,53–55 These mutations are discussed in detail in Chapter 57 but are useful to consider here as a basis for an evolving paradigm of pathogenesis This paradigm has been labeled SAPE—sentinal acute pancreatitis event—and conceptualizes a complex genetic background that provides the predisposition to disease.1,29,36,37,41,46,47 This background may include mutations in genes that code for digestive enzymes, protease-enzyme inhibitors, ion channels, and a variety of others including genes that affect the metabolism of environmental toxins (e.g., tobacco or alcohol), genes that have a role in inflammation or fibrosis, and others yet to be discovered. Although a few mutations may be sufficient to produce pancreatic damage in most or all who carry it, the majority of mutations identified to date are not that dominant. On this complex polygenic background is overlaid some environmental insult, such as chronic alcohol use, that applies physiologic stress to the acinar, ductal, and stellate cells. This stress may be insufficient to produce injury or may produce cellular necrosis or apoptosis. The initiating event for necrosis is the activation of digestive enzymes within the acinar cell, either by the toxic effect of the environmental insult or because of some underlying mutation that leads to excessive activation of trypsin (see Fig. 57-3). Inflammation follows the necrosis, and this necroinflammatory process may either progress or resolve. In some individuals, the situation may never progress beyond this stage. In others, continued cell metabolic and oxidative stresses or some other trigger could produce continuing or repeated acinar cell injury with necrosis. This process, as is believed to be the case in other organs, could be associated with the activation of stellate cells. The acute inflammatory response could resolve, or, alternatively, the inflammatory response might switch to an anti-inflammatory response, with resident macrophages producing cytokines and activated stellate cells laying down extracellular matrix, with the formation of fibrosis. Fibrosis would start a vicious circle by causing additional acinar cell ischemia and continuing to drive the inflammatory–anti-inflammatory cycle. This type of hypothesis could theoretically explain many forms of chronic pancreatitis. This framework seems to fit all the developing experimental and clinical data and is a useful way in which to think about the pathophysiology of chronic pancreatitis: as a disease associated with a variety of different genetic predispositions, a variety of disease triggers, and a similar final common pathway producing pancreatic injury and fibrosis, ultimately with organ failure.
ETIOLOGY (Table 59-1)
ALCOHOL
In Western countries, alcohol is the cause of at least 70% of all cases of chronic pancreatitis.8–10,14,56–60 The risk of alcoholic chronic pancreatitis increases logarithmically with rising alcohol use, but there is no true threshold value below which the disease does not occur.48,60–63 Therefore, in countries with widespread alcohol consumption, it may be difficult to determine with certainty whether alcohol contributed to or caused the disease. In nearly all patients, at least 5 years (and in most patients more than 10 years) of intake exceeding 150 g/day (a standard drink contains about 14 g of alcohol) are required before the development of chronic pancreatitis. Only 3% to 15% of heavy drinkers ultimately develop chronic pancreatitis, suggesting an important cofactor.56,61,62,64 Potential cofactors include genetic polymorphisms and mutations,27–29,47,48,64,65 a diet high in fat and protein,47,61,64 the type of alcohol or manner of ingestion,62 an associated relative deficiency of antioxidants or trace elements,66,67 and smoking.42,47,56,61,64,68,69 Of these, smoking appears to be the strongest association. In some studies, 90% of those who develop alcoholic chronic pancreatitis are also chronic smokers.64,68,69 Smoking also appears to predispose to more rapid development of pancreatic calcification.42,43,62,70,71 There are also racial differences in the risk for development of alcoholic pancreatitis, perhaps suggesting some difference in the ability to detoxify environmental toxins or alcohol or other genetic or epigenetic factors.44,47,62,64 Although the risk of alcoholic chronic pancreatitis is higher in blacks,44 data from self-reported surveys of alcohol use demonstrate that the proportion of blacks who drink alcohol or smoke is actually lower than that in whites.72
Table 59-1 Classification of Chronic Pancreatitis
CFTR, cystic fibrosis transmembrane conductance regulator gene; PRSS1, protease serine 1 gene; SPINK1, serine protease inhibitor, Kazal type 1 gene.
Many patients with alcoholic chronic pancreatitis have an early phase of recurrent attacks of acute pancreatitis, which may last five or six years, followed by the later development of chronic pain or exocrine or endocrine insufficiency. It has been believed, based on several large natural history studies, that most patients who present with their first attack of acute alcoholic pancreatitis have already developed histologic chronic pancreatitis. Up to 40% of patients presenting with acute alcoholic pancreatitis, however, do not progress to clinically identifiable chronic pancreatitis (calcification and exocrine or endocrine insufficiency) even with very long follow-up, and may not even have recurrent attacks.39,62,64,73–75 Based on autopsy studies6,7 and functional studies,76 however, it is likely these patients actually have histologic evidence of chronic pancreatitis, although they may not have developed a recognizable chronic clinical illness.
Not all patients with alcoholic chronic pancreatitis present with acute episodes of pancreatitis. Less than 10% of patients present with exocrine or endocrine insufficiency in the absence of abdominal pain.57–5964 Some present with chronic pain in the absence of antecedent acute attacks of pain. Cessation of alcohol use after the onset of alcoholic pancreatitis appears to diminish the rate of progression to exocrine insufficiency and endocrine insufficiency but does not halt it.77 Stopping alcohol does seem to reduce the chance of recurrent attacks of acute alcoholic pancreatitis in those who have not yet developed obvious chronic pancreatitis.75
The prognosis of alcoholic chronic pancreatitis is relatively poor,20,21 and mortality is generally greater than that seen in chronic pancreatitis of other etiologies. Quality of life is also substantially diminished. Pain generally continues for years, although it may spontaneously remit. Exocrine and/or endocrine insufficiency develops in many patients, although this process may take several years. In one large natural history study, exocrine insufficiency developed in 48% of patients at a median of 13.1 years after presentation, whereas endocrine insufficiency developed in 38% after a median of 19.8 years after presentation.58 Diffuse pancreatic calcifications developed in 59% at a median of 8.7 years after diagnosis. Other studies have noted more rapid and more frequent development of calcifications, exocrine insufficiency, and endocrine insufficiency.57
TOBACCO
Exposure to tobacco smoke can induce pancreatic damage in animals.78 As mentioned, smoking is common in patients with alcoholic chronic pancreatitis and is associated with an increased risk for pancreatic calcifications, and smoking cessation after the clinical onset of chronic pancreatitis reduces the risk of subsequent calcifications.79 There is also evidence that smoking is an independent risk factor for chronic pancreatitis.80,81 Smoking is also associated with a much higher rate of secondary pancreatic cancer and overall mortality in patients with chronic pancreatitis.11,20
TROPICAL PANCREATITIS
Tropical pancreatitis is one of the most common forms of chronic pancreatitis in certain areas of southwest India. It has been reported from a number of other areas, including Africa, southeast Asia, and Brazil. The disease as initially described is essentially restricted to areas within 30 degrees of latitude from the equator. Tropical pancreatitis is generally a disease of youth and early adulthood, with a mean age at onset of 24 years.82–84 More than 90% of patients have the illness before 40 years. The overall prevalence from surveys in an endemic area (southern India) is 1 in 500 to 1 in 800 population.82,85 Tropical pancreatitis accounts for about 70% of all cases of chronic pancreatitis in southern India, whereas alcohol is a more dominant cause in the north. Surveys in Kerala province in southwest India have noted that there is a shift toward older age at presentation, less malnutrition, and less severe diabetes.86 These later surveys also note higher rates of alcohol abuse in this area and alcohol gradually becoming a more common cause of chronic pancreatitis.
The pathophysiology of tropical pancreatitis is unknown. As discussed in Chapter 57, a number of genetic mutations have been identified, with mutations in the SPINK1 and chymotrypsinogen genes being most common.87 Environmental triggers for the disease that have been proposed include protein-calorie malnutrition, deficiencies of trace elements and micronutrients coupled with oxidative stress (via xenobiotics, industrial pollutants, diet, or nutritional deficiency), cyanogenic glycosides present in cassava (tapioca—a main dietary component), viral and parasitic infections, and others.
GENETIC
Only one type of mutation appears sufficient to cause chronic pancreatitis: mutations in PRSS1 in families with hereditary pancreatitis. All other identified mutations (discussed in Chapter 57) and polymorphisms should be considered as cofactors, mutations that predispose to disease by increasing susceptibility, or as modifier genes, that increase the pace or severity of disease. It is certainly possible that mixtures of polymorphisms and mutations work together to determine the susceptibility to disease. The most commonly identified mutations occur in the PRSS1 (cationic trypsinogen), SPINK1 (trypsin inhibitor), and CFTR genes. Several studies have suggested that certain less severe CFTR gene mutations and SPINK1 mutations may be associated with idiopathic chronic pancreatitis. CFTR gene mutations have been identified in up to half of patients with idiopathic chronic pancreatitis.46,49–52 This proportion is far greater than expected within this population. Analysis of these data has suggested that the combination of a more severe CFTR mutation on one chromosome with a mild mutation on the other is particularly associated with chronic pancreatitis (see Chapter 57). These studies examined only the most common CFTR mutations. There are now more than 1200 known CFTR mutations, and further studies are needed to define their contribution to idiopathic and other forms of chronic pancreatitis. SPINK1 mutations have also been identified with increased frequency in patients with unexplained chronic pancreatitis, including some mixed heterozygotes with combined CFTR and SPINK1 mutations. Genetic testing for these mutations is commercially available.
AUTOIMMUNE PANCREATITIS
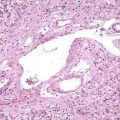
Autoimmune pancreatitis refers to a distinct chronic inflammatory and sclerosing disease of the pancreas. The distinct characteristic of the disease is a dense infiltration of the pancreas, and often other organs, with lymphocytes and plasma cells (Fig. 59-2A), many of which express IgG4 on their surface. The autoimmune target of this IgG4 and the trigger for disease are unknown. However, recently a protein expressed in pancreatic acinar cells, UBR2 (ubiquitin-protein ligase E3 component n-recognin 2) has been proposed to be the target of the antibody.87a Of interest, the antibodies also react to a protein of Helicobacter pylori, PBP (plasminogen-binding protein), suggesting a role for H. pylori infection in autoimmune pancreatitis.
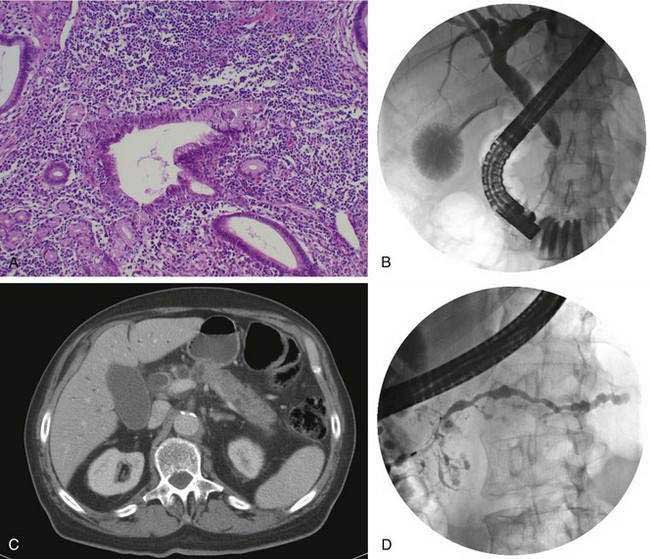
Fibrosis, sclerosis, and obliterative phlebitis are characteristically seen in association with the chronic inflammatory infiltrate. Although this inflammatory infiltrate is present in the pancreas, similar infiltrates may be seen in the bile duct, salivary glands, retroperitoneum, lymph nodes, kidney, prostate, ampulla, and occasionally other organs.24 An equally characteristic feature of this disease is the often rapid response to glucocorticoid therapy. Much of the information on this disease comes from a series of studies from Japan and other Asian countries. Early reports noted chronic pancreatitis characterized by the presence of autoantibodies, elevated levels of serum immunoglobulins, enlargement of the pancreas (diffuse or focal), pancreatic duct strictures, and pathologic features of a dense lymphocytic infiltrate.88
Studies from Japan note that up to 6% of all patients evaluated for chronic pancreatitis have autoimmune pancreatitis, and the overall prevalence is estimated to be 0.82 per 100,000 persons.24,88–92 The disease may occur in an isolated form or may be associated with extrapancreatic manifestations. The most common extrapancreatic conditions identified include biliary strictures, hilar lymphadenopathy, sclerosing sialadenitis, retroperitoneal fibrosis, and tubulointerstitial nephritis.24,88–93 Autoimmune pancreatitis may therefore be one manifestation of what has been termed IgG4-related sclerosing disease or IgG4-related systemic disease. There also appears to be some overlap with an unusual variant of Sjögren’s disease termed Mikulicz’s disease, in which a massive IgG4-positive mononuclear infiltrate is seen in the salivary and lacrimal glands.94
The disease is seen more commonly in men (2 : 1) and usually manifests in middle age. More than 85% of patients present after the age of 50 years. One of the most common presentations is painless obstructive jaundice due to obstruction of the intrapancreatic bile duct (see Fig. 59-2B). Jaundice may occur from compression of the bile duct by the enlarged pancreas or by infiltration of the biliary tree by the chronic inflammatory process. Additional symptoms may include weight loss, vomiting, and glucose intolerance.24 Although pain is not frequently present, abdominal and referred back pain may occur. These clinical features, coupled with imaging studies demonstrating diffuse or focal pancreatic enlargement (see Fig. 59-2C), often raise the suspicion of pancreatic adenocarcinoma. In studies in the United States in patients who underwent pancreatic resection for presumed pancreatic carcinoma but were found to have no malignancy in the resected specimen, up to 11% show evidence of autoimmune pancreatitis.95 Jaundice or cholestasis may also occur due to strictures of the biliary tree in other locations. A pattern similar to that seen in primary sclerosing cholangitis (PSC) is seen, with a predilection for involvement of the hilar region. The pattern may mimic not only PSC but also cholangiocarcinoma. The disease, unlike classic PSC, is not associated with inflammatory bowel disease and is steroid-responsive (see Chapter 68). Additional clinical manifestations include a sclerosing sialadenitis (usually presenting as bilateral symmetrical swelling of the salivary glands), retroperitoneal fibrosis (most commonly presenting as hydronephrosis due to entrapment of the ureters), tubulointerstitial nephritis, lymphadenopathy (particularly mediastinal, cervical, and abdominal), prostatitis, and sclerosing cholecystitis, interstitial pneumonia, pseudotumors of the liver, lung, and pituitary.
Abdominal ultrasonography usually shows a diffusely enlarged and hypoechoic pancreas. The appearance on EUS is similar. CT most commonly reveals a diffusely enlarged sausage-shaped pancreas (see Fig. 59-2C) in which enhancement with the intravenous contrast agent is delayed and prolonged.24,91,96 Some patients may have a capsule-like low-density rim around the pancreas in delayed images. Focal swelling can also occur, mimicking a pancreatic mass.24,91,96–98 Additional CT findings, such as contiguous fibrosis and inflammation extending into the retroperitoneum or surrounding the retroperitoneal vessels, can also raise a suspicion of carcinoma. One of the major clinical challenges in such cases is differentiating these inflammatory pseudotumors from true tumors.97 MRI of the pancreas also may reveal this diffuse pancreatic enlargement, typically with decreased T1-weighted intensity and increased T2-weighted intensity. MRCP can be very helpful in identifying the biliary strictures and in visualizing the pancreatic duct. CT and MRI may also reveal less common findings of autoimmune pancreatitis including gland atrophy and pancreatic calcifications. Endoscopic ultrasonography may demonstrate a diffusely enlarged and hypoechoic gland. The use of fine-needle aspiration of the gland is usually not diagnostic, although there are reports of Tru-cut needle biopsy being diagnostic.99
One of the hallmarks of autoimmune pancreatitis is diffuse or segmental irregularity and narrowing of the pancreatic duct (see Fig. 59-2D). The duct may be diffusely narrowed and thread-like, or may instead demonstrate alternating areas of stricture and normal caliber or dilated duct.24,91,93,98 MRCP is often able to identify the pancreatic duct abnormalities but may not be able to visualize the duct if it is thread-like and diffusely affected. ERCP is better able to visualize the pancreatic duct, but carries more risk and cost than MRCP. Some patients may have a more focal segmental or isolated area of pancreatic duct narrowing, in a pattern more suggestive of malignancy. Some studies in which a second ERCP has been performed note progression from a segmental form to diffuse form in the absence of glucocorticoid treatment.24,100
The pathologic hallmark in the pancreas is infiltration of inflammatory cells and fibrosis around medium-sized interlobular ducts. Obliterative phlebitis of medium and small veins and a whirling or storiform fibrosis of the pancreas, which may extend to the peripancreatic tissues, is characteristic. The inflammatory infiltrate is composed of T lymphocytes and plasma cells. Interstitial fibrosis with acinar cell atrophy is another common finding. Two histologic variants have been described.24 The first, LPSP, is defined by the histologic features noted earlier (Fig. 59-3). A second variant, termed idiopathic duct centric chronic pancreatitis (IDCP), is less common. This variant has a neutrophilic infiltrate with microabscesses, and obliterative phlebitis is rare relative to the aforementioned LPSP variant. End-stage disease may demonstrate atrophy and calcification in the gland, and at that point it may be indistinguishable from other forms of chronic pancreatitis.
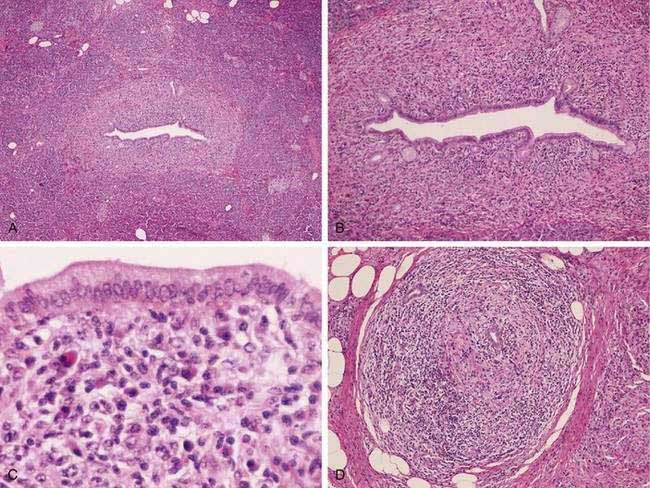
The disease may be suspected from the clinical and imaging features previously noted. Laboratory evaluation may reveal elevations in serum immunoglobulins,24 seen in from one half to two thirds of cases, especially in IgG4. In studies from Japan, an IgG4 level greater than 135 mg/dL has a sensitivity of 90% and a specificity of greater than 95% for autoimmune pancreatitis.88,91 A large study in the United States, using an IgG4 cut-off of 140 mg/dL, found a sensitivity of 76% and a specificity of 93%.101 This study demonstrated that patients with histologically proven autoimmune pancreatitis may have a normal serum level of IgG4, and that up to 10% of patients with pancreatic ductal adenocarcinoma may have elevations in IgG4 levels. A variety of autoantibodies have been reported, including antinuclear antibodies (ANAs), antilactoferrin antibodies, anticarbonic anhydrase II antibodies, antismooth muscle antibodies (ASMs), rheumatoid factor (RF), and antimitochondrial antibody (AMA). These autoantibodies do not have the sensitivity of IgG4 and hence are inferior for diagnostic purposes.
There are several proposals for diagnostic criteria for autoimmune chronic pancreatitis. The first widely used system102 proposed by the Mayo Clinic uses the mnemonic HISORt (histology, imaging, serology, other organ involvement, and response to steroid therapy). A consensus conference led to combined Japan-Korea diagnostic guidelines.91 These systems are presented in Table 59-2.
| FEATURE | MAYO CLINIC HISORt CRITERIA | ASIAN CONSENSUS CRITERIA |
|---|---|---|
| Histology | At least one of the following: |
ANA, antinuclear antibodies; CT, computed tomography; HISORt, histology, imaging, serology, other organ involvement, and response to glucocorticoid therapy; HPF, high-power field; IgG, immunoglobulin G; LPSP, lymphoplasmacytic sclerosing pancreatitis; MRI, magnetic resonance imaging; RF, rheumatoid factor.
Autoimmune chronic pancreatitis may progress rapidly, from the initial symptoms to end-stage chronic pancreatitis, within months. There is evidence that early therapy with glucocorticoids may prevent subsequent disease complications.24,103 Glucocorticoid therapy usually produces rather dramatic improvement with rapid resolution of both symptoms and radiographic abnormalities. There are no clear recommendations for glucocorticoid dose, although 30 to 40 mg of prednisone orally per day for four to eight weeks is reasonable.24 Repeat pancreatic imaging at four weeks is prudent, to assess for clinical response. Once a response is clear-cut, tapering of the prednisone dose at a rate of 5 mg per week is typical. Complete serologic response (normalization of serum IgG4) may not be apparent for several months, although decreases may be seen within four weeks. Prior to initiating glucocorticoid therapy, vigorous efforts should be made to exclude malignancy. In some cases this is not possible, and some possibility of underlying malignancy may remain. A typical scenario might be a middle-aged man with obstructive jaundice and an enlargement of the pancreatic head, in whom ERCP and EUS with tissue sampling have been inconclusive. In this setting, there may be concern in delaying the ultimate diagnosis of malignancy for a therapeutic trial of glucocorticoids. Response to glucocorticoid therapy, however, is usually obvious within two to four weeks and such a trial is not unreasonable if follow-up is intense. Some patients in whom malignancy remains a possibility undergo laparotomy and often resection of the mass.
Between 30% and 40% of patients experience a relapse after glucocorticoid therapy.24,104–106 Relapse may be managed by a repeat course of glucocorticoids followed by maintenance at a low dose of prednisone (e.g., 5 to 10 mg/day). Immunodulators, such as azathioprine, have been used in a few steroid-dependent patients with success.24 Glucocorticoid therapy may improve not only the structural abnormalities of the pancreas but also exocrine and endocrine pancreatic function (and salivary function if it is affected).107 Improvement in function is variable, depending on the level of fibrosis and atrophy already established when therapy is initiated. Clinical relapses after resection (e.g., pancreaticoduodenectomy) are rare.
OBSTRUCTIVE CHRONIC PANCREATITIS
Obstruction of the main pancreatic duct by tumors, scars, ductal stones, duodenal wall cysts, or stenosis of the papilla of Vater or the minor papilla can produce chronic pancreatitis in the parenchyma upstream of the obstruction. Obstruction of the pancreatic ducts may also be an important contributor to other forms of chronic pancreatitis (i.e., obstruction of small or large ductal branches by protein precipitates in alcoholic chronic pancreatitis). Obstructive chronic pancreatitis, however, refers to a distinct entity produced by a (generally) single dominant narrowing or stricture of the main pancreatic duct. It is believed that relief of the obstruction can lead to reversal of pancreatic damage, although there is no direct proof of this belief. A number of distinct entities can produce obstructive chronic pancreatitis. Acquired strictures of the main pancreatic duct can occur as a consequence of tumor obstruction from adenocarcinoma, islet cell tumor, intraductal papillary mucinous tumors, or ampullary neoplasms (see Chapter 60). Benign strictures may also develop after a severe attack of acute pancreatitis, particularly an episode associated with significant pancreatic necrosis and pseudocysts. Blunt and penetrating trauma to the pancreas can also lead to pancreatic duct strictures, most commonly in the midbody of the gland where the duct crosses the spine. Each of these processes can be associated with chronic pancreatitis in the gland upstream from the obstruction. The pathology in obstructive chronic pancreatitis is one of diffuse interlobular and intralobular fibrosis, usually equally and symmetrically distributed in the affected region.
Pancreas divisum is a common normal variant, occurring in approximately 4% to 11% of the population (see Chapter 55). In some patients with this anomaly, the minor papilla may be inadequate to allow free flow of pancreatic juice into the duodenum, possibly causing acute episodes of pancreatitis. Pancreas divisum is not often considered a cause of chronic pancreatitis. Large natural history studies have failed to identify a clear link between pancreas divisum and either acute or chronic pancreatitis. Nonetheless, rare patients may present with pancreas divisum and substantial dilation of the dorsal pancreatic duct, associated with obstructive chronic pancreatitis. These rare patients have strictures of the minor papilla. Most analyses of the effectiveness of minor papilla endoscopic sphincterotomy and stenting, or minor papilla surgical sphincteroplasty, have not specifically examined the response to these therapies in this rare subgroup of patients,108 and the response to these therapies even in the setting of acute relapsing pancreatitis is unknown. Endoscopic therapy or surgical therapy for patients with pancreas divisum, chronic pancreatic type pain, and a nondilated dorsal pancreatic duct is rarely, if ever, effective.
Sphincter of Oddi dysfunction (see Chapter 63), like pancreas divisum, is most often proposed as a cause of acute or recurrent acute pancreatitis rather than chronic pancreatitis. Although abnormalities of sphincter of Oddi manometry may be seen in some patients with chronic pancreatitis,109,110 they are unpredictable and not likely to be playing a role in causing disease. Interestingly, one family in whom sphincter of Oddi dysfunction had been presumed to be causing chronic pancreatitis was actually found to suffer from a mutation of the trypsinogen gene.1 It is much more likely that abnormal sphincter of Oddi pressures are a consequence of chronic pancreatitis rather than a cause. The response to sphincter ablation in patients with chronic pancreatitis and sphincter of Oddi dysfunction is unpredictable but generally poor. Surgical textbooks have long cautioned against sphincteroplasty as therapy for chronic pancreatitis, recognition of the general lack of efficacy of sphincter ablation in this situation.
MISCELLANEOUS
Recurrent or Severe Acute Pancreatitis
Recurrent episodes of acute pancreatitis of any etiology may eventually lead to the development of a chronic inflammatory response within the pancreas, the activation of pancreatic stellate cells, and chronic pancreatitis. This can also happen with even one severe attack, usually with associated pancreatic necrosis or the need for surgical débridement. One more common example of this is hypertriglyceridemia. Elevations of serum triglyceride values greater than 1000 mg/dL can produce acute pancreatitis. Many of these patients will have repeated clinical and subclinical attacks of acute inflammation, and ultimately develop chronic pancreatitis.111
In patients with any etiology of severe acute pancreatitis complicated by substantial pancreatic necrosis, chronic pancreatitis may develop (see Chapter 58). This process occurs most commonly in patients who undergo surgical necrosectomy. In one study, patients with necrotizing biliary pancreatitis who did not undergo necrosectomy had exocrine insufficiency and endocrine insufficiency less commonly than those who did (13% vs. 58% and 26% vs. 75%, respectively, for exocrine and endocrine insufficiency).112 Thus, even in the absence of necrosectomy, severe necrotizing gallstone pancreatitis may occasionally lead to chronic pancreatitis.113 Other studies have noted exocrine and endocrine pancreatic insufficiency commonly occur after necrosectomy.114 Persistent decreases in pancreatic function testing may be seen in up to 80% of patients after recovery from necrotizing pancreatitis. Those with acute alcoholic pancreatitis, complicated by significant necrosis, are most likely to develop features of chronic pancreatitis,115 likely because they usually already have underlying chronic pancreatitis at the time of their first severe attack. Residual strictures of the pancreatic duct are also not uncommon after severe acute pancreatitis, and they may also contribute to the development of chronic pancreatitis in this group of patients. In others, a prolonged and smoldering clinical course ultimately leads to chronic pancreatitis.
Asymptomatic Pancreatic Fibrosis
There are several situations in which histologic evidence of chronic pancreatitis, and specifically fibrosis, may be seen in the absence of clinical chronic pancreatitis. Two of these conditions have already been discussed. Older adults commonly develop histologic changes within the pancreas that resemble chronic pancreatitis.3,4,5 ERCP may also demonstrate changes consistent with chronic pancreatitis in these patients.116 These structural changes do not correspond to functional disturbances of pancreatic function or to clinical features of chronic pancreatitis.117 As mentioned, chronic alcoholics who do not have clinical chronic pancreatitis commonly have histologic evidence of chronic pancreatitis.6,7
The incidence of acute pancreatitis is increased in patients undergoing hemodialysis, and some evidence suggests that chronic pancreatitis may also be seen with greater frequency in this population. One ultrasonographic screening study of patients undergoing hemodialysis noted changes consistent with chronic pancreatitis in 20%.118 Autopsy studies also show changes consistent with chronic pancreatitis in the majority of patients,119 although very few of such patients had clinical chronic pancreatitis. Chronic renal failure appears to frequently produce asymptomatic pancreatic fibrosis.120
Diabetes mellitus can occur as a consequence of destruction of islet cells from long-standing chronic pancreatitis. Up to 1% of all diabetes mellitus is believed to be due to chronic pancreatitis. In contrast, changes in pancreatic morphology and function are common in patients with long-standing diabetes. The pancreas is smaller than normal, particularly in patients with type 1 diabetes.121 The pancreatic duct is abnormal in 40% to 50% of diabetic patients when studied by ERCP, with abnormalities suggestive of chronic pancreatitis.122,123 Pancreatic function, as defined by fecal elastase measurements124 or by more formal direct pancreatic function testing,125 is abnormal in 40% to 50% of patients. The abnormalities are much more likely to be seen in type 1 than type 2 diabetes. The reason for this association is not clear, and indeed, it is not clear whether the diabetes is causing the changes in the pancreas or vice versa. Because insulin is a trophic factor for the exocrine pancreas, and because diabetes can produce microvascular angiopathy, insulin deficiency and long-standing diabetes together could explain the pancreatic damage. Although these measures of pancreatic structure and function may be present, they are usually not responsible for symptoms and these patients do not routinely merit treatment with pancreatic enzymes.126 Clinical evidence of chronic pancreatitis and exocrine insufficiency is very rare in diabetic patients.
IDIOPATHIC CHRONIC PANCREATITIS
Idiopathic pancreatitis accounts for 10% to 30% of all cases of chronic pancreatitis. However, many patients are probably mislabeled as having idiopathic disease. Given that there is no threshold of alcohol ingestion for alcohol-induced chronic pancreatitis, some of these patients certainly suffer from alcoholic chronic pancreatitis. Similarly, some of these idiopathic cases occur in patients with genetic abnormalities,45,52,55 and some of what we formerly labeled idiopathic is instead autoimmune. Interpreting the literature on idiopathic chronic pancreatitis is therefore difficult because most studies of this entity are probably dealing with cases with several different etiologies.
Idiopathic chronic pancreatitis appears to occur in two forms, an early-onset type that manifests in the late second or third decade of life and a late-onset form that appears in the sixth or seventh decade of life.58,127
Early-onset idiopathic chronic pancreatitis has a mean age at onset of around 20 years. There appears to be an equal gender distribution,58 although one series found a male preponderance.127 Pain is the predominant feature of this disease, occurring in up to 96% of patients, a higher rate than in either alcoholic or late-onset chronic pancreatitis (see later). Pancreatic calcifications, exocrine insufficiency, and endocrine insufficiency (i.e., diabetes) are extremely rare at presentation (<10%) and develop very slowly thereafter. The mean time to calcification in this group is 25 years, to exocrine insufficiency 26 years, and to endocrine insufficiency 27.5 years.58 Complications of chronic pancreatitis (pseudocyst, abscess, biliary obstruction, and duodenal obstruction) occur in about 20% of patients, whereas surgery (primarily for abdominal pain) is ultimately needed in 60%. Thus, early-onset idiopathic chronic pancreatitis is a disease characterized by severe pain but much delayed development of structural (calcifications) or functional (exocrine or endocrine insufficiency) evidence of chronic pancreatitis. The delay may make diagnosis quite difficult because most available diagnostic tools rely on these structural or functional abnormalities.
Late-onset idiopathic chronic pancreatitis manifests less commonly as pain. In the best-documented series,58 only 54% of patients presented with pain, although 75% ultimately experienced pain. The median age of onset is 56 years, and the disease occurs equally in men and women. Exocrine insufficiency (22%) and endocrine insufficiency (22%) were present not infrequently at the time of diagnosis, ultimately occurring in 46% and 41% of cases, respectively. The median time to development of exocrine insufficiency and endocrine insufficiency was 16.9 and 11.9 years, respectively. Life-table analysis suggested that with very long follow-up (>30 years), exocrine insufficiency will ultimately develop in 75%, endocrine insufficiency in 50% to 60%, and diffuse pancreatic calcifications in 90% of patients. The disease therefore tends to be one of a comparatively painless course associated with the frequent development of pancreatic calcifications, exocrine insufficiency, and endocrine insufficiency. Aging itself can be associated with the development of structural changes within the pancreatic parenchyma and duct that are indistinguishable from those seen in late-onset chronic pancreatitis,117 so the distinction between normal aging and late-onset idiopathic chronic pancreatitis may not always be clear.
CLINICAL FEATURES
ABDOMINAL PAIN
Abdominal pain is the most common clinical problem in patients with chronic pancreatitis, and the symptom that most detracts from quality of life. Severe pain decreases appetite and limits food consumption, contributing to weight loss and malnutrition. Chronic severe pain leads to a dramatic reduction in quality of life,16–19 loss of social functioning, and the all too frequent addiction to narcotic analgesics. Intractable pain is the most common reason for hospitalization and for surgery in patients with chronic pancreatitis. There is no single characteristic pain pattern. Pain is most commonly described as being felt in the epigastrium, often with radiation to the back. Pain is usually described as boring, deep, and penetrating and is often associated with nausea and vomiting. Pain may be relieved by sitting forward or leaning forward, by assuming the knee-chest position on one side, or by squatting and clasping the knees to the chest. Pain may worsen after a meal and often is nocturnal.
The natural history of abdominal pain varies and is difficult to predict. As an example, many patients with alcoholic chronic pancreatitis initially present with episodes of pain interspersed with periods of feeling relatively well. During these more acute episodes of pain, such a patient may be labeled as having acute pancreatitis. As time passes, pain may become more continuous, and the diagnosis of chronic pancreatitis more obvious. Some patients may present with the more gradual onset of constant abdominal pain, and some may have no pain. Once pain develops, it commonly changes over time in character, severity, and timing. Depending on the etiology of chronic pancreatitis, 50% to 90% of patients experience pain during the course of the disease.40,57–59,127–129
Some of these same observational studies document a decrease in pain over time in the majority of patients, although the timing and the magnitude of this decrease vary among the studies. In one study, pain relief appeared to occur most commonly at the time of development of diffuse pancreatic calcifications, exocrine insufficiency, and endocrine insufficiency.57 Other studies have not found this same correlation, but many have noted a less pronounced tendency for pain to “burn out” over time.58,59,129–131 Pain seems to eventually decrease in at least half of patients. Some of the pain relief is due to surgery for pain or complications, but pain relief over very long follow-up is also seen in medically treated patients in approximately similar proportions.58,59,129,132,133 The pain pattern in an individual patient, however, is not accurately predictable, and the pain may worsen, stabilize, or improve over time. The judgment of therapeutic efficacy for any treatment for chronic pancreatitis must take into account this extremely varying natural history of pain.
Increased Pressure and Ischemia
One commonly invoked mechanism of pain is tissue ischemia, driven by increased pressure within the pancreatic duct or parenchyma. Several lines of clinical and experimental evidence point to increased pressure within the pancreatic duct or parenchyma as being important in the genesis of pancreatic pain. Pancreatic ductal and tissue pressures are usually elevated in patients with chronic pancreatitis undergoing surgery for chronic pain.134–136 Elevations in pancreatic ductal pressure measured during ERCP have also been documented in some but not all patients with painful chronic pancreatitis.110,137,138 Surgical drainage of the pancreatic duct leads to an immediate reduction in pressure to normal levels and is associated with pain relief.134,135 However, one study of ERCP-measured pancreatic duct pressures found no difference in patients with chronic pancreatitis between those who had pain and those who did not,137 and one small study of endoscopic stenting found that a reduction in pressure after stenting was not correlated with pain relief.138
One would assume that increased pressure within the pancreatic duct would be related to obstruction of the pancreatic duct, either in the main duct or in side branches. The presence of a pancreatic duct stricture and upstream pancreatic duct dilation might be an accurate indicator of a group of patients with increased pressure and therefore pain. However, there is often not a clear-cut relationship between pancreatic duct strictures or ductal dilation and pain.40,57–59,130,137–139 In many studies, pancreatic duct dilation and strictures are equally common in those with and without pain. Nonetheless, patients with a dilated pancreatic duct or pancreatic duct stricture are most likely to experience pain relief from endoscopic or surgical therapy.
The mechanism by which increased pressure could cause pain is speculative but is likely related to pancreatic tissue ischemia. In animal models of chronic pancreatitis, increased pancreatic pressure is associated with reductions in pancreatic blood flow, tissue oxygen tension, and interstitial pH.140 In these models pancreatic secretagogues lead to a further decrease in pancreatic blood flow (rather than the normally expected increase), decreased capillary filling, and worsening tissue ischemia. These observations are consistent with those seen in a compartment type syndrome. Small studies in humans with chronic pancreatitis undergoing surgery also demonstrate lower pancreatic tissue pH than patients without chronic pancreatitis.140 Pancreatic blood flow, measured at ERCP with the use of platinum electrodes, is also lower in patients with chronic pancreatitis compared with controls.141 Tissue ischemia that is worsened by secretory stimulation of the pancreas may therefore be the mechanism by which elevations in tissue pressure cause pain.
Alterations in Peripheral and Central Nociceptive Nerves
Morphologic studies in patients with chronic pancreatitis demonstrate increases in the diameter and number of intrapancreatic nerves, foci of inflammatory cells associated with nerves and ganglia, and damage to the perineural sheath.142,143 The disruption of the perineural sheath may allow inflammatory mediators to gain access to the neural elements. Regardless of the local events in and around the pancreas causing pain, perception of the pain message requires communication with the central nervous system. The innervation of the pancreas is complex, with both visceral somatic and autonomic nerves (see Chapter 55). Dendrites of the pancreatic nociceptive sensory afferents travel with sympathetic nerves from the pancreas and reach the celiac ganglia, although no synapse is made there. These dendritic fibers continue, bundled as the left and right greater splanchnic nerves, to the sympathetic trunk ganglia, before reaching the first cell body, located in the dorsal root ganglia in spinal cord segments T5 through T9-T10. Projections of these dorsal root neurons often traverse upward and downward for several spinal segments before entering the dorsal horn of the spinal cord. Afferent pain fibers may cross the midline in several of these connections, accounting for the midline perception of pancreatic pain. Axons from the first-order dorsal root ganglion cell bodies have two distinct pathways. Some project to the dorsal horn of the spinal cord and may release a variety of mediators including substance P, calcitonin gene-related peptide, and glutamate onto second-order neurons that project to the thalamus via the spinothalamic white matter columns. These may then synapse with third-order neurons that project to the somatosensory cortex (for cognitive integration of pain) and to the limbic system and hypothalamus (for affective and autonomic integration of pain). A second pathway for projections involves synapses within the same level of the spinal cord with sympathetic efferent cell bodies that project back down the splanchnic nerves to the celiac plexus, with second-order sympathetic neurons projecting back to the pancreas. Vagal afferents may also carry noxious stimuli from the pancreas (especially for stretch).
Noxious stimulation of these pathways can occur through a variety of mechanisms. Pressure, ischemia, inflammation, heat, and other classic stimuli can activate these pathways. The accumulation of inflammatory mediators and nerve injury can sensitize the nerve, making it hyper-responsive.144 At the periphery is an increase in substance P and calcitonin gene-related peptide in interlobular and intralobular nerve bundles in patients with chronic pancreatitis.145 Both of these neurotransmitters are involved in pain transmission. The close spatial relationship between intrapancreatic nerves and inflammatory cells strongly supports the concept of neuroimmune interaction. Growth-associated protein 43 (GAP-43), a marker of neuronal plasticity and remodeling, is seen in pancreatic nerves infiltrated with lymphocytes, and its presence correlates with pain.146,147 Additional studies have demonstrated expression of nerve growth factor (NGF) and one of its receptors (TrkA) in patients with painful chronic pancreatitis and in animal models of chronic pancreatitis.147,148 NGF is one of the key molecules involved in sensitization.144 Endogenous proteases, like trypsin, can also activate and sensitize sensory neurons in the pancreas, a process mediated through the protease-activated receptor-2 (PAR-2).149 Another activator of PAR-2 is tryptase, a mast cell product. Interestingly, mast cells are seen commonly in pancreatic tissue specimens from patients with chronic pancreatitis. The exact mechanisms by which the inflammatory cells and their products and intrapancreatic neurons interact in chronic pancreatitis remain to be clarified, although the data suggest that the production of sensitizing factors in the vicinity of pancreatic nerves alters sensory neuron form and function.
In addition, there is substantial evidence from studies of other types of chronic pain that chronic peripheral nerve injury or inflammation leads to changes in nociceptive processing that involve both the spinal cord and central nervous system. Chronic pain can produce a centrally sensitized pain state in which elimination of the original source of pain does not relieve pain.144,150 In this situation, pain may occur in response to innocuous or physiologic stimuli (allodynia) or may respond in an exaggerated fashion to stimuli that are painful (hyperalgesia). These phenomena depend on changes at both the spinal cord level and the brain. One study monitoring EEG activity of patients with chronic pancreatitis noted increased areas of referred pain to electrical stimulation of the esophagus, stomach, or duodenum.151 The central nervous system reorganization and plasticity underlying hyperalgesia and allodynia are likely major factors limiting the effectiveness of treatments for pain. Nowhere is this fact made more obvious than in the patient who continues to have pancreatic pain after a total pancreatectomy. Pain is complex, and no single mechanism is likely to be present in all patients, implying that no single therapy will be effective.
STEATORRHEA
The human pancreas has substantial exocrine reserve. Steatorrhea does not occur until pancreatic lipase secretion is reduced to less than 10% of the maximum output.152 Steatorrhea is therefore a feature of far-advanced chronic pancreatitis, in which most of the acinar cells have been injured or destroyed, but may also be seen with complete blockage of the pancreatic duct. With advanced chronic pancreatitis, maldigestion of fat, protein, and carbohydrates will occur. Azotorrhea (protein maldigestion) also occurs when secretion of proteases is less than 10% of normal.152 Affected patients may present with diarrhea and weight loss. Some patients may note bulky foul-smelling stools or may even note the passage of frank oil droplets. Unlike in small bowel diseases associated with malabsorption, watery diarrhea, excess gas, and abdominal cramps are uncommon in the steatorrhea seen in patients with chronic pancreatitis. This difference may be due to better-preserved carbohydrate absorption and small bowel and colonic function in patients with chronic pancreatitis and exocrine insufficiency than in those with small intestinal diseases such as celiac disease. Even when there is significant loss of fat in stool, most patients pass only three or four stools daily and some may pass only one.
In general, fat maldigestion occurs earlier and is more severe than protein or carbohydrate maldigestion. There are several explanations for this phenomenon. First, fat digestion depends primarily on pancreatic lipase and colipase, although gastric lipase is able to hydrolyze up to 20% of dietary fat (see Chapter 49). Second, lipase output decreases earlier and more substantially as chronic pancreatitis progresses compared with the secretion of other pancreatic enzymes such as trypsin or amylase. Third, lipase is more sensitive to acid destruction than other pancreatic enzymes. As bicarbonate secretion decreases in chronic pancreatitis and duodenal pH drops, lipase in particular is inactivated. Fourth, in addition to lipase inactivation, the low duodenal pH also predisposes to precipitation of bile salts, thereby preventing the formation of mixed micelles and further interfering with lipid digestion and absorption. Fifth, lipase is more sensitive to digestion and degradation by pancreatic proteases than other digestive enzymes.
The median time to development of exocrine insufficiency in chronic pancreatitis has been reported to be as low as 5.6 years,57 but most studies report longer duration of disease prior to development of steatorrhea. In one large natural history study, the median time to development of exocrine insufficiency was 13.1 years in patients with alcoholic chronic pancreatitis, 16.9 years in patients with late-onset idiopathic chronic pancreatitis, and 26.3 years in patients with early-onset idiopathic chronic pancreatitis.58 With very long follow-up, approximately 50% to 80% of patients with chronic pancreatitis eventually have exocrine insufficiency.57–59
Significant weight loss is uncommon despite maldigestion. Patients generally increase their caloric intake to compensate for stool losses. Weight is usually maintained despite the fact that the resting energy expenditure is generally increased in patients with chronic pancreatitis. Weight loss is most commonly seen during painful flares that prevent adequate oral intake because of pain, nausea, or vomiting. Weight loss may also occur as a result of the development of a concomitant disease such as small bowel bacterial overgrowth (SBBO) or pancreatic or extrapancreatic malignancy. Finally, weight loss may occur in patients who develop financial difficulties, suffer from chronic severe alcoholism, or lose social support because these may contribute to inadequate caloric and protein intake. Substantial weight loss should lead to an investigation of these potential causes. More subtle weight loss may be more common than is appreciated.153,154
Deficiencies of fat-soluble vitamins may develop in patients with chronic pancreatitis and steatorrhea.155,156 Significant vitamin D deficiency and osteopenia and osteoporosis occur in patients with chronic pancreatitis.154,157–160 These studies demonstrate osteopenia in 50% to 70% of patients and osteoporosis in up to 20% of patients with chronic pancreatitis and steatorrhea. Levels of the active forms of vitamin D [1,25-(OH)2-D3 and 25-(OH)D3] are generally below the normal range in these patients. Deficiencies of water-soluble vitamins and micronutrients are rare and generally seen only as a consequence of inadequate intake in chronic alcoholics. Despite the fact that vitamin B12 absorption requires intact pancreatic function to degrade R-factor from dietary cobalamin (see Chapter 49), vitamin B12 deficiency is uncommon in patients with chronic pancreatitis in the absence of ongoing alcohol abuse.
DIABETES MELLITUS
Like exocrine insufficiency, endocrine insufficiency is a consequence of long-standing chronic pancreatitis and is especially common after pancreatic resection and in tropical (fibrocalcific) pancreatitis. Islet cells appear to be relatively resistant to destruction in chronic pancreatitis (see Fig. 59-1).161,162 When diabetes develops, the mechanism is more complex than just a simple loss of beta cells due to the progressive destruction of islets.162 Infusion of glucagon-like peptide 1 (GLP-1) in patients with diabetes due to chronic pancreatitis stimulates release of endogenous insulin, confirming that some residual functional beta cells remain.162–164 In addition, levels of islet amyloid polypeptide (amylin) may be elevated in patients with diabetes due to chronic pancreatitis, a feature most commonly associated with insulin resistance in type 2 diabetes mellitus.165,166 Several studies in animal models and humans also demonstrate a loss of hepatic insulin receptor expression and an impairment in hepatic insulin receptor function, a change that can produce decreased hepatic glucose output, and that appears to be due to decreased pancreatic polypeptide secretion from islet cells.162 There is also a relative decrease in stimulated glucagon secretion from these damaged islets, although basal levels may remain normal.163 These various factors make diabetes due to chronic pancreatitis different than either type 1 or type 2 diabetes. About half of patients with chronic pancreatitis who develop diabetes will require insulin.163 Unlike type 1 diabetes, insulin-producing beta cells and glucagon-producing alpha cells are injured. This combination increases the risk of prolonged and severe hypoglycemia with overvigorous insulin treatment, owing to the lack of a compensatory release of glucagon.162,167,168
Diabetes mellitus appears to be nearly as common as steatorrhea in patients with far-advanced chronic pancreatitis. In one study the median times to development of diabetes in patients with alcoholic, late-onset idiopathic, and early-onset idiopathic chronic pancreatitis were 19.8 years, 11.9 years, and 26.3 years, respectively.58 Other studies have noted shorter median times of 6 to 10 years.57,129 Ultimately, 40% to 80% of patients with chronic pancreatitis have diabetes after long follow-up, depending on etiology.162,163 In one large cohort study, 83% of patients had diabetes 25 years after the clinical onset of chronic pancreatitis.169 In that study, risk factors for the development of diabetes included early onset of pancreatic calcifications and resection of the pancreatic tail. The latter risk has been seen in other studies170 and is probably explained by the observation that the islets may be concentrated in the body and tail of the pancreas. Microangiopathic complications are as common in patients with diabetes associated with chronic pancreatitis as in patients with type 1 diabetes with similar duration of disease.171,172
DIAGNOSIS
An impressive number and variety of diagnostic tests for chronic pancreatitis have been developed over the last few decades. This bewildering array is not only confusing but serves to point out that no single test is so accurate that it can replace all others. Many of these diagnostic tests have not been studied in a sufficiently wide spectrum of disease to define their sensitivity and specificity. These diagnostic tests are usually separated into those that are designed to detect abnormalities of pancreatic function (discussed in Chapter 56) and those that detect abnormalities of pancreatic structure (Table 59-3). Before considering each of these tests in more detail, it is useful to remember that in almost all patients, chronic pancreatitis is a slowly progressive disease. In the early stages within the pancreas, chronic inflammation, cellular necrosis and apoptosis, and activation of pancreatic stellate cells have all developed, but these features of chronic pancreatitis remain visible only on histology. With progressive fibrosis and loss and destruction of tissue, the disease becomes more evident. Abnormalities of pancreatic structure or function may take years or even decades to develop, or may not develop at all.1,57,58 All available diagnostic tests are most accurate in far-advanced disease, when obvious functional or structural abnormalities have developed. Conversely, to greater or lesser degrees, all diagnostic tests are less accurate in less advanced or early chronic pancreatitis.
Table 59-3 Available Diagnostic Tests for Chronic Pancreatitis*
| Tests of function | Direct hormonal stimulation (with pancreatic stimulation by secretin or cholecystokinin or both): |
| Using oroduodenal tube† | |
| Using endoscopy† | |
| Magnetic resonance cholangiopancreatography with secretin stimulation | |
| Fecal elastase | |
| Fecal chymotrypsin | |
| Serum trypsinogen (trypsin) | |
| Fecal fat | |
| Blood glucose level | |
| Tests of structure | Endoscopic ultrasonography |
| Endoscopic retrograde cholangiopancreatography | |
| Magnetic resonance imaging with magnetic resonance cholangiopancreatography | |
| Computed tomography | |
| Abdominal ultrasonography | |
| Plain abdominal film |
* Ranked in estimated order of decreasing sensitivity for each category.
Functional abnormalities in chronic pancreatitis include exocrine insufficiency (maldigestion and steatorrhea), and endocrine insufficiency (diabetes mellitus). In addition, some diagnostic tests measure maximum stimulated secretory capacity of the pancreas, which becomes abnormal before there is failure of exocrine or endocrine function. Structural abnormalities that can be diagnostic include changes within the main pancreatic duct (dilation, strictures, irregularity, pancreatic ductal stones), side branches of the pancreatic duct (dilation, irregularity), or pancreatic parenchyma (lobularity of the gland, alterations in echogenicity, cysts, enlargement or atrophy, and others). Patients with alcoholic chronic pancreatitis, hereditary chronic pancreatitis, tropical pancreatitis, and late-onset idiopathic chronic pancreatitis are most prone to development of these abnormalities of function or structure, although the process may still take several years. These changes develop particularly slowly, and sometimes not at all, in patients with early-onset idiopathic chronic pancreatitis.58
The determination of the sensitivity, specificity, and accuracy of any of these diagnostic tests requires that the test result be compared with some gold standard, a test that gives reliable and certain evidence as to the presence or absence of disease. In the case of chronic pancreatitis, this gold standard is pancreatic histology (see Fig. 59-1). Unfortunately, the histologic changes are not uniform throughout the gland2,22,23 so that findings in a biopsy specimen may be misleading. Even more important, obtaining pancreatic tissue carries risk and is seldom performed solely for diagnosis.
Given the lack of a useful gold standard, one is often left with comparing a new diagnostic test with some substitute for the gold standard. One such substitute is long-term follow-up. Most series have not monitored patients diagnosed with early chronic pancreatitis or possible early chronic pancreatitis (patients in whom diagnostic tests are not unequivocally positive) for long enough to establish the presence or absence of chronic pancreatitis with certainty. The second potential substitute for the gold standard is some other diagnostic test, and in fact, new diagnostic tests are often compared with such modalities as ERCP, CT, and pancreatic function tests, and composites of these.173
TESTS OF PANCREATIC FUNCTION
Tests of pancreatic function can be divided into those that directly measure pancreatic function by measuring the output of enzymes or bicarbonate from the pancreas and those that measure the released enzymes indirectly (through its action on a substrate or its level in blood or stool) (see also Chapter 56).
Direct Tests
Direct hormonal stimulation tests are believed to be the most sensitive function test for chronic pancreatitis.1,174–176 A few studies have compared the results of these hormonal stimulation tests with pancreatic histology,177–179 with overall sensitivities ranging from 67% to 88%. The largest study compared histology with combined secretin-cholecystokinin testing in 108 patients.177 There was a linear correlation of stimulated bicarbonate output with histologic severity of chronic pancreatitis. Although mean peak bicarbonate concentration was in the normal range (>80 mEq/L) in 69 patients with normal or equivocal histology, mean bicarbonate concentration was 70, 63, and 50 mEq/L in those with mild, moderate, and severe histologic chronic pancreatitis, respectively. The overall sensitivity of hormonal stimulation testing in this study was 67%, with a specificity of 90% and overall accuracy of 81%. When the analysis was restricted to the 29 patients with moderate or severe histologic changes of chronic pancreatitis, the sensitivity of hormonal stimulation testing rose to 79%. In this same group of 29 patients, the sensitivity of ERCP was 66%.
In comparisons with ERCP, direct hormonal stimulation tests appear to be on average somewhat more sensitive for the diagnosis of chronic pancreatitis. The values for sensitivity of pancreatic function testing range from 74% to 97%, with specificity ranging from 80% to 90%.174,175,180–185 In these studies the two tests agree in about three quarters of patients, although some studies note higher rates of concordance. Most studies also note a general correlation between increasing structural abnormalities and progressive abnormalities of hormone stimulation test results, although the relationship is not exact.
Most of these studies also identify patients with discordant results—patients with abnormal ERCPs and normal hormonal stimulation test results as well as those with normal ERCPs and abnormal hormonal stimulation test results. In four studies the percentage of patients with an abnormal hormonal stimulation test result and a normal ERCP ranged from 3% to 20%.180–184 Two small studies have followed such patients whose diagnosis was based solely on an abnormal hormonal stimulation test result, and both found development of chronic pancreatitis on follow-up in 90% of patients.184,186 These data point out that direct pancreatic function testing appears to be able to identify a group of patients with chronic pancreatitis who have functional abnormalities of stimulated secretion but who do not (yet) have ERCP-identifiable structural abnormalities. Conversely, most of these studies also report patients with normal hormonal stimulation test results and abnormal ERCPs. This group of patients is generally less common, averaging less than 10% in several studies.180–186 Long-term follow-up in a small group of such patients noted development of chronic pancreatitis in 0% to 26%.184,186 These studies point out that in situations in which results of the two tests disagree, hormonal stimulation testing appears to be somewhat more sensitive and specific than ERCP.
Some experts have suggested that the pancreas has such reserve that 30% to 50% damage to the gland is necessary before direct pancreatic function tests yield reliably positive results. Despite their theoretical advantages, direct pancreatic function tests have a number of limitations.175,176 First, they have not been well standardized across institutions offering the test. Second, they are available at only a very few referral centers and so are not available to the majority of clinicians seeing patients with chronic pancreatitis. Third, it can be difficult for patients to tolerate unsedated placement of an oroduodenal tube for the hour or more required for the test. Fourth, accurate measurement of bicarbonate concentrations or enzyme output may be challenging. False-positive results have been reported in patients who have undergone Billroth II gastrectomy, in patients with diabetes, celiac disease, and cirrhosis, and in patients recovering from a recent attack of acute pancreatitis.
There are variations of direct pancreatic function tests that may be easier for patients to tolerate (by sedating them) and might be able to be made more widely available. One proposed variation is to collect pancreatic secretions at the time of ERCP by placement of a catheter directly in the pancreatic duct (the so-called intraductal secretin test). This test typically samples pancreatic output for only 15 minutes, to minimize the likelihood of ERCP-induced pancreatitis. It is not standardized and does not appear to be as accurate as standard direct pancreatic function testing,175,187,188 probably because of the rather brief collection time. Another variation of pancreatic function testing is to use sedation and a standard upper endoscope to take the place of the usual oroduodenal tube, with analysis of bicarbonate output using the regular hospital laboratory. This variation attempts to bypass the difficulties limiting the widespread application of standard direct pancreatic function tests, such as passage of the collection tube in unanesthetized patients and need for a dedicated laboratory to measure the bicarbonate concentration by back-titration. Unlike the intraductal secretin test, this endoscopic variation appears to be nearly, although not quite, as accurate as standard direct pancreatic function testing.189–191 The initial descriptions of this test used a 60-minute collection with timed aspirates of duodenal fluid every 15 minutes. This is a long time to keep a patient sedated and an endoscopy room occupied, although it may be possible to shorten the test and only collect samples at 30 and 45 minutes after secretin injection192 with only moderate loss of sensitivity. Another variation of the test is to measure lipase output rather than bicarbonate output, with secretin193 or CCK194 as the secretagogue. These variations appear to be less accurate than standard direct pancreatic function testing. Like traditional pancreatic function testing, endoscopic-based pancreatic function tests have been compared to alternative diagnostic tests like ERCP,195 with an overall agreement in 86% of patients and a high negative predictive value for endoscopic pancreatic function testing. The value of pancreatic function tests, whether traditional or endoscopic, lies in their high sensitivity and consequent ability to rule out chronic pancreatitis.196
Indirect Tests
The desire to develop indirect tests of pancreatic function is an outgrowth of the complexity, discomfort, and limited availability of direct pancreatic function testing. Indirect tests can generally measure pancreatic enzymes in blood or stool. Tests that measure the effect of pancreatic enzymes on an orally administered substrate with collection of metabolites in blood, breath, or urine (see Chapter 56) are of historical interest only.
Serum Trypsinogen
Serum trypsinogen (often called serum trypsin) can be measured in blood and provides a rough estimation of pancreatic function. Very low levels of serum trypsinogen (<20 ng/mL) can be seen in patients with advanced chronic pancreatitis with steatorrhea.197 Serum trypsin is not decreased in patients with other forms of steatorrhea, but low levels of serum trypsinogen may be seen in patients with pancreatic ductal obstruction, including malignant obstruction. The test is available through commercial laboratories.
Pancreatic Enzymes in Stool
Low concentrations of chymotrypsin or elastase in stool can reflect inadequate delivery of these pancreatic enzymes to the duodenum. Both can be measured on random samples of stool. Fecal chymotrypsin is low in most patients with chronic pancreatitis and steatorrhea.174 Fecal chymotrypsin is available from commercial laboratories. False-positive results have been reported in other malabsorptive conditions (celiac disease, Crohn’s disease), in diarrheal diseases in which the stool is diluted, and in severe malnutrition. Because the test is usually normal in patients without steatorrhea, it is reliably positive only in advanced chronic pancreatitis.
Fecal elastase has significant advantages over fecal chymotrypsin in that it is much more stable in passage through stool and is easier to measure. Levels less than 200 µg per gram of stool are considered abnormal. The test is reasonably accurate in more advanced chronic pancreatitis.198–201 Fecal elastase can be low in other diseases causing diarrhea, such as short bowel syndrome and small bowel bacterial overgrowth. Fecal elastase measurement is available through reference laboratories in the United States.
TESTS OF PANCREATIC STRUCTURE (IMAGING)
Plain Abdominal Radiography
The finding of diffuse (but not focal) pancreatic calcifications on plain abdominal films is quite specific for chronic pancreatitis. Focal calcifications may be seen in cystic and islet cell tumors of the pancreas, and in peripancreatic vascular calcifications. Calcifications occur late in the natural history of chronic pancreatitis and may take from 5 to 25 years to develop.57,58 Calcifications are most common in alcoholic, late-onset idiopathic, hereditary, and tropical pancreatitis and far less common in early-onset idiopathic pancreatitis. Acceleration of the clinical course of chronic pancreatitis and subsequent calcifications are particularly common in patients who smoke.42,43,202,203 Calcifications are not static once they develop and may in fact wax and wane over time.204
Abdominal Ultrasonography
Ultrasonography has been widely studied as a diagnostic tool for chronic pancreatitis.205 This modality is limited in that the pancreas (and particularly the pancreatic head) cannot be adequately visualized in some patients owing to overlying bowel gas or body habitus. Ultrasonographic findings indicative of chronic pancreatitis include dilation of the pancreatic duct, shadowing pancreatic ductal stones, gland atrophy or enlargement, irregular gland margins, pseudocysts, and changes in the parenchymal echotexture (Table 59-4). Most studies suggest a sensitivity of 50% to 80% with a specificity of 80% to 90%.174 The true sensitivity and specificity may be different because most of these studies are older and did not use modern state-of-the-art equipment.
Table 59-4 Grading of Chronic Pancreatitis by Ultrasonography (US) or Computed Tomography (CT)
| GRADE | US OR CT FINDINGS |
|---|---|
| Normal | No abnormal findings on a good-quality study visualizing the entire gland |
| Equivocal | One of the following: |
| Mild dilatation of the pancreatic duct (2-4 mm) in the body of the gland | |
| Gland enlargement ≤2-fold normal | |
| Mild-moderate | One of the preceding findings plus at least one of the following: |
| Pancreatic duct dilatation (>4 mm) | |
| Pancreatic duct irregularity | |
| Cavities <10 mm | |
| Parenchymal heterogeneity | |
| Increased echogenicity of duct wall | |
| Irregular contour of the head or body | |
| Focal necrosis of parenchyma | |
| Severe | Mild/moderate features plus one or more of the following: |
| Cavity >10 mm | |
| Intraductal filling defects | |
| Calculi/pancreatic calcification | |
| Ductal obstruction (stricture) | |
| Severe duct dilatation or irregularity | |
| Contiguous organ invasion |
Adapted from Sarner M, Cotton PB: Classification of pancreatitis. Gut 1984; 25:756.
In one more recent study comparing transabdominal ultrasonography with CT, ERCP, and EUS, the accuracy of ultrasonography was 56%.206 In this study, some abnormality (such as changes in parenchymal echotexture) was noted on ultrasonography in 40% of patients who had a normal pancreas as defined by the other diagnostic tests. A large screening study of transabdominal ultrasonography in Japan encompassing 130,000 examinations found increased echogenicity, mild dilation of the pancreatic duct, small cystic cavities, and even ductal calcification in the absence of clinical features of chronic pancreatitis.207 The majority of these abnormalities could not be attributed to chronic pancreatitis and were instead attributed to aging. These studies would suggest that there is a large spectrum of ultrasonographic findings in normal individuals and that it can be difficult to distinguish normal (or age-related) variability from chronic pancreatitis if the visualized changes are mild. Thus, transabdominal ultrasonography is often not useful in the evaluation of patients with suspected chronic pancreatitis. The finding of a normal pancreas or moderate to marked changes of advanced chronic pancreatitis is generally definitive. Mild changes of chronic pancreatitis are less specific and must be interpreted in light of the clinical history and the patient’s age. Ultrasonography can be useful in screening for complications of chronic pancreatitis (e.g., pseudocyst or bile duct obstruction) and in evaluating for other conditions that might mimic the symptoms of chronic pancreatitis (i.e., biliary tract disease).
Computed Tomography
The overall sensitivity of CT for chronic pancreatitis is between 75% and 90%, with a specificity of 85% or more.208–210 CT is able to image the pancreas in essentially all patients and hence has a substantial advantage over ultrasonography. Table 59-4 outlines the diagnostic abnormalities seen on CT in chronic pancreatitis. Most studies of diagnostic CT in chronic pancreatitis have not used state-of-the-art CT technology. One study using a 64 multi-row detector CT demonstrated the ability to accurately image the pancreatic duct and an overall sensitivity or more than 80% and specificity of near 100% when compared with MRCP or ERCP.211 It is almost certain that modern multidetector scanners using a pancreas protocol have better sensitivity than these older studies suggest, although the magnitude of the greater sensitivity is not known. Like all diagnostic tests, CT is most accurate in advanced chronic pancreatitis after substantial structural changes have developed (Fig. 59-4). Although CT is more expensive than ultrasonography and exposes the patient to ionizing radiation, it is more sensitive and more specific.
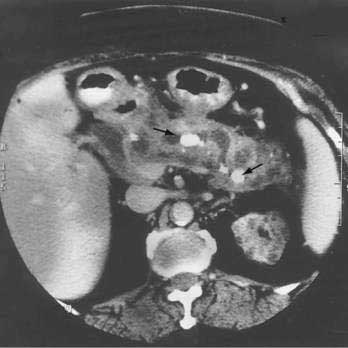
Magnetic Resonance Imaging
MRI, coupled with MRCP is as accurate, and probably more so, than CT in patients with chronic pancreatitis.208–212 MRCP results agree with ERCP results in about 90% of cases.212–213 Agreement between MRCP and ERCP is less common in areas where the pancreatic duct is small (tail of pancreas and side branches) or when the ductal changes are more subtle.214 Improved visualization of the pancreatic duct can be achieved by administering secretin.212–216 In addition, signal intensity and arterial enhancement ratios can be obtained, using gadolinium as a contrast agent, which may improve the ability to image the gland.212,216 Finally, a qualitative or semi-quantitative assessment of fluid output from the pancreas to the duodenum can be made during MRCP after secretin injection, which may allow additional insights into pancreatic function.217–219 The accuracy of this type of pancreatic function test has been compared with the intraductal secretin test217 and with low-sensitivity tests such as fecal elastase,220 but not with a more formal hormonal-stimulation pancreatic function test. Some analyses suggest that just measuring volume after secretin stimulation, instead of bicarbonate concentration, is too inaccurate to be clinically useful.221 Advancements in MR image analysis will continue to improve the image quality of MRCP, which in the future will equal ERCP in accuracy. Like ERCP, however, MRCP will be inaccurate in patients without significant ductal abnormalities. Although MRI is widely available, not all centers have the capacity to perform high-quality MRCP.
Endoscopic Retrograde Cholangiopancreatography
Pancreatography has been considered the most specific and sensitive test of pancreatic structure, and many consider it the de facto gold standard. It also has the advantage over all previously discussed tests in that therapy (e.g., pancreatic duct stenting or stone extraction) may be administered during its performance. The disadvantage, however, is that ERCP is the riskiest diagnostic test, with complications occurring in at least 5% of patients (in as many as 20% of certain subgroups) and a mortality rate of 0.1% to 0.5%. In most studies in patients with chronic pancreatitis, the sensitivity of ERCP is between 70% and 90%, with a specificity of 80% to 100%.173,174,181–185,222,223 Thus, chronic pancreatitis can exist in the absence of any visible changes within the pancreatic duct.177,222–224
The diagnostic features of chronic pancreatitis on ERCP are listed in Table 59-5. These were developed at an international consensus conference held more than 20 years ago.225 The diagnosis is based on abnormalities seen in the main pancreatic duct and the side branches. ERCP is highly sensitive and specific in patients with advanced disease. The appearance of a massively dilated pancreatic duct with alternating strictures (the chain-of-lakes appearance) is characteristic of the most advanced chronic pancreatitis (Fig. 59-5). Less dramatic pancreatographic changes are less definitive and specific (Fig. 59-6).222,223
Table 59-5 Cambridge Grading of Chronic Pancreatitis on Endoscopic Retrograde Pancreatography
| GRADE | MAIN PANCREATIC DUCT | SIDE BRANCHES |
|---|---|---|
| Normal | Normal | Normal |
| Equivocal | Normal | <3 Abnormal |
| Mild | Normal | ≥3 Abnormal |
| Moderate | Abnormal | ≥3 Abnormal |
| Severe | Abnormal with at least 1 of the following: | ≥3 Abnormal |
| Large cavity (>10 mm) | ||
| Obstruction | ||
| Filling defects | ||
| Severe dilatation or irregularity |
Adapted from Axon ATR, Classen M, Cotton PB, et al. Pancreatography in chronic pancreatitis: International definitions. Gut 1984; 25:1107-12.
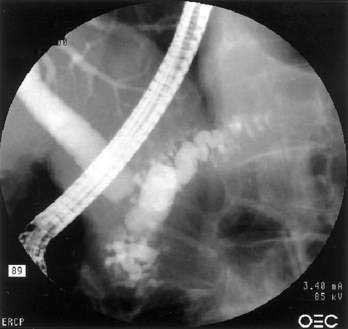
The accurate interpretation of an ERCP requires a study of adequate quality (filled to the second generation of the side branches and without significant movement artifact) and the capability to obtain radiographic images of high resolution. Many pancreatograms do not meet these criteria for an adequate study.223 An underfilled pancreatic duct can appear to have an irregular duct margin (leading to overdiagnosis of chronic pancreatitis) or might not delineate changes within the inadequately filled side branches (leading to underdiagnosis of chronic pancreatitis).
The pancreatic duct abnormalities characteristic of chronic pancreatitis can be seen in other conditions as well. The most common is the effect of aging on the pancreatic duct. Although pancreatic function is well preserved in aging, impressive abnormalities may develop in the pancreatic duct. They include focal or diffuse dilatation of the main pancreatic duct and its side branches, the development of cystic cavities, and even ductal calculi.117,222,223,226,227 In the large screening ultrasonography study mentioned previously, 50% of all calcification and more than 80% of ductal dilation and cystic lesions seen were believed to be attributable to aging, not chronic pancreatitis.207 Temporary changes in the pancreatic duct may also occur after an episode of acute pancreatitis and may take months to resolve.222,223,225 Pancreatic carcinoma may produce changes within the pancreatic duct that resemble those of chronic pancreatitis. Finally, the placement of pancreatic duct stents can produce new abnormalities within the pancreatic duct that mimic chronic pancreatitis and that may not entirely resolve after stent removal.222,223,228,229 Pancreatic stents are being placed with much more frequency to prevent post-ERCP pancreatitis. These temporary, very small-caliber stents used for these purposes appear to less commonly produce these ductal changes.230
There is significant potential for substantial interobserver and intraobserver variability in the interpretation of ERCP.222,223 The initial consensus conference225 identified some abnormalities such as a dilated duct, abnormal duct contour, and abnormal side branches but did not define absolute criteria to differentiate normal from abnormal or normal variant. In one study, 74 postmortem pancreatograms were submitted to six experienced endoscopists.116 They were asked to judge whether the pancreatogram demonstrated chronic pancreatitis and the severity of the abnormalities. The pancreas was then examined for histologic correlation. All six endoscopists correctly identified the 5 subjects with chronic pancreatitis. In the remaining 69 subjects, there was no histologic evidence of chronic pancreatitis. Depending on the observer, between 42% and 98% of these pancreatograms were read as demonstrating chronic pancreatitis, largely based on mild abnormalities within the main duct and side branches. The mistaken interpretations were felt to be due to age-related changes within the pancreas. Another study attempted to estimate intraobserver variability by submitting 51 pancreatograms to four expert endoscopists on three separate occasions.231 Each endoscopist was consistent in his or her own three reports in 47% to 95% of cases (yielding a rate of intraobserver variability as high as 53%). Much of the intraobserver and interobserver variability in ERCP evaluations is related to the interpretation of mild or subtle pancreatographic changes rather than dramatic abnormalities. This is the most substantial clinical problem related to ERCP as a diagnostic tool; subtle or minor abnormalities of the pancreatic duct are quite nonspecific and are not reliable markers of chronic pancreatitis.
Endoscopic Ultrasonography
EUS allows a highly detailed examination of pancreatic parenchyma and the pancreatic duct by overcoming the imaging problems in transabdominal ultrasonography (such as intervening gas in the bowel lumen). The diagnosis of chronic pancreatitis on EUS is based on the presence of abnormalities in the pancreatic duct and the parenchyma (Table 59-6). These features may be individually classified as none, minimal, moderate, or extensive but in practice are generally only graded as present or absent, and the total number of features is used as the score. The sensitivity and specificity of the test is determined by the threshold total score used to define chronic pancreatitis. A large range of threshold scores have been used, ranging from 1 to 6. Most studies have used the presence of 3 or more features to define a positive result.222
Table 59-6 Diagnosis of Chronic Pancreatitis on Endoscopic Ultrasonography
| Parenchymal abnormalities | Hyperechoic foci |
| Hyperechoic strands | |
| Lobularity of contour | |
| Cysts | |
| Ductal abnormalities | Main duct dilatation |
| Main duct irregularity | |
| Hyperechoic ductal walls | |
| Visible side branches | |
| Calcification |
EUS has been compared with pancreatic histology in a limited number of patients. One study compared EUS features with histology in 71 patients who underwent surgical therapy for presumed chronic pancreatitis.232 Utilizing a cut-off of more than three EUS criteria, the sensitivity of EUS was 83% and the specificity was 57%. In a subgroup with more advanced histologic evidence of chronic pancreatitis by histology, the sensitivity was 83% and specificity 80%. No single EUS criteria predicted fibrosis better than any other, but there was a general correlation between the number of EUS criteria and the histologic severity of disease. Another study in 42 patients who underwent EUS prior to pancreatic surgery (largely for carcinoma) determined a cut-off of more than four EUS criteria had a sensitivity of 90% and specificity of 86%.233 A study using EUS-guided Tru-cut biopsy of the pancreas234 noted poor agreement between EUS and histology, but the histologic specimens obtained were small and likely to not be representative of the entire gland.
EUS and ERCP agree in about 80% of patients.222,223,225–227 EUS has also been compared with standard and intraductal direct pancreatic function testing. The agreement between these tests varies widely in these studies, ranging from 10% to 90%.222,223,235,238–240 This variability is partly related to the severity of disease. The agreement is best in those with more advanced disease. In one study239 the sensitivity of EUS for advanced chronic pancreatitis (classic findings on ERCP and an abnormal pancreatic function test) was good (sensitivity of 73% and specificity of 81% using more than three criteria), but the sensitivity for less advanced chronic pancreatitis was only 10%.
In the majority of instances in which EUS findings disagree with results of other diagnostic tests, it is the EUS findings that are abnormal when the other tests are normal. This speaks to the question of the specificity of EUS. Specificity is determined by the cut-off chosen. Specificity is degraded by other conditions that can mimic the EUS findings of chronic pancreatitis. This may be a substantial conundrum, because EUS changes of chronic pancreatitis are frequently encountered. The edema associated with a recent episode of acute pancreatitis can make duct margins and intralobular septa more apparent, which will reduce specificity.222 Age-related changes can be observed in the pancreas by EUS that mimic the changes of chronic pancreatitis.241 Chronic alcohol and tobacco use can produce similar changes, in the absence of clinical chronic pancreatitis.222,242–244 In one study 39% of patients with unexplained dyspepsia had five or more EUS features of chronic pancreatitis, and 34% of controls had three or more features.245 It is unlikely that chronic pancreatitis is present or will develop in all or even most of these patients.
The diagnosis of chronic pancreatitis by EUS is based on the interpretation of sonographic images. There is moderate interobserver variation in this interpretation.246 An international consensus conference met in 2007 to try to develop more reliable EUS criteria for chronic pancreatitis.247 This new system (called the Rosemont criteria) uses major and minor criteria and attempts to provide semiquantification of severity. Whether it is more specific or less prone to interobserver variability remains to be determined. What also remains to be determined is whether the presence of three or four (or fewer) EUS features, in the absence of corroborating information from other diagnostic tests, is adequate for a conclusive diagnosis of chronic pancreatitis to be made.196,222,248,249 Resolution of this issue will require very long follow-up of patients, given the lack of a useful gold standard. Other advances in EUS imaging including the use of contrast, digital image analysis, and EUS elastography are just beginning to be studied, but may improve specificity in the future.
EUS is highly accurate in patients with advanced chronic pancreatitis (Fig. 59-7). An EUS score of greater than 5 is highly specific for chronic pancreatitis. A completely normal or near normal EUS (0 to 2 criteria) essentially rules out chronic pancreatitis.222,248,249 EUS scores of 3 or 4 should be considered indeterminate and should be interpreted in light of the patients clinical features and with recognition of the effect of alcohol, tobacco, age, and other associated conditions (diabetes, chronic kidney disease) that can mimic the changes of chronic pancreatitis.