Chapter 348 Cholestasis
348.1 Neonatal Cholestasis
H. Hesham A-kader and William F. Balistreri
Neonatal cholestasis is defined biochemically as prolonged elevation of the serum levels of conjugated bilirubin beyond the 1st 14 days of life. Jaundice that appears after 2 wk of age, progresses after this time, or does not resolve at this time should be evaluated and a conjugated bilirubin level determined. Cholestasis in a newborn can be due to infectious, genetic, metabolic, or undefined abnormalities giving rise to mechanical obstruction of bile flow or to functional impairment of hepatic excretory function and bile secretion (see Table 347-3). Mechanical lesions include stricture or obstruction of the common bile duct; biliary atresia is the prototypic obstructive abnormality. Functional impairment of bile secretion can result from congenital defects or damage to liver cells or to the biliary secretory apparatus.
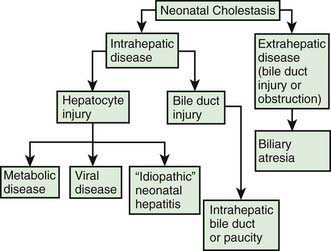
Neonatal cholestasis can be divided into extrahepatic and intrahepatic disease (Fig. 348-1). The clinical features of any form of cholestasis are similar. In an affected neonate, the diagnosis of certain entities, such as galactosemia, sepsis, or hypothyroidism, is relatively simple and a part of most neonatal screening programs. In most cases, the cause of cholestasis is more obscure. Differentiation among biliary atresia, idiopathic neonatal hepatitis, and intrahepatic cholestasis is particularly difficult.
Mechanisms
Functional abnormalities in the generation of bile flow can also have a role in neonatal cholestasis. Bile flow is directly dependent on effective hepatic bile acid excretion by the hepatocytes. During the phase of relatively inefficient liver cell transport and metabolism of bile acids in early life, minor degrees of hepatic injury can further decrease bile flow and lead to production of atypical and potentially toxic bile acids. Selective impairment of a single step in the series of events involved in hepatic excretion produces the full expression of a cholestatic syndrome. Specific defects in bile acid synthesis are found in infants with various forms of intrahepatic cholestasis (Table 348-1). Severe forms of familial cholestasis have been associated with neonatal hemochromatosis and an aberration in the contractile proteins that compose the cytoskeleton of the hepatocyte. Neonatal hemochromatosis can also be an alloimmune-mediated gestational (maternal antibodies against fetal hepatocytes) disease responsive to maternal intravenous immunoglobulin (IVIG). Sepsis is known to cause cholestasis, presumably mediated by an endotoxin produced by Escherichia coli.
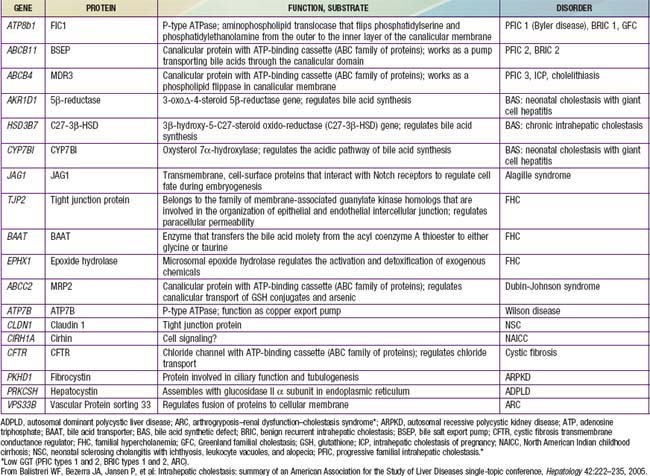
Table 348-1 PROPOSED SUBTYPES OF INTRAHEPATIC CHOLESTASIS
Note: FIC1 deficiency, BSEP deficiency, and some of the disorders of bile acid biosynthesis are characterized clinically by low levels of serum GGT despite the presence of cholestasis. In all other disorders listed, the serum GGT level is elevated.
ADPLD, autosomal dominant polycystic liver disease (cysts in liver only); ARPKD, autosomal recessive polycystic kidney disease (cysts in liver and kidney); BAAT, bile acid transporter; BRIC, benign recurrent intrahepatic cholestasis; BSEP, bile salt export pump in; GGT, γ-glutamyl transpeptidase; PFIC, progressive familial intrahepatic cholestasis.
From Balistreri WF, Bezerra JA, Jansen P, et al: Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference, Hepatology 42:222–235, 2005.
Evaluation
The evaluation of the infant with jaundice should follow a logical, cost-effective sequence in a multistep process (Table 348-2). Although cholestasis in the neonate may be the initial manifestation of numerous and potentially serious disorders, the clinical manifestations are usually similar and provide very few clues about etiology. Affected infants have icterus, dark urine, light or acholic stools, and hepatomegaly, all resulting from decreased bile flow due to either hepatocyte injury or bile duct obstruction. Hepatic synthetic dysfunction can lead to hypoprothrombinemia and bleeding. Administration of vitamin K should be included in the initial treatment of cholestatic infants to prevent hemorrhage.
Table 348-2 VALUE OF SPECIFIC TESTS IN THE EVALUATION OF PATIENTS WITH SUSPECTED NEONATAL CHOLESTASIS
| TEST | RATIONALE |
|---|---|
| Serum bilirubin fractionation (i.e., assessment of the serum level of conjugated bilirubin) | Indicates cholestasis |
| Assessment of stool color (does the baby have pigmented or acholic stools?) | Indicates bile flow into intestine |
| Urine and serum bile acids measurement | Confirms cholestasis; might indicate inborn error of bile acid biosynthesis |
| Hepatic synthetic function (albumin, coagulation profile) | Indicates severity of hepatic dysfunction |
| α1-Antitrypsin phenotype | Suggests (or excludes) PiZZ |
| Thyroxine and TSH | Suggests (or excludes) endocrinopathy |
| Sweat chloride and mutation analysis | Suggests (or excludes) cystic fibrosis |
| Urine and serum amino acids and urine reducing substances | Suggests (or excludes) metabolic liver disease |
| Ultrasonography | Suggests (or excludes) choledochal cyst; might detect the triangular cord (TC) sign, suggesting biliary atresia |
| Hepatobiliary scintigraphy | Documents bile duct patency or obstruction |
| Liver biopsy | Distinguishes biliary atresia; suggests alternative diagnosis |
PiZZ, protease inhibitor ZZ phenotype; TSH, thyroid-stimulating hormone.
Intrahepatic Cholestasis
Neonatal Hepatitis
The term neonatal hepatitis implies intrahepatic cholestasis (see Fig. 348-1), which has various forms (Tables 348-1 and 348-3).
Zellweger (cerebrohepatorenal) syndrome is a rare autosomal recessive genetic disorder marked by progressive degeneration of the liver and kidneys (Chapter 80.2). The incidence is estimated to be 1/100,000 births; the disease is usually fatal in 6-12 mo. Affected infants have severe, generalized hypotonia and markedly impaired neurologic function with psychomotor retardation. Patients have an abnormal head shape and unusual facies, hepatomegaly, renal cortical cysts, stippled calcifications of the patellas and greater trochanter, and ocular abnormalities. Hepatic cells on ultrastructural examination show an absence of peroxisomes. MRI performed in the 3rd trimester can allow analysis of cerebral gyration and myelination, facilitating the prenatal diagnosis of Zellweger syndrome.
Disorders of Transport, Secretion, Conjugation, and Biosynthesis of Bile Acids
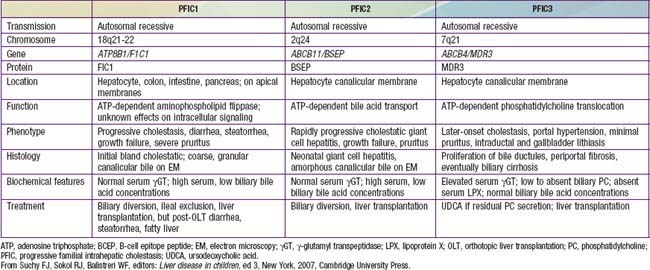
PFIC 1 (FIC-1 deficiency) has been mapped to chromosome 18q12 and results from defect in the gene for F1C1 (ATP8B1; Tables 348-3 and 348-4). F1C1 is a P-type adenosine triphosphatase (ATPase) that functions as aminophospholipid flippase, facilitating the transfer of phosphatidyl serine and phosphatidyl ethanolamine from the outer to inner hemileaflet of the cellular membrane. F1C1 might also play a role in intestinal bile acid absorption, as suggested by the high level of expression in the intestine. Defective F1C1 might also result in another form of intrahepatic cholestasis: benign recurrent intrahepatic cholestasis (BRIC) type I. The disease is characterized by recurrent bouts of cholestasis, jaundice, and severe pruritus lasting from a 2 wk to 6 mo period; it can last up to 5 yr. The episodes vary from few episodes per year to 1 episode per decade and can profoundly affect the quality of life. Nonsense, frame shift, and deletional mutations cause PFIC type I; missense and split type mutations result in BRIC type I. Typically, patients with BRIC type I have normal cholesterol and GGT levels.
 months of age but subsequently died from disseminated Epstein-Barr virus–related lymphoproliferative disease.
months of age but subsequently died from disseminated Epstein-Barr virus–related lymphoproliferative disease.Biliary Atresia
The term biliary atresia is imprecise because the anatomy of abnormal bile ducts in affected patients varies markedly. A more appropriate terminology would reflect the pathophysiology, namely, progressive obliterative cholangiopathy. Patients can have distal segmental bile duct obliteration with patent extrahepatic ducts up to the porta hepatis. This is a surgically correctable lesion, but it is uncommon. The most common form of biliary atresia, accounting for ∼85% of the cases, is obliteration of the entire extrahepatic biliary tree at or above the porta hepatis. This presents a much more difficult problem in surgical management. Most patients with biliary atresia (85-90%) are normal at birth and have a postnatal progressive obliteration of bile ducts; the embryonic or fetal-onset form manifests at birth and is associated with other congenital anomalies (situs inversus, polysplenia, intestinal malrotation, complex congenital heart disease) within the polysplenia spectrum (biliary atresia splenic malformation [BASM]) (Fig. 348-2) (Chapter 425.11). The postnatal onset may be an immune- or infection-mediated process.
Biliary atresia has been detected in 1/10,000-15,000 live births.
Differentiation of Idiopathic Neonatal Hepatitis from Biliary Atresia
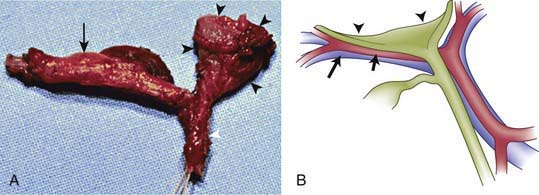
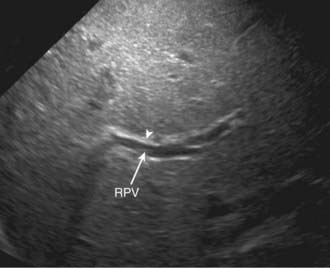
Abdominal ultrasound is a helpful diagnostic tool in evaluating neonatal cholestasis because it identifies choledocholithiasis, perforation of the bile duct, or other structural abnormalities of the biliary tree such as a choledochal cyst. In patients with biliary atresia, ultrasound can detect associated anomalies such as abdominal polysplenia and vascular malformations. The gallbladder either is not visualized or is a microgallbladder in patients with biliary atresia. Children with intrahepatic cholestasis caused by idiopathic neonatal hepatitis, cystic fibrosis, or total parenteral nutrition can have similar ultrasonographic findings. Ultrasonographic triangular cord (TC) sign, which represents a cone-shaped fibrotic mass cranial to the bifurcation of the portal vein, may be seen in patients with biliary atresia (Figs. 348-3 and 348-4). The echogenic density, which represents the fibrous remnants at the porta hepatis of biliary atresia cases at surgery, may be a helpful diagnostic tool in evaluating patients with neonatal cholestasis.
Management of Patients with Suspected Biliary Atresia
Some patients with biliary atresia, even of the “noncorrectable” type, derive long-term benefits from interventions such as the Kasai procedure. In most, a degree of hepatic dysfunction persists. Patients with biliary atresia usually have persistent inflammation of the intrahepatic biliary tree, which suggests that biliary atresia reflects a dynamic process involving the entire hepatobiliary system. This might account for the ultimate development of complications such as portal hypertension. The short-term benefit of hepatoportoenterostomy is decompression and drainage sufficient to forestall the onset of cirrhosis and sustain growth until a successful liver transplantation can be done (Chapter 360).
Management of Chronic Cholestasis
Treatment of such patients is empirical, and is guided by careful monitoring (Table 348-5). No therapy is known to be effective in halting the progression of cholestasis or in preventing further hepatocellular damage and cirrhosis.
Table 348-5 SUGGESTED MEDICAL MANAGEMENT OF PERSISTENT CHOLESTASIS
| CLINICAL IMPAIRMENT | MANAGEMENT |
|---|---|
| Malnutrition resulting from malabsorption of dietary long-chain triglycerides | Replace with dietary formula or supplements containing medium-chain triglycerides |
| Fat-soluble vitamin malabsorption: | |
| Vitamin A deficiency (night blindness, thick skin) | Replace with 10,000-15,000 IU/day as Aquasol A |
| Vitamin E deficiency (neuromuscular degeneration) | Replace with 50-400 IU/day as oral α-tocopherol or TPGS |
| Vitamin D deficiency (metabolic bone disease) | Replace with 5,000-8,000 IU/day of D2 or 3-5 µg/kg/day of 25-hydroxycholecalciferol |
| Vitamin K deficiency (hypoprothrombinemia) | Replace with 2.5-5.0 mg every other day as water-soluble derivative of menadione |
| Micronutrient deficiency | Calcium, phosphate, or zinc supplementation |
| Deficiency of water-soluble vitamins | Supplement with twice the recommended daily allowance |
| Retention of biliary constituents such as cholesterol (itch or xanthomas) | Administer choleretic bile acids (ursodeoxycholic acid, 15-30 mg/kg/day) |
| Progressive liver disease; portal hypertension (variceal bleeding, ascites, hypersplenism) | Interim management (control bleeding; salt restriction; spironolactone) |
| End-stage liver disease (liver failure) | Transplantation |
TPGS, D-tocopherol polyethylene glycol 1000 succinate.
Pruritus is a particularly troublesome complication of chronic cholestasis, often with the appearance of xanthomas. Both features seem to be related to the accumulation of cholesterol and bile acids in serum and in tissues. Elimination of these retained compounds is difficult when bile ducts are obstructed, but if there is any degree of bile duct patency, administration of ursodeoxycholic acid can increase bile flow or interrupt the enterohepatic circulation of bile acids and thus decrease the xanthomas and ameliorate the pruritus (see Table 348-5). Ursodeoxycholic acid therapy can also lower serum cholesterol levels. The recommended initial dose is 15 mg/kg/24 hr.
Progressive fibrosis and cirrhosis lead to the development of portal hypertension and consequently to ascites and variceal hemorrhage. The presence of ascites is a risk factor for the development of spontaneous bacterial peritonitis (SBP). The first step in the management of patients with ascites is to rule out SBP and restrict sodium intake to 0.5 g (∼1-2 mEq/kg/24 hr). There is no need for fluid restriction in patients with adequate renal output. Should this be ineffective, diuretics may be helpful. The diuretic of choice is spironolactone (3-5 mg/kg/24 hr in 4 doses). If spironolactone alone does not control ascites, the addition of another diuretic such as thiazide or furosemide may be beneficial. Patients with ascites but without peripheral edema are at risk for reduced plasma volume and decreased urine output during diuretic therapy. Tense ascites alters renal blood flow and systemic hemodynamics. Paracentesis and intravenous albumin infusion can improve hemodynamics, renal perfusion, and symptoms. Follow-up includes dietary counseling and monitoring of serum and urinary electrolyte concentrations (Chapters 356 and 359).
In patients with portal hypertension, variceal hemorrhage and the development of hypersplenism are common. It is important to ascertain the cause of bleeding because episodes of gastrointestinal hemorrhage in patients who have chronic liver disease may be due to gastritis or peptic ulcer disease. Because the management of these various complications differs, differentiation, perhaps via endoscopy, is necessary before treatment is initiated (Chapter 359). If the patient is volume depleted, blood transfusion should be carefully administered, avoiding overtransfusion, which can precipitate further bleeding. Balloon tamponade is not recommended in children because it can be associated with significant complications. Sclerotherapy or endoscopic variceal ligation may be useful palliative measures in the management of bleeding varices and may be superior to surgical alternatives.
For patients with advanced liver disease, hepatic transplantation has a success rate >85% (Chapter 360). If the operation is technically feasible, it will prolong life and might correct the metabolic error in diseases such as α1-antitrypsin deficiency, tyrosinemia, and Wilson disease. Success depends on adequate intraoperative, preoperative, and postoperative care and on cautious use of immunosuppressive agents. Scarcity of donors of small livers severely limits the application of liver transplantation for infants and children. The use of reduced-size transplants and living donors increases the ability to treat small children successfully.
348.2 Cholestasis in the Older Child
Cholestasis with onset after the neonatal period is most often caused by acute viral hepatitis or exposure to hepatotoxic drugs. However, many of the conditions causing neonatal cholestasis can also cause chronic cholestasis in older patients. Therefore, older children and adolescents with conjugated hyperbilirubinemia should be evaluated for acute and chronic viral hepatitis, α1-antitrypsin deficiency, Wilson disease, liver disease associated with inflammatory bowel disease, autoimmune hepatitis, and the syndromes of intrahepatic cholestasis. Other causes include obstruction caused by cholelithiasis, abdominal tumors, enlarged lymph nodes, or hepatic inflammation resulting from drug ingestion. Management of cholestasis in the older child is similar to that proposed for neonatal cholestasis (see Table 348-5).
Aagenaes O. Hereditary recurrent cholestasis with lymphedema: two new families. Acta Pediatr Scand. 1974;63:465-471.
Carlton VE, Harris BZ, Puffenberger EG, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91-96.
Chardot C, Serinet MO. Prognosis of biliary atresia: what can be further improved. J Pediatr. 2006;148:432-435.
Chen HL, Liu YJ, Su YN, et al. Diagnosis of BSEP/ABCB11 mutation in Asian patients with cholestasis using denaturing high performance liquid chromatography. J Pediatr. 2008;153:825-832.
Chen ST, Chen HL, Su YN, et al. Prenatal diagnosis of progressive familial intrahepatic cholestasis type 2. J Gastroenterol Hepatol. 2008;23:1390-1393.
Davenport M, Tizzard SA, Underhill J, et al. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr. 2006;149:393-400.
Debray D, Pariente D, Gauthier F, et al. Cholelithiasis in infancy: a study of 40 cases. J Pediatr. 1993;122:385.
Ekong UD, Melin-Aldana H, Whitington PF. Regression of severe fibrotic liver disease in 2 children with neonatal hemochromatosis. Pediatr Gastroenterol Nutr. 2008;46:329-333.
Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374:1704-1713.
Heubi JE, Setchell KDR, Bove KE. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27:282-294.
Hinds R, Davenport M, Mieli-Vergani G, et al. Antenatal presentation of the biliary atresia. J Pediatr. 2004;144:43-46.
Hoffenberg EJ, Narkewicz MR, Sondheimer JM, et al. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr. 1995;127:220.
Imanieh MH, Dehghani SM, Bagheri MH, et al. Triangular cord sign in detection of biliary atresia: is it a valuable sign? Dig Dis Sci. 2010;55:172-175.
Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004;40:27-38.
Mizuochi T, Kimura A, Ueki I, et al. Molecular genetic and bile acid profiles in two Japanese patients with 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase deficiency. Pediatr Res. 2010;68:258-263.
Muraji T, Hosala N, Irie N, et al. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes os maternal cells in the liver. Pediatrics. 2008;121:517-531.
Scheimann AO, Strautnieks SS, Kinsley AS, et al. Mutation in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr. 2007;150:556-559.
Schreiber RA, Barker CC, Roberts EA, et al. Biliary atresia: the Canadian experience. J Pediatr. 2007;151:659-665.
Serinet MO, Wildhaber BE, Broue P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123:1280-1286.
Setchell KD, Schwarz M, O’Connell NC, et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7α-hydroxylase gene causes severe neonatal liver disease. J Clin Invest. 1998;102:1690-1703.
Shih HH, Lin TM, Chuang JH, et al. Promotor polymorphism of the CD14 endotoxin receptor gene is associated with biliary atresia and idiopathic neonatal cholestasis. Pediatrics. 2005;116:437-441.
Shneider BL, Brown MB, Haver B, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467-474.
Sokol RJ. Biliary atresia screening: why, when, and how? Pediatrics. 2009;123:e951-e952.
Stringer MD, Dhawan A, Davenport M, et al. Choledochal cysts: lessons from a 20 year experience. Arch Dis Child. 1995;73:528-531.
Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217.
Utterson EC, Shepherd RW, Sokol RJ, et al. Biliary atresia: clinical profiles, risk factors, and outcomes of 755 patients listed for liver transplantation. J Pediatr. 2005;147:180-185.
van Mil SW, van der Woerd WL, van der Brugge G, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379-384.
Wadhwani SI, Turmelle YP, Nagy R, et al. Prolonged neonatal jaundice and the diagnosis of biliary atresia: a single-center analysis of trends in age at diagnosis and outcomes. Pediatrics. 2008;1221:e1438-e1440.
Whitington PF, Kelly S. Outcome of pregnancies at risk for neonatal hemochromatosis is improved by treatment with high-dose intravenous immunoglobulin. Pediatrics. 2008;121:e1615-e1621.
Willot S, Uhlein S, Michaud L, et al. Effect of ursodeoxycholic acid on liver function in children after successful surgery for biliary atresia. Pediatrics. 2008;122:e1236-e1241.
Yeh JN, Jeng YM, Chen HL, et al. Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants. J Pediatr. 2006;148:642-646.





