[level-membership-for-critical-care-medicine-category]39
Chronic Obstructive Pulmonary Disease

In 1997, the U.S. National Heart Lung and Blood Institute and the World Health Organization held an international workshop that led to the first Global Initiative on Obstructive Lung Disease (GOLD) report. The most recent iteration of the GOLD guidelines defines chronic obstructive pulmonary disease (COPD) as a preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by chronic airflow limitation that is not fully reversible. The airflow limitation is usually both progressive and associated with abnormal inflammatory response of the lungs to noxious particles and gases.1
Definitions
Although controversies remain over the definition of exacerbation, how they should be monitored, and their underlying mechanisms, acute exacerbations of COPD (AECOPD) are major and increasingly recognized events in the disease course. They typically occur one to three times per year. Exacerbations are associated with an increase in economic burden2 and a decline in health-related quality of life.3 Those patients with more than two exacerbations per year have significantly worse health-related complications and decline in lung function than those with two or fewer exacerbations.4 As mentioned before, there is no general agreement on the definition of AECOPD, but it has been defined according to the presence of specific signs and symptoms, worsening in symptoms, and the need for medical intervention, and each of these approaches has positive and negative connotations. It is well recognized, however, that the presence of increased shortness of breath, increased sputum volume, and increased purulence are the three specific symptoms that represent an exacerbation.5
Background
COPD affects more than 200 million people worldwide and is the fourth leading cause of death. COPD is a disease that is both preventable and treatable. COPD is a major cause of morbidity and death in the world, with an increasing burden due to epidemiologic changes that expose more of the population to COPD risk factors. Although the major environmental risk for COPD is tobacco smoking, only 20% of smokers develop COPD. Indoor air pollution from burning biomass fuel is associated with increased risk of COPD in developing countries. In the United States, COPD is the fourth leading cause of death6 and is exceeded only by heart attacks, cancer, and stroke.
COPD has had a similar effect on health and mortality rate throughout the developed and underdeveloped sectors of the world, and many of the important issues surrounding COPD in the United States apply elsewhere.7
The real prevalence is masked by the burden of undiagnosed COPD; when the British Lung Foundation “missing millions” campaign performed spirometry screening in 3802 adults, they found that the prevalence of COPD was 10.2% (4.4% having GOLD II or worse) with only a quarter having a prior diagnosis.8
Pathophysiology
The main pathophysiologic feature in COPD is the limitation to expiratory flow.9 Chronic expiratory flow limitation and hyperinflation are the mechanical hallmarks of COPD.10 Expiratory airflow limitation results from many factors; among them, narrowing of the peripheral airways,11 mucus hypersecretion,12 and impaired ciliary clearance13 are the most important factors.
Several mechanistic concepts have been implicated in the pathogenesis of COPD. First, the hallmark of COPD is development of exaggerated chronic inflammation in the lung in response to inhalation of cigarette smoke compared with smokers without lung disease.14 Host factors including genetic susceptibility, epigenetic changes, and oxidative stress contribute by amplifying inflammation induced by cigarette smoke. Second, patients with deficiency of α1-antitrypsin, the main inhibitor of neutrophil elastase, develop emphysema early in life owing to an increase in proteolytic activity.15 Third, an imbalance between oxidants and antioxidants in the lungs of patients with COPD, resulting in excessive oxidative stress, not only amplifies airway inflammation in smokers but also induces cell death of structural cells in the lung. Disruption of the balance between cell death and replenishment of structural cells in the lung contributes to the destruction of alveolar septa, leading to emphysema.16 Autoimmunity has been proposed as a late pathogenic event in the progressive course of the disease.
COPD is characterized by a specific pattern of inflammation involving increased numbers of CD8+ TC lymphocytes present only in smokers who develop the disease. These cells as well as neutrophils and macrophages release inflammatory mediators and interact with epithelial cells in the airways, lung parenchyma, and pulmonary endothelium; all these relationships and interactions produce structural changes in the lungs by activation of growth factors.17 Changes in the airway architecture, endothelium, and lung parenchyma lead to the characteristic physiologic abnormalities and symptoms of COPD.
In acute exacerbations there is an increase in sputum neutrophil numbers as well as an increase in neutrophils in bronchial biopsies, which rarely are seen in the stable state.18 Interestingly virally induced exacerbations are associated with increased expression of eosinophils in sputum. Viral infections induce the expression of chemokine (C-C motif) ligand 5 (CCL5) in airway epithelial cells,19 CCL5 may act synergistically with CD8+ cells to enhance the apoptosis of virally infected cells, thus leading to increased tissue destruction.20 Also, an increase in the concentration of the elastolytic enzyme matrix metalloproteinase-1 during exacerbations is consistent with evidence of elastolysis, which may provide a causal link between exacerbations and accelerated decline in lung function.21
Data from patients with AECOPD that required mechanical ventilation indicates the presence of increased central drive, dyspnea, tachypnea, reduced tidal volume, and development of hypercapnic respiratory failure, but ventilation/perfusion match remains relatively preserved.22 AECOPD appears to be characterized by increased central drive, decreased inspiratory capacity, and decreased inspiratory muscle force, perhaps secondary to dynamic hyperinflation. There is an association between increased serum levels of interleukin 6 (IL-6) and leukotriene B4 (LTB4) and the magnitude of dyspnea, respiratory rate, and inspiratory capacity, suggesting that it may be possible to detect serum changes that reflect the inflammatory burden of the exacerbation.23
Although hypercapnia depends on the severity of airflow limitation, there is considerable variability in the relationship of PaCO2 to forced expiratory volume in 1 second (FEV1) and total lung resistance, best explained by contribution of dead space and minute ventilation. In stable COPD patients with severe airflow obstruction, shallow breathing is the main factor associated with CO2 retention,24 the diaphragm is less effective than in normal subjects, and with increasing airflow obstruction and hyperinflation, the contribution of the rib cage muscle to the generation of ventilatory pressure increases.25 Abdominal muscles are recruited during expiration in patients with severe COPD, and the expiratory rise in gastric pressure is directly related to intrinsic positive end-expiratory pressure (PEEPi). During acute exacerbation, patients with severe airflow obstruction increase the inspiratory recruitment of the rib cage muscles relative to the diaphragm. This recruitment is associated with abdominal muscle contraction and a reduction in abdominal volume at end expiration, which contributes to PEEPi. Dynamic hyperinflation can be overestimated during chronic and acute airway obstruction if abdominal muscle function is not evaluated.26
The worsening gas exchange and the deterioration of the arterial blood gas values during acute exacerbations in patients with severe COPD can be explained by several factors. These factors are, in no particular order, respiratory muscle fatigue,27 increases in dead space ventilation, and alveolar hypoventilation.28 Minute ventilation may be normal early in an exacerbation, but the respiratory rate is generally increased. There is an associated increase in physiologic dead space that impairs CO2 elimination and may result in acidemia.29 The hypoxemia seen during exacerbations results from the combination of two factors: alveolar hypoventilation and, later, worsening of ventilation/perfusion matching. Increases in ventilation/perfusion heterogeneity are attributed to (1) a reduction in the effectiveness of hypoxic vasoconstriction as a protective mechanism as pulmonary artery pressure rises and vasodilatory inflammatory mediators are released, and (2) the failure to redirect perfusion away from inadequately ventilated regions because of the reduction in cross-sectional area of the pulmonary vascular bed.
Clinical Manifestations
Most COPD patients with acute exacerbations initially demonstrate some combination of increasing cough, worsening of dyspnea, increased sputum production, purulent sputum, or increase in viscosity of the sputum, rather than a deterioration noted by laboratory or respiratory function parameters (Box 39.1). Symptoms may come on slowly over several days or acutely, depending somewhat on the severity of the underlying disease. Often, patients have a history of upper respiratory tract infection. Patients generally appear in acute distress. Vital signs typically demonstrate tachycardia and tachypnea, and blood pressure can be reduced in response to the effect of PEEPi. Use of accessory inspiratory muscles may be seen with increasing severity of exacerbations. With inspiration, the diaphragm normally moves down as it contracts, forcing the abdominal contents out. With diaphragmatic fatigue, the diaphragm no longer functions as a primary muscle of inspiration but instead assists the intercostal muscles’ inspiratory effort by fixing the rib cage. This action is associated with a rise in the diaphragm, and the abdomen moves in instead of out, as it does with normal inspiration. This sign is called paradoxical breathing28 and it implies respiratory muscle fatigue and often imminent ventilatory failure and respiratory arrest.29 Wheezing and other auscultatory findings of obstruction are also present. Cyanosis is an insensitive manifestation, but when seen, it denotes severe hypoxemia. Patients with severe acute CO2 retention may present in coma.
Precipitating Factors
The most common precipitant factor is respiratory infection, either bacterial or viral.30,31 Air pollution can also precipitate exacerbations of COPD32,33; however, the cause of one third of AECOPD events cannot be identified. Some other conditions that can ether mimic or induce an exacerbation include pneumonia, arrhythmia, pneumothorax, pulmonary embolism,34 and pleural effusion. Medication failure and lack of compliance have been shown also to lead to exacerbations.
Infections
Bacteria and viruses account for the vast majority of episodes of exacerbation. Respiratory viruses are associated with 30% of exacerbations with or without a superimposed bacterial infection.35 Another study reported that up to 78% of the patients admitted into the hospital with severe AECOPD had evidence of viral or bacterial infection.19
Several studies have been conducted to investigate airway bacterial infections as etiologic factors involved in COPD exacerbations.36–38 Bronchoscopic sampling of the distal airway has demonstrated the presence of pathogenic bacteria in 50% of exacerbations. Acquisition of new strains of bacterial pathogens has been associated with more than twofold increase in the risk of AECOPD.39 At present, there appears to be an agreement that the major pathogens isolated from sputum during acute exacerbation are Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis40; however, all of these bacteria can be isolated in patients during the stable phases of COPD.41,42 Atypical bacteria, mostly Chlamydia pneumoniae, have been implicated in approximately 10% of acute exacerbations.43,44 Other potential microorganisms that should be considered include other Streptococcus species, enteric gram-negative bacilli, and Legionella.45 Bacterial colonization may be a factor increasing airway inflammation.46 There is an association between bacterial colonization and increased markers of inflammation in sputum and in the frequency of exacerbations.47
Approximately 50% of AECOPD events are associated with upper respiratory tract virus infections. Infections with rhinovirus, respiratory syncytial virus, and influenza virus have been associated with AECOPD.48 COPD patients with a history of frequent exacerbations may be more susceptible to respiratory viral infections. There is increasing recognition that many patients with exacerbations have concomitant viral and bacterial infection. Approximately a quarter of patients admitted to hospital with AECOPD have coinfection with viruses and bacteria, and those patients have more severe exacerbations.48
Environmental Factors
Environmental factors are among noninfectious causes of COPD exacerbation that should be investigated as precipitating causes.32,49,50 Epidemiologic studies have shown that hospital admissions with AECOPD increase slightly with a rise in atmospheric levels of sulfur dioxide, ozone, nitrogen dioxide, and particulates. There is convincing evidence that exposure to particulates with a 50% cutoff aerodynamic diameter of 10 µm is associated with increased hospital admissions for AECOPD.51 Air pollution is implicated as a trigger of exacerbations52; however, a direct cause-and-effect relationship has been difficult to establish over the last 50 years.53 From an epidemiologic viewpoint, definitive evidence exists regarding a role of air pollutants in the increased death rates seen in cities during periods of heavy pollution.54 The recent dramatic increase in motor vehicle traffic has produced a relative increase in the levels of newer pollutants, such as ozone and fine-particulate air pollution. Elucidation of the mechanisms of the harmful effects of these pollutants should allow improved risk assessment for the patients with airway diseases who are susceptible to the effects of these air pollutants.
Pulmonary Thromboembolism
Pulmonary thromboembolism (PTE) can precipitate acute COPD exacerbations through either impairment of gas exchange or increases in pulmonary vascular pressures.55,56 Some evidence suggests that deep venous thrombosis occurs in more than 5 million people each year. More than 500,000 people eventually develop PTE, which is the primary cause of death in more than 100,000 patients annually in the United States.57 The precise incidence of PTE in COPD is unknown. Studies in COPD patients have found pulmonary embolus in up to 50% of autopsies and in patients admitted to hospital with severe AECOPD of unknown cause; 25% had pulmonary embolism confirmed by spiral computed tomography (CT).58 PTE risk factors inherent to COPD are sedentary lifestyle, right ventricular failure, right ventricular mural thrombi, and secondary polycythemia.59 Patients with COPD have also been shown to have increased platelet aggregation and increased plasma β-thromboglobulin.60
Up to 30% of untreated thromboembolic patients die. The necessity of diagnosing PTE as the precipitating factor in acute COPD exacerbation is crucial. However, the diagnosis of PTE is extremely difficult in COPD exacerbation. Nonetheless, the approach to diagnosis is similar to that used with other patients.61 Unfortunately, most patients with COPD have an indeterminate ventilation/perfusion scan, usually making this scan unhelpful in evaluation for PTE.62 For patients with such indeterminate results (low or intermediate probability), noninvasive testing of the lower extremity should be conducted.63–65 If positive results for deep venous thrombosis are obtained, anticoagulation therapy must be initiated. It has been proposed that use of newer D-dimer assays may also have a role as a diagnostic tool; however, even with a sensitivity of 98%, specificity is problematic, with a value of 39%.66 When the diagnosis is still in doubt (intermediate-probability scan and negative leg study or low-probability scan and negative leg study with intermediate clinical probability of PTE), helical CT or conventional pulmonary angiography may be required. Even though the safety of pulmonary angiography in patients with cor pulmonale has been questioned, data from the Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED) study support both safety and accuracy in patients with COPD.61 Helical CT has become an increasingly accepted technique and is the method of choice for direct visualization of pulmonary emboli (PE). The quantitative assessment of tissue perfusion may yield more important information for patient management than the direct visualization of emboli by CT alone. Enhanced multislice helical CT with thin collimation can be used to analyze precisely the subsegmental pulmonary arteries and may identify even more distal pulmonary arteries. Recent data suggest that spiral CT may be an alternative to angiography, particularly when results are not discordant with pretest clinical probability of PE and when combined with other tests that support CT findings (D-dimer, leg ultrasound, lung scanning), and may have adequate sensitivity and specificity in the COPD population.62
Medication Failure or Noncompliance
Many acute COPD exacerbations can be explained in terms of inadequate pharmacologic therapy or noncompliance with pharmacologic therapy. These factors are likely underestimated causes of exacerbation in COPD patients. It is also possible that patients have toxic drug effects of cardiac, gastrointestinal, or metabolic nature. Certain pharmacologic drug interactions can precipitate toxicity or loss of effect of one drug, and the physician must ascertain whether newer medications for other conditions have recently been added to the patient’s therapeutic regimen.67,68
Other Causes
Clinical decompensation in patients with stable COPD may also occur as a result of acute congestive heart failure (CHF) or cardiac arrhythmia. One study found that 27% of COPD patients die as a result of coronary disease,69 likely related to risk factors such as smoking, diabetes mellitus, and vasculopathy. Heart failure may also lead to a symptomatic exacerbation of COPD; however, it may be difficult to differentiate the symptoms of CHF and those of AECOPD.70
Other causes of exacerbation include sleep-disordered breathing,71–73 vocal cord paralysis, tumor or scarring from prior intubations, and the development of spontaneous pneumothorax.74,75 Finally, pleural effusion can also produce respiratory deterioration, especially in patients with poor respiratory reserve.76
Initial Management
Because chronic airflow obstruction cannot be reversed, acute management of COPD is directed at reversible pathogenic mechanisms, including pulmonary infection, airway tissue inflammation, bronchoconstriction, and support of failing muscular function. Box 39.2 summarizes initial general management.
Oxygen
Long-term oxygen treatment has been demonstrated to significantly reduce mortality rate in patients with COPD and severe resting arterial hypoxemia.77 Oxygen therapy is indicated for patients with an arterial oxygen tension (PaO2) of less than 55 mm Hg. The therapeutic goal is to maintain oxygen saturation greater than 90% during rest, sleep, and exertion, as it is now well established that such measures increase survival and that 24 hours is more effective than 12 hours. There is a clear rationale for the use of oxygen in severe COPD. Enhancing blood oxygenation by increasing the concentration of inspired oxygen compensates for a major physiologic consequence of COPD with hypoxemia. Oxygen remains the mainstay of initial therapy in most COPD exacerbations. Relief of hypoxemia, and consequently of hypoxemic pulmonary vasoconstriction, decreases pulmonary vascular resistance, with variable effects on the ventilation/perfusion ratio.78,79 Oxygen delivery may increase as a result of increases in oxygen arterial content and anticipated improved right-sided heart function. However, one study demonstrated that relief of hypoxemia did not increase cardiac output.80
Hypercapnia is well tolerated when it is chronic.81–83 However, oxygen should be administered cautiously in patients who are chronically hypercapnic because it is known to lead to clinically significant rises in PaCO2 in select COPD patients as a result of changes in the physiologic dead space and perhaps suppression of the respiratory drive.84,85 Acute increases in PaCO2 are more likely to occur in patients with elevated baseline PaCO2. A randomized study has shown that, although oxygen administration worsened hypercapnia and respiratory acidosis, these changes were well tolerated in most patients.86 Oxygen therapy should not be withheld in acutely ill hypoxemic patients because tissue hypoxia can lead to acute organ dysfunction. However, oxygen should be initiated at a low FIO2 and slowly titrated up as necessary with vigilant monitoring to document improvement and stabilization in PaO2, with special attention paid to maintaining the oxyhemoglobin between 88% and 92% or greater without producing dangerous falls in pH as a result of rises in CO2. These dangerous rises in PaCO2 are typically associated with worsening mental status. Nasal cannulas or Venturi masks can be used to initiate a low FIO2. Either nasal cannula at a flow rate of 1 L/minute or a Venturi mask initially at the lowest setting (25%) is appropriate for initiating oxygen in patients known or suspected to be chronic CO2 retainers. However, in the presence of acute severe hypoxemia in patients with impending respiratory failure, high-flow oxygen therapy may be in the patient’s best interest, regardless of the risk for CO2 retention.87
Drug Treatment
Bronchodilators
Bronchodilator therapy has important roles in both the prevention and treatment of AECOPD. Bronchodilators are the primary treatment to alleviate patient symptoms, improve physiologic state, and prevent or reverse respiratory failure; however, its use has not been shown to improve survival. Systematic reviews have demonstrated that inhaled delivery of short-acting β2-selective agonists and anticholinergic agents have greater effect on spirometry and is the therapy of choice for AECOPD over parenteral bronchodilators.88 Bronchodilator treatment in acutely ill COPD patients has been shown to decrease inspiratory muscle loading, with an increase in FEV1 and a decrease in functional residual capacity (FRC) and dynamic hyperinflation.89 In mechanically ventilated patients, a reduction in expiratory resistance and dynamic hyperinflation (measured as a decrease in PEEPi) has been described.90
There is no evidence supporting the use of one inhaled β2-selective agonist over another. There is no difference in outcome between β2-agonist compared with ipratropium bromide and no evidence that the combination of these two drugs is any more effective in AECOPD; these results are in contrast with the greater efficacy of these combinations in stable COPD.88 The widespread use of inhaled β-agonists has been accompanied by clinical concern of cardiac complications in elderly patients and those with coronary artery disease. However, in a study performed on clinically stable COPD or asthma patients with a history of myocardial ischemia, no ischemic events, or dysrhythmias, were observed when commonly used doses of salbutamol were administered.91 To date, no clinical studies have evaluated the use of inhaled long-acting bronchodilators (either β2-agonist or anticholinergic agents) with or without inhaled corticosteroids during AECOPD.
Several studies have suggested that the combination of β2-agonists and anticholinergic agents prevent exacerbations, particularly long-acting agents.92–96 It is possible that these agents reduce exacerbation frequency as a result of the effect on reduction of dynamic hyperinflation at rest and exercise,97 as well as nonbronchodilator mechanisms, such as anti-inflammatory effect.98
Corticosteroids
COPD is recognized as an inflammatory disorder, and the severity of airway inflammation correlates with the severity of the underlying COPD.99 During exacerbations, there is a large increase in concentration of proinflammatory cells, including neutrophils. Systemic corticosteroids improve lung function significantly, shorten hospital stay, and reduce the risk for relapses as compared with placebo in patients with AECOPD.100 In the ISOLDE trial, the median exacerbation rate was reduced by 25%, and there was also a reduction in the health status deterioration.101 Systemic corticosteroids have been demonstrated to improve respiratory mechanics in mechanically ventilated patients, with a decrease in airway resistance and dynamic air-trapping.102 A meta-analysis demonstrated that the use of systemic corticosteroids was associated with significant reduction in treatment failure, defined as either clinical deterioration, withdrawal from the study due to unsatisfactory clinical improvement, or relapse of exacerbation symptoms during the follow-up period. It also showed beneficial effects in reducing the length of hospitalization by a weighted mean of 1.42 days.103
Oral corticosteroids have beneficial effects in the management of AECOPD. Prednisolone, administered at 30 mg/day for 14 days, shortened the length of hospitalization by 2 days, improved FEV1 by 60 mL/day, and accelerated recovery from symptoms.104 The majority of patients with COPD probably requires only 2 weeks with oral corticosteroids and therapy for 8 weeks produced no incremental benefits above those achieved at 2 weeks.105 One of the most important concerns regarding the use of corticosteroids in AECOPD is the possibility of confusing it with community-acquired pneumonia. Nevertheless, there is no evidence that corticosteroid use worsened the prognosis of community-acquired pneumonia if appropriate antibiotics are used; moreover, systemic corticosteroids may reduce morbidity and mortality rates in community-acquired pneumonia.106
The use of inhaled corticosteroids is associated with decrease in exacerbation events by 12% to 25%.107 Studies involving inhaled corticosteroids and long-acting β2-agonist showed reduction in exacerbation frequency to a greater extent than using either corticosteroid or long-acting β2-agonist alone.108,109 There is also a trend toward mortality rate reduction over 3 years of 17.5%, although without statistical significance.110 There is no evidence that the addition of inhaled corticosteroids to systemic corticosteroids in AECOPD has any impact either in recovery or in mortality rate.
Antibiotics
Use of antibiotics in AECOPD remains a controversial topic, with some authors recommending antibiotic therapy and others not, maybe in part because of the heterogeneity of the population studied.111,112 Compared with placebo, antibiotic use during AECOPD reduced treatment failures by 46%, defined as requiring additional antibiotics within the first 7 days or unchanged or deteriorated symptoms within 21 days. Antibiotics reduced treatment failures particularly in those patients who were hospitalized but not when they were used in ambulatory patients.113 Three clinical trials involving 181 patients demonstrated that in-hospital mortality rate can be reduced by 78% with the use of antibiotics during AECOPD.114,115 Patients who present with dyspnea and increased sputum volume or purulence or patients who require mechanical ventilation benefit from a 3- to 7-day course of oral or parenteral antibiotics.116
Although routine sputum cultures are recommended in all patients with AECOPD, invasive techniques (transtracheal aspirates,117,118 bronchoscopic aspirates, or protected specimen brushing5,119–123) are not indicated. Exceptions include culture-negative community-acquired pneumonia not responding to therapy and ventilator-associated pneumonia (VAP).124,125 Because many COPD patients have airway colonization by bacteria, without clinical signs of infection and exacerbation, there is no clear significance of a positive culture in a COPD patient; however, in the presence of exacerbation associated with an alteration in sputum character or quantity, potentially pathogenic organisms grown from sputum should be covered with an appropriate antibiotic.126 Thus, the selection of empiric antibiotics to treat AECOPD should depend on the severity of underlying disease and severity of COPD exacerbation. Therapy for more severe exacerbations should include coverage for antibiotic-resistant bacteria, such as Pseudomonas or methicillin-resistant Staphylococcus aureus.
Hemodynamic Support
Fluid Management
COPD patients often have chronic pulmonary hypertension, which may worsen with COPD exacerbation because of hypoxic vasoconstriction, dynamic lung hyperinflation, and in mechanically ventilated patients, PEEPi. This may lead to acute or worsening right ventricular failure. In one study, the prevalence of right ventricular failure in terminal COPD patients was 66% and the prevalence of left ventricular failure was only 6%.127 As in other patients with right ventricular failure, hemodynamic stability is related to maintenance of mean arterial pressure. Mean systemic pressure can potentially be increased in these patients by increasing intravascular volume or by selectively improving compliance of the pulmonary vascular bed.128 Intravenous fluid challenge is the initial step in hemodynamic support in the presence of hemodynamic compromise. Despite the presence of peripheral edema, diuretics should be avoided, given the potential to induce loss of intravascular volume, decreased venous return, decreased cardiac output, and hypotension.
Inotropes and Vasodilators
In patients who remain hemodynamically unstable despite fluid therapy, adrenergic therapy is advised. Although some literature recommends adrenergic therapy for right ventricular failure using norepinephrine129,130 or dobutamine,131 we could find no controlled trials of adrenergic drug therapy in hemodynamically unstable COPD patients. However, pulmonary embolism patients have a hemodynamic derangement similar to that of COPD patients with right ventricular failure, and as such, dobutamine seems a reasonable choice in attempts to increase right ventricular function and increase cardiac output in the normotensive patient with decreased tissue perfusion. In the presence of hypotension, dobutamine would be used in combination with a combined inotrope vasopressor such as norepinephrine or dopamine. Nitroglycerine has been shown in one study to enhance right ventricular performance when added to dobutamine.132 Nitroglycerine should be administered cautiously in patients who may have suboptimal right ventricle filling. Digoxin has no anticipated clinical utility in right ventricular failure, unless it is associated with left ventricular failure or arrhythmias that respond to digoxin.133–135
Although vasodilators have been used in clinical trials of COPD with right ventricular failure,136–143 there is no consistent evidence of clinical outcome benefit. Inhaled nitric oxide, a selective pulmonary vasodilator with a short half-life and no systemic vasodilator properties, has shown variable results in patients with AECOPD.144–148
Nutritional Support
Chapter 82 deals with overall nutritional support of the critically ill patient. Highlights of some issues directly related to the COPD patient follow.
Malnutrition has been recognized as a factor that increases mortality rate149,150 in COPD. Weight gain has been associated with decreased mortality rate.151 Nutritional status tends to decline markedly during acute illness in COPD patients,152–154 and patients may not recover to their previous nutritional state during convalescence, with recurrent exacerbations leading to a stepwise decline in nutritional status over time.155 Recent studies have shown that skeletal muscle mass is directly related to muscle dysfunction in COPD patients.156 Nutritional status has been correlated with weaning outcome.157 Short-term studies of oral supplemental feeding or enteral feeding have demonstrated increases in body weight and improvement in immunologic markers and respiratory muscle function.158–160 Nutritional support has been demonstrated to produce improvement in lung function in hospitalized patients with acute exacerbation of COPD.161
Special care must be taken to avoid overfeeding patients with COPD exacerbation because excess calories, particularly if carbohydrate rich, elevate total oxygen consumption and CO2 production, which may complicate management in patients with hypercapnia.162 In one study of mechanically ventilated patients, enteral nutrition was administered with a fixed carbohydrate content and an association between total caloric intake and CO2 production when providing nutritional support to the COPD patient with acute respiratory failure (ARF) was noted. It has been recommended that enteral alimentation provide total calories from 1.25 to 1.3 times the resting energy expenditure of the patient with a respiratory quotient target of 0.7 to 0.8 and that carbohydrate calories be limited to 40% of total calories.163
Noninvasive Mechanical Ventilation
ARF in the setting of AECOPD is characterized by the worsening of hypoxemia and a variable degree of carbon dioxide retention and acidemia. Worsening in ventilation/perfusion ratio (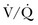 ) mismatching is probably the leading mechanism in the occurrence of the hypoxemia by the enlargement of physiologic dead space and the rise of wasted ventilation.164 The increase in airway resistance and the need of a higher minute ventilation may result in expiratory flow limitation, dynamic hyperinflation, and related PEEPi with subsequent increased inspiratory threshold load and dysfunction of the respiratory muscles, which may lead to their fatigue.165 Dyspnea, right ventricular failure, and encephalopathy characterize severe AECOPD complicated by ARF. Arterial pH reflects the acute worsening of the alveolar ventilation, and, regardless of the chronic level of PaCO2, it represents the best marker of the ARF severity.166
) mismatching is probably the leading mechanism in the occurrence of the hypoxemia by the enlargement of physiologic dead space and the rise of wasted ventilation.164 The increase in airway resistance and the need of a higher minute ventilation may result in expiratory flow limitation, dynamic hyperinflation, and related PEEPi with subsequent increased inspiratory threshold load and dysfunction of the respiratory muscles, which may lead to their fatigue.165 Dyspnea, right ventricular failure, and encephalopathy characterize severe AECOPD complicated by ARF. Arterial pH reflects the acute worsening of the alveolar ventilation, and, regardless of the chronic level of PaCO2, it represents the best marker of the ARF severity.166
Those patients with severe AECOPD develop ARF requiring ventilatory support. The frequency of this support varies among series, being as high as 74%.167 Ventilatory support may be of two types: invasive and noninvasive. Noninvasive ventilation (NIV) using nasal masks or facemasks has proved efficacious in ARF caused by COPD in several clinical studies, with success rates as high as 65% in some series.168–170 As with invasive mechanical ventilation, the goals of NIV include respiratory muscle rest, ventilatory support during treatment of reversible conditions, and the correction of severe hypoxemia and hypercarbia.
Several prospective, randomized, controlled studies confirmed the clinical efficacy of NIV in the treatment of the ARF during AECOPD. Compared to standard medical therapy alone the application of NIV improves survival, reduces the need for endotracheal intubation and the rate of complications, and shortens length of stay in hospital and intensive care unit (ICU).167,170,171
Indications
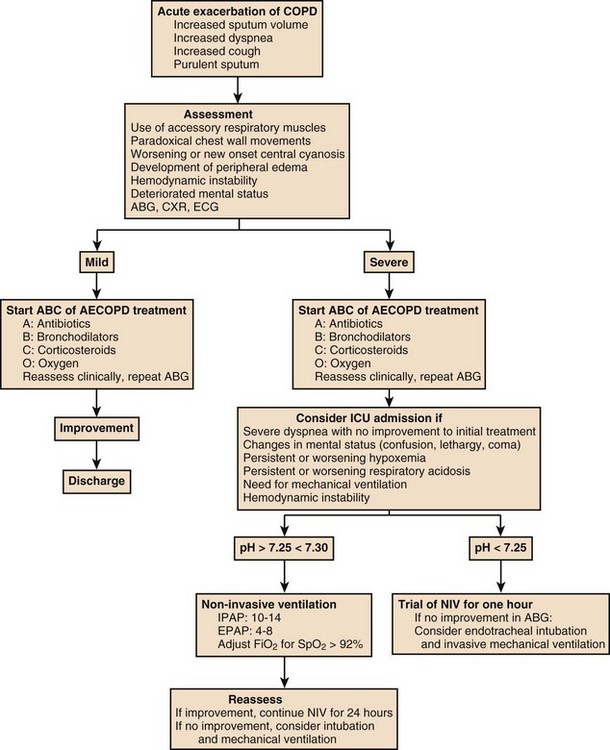
Patients with mild exacerbation, with normal or mildly reduced pH (pH > 7.35) and without clinical signs of respiratory failure, do not appear to benefit from a trial of NIV.172
In patients with moderate ARF, characterized by pH levels between 7.25 and 7.30, NIV is indicated for a few hours per day (<12 hours/day) in order to prevent endotracheal intubation, with low failure rates (15% to 20%), and appear to have the greater benefit.173
In more severely ill patients (pH < 7.25), the rate of NIV failures is inversely related to respiratory acidosis. In this population, the use of NIV as an alternative to invasive ventilation does not affect the mortality rate or the duration of ventilatory support, but it is associated with a lower rate of complications (VAP, difficult weaning). Despite the elevated rate of failure, it is worth a short trial of NIV if no contraindication is present (decreased level of consciousness, cardiac or respiratory arrest, severe gastrointestinal bleeding, severe hemodynamic instability, facial surgery or trauma, upper airway obstruction, inability to clear secretions).174
Implementation of Noninvasive Positive-Pressure Ventilation
NIV is usually delivered in assisted ventilation modality but no differences in success rate were found when applied in controlled ventilatory modality.175 Leak is a constant feature of NIV and may affect triggering of the ventilator, delivered FIO2, and air humidification. As such, the choice of the interface is one of the crucial issues affecting NIV outcome; although facemask is the standard interface to deliver NIV in patients with ARF, poor mask tolerance, skin lesions, and leaks are reported among factors causing NIV failure and intubation requirement.176
The use of pressure support ventilation (PSV) is the most common mode of ventilation for application of NIV. Nonetheless, bilevel positive airway pressure has become the preferred mode of NIV administration in COPD patients because it is generally as comfortable as PSV but produces greater improvement in gas exchange and reduces the work of breathing more effectively than PSV alone.177–179 Specifically designed noninvasive bilevel ventilators will have a better leak tolerance and adjust trigger and cycle sensitivity to improve patient-ventilator synchrony.180
The goal of NIV is to provide ventilatory support to rest fatigued respiratory muscles while the underlying illness is treated. The use of adequate ventilatory support is important regardless of the ventilator and modality chosen to deliver NIV. When using pressure-cycled modes, the recommended approach is to start at low pressures for patient comfort and titrate the pressure support or inspiratory positive airway pressure (IPAP) to achieve a tidal volume of at least 6 mL/kg, improvement in clinical signs (RR < 25 beats per minute, reduced accessory muscle use), and patient comfort.181 External PEEP, referred to as expiratory positive airway pressure (EPAP) in bilevel ventilators, may counteract PEEPi, and thus aid triggering, reduce oxygen consumption, and improve comfort. Although PEEPi can be as high as 15 cm H2O in patients with severe AECOPD, EPAP levels higher than 5 cm H2O are rarely tolerated or necessary. Therefore, slow titration to patient comfort is important to maximize the likelihood of NIV success. Improvements in pH, PaCO2, and level of consciousness at 60 minutes are predictors of success in patients on AECOPD who respond to NIV.178
The British Thoracic Society recommended that patients with ARF who respond to NIV be maintained as continuously as possible during the first 24 hours. If the respiratory status has stabilized, then the duration of ventilatory support can be reduced on subsequent days. Masks are removed for meals, conversation, comfort, and respiratory treatment as necessary.182
Weaning
Weaning from NIV may be accomplished either by progressively decreasing the levels of inspiratory positive-pressure support or by permitting the patient to be intermittently off NIV for increasing lengths of time. A combination of both strategies can also be used. In general, it is useful to wean patients by progressively increasing the period of spontaneous breathing without NIV. Once the acute process improves, many patients can be weaned relatively quickly. Unlike invasive ventilation, NIV can be reinstituted easily and quickly if the patient shows signs of fatigue or intolerance to spontaneous breathing. The use of nocturnal NIV may be needed during the early weaning period and may be continued at home in some patients.183
Some studies have analyzed weaning outcome in intubated patients with AECOPD, comparing NIV with conventional weaning methods. Results showed that NIV reduced weaning time, shortened intensive care days, decreased the incidence of nosocomial pneumonia, and improved 60-day survival rates.184,185 NIV has also been used successfully in another study on patients with hypercarbia after extubation.186 A meta-analysis concluded that, notwithstanding, the use of NIV to facilitate weaning, in mechanically ventilated patients, with predominantly COPD, is associated with promising but insufficient evidence of clinical benefit at present.187
Invasive Mechanical Ventilation
Invasive ventilation is indicated for patients who are not suited for NIV or who failed NIV, respiratory or cardiac arrest, respiratory pauses with loss of consciousness or gasping for air, altered level of consciousness (from psychomotor agitation to coma), massive aspiration, persistent inability to remove respiratory secretions, severe hemodynamic instability without response to fluids and vasopressors, severe ventricular arrhythmias, and life-threatening hypoxemia in patients unable to tolerate NIV. Patients with AECOPD requiring invasive mechanical ventilation have a higher ICU mortality rate and in-hospital mortality rate when compared with nonventilated patients.188
Initial Approach and Maintenance
Ventilation Modes
Mechanical ventilation of patients with AECOPD should be targeted to decrease excessive respiratory work. Patient-triggered ventilation modes, either assist-control or synchronized intermittent mandatory ventilation (SIMV), typically accomplish this goal. Special care must be taken; however, if these modes are not adequately adjusted to fit the characteristics of the patient, an increase in respiratory work will result.189
PSV has been extensively used and reviewed in the literature.190,191 In AECOPD patients, PSV has been shown to decrease inspiratory effort as the applied pressure is increased; however, the response among patients varied significantly.180 At higher levels of support pressure, many patients show an activation of the respiratory muscles during the late phase of inflation, with the potential to produce ventilation dyssynchrony. This may be more common in patients with longer time constants and those who require higher inspiratory flows delivered for longer periods.192
Inspired Oxygen
The goal of oxygenation should be to maintain an oxyhemoglobin saturation of at least 90% to 92%. In dark-skinned patients, pulse oximetry may overestimate oxyhemoglobin saturation, and in these patients, we recommend targeting pulse oximetry values of 95% to ensure adequate oxygenation. Although there is no clear-cut clinical evidence that allows determination of the FIO2 threshold of concern for oxygen toxicity, based on animal studies of oxygen toxicity in normal lungs, attempts to lower FIO2 to 0.6 or less by the end of the first 24 hours of mechanical ventilation is a reasonable goal.193
Intrinsic Positive End-Expiratory Pressure
Before discussing ventilation settings in AECOPD complicated with ARF, a review of intrinsic PEEP (PEEPi, auto-PEEP, dynamic hyperinflation) is in order. The two primary pathophysiologic changes that contribute to the development of respiratory distress and ARF in patients with obstructive lung disease are increased expiratory airflow resistance and dynamic hyperinflation. In COPD, the alveolar attachments that normally keep the smaller airway open via radial traction are lost. This leads to airway narrowing and collapse especially during expiration. In AECOPD the already narrowed airways may be further compromised by increased secretions, mucosal swelling, and peribronchial inflammation. The time constant for lung emptying is prolonged and end-expiratory lung volume is dynamically increased. Therefore, the next inspiratory effort occurs before the expiratory phase is completed, leading to air-trapping, and the respiratory system is prevented from returning to its resting state at the end of the expiration and thus dynamic hyperinflation develops. Dynamic hyperinflation results in PEEPi.194 Initially, PEEP is beneficial because it maintains the airways open, thereby reducing the airway resistance.
PEEPi is typically not detected on the pressure gauge of the ventilator because it is open to the atmosphere except for a very brief moment at the end of expiration. If the expiration port of the circuit is occluded at end expiration in a relaxed patient with a delay of next inspiration, the pressure inside the lungs and in the circuit will begin to equilibrate; if occlusion is sufficiently prolonged, PEEPi may be recorded.195
Inspiratory Triggering Sensitivity Settings
The setting of the optimal triggering threshold is more difficult in COPD patients, especially if dynamic lung hyperinflation exists. This is because the patient needs to generate a negative pressure equal to PEEPi before interfacing with the preset sensitivity on the ventilator. When PEEPi is high, the patient may exert significant inspiratory effort before the triggering threshold is reached, another cause of dyssynchrony. On the other hand, if the sensitivity of the ventilator has been placed at a very sensitive level, the ventilator may cycle inappropriately and can cause serious respiratory alkalosis, especially in the absence of significant PEEPi. Many ventilators have flow-triggered options, and although theoretically flow triggering may reduce patient effort, some reports failed to show differences when flow triggering is compared with newer pressure-triggering devices, even in COPD patients, although it is known that flow-triggered ventilators work better when the patient has elevated requirements of inspiratory flow.196,197
Inspiratory Flow Rate
High inspiratory flow rate helps satisfy the demands of most dyspneic or tachypneic COPD patients; moreover, it decreases the likelihood of dynamic hyperinflation and PEEPi. This decreases inspiratory time and increases expiratory time, minimizing PEEPi. An improvement in gas exchange has been found when inspiratory flows were increased from 40 to 60 L/minute in COPD patients.198
Positive End-Expiratory Pressure
In the past, PEEP was avoided in patients with AECOPD because of the concern of worsening dynamic hyperinflation. It is now recognized that the application of extrinsic PEEP below 75% to 85% of PEEPi may facilitate ventilator triggering, because alveolar pressure now needs to be decreased to only below the level of the external PEEP, instead of below the level of atmospheric pressure, decreasing the work required to trigger the inspiration.199,200
Weaning
In general, restoration of respiratory muscle function requires approximately 24 to 48 hours of mechanical ventilation.201–203 Before weaning patients with AECOPD from mechanical ventilation, the premorbid condition that triggered ARF should be corrected and an adequate neuromuscular competency/workload ratio achieved. Therefore, the strategy for facilitating weaning from the ventilator should include an increase in inspiratory force and a decrease in the load on the respiratory system. See Chapter 43 for a comparison of various weaning techniques.
Prognosis
The outcome for the patients with AECOPD with ARF depends on the severity of the COPD, the trigger for ARF, and avoidance of ICU complications. In patients who require mechanical ventilation for acute exacerbation, the overall in-hospital mortality rate is greater than 20%.204 For elderly patients, the mortality rate is greater than 50%.205 In a multicenter study of patients aged 65 or older, the mortality rate was 30% at hospital discharge, 41% at 90 days, 47% at 180 days, and 59% at 1 year. The time course and recovery following COPD exacerbation in a cohort of 101 patients with a mean FEV1 of 41.9% has been reported.206 Patients recorded daily morning peak expiratory flow rate (PEFR) following the onset of the exacerbation. Median recovery time for PEFR was 6 days. Recovery of PEFR to baseline values was complete in only 75.2% of exacerbations at 35 days, and 7.1% of exacerbations had still not returned to baseline at 91 days.206
References
1. Global Initiative for Chronic Obstructive Pulmonary Disease (GOLD). Global strategy for diagnosis, management and prevention of COPD. http://www.goldcopd.org/, 2010. [available at].
2. Celli, BR, Barnes, PJ. Exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 2007; 29:1224–1238.
3. Seemungal, TA, Donaldson, GC, Paul, EA, et al. Effect of exacerbation in quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998; 157:1418–1422.
4. Donaldson, GC, Seemungal, TA, Bhowmik, A, et al. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002; 57:847–852.
5. Anthonisen, NR, Manfreda, J, Warren, CP, et al. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med. 1987; 106:196–204.
6. Pauwels, R, Rabe, K. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004; 364:613–620.
7. Lopez, AD, Mathers, CD, Ezzati, M, et al. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet. 2006; 367:1747–1757.
8. Miravitlles, M, Soriano, JB, Garcia-Rio, F, et al. Prevalence of COPD in Spain: Impact of undiagnosed COPD on quality of life and daily life activities. Thorax. 2009; 64:863–868.
9. Thurlbeck, WM. Pathophysiology of chronic obstructive pulmonary disease. Clin Chest Med. 1990; 11:389.
10. Gottfried, SB. The role of PEEP in mechanically ventilated patients. In: Roussos C, Marini JJ, eds. Ventilatory Failure. Berlin: Springer-Verlag, 1991.
11. Hogg, JC, Macklem, PT, Thurlbeck, WM, et al. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med. 1968; 278:1355.
12. Wanner, A. The role of mucus in chronic obstructive pulmonary disease. Chest. 1990; 97(Suppl):11S.
13. Brusselle, GG, Joos, GF, Bracke, KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011; 378:1015–1026.
14. Scano, G, Gorini, M, Duranti, R, et al. Physiological changes during severe airflow obstruction in chronic obstructive pulmonary disease. Heart Lung. 2000; 29:124.
15. American Thoracic Society. Skeletal muscle function in chronic obstructive pulmonary disease: A statement of the American Thoracic Society and European Respiratory Society. Am J Respir Crit Care Med. 1999; 159:S1.
16. Sinden, NJ, Stockley, RA. Systemic inflammation and comorbidity in COPD: A result of overspill of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010; 65:930–936.
17. Wouters, EF, Reynaert, NL, Dentener, MA, et al. Systemic and local inflammation in asthma and chronic obstructive pulmonary disease: Is there a connection? Proc Am Thorac Soc. 2009; 6:638–647.
18. Qiu, Y, Zhu, J, Bandi, V, et al. Biopsy neutrophilia, chemokine and receptor gene expression in severe exacerbations of COPD. Am J Respir Crit Care Med. 2003; 168:968–975.
19. Papi, A, Bellettato, CM, Braccioni, F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006; 173:1114–1121.
20. Gern, JE, French, DA, Grindle, KA, et al. Double-stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2003; 28:731–737.
21. Mercer, PF, Shute, JK, Bhowmik, A, et al. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005; 6:151.
22. Cohen, CA, Zagelbaum, G, Gross, D, et al. Clinical manifestation of inspiratory muscles fatigue. Am J Med. 1982; 73:308.
23. Pinto-Plata, V, Livnat, G, Girish, M, et al. Systemic cytokines, clinical and physiological changes in patients hospitalized for COPD exacerbations. Chest. 2006; 131:37–43.
24. Aubier, M, Murciano, D, Milic-Emili, J. Effects of administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis. 1980; 122:747.
25. Juan, G, Calverley, P, Talamo, C. Effect of carbon dioxide on diaphragmatic function in humans beings. N Engl J Med. 1984; 310:874.
26. Derenne, JP, Fleury, B, Pariente, R. Acute respiratory failure of chronic obstructive disease. Am Rev Respir Dis. 1988; 138:1006.
27. Orozco-Levi, M. Structure and function on the respiratory muscles in patients with COPD: Impairment or adaptation. Eur Respir J. 2003; 22:41S–51S.
28. Barbera, JA, Roca, J, Ferrer, A, et al. Mechanisms of worsening gas exchange during acute exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 1997; 10:1285.
29. Yanos, J, Keamy, MF, Leisk, L. The mechanisms of respiratory arrest in inspiratory loading and hypoxemia. Am Rev Respir Dis. 1990; 141:933.
30. McHardy, VU, Inglis, JM, Calder, MA, et al. A study of infective and other factors in exacerbations of chronic bronchitis. Br J Dis Chest. 1980; 74:228.
31. Soler, N, Torres, A, Ewig, S, et al. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998; 157:1498.
32. Dockery, DW, Pope, AC, Xu, X, et al. An association between air pollution and mortality in six U. S. cities. N Engl J Med. 1993; 329:1753.
33. Thurson, GD, Kazuhiko, I. Epidemiological studies of acute ozone exposures and mortality. Journal of Exposure Analysis and Enviromental Epidemiology. 2011; 11:286–294.
34. Lippmann, M, Fein, A. Pulmonary embolism in the patients with chronic obstructive disease. Chest. 1981; 79:39.
35. Sethi, S. Infectious etiology of acute exacerbations of chronic bronchitis. Chest. 2000; 117:380S.
36. Gump, DW, Phillips, CA, Forsyth, BR, et al. Role of infection in chronic bronchitis. Am Rev Respir Dis. 1976; 113:465.
37. Buscho, RO, Saxta, D, Shultz, PS, et al. Infections with viruses and Mycoplasma pneumoniae during exacerbation of chronic bronchitis. J Infect Dis. 1978; 137:377.
38. Smith, CB, Golden, C, Kanner, R, et al. Association of viral and Mycoplasma pneumoniae infections with acute respiratory illness in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1980; 121:225.
39. Sethi, S, Evans, N, Grant, BJ, et al. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002; 347:465.
40. Murphy, TS, Sethi, S. Bacterial infection in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1992; 146:1067.
41. Tager, I, Speizer, FE. Role of infection in chronic bronchitis. N Engl J Med. 1975; 292:563.
42. Schreiner, A, Bjerkestrand, G, Digranes, A, et al. Bacteriological findings in the transtracheal aspirate from patients with acute exacerbation of chronic bronchitis. Infection. 1978; 6:54.
43. Blasi, F, Legnani, D, Lombardo, VM, et al. Chlamydia pneumoniae infection in acute exacerbations of COPD. Eur Respir J. 1993; 6:19.
44. Miyashita, N, Niki, Y, Nakajima, M, et al. Chlamydia pneumoniae infections in patients with diffuse panbronchiolitis and COPD. Chest. 1998; 114:969.
45. Eller, J, Ede, A, Schaberg, T, et al. Infective exacerbation of chronic bronchitis: Relation between bacteriologic etiology and lung function. Chest. 1998; 113:1542.
46. Hill, AT, Campbell, EJ, Hill, SL, et al. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000; 109:288.
47. Patel, IS, Seemungal, TA, Wilks, M, et al. Relationship between bacterial colonization and the frequency, character, and severity of COPD exacerbations. Thorax. 2005; 60:925.
48. Wedzicha, JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004; 1:115–120.
49. California Department of Public Health. Clean air for California: Initial report of the air pollution study project. San Francisco: State of California Department of Public Health; 1955.
50. Delfino, RJ, Becklake, MR, Hanley, JA. The relationship of urgent hospital admission for respiratory illness to photochemical air pollution levels in Montreal. Environ Res. 1994; 67:1.
51. Wordley, J, Walters, S, Ayres, JG. Short term variations in hospital admissions and mortality and particulate air pollution. Occup Environ Med. 1997; 54:108.
52. Thurston, GD, Ito, K. Epidemiological studies of ozone exposure effects. In: Holgate ST, Samet JM, Koren HS, et al, eds. Air Pollution and Health. London: Academic Press, 1999.
53. Schwartz, J. PM10, ozone and hospital admissions for the elderly in Minneapolis-St. Paul, Minnesota. Arch Environ Health. 1994; 49:366.
54. Bates, DV. Setting the stage: Critical risks. In: Bates DV, ed. Environmental Health Risks and Public Policy: Decision Making in Free Societies. Seattle: University of Washington Press, 1994.
55. Baum, GL, Fisher, FD. The relationship of fatal pulmonary insufficiency with cor pulmonale, right-sided mural thrombi and pulmonary emboli: A preliminary report. Am J Med Sci. 1960; 240:609.
56. Moser, K, Lemoine, J, Natchtwey, R, et al. Deep venous thrombosis a pulmonary embolism: Frequency in a respiratory intensive unit. JAMA. 1981; 246:1422.
57. Dalen, JE, Alpert, JS. Natural history of pulmonary embolism. In: Sasahara AA, Sonnenblick EH, Lesch M, eds. Pulmonary Embolism. New York: Grune & Stratton, 1975.
58. Tillie-Leblond, I, Marqette, CH, Perez, T, et al. Pulmonary embolism in patients with unexplained exacerbations of chronic obstructive pulmonary disease: Prevalence and risk factors. Ann Intern Med. 2006; 144:390.
59. Cordova, C, Musca, A, Violi, F, et al. Platelet hyperfunction in patients with chronic airway obstruction. Eur J Respir Dis. 1985; 66:9.
60. Fanta, CH, Wright, TC, McFaden, ER, Jr. Differentiation of recurrent pulmonary emboli from chronic obstructive pulmonary disease as a cause of cor pulmonale. Chest. 1981; 79:92.
61. PIOPED Investigators. Value of ventilation/perfusion scan in acute pulmonary embolism: Results of the PIOPED. JAMA. 1990; 263:2753.
62. Wildberger, JE, Schoepf, UJ, Mahnken, AH, et al. Approaches to CT perfusion imaging in pulmonary embolism. Semin Roentgenol. 2005; 40:64–73.
63. Coche, E, Pawlak, S, Dechambre, S, Maldague, B. Peripheral pulmonary arteries: Identification at multi-slice spiral CT with 3D reconstruction. Eur Radiol. 2003; 13:815–822.
64. Lensing, AW, Pradoni, P, Brandjes, D, et al. Detection of deep venous thrombosis by real time B-mode ultrasonography. N Engl J Med. 1989; 320:342.
65. Wheeler, HB, O’Donnell, JA, Anderson, FA, et al. Occlusive impedance phlebography: A diagnostic procedure for venous thrombosis and pulmonary embolism. Prog Cardiovasc Dis. 1974; 17:199.
66. Bounameaux, H, Cirafici, D, DeMoerloose, P, et al. Measurement of the D-dimer in patients as a diagnostic aid in suspected pulmonary embolism. Lancet. 1991; 37:196.
67. Barnes, PB. Strategies for novel COPD therapies. Pulm Pharmacol Ther. 1999; 12:67.
68. Siafakas, NM, Bouros, D, Management of acute exacerbation of chronic obstructive pulmonary disease. Management of Chronic Obstructive Pulmonary Disease. Postma, DS, Siafakas, NM, eds. Management of Chronic Obstructive Pulmonary Disease; Vol. 3. European Respiratory Society Journals, Sheffield, UK, 1998.
69. Kuller, L, Ockene, J, Townsend, M, et al. The epidemiology of pulmonary function and COPD mortality in the multiple risk factor intervention trial. Am Rev Respir Dis. 1989; 140:76S.
70. Rutten, FH, Cramer, MJ, Lammers, JW, et al. Heart failure and chronic obstructive pulmonary disease: An ignored combination? Eur J Heart Fail. 2006; 8:706.
71. Douglas, NJ, White, DP, Weil, JV, et al. Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis. 1982; 126:758.
72. Gould, GA, Gugger, M, Molloy, J, et al. Breathing pattern and eye movement density during REM sleep in man. Am Rev Respir Dis. 1988; 138:874.
73. Catterall, JR, Calverley, PMA, MacNee, W, et al. Mechanism of transient nocturnal hypoxemia in hypoxic chronic bronchitis and emphysema. J Appl Physiol. 1985; 59:1698.
74. Videm, V, Pillgram-Larsen, J, Ellingsen, O, et al. Spontaneous pneumothorax in chronic obstructive pulmonary disease: Complications, treatment and recurrences. Eur J Respir Dis. 1987; 71:365.
75. Chistensen, EE, Dietz, GW. Subpulmonic pneumothorax in patients with chronic obstructive pulmonary disease. Radiology. 1976; 121:33.
76. Estenne, M, Yernault, JC, DeTrayer, A. Mechanism of relief of dyspnea after thoracentesis in patients with large pleural effusions. Am J Med. 1983; 74:813.
77. Eaton, T, Lewis, C, Young, P, et al. Long term oxygen therapy improves health-related quality of life. Respir Med. 2004; 98:285.
78. Hunt, JM, Copland, J, McDonald, CF, et al. Cardiopulmonary response to oxygen therapy in hypoxaemic chronic airflow obstruction. Thorax. 1989; 44:930.
79. MacNee, W, Wathen, CG, Flenley, DC, et al. The effects of controlled oxygen therapy on ventricular function in patients with stable and decompensated cor pulmonale. Am Rev Respir Dis. 1988; 137:1289.
80. Esteban, A, Cerda, E, De La Cal, MA, et al. Hemodynamic effects of oxygen therapy in patients with acute exacerbations of chronic obstructive pulmonary disease. Chest. 1993; 104:471.
81. Meissner, HH, Franklin, C. Extreme hypercapnia in a fully alert patient. Chest. 1992; 102:1298.
82. Potkin, RT, Swenson, ER. Resuscitation from severe acute hypercapnia. Chest. 1992; 102:1742.
83. Carroll, GC, Rothenberg, DM. Carbon dioxide narcosis. Chest. 1992; 102:986.
84. Robinson, TD, Freiberg, DB, Regnis, JA, et al. The role of hypoventilation and ventilation-perfusion redistribution in oxygen-induced hypercapnia during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000; 161:1524.
85. Hanson, CW, III., Marshall, BE, Frasch, HF, et al. Causes of hypercarbia with oxygen therapy in patients with chronic obstructive pulmonary disease. Crit Care Med. 1996; 24:23.
86. Agusti, AG, Carrera, M, Barbe, F, et al. Oxygen therapy during exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 1999; 14:934.
87. Wasserman, K. Uses of oxygen in the treatment of acute respiratory failure secondary to obstructive lung disease. Monaldi Arch Chest Dis. 1993; 48:509.
88. McCrory, DC, Brown, CD. Anti-cholinergic bronchodilators versus beta2-sympathomimetic agents for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (4):2002.
89. Duranti, R, Misuri, G, Gorini, M, et al. Mechanical loading and control of breathing in patients with severe chronic obstructive pulmonary disease. Thorax. 1995; 50:127.
90. Bernasconi, M, Brandolese, R, Poggi, R, et al. Dose-response effects and time course of effects of inhaled fenoterol on respiratory mechanics and arterial oxygen tension in mechanically ventilated patients with chronic airflow obstruction. Intensive Care Med. 1990; 16:108.
91. Koutsogiannis, Z, Kelly, AM. Does high dose ipratropium bromide added to salbutamol improve pulmonary function for patients with chronic obstructive airways disease in the emergency department? Aust N Z J Med. 2000; 30:38.
92. Niewoehner, DE, Rice, K, Cote, C, et al. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator: A randomized trial. Ann Intern Med. 2005; 6(143):317–326.
93. Mahler, DA, Donohoue, JF, Barbee, RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest. 1999; 115:957–965.
94. Rennard, SI, Anderson, W, ZuWallack, R, et al. Use of long acting inhaled beta2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001; 163:1087–1092.
95. Vincken, W, van Noord, JA, Greefhorst, AP, et al. Improved health outcomes in patients with COPD during 1 year’s treatment with tiotropium. Eur Respir J. 2002; 19:209–216.
96. Casaburi, R, Mahler, DA, Jones, PW, et al. A long term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J. 2002; 19:217–224.
97. The COMBIVENT Inhalation Solution Study Group. Routine nebulized ipratropium and albuterol together are better than either alone in COPD. Chest. 1997; 112:1514.
98. Buhling, F, Lieder, N, Reisenauer, A, et al. Anti-inflammatory effect of tiotropium mediated by suppression of acetylcholine-induced release of chemotactic activity. Eur Respir J. 2004; 24:318S.
99. Aaron, SD, Vandemheen, KL, Hebert, P, et al. Outpatient oral prednisone after emergency treatment of chronic obstructive pulmonary disease. N Engl J Med. 2003; 348:2618–2625.
100. Albert, RK, Martin, TR, Lewis, SW. Controlled clinical trial of methylprednisolone in patients with chronic bronchitis and acute respiratory insufficiency. Ann Intern Med. 1980; 92:753–758.
101. Bullard, MJ, Liaw, SJ, Tsai, YH, et al. Early corticosteroid use in acute exacerbations of chronic airflow obstruction. Am J Emerg Med. 1996; 14:139–143.
102. Wood-Baker, R, Walters, EH. Corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (2):2000.
103. Bradley, SQ, Wen, QG, Don, DS. Contemporary management of acute exacerbations of COPD: A systematic review and meta-analysis. Chest. 2008; 133:756–766.
104. Davies, L, Angus, RM, Calverley, PM. Oral corticosteroids in patients admitted to hospital with exacerbations of chronic obstructive pulmonary disease: A prospective randomized controlled trial. Lancet. 1999; 354:456–460.
105. Vestbo, J, Sorensen, T, Lange, P, et al. Long term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: A randomized controlled trial. Lancet. 1999; 353:1819–1823.
106. Confalonieri, M, Urbino, R, Potena, A, et al. Hydrocortisone infusion for severe community acquired pneumonia: A preliminary randomized study. Am J Respir Crit Care Med. 2005; 171:242–248.
107. Pauwels, RA, Lofdahl, CG, Laitinen, LA, et al. Long-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. N Engl J Med. 1999; 340:1948–1953.
108. Niewoehner, DE, Erbland, ML, Deupree, RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 1999; 340:1941–1947.
109. Szafranski, W, Cukier, A, Ramirez, A, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J. 2003; 21:74–81.
110. Calverley, PM, Anderson, JA, Celli, B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007; 356:775–789.
111. Hirschmann, JV. Do bacteria cause exacerbations of COPD? Chest. 2000; 118:193.
112. Murphy, TF, Sethi, S, Niederman, MS. The role of bacteria in exacerbations of COPD: A constructive view. Chest. 2000; 118:204.
113. Stockley, RA, O’Brien, C, Pye, A, et al. Relationship of sputum color to nature and outpatient management of acute exacerbations of COPD. Chest. 2000; 117:1638–1645.
114. Hansen, M, Evald, T, Balslov, S, et al. A randomized controlled trial between amoxicillin and placebo in the treatment of acute exacerbations of chronic bronchitis. Eur Respir J. 1990; 3:89.
115. Jorgensen, AF, Coolidge, J, Pedersen, PA, et al. Amoxicillin in treatment of acute uncomplicated exacerbations of chronic bronchitis: A double blind, placebo-controlled multicentre study in general practice. Scand J Prim Health Care. 1992; 10:7–11.
116. Nouira, S, Marghli, S, Belghith, M, et al. Once daily oral ofloxacin in chronic obstructive pulmonary disease exacerbation requiring mechanical ventilation: A randomized controlled trial. Lancet. 2001; 358:2020–2025.
117. Irwin, RS, Erickson, AD, Pratter, ME, et al. Prediction of tracheobronchial colonization in current cigarette smokers with chronic obstructive bronchitis. J Infect Dis. 1982; 145:234.
118. Bjerkestrand, G, Digranes, A, Schreiner, A. Bacteriologic findings in transtracheal aspirates from patients with chronic bronchitis and bronchiectasis. Scand J Respir Dis. 1975; 56:201.
119. Destache, CJ, Dewan, N, O’Donohue, WJ, et al. Clinical and economic considerations in the treatment of acute exacerbations on chronic bronchitis. J Antimicrob Chemother. 1999; 43:107.
120. Monso, E, Rosell, A, Bonet, G, et al. Risk factors for lower airway bacterial colonization in chronic bronchitis. Eur Respir J. 1999; 13:338.
121. Sánchez Nieto, JM, Torres, A, García Córdoba, F, et al. Impact of invasive and noninvasive quantitative culture sampling on outcome of ventilator-associated pneumonia: A pilot study. Am J Respir Crit Care Med. 1998; 157:371.
122. Jourdain, B, Novara, A, Jolly Guillou, ML, et al. Role of quantitative cultures of endotracheal aspirates in the diagnosis of nosocomial pneumonia. Am J Respir Crit Care Med. 1995; 152:241.
123. Meduri, GU, Chastre, J. The standardization of bronchoscopic techniques for ventilator-associated pneumonia. Chest. 1992; 102:557S.
124. Adams, S, Melo, J, Anzueto, A. Effect of antibiotics on the recurrence rates of chronic obstructive pulmonary disease. Chest. 1997; 112:22S.
125. Baughman, RP, Pina, E. Infections in acute exacerbation of chronic bronchitis: What are they and how do we know? Semin Respir Crit Care Med. 2000; 21:87.
126. Ram, F, Rodriguez-Roisin, R, Granados-Navarrete, A, et al. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. (19):2006.
127. Vizza, CD, Lynch, JP, Ochoa, LL, et al. Right and left ventricular dysfunction in patients with severe pulmonary disease. Chest. 1998; 113:576.
128. Goldberg, HS, Rabson, J. Control of cardiac output by systemic vessels. Am J Cardiol. 1981; 47:696.
129. Martin, C, Perrin, G, Saux, P, et al. Effects of norepinephrine on right ventricular function in septic shock patients. Intensive Care Med. 1994; 20:444.
130. Hirsch, LJ, Rooney, MW, Wat, SS, et al. Norepinephrine and phenylephrine effects on right ventricular function in experimental canine pulmonary embolism. Chest. 1991; 100:796.
131. Angle, MR, Molloy, DW, Penner, B, et al. The cardiopulmonary and renal hemodynamic effects of norepinephrine in canine pulmonary embolism. Chest. 1989; 95:1333.
132. Juilliere, Y, Feldmann, L, Perrin, O, et al. Beneficial cumulative role of both nitroglycerin and dobutamine on right ventricular systolic function in congestive heart failure patients awaiting heart transplantation. Int J Cardiol. 1995; 52:17.
133. Mathur, PN, Powles, ACP, Pugsley, SO, et al. Effect of long-term administration of digoxin on exercise performance in chronic airflow obstruction. Eur J Respir Dis. 1985; 66:273.
134. Brown, SE, Pakron, FJ, Milne, N, et al. Effects of digoxin on exercise capacity and right ventricular function during exercise in chronic airflow obstruction. Chest. 1984; 85:187.
135. Mathur, PN, Powles, ACP, Pugsley, SO, et al. Effect of digoxin on right ventricular function in severe chronic airflow obstruction. Ann Intern Med. 1981; 95:283.
136. Mols, P, Huynh, CH, Dechamps, P, et al. Acute effects of nifedipine on systolic and diastolic ventricular function in patients with chronic obstructive pulmonary disease. Chest. 1993; 103:1381.
137. Kalra, L, Bone, MF. Effect of nifedipine on physiologic shunting and oxygenation in chronic obstructive pulmonary disease. Am J Med. 1993; 94:419.
138. Gassner, A, Sommer, G, Fridrich, L, et al. Differential therapy with calcium antagonists in pulmonary hypertension secondary to COPD: Hemodynamic effects of nifedipine, diltiazem, and verapamil. Chest. 1990; 98:829.
139. Agostoni, P, Doria, E, Galli, C, et al. Nifedipine reduces pulmonary pressure and vascular tone during short- but not long-term treatment of pulmonary hypertension in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1989; 139:120.
140. Corriveau, ML, Rosen, BJ, Keller, CA, et al. Effect of posture, hydralazine, and nifedipine on hemodynamics, ventilation, and gas exchange in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1988; 138:1494.
141. Corriveau, ML, Vu-Dinh, Minh, Dolan, GF. Long-term effects of hydralazine on ventilation and blood gas values in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Med. 1987; 83:886.
142. Keller, CA, Shepard, JW, Jr., Chun, DS, et al. Effects of hydralazine on hemodynamics, ventilation, and gas exchange in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am Rev Respir Dis. 1984; 130:606.
143. Brent, BN, Matthay, RA, Mahler, DA, et al. Relationship between oxygen uptake and oxygen transport in stable patients with chronic obstructive pulmonary disease: Physiologic effects of nitroprusside and hydralazine. Am Rev Respir Dis. 1984; 129:682.
144. Brent, BN, Berger, HJ, Matthay, RA, et al. Contrasting acute effects of vasodilators (nitroglycerin, nitroprusside, and hydralazine) on right ventricular performance in patients with chronic obstructive pulmonary disease and pulmonary hypertension: A combined radionuclide-hemodynamic study. Am J Cardiol. 1983; 51:1682.
145. Baigorri, F, Joseph, D, Artigas, A, et al. Inhaled nitric oxide does not improve cardiac or pulmonary function in patients with an exacerbation of chronic obstructive pulmonary disease. Crit Care Med. 1999; 27:215.
146. Germann, P, Ziesche, R, Leitner, C, et al. Addition of nitric oxide to oxygen improves cardiopulmonary function in patients with severe COPD. Chest. 1998; 114:29.
147. Yoshida, M, Taguchi, O, Gabazza, EC, et al. Combined inhalation of nitric oxide and oxygen in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1997; 155:526.
148. Blanch, L, Joseph, D, Fernandez, R, et al. Hemodynamic and gas exchange responses to inhalation of nitric oxide in patients with the acute respiratory distress syndrome and in hypoxemic patients with chronic obstructive pulmonary disease. Intensive Care Med. 1997; 23:51.
149. Gray Donald, K, Gibbons, L, Shapiro, SH, et al. Nutritional status and mortality in chronic obstructive pulmonary diseases. Am J Respir Crit Care Med. 1996; 153:961.
150. Donahoe, M, Rogers, RM. Nutritional assessment and support in chronic obstructive pulmonary disease. Clin Chest Med. 1990; 11:487.
151. Wilson, DO, Rogers, RM, Wright, EC, et al. Body weight in chronic obstructive pulmonary disease. The National Institutes of Health Intermittent Positive-Pressure Breathing Trial. Am Rev Respir Dis. 1989; 139:1435.
152. Rochester, DF. Body weight and respiratory muscle function in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1986; 134:446.
153. Vandenbergh, E, Woestijne, vdKP, Gyselen, A. Weight changes in the terminal stages of chronic obstructive pulmonary disease. Am Rev Respir Dis. 1967; 95:556.
154. Schols, A, Slangen, J, Volovics, L, et al. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998; 157:1791.
155. Driver, AG, McAlevy, MT, Smith, JL. Nutritional assessment of patients with chronic obstructive pulmonary disease and acute respiratory failure. Chest. 1982; 82:568.
156. Hunter, AMB, Carey, MA, Larsh, HW. The nutritional status of patients with chronic obstructive pulmonary disease. Am Rev Respir Dis. 1981; 124:376.
157. Bernard, S, LeBlanc, P, Whittom, F, et al. Peripheral muscle weakness in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998; 158:629.
158. Bassili, HR, Deitel, M. Effect of nutritional support on weaning patients off mechanical ventilators. J Parenter Enteral Nutr. 1981; 5:161.
159. Whittaker, JS, Ryan, CF, Buckley, PA, et al. The effects of refeeding on peripheral and respiratory muscle function in malnourished chronic pulmonary disease patients. Am Rev Respir Dis. 1990; 142:283.
160. Fuenzalida, CE, Petty, TL, Jones, ML, et al. The immune response to short-term nutritional intervention in advanced chronic obstructive pulmonary disease. Am Rev Respir Dis. 1990; 142:49.
161. Wilson, DO, Rogers, RM, Sanders, MH, et al. Nutritional intervention in malnourished patients with emphysema. Am Rev Respir Dis. 1986; 134:672.
162. Saudny, UH, Martin, JG, Gray, DK. Impact of nutritional support on functional status during an acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1997; 156:794.
163. Rose, W. Total parenteral nutrition and the patient with chronic obstructive pulmonary disease. J Intraven Nurs. 1992; 15:18.
164. Calverley, PM. Respiratory failure in chronic obstructive pulmonary disease. Eur Respir J. 2003; 22:26–30.
165. O’Donnell, DE, Parker, CM. COPD exacerbations—3: Pathophysiology. Thorax. 2006; 61:354–361.
166. Plant, PK, Elliott, MW. Chronic obstructive pulmonary disease—9: Management of ventilatory failure in COPD. Thorax. 2003; 58:537–542.
167. Brochard, L, Mancebo, J, Wysocki, M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 1995; 333:817–822.
168. Bott, J, Carroll, MP, Conway, JH, et al. Randomized controlled trial of nasal ventilation in acute respiratory failure due to chronic obstructive airways disease. Lancet. 1993; 341:1555.
169. Collaborative Research Group of Noninvasive Mechanical Ventilation for Chronic Obstructive Pulmonary Disease. Early use of non-invasive positive pressure ventilation for acute exacerbations of chronic pulmonary disease: A multicentre randomized controlled trial. Chin Med J. 2005; 118:2034–2040.
170. Kramer, N, Meyer, TJ, Meharg, J, et al. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med. 1995; 151:1799–1806.
171. Lightlower, JV, Wedzicha, JA, Elliott, MW, et al. Noninvasive positive pressure ventilation to treat respiratory failure resulting from exacerbation of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ. 2003; 326:185–190.
172. Elliott, MW. Noninvasive ventilation in acute exacerbations of chronic obstructive pulmonary disease: A new gold standard? Intensive Care Med. 2002; 28:1691–1694.
173. Squadrone, E, Frigerio, P, Fogliati, C, et al. Noninvasive vs. invasive ventilation in COPD patients with acute respiratory failure deemed to require ventilatory assistance. Intensive Care Med. 2004; 30:1303–1310.
174. Nava, S, Navalesi, P, Conti, G. Time of non-invasive ventilation. Intensive Care Med. 2006; 32:361–370.
175. Lellouche, F, Maggiore, SM, Deye, N, et al. Effect of the humidification device on the work of breathing during noninvasive ventilation. Intensive Care Med. 2002; 28:1582–1589.
176. Costa, R, Navalesi, P, Antonelli, M, et al. Physiologic evaluation of different levels of assistance during noninvasive ventilation delivered through a helmet. Chest. 2005; 128:2984–2990.
177. Girault, C, Richard, J-C, Chevron, V, et al. Comparative physiologic effects of noninvasive assist-control and pressure support ventilation in acute hypercapnic respiratory failure. Chest. 1997; 111:1639.
178. Appendini, L, Patessio, A, Zanaboni, S, et al. Physiologic effects of positive end-expiratory pressure and mask pressure support during exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994; 149:1069–1076.
179. Kacmarek, RM. Characteristics of pressure-targeted ventilators used for noninvasive positive pressure ventilation. Respir Care. 1997; 42:380.
180. Jubran, A, van de Graaff, WB, Tobin, MJ. Variability of patient-ventilator interaction with pressure support ventilation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995; 152:129–136.
181. Plant, PK, Owen, JL, Elliott, MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: A multicentre randomized controlled trial. Lancet. 2000; 355:1931–1935.
182. Conti, G, Antonelli, M, Navalesi, P, et al. Noninvasive vs. conventional mechanical ventilation in patients with chronic obstructive pulmonary disease after failure of medical treatment in the ward; a randomized trial. Intensive Care Med. 2002; 28:1701–1707.
183. Girault, C, Daudenthun, I, Chevron, V, et al. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: A prospective, randomized controlled study. Am J Respir Crit Care Med. 1999; 160:86.
184. Nava, S, Ambrosino, N, Clini, E, et al. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease: A randomized, controlled trial. Ann Intern Med. 1998; 128:721.
185. Hilbert, G, Gruson, D, Portel, L, et al. Noninvasive pressure support ventilation in COPD patients with postextubation hypercapnic respiratory insufficiency. Eur Respir J. 1998; 11:1349.
186. Confalonieri, M, Gazzaniga, P, Gandola, L, et al. Haemodynamic response during initiation of non-invasive positive pressure ventilation in COPD patients with acute respiratory failure. Respir Med. 1998; 92:331.
187. Burns, KE, Adhikari, NK, Meade, MO. A meta-analysis of noninvasive weaning to facilitate liberation from mechanical ventilation. Can J Anaesth. 2006; 53:305–315.
188. Seneff, MG, Wagner, DP, Wagner, RP, et al. Hospital and 1-year survival of patients admitted to intensive care units with acute exacerbation of chronic obstructive pulmonary disease. JAMA. 1995; 274:1852.
189. Braschi, A, Iotti, G, Rodi, G, et al. Dynamic pulmonary hyperinflation (DPH) during intermittent mandatory ventilation (IMV). Intensive Care Med. 1988; 14:589.
190. Brochard, L. Pressure support ventilation. In: Tobin MJ, ed. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill, 1994.
191. MacIntyre, NR. Respiratory function during pressure support ventilation. Chest. 1986; 89:117.
192. Jubran, A, Tobin, MJ. Reliability of pulse oximetry in titrating supplemental oxygen therapy in ventilator-dependent patients. Chest. 1990; 97:1420.
193. Slutsky, AS. Mechanical ventilation. American College of Chest Physicians’ Consensus Conference. Chest. 1993; 104:1833.
194. Ranieri, VM, Grasso, S, Fiore, T, et al. Auto-positive end-expiratory pressure and dynamic hyperinflation. Clin Chest Med. 1996; 17:379–394.
195. Pepe, PE, Marini, JJ. Occult positive end-expiratory pressure in mechanically ventilated patients with airflow obstruction. Am Rev Respir Dis. 1982; 126:166.
196. Sydow, M, Golisch, W, Buscher, H, et al. Effect of low-level PEEP on inspiratory work of breathing in intubated patients, both with healthy lungs and with COPD. Intensive Care Med. 1995; 21:887.
197. Sassoon, C, Del Rosario, N, Fei, R, et al. Influence of pressure- and flow-triggered synchronous intermittent mandatory ventilation on inspiratory muscle work. Crit Care Med. 1994; 22:1933.
198. Connors, AF, McCaffree, DR, Gray, BA. Effect of inspiratory flow rate on gas exchange during mechanical ventilation. Am Rev Respir Dis. 1981; 124:537.
199. Petrof, BJ, Legaré, M, Goldberg, P, et al. Continuous positive airway pressure reduces work of breathing and dyspnea during weaning from mechanical ventilation in severe chronic obstructive pulmonary disease. Am Rev Respir Dis. 1990; 141:281.
200. Smith, TC, Marini, JJ. Impact of PEEP on lung mechanics and work of breathing in severe airflow obstruction. J Appl Physiol. 1988; 65:1488.
201. Grassino, A, Macklem, PT. Respiratory muscle fatigue and ventilator failure. Ann Rev Med. 1984; 35:625.
202. Braun, NMT, Faulkner, J, Hughes, RL, et al. When should respiratory muscles be exercised? Chest. 1983; 84:76.
203. Kaelin, RM, Assimacopoulos, A, Chevrolet, JC. Failure to predict six-month survival of patients with COPD requiring mechanical ventilation by analysis of simple indices. Chest. 1987; 92:971.
204. Petty, TL. Acute respiratory failure in COPD. In Petty TL, ed. : Chronic Obstructive Pulmonary Disease, 2nd ed, New York: Marcel Dekker, 1985.
205. Swinburne, AJ, Fedullo, AJ, Bixby, K, et al. Respiratory failure in the elderly: Analysis of outcome after treatment with mechanical ventilation. Arch Intern Med. 1993; 153:1657.
206. Seemungal, TA, Donaldson, GC, Bhowmik, A, et al. Time course and recovery of exacerbations in patients with chronic obstructive disease. Am J Respir Crit Care Med. 2000; 161:1608.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]39
Chronic Obstructive Pulmonary Disease
In 1997, the U.S. National Heart Lung and Blood Institute and the World Health Organization held an international workshop that led to the first Global Initiative on Obstructive Lung Disease (GOLD) report. The most recent iteration of the GOLD guidelines defines chronic obstructive pulmonary disease (COPD) as a preventable and treatable disease with some significant extrapulmonary effects that may contribute to the severity in individual patients. Its pulmonary component is characterized by chronic airflow limitation that is not fully reversible. The airflow limitation is usually both progressive and associated with abnormal inflammatory response of the lungs to noxious particles and gases.1
Definitions
Although controversies remain over the definition of exacerbation, how they should be monitored, and their underlying mechanisms, acute exacerbations of COPD (AECOPD) are major and increasingly recognized events in the disease course. They typically occur one to three times per year. Exacerbations are associated with an increase in economic burden2 and a decline in health-related quality of life.3 Those patients with more than two exacerbations per year have significantly worse health-related complications and decline in lung function than those with two or fewer exacerbations.4 As mentioned before, there is no general agreement on the definition of AECOPD, but it has been defined according to the presence of specific signs and symptoms, worsening in symptoms, and the need for medical intervention, and each of these approaches has positive and negative connotations. It is well recognized, however, that the presence of increased shortness of breath, increased sputum volume, and increased purulence are the three specific symptoms that represent an exacerbation.5
Background
COPD affects more than 200 million people worldwide and is the fourth leading cause of death. COPD is a disease that is both preventable and treatable. COPD is a major cause of morbidity and death in the world, with an increasing burden due to epidemiologic changes that expose more of the population to COPD risk factors. Although the major environmental risk for COPD is tobacco smoking, only 20% of smokers develop COPD. Indoor air pollution from burning biomass fuel is associated with increased risk of COPD in developing countries. In the United States, COPD is the fourth leading cause of death6 and is exceeded only by heart attacks, cancer, and stroke.
COPD has had a similar effect on health and mortality rate throughout the developed and underdeveloped sectors of the world, and many of the important issues surrounding COPD in the United States apply elsewhere.7
The real prevalence is masked by the burden of undiagnosed COPD; when the British Lung Foundation “missing millions” campaign performed spirometry screening in 3802 adults, they found that the prevalence of COPD was 10.2% (4.4% having GOLD II or worse) with only a quarter having a prior diagnosis.8
Pathophysiology
The main pathophysiologic feature in COPD is the limitation to expiratory flow.9 Chronic expiratory flow limitation and hyperinflation are the mechanical hallmarks of COPD.10 Expiratory airflow limitation results from many factors; among them, narrowing of the peripheral airways,11 mucus hypersecretion,12 and impaired ciliary clearance13 are the most important factors.
Several mechanistic concepts have been implicated in the pathogenesis of COPD. First, the hallmark of COPD is development of exaggerated chronic inflammation in the lung in response to inhalation of cigarette smoke compared with smokers without lung disease.14 Host factors including genetic susceptibility, epigenetic changes, and oxidative stress contribute by amplifying inflammation induced by cigarette smoke. Second, patients with deficiency of α1-antitrypsin, the main inhibitor of neutrophil elastase, develop emphysema early in life owing to an increase in proteolytic activity.15 Third, an imbalance between oxidants and antioxidants in the lungs of patients with COPD, resulting in excessive oxidative stress, not only amplifies airway inflammation in smokers but also induces cell death of structural cells in the lung. Disruption of the balance between cell death and replenishment of structural cells in the lung contributes to the destruction of alveolar septa, leading to emphysema.16 Autoimmunity has been proposed as a late pathogenic event in the progressive course of the disease.
COPD is characterized by a specific pattern of inflammation involving increased numbers of CD8+ TC lymphocytes present only in smokers who develop the disease. These cells as well as neutrophils and macrophages release inflammatory mediators and interact with epithelial cells in the airways, lung parenchyma, and pulmonary endothelium; all these relationships and interactions produce structural changes in the lungs by activation of growth factors.17 Changes in the airway architecture, endothelium, and lung parenchyma lead to the characteristic physiologic abnormalities and symptoms of COPD.
In acute exacerbations there is an increase in sputum neutrophil numbers as well as an increase in neutrophils in bronchial biopsies, which rarely are seen in the stable state.18 Interestingly virally induced exacerbations are associated with increased expression of eosinophils in sputum. Viral infections induce the expression of chemokine (C-C motif) ligand 5 (CCL5) in airway epithelial cells,19 CCL5 may act synergistically with CD8+ cells to enhance the apoptosis of virally infected cells, thus leading to increased tissue destruction.20 Also, an increase in the concentration of the elastolytic enzyme matrix metalloproteinase-1 during exacerbations is consistent with evidence of elastolysis, which may provide a causal link between exacerbations and accelerated decline in lung function.21
Data from patients with AECOPD that required mechanical ventilation indicates the presence of increased central drive, dyspnea, tachypnea, reduced tidal volume, and development of hypercapnic respiratory failure, but ventilation/perfusion match remains relatively preserved.22 AECOPD appears to be characterized by increased central drive, decreased inspiratory capacity, and decreased inspiratory muscle force, perhaps secondary to dynamic hyperinflation. There is an association between increased serum levels of interleukin 6 (IL-6) and leukotriene B4 (LTB4) and the magnitude of dyspnea, respiratory rate, and inspiratory capacity, suggesting that it may be possible to detect serum changes that reflect the inflammatory burden of the exacerbation.23
Although hypercapnia depends on the severity of airflow limitation, there is considerable variability in the relationship of PaCO2 to forced expiratory volume in 1 second (FEV1) and total lung resistance, best explained by contribution of dead space and minute ventilation. In stable COPD patients with severe airflow obstruction, shallow breathing is the main factor associated with CO2 retention,24 the diaphragm is less effective than in normal subjects, and with increasing airflow obstruction and hyperinflation, the contribution of the rib cage muscle to the generation of ventilatory pressure increases.25 Abdominal muscles are recruited during expiration in patients with severe COPD, and the expiratory rise in gastric pressure is directly related to intrinsic positive end-expiratory pressure (PEEPi). During acute exacerbation, patients with severe airflow obstruction increase the inspiratory recruitment of the rib cage muscles relative to the diaphragm. This recruitment is associated with abdominal muscle contraction and a reduction in abdominal volume at end expiration, which contributes to PEEPi. Dynamic hyperinflation can be overestimated during chronic and acute airway obstruction if abdominal muscle function is not evaluated.26
The worsening gas exchange and the deterioration of the arterial blood gas values during acute exacerbations in patients with severe COPD can be explained by several factors. These factors are, in no particular order, respiratory muscle fatigue,27 increases in dead space ventilation, and alveolar hypoventilation.28 Minute ventilation may be normal early in an exacerbation, but the respiratory rate is generally increased. There is an associated increase in physiologic dead space that impairs CO2 elimination and may result in acidemia.29 The hypoxemia seen during exacerbations results from the combination of two factors: alveolar hypoventilation and, later, worsening of ventilation/perfusion matching. Increases in ventilation/perfusion heterogeneity are attributed to (1) a reduction in the effectiveness of hypoxic vasoconstriction as a protective mechanism as pulmonary artery pressure rises and vasodilatory inflammatory mediators are released, and (2) the failure to redirect perfusion away from inadequately ventilated regions because of the reduction in cross-sectional area of the pulmonary vascular bed.
Clinical Manifestations
Most COPD patients with acute exacerbations initially demonstrate some combination of increasing cough, worsening of dyspnea, increased sputum production, purulent sputum, or increase in viscosity of the sputum, rather than a deterioration noted by laboratory or respiratory function parameters (Box 39.1). Symptoms may come on slowly over several days or acutely, depending somewhat on the severity of the underlying disease. Often, patients have a history of upper respiratory tract infection. Patients generally appear in acute distress. Vital signs typically demonstrate tachycardia and tachypnea, and blood pressure can be reduced in response to the effect of PEEPi. Use of accessory inspiratory muscles may be seen with increasing severity of exacerbations. With inspiration, the diaphragm normally moves down as it contracts, forcing the abdominal contents out. With diaphragmatic fatigue, the diaphragm no longer functions as a primary muscle of inspiration but instead assists the intercostal muscles’ inspiratory effort by fixing the rib cage. This action is associated with a rise in the diaphragm, and the abdomen moves in instead of out, as it does with normal inspiration. This sign is called paradoxical breathing28 and it implies respiratory muscle fatigue and often imminent ventilatory failure and respiratory arrest.29 Wheezing and other auscultatory findings of obstruction are also present. Cyanosis is an insensitive manifestation, but when seen, it denotes severe hypoxemia. Patients with severe acute CO2 retention may present in coma.
Precipitating Factors
The most common precipitant factor is respiratory infection, either bacterial or viral.30,31 Air pollution can also precipitate exacerbations of COPD32,33; however, the cause of one third of AECOPD events cannot be identified. Some other conditions that can ether mimic or induce an exacerbation include pneumonia, arrhythmia, pneumothorax, pulmonary embolism,34 and pleural effusion. Medication failure and lack of compliance have been shown also to lead to exacerbations.
Infections
Bacteria and viruses account for the vast majority of episodes of exacerbation. Respiratory viruses are associated with 30% of exacerbations with or without a superimposed bacterial infection.35 Another study reported that up to 78% of the patients admitted into the hospital with severe AECOPD had evidence of viral or bacterial infection.19
Several studies have been conducted to investigate airway bacterial infections as etiologic factors involved in COPD exacerbations.36–38 Bronchoscopic sampling of the distal airway has demonstrated the presence of pathogenic bacteria in 50% of exacerbations. Acquisition of new strains of bacterial pathogens has been associated with more than twofold increase in the risk of AECOPD.39 At present, there appears to be an agreement that the major pathogens isolated from sputum during acute exacerbation are Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis40; however, all of these bacteria can be isolated in patients during the stable phases of COPD.41,42 Atypical bacteria, mostly Chlamydia pneumoniae, have been implicated in approximately 10% of acute exacerbations.43,44 Other potential microorganisms that should be considered include other Streptococcus species, enteric gram-negative bacilli, and Legionella.45 Bacterial colonization may be a factor increasing airway inflammation.46 There is an association between bacterial colonization and increased markers of inflammation in sputum and in the frequency of exacerbations.47
Approximately 50% of AECOPD events are associated with upper respiratory tract virus infections. Infections with rhinovirus, respiratory syncytial virus, and influenza virus have been associated with AECOPD.48 COPD patients with a history of frequent exacerbations may be more susceptible to respiratory viral infections. There is increasing recognition that many patients with exacerbations have concomitant viral and bacterial infection. Approximately a quarter of patients admitted to hospital with AECOPD have coinfection with viruses and bacteria, and those patients have more severe exacerbations.48
Environmental Factors
Environmental factors are among noninfectious causes of COPD exacerbation that should be investigated as precipitating causes.32,49,50 Epidemiologic studies have shown that hospital admissions with AECOPD increase slightly with a rise in atmospheric levels of sulfur dioxide, ozone, nitrogen dioxide, and particulates. There is convincing evidence that exposure to particulates with a 50% cutoff aerodynamic diameter of 10 µm is associated with increased hospital admissions for AECOPD.51 Air pollution is implicated as a trigger of exacerbations52; however, a direct cause-and-effect relationship has been difficult to establish over the last 50 years.53 From an epidemiologic viewpoint, definitive evidence exists regarding a role of air pollutants in the increased death rates seen in cities during periods of heavy pollution.54 The recent dramatic increase in motor vehicle traffic has produced a relative increase in the levels of newer pollutants, such as ozone and fine-particulate air pollution. Elucidation of the mechanisms of the harmful effects of these pollutants should allow improved risk assessment for the patients with airway diseases who are susceptible to the effects of these air pollutants.
Pulmonary Thromboembolism
Pulmonary thromboembolism (PTE) can precipitate acute COPD exacerbations through either impairment of gas exchange or increases in pulmonary vascular pressures.55,56 Some evidence suggests that deep venous thrombosis occurs in more than 5 million people each year. More than 500,000 people eventually develop PTE, which is the primary cause of death in more than 100,000 patients annually in the United States.57 The precise incidence of PTE in COPD is unknown. Studies in COPD patients have found pulmonary embolus in up to 50% of autopsies and in patients admitted to hospital with severe AECOPD of unknown cause; 25% had pulmonary embolism confirmed by spiral computed tomography (CT).58 PTE risk factors inherent to COPD are sedentary lifestyle, right ventricular failure, right ventricular mural thrombi, and secondary polycythemia.59 Patients with COPD have also been shown to have increased platelet aggregation and increased plasma β-thromboglobulin.60
Up to 30% of untreated thromboembolic patients die. The necessity of diagnosing PTE as the precipitating factor in acute COPD exacerbation is crucial. However, the diagnosis of PTE is extremely difficult in COPD exacerbation. Nonetheless, the approach to diagnosis is similar to that used with other patients.61 Unfortunately, most patients with COPD have an indeterminate ventilation/perfusion scan, usually making this scan unhelpful in evaluation for PTE.62 For patients with such indeterminate results (low or intermediate probability), noninvasive testing of the lower extremity should be conducted.63–65 If positive results for deep venous thrombosis are obtained, anticoagulation therapy must be initiated. It has been proposed that use of newer D-dimer assays may also have a role as a diagnostic tool; however, even with a sensitivity of 98%, specificity is problematic, with a value of 39%.66 When the diagnosis is still in doubt (intermediate-probability scan and negative leg study or low-probability scan and negative leg study with intermediate clinical probability of PTE), helical CT or conventional pulmonary angiography may be required. Even though the safety of pulmonary angiography in patients with cor pulmonale has been questioned, data from the Prospective Investigation of Pulmonary Embolism Diagnosis (PIOPED) study support both safety and accuracy in patients with COPD.61 Helical CT has become an increasingly accepted technique and is the method of choice for direct visualization of pulmonary emboli (PE). The quantitative assessment of tissue perfusion may yield more important information for patient management than the direct visualization of emboli by CT alone. Enhanced multislice helical CT with thin collimation can be used to analyze precisely the subsegmental pulmonary arteries and may identify even more distal pulmonary arteries. Recent data suggest that spiral CT may be an alternative to angiography, particularly when results are not discordant with pretest clinical probability of PE and when combined with other tests that support CT findings (D-dimer, leg ultrasound, lung scanning), and may have adequate sensitivity and specificity in the COPD population.62
Medication Failure or Noncompliance
Many acute COPD exacerbations can be explained in terms of inadequate pharmacologic therapy or noncompliance with pharmacologic therapy. These factors are likely underestimated causes of exacerbation in COPD patients. It is also possible that patients have toxic drug effects of cardiac, gastrointestinal, or metabolic nature. Certain pharmacologic drug interactions can precipitate toxicity or loss of effect of one drug, and the physician must ascertain whether newer medications for other conditions have recently been added to the patient’s therapeutic regimen.67,68
Other Causes
Clinical decompensation in patients with stable COPD may also occur as a result of acute congestive heart failure (CHF) or cardiac arrhythmia. One study found that 27% of COPD patients die as a result of coronary disease,69 likely related to risk factors such as smoking, diabetes mellitus, and vasculopathy. Heart failure may also lead to a symptomatic exacerbation of COPD; however, it may be difficult to differentiate the symptoms of CHF and those of AECOPD.70
Other causes of exacerbation include sleep-disordered breathing,71–73 vocal cord paralysis, tumor or scarring from prior intubations, and the development of spontaneous pneumothorax.74,75 Finally, pleural effusion can also produce respiratory deterioration, especially in patients with poor respiratory reserve.76
Initial Management
Because chronic airflow obstruction cannot be reversed, acute management of COPD is directed at reversible pathogenic mechanisms, including pulmonary infection, airway tissue inflammation, bronchoconstriction, and support of failing muscular function. Box 39.2 summarizes initial general management.
Oxygen
Long-term oxygen treatment has been demonstrated to significantly reduce mortality rate in patients with COPD and severe resting arterial hypoxemia.77 Oxygen therapy is indicated for patients with an arterial oxygen tension (PaO2) of less than 55 mm Hg. The therapeutic goal is to maintain oxygen saturation greater than 90% during rest, sleep, and exertion, as it is now well established that such measures increase survival and that 24 hours is more effective than 12 hours. There is a clear rationale for the use of oxygen in severe COPD. Enhancing blood oxygenation by increasing the concentration of inspired oxygen compensates for a major physiologic consequence of COPD with hypoxemia. Oxygen remains the mainstay of initial therapy in most COPD exacerbations. Relief of hypoxemia, and consequently of hypoxemic pulmonary vasoconstriction, decreases pulmonary vascular resistance, with variable effects on the ventilation/perfusion ratio.78,79 Oxygen delivery may increase as a result of increases in oxygen arterial content and anticipated improved right-sided heart function. However, one study demonstrated that relief of hypoxemia did not increase cardiac output.80
Hypercapnia is well tolerated when it is chronic.81–83 However, oxygen should be administered cautiously in patients who are chronically hypercapnic because it is known to lead to clinically significant rises in PaCO2 in select COPD patients as a result of changes in the physiologic dead space and perhaps suppression of the respiratory drive.84,85 Acute increases in PaCO2 are more likely to occur in patients with elevated baseline PaCO2. A randomized study has shown that, although oxygen administration worsened hypercapnia and respiratory acidosis, these changes were well tolerated in most patients.86 Oxygen therapy should not be withheld in acutely ill hypoxemic patients because tissue hypoxia can lead to acute organ dysfunction. However, oxygen should be initiated at a low FIO2 and slowly titrated up as necessary with vigilant monitoring to document improvement and stabilization in PaO2, with special attention paid to maintaining the oxyhemoglobin between 88% and 92% or greater without producing dangerous falls in pH as a result of rises in CO2. These dangerous rises in PaCO2 are typically associated with worsening mental status. Nasal cannulas or Venturi masks can be used to initiate a low FIO2. Either nasal cannula at a flow rate of 1 L/minute or a Venturi mask initially at the lowest setting (25%) is appropriate for initiating oxygen in patients known or suspected to be chronic CO2 retainers. However, in the presence of acute severe hypoxemia in patients with impending respiratory failure, high-flow oxygen therapy may be in the patient’s best interest, regardless of the risk for CO2 retention.87
Drug Treatment
Bronchodilators
Bronchodilator therapy has important roles in both the prevention and treatment of AECOPD. Bronchodilators are the primary treatment to alleviate patient symptoms, improve physiologic state, and prevent or reverse respiratory failure; however, its use has not been shown to improve survival. Systematic reviews have demonstrated that inhaled delivery of short-acting β2-selective agonists and anticholinergic agents have greater effect on spirometry and is the therapy of choice for AECOPD over parenteral bronchodilators.88 Bronchodilator treatment in acutely ill COPD patients has been shown to decrease inspiratory muscle loading, with an increase in FEV1 and a decrease in functional residual capacity (FRC) and dynamic hyperinflation.89 In mechanically ventilated patients, a reduction in expiratory resistance and dynamic hyperinflation (measured as a decrease in PEEPi) has been described.90
There is no evidence supporting the use of one inhaled β2-selective agonist over another. There is no difference in outcome between β2-agonist compared with ipratropium bromide and no evidence that the combination of these two drugs is any more effective in AECOPD; these results are in contrast with the greater efficacy of these combinations in stable COPD.88 The widespread use of inhaled β-agonists has been accompanied by clinical concern of cardiac complications in elderly patients and those with coronary artery disease. However, in a study performed on clinically stable COPD or asthma patients with a history of myocardial ischemia, no ischemic events, or dysrhythmias, were observed when commonly used doses of salbutamol were administered.91 To date, no clinical studies have evaluated the use of inhaled long-acting bronchodilators (either β2-agonist or anticholinergic agents) with or without inhaled corticosteroids during AECOPD.
Several studies have suggested that the combination of β2-agonists and anticholinergic agents prevent exacerbations, particularly long-acting agents.92–96 It is possible that these agents reduce exacerbation frequency as a result of the effect on reduction of dynamic hyperinflation at rest and exercise,97 as well as nonbronchodilator mechanisms, such as anti-inflammatory effect.98
Corticosteroids
COPD is recognized as an inflammatory disorder, and the severity of airway inflammation correlates with the severity of the underlying COPD.99 During exacerbations, there is a large increase in concentration of proinflammatory cells, including neutrophils. Systemic corticosteroids improve lung function significantly, shorten hospital stay, and reduce the risk for relapses as compared with placebo in patients with AECOPD.100 In the ISOLDE trial, the median exacerbation rate was reduced by 25%, and there was also a reduction in the health status deterioration.101 Systemic corticosteroids have been demonstrated to improve respiratory mechanics in mechanically ventilated patients, with a decrease in airway resistance and dynamic air-trapping.102 A meta-analysis demonstrated that the use of systemic corticosteroids was associated with significant reduction in treatment failure, defined as either clinical deterioration, withdrawal from the study due to unsatisfactory clinical improvement, or relapse of exacerbation symptoms during the follow-up period. It also showed beneficial effects in reducing the length of hospitalization by a weighted mean of 1.42 days.103
Oral corticosteroids have beneficial effects in the management of AECOPD. Prednisolone, administered at 30 mg/day for 14 days, shortened the length of hospitalization by 2 days, improved FEV1 by 60 mL/day, and accelerated recovery from symptoms.104 The majority of patients with COPD probably requires only 2 weeks with oral corticosteroids and therapy for 8 weeks produced no incremental benefits above those achieved at 2 weeks.105 One of the most important concerns regarding the use of corticosteroids in AECOPD is the possibility of confusing it with community-acquired pneumonia. Nevertheless, there is no evidence that corticosteroid use worsened the prognosis of community-acquired pneumonia if appropriate antibiotics are used; moreover, systemic corticosteroids may reduce morbidity and mortality rates in community-acquired pneumonia.106
The use of inhaled corticosteroids is associated with decrease in exacerbation events by 12% to 25%.107 Studies involving inhaled corticosteroids and long-acting β2-agonist showed reduction in exacerbation frequency to a greater extent than using either corticosteroid or long-acting β2-agonist alone.108,109 There is also a trend toward mortality rate reduction over 3 years of 17.5%, although without statistical significance.110 There is no evidence that the addition of inhaled corticosteroids to systemic corticosteroids in AECOPD has any impact either in recovery or in mortality rate.
Antibiotics
Use of antibiotics in AECOPD remains a controversial topic, with some authors recommending antibiotic therapy and others not, maybe in part because of the heterogeneity of the population studied.111,112 Compared with placebo, antibiotic use during AECOPD reduced treatment failures by 46%, defined as requiring additional antibiotics within the first 7 days or unchanged or deteriorated symptoms within 21 days. Antibiotics reduced treatment failures particularly in those patients who were hospitalized but not when they were used in ambulatory patients.113 Three clinical trials involving 181 patients demonstrated that in-hospital mortality rate can be reduced by 78% with the use of antibiotics during AECOPD.114,115 Patients who present with dyspnea and increased sputum volume or purulence or patients who require mechanical ventilation benefit from a 3- to 7-day course of oral or parenteral antibiotics.116
Although routine sputum cultures are recommended in all patients with AECOPD, invasive techniques (transtracheal aspirates,117,118 bronchoscopic aspirates, or protected specimen brushing5,119–123) are not indicated. Exceptions include culture-negative community-acquired pneumonia not responding to therapy and ventilator-associated pneumonia (VAP).124,125 Because many COPD patients have airway colonization by bacteria, without clinical signs of infection and exacerbation, there is no clear significance of a positive culture in a COPD patient; however, in the presence of exacerbation associated with an alteration in sputum character or quantity, potentially pathogenic organisms grown from sputum should be covered with an appropriate antibiotic.126 Thus, the selection of empiric antibiotics to treat AECOPD should depend on the severity of underlying disease and severity of COPD exacerbation. Therapy for more severe exacerbations should include coverage for antibiotic-resistant bacteria, such as Pseudomonas or methicillin-resistant Staphylococcus aureus.
Hemodynamic Support
Fluid Management
COPD patients often have chronic pulmonary hypertension, which may worsen with COPD exacerbation because of hypoxic vasoconstriction, dynamic lung hyperinflation, and in mechanically ventilated patients, PEEPi. This may lead to acute or worsening right ventricular failure. In one study, the prevalence of right ventricular failure in terminal COPD patients was 66% and the prevalence of left ventricular failure was only 6%.127 As in other patients with right ventricular failure, hemodynamic stability is related to maintenance of mean arterial pressure. Mean systemic pressure can potentially be increased in these patients by increasing intravascular volume or by selectively improving compliance of the pulmonary vascular bed.128
[/not-level-membership-for-critical-care-medicine-category]
