[level-membership-for-basic-science-category]
CHAPTER 18 Chromosome Disorders
The development of a reliable technique for chromosome analysis in 1956 soon led to the discovery that several previously described conditions were due to an abnormality in chromosome number. Within 3 years, the causes of Down syndrome (47,XX/XY, +21), Klinefelter syndrome (47,XXY), and Turner syndrome (45,X) had been established. Shortly after, other autosomal trisomy syndromes were recognized, and over the ensuing years many other multiple malformation syndromes were described in which there was loss or gain of chromosome material.
In Chapter 3, the basic principles of chromosome structure, function, and behavior during cell division were described, together with an account of chromosome abnormalities and how they can arise and be transmitted in families. In this chapter, the medical aspects of chromosome abnormalities, and some of their specific syndromes, are described.
Incidence of Chromosome Abnormalities
Chromosome abnormalities are present in at least 10% of all spermatozoa and 25% of mature oocytes. Some 15% to 20% of all recognized pregnancies end in spontaneous miscarriage, and many more zygotes and embryos are so abnormal that survival beyond the first few days or weeks after fertilization is not possible. Approximately 50% of all spontaneous miscarriages have a chromosome abnormality (Table 18.1) and the incidence of chromosomal abnormalities in morphologically normal embryos is around 20%. Chromosome abnormalities therefore account for the spontaneous loss of a very high proportion of all human conceptions.
Table 18.1 Chromosome Abnormalities in Spontaneous Abortions (Percentage Values Relate to Total of Chromosomally Abnormal Abortuses)
| Abnormality | Incidence (%) |
|---|---|
| Trisomy 13 | 2 |
| Trisomy 16 | 15 |
| Trisomy 18 | 3 |
| Trisomy 21 | 5 |
| Trisomy other | 25 |
| Monosomy X | 20 |
| Triploidy | 15 |
| Tetraploidy | 5 |
| Other | 10 |
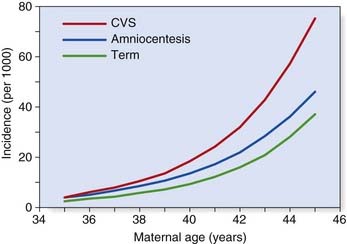
From conception onward, the incidence of chromosome abnormalities falls rapidly. By birth it has declined to a level of 0.5% to 1%, although the total is higher (5%) in stillborn infants. Table 18.2 lists the incidence figures for chromosome abnormalities based on newborn surveys. It is notable that among the commonly recognized aneuploidy syndromes, there is also a high proportion of spontaneous pregnancy loss (Table 18.3). This is illustrated by comparison of the incidence of conditions such as Down syndrome at the time of chorionic villus sampling (11 to 12 weeks), amniocentesis (16 weeks), and term (Figure 18.1).
Table 18.2 Incidence of Chromosome Abnormalities in the Newborn
| Abnormality | Incidence per 10,000 Births |
|---|---|
| Autosomes | |
| Trisomy 13 | 2 |
| Trisomy 18 | 3 |
| Trisomy 21 | 15 |
| Sex Chromosomes | |
| female births | |
| 45,X | 1–2 |
| 47,XXX | 10 |
| male births | |
| 47,XXY | 10 |
| 47,XYY | 10 |
| Other unbalanced rearrangements | 10 |
| Balanced rearrangements | 30 |
| Total | 90 |
Table 18.3 Spontaneous Pregnancy Loss in Commonly Recognized Aneuploidy Syndromes
| Disorder | Proportion Undergoing Spontaneous Pregnancy Loss (%) |
|---|---|
| Trisomy 13 | 95 |
| Trisomy 18 | 95 |
| Trisomy 21 | 80 |
| Monosomy X | 98 |
Down Syndrome (Trisomy 21)
Incidence
The overall birth incidence, when adjusted for the increasingly widespread impact of antenatal screening, is approximately 1 : 1000 in the United Kingdom, which has a national register. In the United States, the birth incidence has been estimated at approximately 1 : 800. In the United Kingdom, approximately 60% of Down syndrome cases are detected prenatally. There is a strong association between the incidence of Down syndrome and advancing maternal age (Table 18.4).
Table 18.4 Incidence of Down Syndrome in Relation to Maternal Age
| Maternal Age at Delivery (Years) | Incidence of Down Syndrome |
|---|---|
| 20 | 1 in 1500 |
| 25 | 1 in 1350 |
| 30 | 1 in 900 |
| 35 | 1 in 400 |
| 36 | 1 in 300 |
| 37 | 1 in 250 |
| 38 | 1 in 200 |
| 39 | 1 in 150 |
| 40 | 1 in 100 |
| 41 | 1 in 85 |
| 42 | 1 in 65 |
| 43 | 1 in 50 |
| 44 | 1 in 40 |
| 45 | 1 in 30 |
Adapted from Cuckle HS, Wald NJ, Thompson SG 1987 Estimating a woman’s risk of having a pregnancy associated with Down syndrome using her age and serum alpha-fetoprotein level. Br J Obstet Gynaecol 94:387–402.
Clinical Features
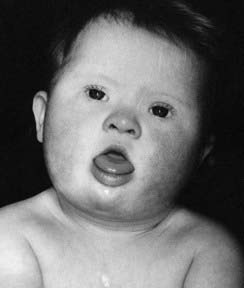
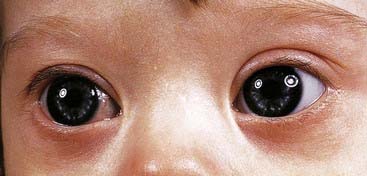
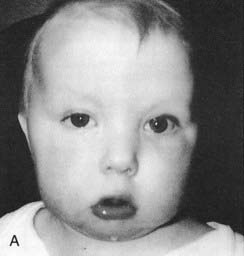
These are summarized in Box 18.1. The most common finding in the newborn period is severe hypotonia. Usually the facial characteristics of upward sloping palpebral fissures, small ears, and protruding tongue (Figures 18.2 and 18.3) prompt rapid suspicion of the diagnosis, although this can be delayed in very small or premature babies. Single palmar creases are found in 50% of children with Down syndrome (Figure 18.4), in contrast to 2% to 3% of the general population. Congenital cardiac abnormalities are present in 40% to 45% of babies with Down syndrome, with the three most common lesions being atrioventricular canal defects, ventricular septal defects, and patent ductus arteriosus.
Box 18.1
Common Findings in Down Syndrome
Craniofacial
Brachycephaly, epicanthic folds, protruding tongue, small ears, upward sloping palpebral fissures
Limbs
Single palmar crease, small middle phalanx of fifth finger, wide gap between first and second toes

Natural History
Affected children show a broad range of intellectual ability with IQ scores ranging from 25 to 75. The average IQ of young adults is around 40 to 45. Social skills are relatively well-advanced and most children are happy and very affectionate. Adult height is usually around 150 cm. In the absence of a severe cardiac anomaly, which leads to early death in 15% to 20% of cases, average life expectancy is 50 to 60 years. Most affected adults develop Alzheimer disease in later life, possibly because of a gene dosage effect—the amyloid precursor protein gene is on chromosome 21. This gene is known to be implicated in some familial cases of Alzheimer disease (p. 243).
Chromosome Findings
These are listed in Table 18.5. In cases resulting from trisomy 21, the additional chromosome is maternal in origin in more than 90% of cases, and DNA studies have shown that this arises most commonly as a result of non-disjunction in maternal meiosis I (p. 39). Robertsonian translocations (p. 47) account for approximately 4% of all cases, in roughly one-third of which a parent is found to be a carrier. Children with mosaicism are often less severely affected than those with the full syndrome.
| Abnormality | Frequency (%) |
|---|---|
| Trisomy | 95 |
| Translocation | 4 |
| Mosaicism | 1 |
Efforts have been made to correlate the various clinical features in trisomy Down syndrome with specific regions of chromosome 21, by studying children with partial trisomy for different regions. There is some support for a Down syndrome ‘critical region’ at the distal end of the long arm (21q22), because children with trisomy for this region alone usually have typical Down syndrome facial features. Chromosome 21 is a ‘gene-poor’ chromosome with a high ratio of AT to GC sequences (p. 69). At present the only reasonably well-established genotype-phenotype correlation in trisomy 21 is the high incidence of Alzheimer disease.
Recurrence Risk
For straightforward trisomy 21, the recurrence risk is related to maternal age (variable) and the simple fact that trisomy has already occurred (∼1%). The combined recurrence risk is usually between 1 : 200 and 1 : 100. In translocation cases, similar figures apply if neither parent is a carrier. In familial translocation cases, the recurrence risks vary from around 1% to 3% for male carriers up to 10% to 15% for female carriers, with the exception of very rare carriers of a 21q21q translocation, for whom the recurrence risk is 100% (p. 48).
Prenatal diagnosis can be offered based on analysis of chorionic villi or cultured amniotic cells. Prenatal screening programs have been introduced based on the so-called triple or quadruple tests of maternal serum at 16 weeks’ gestation (p. 328).
Patau Syndrome (Trisomy 13) and Edwards Syndrome (Trisomy 18)
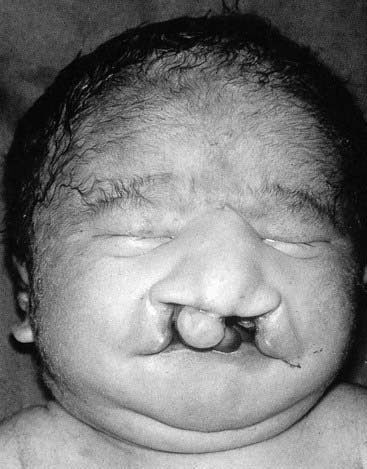
These very severe conditions were first described in 1960 and share many features in common (Figures 18.5 and 18.6). The incidence for both is approximately 1 : 5000 and prognosis is very poor, with most infants dying during the first days or weeks of life, though most cases are detected prenatally, often leading to termination. In the unusual event of longer term survival, there are severe learning difficulties. Cardiac abnormalities occur in at least 90% of cases.
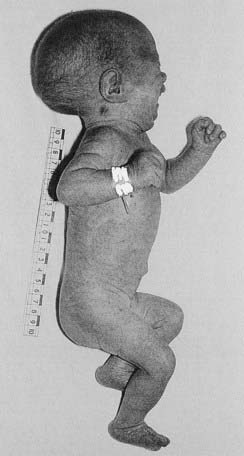
Chromosome analysis usually reveals straightforward trisomy. Both disorders occur more frequently with advanced maternal age, the additional chromosome being of maternal origin (see Table 3.4, p. 43). Approximately 10% of cases are caused by mosaicism or unbalanced rearrangements, particularly Robertsonian translocations in Patau syndrome.
Triploidy
Triploidy (69,XXX, 69,XXY, 69,XYY) is a relatively common finding in material cultured from spontaneous abortions, but is seen only rarely in a liveborn infant. Such a child almost always shows severe intrauterine growth retardation with relative preservation of head growth at the expense of a small thin trunk. Syndactyly involving the third and fourth fingers and/or the second and third toes is a common finding. Cases of triploidy resulting from a double paternal contribution usually miscarry in early to mid-pregnancy and are associated with partial hydatidiform changes in the placenta (p. 101). Cases with a double maternal contribution survive for longer but rarely beyond the early neonatal period.
Hypomelanosis of Ito
Several children with mosaicism for diploidy/triploidy have been identified. These can demonstrate the clinical picture seen in full triploidy but in a milder form. An alternative presentation occurs as the condition known as hypomelanosis of Ito. In this curious disorder, the skin shows alternating patterns of normally pigmented and depigmented streaks that correspond to the embryological developmental lines of the skin known as Blaschko’s lines (see Figure 18.7). Most children with hypomelanosis of Ito have moderate learning difficulties and convulsions that can be particularly difficult to treat. There is increasing evidence that this clinical picture represents a non-specific embryological response to cell or tissue mosaicism. A similar pattern of skin pigmentation is sometimes seen in women with one of the rare X-linked dominant disorders (p. 117) with skin involvement, such as incontinentia pigmenti (see Figure 7.18, p. 118). Such women can be considered as being mosaic, as some cells express the normal gene, whereas others express only the mutant gene.
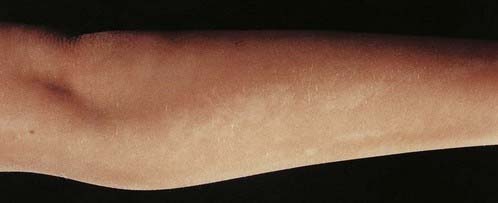
FIGURE 18.7 Mosaic pattern of skin pigmentation on the arm of a child with hypomelanosis of Ito.
(Reproduced with permission from Jenkins D, Martin K, Young ID 1993 Hypomelanosis of Ito associated with mosaicism for trisomy 7 and apparent ‘pseudomosaicism’ at amniocentesis. J Med Genet 1993; 30:783–784.)
Disorders of the Sex Chromosomes
Turner Syndrome (45,X)
This condition was first described clinically in 1938. The absence of a Barr body, consistent with the presence of only one X chromosome, was noted in 1954 and cytogenetic confirmation was forthcoming in 1959. Although common at conception and in spontaneous abortions (see Table 18.1), the incidence in liveborn female infants is low, with estimates ranging from 1 : 5000 to 1 : 10,000.
Clinical Features
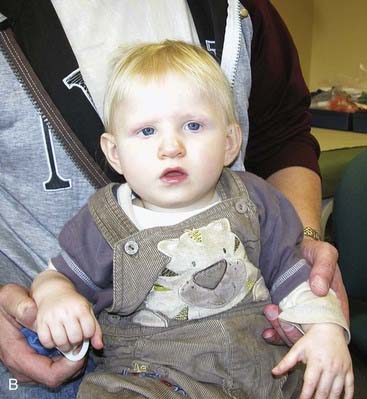
Presentation can be at any time from pregnancy to adult life. Increasingly, Turner syndrome is being detected during the second trimester as a result of routine ultrasonography, showing either generalized edema (hydrops) or swelling localized to the neck (nuchal cyst or thickened nuchal pad) (Figure 18.8). At birth many babies with Turner syndrome look entirely normal. Others show the residue of intrauterine edema with puffy extremities (Figure 18.9) and neck webbing. Other findings may include a low posterior hairline, increased carrying angles at the elbows, short fourth metacarpals, widely spaced nipples, and coarctation of the aorta, which is present in 15% of cases.
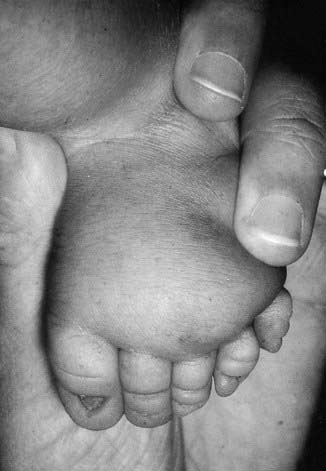
Intelligence in Turner syndrome is normal. However, studies have shown some differences in social cognition and higher order executive function skills according to whether the X chromosome was paternal or maternal in origin (p. 105). The two main medical problems are short stature and ovarian failure. The short stature becomes apparent by mid-childhood, and without growth hormone treatment the average adult height is 145 cm. This short stature is due, at least in part, to haploinsufficiency for the SHOX gene, which is located in the pseudoautosomal region (p. 118). Ovarian failure commences during the second half of intrauterine life and almost invariably leads to primary amenorrhea and infertility. Estrogen replacement therapy should be initiated at adolescence for the development of secondary sexual characteristics and long-term prevention of osteoporosis. In vitro fertilization using donor eggs offers the prospect of pregnancy for women with Turner syndrome.
Chromosome Findings
These are summarized in Table 18.6. The most common finding is 45,X (sometimes erroneously referred to as 45,XO). In 80% of cases, it arises through loss of a sex chromosome (X or Y) paternal meiosis. In a significant proportion of cases, there is chromosome mosaicism and those with a normal cell line (46,XX) have a chance of being fertile. Some cases with a 46,XY cell line are phenotypically male, and all cases with some Y-chromosome material in their second cell line must be investigated for possible gonadal dysgenesis—intracellular male gonads can occasionally become malignant and require surgical removal.
| Karyotype | Frequency (%) |
|---|---|
| Monosomy X—45,X | 50 |
| Mosaicism (e.g., 45,X/46,XX) | 20 |
| Isochromosome—46,X,i(Xq) | 15 |
| Ring—46,X,r(X) | 5 |
| Deletion—46,X,del(Xp) | 5 |
| Other | 5 |
Fragile X Syndrome
This condition, which could equally well be classified as a single gene disorder rather than a chromosome abnormality, has the unique distinction of being both the most common inherited cause of learning difficulties and the first disorder in which a dynamic mutation (triplet repeat expansion) was identified (p. 24) in 1991. It affects approximately 1 : 5000 males and accounts for 4% to 8% of all males with learning difficulties. Martin and Bell described the condition in the 1940s before the chromosome era, and it has also been known as Martin-Bell syndrome. The chromosomal abnormality was first described in 1969 but the significance not fully realized until 1977.
Clinical Features
Older boys and adult males usually have a recognizable facial appearance with high forehead, large ears, long face, and prominent jaw (Figure 18.10). After puberty most affected males have large testes (macro-orchidism). There may also be evidence of connective tissue weakness, with hyperextensible joints, stretch marks on the skin (striae) and mitral valve prolapse. The learning difficulties are moderate to severe and many show autistic features and/or hyperactive behavior. Speech tends to be halting and repetitive. Female carriers can show some of the facial features, and approximately 50% of women with the full mutation show mild-to-moderate learning difficulties.

The Fragile X Chromosome
The fragile X syndrome takes its name from the appearance of the X chromosome, which shows a fragile site close to the telomere at the end of the long arm at Xq27.3 (Figure 18.11). A fragile site is a non-staining gap usually involving both chromatids at a point at which the chromosome is liable to break. In this condition, detection of the fragile site involves the use of special culture techniques such as folate or thymidine depletion, which can result in the fragile site being detectable in up to 50% of cells from affected males. Demonstration of the fragile site in female carriers is much more difficult and cytogenetic studies alone are not a reliable means of carrier detection, in that, although a positive result confirms carrier status, the absence of the fragile site does not exclude a woman from being a carrier.
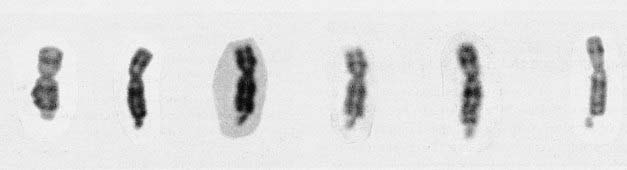
The Molecular Defect
The full mutation is unstable not only during female meiosis but also in somatic mitotic divisions. Consequently, in an affected male gel electrophoresis shows a ‘smear’ of DNA consisting of many different-sized alleles rather than a single band (Figure 18.12). Note that a normal allele and premutation can be identified by polymerase chain reaction (PCR), whereas Southern blotting is necessary to detect full mutations as the long GCC expansion is often refractory to PCR amplification. At the molecular level, a full mutation suppresses transcription of the FMR-1 gene by hypermethylation, and this in turn is thought to be responsible for the clinical features seen in males, and in some females with a large expansion (Table 18.7). The FMR-1 gene contains 17 exons encoding a cytoplasmic protein that plays a crucial role in the development and function of cerebral neurons. The FMR-1 protein can be detected in blood using specific monoclonal antibodies.
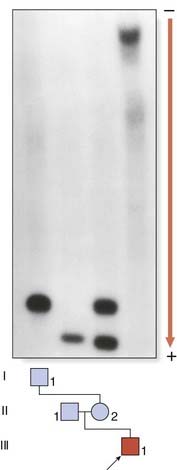
Table 18.7 Fragile X Syndrome: Genotype-Phenotype Correlations
| Number of Triplet Repeats (Normal Range 10–50) | Fragile Site | Intelligence Detectable |
|---|---|---|
| Males | ||
| 51–58 (intermediate alleles) | ||
| 59–200 (premutation) | No | Normal (normal transmitting male) |
| 200–2000 (full mutation) | Yes (in up to 50% of cells) | Moderate-to-severe learning difficulties |
| Females | ||
| 51–58 (intermediate alleles) | ||
| 59–200 (premutation) | No | Normal |
| 200–2000 (full mutation) | Yes (usually <10% of cells) | 50% normal, 50% mild learning difficulties |
Genetic Counseling and the Fragile X Syndrome
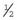 ×
× 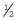 (i.e.,
(i.e., 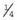 ). Prenatal diagnosis can be offered based on analysis of DNA from chorionic villi, but in the event of a female fetus with a full mutation accurate prediction of phenotype cannot be made.
). Prenatal diagnosis can be offered based on analysis of DNA from chorionic villi, but in the event of a female fetus with a full mutation accurate prediction of phenotype cannot be made.The fragile X syndrome is a condition for which population screening could be offered, either among selected high-risk groups such as males with learning difficulties or on a widespread general population basis. Such programs will have to surmount major ethical, financial, and logistical concerns if they are to achieve widespread acceptance (p. 317).
Chromosome Deletion and Microdeletion Syndromes
Microscopically visible deletions of the terminal portions of chromosomes 4 and 5 cause the Wolf-Hirschhorn (4p–) (Figure 18.13) and cri-du-chat (5p–) (Figure 18.14) syndromes respectively. In both conditions severe learning difficulties is usual, often with failure to thrive. However, there is considerable variability, particularly in Wolf-Hirschhorn syndrome, and no clear correlation of the phenotype with the precise loss of chromosomal material. Cri-du-chat syndrome derives its name from the characteristic cat-like cry of affected neonates—a consequence of underdevelopment of the larynx. Both conditions are rare, with estimated incidences of approximately 1 : 50,000 births. Not all cases have cytogenetically visible chromosome deletions, and clinicians can ask for specific fluorescent in-situ hybridization (FISH; see p. 34) analysis if microarray-CGH (pp. 36, 281) is not available.

Microdeletions
Through high-resolution prometaphase banding (p. 33) and FISH (p. 34), several previously unexplained syndromes have been shown to be due to submicroscopic or micro-deletions. Some microdeletions involve loss of only a few genes at closely adjacent loci, resulting in contiguous gene syndromes. For example, several boys with Duchenne muscular dystrophy (DMD) have been described who also have other X-linked disorders, such as retinitis pigmentosa and glycerol kinase deficiency. The loci for these disorders are very close to the DMD locus on Xp21. Examples of well known microdeletion syndromes are given in Table 18.8, all of them relatively rare.
| Syndrome | Chromosome |
|---|---|
| Deletion 1p36 | 1 |
| Williams | 7 |
| Langer-Giedion | 8 |
| WAGR | 11 |
| Angelman | 15 |
| Prader-Willi | 15 |
| Rubinstein-Taybi | 16 |
| Miller-Dieker | 17 |
| Smith-Magenis | 17 |
| DiGeorge/Sedláčková/velocardiofacial | 22 |
WAGR, Wilms’ tumor, aniridia, genitourinary malformations, and retardation of growth and development.
Microarray-CGH
The 1990s witnessed the development of FISH-based analysis of all chromosome telomeres using subtelomeric probes. This led to the diagnosis of some cases of learning difficulty/dysmorphic patients, not detected by multiplex ligation-dependent probe amplification (p. 66). In recent few years this has been rapidly superseded by extensive microarray comparative genomic hybridization (CGH) testing (p. 36) and a number of new microdeletion (and to a lesser extent microduplication) syndromes have emerged. At high resolution, this new technology is yielding significant results in about 20% of cases of well selected, previously unknown dysmorphic patients with developmental delay/learning disability. This compares to a positive pick-up rate of 4% to 5% from standard karyotyping on patients considered likely to have a chromosome disorder. Examples of these new and emerging syndromes are shown in the following section.
Lessons from Microdeletion Syndromes
Retinoblastoma
It was originally observed that approximately 5% of children presenting with retinoblastoma had other abnormalities, including learning difficulties. In some of these, a constitutional interstitial deletion of a region of chromosome 13q was identified. The smallest region of overlap was 13q14, which was subsequently shown to be the position of the locus for the autosomal dominant form of retinoblastoma due to mutations in the RB1 gene (p. 216).
Wilms’ Tumor
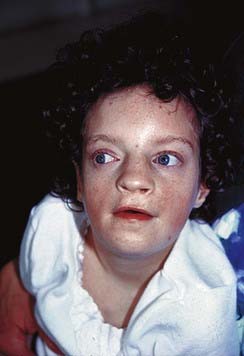
Some children with the rare renal embryonal neoplasm known as Wilms’ tumor (or hypernephroma) also have aniridia, genitourinary abnormalities, and retardation of growth and development. This combination is referred to as the WAGR syndrome. Chromosome analysis in these children often reveals an interstitial deletion of 11p13 (Figure 18.15). The deletion genes include PAX6, which is responsible for the aniridia (Figure 18.16). Confirmation is made by FISH probe analysis (or direct gene mutation analysis in cases of pure aniridia). Loss of the WT1 gene causes the development of Wilms’ tumor (see also p. 219). Knowing this, it can now be predicted whether a newly diagnosed child with deletion 11p13 is at high risk of developing a Wilms’ tumor, using a separate FISH analysis at the WT1 locus. It is important to note that, however, that the genetics of Wilms’ tumor is complex, with other loci sometimes involved.
FIGURE 18.15 A, Metaphase spread showing the number 11 chromosomes (double arrows). The chromosome indicated by the single arrow has an interstitial deletion in the short arm. See Figures 18.11 and 18.12.
(Courtesy Meg Heath, City Hospital, Nottingham, UK)
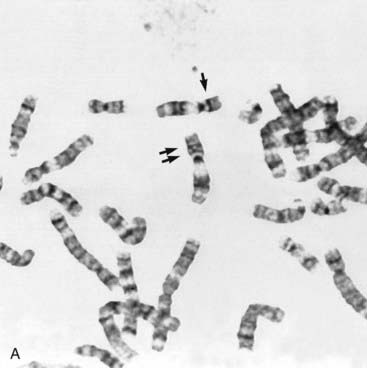

(Courtesy Dr. John Crolla, Salisbury and Dr. Veronica van Heyningen, Edinburgh, UK.)
Angelman and Prader-Willi Syndromes
These two conditions have special place in medical genetics as paradigms for genomic imprinting. Children with Angelman syndrome (see Figure 7.24; p. 123) have inappropriate laughter, convulsions, poor coordination (ataxia), and severe learning difficulties. Children with Prader-Willi syndrome (see Figure 7.22; p. 122) are extremely floppy (hypotonic) with poor feeding in infancy, and later develop hyperphagia and obesity, with mild-to-moderate learning difficulties. A large proportion of children with these disorders have a microdeletion involving 15q11-13.
In contrast, a deletion occurring at the same region on the maternally inherited chromosome 15 causes Angelman syndrome. Non-deletion cases also exist and are often due to uniparental disomy (p. 121), with both number 15 chromosomes being paternal in origin in Angelman syndrome, and maternal in origin in Prader-Willi syndrome. These parent-of-origin effects are explained by imprinting (see Figure 7.23; p. 123).
DiGeorge/Sedláčková/Velocardiofacial Syndrome
DiGeorge syndrome affects approximately 1 : 4000 births, is usually sporadic, and is characterized by heart malformations (particularly those involving the cardiac outflow tract), thymic and parathyroid hypoplasia, cleft palate and typical facies. The molecular defect is a 3-Mb microdeletion on chromosome 22 (22q11.2). Dr Eva Sedláčková from Prague reported a large series of children with a congenitally short palate in 1955, 10 years earlier than DiGeorge, and these patients clearly had the same condition. A similar phenotype was described by Shprintzen and referred to as velocardiofacial syndrome. Because of the confusion of eponyms and other terms given to this condition over the years, ‘deletion 22q11 syndrome’ now has the most widespread acceptance (at the molecular level the deleted DNA segment is still called the DiGeorge Critical Region). Figure 18.17 shows individuals with deletion 22q11.2 at different ages. Because it is the most common of the microdeletion syndromes, it has been intensely researched. It is variable and many affected individuals are able to reproduce, so the condition follows autosomal dominant inheritance in some families. The 3 Mb deletion occurs because this region is flanked by two identical sequences of DNA, known as low-copy repeats (LCRs), of the type that occur frequently throughout the genome. At meiosis the chromosomes can be ‘confused’ when they align, such that the downstream DNA sequence aligns with the upstream. If recombination occurs between these two flanking regions, a deletion of 3 Mb results on one chromosome 22. It is possible that the phenotypic features may be due largely to haploinsufficiency for the TBX1 gene that lies within the region.


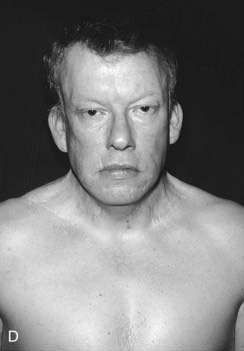
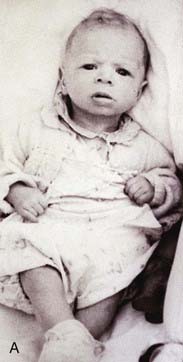
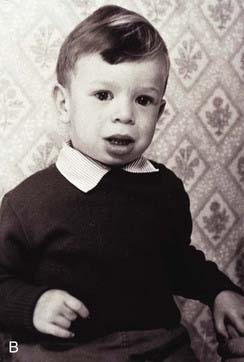
Williams Syndrome
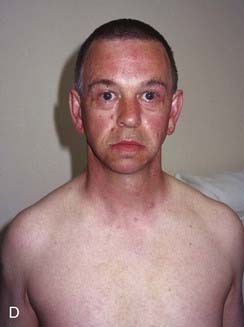
Williams syndrome occurs because of a microdeletion at chromosome 7q11 and diagnosis can be confirmed by FISH. The clinical phenotype was first reported by Williams in 1961 and later expanded by Beuren (hence, sometimes, Williams-Beuren syndrome). Hypocalcaemia is a variable feature in childhood and sometimes persists, whilst supravalvular aortic stenosis (SVAS) and peripheral pulmonary artery stenosis are congenital abnormalities of the great vessels. Haploinsufficiency at 7q11 leads to loss of one copy of the gene that encodes elastin, a component of connective tissue. This is probably the key factor causing SVAS and the vascular problems that are more common in later life. Individuals have a characteristic appearance (Figure 18.18) with mild short stature, a full lower lip, and sloping shoulders. Equally characteristic is their behavior. They are typically very outgoing in childhood—having a ‘cocktail party manner’—but become withdrawn and sensitive as adults. All are intellectually impaired to the extent that they cannot lead independent lives, and the majority do not reproduce, although parent-child transmission has been reported.

Smith-Magenis Syndrome
This microdeletion syndrome is due to loss of chromosome material at 17p.11.2, often visible cytogenetically. As with DiGeorge syndrome, the mechanism of deletion in many cases involves homologous recombination between flanking LCRs. The physical characteristics are not highly distinctive (Figure 18.19), but congenital heart disease occurs in one-third, scoliosis develops in late childhood in more than half, and hearing impairment in about two-thirds. The syndrome is most likely to be recognized by the behavioral characteristics: as children, patients exhibit self-harming (head-banging, pulling out nails, and inserting objects into orifices), a persistently disturbed sleep pattern, and characteristic ‘self-hugging’. Some degree of learning difficulty is the norm. The sleep pattern can often be managed by judicious use of melatonin.

Deletion 1p36 Syndrome
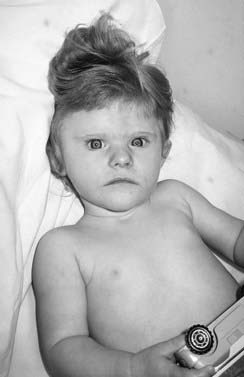
A new microdeletion syndrome that emerged through improved cytogenetic techniques and the use of FISH in the 1990s is 1p36 syndrome which, in keeping with today’s approach to nomenclature, has no eponym. The features are hypotonia, microcephaly, growth delay, severe learning difficulties, epilepsy (including infantile spasms), characteristically straight eyebrows with slightly deep-set eyes, and midface hypoplasia (Figure 18.20). Some cases developed dilated cardiomyopathy.
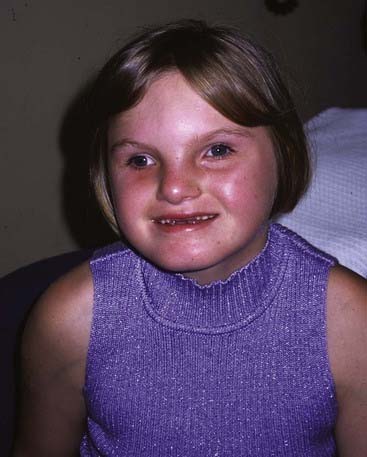
Deletion 9q34 Syndrome
Another of the relative new microdeletion syndromes, this was first reported as a condition featuring significant learning difficulties, hypotonia, obesity, brachycephaly, arched eyebrows, synophrys, anteverted nostrils, prognathism, sleep disturbances, and behavioural problems. Many patients have severe speech delay and not all manifest obesity. The case pictured here (Figure 18.21) bears a passing resemblance to Angelman syndrome. Some patients with the phenotypic features but no microdeletion have been shown to have mutations in the Euchromatin histone methyl transferase 1 (EHMT1) gene, which lies within the region. The syndrome might therefore be mainly due to haploinsufficiency for this gene.
Deletion 17q21.31 Syndrome
This new condition has a prevalence of approximately 1 : 16,000 and is probably significantly underdiagnosed. The main features are severe developmental delay, hypotonia, and characteristic facial dysmorphisms including a long face with a high forehead and tubular or pear-shaped nose, a bulbous nasal tip, large ears, and everted lower lip (Figure 18.22). Individuals tend to be friendly. Other clinically important features include epilepsy, heart defects, kidney anomalies, and long slender fingers.

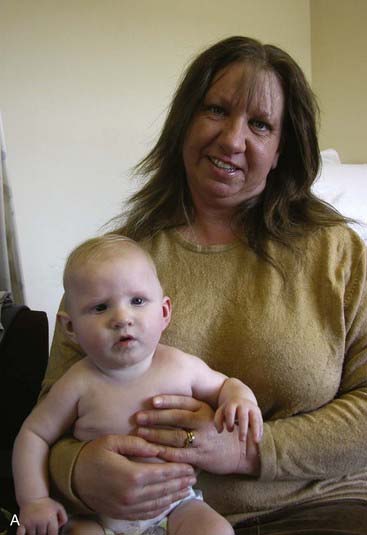
Deletion 1q21.1 Syndrome
This condition was first identified in three individuals from a cohort of 505 with congenital heart disease. The phenotype is broad and includes mild-moderate mental retardation, small head size, growth retardation, heart defects, cataracts, hand deformities and skeletal problems, learning disabilities, seizures, and autism. On the other hand, however, some individuals with the aberration are only slightly affected, or apparently unaffected. A mother and her child, both with the deletion, are shown in Figure 18.23. Variable penetrance and lack of highly distinctive features make genetic counseling for this aberration highly problematic. Some experts regard it as a susceptibility locus rather than a clinically distinct syndrome.
Chromosome disorders and behavioral phenotypes. The distinctive behavior of children with Williams syndrome—their outgoing ‘cocktail party manner’—has been recognized as part of the condition for a long time. As the microdeletion conditions have emerged, it has been increasingly clear that patterns of behavior can reliably be attributed to certain disorders. This is very striking in Smith-Magenis syndrome, but also apparent to a lesser extent in deletion 22q11, cri-du-chat, and Angelman and Prader-Willi syndromes. It is also apparent in the aneuploidies (Down and Klinefelter syndromes), as well as in 47,XXX and 47,XYY and fragile X syndromes. Behavioral phenotypes have therefore become an area of considerable interest to clinical scientists and the observations lend support to the belief that behavior, to some extent at least, is genetically determined. In studying chromosome disorders we are of course looking at genetically abnormal situations, and from this we cannot necessarily extrapolate directly to ‘normal’ situations. For the latter, twin studies have provided substantial and valuable information. This field of study remains complex and understandably controversial. However, most now accept that behavior is a complex interaction of genetic background, physical influences during early development (e.g., fetal well-being), nurturing experiences, family size, culture, and belief systems.
Disorders of Sexual Differentiation
The process of sexual differentiation has been described in Chapter 6 (p. 101). Given the complexity of the sequential cascade of events that takes place between 6 and 14 weeks of embryonic life, it is not surprising that errors can occur. Many of these errors can lead to sexual ambiguity or to discordance between the chromosomal sex and the appearance of the external genitalia. These disorders are also sometimes referred to as various forms of intersex (Box 18.2).
True Hermaphroditism
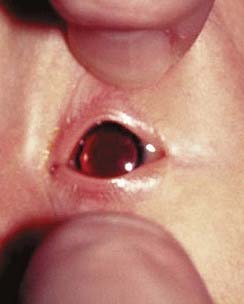
In this extremely rare condition, an individual has both testicular and ovarian tissue, often in association with ambiguous genitalia. When an exploratory operation is carried out in these patients, an ovary can be found on one side and a testis on the other. Alternatively, there can be a mixture of ovarian and testicular tissue in the gonad, which is known as an ovotestis. Most patients with true hermaphroditism have a 46,XX karyotype, and in many of these individuals the paternally derived X chromosome carries Y chromosome–specific DNA sequences as a result of illegitimate crossing over between the X and Y chromosomes during meiosis I in spermatogenesis (Figure 18.24).
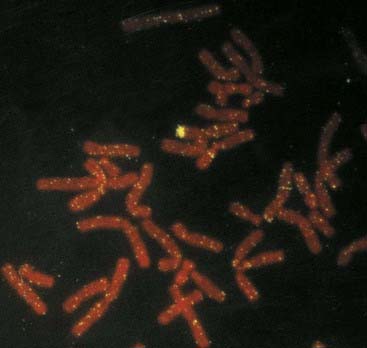
FIGURE 18.24 FISH showing hybridization of a Y chromosome paint to the short arm of an X chromosome in a 46,XX male.
(Courtesy Nigel Smith, City Hospital, Nottingham, UK.)
A small proportion of patients with true hermaphroditism are found to be chimeras with both 46,XX and 46,XY cell lines, a situation analogous to freemartins in cattle (p. 51).
Male Pseudohermaphroditism
The most widely recognized cause of male pseudohermaphroditism is androgen insensitivity (p. 175). In this condition, which is also known as testicular feminization syndrome, the karyotype is normal male and the external phenotype is essentially that of a normal female. Internally the vagina ends blindly and the uterus and fallopian tubes are absent. Testes are located in the abdomen or in the inguinal canal, where they can be mistaken for inguinal herniae. This condition is caused by the absence of androgen receptors in the target organs, so that, although testosterone is formed normally, its peripheral masculinizing effects are blocked. Androgen receptors are encoded by a gene on the X chromosome in which both deletions and point mutations have been identified. Curiously, expansion of a CAG repeat in the first exon of this androgen receptor gene causes a neurological disorder known as Kennedy disease, or spino-bulbar muscular atrophy. This is a rare example of the phenomenon sometimes called a ‘gene within a gene’.
Other causes of male pseudohermaphroditism include:
Female Pseudohermaphroditism
Congenital adrenal hyperplasia (CAH) is by far the most important cause of female pseudohermaphroditism (p. 174). This can be caused by several different enzyme defects in the adrenal cortex, all of which show autosomal recessive inheritance. Reduced cortisol production leads to an increase in adrenocorticotrophic hormone secretion, which in turn causes hyperplasia of the adrenal glands. In the most common form of CAH, because of a 21-hydroxylase deficiency, hormone synthesis switches from the manufacture of cortisol and aldosterone to the androgen pathway (see Figure 11.5; p. 174), leading to striking virilization of a female fetus (see Figure 11.6; p. 175). The lack of cortisol and aldosterone usually leads to rapid collapse within days of birth, which can prove fatal unless appropriate hormone and electrolyte supplementation is initiated.
Chromosomal Breakage Syndromes
Constitutional and acquired chromosome abnormalities that predispose to malignancy are considered in Chapter 14. In addition to these conditions, it is recognized that a small number of hereditary disorders are characterized by an excess of chromosome breaks and gaps as well as an increased susceptibility to neoplasia.
Ataxia Telangiectasia
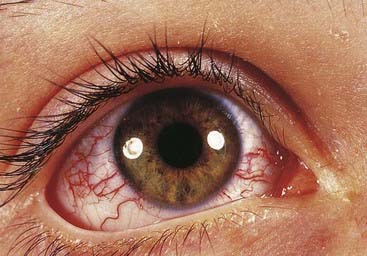
This is an autosomal recessive disorder that presents in early childhood with ataxia, oculocutaneous telangiectasia (Figure 18.25), radiation sensitivity, and susceptibility to sinus and pulmonary infection (p. 204). There is a 10% to 20% risk of leukemia or lymphoma. Cells from patients show an increase in spontaneous chromosome abnormalities, such as chromatid gaps and breaks, which are enhanced by radiation. The gene for ataxia telangiectasia is called ATM and maps to chromosome 11q23. The protein product is thought to act as a ‘checkpoint’ protein kinase, which interacts with the TP53 and BRCA1 gene products to arrest cell division and thereby allow repair of radiation-induced chromosome breaks before the S phase in the cell cycle (p. 39).
Bloom Syndrome
Children with this autosomal recessive disorder are small with a light-sensitive facial rash and reduced immunoglobulin levels (IgA and IgM). The risk of lymphoreticular malignancy is approximately 20%. Cultured cells show an increased frequency of chromosome breaks, particularly if they are exposed in vitro to ultraviolet light. The gene for Bloom syndrome maps to chromosome 15q26, where it encodes one member of a group of enzymes called the DNA helicases (p. 14). These are responsible for unwinding double-stranded DNA before replication, repair, and recombination. Normally the Bloom syndrome gene plays a major role in maintaining genome stability. When defective in the homozygous state, DNA repair is impaired and the rate of recombination between sister chromatids is increased dramatically. This can be demonstrated by looking for sister chromatid exchanges (see the following section).
Fanconi Anemia
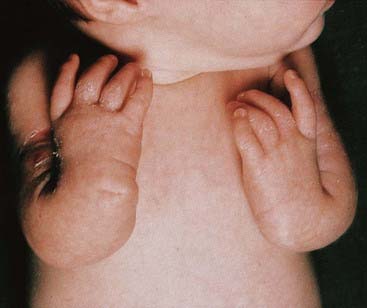
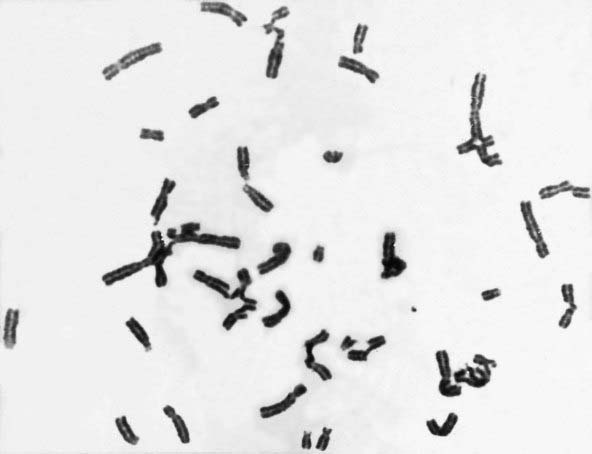
This autosomal recessive disorder is associated with upper limb abnormalities involving the radius and thumb (Figure 18.26), increased pigmentation, and bone marrow failure leading to deficiency of all types of blood cells (i.e., pancytopenia). There is also an increased risk of neoplasia, particularly leukemia, lymphoma, and hepatic carcinoma. Multiple chromosomal breaks are observed in cultured cells (Figure 18.27) and the basic defect lies in the repair of DNA strand cross-links. There are five known subtypes of Fanconi anemia, each caused by recessive mutations at different autosomal loci. The most common, type A, maps to chromosome 16q24.
Xeroderma Pigmentosa
Chromosome Breakage and Sister Chromatid Exchange
Strong evidence of increased chromosome instability is provided by the demonstration of an increased number of sister chromatid exchanges (SCEs) in cultured cells. An SCE is an exchange (crossing over) of genetic material between the two chromatids of a chromosome in mitosis, in contrast to recombination in meiosis I, which is between homologous chromatids. SCEs can be demonstrated by differences in the uptake of certain stains by the two chromatids of each metaphase chromosome after two rounds of cell division in the presence of the thymidine analogue 5-bromodeoxyuridine (BUdR), which becomes incorporated in the newly synthesized DNA (Figure 18.28). There are normally about 10 SCEs per cell, but the number is greatly increased in cells from patients with Bloom syndrome and xeroderma pigmentosa. In the latter condition, this is apparent only after the cells have been exposed to ultraviolet light.
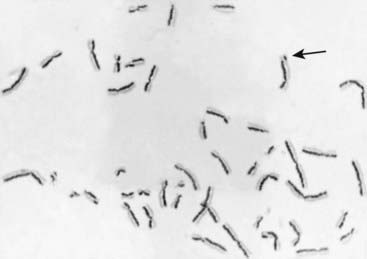
It is not clear how SCEs relate to the increased chromosome breakage observed in these two disorders, but it is thought that the explanation could involve one of the steps in DNA replication. It is also of interest that the number of SCEs in normal cells is increased on exposure to certain carcinogens and chemical mutagens. For this reason the frequency of SCEs in cells in culture has been suggested as a useful in vitro test of the carcinogenicity and mutagenicity of chemical compounds (p. 26).
Indications for Chromosomal/Microarray-Cgh Analysis
It should be apparent from the contents of this chapter that chromosome abnormalities can present in many different ways. Consequently it is appropriate to consider the indications for chromosome analysis, which increasingly means microarray-CGH, under a number of different headings (Box 18.3).
Multiple Congenital Abnormalities
Unexplained Learning Difficulties
Chromosome abnormalities cause at least one-third of the 50% of learning difficulties that are attributable to genetic factors. Although most children with a chromosome abnormality have other features such as growth retardation and physical anomalies, this is not always so. If the fragile X syndrome is a possibility, it is important that the cytogenetics laboratory is informed so that the correct culture conditions are used (p. 279), although in most centers the fragile X syndrome is now diagnosed by molecular methods rather than chromosome analysis. If standard karyotyping and fragile X syndrome testing is negative, great reliance will increasingly be placed on microarray CGH.
Sexual Ambiguity
The birth of a child with ambiguous genitalia should be regarded as a medical emergency, not only because of the inevitable parental anxiety, but also because of the importance of ruling out the potentially life-threatening diagnosis of salt-losing congenital adrenal hyperplasia (p. 174). A chromosome analysis should be among the first investigations undertaken.
Infertility and Recurrent Miscarriage
At least 15% of all recognized pregnancies end in spontaneous miscarriage; in 50% of cases this is because of a chromosome abnormality (p. 273). Unfortunately, some couples experience recurrent pregnancy loss—usually defined as more than three spontaneous miscarriages. Often no explanation is found and many such couples go on to have successful pregnancies. However, in 3% to 6% one partner is found to carry a chromosome rearrangement that predisposes to severe imbalance through malsegregation at meiosis (p. 39). Consequently it is now standard practice to offer chromosome analysis to all such couples.
Malignancy and Chromosome Breakage Syndromes
Certain types of leukemia and many solid tumors, such as retinoblastoma (p. 215) and Wilms’ tumor (p. 282), are associated with specific chromosomal abnormalities that can be of both diagnostic and prognostic value. Clinical features suggestive of a chromosome breakage syndrome (p. 288), such as a combination of photosensitivity and short stature, should also lead to appropriate chromosome fragility studies, such as analysis of sister chromatid exchanges.
De Grouchy J, Turleau C. Clinical atlas of human chromosomes, 2nd ed. Chichester: John Wiley; 1984.
A lavishly illustrated atlas of known chromosomal syndromes.
Donnai D, Karmiloff-Smith A. Williams syndrome: from genotype through to the cognitive phenotype. Am J Med Genet (Semin Med Genet). 2000;97:164-171.
Gardner RJM, Sutherland GR. Chromosome abnormalities and genetic counseling, 2nd ed. Oxford: Oxford University Press; 1996.
A useful updated guide to genetic counseling in families with a chromosome disorder.
Hagerman RJ, Silverman AC, editors. Fragile X syndrome. Diagnosis, treatment and research. Baltimore: Johns Hopkins University Press, 1991.
A detailed account of the clinical and genetic aspects of the fragile X syndrome.
Jacobs PA, Browne C, Gregson N, Joyce C, White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29:103-108.
Ratcliffe S. Long term outcome of children of sex chromosome abnormalities. Arch Dis Child. 1999;80:192-195.
Schinzel A. Human cytogenetics database. Oxford: Oxford University Press; 1994.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 18 Chromosome Disorders
The development of a reliable technique for chromosome analysis in 1956 soon led to the discovery that several previously described conditions were due to an abnormality in chromosome number. Within 3 years, the causes of Down syndrome (47,XX/XY, +21), Klinefelter syndrome (47,XXY), and Turner syndrome (45,X) had been established. Shortly after, other autosomal trisomy syndromes were recognized, and over the ensuing years many other multiple malformation syndromes were described in which there was loss or gain of chromosome material.
In Chapter 3, the basic principles of chromosome structure, function, and behavior during cell division were described, together with an account of chromosome abnormalities and how they can arise and be transmitted in families. In this chapter, the medical aspects of chromosome abnormalities, and some of their specific syndromes, are described.
Incidence of Chromosome Abnormalities
Chromosome abnormalities are present in at least 10% of all spermatozoa and 25% of mature oocytes. Some 15% to 20% of all recognized pregnancies end in spontaneous miscarriage, and many more zygotes and embryos are so abnormal that survival beyond the first few days or weeks after fertilization is not possible. Approximately 50% of all spontaneous miscarriages have a chromosome abnormality (Table 18.1) and the incidence of chromosomal abnormalities in morphologically normal embryos is around 20%. Chromosome abnormalities therefore account for the spontaneous loss of a very high proportion of all human conceptions.
Table 18.1 Chromosome Abnormalities in Spontaneous Abortions (Percentage Values Relate to Total of Chromosomally Abnormal Abortuses)
| Abnormality | Incidence (%) |
|---|---|
| Trisomy 13 | 2 |
| Trisomy 16 | 15 |
| Trisomy 18 | 3 |
| Trisomy 21 | 5 |
| Trisomy other | 25 |
| Monosomy X | 20 |
| Triploidy | 15 |
| Tetraploidy | 5 |
| Other | 10 |
From conception onward, the incidence of chromosome abnormalities falls rapidly. By birth it has declined to a level of 0.5% to 1%, although the total is higher (5%) in stillborn infants. Table 18.2 lists the incidence figures for chromosome abnormalities based on newborn surveys. It is notable that among the commonly recognized aneuploidy syndromes, there is also a high proportion of spontaneous pregnancy loss (Table 18.3). This is illustrated by comparison of the incidence of conditions such as Down syndrome at the time of chorionic villus sampling (11 to 12 weeks), amniocentesis (16 weeks), and term (Figure 18.1).
Table 18.2 Incidence of Chromosome Abnormalities in the Newborn
| Abnormality | Incidence per 10,000 Births |
|---|---|
| Autosomes | |
| Trisomy 13 | 2 |
| Trisomy 18 | 3 |
| Trisomy 21 | 15 |
| Sex Chromosomes | |
| female births | |
| 45,X | 1–2 |
| 47,XXX | 10 |
| male births | |
| 47,XXY | 10 |
| 47,XYY | 10 |
| Other unbalanced rearrangements | 10 |
| Balanced rearrangements | 30 |
| Total | 90 |
Table 18.3 Spontaneous Pregnancy Loss in Commonly Recognized Aneuploidy Syndromes
| Disorder | Proportion Undergoing Spontaneous Pregnancy Loss (%) |
|---|---|
| Trisomy 13 | 95 |
| Trisomy 18 | 95 |
| Trisomy 21 | 80 |
| Monosomy X | 98 |
Down Syndrome (Trisomy 21)
Incidence
The overall birth incidence, when adjusted for the increasingly widespread impact of antenatal screening, is approximately 1 : 1000 in the United Kingdom, which has a national register. In the United States, the birth incidence has been estimated at approximately 1 : 800. In the United Kingdom, approximately 60% of Down syndrome cases are detected prenatally. There is a strong association between the incidence of Down syndrome and advancing maternal age (Table 18.4).
Table 18.4 Incidence of Down Syndrome in Relation to Maternal Age
| Maternal Age at Delivery (Years) | Incidence of Down Syndrome |
|---|---|
| 20 | 1 in 1500 |
| 25 | 1 in 1350 |
| 30 | 1 in 900 |
| 35 | 1 in 400 |
| 36 | 1 in 300 |
| 37 | 1 in 250 |
| 38 | 1 in 200 |
| 39 | 1 in 150 |
| 40 | 1 in 100 |
| 41 | 1 in 85 |
| 42 | 1 in 65 |
| 43 | 1 in 50 |
| 44 | 1 in 40 |
| 45 | 1 in 30 |
Adapted from Cuckle HS, Wald NJ, Thompson SG 1987 Estimating a woman’s risk of having a pregnancy associated with Down syndrome using her age and serum alpha-fetoprotein level. Br J Obstet Gynaecol 94:387–402.
Clinical Features
These are summarized in Box 18.1. The most common finding in the newborn period is severe hypotonia. Usually the facial characteristics of upward sloping palpebral fissures, small ears, and protruding tongue (Figures 18.2 and 18.3) prompt rapid suspicion of the diagnosis, although this can be delayed in very small or premature babies. Single palmar creases are found in 50% of children with Down syndrome (Figure 18.4), in contrast to 2% to 3% of the general population. Congenital cardiac abnormalities are present in 40% to 45% of babies with Down syndrome, with the three most common lesions being atrioventricular canal defects, ventricular septal defects, and patent ductus arteriosus.
Box 18.1
Common Findings in Down Syndrome
Craniofacial
Brachycephaly, epicanthic folds, protruding tongue, small ears, upward sloping palpebral fissures
Limbs
Single palmar crease, small middle phalanx of fifth finger, wide gap between first and second toes
Natural History
Affected children show a broad range of intellectual ability with IQ scores ranging from 25 to 75. The average IQ of young adults is around 40 to 45. Social skills are relatively well-advanced and most children are happy and very affectionate. Adult height is usually around 150 cm. In the absence of a severe cardiac anomaly, which leads to early death in 15% to 20% of cases, average life expectancy is 50 to 60 years. Most affected adults develop Alzheimer disease in later life, possibly because of a gene dosage effect—the amyloid precursor protein gene is on chromosome 21. This gene is known to be implicated in some familial cases of Alzheimer disease (p. 243).
Chromosome Findings
These are listed in Table 18.5. In cases resulting from trisomy 21, the additional chromosome is maternal in origin in more than 90% of cases, and DNA studies have shown that this arises most commonly as a result of non-disjunction in maternal meiosis I (p. 39). Robertsonian translocations (p. 47) account for approximately 4% of all cases, in roughly one-third of which a parent is found to be a carrier. Children with mosaicism are often less severely affected than those with the full syndrome.
| Abnormality | Frequency (%) |
|---|---|
| Trisomy | 95 |
| Translocation | 4 |
| Mosaicism | 1 |
Efforts have been made to correlate the various clinical features in trisomy Down syndrome with specific regions of chromosome 21, by studying children with partial trisomy for different regions. There is some support for a Down syndrome ‘critical region’ at the distal end of the long arm (21q22), because children with trisomy for this region alone usually have typical Down syndrome facial features. Chromosome 21 is a ‘gene-poor’ chromosome with a high ratio of AT to GC sequences (p. 69). At present the only reasonably well-established genotype-phenotype correlation in trisomy 21 is the high incidence of Alzheimer disease.
Recurrence Risk
For straightforward trisomy 21, the recurrence risk is related to maternal age (variable) and the simple fact that trisomy has already occurred (∼1%). The combined recurrence risk is usually between 1 : 200 and 1 : 100. In translocation cases, similar figures apply if neither parent is a carrier. In familial translocation cases, the recurrence risks vary from around 1% to 3% for male carriers up to 10% to 15% for female carriers, with the exception of very rare carriers of a 21q21q translocation, for whom the recurrence risk is 100% (p. 48).
Prenatal diagnosis can be offered based on analysis of chorionic villi or cultured amniotic cells. Prenatal screening programs have been introduced based on the so-called triple or quadruple tests of maternal serum at 16 weeks’ gestation (p. 328).
Patau Syndrome (Trisomy 13) and Edwards Syndrome (Trisomy 18)
These very severe conditions were first described in 1960 and share many features in common (Figures 18.5 and 18.6). The incidence for both is approximately 1 : 5000 and prognosis is very poor, with most infants dying during the first days or weeks of life, though most cases are detected prenatally, often leading to termination. In the unusual event of longer term survival, there are severe learning difficulties. Cardiac abnormalities occur in at least 90% of cases.
Chromosome analysis usually reveals straightforward trisomy. Both disorders occur more frequently with advanced maternal age, the additional chromosome being of maternal origin (see Table 3.4, p. 43). Approximately 10% of cases are caused by mosaicism or unbalanced rearrangements, particularly Robertsonian translocations in Patau syndrome.
Triploidy
Triploidy (69,XXX, 69,XXY, 69,XYY) is a relatively common finding in material cultured from spontaneous abortions, but is seen only rarely in a liveborn infant. Such a child almost always shows severe intrauterine growth retardation with relative preservation of head growth at the expense of a small thin trunk. Syndactyly involving the third and fourth fingers and/or the second and third toes is a common finding. Cases of triploidy resulting from a double paternal contribution usually miscarry in early to mid-pregnancy and are associated with partial hydatidiform changes in the placenta (p. 101). Cases with a double maternal contribution survive for longer but rarely beyond the early neonatal period.
Hypomelanosis of Ito
Several children with mosaicism for diploidy/triploidy have been identified. These can demonstrate the clinical picture seen in full triploidy but in a milder form. An alternative presentation occurs as the condition known as hypomelanosis of Ito. In this curious disorder, the skin shows alternating patterns of normally pigmented and depigmented streaks that correspond to the embryological developmental lines of the skin known as Blaschko’s lines (see Figure 18.7). Most children with hypomelanosis of Ito have moderate learning difficulties and convulsions that can be particularly difficult to treat. There is increasing evidence that this clinical picture represents a non-specific embryological response to cell or tissue mosaicism. A similar pattern of skin pigmentation is sometimes seen in women with one of the rare X-linked dominant disorders (p. 117) with skin involvement, such as incontinentia pigmenti (see Figure 7.18, p. 118). Such women can be considered as being mosaic, as some cells express the normal gene, whereas others express only the mutant gene.
FIGURE 18.7 Mosaic pattern of skin pigmentation on the arm of a child with hypomelanosis of Ito.
(Reproduced with permission from Jenkins D, Martin K, Young ID 1993 Hypomelanosis of Ito associated with mosaicism for trisomy 7 and apparent ‘pseudomosaicism’ at amniocentesis. J Med Genet 1993; 30:783–784.)
Disorders of the Sex Chromosomes
Turner Syndrome (45,X)
This condition was first described clinically in 1938. The absence of a Barr body, consistent with the presence of only one X chromosome, was noted in 1954 and cytogenetic confirmation was forthcoming in 1959. Although common at conception and in spontaneous abortions (see Table 18.1), the incidence in liveborn female infants is low, with estimates ranging from 1 : 5000 to 1 : 10,000.
Clinical Features
Presentation can be at any time from pregnancy to adult life. Increasingly, Turner syndrome is being detected during the second trimester as a result of routine ultrasonography, showing either generalized edema (hydrops) or swelling localized to the neck (nuchal cyst or thickened nuchal pad) (Figure 18.8). At birth many babies with Turner syndrome look entirely normal. Others show the residue of intrauterine edema with puffy extremities (Figure 18.9) and neck webbing. Other findings may include a low posterior hairline, increased carrying angles at the elbows, short fourth metacarpals, widely spaced nipples, and coarctation of the aorta, which is present in 15% of cases.
Intelligence in Turner syndrome is normal. However, studies have shown some differences in social cognition and higher order executive function skills according to whether the X chromosome was paternal or maternal in origin (p. 105). The two main medical problems are short stature and ovarian failure. The short stature becomes apparent by mid-childhood, and without growth hormone treatment the average adult height is 145 cm. This short stature is due, at least in part, to haploinsufficiency for the SHOX gene, which is located in the pseudoautosomal region (p. 118). Ovarian failure commences during the second half of intrauterine life and almost invariably leads to primary amenorrhea and infertility. Estrogen replacement therapy should be initiated at adolescence for the development of secondary sexual characteristics and long-term prevention of osteoporosis. In vitro fertilization using donor eggs offers the prospect of pregnancy for women with Turner syndrome.
Chromosome Findings
These are summarized in Table 18.6. The most common finding is 45,X (sometimes erroneously referred to as 45,XO). In 80% of cases, it arises through loss of a sex chromosome (X or Y) paternal meiosis. In a significant proportion of cases, there is chromosome mosaicism and those with a normal cell line (46,XX) have a chance of being fertile. Some cases with a 46,XY cell line are phenotypically male, and all cases with some Y-chromosome material in their second cell line must be investigated for possible gonadal dysgenesis—intracellular male gonads can occasionally become malignant and require surgical removal.
| Karyotype | Frequency (%) |
|---|---|
| Monosomy X—45,X | 50 |
| Mosaicism (e.g., 45,X/46,XX) | 20 |
| Isochromosome—46,X,i(Xq) | 15 |
| Ring—46,X,r(X) | 5 |
| Deletion—46,X,del(Xp) | 5 |
| Other | 5 |
