118 Chromosomal Abnormalities
Trisomy 18 (Edwards Syndrome)
Clinical Presentation
Trisomy 18 is one of the more common malformation syndromes, with an incidence of 0.3 in 100 newborn infants. On prenatal ultrasound, increased nuchal translucency may be seen early in gestation. At birth, reduced birth weight, a small placenta, and fetal distress may be noted along with multiple other clinical features (Table 118-1).
Table 118-1 Frequent Clinical Features in Trisomies 18 and 13
| Trisomy 18 | Trisomy 13 | |
|---|---|---|
| Head and CNS | Microcephaly, prominent occiput | Microcephaly, parieto-occipital scalp defect, holoprosencephaly (≈66%) |
| Eyes | Short palpebral fissures | Anophthalmia or microphthalmia |
| Ears | Low set, posteriorly rotated | Abnormal helices |
| Mouth | Small, micrognathia | Cleft lip or palate |
| Hands |
ASD, atrial septal defect; CNS, central nervous system; PDA, patent ductus arteriosus; VSD, ventricular septal defect.
Trisomy 13 (Patau Syndrome)
Clinical Presentation
Trisomy 13 occurs with an incidence of approximately 1 in 5000 births. Early prenatal detection is possible with a combined screening protocol that includes maternal age, nuchal translucency, and maternal serum markers. The mean birth weight is reduced, and multiple dysmorphic features may be observed (see Table 118-1).
Turner’s Syndrome (45,X)
Clinical Presentation
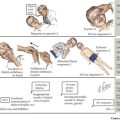
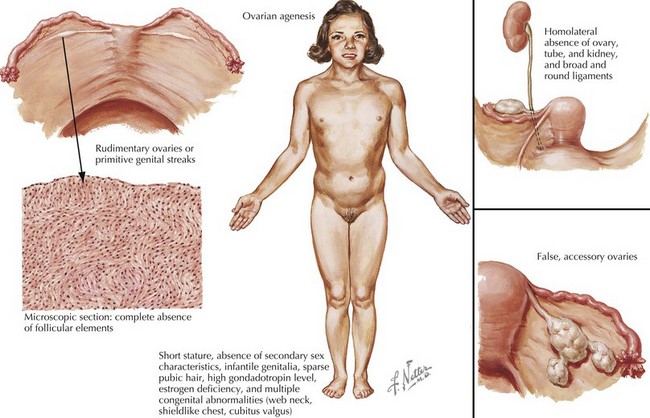
Females with Turner syndrome may present prenatally with features of lymphedema/hydrops and/or a congenital heart defect, in puberty due to short stature or in adulthood with premature ovarian failure (Figure 118-1). Multiple organ systems may be involved, but no single feature is pathognomonic or universally present.
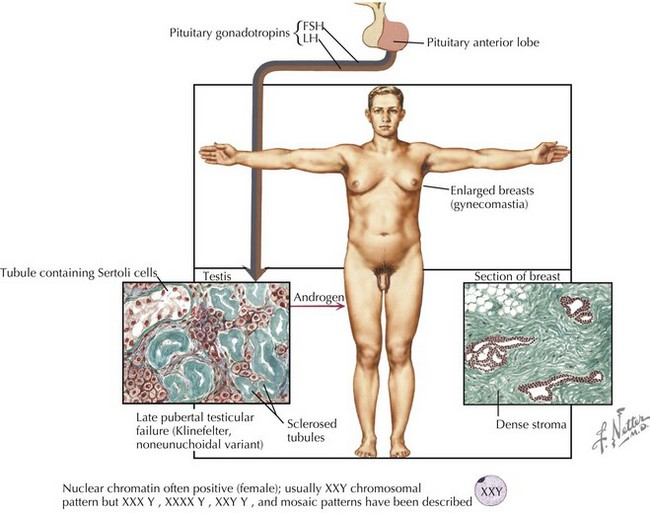
Klinefelter Syndrome
Clinical Presentation
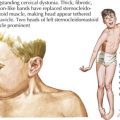
Although the age and onset of puberty is unchanged in boys with Klinefelter syndrome, testicular growth is arrested, leading to oligospermia or azoospermia and low testosterone levels. A small fraction of men present with gynecomastia, eunuchoid habitus, and decreased hair (Figure 118-2). Muscle strength may be decreased. Testosterone replacement therapy aims at increasing muscle mass, libido, energy, and improving mood through support of androgen-dependent processes. Fertility is rarely preserved except in mosaic men.
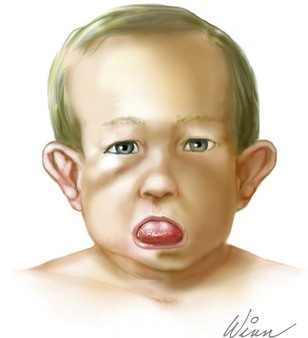
22q11.2 Deletion Syndrome
Clinical Presentation
The suspicion for del22q11.2 in a newborn is often raised by the presence of one or more of a constellation of physical findings that include congenital heart disease, especially conotruncal malformations, hypocalcemia, immune deficiency, and characteristic facial features. Frequently observed dysmorphisms include overfolded or squared-off ear helices, small and protuberant ears with preauricular pits or tags, a prominent nasal root and bulbous nasal tip, hooded eyelids, ocular hypertelorism, cleft lip and palate, asymmetric crying facies, and craniosynostosis (Figure 118-3). Infants often have feeding difficulties secondary to severe dysphagia and palatal abnormalities. In older patients, learning difficulties, growth hormone deficiency, autoimmune disease, and psychiatric illness may become apparent. Other organ systems can be involved, and a renal ultrasound to evaluate for renal abnormalities and chest radiography to assess for vertebral malformations should be performed.
Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008;148C(4):246-251.
Loscalzo M. Turner syndrome. Pediatr Rev. 2008;29(7):219-227.
Mainardi PC. Cri du chat syndrome. Orphanet J Rare Dis. 2006;1:33.
National Center for Biotechnology Information. GeneReviews. Available at http://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests