[level-membership-for-basic-science-category]
18 Chronic kidney disease and end’stage renal disease
Chronic kidney disease (CKD) is defined by a reduction in the glomerular filtration rate (GFR) and/or urinary abnormalities or structural abnormalities of the renal tract. The severity of CKD is classified from 1 to 5 depending upon the level of GFR (Table 18.1). It is a common condition affecting up to 10% of the population in Western societies and is more common in some ethnic minority populations and in females. The incidence increases exponentially with age such that some degree of CKD is almost inevitable in persons over 80 years of age. Social deprivation is also associated with a higher prevalence of CKD. The scale of CKD and the consequences for the health service has been appreciated only in the last few years.
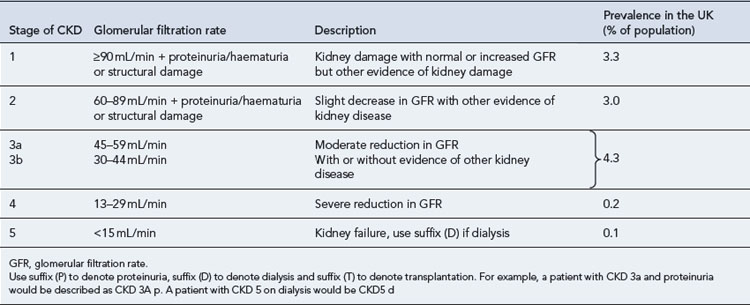
Estimates for the incidence of the various grades of CKD are shown in Table 18.1 and have been derived from large American studies, although data suggests the rates in the UK are similar (UK Renal Registry, 2008). In the past, patients with CKD were often unrecognised owing to difficulties in measuring or estimating the GFR and their health needs were largely unmet. The recent development of simple methods to estimate GFR has revealed a huge population of patients with significant kidney disease. This will pose a considerable challenge to health services in the future. National guidance on the management of CKD has been published (NICE, 2008) and includes management in primary and secondary care.
Renin-angiotensin-aldosterone system
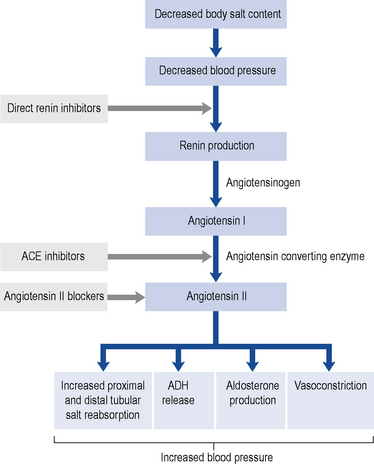
Renin promotes cleavage of the protein angiotensinogen, which is produced by the liver, to produce angiotensin I. Angiotensin I is converted to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II has two major physiological effects. First, it acts on the zona glomerulosa of the adrenal cortex to promote production of the mineralocorticoid hormone aldosterone, with resultant increased distal tubular salt and water reabsorption. Furthermore, it promotes antidiuretic hormone (ADH) release, which increases proximal tubular sodium reabsorption and promotes thirst. In combination, these lead to salt and fluid retention, high intravascular volumes, hypertension and oedema. Second, it is a direct vasoconstrictor and promotes systemic and (preferential) renal hypertension. The renal effects are predominantly on the efferent glomerular arteriole. Vasoconstriction at this site is mediated by a high density of angiotensin II receptors. When these receptors are ligated by angiotensin II, there is increased intra-glomerular pressures. Whilst this leads to an overall increase in GFR in the short-term, over a longer period glomerular hypertension promotes accelerated glomerular scarring and worsening CKD. In addition to the vascular and endocrine effects of the RAAS, it is now recognised that there is a local immune modulatory role for this system. Both resident (e.g. tubular epithelial) cells and inflammatory (monocytes and macrophages) cells synthesise components of the RAAS and are themselves targeted by the system. For example, monocytes and macrophages express the angiotensin II receptor and activation through this receptor leads to an enhanced inflammatory and fibrotic phenotype of the cell. This raises the intriguing concept that some of the effects of blocking the RAAS are due to direct anti-inflammatory and anti-fibrotic effects. Figure 18.1 shows this pathway and identifies the points at which pharmacological interventions targeted for a biological effect translates into clinical outcomes.
Measurement of renal function
MDRD glomerular filtration rate equation
Eight eGFR equations were validated for the MDRD study (Levey et al., 1999). These used demographic and serum variables (including serum creatinine level, age, gender, non-black ethnicity, higher serum urea levels, and lower serum albumin levels) in a series of equations. The four-variable equation (also known as the abbreviated (a)MDRD equation) has been adopted into clinical practice and incorporates age, creatinine, gender and ethnicity (Fig. 18.2).

Other estimates of kidney function
Creatinine clearance
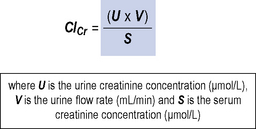
This is similar to the GFR as nearly all the filtered creatinine appears in the urine. It is a measurement of the volume of blood that is cleared of creatinine with time. Measurements of creatinine clearance (ClCr) require accurate collection of 24 h urine samples with a serum creatinine sample midway through this period. This is time-consuming, inconvenient and prone to inaccuracy and as such is now rarely used in clinical practice. Figure 18.3 shows the equation for measuring creatinine clearance.
Cockroft–Gault equation
The Cockroft–Gault equation uses weight, sex and age to estimate creatinine clearance and was derived using average population data (Cockroft and Gault, 1976). The equation is shown in Fig. 18.4.

Estimates of glomerular filtration rate in paediatric patients
Estimates of GFRs in paediatric patients can be made using the Schwartz formula (Schwartz, 1985) or the Counahan–Barratt method (Counahan et al., 1976) which both rely upon inclusion of the height of the child in estimating creatinine clearance, since height correlates with muscle mass.
Significance of CKD
Patients with CKD 1–3 (Table 18.1) are frequently asymptomatic. The reduction of GFR is insufficient to cause uraemic symptoms and any minor abnormalities in the urine such as proteinuria or haematuria are usually not noticed by patients. There is a frequent association with high blood pressure which may be the cause or a consequence of renal damage. Recognition of these patients is important as it allows early modification of traditional cardiovascular risk factors. These patients should be investigated to determine if there is a treatable cause for their CKD and followed up to identify those individuals with progressive disease.
Patients with CKD stages 4 and 5 (Table 18.1) should usually be followed up in a nephrology clinic because they will require specialist management of the complications of CKD such as anaemia and bone disease, whilst many will also be undergoing preparation for renal replacement therapy.
Causes of CKD
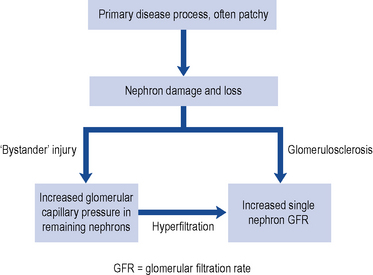
The reduction in renal function observed in CKD results from damage to the infrastructure of the kidney in discrete areas rather than throughout the kidney. The nephron is the functional unit of the kidney and while the mechanism of damage depends on the underlying cause of renal disease, as nephrons become damaged and fail, remaining nephrons compensate for loss of function by hyperfiltration secondary to raised intra-glomerular pressure. This causes ‘bystander’ damage with secondary nephron loss. This vicious cycle is illustrated in Fig. 18.5. The patient remains well until so many nephrons are lost that the GFR can no longer be maintained despite activation of compensatory mechanisms. As a consequence there is a progressive decline in kidney function.
CKD arises from a variety of causes (Table 18.2), although by the time a patient has established CKD it may not be possible to identify the exact cause. However, attempting to establish the cause is useful in the identification and elimination of reversible factors, to plan for likely outcomes and treatment needs, and for appropriate counselling when a genetic basis is established. The causes of CKD listed in Table 18.2 are ordered according to prevalence. It is important to note the prevalence of these factors is different in CKD and end stage renal disease. In end stage renal disease, diseases such as adult polycystic kidney disease (APKD) are overrepresented and ischaemic/hypertensive nephropathy underrepresented. The reasons for this are that individuals with APKD are likely to survive to reach end stage renal disease while those with diabetes or ischaemic renal damage may succumb to cardiovascular disease before end stage renal disease is reached.
Table 18.2 Primary diagnosis in renal replacement therapy patients by age and gender (Renal Registry, 2008)
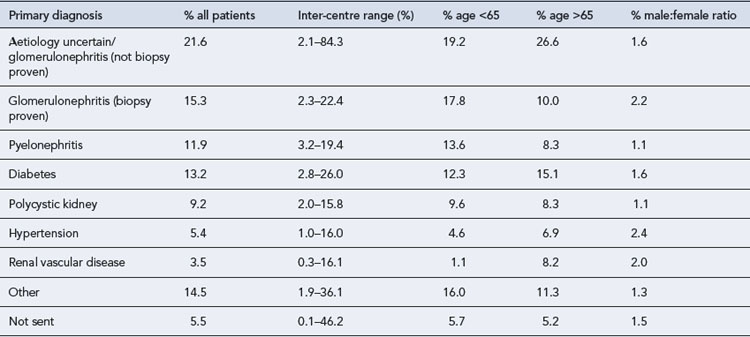
Ischaemic/hypertensive renal disease
Ischaemic nephropathy has traditionally been referred to under perfusion of the kidneys caused by renal artery stenosis. This explanation has fallen from favour recently and this term is now taken to mean impairment of renal function beyond occlusion of the main renal arteries. Hypertension results in atherosclerosis which can cause occlusive renovascular disease and small vessel damage. In patients with significant large vessel occlusive disease arteriolar nephrosclerosis, interstial fibrosis and glomerular collapse may be present (Lerman and Textor, 2001). These diagnoses account for around 30% of CKD and a smaller proportion of end stage renal disease. The effective management of hypertension is crucial to reduce renal damage.
Metabolic diseases
Diabetes mellitus is the most common metabolic disease that leads to CKD, whilst the predominant lesion is glomerular and referred to as diabetic nephropathy. Diabetes accounts for around 13% of CKD (see Table 18.2) and is associated with faster renal deterioration than other pathologies: these patients are at very significant cardiovascular risk by virtue of both CKD and diabetes. Both type 1 and 2 diabetes can result in diabetic nephrophy, patients with type 1 diabetes usually present with renal complications at a younger age and may benefit from combined kidney and pancreas transplantation. Patients with diabetes may present with no proteinuria, micro albuminuria or overt proteinuria, though as the level of proteinuria increases the GFR usually declines and in many patients this represents an inexorable decline towards end stage renal disease.
Clinical manifestations
End stage renal disease is characterised by the requirement of renal replacement therapy to sustain life and it is often accompanied by uraemia, anaemia, acidosis, osteodystrophy, neuropathy and is frequently accompanied by hypertension, fluid retention and susceptibility to infection (Fig. 18.6). It results from a significant reduction in the excretory, homeostatic, metabolic and endocrine functions of the kidney that occur over a period of months or years.
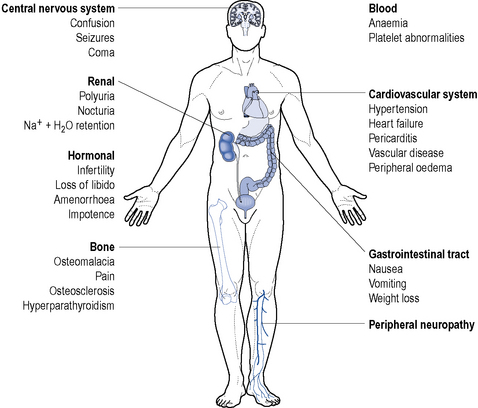
In the following section, the clinical features of CKD are described, along with the pathogenesis.
Bone disease (renal osteodystrophy)
Renal osteodystrophy describes the four types of bone disease associated with CKD:
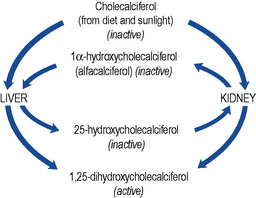
Cholecalciferol, the precursor of active vitamin D, is both absorbed from the gastro-intestinal tract and produced in the skin by the action of sunlight. Production of active vitamin D, 1,25-dihydroxycholecalciferol (calcitriol), requires the hydroxylation of the colecalciferol molecule at both the 1α and the 25 position (Fig. 18.7).
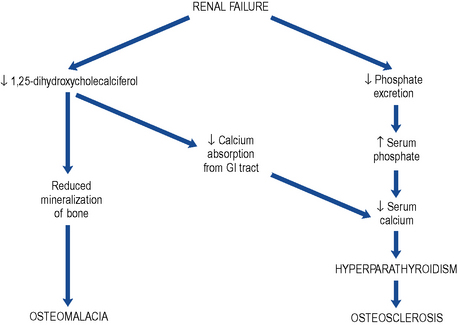
The deficiency in vitamin D with the consequent reduced calcium absorption from the gut in combination with the reduced renal tubular reabsorption results in hypocalcaemia (Fig. 18.8).
Electrolyte disturbances
Sodium
Serum sodium levels can be relatively normal even when creatinine clearance is very low. However, patients may exhibit hypo- or hypernatraemia depending upon the condition and therapy employed (see Table 18.3).
Table 18.3 Causes and mechanism of serum sodium abnormalities in chronic kidney disease
| Mechanism | Cause/effect | |
|---|---|---|
| Hypernatraemia | Sodium overload | Drugs, for example, antibiotic sodium salts |
| Hypotonic fluid loss | Osmotic diuresis | |
| Sweating | ||
| ↓ Water intake | Unconsciousness | |
| Hyponatraemia | Dilution by intracellular water movement | Mannitol |
| Hyperglycaemia | Water overload | Acute dilution by intravenous fluids, for example, 5% dextrose infusion |
| Excessive intake | ||
| Congestive cardiac failure | ||
| Nephrotic syndrome |
Potassium
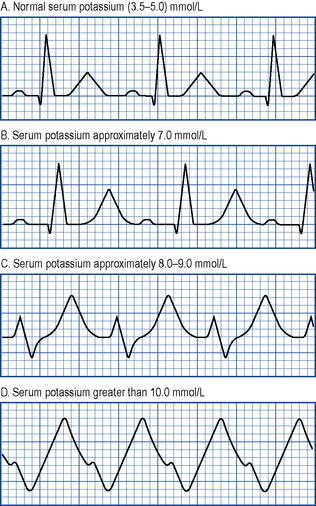
ECG changes accompany any rise in serum potassium and become more pronounced as levels increase. T waves peak (‘tenting’), there is a reduction in the size of P waves, an increase in the PR interval and a widening of the QRS complex. P waves eventually disappear and the QRS complex becomes even wider. Ultimately, the ECG assumes a sinusoidal appearance prior to cardiac arrest (see Fig. 18.10).
Diagnosis, investigations and monitoring
Intravenous urography
Graphical plots of glomerular filtration rate
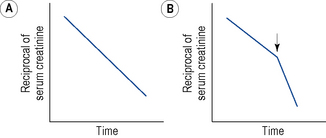
All patients with CKD should have their serum biochemistry and haematology monitored regularly to detect any of the sequelae of the disease. In some patients with CKD, the decline in renal function progresses at a constant rate and may be monitored by plotting the estimated GFR against time (Fig. 18.11). The intercept with the x-axis indicates the time at which renal function will fall to zero and can be used to predict when the GFR will reach approximately 10 mL/min, that is, the level at which renal replacement therapy should be initiated (Fig. 18.11A). If an abrupt decline in the slope of the reciprocal plot is noted (Fig. 18.11B), this indicates a worsening of the condition or the presence of an additional renal insult. The cause should be detected and remedied if possible. It is, however, increasingly recognised that most patients with CKD do not sustain a predictable decline. Many stabilise or follow a path of episodes of accelerated decline followed by months or years of stability. These observations are of great interest and are an increasing focus of clinical research.
Treatment
The aims of the treatment of CKD can be summarised as follows:
Hypertension
The drugs used to treat hypertension in renal disease are generally the same as those used in other forms of hypertension, although allowance must be made for the effect of renal failure on drug disposition (NICE, 2008).
Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers
Diuretics
Diuretics are of use in patients with salt and volume overload, which is usually indicated by the presence of oedema. This type of hypertension may be particularly difficult to treat. The choice of agent is generally limited to a loop diuretic. Potassium sparing diuretics are usually contraindicated owing to the risks of developing hyperkalaemia, and thiazides become ineffective as renal failure progresses. In combination with ACE inhibitors, spironolactone can significantly reduce proteinuria; however, the combination of these agents clearly raises the risk of significant hyperkalaemia and care must be taken (Bianchi et al., 2005). The combination should be avoided when the eGFR falls to <30 mL/min.
Management of symptoms associated with CKD
Dietary modifications in CKD
Fluid retention
Potassium restriction
Hyperkalaemia often occurs in CKD and may cause life-threatening cardiac arrhythmias. If untreated, asystolic cardiac arrest and death may result. Patients are often put on a potassium-restricted diet by avoiding potassium-rich foods such as fruit and fruit drinks, vegetables, chocolate, beer, instant coffee and ice cream. Many medicines have a high potassium content, for example, potassium citrate mixture, some antibiotics and ispaghula husk sachets. The use of these drugs is less of a problem in dialysed patients. Emergency treatment is necessary if the serum potassium level is above 7.0 mmol/L or if there are ECG changes. The most effective treatment is dialysis but if this is not available other measures may be tried (see Chapter 17).
Anaemia
There have been several studies of ESAs which have shown an increased risk of cardiovascular morbidity and overall mortality in people treated to a target >12.5 g/dL (Phrommintikul et al., 2007). This has lead to more conservative dosing strategies and prompt discontinuation or reduction of dose in patients with Hb >12.5 g/dL.
Osteodystrophy
Vitamin D deficiency and hyperparathyroidism
Cinacalcet is a calcimimetic which increases the sensitivity of calcium sensing receptors to extracellular calcium ion, this results in reduced PTH production. The benefit of this treatment is the suppression of PTH without resultant hypercalcaemia. It is recommended (NICE, 2007) for use as an alternative to parathyroidectomy for patients who are not fit enough to undergo this procedure. Common therapeutic problems in chronic renal failure are summarised in Table 18.4.
Table 18.4 Common therapeutic problems in chronic renal failure
| Problem | Comment |
|---|---|
| Drug choice | Care with choice/dose of all drugs. Care to avoid renotoxic agents pre-dialysis to preserve function. Beware herbal therapies as some contain immune system boosters (reverse immunosuppressant effects) and some are nephrotoxic |
| Drug excretion | CKD will lead to accumulation of drugs and their active metabolites if they are normally excreted by the kidney |
| Dietary restrictions | Restrictions on patient often severe. Fluid allowance includes foods with high water content, for example, gravy, custard, and fruit |
| Hypertension | Frequently requires complex multiple drug regimens. CCBs can cause oedema that might be confused with fluid overload |
| Analgesia | Side-effects are increased. Initiate with low doses and gradually increase. Avoid pethidine as metabolites accumulate. Avoid NSAIDs unless specialist advice available |
| Anaemia | Epoetin requires sufficient iron stores to be effective. Absorption from oral iron supplements may be poor and i.v. iron supplementation might be required. Care required to make sure that epoetin use does not produce hypertension |
| Immunosuppression | Use of live vaccines should be avoided (BCG, MMR, mumps, oral polio, oral typhoid, smallpox, yellow fever) |
| Pruritis (itching) | Can be severe. Treat with chlorphenamine; less sedating antihistamines often less effective. Some relief with topical agents, for example, crotamiton |
| Restless legs | Involuntary jerks can prevent sleep. Clonazepam 0.5–1 mg at night may help |
Renal transplantation
Immunosuppressants
The major pharmacological groups of immunosuppressive agents are summarised in Table 18.5.
Table 18.5 Mechanism of action of immunosuppressants commonly used following renal transplantation
| Drug | Mechanism | Comment |
|---|---|---|
| Steroids | Bind to steroid receptors and inhibit gene transcription and function of T-cells, macrophages and neutrophils | Prophylaxis against and reversal of rejection |
| Ciclosporin | Forms complex with intracellular protein cyclophilin → inhibits calcineurin. Ultimately inhibits interleukin-2 synthesis and T-cell activation | Long-term maintenance therapy against rejection |
| Tacrolimus | Forms complex with an intracellular protein → inhibits calcineurin | Long-term maintenance therapy against rejection Rescue therapy in severe or refractory rejection |
| Sirolimus | Inhibits interleukin-2 cell signalling → blocks T-cell cycling and inhibits B-cells | Usually used in combination with ciclosporin ± steroids |
| Mycophenolate | Inhibits inosine monophosphate dehydrogenase → reduces nucleic acid synthesis → inhibits T- and B-cell function | Usually used in combination with ciclosporin/tacrolimus ± steroids |
| Azathioprine | Incorporated as a purine in DNA → inhibits lymphocyte and neutrophil proliferation | Usually used in combination with ciclosporin/tacrolimus ± steroids |
| Muromonab (OKT3, mouse monoclonal anti-CD3) | Binds to CD3 complex → blocks, inactivates or kills T-cell. Short t1/2 | Prophylaxis against rejection Reversal of severe rejection |
| Polyclonal horse/rabbit antithymocyte or antilymphocyte globulin (ATG, ALG) | Antibodies against lymphocycte proteins → alter T- and B-cell activity | Prophylaxis against rejection Reversal of severe rejection |
| Humanised or chimaeric anti-CD25 (basiliximab and daclizumab) | Monoclonal antibodies that bind CD25 in interleukin-2 complex → prevent T-cell proliferation | Prophylaxis against acute rejection in combination with ciclosporin and steroids |
The commonest combination used at induction is the calcineurin inhibitor (CNI) tacrolimus, the anti-proliferative agent mycophenolate mofetil (MMF) and corticosteroids. Most patients also receive antibody induction. The antibody that is most commonly used is a monoclonal anti-CD25 antibody for people at low or medium immunological risk and anti-T-cell polyclonal antibodies (thymoglobulin or ATG) for people at high immunological risk. Patients at high immunological risk include: those who have lost a previous transplant because of rejection; the presence of preformed circulating anti-HLA antibodies at the time of transplantation (sensitisation); and major HLA mismatches (particularly at HLA-DR) between donor and recipient. Guidelines for the use of immunosuppressive therapy in kidney transplant patients have been issued (NICE, 2004). More recent international consensus guidelines recommend use of newer agents such as MMF and emphasise the use of tacrolimus (rather than ciclosporin) as the CNI of choice. Tacrolimus is associated with less acute rejection than ciclosporin and may be associated with better graft function at one year and less graft loss (Knoll and Bell, 1999). However, there is no overwhelming evidence as yet that patients who receive tacrolimus as a CNI from induction have a survival benefit compared to patients who receive ciclosporin. It should be noted that generic/proprietary formulations of some drugs (e.g. tacrolimus and ciclosporin) are not interchangeable.
Ciclosporin
Ciclosporin interacts with a number of drugs that either lead to a reduction in ciclosporin levels, increase the risk of rejection or cause an elevation in ciclosporin levels leading to increased toxicity. Some drugs enhance the nephrotoxicity of ciclosporin (Box 18.2).
Monoclonal antibodies
The humanised or chimeric anti-CD25 monoclonal antibodies basiliximab and daclizumab are clinically similar and bind to CD25 in the IL-2 complex of activated T-lymphocytes. This renders all T-cells resistant to IL-2 and therefore prevents T-cell proliferation. They are used as prophylaxis against acute rejection in combination with CNIs and steroids (NICE, 2004). Daclizumab is currently not available in the UK.
Implementation of regular dialysis treatment
The principle of dialysis is simple. The patient’s blood and a dialysis solution are positioned on opposing sides of a semi-permeable membrane across which exchange of metabolites occurs. The two main types of dialysis used in CKD are haemodialysis and peritoneal dialysis. Neither has been shown to be superior to the other in any particular group of patients and so the personal preference of the patient is important when selecting dialysis modality. Haemodialysis and acute peritoneal dialysis are discussed in Chapter 17.
The various techniques of haemofiltration, a technique rela- ted to haemodialysis, are also discussed in detail in Chapter 17.
Case 18.1
| Reference range | ||
|---|---|---|
| Sodium | 137 mmol/L | (135–145) |
| Potassium | 4.8 mmol/L | (3.5–5.0) |
| Phosphate | 2.5 mmol/L | (0.9–1.5) |
| Calcium | 1.6 mmol/L | (2.20–2.55) |
| Urea | 52 mmol/L | (3.0–6.5) |
| Creatinine | 620 μmol/L | (50–120) |
| Haemoglobin | 7.5 g/dL | (13.5–18.0) |
Case 18.3
| Reference range | ||
|---|---|---|
| Haemoglobin | 5.6 g/dL | (13.5–17.5) |
| Red cell count | 2.92 × 109 L−1 | (4.5–6.5 × 109 L−1) |
| Haematocrit | 0.208 | (0.40–0.54) |
| Serum ferritin | 88.0 μcg/L | (15–300) |
Bianchi S., Bigazzi R., Campese V.M. Antagonists of aldosterone and proteinuria in patients with chronic kidney disease: a controlled pilot study. Am. J. Kidney Dis.. 2005;46:45-51.
Cockroft D., Gault M. Predication of creatinine clearance from serum creatinine. Nephron. 1976;16:31-34.
Counahan, R. Chantler C., Ghazali S., et al. Estimation of glomerular filtration from plasma creatinine concentration in children. Arch. Dis. Child. 1976;51:875-878.
Knoll G.A., Bell R.C. Tacrolimus versus ciclosporin for immunosuppression in renal transplant: meta-analysis of randomized trials. Br. Med. J.. 1999;318:1104-1107.
Lerman L., Textor S.C. Pathophysiology of ischaemic nephropathy. Urol. Clin. North Am.. 2001;28:793-803.
Levey A.S., Bosch J.P., Lewis J.B., et al. A more accurate method to estimate glomerular function rate from serum creatinine: a new prediction equation. Ann. Intern. Med.. 1999;130:461-470.
National Institute for Health and Clinical Excellence. Immunosuppressive Therapy for Renal Transplantation in Adults. London: NICE, 2004. Technology Appraisal 85. Available at: http://guidance .nice.org.uk/TA85
National Institute for Health and Clinical Excellence. Cinacalcet for the Treatment of Secondary Hyperparathyroidism in Patients with End Stage Renal Disease on Maintenance Dialysis Therapy, Technology Appraisal 117. London: NICE. 2007. Available at: http://guidance.nice.org.uk/TA117/Guidance/doc/English
National Institute for Health and Clinical Excellence. Chronic Kidney Disease: Early Identification and Management of Chronic Kidney Disease in Adults in Primary and Secondary Care. London: NICE, 2008. Clinical Guideline 73 Available at http://www.nice.org.uk/nicemedia/pdf/CG073NICEGuideline.pdf
Phrommintikul A., Haas S.J., Elsik M., et al. Mortality and target haemoglobin concentrations in anaemic patients with chronic kidney disease treated with erythropoietin: a meta-analysis. Lancet. 2007;369:381-388.
Schwartz G.J. A simple estimate of glomerular filtration rate in adolescent boys. J. Pediatr.. 1985;106:522-526.
UK Renal Registry. The 11th Annual Report. Bristol: Renal Association, 2008.
Daugirdas, J.T., Blake P.G., Ing S.T. Handbook of Dialysis, fourth ed. Philadelphia: Lippincott Williams & Wilkins, 2007.
Feehally J., Floege J., Johnson R.J. Comprehensive Clinical Nephrology, third ed. London: Mosby, 2007.
Go A.S., Chertow G.M., Fan D., et al. Chronic kidney disease and the risks of death, cardiovascular events and hospitalisation. N. Engl. J. Med.. 2004;351:1296-1305.
Steddon S., Ashman N., Chesser A., et al. Oxford Handbook of Nephrology and Hypertension. Oxford: Oxford University Press, 2007.
The ONTARGET investigators. Telmisartan, ramipril, or both in patients at high risk of vascular events. N. Engl. J. Med.. 2008;358:1547-1559.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
18 Chronic kidney disease and end’stage renal disease
Chronic kidney disease (CKD) is defined by a reduction in the glomerular filtration rate (GFR) and/or urinary abnormalities or structural abnormalities of the renal tract. The severity of CKD is classified from 1 to 5 depending upon the level of GFR (Table 18.1). It is a common condition affecting up to 10% of the population in Western societies and is more common in some ethnic minority populations and in females. The incidence increases exponentially with age such that some degree of CKD is almost inevitable in persons over 80 years of age. Social deprivation is also associated with a higher prevalence of CKD. The scale of CKD and the consequences for the health service has been appreciated only in the last few years.
Estimates for the incidence of the various grades of CKD are shown in Table 18.1 and have been derived from large American studies, although data suggests the rates in the UK are similar (UK Renal Registry, 2008). In the past, patients with CKD were often unrecognised owing to difficulties in measuring or estimating the GFR and their health needs were largely unmet. The recent development of simple methods to estimate GFR has revealed a huge population of patients with significant kidney disease. This will pose a considerable challenge to health services in the future. National guidance on the management of CKD has been published (NICE, 2008) and includes management in primary and secondary care.
Renin-angiotensin-aldosterone system
Renin promotes cleavage of the protein angiotensinogen, which is produced by the liver, to produce angiotensin I. Angiotensin I is converted to angiotensin II by angiotensin-converting enzyme (ACE). Angiotensin II has two major physiological effects. First, it acts on the zona glomerulosa of the adrenal cortex to promote production of the mineralocorticoid hormone aldosterone, with resultant increased distal tubular salt and water reabsorption. Furthermore, it promotes antidiuretic hormone (ADH) release, which increases proximal tubular sodium reabsorption and promotes thirst. In combination, these lead to salt and fluid retention, high intravascular volumes, hypertension and oedema. Second, it is a direct vasoconstrictor and promotes systemic and (preferential) renal hypertension. The renal effects are predominantly on the efferent glomerular arteriole. Vasoconstriction at this site is mediated by a high density of angiotensin II receptors. When these receptors are ligated by angiotensin II, there is increased intra-glomerular pressures. Whilst this leads to an overall increase in GFR in the short-term, over a longer period glomerular hypertension promotes accelerated glomerular scarring and worsening CKD. In addition to the vascular and endocrine effects of the RAAS, it is now recognised that there is a local immune modulatory role for this system. Both resident (e.g. tubular epithelial) cells and inflammatory (monocytes and macrophages) cells synthesise components of the RAAS and are themselves targeted by the system. For example, monocytes and macrophages express the angiotensin II receptor and activation through this receptor leads to an enhanced inflammatory and fibrotic phenotype of the cell. This raises the intriguing concept that some of the effects of blocking the RAAS are due to direct anti-inflammatory and anti-fibrotic effects. Figure 18.1 shows this pathway and identifies the points at which pharmacological interventions targeted for a biological effect translates into clinical outcomes.
Measurement of renal function
MDRD glomerular filtration rate equation
Eight eGFR equations were validated for the MDRD study (Levey et al., 1999). These used demographic and serum variables (including serum creatinine level, age, gender, non-black ethnicity, higher serum urea levels, and lower serum albumin levels) in a series of equations. The four-variable equation (also known as the abbreviated (a)MDRD equation) has been adopted into clinical practice and incorporates age, creatinine, gender and ethnicity (Fig. 18.2).
Other estimates of kidney function
Creatinine clearance
This is similar to the GFR as nearly all the filtered creatinine appears in the urine. It is a measurement of the volume of blood that is cleared of creatinine with time. Measurements of creatinine clearance (ClCr) require accurate collection of 24 h urine samples with a serum creatinine sample midway through this period. This is time-consuming, inconvenient and prone to inaccuracy and as such is now rarely used in clinical practice. Figure 18.3 shows the equation for measuring creatinine clearance.
Cockroft–Gault equation
The Cockroft–Gault equation uses weight, sex and age to estimate creatinine clearance and was derived using average population data (Cockroft and Gault, 1976). The equation is shown in Fig. 18.4.
Estimates of glomerular filtration rate in paediatric patients
Estimates of GFRs in paediatric patients can be made using the Schwartz formula (Schwartz, 1985) or the Counahan–Barratt method (Counahan et al., 1976) which both rely upon inclusion of the height of the child in estimating creatinine clearance, since height correlates with muscle mass.
Significance of CKD
Patients with CKD 1–3 (Table 18.1) are frequently asymptomatic. The reduction of GFR is insufficient to cause uraemic symptoms and any minor abnormalities in the urine such as proteinuria or haematuria are usually not noticed by patients. There is a frequent association with high blood pressure which may be the cause or a consequence of renal damage. Recognition of these patients is important as it allows early modification of traditional cardiovascular risk factors. These patients should be investigated to determine if there is a treatable cause for their CKD and followed up to identify those individuals with progressive disease.
Patients with CKD stages 4 and 5 (Table 18.1) should usually be followed up in a nephrology clinic because they will require specialist management of the complications of CKD such as anaemia and bone disease, whilst many will also be undergoing preparation for renal replacement therapy.
Causes of CKD
The reduction in renal function observed in CKD results from damage to the infrastructure of the kidney in discrete areas rather than throughout the kidney. The nephron is the functional unit of the kidney and while the mechanism of damage depends on the underlying cause of renal disease, as nephrons become damaged and fail, remaining nephrons compensate for loss of function by hyperfiltration secondary to raised intra-glomerular pressure. This causes ‘bystander’ damage with secondary nephron loss. This vicious cycle is illustrated in Fig. 18.5. The patient remains well until so many nephrons are lost that the GFR can no longer be maintained despite activation of compensatory mechanisms. As a consequence there is a progressive decline in kidney function.
CKD arises from a variety of causes (Table 18.2), although by the time a patient has established CKD it may not be possible to identify the exact cause. However, attempting to establish the cause is useful in the identification and elimination of reversible factors, to plan for likely outcomes and treatment needs, and for appropriate counselling when a genetic basis is established. The causes of CKD listed in Table 18.2 are ordered according to prevalence. It is important to note the prevalence of these factors is different in CKD and end stage renal disease. In end stage renal disease, diseases such as adult polycystic kidney disease (APKD) are overrepresented and ischaemic/hypertensive nephropathy underrepresented. The reasons for this are that individuals with APKD are likely to survive to reach end stage renal disease while those with diabetes or ischaemic renal damage may succumb to cardiovascular disease before end stage renal disease is reached.
Table 18.2 Primary diagnosis in renal replacement therapy patients by age and gender (Renal Registry, 2008)
Ischaemic/hypertensive renal disease
Ischaemic nephropathy has traditionally been referred to under perfusion of the kidneys caused by renal artery stenosis. This explanation has fallen from favour recently and this term is now taken to mean impairment of renal function beyond occlusion of the main renal arteries. Hypertension results in atherosclerosis which can cause occlusive renovascular disease and small vessel damage. In patients with significant large vessel occlusive disease arteriolar nephrosclerosis, interstial fibrosis and glomerular collapse may be present (Lerman and Textor, 2001). These diagnoses account for around 30% of CKD and a smaller proportion of end stage renal disease. The effective management of hypertension is crucial to reduce renal damage.
Metabolic diseases
Diabetes mellitus is the most common metabolic disease that leads to CKD, whilst the predominant lesion is glomerular and referred to as diabetic nephropathy. Diabetes accounts for around 13% of CKD (see Table 18.2) and is associated with faster renal deterioration than other pathologies: these patients are at very significant cardiovascular risk by virtue of both CKD and diabetes. Both type 1 and 2 diabetes can result in diabetic nephrophy, patients with type 1 diabetes usually present with renal complications at a younger age and may benefit from combined kidney and pancreas transplantation. Patients with diabetes may present with no proteinuria, micro albuminuria or overt proteinuria, though as the level of proteinuria increases the GFR usually declines and in many patients this represents an inexorable decline towards end stage renal disease.
Clinical manifestations
End stage renal disease is characterised by the requirement of renal replacement therapy to sustain life and it is often accompanied by uraemia, anaemia, acidosis, osteodystrophy, neuropathy and is frequently accompanied by hypertension, fluid retention and susceptibility to infection (Fig. 18.6). It results from a significant reduction in the excretory, homeostatic, metabolic and endocrine functions of the kidney that occur over a period of months or years.
In the following section, the clinical features of CKD are described, along with the pathogenesis.
Bone disease (renal osteodystrophy)
Renal osteodystrophy describes the four types of bone disease associated with CKD:
Cholecalciferol, the precursor of active vitamin D, is both absorbed from the gastro-intestinal tract and produced in the skin by the action of sunlight. Production of active vitamin D, 1,25-dihydroxycholecalciferol (calcitriol), requires the hydroxylation of the colecalciferol molecule at both the 1α and the 25 position (Fig. 18.7).
The deficiency in vitamin D with the consequent reduced calcium absorption from the gut in combination with the reduced renal tubular reabsorption results in hypocalcaemia (Fig. 18.8).
Electrolyte disturbances
Sodium
Serum sodium levels can be relatively normal even when creatinine clearance is very low. However, patients may exhibit hypo- or hypernatraemia depending upon the condition and therapy employed (see Table 18.3).
Table 18.3 Causes and mechanism of serum sodium abnormalities in chronic kidney disease
| Mechanism | Cause/effect | |
|---|---|---|
| Hypernatraemia | Sodium overload | Drugs, for example, antibiotic sodium salts |
| Hypotonic fluid loss | Osmotic diuresis | |
| Sweating | ||
| ↓ Water intake | Unconsciousness | |
| Hyponatraemia | Dilution by intracellular water movement | Mannitol |
| Hyperglycaemia | Water overload | Acute dilution by intravenous fluids, for example, 5% dextrose infusion |
| Excessive intake | ||
| Congestive cardiac failure | ||
| Nephrotic syndrome |
