[level-membership-for-emergency-medicine-category]Chapter 74
Chronic Obstructive Pulmonary Disease
Perspective
Chronic obstructive pulmonary disease (COPD) is one of the most common causes of death worldwide. Although prevalence estimates vary by measurement methods and by population studied, there is general agreement that COPD is underdiagnosed and under-reported. Regardless of the success of smoking cessation programs, smoking behavior in the past several decades and the delay of the appearance of symptoms in an aging population virtually guarantee an increase in prevalence, especially in developed nations. In addition, it is becoming increasingly clear that other risk factors besides cigarette smoking contribute significantly to the worldwide burden of COPD.1
It is estimated that COPD will be the fifth leading cause of lost disability-adjusted life years worldwide by 2030.2 The financial burden of COPD is enormous, accounting for billions of dollars every year for treatment and lost productivity. The majority of these costs are related to hospitalization for acute exacerbations.3 Despite its enormous effects, COPD has received relatively less attention from basic medical researchers and clinicians than other diseases. Large multinational collaborations, such as the Global Initiative for Chronic Obstructive Lung Disease (GOLD), sponsored jointly by the National Heart, Lung, and Blood Institute and the World Health Organization, have reinvigorated the scientific and medical communities frustrated by the unrelenting progressive nature of COPD and its poor response to existing therapies.
The definition of COPD is imprecise and incorporates advances in our understanding of its underlying mechanisms and natural history. In their consensus statement, the GOLD collaborators define COPD as “a preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by airflow limitation that is not fully reversible.” They also state that “the airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases.”4 This definition reflects new data underscoring the systemic nature of the disease, as well as a deliberate optimism with respect to new prevention and treatment strategies. It specifically avoids mention of chronic bronchitis and emphysema, two entities that have been traditionally included in the definition of COPD. Chronic bronchitis, defined as the presence of cough and sputum production for at least 3 months in each of 2 consecutive years, can occur without airflow limitation. Emphysema, the destruction of alveoli, is a pathologic term, not one that pertains to clinical diagnosis. Unlike with many earlier definitions of COPD, the GOLD collaborators also specifically exclude asthma, which is reversible airflow limitation. Whether reversible airflow limitation is considered to be part of COPD itself or caused by coexisting asthma is of limited significance to the emergency physician, who will continue to make every attempt to identify and reverse airflow limitation.
As many as 50% of all acute COPD exacerbations are not reported to physicians. In addition, not all reported exacerbations necessitate hospitalization.5 Nonetheless, in 2007 almost 2% of all hospital admissions in the United States were directly attributed to COPD, and it was considered a contributory factor in another 9%. In patients older than 65 years, the percentage of all hospitalizations related to COPD approaches 20%.6 As the severity of the underlying disease progresses, so does the frequency of exacerbations.7 Moreover, in a subset of patients, incomplete recovery from acute exacerbations may reflect a contribution of exacerbations to the pathophysiology of relentless disease progression.
Principles of Disease
In the past several decades the discovery that chronic airway inflammation plays a central role in the pathophysiology of asthma has led to an important change in its management, specifically, the liberal use of corticosteroids for treating moderate to severe disease. Airway inflammation is also at the center of the pathophysiology of COPD, but the inflammatory process of COPD differs from that of asthma. In COPD, neutrophils, CD8+ lymphocytes, and macrophages predominate in bronchial washings, whereas in asthma the cellular response is characterized by the presence of eosinophils.8 The inflammatory mediators differ in COPD, and several mediators, such as tumor necrosis factor, leukotriene B4, and interleukin-8, are linked to the destruction of parenchyma. These differences in the nature of the inflammatory response in COPD may account for its relatively poor response to current anti-inflammatory treatment compared with asthma.
Cigarette smoking, the most significant risk factor for the development of COPD, exerts its effects at multiple points in the inflammatory cascade of COPD, negatively affecting both the protease to antiprotease and oxidant to antioxidant balances. Although smoking cessation slows the progression of the disease, it does not end the chronic inflammatory process within the airways, indicating that mechanisms independent of smoking are involved.9 Moreover, although a majority of COPD patients have a significant smoking history, only a minority of smokers ever develop airflow limitation. This suggests the importance of other factors, both environmental and genetic.1 Other identified causative factors include heavy occupational exposure to dusts and air pollution from indoor cooking, particularly in the developing world.10–12 Long-term passive exposure to tobacco smoke and urban air pollution also appear to be contributory.13,14 Although there is an association between early childhood lower respiratory tract infections and later development of COPD, a causal relationship is less certain.
Staging the Severity of Disease
Most classifications of disease severity are based on quantitative measurements of airflow limitation, such as the forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity (FVC) ratio. These indices are measured after any reversible airflow limitation is addressed by treatment with bronchodilator medications. The GOLD collaborators define four stages, beginning with a mild stage (stage I), when spirometry is abnormal but symptoms may not yet be apparent, and ending in very severe COPD (stage IV), when FEV1 is less than 30% of predicted (Table 74-1). Frequent exacerbations are usually seen when the FEV1 falls below 50% predicted (stages III and IV).4,15,16
Table 74-1
The GOLD Classification of Severity of Chronic Obstructive Pulmonary Disease (COPD)
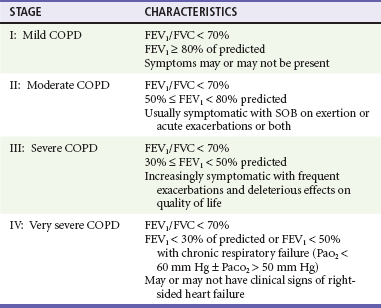
Adapted from Rabe K, Hurd S, Anzueto A, et al: Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532, 2007.
Acute Exacerbations
As with asthma, viral infection appears to be a frequent inciting agent in COPD exacerbations. Commonly implicated viruses include rhinovirus, respiratory syncytial virus, coronavirus, and influenza virus.17–19 Exacerbations associated with a viral cause are longer and more severe than those without an apparent inciting agent.18,20
Controversy remains regarding the role of bacterial pathogens in acute exacerbations of COPD, because the evidence for the role of bacteria in the pathogenesis remains indirect. Almost half of all exacerbations are associated with negative cultures for the typical respiratory pathogens, such as Haemophilus influenzae, Streptococcus pneumoniae, Moraxella (Branhamella) catarrhalis, and Pseudomonas aeruginosa. In addition, these organisms are recovered from the tracheobronchial tree of patients in their chronic, steady state, suggesting that bacteria may play a more important role in the pathogenesis of chronic COPD than in acute exacerbations.21 Although molecular typing shows that recent colonization with new serotypes of the common pathogens is associated with an exacerbation, there is a possibility that this relationship is not causal. Experimental evidence of specific immunologic responses to bacteria in exacerbations suggests, however, that they do play a significant role.22,23
Environmental factors, such as air pollution, are also implicated in COPD exacerbations. Indirect evidence for this relationship is largely derived from hospitalization rates for exacerbations during periods of increased air pollution.24 Finally, in as many as one third of all COPD exacerbations, no specific cause can be identified.
Clinical Features
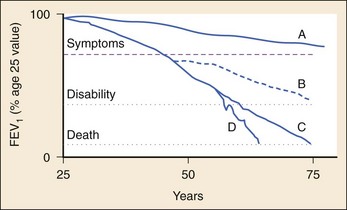
COPD patients have a long premorbid course during which decreases in air flow indices can be measured in the absence of symptoms. Intermittent cough or shortness of breath on exertion may be easily misattributed to poor physical conditioning. Moreover, patients may remain asymptomatic for many years by gradually limiting their activities in proportion to their pulmonary reserve. After several years, a daily productive cough frequently develops, and periods of dyspnea, the cardinal symptom of airflow limitation, increase. The clinical progression of COPD is slow and insidious, with gradual decreases in airflow punctuated by increasingly frequent and debilitating exacerbations. Eventually the patient becomes truly incapacitated by dyspnea on minimal or no exertion. Profound muscle wasting and weight loss and the emergence of cor pulmonale or chronic ventilatory failure are characteristic of end-stage disease. Figure 74-1 depicts the progression of COPD over time.
Diagnostic Strategies
Pulse Oximetry, Arterial Blood Gas Analysis, and Waveform Capnography
ABG values, once a mainstay of ED evaluation of COPD patients, are of limited value in management of acute COPD episodes. Status and response to therapy often are monitored noninvasively by capnography and pulse oximetry. The presence of respiratory failure unresponsive to therapy (defined as PaO2 <40 mm Hg, PaCO2 >60 mm Hg, and pH <7.25 mm Hg) warrants consideration of admission to an intensive care unit, but clinical evaluation is much more important than any particular blood gas values.4 When baseline blood gas levels are not available, the usefulness of ABGs is even more limited, and interpretation should be based on the degree of acidemia present, which likely represents the extent of acute CO2 retention. ABGs should not be used to determine whether a patient requires intubation or noninvasive ventilatory support (NIVS). These decisions should rather be guided by the overall state of the patient, progression of fatigue, comorbid illness, and response to therapy. Patients with very poor blood gas values may do well without intubation or NIVS, but others with mildly disturbed values may require urgent airway intervention. Thus measurement of ABGs should not be performed routinely in the ED and should be undertaken only in response to specific circumstances, such as irregular or apparently unreliable pulse oximetry values or when a single baseline correlation with end-tidal carbon dioxide (ETCO2) is desired.
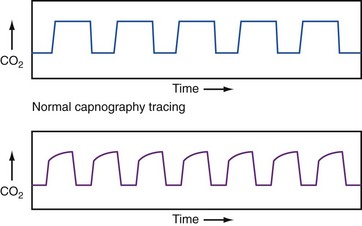
Waveform capnography represents the continuous quantitative measurement of exhaled CO2. It has emerged as a potential diagnostic and monitoring tool in patients with acute respiratory distress. The appearance of the waveform may assist the clinician in differentiating acute exacerbation of COPD from other causes of acute dyspnea, such as CHF. In patients with obstructed airways, the plateau phase of the waveform typically steepens in proportion to the severity of the obstruction, as depicted in Figure 74-2. Unfortunately, in patients with COPD, the actual end-tidal PCO2 measurements obtained from capnography do not correlate well with arterial PCO2 measurements, especially in more severe disease. Nonetheless, the patterns and trends from capnography may be helpful in guiding management and assessing response to therapy.25
Chest Radiography
In patients who are known to have COPD, the primary role of the chest radiograph is to determine whether there is an acute, treatable cause for clinical deterioration, especially pneumothorax or parenchymal consolidation (atelectasis secondary to mucous plugging, pneumonia, or obstruction by tumor). Otherwise, the chest radiograph is of limited use and may exhibit a range of chronic changes, depending on disease severity and the relative degree of the various pathologic processes. Findings may include hyperinflated lung fields, decreased vascular markings, and a small cardiac silhouette or, in contrast, normal inflation with increased vascular markings and an enlarged heart.26,27 In cor pulmonale, impingement on the retrosternal airspace by the enlarged right ventricle can be seen on the lateral film.
Electrocardiogram and Cardiac Monitoring
The classic descriptions of P pulmonale (peaked P waves in leads II, III, and aVF), low QRS voltage, clockwise rotation, and poor R wave progression in the precordial leads are interesting correlates of COPD but are both insensitive and nonspecific. The presence of electrocardiogram (ECG) criteria for RVH suggests established cor pulmonale. These findings, however, can be easily obscured on the ECG by other processes, and the absence of criteria for RVH cannot be relied on to rule out cor pulmonale.28
In severely ill patients or those with concomitant chest pain, continuous ECG monitoring may be helpful, at least for the initial phase of the patient’s evaluation and treatment. ECG monitoring can detect dysrhythmias associated with COPD exacerbations and changes of rate and rhythm in response to therapy. The most common dysrhythmias associated with COPD are atrial tachydysrhythmias, such as atrial fibrillation and multifocal atrial tachycardia. Although atrial fibrillation may require treatment with rate control or conversion, multifocal atrial tachycardia often resolves with the treatment of the COPD exacerbation itself.29
Blood Tests
The measurement of B-type natriuretic peptide (BNP) may be useful as a diagnostic adjunct in patients with acute dyspnea. BNP is a naturally occurring peptide that is released by the ventricles in response to volume expansion and stretch. It plays a central role in the neurohormonal response to “unload” the ventricles in CHF through natriuresis, diuresis, vasodilation, and suppression of the renin-angiotensin system. BNP values above 500 pg/mL are suggestive of decompensated heart failure, and levels below 100 pg/mL are very suggestive of the absence of CHF.30
Despite the sensitivity of BNP in detecting the physiologic presence of heart failure, its utility as a routine test in the assessment of dyspneic patient is debated. Some data indicate that routine use reduces evaluation times, total costs, and diagnostic accuracy.31–33 Data on the clinical usefulness of BNP, however, are derived from studies that include patients with clear clinical features of either pulmonary or cardiac disease, casting doubt on its usefulness in the subset of patients who present a true diagnostic dilemma.34,35 BNP measurements may be helpful in revealing patients with unsuspected CHF, but caution should be exercised to prevent over-reliance on this test (see the discussion of differential considerations).
Differential Considerations
In most cases, differentiation of COPD exacerbation from acute CHF can be made on clinical grounds. Nonetheless, a significant percentage of patients who come to the ED with acute dyspnea and an established diagnosis of COPD are ultimately diagnosed with acute CHF despite having no prior history of heart failure. The addition of a BNP assay to the ED evaluation of such patients identifies some in whom a new diagnosis of CHF is not suspected.33
Because BNP can be elevated in association with right ventricular stretch, incautious interpretation of an elevated BNP may lead the clinician to favor the diagnosis of acute left-sided CHF and overlook cor pulmonale and PE, both critical considerations in the patient with COPD.36 Moreover, acute CHF and COPD often coexist, and even severe elevations in BNP do not obviate the identification and treatment of acute pulmonary pathology. Thus, although BNP measurement may be helpful in the evaluation of the acutely dyspneic patient, it cannot be interpreted in isolation and must not supplant clinical judgment.
Acute pneumothorax can occur with COPD. This diagnosis should be actively pursued in patients with worsening respiratory status, especially when its onset is abrupt. In older patients with COPD, chest pain is often absent. A small pneumothorax cannot be excluded by physical examination and can be very difficult to detect on inspiratory chest films, especially in patients with bullous emphysema. Bedside ultrasound is a useful modality for diagnosing pneumothorax in the ED, particularly in major trauma, and may have a future role in diagnosing pulmonary edema.37,38 In assessing for pneumothorax, however, the presence of COPD may result in false-positive results, and data in this setting are limited.39 Ultimately, it is appropriate to perform chest computed tomography (CT) when clinical suggestion of a pneumothorax remains high and results of plain films and ultrasound are nondiagnostic.
Patients with COPD are often sedentary and consequently at increased risk for venous thromboembolic disease.40 The patient with cor pulmonale is at even higher risk because of increased blood viscosity, high peripheral venous pressure, and venous stasis. PE should be considered when an acute exacerbation is more severe than prior episodes, particularly if deterioration occurs quickly with no other apparent cause.41 Unfortunately, because there is significant overlap in their patterns of presentation, differentiating PE from a COPD exacerbation can be extremely difficult. Prophylactic measures to prevent venous thromboembolic disease are important considerations in the inpatient management of an acute exacerbation of COPD.
A negative screening with a sufficiently sensitive D-dimer assay (enzyme-linked immunosorbent assay [ELISA] or whole-blood agglutination) excludes venous thromboembolic disease in all but high pretest likelihood situations.42,43 Pretest probability can be assessed via any one of several structured, validated scoring systems. If either the D-dimer is elevated or the pretest probability for PE is high, CT pulmonary angiography should be performed.44 This requires multislice CT capability and may include indirect CT venography to evaluate the lower extremities for deep vein thrombosis or lower limb duplex ultrasonography.45–48 A more detailed discussion of the diagnostic evaluation and treatment of PE can be found in Chapter 88.
Box 74-1 summarizes the causes of acute decompensation in the patient with COPD.
Management
Until recently, the only modalities found to alter the progression of COPD and reduce mortality rates were smoking cessation and, for those with severe disease, chronic oxygen therapy.49,50 Outpatient therapy with long-acting bronchodilators and inhaled steroids may slow the progression of COPD51 and reduce exacerbation frequency,52 although the effects are modest. Vaccines are another important aspect of ongoing outpatient care. Although only influenza vaccines reduce mortality,53 both influenza and pneumococcal vaccinations are recommended for COPD patients.4 An overview of the emergency assessment and management of COPD exacerbations is provided in Table 74-2.
Table 74-2
General Therapeutic Guidelines for Chronic Obstructive Pulmonary Disease Exacerbations
| LIFE-THREATENING | MODERATE OR SEVERE | MILD |
| Address ABCs | Oxygen to maintain O2 saturation near 90% | Oxygen to maintain O2 saturation near 90% |
| Bag-valve ventilation, preoxygenation | Nebulized beta-agonist, anticholinergic | MDI or nebulized beta-agonist, anticholinergic |
| Intubation with or without rapid sequence technique | Noninvasive ventilation if severe | Consider oral or intravenous corticosteroid |
| In-line beta-agonist, anticholinergic | Intravenous corticosteroid | Consider oral antibiotic on discharge |
| Intravenous corticosteroid | Intravenous antibiotic | |
| Intravenous antibiotic |
ABC, airway, breathing, and circulation; MDI, metered dose inhaler.
Oxygenation and Ventilation
All COPD patients in acute respiratory distress need continuous ECG and pulse oximetry monitoring. Patients who are hypoxemic should receive controlled oxygen therapy, with a goal of oxygen saturation as measured by pulse oximetry (SpO2) of 88 to 92% or PaO2 above 60 mm Hg.54 The method of administration should be tailored to the clinical circumstances: nasal cannulae are well tolerated but are less useful for patients who are markedly tachypneic or mouth-breathing. Venturi masks are excellent for delivering a more reliable, high-flow, and adjustable concentration of supplemental oxygen but may be difficult for a distressed patient to tolerate.
Some patients with moderate to severe COPD and other chronic pulmonary diseases will respond to high-flow supplemental oxygen by developing hypercapnic respiratory failure. The mechanism for this response is debated but may be a result of several factors, including the suppression of hypoxic respiratory drive in patients accustomed to hypercapnia and the release of hypoxic vasoconstriction in poorly oxygenated areas of the lung, worsening ventilation-perfusion mismatch.55,56 Data from a prospective, randomized clinical trial clearly support setting a specific target of 88 to 92% and avoiding uncontrolled high-flow oxygen in dyspneic COPD patients, including in the prehospital environment.57
The patient in terminal ventilatory failure is cyanotic, speechless, lethargic, usually and confused and has gasping, ineffective respirations. Such patients require immediate endotracheal intubation and mechanical ventilation. Because these patients have exhausted all pulmonary reserve, rapid sequence intubation should be performed with the goal of rapid paralysis and unconsciousness (see Chapter 1). For induction and paralysis, a combination of a hemodynamically stable sedative-hypnotic such as etomidate and a rapidly acting paralytic such as succinylcholine is an appropriate regimen.
Initial ventilator settings should include a fraction of inspired oxygen (FIO2) of 100%, tidal volume in the 6- to 8-mL/kg range, and respiratory rate of 8 to 10 breaths/min in an assist-control mode with an inspiratory flow rate of 80 to 100 L/min. Sedation and analgesia are indicated to facilitate ventilation. Neuromuscular blockade is not routinely required and should be avoided when possible (see Chapter 1). Increased air trapping and resultant high intra-alveolar pressures induce intrinsic positive end-expiratory pressure (iPEEP) that can cause barotrauma. In addition, increased intrathoracic pressure decreases cardiac filling and output; therefore peak flow pressures and systemic blood pressures must be carefully monitored. If continuous capnography is not available, ABGs should be drawn after 15 to 20 minutes to ensure that ventilation is appropriate. In some settings, placement of an arterial line is helpful for monitoring blood pressure and ABG tensions. After intubation, permissive hypercapnia is essential to the ventilatory treatment of these patients, and subsequent normalization of pH and PCO2 should be gradual over many hours. Low volume and rate settings will result in hypercapnia and respiratory acidosis, but this approach helps prevent associated barotrauma often seen in treating these patients. Moreover, hyperventilation alkalosis must be scrupulously avoided, particularly because patients may have preexisting chronic metabolic alkalosis. This alkalosis can result in seizures and dysrhythmias, especially with coexisting hypokalemia.
NIVS is an accepted alternative to invasive ventilation in many patients with ventilatory failure (see Chapter 2). NIVS can be highly effective in avoiding intubation, increasing pH, reducing PCO2 and dyspnea in the first 4 hours of treatment, and reducing mortality rates.58,59 If the patient fails NIVS and requires intubation, NIVS, at the least, optimizes preoxygenation for the intubation. Patients likely to benefit from NIVS are those with moderate to severe ventilatory failure and elevated PCO2, but without marked hypoxemia.60 NIVS cannot substitute for invasive ventilation in patients who are hemodynamically unstable or in whom respiratory arrest appears inevitable. On the opposite end of the spectrum, it remains unclear whether NIVS should be instituted in patients with mild to moderate exacerbations. Although the Cochrane systematic review stresses early NIVS therapy to prevent the development of worsening acidosis and need for intubation, there is insufficient evidence to recommend the routine use of NIVS in mild exacerbations.59,61 Table 74-3 outlines inclusion and exclusion criteria for the use of NIVS.
Table 74-3
Suggested Selection and Exclusion Criteria for the Use of Noninvasive Ventilatory Support
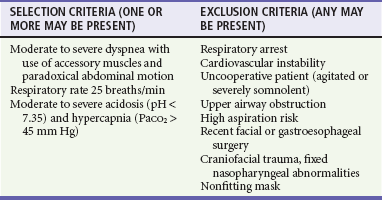
PaCO2, arterial partial pressure of carbon dioxide.
Adapted from Rabe K, Hurd S, Anzueto A, et al: Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532, 2007; and Soto F, Varkey B: Evidence-based approach to acute exacerbations of COPD. Curr Opin Pulm Med 9:117, 2003.
The most important factor in the decision to intubate is the patient’s clinical status, not ABG measurements. Even in the face of a significant rise in PCO2 with oxygen administration, intubation may be unnecessary if the patient’s clinical status has stabilized. Similarly, improving ABG values should not overrule the clinical impression of deterioration. Temporary improvement may be followed by exhaustion and respiratory failure. Box 74-2 outlines indications for invasive mechanical ventilation. Several of these criteria, adapted from the GOLD collaborators, are subject to interpretation, underscoring the critical role of clinical judgment in airway management decisions.
General Drug Therapy
Although bronchospasm is not the primary inciting event in acute COPD exacerbation, both beta-agonists and anticholinergic agents are considered first-line agents. The choice of agent for treating a given patient may depend on the respective side effect profiles of these two classes of drugs.62,63
Anticholinergic agents block muscarinic receptors and prevent smooth muscle contraction while decreasing the release of secretions from submucosal glands. Inhaled anticholinergic agents are as effective as beta2-agonists in COPD and can be used alone or in conjunction with beta2-agonists as first-line therapy in acute exacerbations.64 Although evidence regarding the efficacy of their coadministration is controversial, for moderate to severe exacerbations in the ED, these drugs should be given together for their possible synergistic effects.4 Anticholinergics can be administered by nebulization or metered dose inhaler (MDI) and are also effective for intubated patients. Ipratropium bromide, a quaternary ammonium compound, has been extensively studied in COPD. It has few systemic effects, yet has powerful bronchodilating properties. The nebulization dose is 0.5 mg, which may be repeated every half hour for a total of three doses and subsequently every 4 hours.
Long-acting bronchodilator agents, such as salmeterol (beta2-agonist) and tiotropium (anticholinergic), are added to treatment recommendations for chronic stable COPD.4 Although these agents may reduce the frequency of COPD exacerbations, no role for their use in ED management of acute exacerbation has been defined.
Methylxanthines, principally aminophylline, were once commonly used in COPD exacerbations. Evidence is not supportive of their use, however, because they do not improve outcomes, even when combined with beta-agonists and anticholinergic agents. Recommendations that include methylxanthines as a second-line therapy when other modalities have failed are tempered by concerns about toxicity, which may outweigh the positive effects of these agents.65 If a rare patient is encountered who is taking methylxanthines on a chronic, ambulatory basis, additional methylxanthines should not be used in the course of treatment in the ED.
Corticosteroids
The anti-inflammatory effects of steroids provide a strong rationale for their use in acutely ill patients with COPD. Although steroids may not alter the immediate ED course, evidence points to a modest decrease in the relapse rate of acute exacerbations and improvement of dyspnea.66–68 For severe exacerbations necessitating admission, intravenous methylprednisolone or oral prednisone (1-2 mg/kg) is given early in treatment and continued daily.69 Oral prednisone (40 mg once daily) is appropriate for discharged patients. Recommended doses of corticosteroids vary widely in the published studies. Consensus guidelines emphasize use of doses on the lower end of the accepted range to minimize common adverse effects, such as hyperglycemia, myopathy, and immunosuppression.4,70
Antibiotics
In contrast to asthma exacerbation and acute bronchitis in the setting of normal lung function, in which antibiotics are of no benefit, some COPD patients with an acute exacerbation appear to benefit from antibiotic therapy.71–73 The GOLD collaborators recommend administering antibiotics to patients with an increase in sputum purulence and either increased dyspnea or increased sputum volume, as well as to any patients requiring invasive or noninvasive ventilation (see later discussion of pneumonia).4 Furthermore, some patients may have clinical pneumonia without radiographic evidence, which may also warrant antibiotic therapy. Because the potential benefits of antibiotics outweigh their adverse effects in most patients with acute exacerbations, these agents should be considered in patients in the ED.
Most of the randomized, controlled trials suggesting a treatment benefit with antibiotics were conducted before the emergence of widespread resistance. Amoxicillin, tetracycline, and trimethoprim-sulfamethoxazole were the most common antibiotics used in these studies. Although antibiotics with broader-spectrum coverage, such as the fluoroquinolones and third-generation cephalosporins, are commonly prescribed, the evidence for the superiority of these newer agents is indirect. The GOLD collaborators recommend use of antibiotics that reflect local patterns of antibiotic sensitivity to S. pneumoniae, H. influenzae, and M. catarrhalis.4 Studies suggest that short (3- to 5-day) courses of antibiotics, such as respiratory fluoroquinolones or macrolides, may be as effective as more traditional, longer (7- to 14-day) courses of β-lactams and tetracyclines.74–76
Other Therapeutic Agents
Respiratory Stimulants
Several respiratory stimulants have been studied in patients with COPD, including opioid antagonists, progesterone, acetazolamide, doxapram, and almitrine. Doxapram appears to be the most effective of these agents. Although doxapram can effect small, temporary improvements in blood gas exchange in the first hours of treatment, it is less effective than other techniques, such as NIVS.77 Respiratory stimulants are therefore not recommended for routine use in the ED.
Heliox
Helium-oxygen mixtures decrease the work of breathing and improve airflow by virtue of their low density. Such mixtures, however, fail to demonstrate a benefit in either ventilated or nonventilated patients with COPD exacerbations.78,79
Other Anti-inflammatory Therapy
Preliminary evidence points to the possibility that other therapeutic strategies may reduce exacerbation frequency via immunomodulatory effects, such as long-term macrolide administration,80–82 or the use of roflumilast.83,84 As of yet, however, there does not appear to be any role for roflumilast in the management of acute exacerbations.
Disposition
Significant deterioration from baseline is the general guideline for admission of patients with COPD. Important factors in the decision include the presence of coexisting conditions, failed outpatient management for the current exacerbation, and lack of improvement while in the ED.85,86 The GOLD collaborators propose guidelines for admission, and these are adapted in Box 74-3. If the decision is made to discharge the patient, attention should also be directed to the patient’s vaccination status, proper technique of inhaler use, evaluation of outpatient support systems, appropriate referrals and, perhaps most important, smoking cessation.4,72




References
1. Eisner, MD, et al. An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:693–718.
2. Mathers, C, Boerma, T, Ma Fate, D. The Global Burden of Disease: 2004 Update. Geneva: World Health Organization; 2008.
3. Wilson, L, Devine, E, So, K. Direct medical costs of chronic obstructive pulmonary disease: Chronic bronchitis and emphysema. Respir Med. 2000;94:204–213.
4. Global Strategy for the Diagnosis, Management and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2010. www.goldcopd.org.
5. Seemungal, TA, Donaldson, GC, Bhowmik, A, Jeffries, DJ, Wedzicha, JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608–1613.
6. Centers for Disease Control and Prevention, National Center for Health Statistics. Health Data Interactive. www.cdc.gov/nchs/hdi.htm.
7. Hoogendoorn, M, Feenstra, TL, Hoogenveen, RT, Al, M, Mölken, MR. Association between lung function and exacerbation frequency in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2010;5:435–444.
8. Barnes, PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192.
9. Barnes, PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280.
10. Balmes, J, et al. American Thoracic Society Statement: Occupational contribution to the burden of airway disease. Am J Respir Crit Care Med. 2003;167:787–797.
11. Sezer, H, Akkurt, I, Guler, N, Marakoglu, K, Berk, S. A case-control study on the effect of exposure to different substances on the development of COPD. Ann Epidemiol. 2006;16:59–62.
12. Orozco-Levi, M, et al. Wood smoke exposure and risk of chronic obstructive pulmonary disease. Eur Respir J. 2006;27:542–546.
13. Andersen, ZJ, et al. Chronic obstructive pulmonary disease and long-term exposure to traffic-related air pollution: A cohort study. Am J Respir Crit Care Med. 2011;183:455–461.
14. Eisner, MD, et al. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ Health. 2005;4:7.
15. Hurst, JR, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363:1128–1138.
16. Celli, BR, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:1005–1012.
17. Kherad, O, et al. Upper-respiratory viral infection, biomarkers, and COPD exacerbations. Chest. 2010;138:896–904.
18. Seemungal, TA, Harper-Owen, R, Bhowmik, A, Jeffries, DJ, Wedzicha, JA. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur Respir J. 2000;16:677–683.
19. Greenberg, SB, Allen, M, Wilson, J, Atmar, RL. Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:167–173.
20. Steer, J, Gibson, GJ, Bourke, SC. Predicting outcomes following hospitalization for acute exacerbations of COPD. QJM. 2010;103:817–829.
21. Sethi, S, Evans, N, Grant, BJ, Murphy, TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471.
22. Sethi, S, Wrona, C, Grant, BJ, Murphy, TF. Strain-specific immune response to Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;169:448–453.
23. White, AJ, et al. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax. 2003;58:680–685.
24. Anderson, HR, et al. Air pollution and daily admissions for chronic obstructive pulmonary disease in 6 European cities: Results from the APHEA project. Eur Respir J. 1997;10:1064–1071.
25. Lujan, M, et al. Capnometry in spontaneously breathing patients: The influence of chronic obstructive pulmonary disease and expiration maneuvers. Med Sci Monit. 2008;14:CR485–CR492.
26. Washko, GR. Diagnostic imaging in COPD. Semin Respir Crit Care Med. 2010;31:276–285.
27. Takasugi, J, Godwin, J. Radiology of chronic obstructive pulmonary disease. Radiol Clin North Am. 1998;36:29–55.
28. Harrigan, R, Jones, K. ABC of clinical electrocardiography. Conditions affecting the right side of the heart. BMJ. 2002;324:1201–1204.
29. McCord, J, Borzak, S. Multifocal atrial tachycardia. Chest. 1998;113:203–209.
30. Abroug, F, et al. Association of left-heart dysfunction with severe exacerbation of chronic obstructive pulmonary disease: diagnostic performance of cardiac biomarkers. Am J Respir Crit Care Med. 2006;174:990–996.
31. Mueller, C, et al. Use of B-type natriuretic peptide in the management of acute dyspnea in patients with pulmonary disease. Am Heart J. 2006;151:471–477.
32. Steg, PG, et al. B-type natriuretic peptide and echocardiographic determination of ejection fraction in the diagnosis of congestive heart failure in patients with acute dyspnea. Chest. 2005;128:21–29.
33. McCullough, PA, et al. Uncovering heart failure in patients with a history of pulmonary disease: Rationale for the early use of B-type natriuretic peptide in the emergency department. Acad Emerg Med. 2003;10:198–204.
34. Hohl, C, Mitelman, B, Wyer, P, Lang, E. Should emergency physicians use B-type natriuretic peptide testing in patients with unexplained dyspnea? CJEM. 2003;5:162–165.
35. Hohl, C. Should natriuretic peptide testing be incorporated into emergency medicine practice? CJEM. 2006;8:259–261.
36. Collins, SP, Ronan-Bentle, S, Storrow, AB. Diagnostic and prognostic usefulness of natriuretic peptides in emergency department patients with dyspnea. Ann Emerg Med. 2003;41:532–545.
37. Volpicelli, G, Cardinale, L, Garofalo, G, Veltri, A. Usefulness of lung ultrasound in the bedside distinction between pulmonary edema and exacerbation of COPD. Emerg Radiol. 2008;15:145–151.
38. Lichtenstein, DA, Mezière, GA. Relevance of lung ultrasound in the diagnosis of acute respiratory failure: The BLUE protocol. Chest. 2008;134:117–125.
39. Slater, A, Goodwin, M, Anderson, K, Gleeson, F. COPD can mimic the appearance of pneumothorax on thoracic ultrasound. Chest. 2006;129:545–550.
40. Ambrosetti, M, Ageno, W, Spanevello, A, Salerno, M, Pedretti, RF. Prevalence and prevention of venous thromboembolism in patients with acute exacerbations of COPD. Thromb Res. 2003;112:203–207.
41. Erelel, M, Cuhadaroglu, C, Ece, T, Arseven, O. The frequency of deep venous thrombosis and pulmonary embolus in acute exacerbation of chronic obstructive pulmonary disease. Respir Med. 2002;96:515–518.
42. Gunen, H, Gulbas, G, In, E, Yetkin, O, Hacievliyagil, SS. Venous thromboemboli and exacerbations of COPD. Eur Respir J. 2010;35:1243–1248.
43. Frost, SD, Brotman, DJ, Michota, FA. Rational use of D-dimer measurement to exclude acute venous thromboembolic disease. Mayo Clin Proc. 2003;78:1385–1391.
44. van Belle, A, et al. Effectiveness of managing suspected pulmonary embolism using an algorithm combining clinical probability, D-dimer testing, and computed tomography. JAMA. 2006;295:172–179.
45. Kavanagh, EC, O’Hare, A, Hargaden, G, Murray, JG. Risk of pulmonary embolism after negative MDCT pulmonary angiography findings. AJR Am J Roentgenol. 2004;182:499–504.
46. Remy-Jardin, M, et al. CT angiography of pulmonary embolism in patients with underlying respiratory disease: Impact of multislice CT on image quality and negative predictive value. Eur Radiol. 2002;12:1971–1978.
47. Cham, MD, et al. Deep venous thrombosis: Detection by using indirect CT venography. The Pulmonary Angiography–Indirect CT Venography Cooperative Group. Radiology. 2000;216:744–751.
48. Ghaye, B, Szapiro, D, Willems, V, Dondelinger, RF. Combined CT venography of the lower limbs and spiral CT angiography of pulmonary arteries in acute pulmonary embolism: Preliminary results of a prospective study. JBR-BTR. 2000;83:271–278.
49. Stoller, JK, et al. Oxygen therapy for patients with COPD: Current evidence and the Long-term Oxygen Treatment Trial. Chest. 2010;138:179–187.
50. Anthonisen, NR, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA. 1994;272:1497–1505.
51. Celli, BR, et al. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: Results from the TORCH study. Am J Respir Crit Care Med. 2008;178:332–338.
52. Niewoehner, DE, et al. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator: A randomized trial. Ann Intern Med. 2005;143:317–326.
53. Schembri, S, Morant, S, Winter, JH, MacDonald, TM. Influenza but not pneumococcal vaccination protects against all-cause mortality in patients with COPD. Thorax. 2009;64:567–572.
54. O’Driscoll, BR, Howard, LS, Davison, AG, British Thoracic Society. BTS guideline for emergency oxygen use in adult patients. Thorax. 2008;63(Suppl 6):vi1–68.
55. Dick, C, Liu, Z, Sassoon, C, Berry, R, Mahutte, C. O2-induced change in ventilation and ventilatory drive in COPD. Am J Respir Crit Care Med. 1997;155:609–614.
56. Robinson, T, Freiberg, D, Regnis, J, Young, I. The role of hypoventilation and ventilation-perfusion redistribution in oxygen-induced hypercapnia during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1524–1529.
57. Austin, MA, Wills, KE, Blizzard, L, Walters, EH, Wood-Baker, R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: Randomised controlled trial. BMJ. 2010;341:c5462.
58. Berkius, J, Sundh, J, Nilholm, L, Fredrikson, M, Walther, SM. Long-term survival according to ventilation mode in acute respiratory failure secondary to chronic obstructive pulmonary disease: A multicenter, inception cohort study. J Crit Care. 2010;25:e13–18.
59. Ram, F, Picot, J, Lightowler, J, Wedzicha, J. Non-invasive positive pressure ventilation for treatment of respiratory failure due to exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (1):2004.
60. Crummy, F, Buchan, C, Miller, B, Toghill, J, Naughton, M. The use of noninvasive mechanical ventilation in COPD with severe hypercapnic acidosis. Respir Med. 2007;101:53–61.
61. Keenan, S, Sinuff, T, Cook, D, Hill, N. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive-pressure ventilation? A systematic review of the literature. Ann Intern Med. 2003;138:861–870.
62. McCrory, DC, Brown, CD. Inhaled short-acting beta2-agonists versus ipratropium for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (2):2001.
63. McCrory, DC, Brown, CD. Anti-cholinergic bronchodilators versus beta2-sympathomimetic agents for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (4):2002.
64. Campbell, S. For COPD a combination of ipratropium bromide and albuterol sulfate is more effective than albuterol base. Arch Intern Med. 1999;159:156–160.
65. Barr, R, Rowe, B, Camargo, C. Methylxanthines for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (2):2003.
66. Walters, JA, Gibson, PG, Wood-Baker, R, Hannay, M, Walters, EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (1):2009.
67. Niewoehner, D, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340:1941–1947.
68. Aaron, SD, et al. Outpatient oral prednisone after emergency treatment of chronic obstructive pulmonary disease. N Engl J Med. 2003;348:2618–2625.
69. Lindenauer, PK, et al. Association of corticosteroid dose and route of administration with risk of treatment failure in acute exacerbation of chronic obstructive pulmonary disease. JAMA. 2010;303:2359–2367.
70. Quon, BS, Gan, WQ, Sin, DD. Contemporary management of acute exacerbations of COPD: A systematic review and metaanalysis. Chest. 2008;133:756–766.
71. Daniels, JM, et al. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:150–157.
72. Celli, B, MacNee, W. Standards for the diagnosis and treatment of patients with COPD: A summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–946.
73. Rothberg, MB, et al. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA. 2010;303:2035–2042.
74. Anzueto, A, Miravitlles, M. Short-course fluoroquinolone therapy in exacerbations of chronic bronchitis and COPD. Respir Med. 2010;104:1396–1403.
75. El Moussaoui, R, et al. Short-course antibiotic treatment in acute exacerbations of chronic bronchitis and COPD: A meta-analysis of double-blind studies. Thorax. 2008;63:415–422.
76. Grossman, RF, Ambrusz, ME, Fisher, AC, Khashab, MM, Kahn, JB. Levofloxacin 750 mg QD for five days versus amoxicillin/clavulanate 875 mg/125 mg BID for ten days for treatment of acute bacterial exacerbation of chronic bronchitis: A post hoc analysis of data from severely ill patients. Clin Ther. 2006;28:1175–1180.
77. Greenstone, M, Lasserson, TJ. Doxapram for ventilatory failure due to exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (1):2003.
78. Maggiore, SM, et al. A multicenter, randomized trial of noninvasive ventilation with helium-oxygen mixture in exacerbations of chronic obstructive lung disease. Crit Care Med. 2010;38:145–151.
79. Rodrigo, G, Pollack, C, Rodrigo, C, Rowe, B. Heliox for treatment of exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (2):2002.
80. ZY, He, et al. Effect of 6 months of erythromycin treatment on inflammatory cells in induced sputum and exacerbations in chronic obstructive pulmonary disease. Respiration. 2010;80:445–452.
81. Seemungal, TA, et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. 2008;178:1139–1147.
82. Albert, RK, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689–698.
83. Rabe, KF. Roflumilast for the treatment of chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4:543–555.
84. Calverley, PM, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: Two randomised clinical trials. Lancet. 2009;374:685–694.
85. Roche, N, Rabbat, A, Zureik, M, Huchon, G. Chronic obstructive pulmonary disease exacerbations in emergency departments: Predictors of outcome. Curr Opin Pulm Med. 2010;16:112–117.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 74
Chronic Obstructive Pulmonary Disease
Perspective
Chronic obstructive pulmonary disease (COPD) is one of the most common causes of death worldwide. Although prevalence estimates vary by measurement methods and by population studied, there is general agreement that COPD is underdiagnosed and under-reported. Regardless of the success of smoking cessation programs, smoking behavior in the past several decades and the delay of the appearance of symptoms in an aging population virtually guarantee an increase in prevalence, especially in developed nations. In addition, it is becoming increasingly clear that other risk factors besides cigarette smoking contribute significantly to the worldwide burden of COPD.1
It is estimated that COPD will be the fifth leading cause of lost disability-adjusted life years worldwide by 2030.2 The financial burden of COPD is enormous, accounting for billions of dollars every year for treatment and lost productivity. The majority of these costs are related to hospitalization for acute exacerbations.3 Despite its enormous effects, COPD has received relatively less attention from basic medical researchers and clinicians than other diseases. Large multinational collaborations, such as the Global Initiative for Chronic Obstructive Lung Disease (GOLD), sponsored jointly by the National Heart, Lung, and Blood Institute and the World Health Organization, have reinvigorated the scientific and medical communities frustrated by the unrelenting progressive nature of COPD and its poor response to existing therapies.
The definition of COPD is imprecise and incorporates advances in our understanding of its underlying mechanisms and natural history. In their consensus statement, the GOLD collaborators define COPD as “a preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by airflow limitation that is not fully reversible.” They also state that “the airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases.”4 This definition reflects new data underscoring the systemic nature of the disease, as well as a deliberate optimism with respect to new prevention and treatment strategies. It specifically avoids mention of chronic bronchitis and emphysema, two entities that have been traditionally included in the definition of COPD. Chronic bronchitis, defined as the presence of cough and sputum production for at least 3 months in each of 2 consecutive years, can occur without airflow limitation. Emphysema, the destruction of alveoli, is a pathologic term, not one that pertains to clinical diagnosis. Unlike with many earlier definitions of COPD, the GOLD collaborators also specifically exclude asthma, which is reversible airflow limitation. Whether reversible airflow limitation is considered to be part of COPD itself or caused by coexisting asthma is of limited significance to the emergency physician, who will continue to make every attempt to identify and reverse airflow limitation.
As many as 50% of all acute COPD exacerbations are not reported to physicians. In addition, not all reported exacerbations necessitate hospitalization.5 Nonetheless, in 2007 almost 2% of all hospital admissions in the United States were directly attributed to COPD, and it was considered a contributory factor in another 9%. In patients older than 65 years, the percentage of all hospitalizations related to COPD approaches 20%.6 As the severity of the underlying disease progresses, so does the frequency of exacerbations.7 Moreover, in a subset of patients, incomplete recovery from acute exacerbations may reflect a contribution of exacerbations to the pathophysiology of relentless disease progression.
Principles of Disease
In the past several decades the discovery that chronic airway inflammation plays a central role in the pathophysiology of asthma has led to an important change in its management, specifically, the liberal use of corticosteroids for treating moderate to severe disease. Airway inflammation is also at the center of the pathophysiology of COPD, but the inflammatory process of COPD differs from that of asthma. In COPD, neutrophils, CD8+ lymphocytes, and macrophages predominate in bronchial washings, whereas in asthma the cellular response is characterized by the presence of eosinophils.8 The inflammatory mediators differ in COPD, and several mediators, such as tumor necrosis factor, leukotriene B4, and interleukin-8, are linked to the destruction of parenchyma. These differences in the nature of the inflammatory response in COPD may account for its relatively poor response to current anti-inflammatory treatment compared with asthma.
Cigarette smoking, the most significant risk factor for the development of COPD, exerts its effects at multiple points in the inflammatory cascade of COPD, negatively affecting both the protease to antiprotease and oxidant to antioxidant balances. Although smoking cessation slows the progression of the disease, it does not end the chronic inflammatory process within the airways, indicating that mechanisms independent of smoking are involved.9 Moreover, although a majority of COPD patients have a significant smoking history, only a minority of smokers ever develop airflow limitation. This suggests the importance of other factors, both environmental and genetic.1 Other identified causative factors include heavy occupational exposure to dusts and air pollution from indoor cooking, particularly in the developing world.10–12 Long-term passive exposure to tobacco smoke and urban air pollution also appear to be contributory.13,14 Although there is an association between early childhood lower respiratory tract infections and later development of COPD, a causal relationship is less certain.
Staging the Severity of Disease
Most classifications of disease severity are based on quantitative measurements of airflow limitation, such as the forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity (FVC) ratio. These indices are measured after any reversible airflow limitation is addressed by treatment with bronchodilator medications. The GOLD collaborators define four stages, beginning with a mild stage (stage I), when spirometry is abnormal but symptoms may not yet be apparent, and ending in very severe COPD (stage IV), when FEV1 is less than 30% of predicted (Table 74-1). Frequent exacerbations are usually seen when the FEV1 falls below 50% predicted (stages III and IV).4,15,16
Table 74-1
The GOLD Classification of Severity of Chronic Obstructive Pulmonary Disease (COPD)
Adapted from Rabe K, Hurd S, Anzueto A, et al: Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 176:532, 2007.
Acute Exacerbations
As with asthma, viral infection appears to be a frequent inciting agent in COPD exacerbations. Commonly implicated viruses include rhinovirus, respiratory syncytial virus, coronavirus, and influenza virus.17–19 Exacerbations associated with a viral cause are longer and more severe than those without an apparent inciting agent.18,20
Controversy remains regarding the role of bacterial pathogens in acute exacerbations of COPD, because the evidence for the role of bacteria in the pathogenesis remains indirect. Almost half of all exacerbations are associated with negative cultures for the typical respiratory pathogens, such as Haemophilus influenzae, Streptococcus pneumoniae, Moraxella (Branhamella) catarrhalis, and Pseudomonas aeruginosa. In addition, these organisms are recovered from the tracheobronchial tree of patients in their chronic, steady state, suggesting that bacteria may play a more important role in the pathogenesis of chronic COPD than in acute exacerbations.21 Although molecular typing shows that recent colonization with new serotypes of the common pathogens is associated with an exacerbation, there is a possibility that this relationship is not causal. Experimental evidence of specific immunologic responses to bacteria in exacerbations suggests, however, that they do play a significant role.22,23
Environmental factors, such as air pollution, are also implicated in COPD exacerbations. Indirect evidence for this relationship is largely derived from hospitalization rates for exacerbations during periods of increased air pollution.24 Finally, in as many as one third of all COPD exacerbations, no specific cause can be identified.
Clinical Features
COPD patients have a long premorbid course during which decreases in air flow indices can be measured in the absence of symptoms. Intermittent cough or shortness of breath on exertion may be easily misattributed to poor physical conditioning. Moreover, patients may remain asymptomatic for many years by gradually limiting their activities in proportion to their pulmonary reserve. After several years, a daily productive cough frequently develops, and periods of dyspnea, the cardinal symptom of airflow limitation, increase. The clinical progression of COPD is slow and insidious, with gradual decreases in airflow punctuated by increasingly frequent and debilitating exacerbations. Eventually the patient becomes truly incapacitated by dyspnea on minimal or no exertion. Profound muscle wasting and weight loss and the emergence of cor pulmonale or chronic ventilatory failure are characteristic of end-stage disease. Figure 74-1 depicts the progression of COPD over time.
Diagnostic Strategies
Pulse Oximetry, Arterial Blood Gas Analysis, and Waveform Capnography
ABG values, once a mainstay of ED evaluation of COPD patients, are of limited value in management of acute COPD episodes. Status and response to therapy often are monitored noninvasively by capnography and pulse oximetry. The presence of respiratory failure unresponsive to therapy (defined as PaO2 <40 mm Hg, PaCO2 >60 mm Hg, and pH <7.25 mm Hg) warrants consideration of admission to an intensive care unit, but clinical evaluation is much more important than any particular blood gas values.4 When baseline blood gas levels are not available, the usefulness of ABGs is even more limited, and interpretation should be based on the degree of acidemia present, which likely represents the extent of acute CO2 retention. ABGs should not be used to determine whether a patient requires intubation or noninvasive ventilatory support (NIVS). These decisions should rather be guided by the overall state of the patient, progression of fatigue, comorbid illness, and response to therapy. Patients with very poor blood gas values may do well without intubation or NIVS, but others with mildly disturbed values may require urgent airway intervention. Thus measurement of ABGs should not be performed routinely in the ED and should be undertaken only in response to specific circumstances, such as irregular or apparently unreliable pulse oximetry values or when a single baseline correlation with end-tidal carbon dioxide (ETCO2) is desired.
Waveform capnography represents the continuous quantitative measurement of exhaled CO2. It has emerged as a potential diagnostic and monitoring tool in patients with acute respiratory distress. The appearance of the waveform may assist the clinician in differentiating acute exacerbation of COPD from other causes of acute dyspnea, such as CHF. In patients with obstructed airways, the plateau phase of the waveform typically steepens in proportion to the severity of the obstruction, as depicted in Figure 74-2. Unfortunately, in patients with COPD, the actual end-tidal PCO2 measurements obtained from capnography do not correlate well with arterial PCO2 measurements, especially in more severe disease. Nonetheless, the patterns and trends from capnography may be helpful in guiding management and assessing response to therapy.25
Chest Radiography
In patients who are known to have COPD, the primary role of the chest radiograph is to determine whether there is an acute, treatable cause for clinical deterioration, especially pneumothorax or parenchymal consolidation (atelectasis secondary to mucous plugging, pneumonia, or obstruction by tumor). Otherwise, the chest radiograph is of limited use and may exhibit a range of chronic changes, depending on disease severity and the relative degree of the various pathologic processes. Findings may include hyperinflated lung fields, decreased vascular markings, and a small cardiac silhouette or, in contrast, normal inflation with increased vascular markings and an enlarged heart.26,27 In cor pulmonale, impingement on the retrosternal airspace by the enlarged right ventricle can be seen on the lateral film.
Electrocardiogram and Cardiac Monitoring
The classic descriptions of P pulmonale (peaked P waves in leads II, III, and aVF), low QRS voltage, clockwise rotation, and poor R wave progression in the precordial leads are interesting correlates of COPD but are both insensitive and nonspecific. The presence of electrocardiogram (ECG) criteria for RVH suggests established cor pulmonale. These findings, however, can be easily obscured on the ECG by other processes, and the absence of criteria for RVH cannot be relied on to rule out cor pulmonale.28
[/not-level-membership-for-emergency-medicine-category]
