CHAPTER 179 Chiari Malformations
History
In the early 1890s, Dr. Hans Chiari, professor of pathologic anatomy at the German University in Prague, used autopsy specimens to describe four congenital anomalies later termed the Chiari malformations (types I to IV) (Table 179-1). Chiari was not the first to observe and report the type II malformations. In Observationes Medicae, written by the Dutch physician and anatomist Nicholas Tulp (1593-1674), reference is made to hindbrain herniation in a myelodysplastic individual.1 John Cleland (1835-1925) of Scotland reported a single myelodysplastic patient with hindbrain herniation and hydrocephalus in 1883. Temporally, Julius Arnold (1835-1915), professor of anatomy at Heidelberg, described a single myelodysplastic patient with hindbrain herniation and no hydrocephalus. Although the term Arnold-Chiari malformation has been used specifically in reference to hindbrain herniation in myelodysplastic patients, it was Chiari who described and attempted to delineate the pathophysiology of these posterior fossa abnormalities. As such, it is most appropriate to refer to this abnormality as the Chiari II malformation.2
| CHIARI TYPE | FEATURES |
|---|---|
| I |
Terminology
Chiari I Malformation
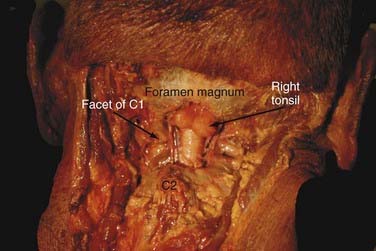
The Chiari I malformation (CIM) consists of caudal displacement of the cerebellar tonsils into the upper cervical spinal canal (Fig. 179-1). The most common associated findings are cervical syringomyelia and on occasion hydrocephalus. Multiple associations have been cited in the medical literature regarding this malformation. The full foramen magnum potentially compresses the herniated cerebellar tissue and restricts normal CSF flow across the craniocervical junction.
Chiari II Malformation
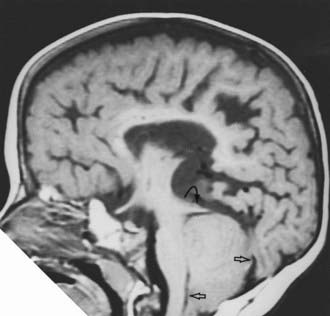
The Chiari II malformation (CIIM) is seen almost exclusively in the setting of myelodysplasia and hydrocephalus. In type II malformations, the structures herniating through the foramen magnum include the cerebellar vermis, brainstem, and fourth ventricle (Fig. 179-2). In addition to these neural structures, the accompanying choroid plexus and the associated basilar artery and posterior inferior cerebellar arteries may also be caudally displaced. The posterior fossa is often small and the foramen magnum expanded, and syringomyelia is seen in many of these patients.
Chiari III Malformation
The Chiari III malformation is the rarest of the Chiari malformations, with herniation of cerebellum and brainstem into a posterior encephalocele. This is the most severe form of hindbrain herniation, and its management is often problematic from both a technical and an ethical point of view. Lesions that prominently involve the posterior fossa contents must be distinguished from high cervical myelomeningoceles, which may look the same superficially but carry a more favorable prognosis.3,4 Patients with a Chiari III malformation generally have a poor prognosis. Severe neurological, developmental, and cranial nerve defects, in conjunction with seizures and respiratory insufficiency, are common. Surgical planning follows the same basic principles of encephalocele closure.
Chiari 0 Malformation
The Chiari 0 malformation is defined as syringomyelia without tonsillar herniation that responds to posterior fossa decompression. Iskandar and associates5 identified five patients with syringomyelia and no evidence of tonsillar herniation. MRI of the entire neuraxis ruled out other causes of a syrinx. Ultimately, abnormal CSF flow at the posterior fossa or foramen magnum was the suspected cause. All five patients underwent a posterior fossa decompression and duraplasty without direct fenestration or management of the syrinx. Significant syrinx and symptom resolution was observed in all patients. Their response to surgery suggests that “Chiari-like” pathophysiology may be present in the absence of tonsillar herniation, which may possibly be intermittent. This contention is supported by the identification of a crowded foramen magnum in two patients, multiple arachnoid adhesions in two others, and a fourth ventricular arachnoid veil in one. Each of these findings can alter CSF flow, although at present the pathophysiology resulting in syrinx formation in this group of patients is poorly understood. As such, a thorough evaluation to exclude other causes of syrinx is necessary before consideration of posterior fossa decompression.
Chiari 1.5 Malformation
Implying that only cerebellar tonsillar tissue is herniated through the foramen magnum, the Chiari I malformation (CIM) does not adequately define varieties of hindbrain herniation that also include descent of the brainstem. From our series of 130 surgical pediatric patients with CIM, we found that 17% had an additional component of brainstem descent.6,7
Signs and Symptoms
Chiari I Malformation
Patients with CIM may present with a variety of symptoms and signs ranging from headache to severe myelopathy and brainstem compromise (Table 179-2). The most common presenting symptom is pain (60% to 70%),8–10 usually occipital and upper cervical in location, and often induced by Valsalva maneuvers such as laughing, sneezing, and coughing. In infants and children who are unable to communicate verbally, headaches may manifest simply as crying and irritability. Other common symptoms include weakness or numbness, loss of temperature sensation, and unsteadiness. Careful investigation reveals that more than 70% to 80% of patients have some type of ophthalmologic or otologic disturbance at diagnosis. Ophthalmologic symptoms include blurry vision, nystagmus, extraocular muscle palsies, diplopia, and visual field deficits.11–14 Otologic complaints consist of tinnitus, fluctuating hearing loss, vertigo, and nausea. Signs at presentation have included weakness, atrophy, hyperreflexia, cape-like sensory loss, ataxia, and lower cranial nerve dysfunction. By stretching the centrally located ventral white commissure through a syrinx, pain and temperature signals cannot cross to the contralateral spinothalamic tract. The loss of pain and temperature sensation occurs only at the levels that are served by stretched spinothalamic fibers. Down-beat nystagmus is reported to be specific for lesions involving the cervicomedullary junction. Abnormal abdominal reflexes are seen in patients with associated syringomyelia. Children younger than 3 years are more likely to present with lower cranial nerve dysfunction.15 This can manifest as poor feeding, failure to thrive, recurrent aspiration pneumonia, dysphagia, choking, or stridor. A diminished gag reflex is common. Vocal cord paralysis with stridor or hoarseness may be present. An inability to maintain airway patency with lower cranial nerve dysfunction may promote sleep apnea, which may be a cause for sudden death in this group.
TABLE 179-2 Clinical Presentation of the Chiari I Malformation
| SYMPTOMS |
Spinal cord dysfunction is the result of direct cord compression or syringomyelia. The incidence of syringomyelia varies between 30% and 70% in CIM. Syrinx location is typically cervical, followed by cervicothoracic. Left untreated, permanent spinal cord damage can result. Progressive scoliosis is a relatively common manifestation (30%) of CIMs when there is coexistent syringomyelia.10,16 Clinical and radiographic signs that raise the suspicion of an underlying neurological defect in a patient with scoliosis include a convexity to the left, leg or foot asymmetry, male gender or prepubertal female, and obviously, any neurological deficit.3,4,16–19 The mechanism by which a syrinx causes scoliosis is not fully understood.
Chiari II Malformations
The CIIM occurs in most (>95%) patients with myelomeningocele and is the leading cause of death in treated myelodysplastic patients today. About one third of these patients develop brainstem symptoms by 5 years of age, and in excess of one third of those die, usually of respiratory failure (Table 179-3). In fact, as many as 20% of patients with symptomatic CIIMs may present as a neurological emergency. When presenting acutely, there is dysfunction of the 9th and 10th cranial nerves, affecting respiration, swallowing, and vocal cord functions; this is often accompanied by stridor, opisthotonos, and nystagmus. Symptomatic deterioration with progressive brainstem dysfunction may be irreversible and lead to death, irrespective of the type of therapy given or the speed with which treatment is instituted.20–23 This potentially catastrophic syndrome occurs most frequently in infants younger than 2 years, particularly younger than 3 months, and has been shown to be unrelated to intracranial pressure and the size or extent of symptoms of the neural tube defect.21 Patients with symptoms who survive the high-risk period (2 to 3 months of age) may improve and become clinically stable. Especially difficult to treat are neonates who fail to initiate adequate ventilation from birth. Although suspected of having inadequate respiratory drive centers and therefore little or no potential of sustained independent ventilation, their management is associated with a poor outcome and difficult ethical decisions.
TABLE 179-3 Clinical Presentation of the Chiari II Malformation
| NEWBORNS |
| (Usually asymptomatic) |
| INFANTS |
Unlike patients with CIM, there is a strong relationship between the type of symptoms and the age of onset. Newborns usually have symptoms. Older children and young adults most commonly display symptoms and signs of spinal cord and cerebellar dysfunction.2–424 A multitude of other symptoms and signs may occur in older patients. Common among these are ophthalmologic findings, which include strabismus, horizontal nystagmus (especially when looking upward), abnormalities of pursuit movements and convergence, and defects of optokinetic movements.25
Diagnostic Studies
Computed Tomography and Magnetic Resonance Imaging
Chiari I Malformations
The diagnosis of CIM should include the absence of an intracranial mass lesion, Dandy-Walker malformation, or hydrocephalus, all of which may cause tonsillar displacement secondary to raised intracranial pressure.26 The true incidence of CIM is not known. However, Meadows and colleagues27 found that of 22,591 patients who underwent MRI of the head, 175 (0.775%) were found to have tonsillar herniation extending more than 5 mm below the foramen magnum.
The position of the cerebellar tonsils in the normal population was studied by Aboulezz and colleagues.28 In a review of 800 MRI examinations, the authors noted that normal or asymptomatic patients may have tonsils that extend 3 mm below the foramen magnum. The tonsillar herniation was noted to be clearly pathologic when it exceeded 5 mm and borderline between 3 and 5 mm. Similarly, Barkovich and colleagues29 studied 200 normal patients and 25 patients with a “firm” diagnosis of CIM. The authors concluded that, in the absence of syringomyelia, 2 mm of tonsillar ectopia was of minimal clinical significance. Recently, Mikulis and associates30 determined that age affects the normal position of the cerebellar tonsils, with ascent of the tonsils with increasing age.
Other associated radiologic anomalies occur infrequently and include most commonly atlanto-occipital assimilation, platybasia, basilar invagination, and fused cervical vertebrae.26 Despite the less frequent occurrence, these changes should be sought because they can lead to cervical instability.
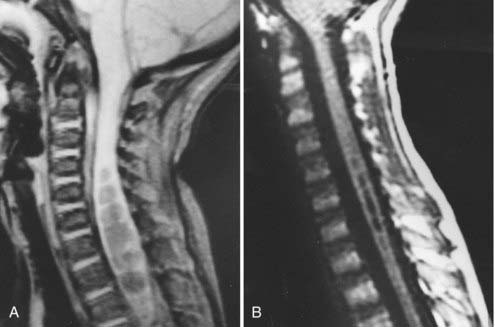
In addition to the absolute position of the tonsillar tips, the configuration of the tonsil is important. The tonsillar tip may be pointed and drawn out and may carry more pathologic significance, or it may be blunt and rounded and be less concerning. Syringomyelia occurs commonly with CIMs and can be seen in 50% to 70% of patients (Fig. 179-3).
Chiari II Malformations
CIIMs are characterized by the elongation and caudal displacement of the cerebellar vermis and brainstem, the presence of a myelomeningocele in virtually all cases and hydrocephalus in most, and the common (40% to 95%) existence of syringomyelia, especially in the lower cervical cord. However, the changes of the CIIM constitute a set of cranial and spinal malformations ranging from the posterior fossa, upper cervical canal, ventricular system, and neural tissues of the brain.31–37
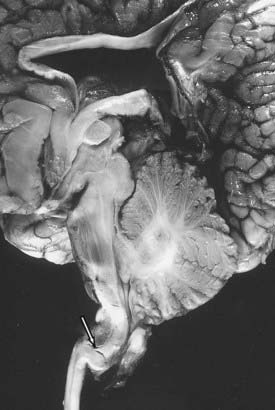
Associated neurological anomalies include tectal beaking, secondary to partial or complete fusion of the colliculi into a single backward pointed peak, and kinking at the level of the cervicomedullary junction (Fig. 179-4). The latter anomaly is caused by caudal displacement of a portion of the medulla in conjunction with a spinal cord that is held in relative immobility by the dentate ligament. The cerebellum is smaller than usual, and upward herniation of the cerebellum may be evident. Finally, callosal agenesis or dysgenesis, as well as abnormalities of the cerebral cortical pattern termed polygyria (not to be confused with the abnormal four-layered cortex seen in polymicrogyria) have frequently been described in Chiari II patients.34
In addition to hydrocephalus, the ventricular system displays multiple abnormalities. The third ventricle may be only mildly dilated and contains a large massa intermedia. The fourth ventricle is typically small or nonvisualized and often flattened and elongated, and it extends into the cervical canal. The lateral ventricles may be asymmetrically dilated, with prominence of the atria and occipital horns (colpocephaly), and the septum pellucidum is frequently absent. This sharpness of the frontal horn (lemon sign) and the caudal displacement of the fourth ventricle (banana sign) are relatively easily seen on in utero ultrasound examinations.38,39
The upper cervical canal also displays several bony and spinal cord anomalies in association with the CIIM. The posterior C1 arch is often missing. Klippel-Feil fusion anomalies of the cervical spine are present in a small group of patients. Basilar impression and C1 assimilation are uncommon in CIIM. Significant shortening and scalloping of the clivus can be seen.40 Other radiographic signs of CIIM include Lückenschädel, scalloping of the posterior surface of the petrous pyramid, falx hypoplasia, falx fenestration, tentorial hypoplasia with a wide incisura and tiny posterior fossa, and enlargement of the foramen.35,36
Cerebrospinal Fluid Flow Studies and Cine-Mode Magnetic Resonance Imaging
One technique to assist in the surgical decision making of patients with Chiari malformations is motion-sensitive MRI, or cine-mode MRI. As opposed to static MRI scanning, cine MRI41–44 may demonstrate lack of CSF flow patterns that can occur in the setting of CIM. The presence of a CIM and a syrinx resulting from disturbed CSF flow, which can be demonstrated by this technique, adds helpful information to the clinical assessment.45 Repeating the study postoperatively may help in evaluating the adequacy of the decompression. Having said this, we find cine MRI of minimal utility in clinical practice and especially in patient selection for operation.
Electrophysiologic Studies
The role of brainstem auditory evoked potentials (BAEPs) in Chiari patients has been investigated, especially in the Chiari II population. Worley and coworkers24 studied 37 newborn infants with myelomeningoceles at a median age of 8 days and followed them for 30 months. None of the infants clinically had brainstem dysfunction at initial testing. Of 12 infants who subsequently developed brainstem dysfunction at a median age of 3 months, 11 had abnormal BAEPs on the first evaluation. Conversely, of the 25 patients who did not develop clinical signs of brainstem dysfunction, 10 had abnormal BAEPs early on in life. Multiple case reports have noted clinical and electrophysiologic improvement after posterior fossa decompression in symptomatic Chiari II patients, demonstrating objective confirmation of the observed clinical improvement.45–47 The potential uses of BAEPs to evaluate neonatal myelomeningocele patients is still evolving, and this test alone is not a reliable indication for surgical intervention but should only be used as added criteria to a detailed history and physical examination.48
Pathophysiology
The theories of pathogenesis of the Chiari malformations may be grouped into the hindbrain dysgenesis and developmental arrest theory, the caudal traction theory, the hydrocephalus and hydrodynamic theory of Gardner, the small posterior fossa/hindbrain overgrowth theory, and the lack of embryologic ventricular distention theory.49
Chiari I Malformation
Nishikawa and coworkers50 suggested that underdevelopment of occipital somites within the paraxial mesoderm creates a small posterior fossa and CIM. This contention is supported by the association of CIM with other spine, skull, somatic, and craniofacial abnormalities, which are the result of mesodermal maldevelopment. The association of craniosynostosis and CIM is known and appears strongest in cases of syndromic, multisuture, and lambdoid synostosis. Lambdoid suture closure, typical in Crouzon’s syndrome, can directly reduce posterior fossa volume. Cephalocranial disproportion in multisuture synostosis can elevate intracranial pressure and promote herniation of posterior fossa elements.
Other medical conditions may promote formation of an abnormally small posterior fossa. Familial vitamin D–resistant rickets causes bony overgrowth of the posterior fossa, thus reducing its volume. Up to 20% of patients with growth hormone deficiency have CIM. Development of a craniospinal pressure gradient across the foramen magnum may cause or hasten the development of CIM. The gradient results from impaired CSF flow across the foramen magnum. Negative CSF pressure in the spinal compartment relative to the intracranial compartment creates a “sump effect” that forces the tonsils down through the foramen magnum. Once CSF flow is blocked at the foramen magnum, low intraspinal pressures can be accentuated and perpetuated by continuous absorption of CSF through spinal pathways, further worsening the clinical situation. Lumboperitoneal shunting, repetitive lumbar punctures, lumbar drainage, and chronic spinal CSF leaks of an iatrogenic nature are all familiar causes of an acquired CIM.51,52 Small posterior fossae have not been identified in all patients with CIM.53–55
Chiari II Malformation
Because the cerebellar vermis develops before the tonsils, an abnormal pressure differential that develops in utero would cause abnormal displacement of the vermis and brainstem structures, without any tonsillar involvement. Such a pressure differential could occur with fluid leakage from the myelomeningocele.4,56
McLone and Knepper49 have advanced a “unified theory,” which would explain the cause of the CIIM along with most, if not all, of the associated anomalies. This theory is based on the above-mentioned assumption that the neural tube defect occurs first, and all the other manifestations, including the Chiari malformation and hydrocephalus, follow secondarily. Leakage of CSF through the spinal defect causes a lack of distention of the primitive cranial ventricular system. In experimental animals, venting fluid from the embryonic ventricular system causes, for example, disorganization of the developing cerebral cortex and abnormal development of the pontine flexure.
Syringohydromyelia
In the 1960s, Gardner56 presented the hydrodynamic theory. Gardner’s theory stated that in normal embryology, CSF pulsations from the choroid plexus play a significant role in the expansion of the neural tube.57 According to Gardner, these pulsations help with the development of the arachnoid pathways as well as with modeling of the expanding brain. He believed that the balance between the pulsatile flow in the supratentorial and fourth ventricular choroid plexus directed brain growth differentially; therefore, if the fourth ventricular pulsations were overactive, the tentorium would be pushed upward, and a Dandy-Walker malformation could develop. Conversely, if the supratentorial pulsations were overactive, tentorial migration becomes such that the posterior fossa is small, allowing the development of a Chiari malformation; in addition, the CSF outlets of the fourth ventricle would remain closed, directing the CSF into the patent opening at the obex, thus causing syringomyelia.57,58
Based on experimental evidence using manometric measurements in normal and Chiari patients, Williams59 expanded on Gardner’s theory by suggesting that Valsalva maneuvers resulted in epidural venous congestion and intracranial as well as intraspinal pressure elevation, causing fluid to flow both cranially and caudally. Although flow into the cranial compartment meets no resistance, caudal flow is delayed by hindbrain adhesions and outlet obstruction, thus creating a pressure differential between the cranial and spinal compartments. This pressure differential may last a few seconds and cause worsening hindbrain impaction and syringomyelia. Repeat measurements were made after surgical decompression, showing equilibration of the pressures in the two compartments, which, in turn, correlated with clinical improvement. However, spinal cord cavitation is often acquired (as in posttraumatic syringomyelia), and a connection between the cyst and the fourth ventricle is not always present, which raises doubts about the adequacy of this theory.
Oldfield and associates60 investigated the anatomy and dynamics of movement of the cerebellar tonsils and CSF during the respiratory and cardiac cycles to explore the mechanism of syringomyelia progression in patients with CIM. During systole, there is normally movement of CSF in a caudal direction across the foramen magnum to counter the increased intracranial volume of blood and maintain physiologic intracranial pressure. This flow reverses in diastole. Dynamic movement of subarachnoid fluid is mirrored by caudal and cranial pulsations of fluid within the central canal during systole and diastole, respectively. In patients with CIM, the cerebellar tonsils are forced down and obstruct CSF flow across the foramen magnum during systole. This piston-like movement of the cerebellar tonsils imparts a systolic pressure wave in the spinal CSF that acts on the surface of the spinal cord, forcing fluid into the cord through perivascular and interstitial spaces. Oldfield and collaborators60 demonstrated the dynamic CSF flow into the syrinx in patients with CIM preoperatively by dynamic MRI and intraoperatively by ultrasound and further demonstrated the resolution of pathologic flow following bony and dural decompression. Postoperatively, adequate decompression of the foramen magnum allows resolution of the syringomyelia.
Treatment
Chiari I Malformation
There is no effective nonsurgical alternative to operative decompression for patients with symptomatic CIM. However, a detailed understanding of the natural history is not objectively known because the true incidence and prevalence are not well recognized. The challenge is identifying which patients will benefit most from posterior fossa decompression. Appropriate selection of patients likely to respond to surgical therapy can be challenging. Surgical indications vary among surgeons, especially for subjective symptoms such as headache. A 2004 opinion survey of neurosurgeons concluded there was widespread agreement to treat patients with a syrinx and progressive scoliosis or symptoms.61 Opinions were mixed on how to manage individuals with asymptomatic syringes as well as on the most appropriate surgical technique.
Once patients are determined to have idiopathic CIM, they are divided into two broad categories based on the presence or absence of a syrinx. All patients with a syrinx regardless of the size, location, or other associated symptoms are offered surgical intervention. This strategy is based on the belief that a syrinx is indicative of pathologic forces acting on the spinal cord that should be corrected to prevent permanent cord damage. However, a recently published study by Nishizawa and coworkers50 challenges this notion. They followed nine asymptomatic patients who had incidentally discovered syringes for 11 years with serial MRIs and clinical examinations. In this period, only one patient showed any deterioration that required surgery. The syringes in the others did not change. They concluded that small, asymptomatic syringes can be safely followed with serial examinations and imaging.
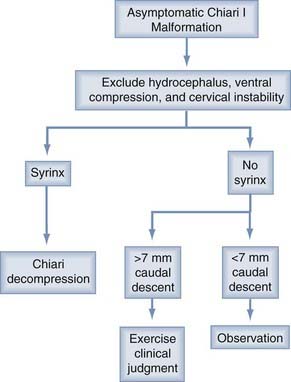
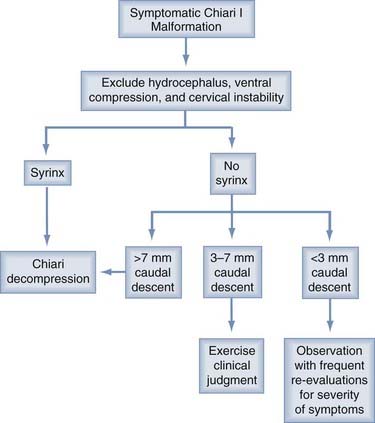
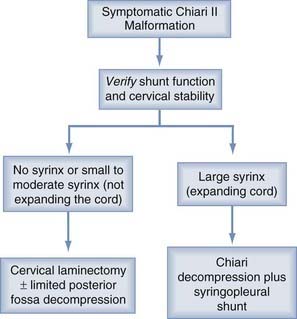
Some basic principals are generally agreed on. If hydrocephalus coexists or any question of raised intracranial pressure is present, this situation should be resolved before consideration is given to a decompression. Only a minority of patients (<10%) fall into this category. After shunting or third ventriculostomy, if symptoms persist or an associated syrinx is large and unchanged for months, then consideration should be given to a Chiari decompression (Figs. 179-5 and 179-6).
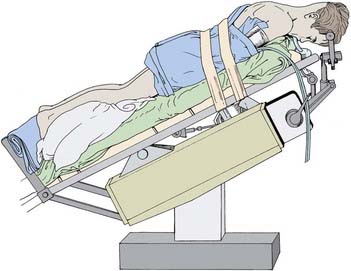
After a decision has been made to do a Chiari decompression, the patient is positioned prone with the head held flexed in a pin fixation device (Fig. 179-7). The midline incision begins in the mid-occiput, just below the inion, and extends to the second vertebra. The soft tissues and occipital musculature are separated in the midline through a relatively avascular plane. The foramen magnum and posterior arch of C1 are exposed the entire width of the dura. There is no reason for exposure more lateral than this because exposure of the vascular structures on each side of C1 carries risk without benefit. The bone of the occiput is removed, followed by the dorsal arch of C1. By leaving the muscle attachments and laminae of C2 intact, the postoperative pain and potential spinal instability are minimized. Rarely, it may be necessary to remove the superior aspect of the C2 laminae. By minimizing the opening and maximizing the decompression, we believe we have enhanced our surgical results and have not seen any cervical kyphosis or postoperative instability.
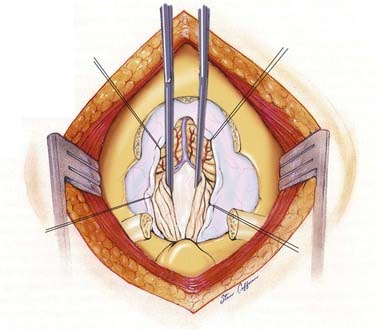
The purpose of the operation is to enlarge the bony area of the craniocervical junction and expand the dura surrounding the brainstem. By doing this, the surgeon wishes to relieve any direct compression and facilitate adequate outflow of CSF from the fourth ventricle. Some have championed the idea of bony decompression alone, and this is being revisited at a number of institutions currently. Alternatively, some surgeons split the dura, opening only the outer layer. Although these methods may be sufficient in many patients, in the more severe forms of tonsillar herniation, especially when a syrinx is also present or intradural arachnoid adhesions are identified preoperatively, an intradural approach is needed. Of help in determining the need for dural grafting and intradural exploration is visualization of the movement of the tonsils seen through the dura or by intraoperative ultrasound. Once the dura is opened in the midline, the tonsils are gently separated to inspect for veils covering the outlets of the fourth ventricle (Fig. 179-8). This re-establishes free flow of CSF from the foramen of Magendie. We consider visualization of the choroid plexus of the fourth ventricle and free flow of CSF into the subarachnoid space as evidence of adequate decompression. Occasionally, in cases with severe tonsillar ectopia, the foramen of Magendie remains occluded, and CSF egress is limited. Extrapial coagulation of one or both tonsillar tips shrinks the tonsils sufficiently to restore CSF flow.
Finally, the need for a dural graft has been debated.62 We believe that such a graft allows for protection of the neural tissues from chemical contamination while providing a capacious dural sac, which probably optimizes the decompression and minimizes postoperative adhesions. We prefer pericranium as the graft. After decompression, the fasciae and skin are closed in a routine manner.
A challenging group of patients includes those in whom there is a significant component of ventral compression. We have defined significant ventral compression as greater than 9 mm of reclination of the odontoid process from a line connecting the basion to the posterior aspect of the body of the axis (Fig. 179-9).63 In these patients, spinal stability should be ensured by performing dynamic cervical radiographs in flexion and extension before decompression. These patients often present with a greater degree of brainstem symptoms and may not benefit from posterior decompression alone. In our hands, dorsal decompression is addressed first with close observation in an intensive care setting postoperatively. Symptoms and signs of respiratory compromise, swallowing difficulty, and hemodynamic instability herald ongoing brainstem compression and warrant occipitocervical stabilization and possibly ventral decompression.
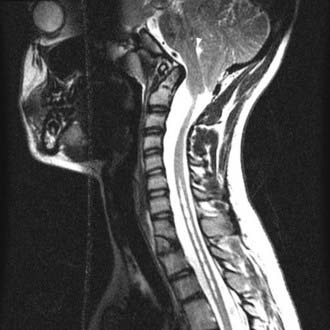
Chiari II Malformation
Early surgical intervention may prove life sustaining in Chiari II patients whose symptoms are referable to the medullary dysfunction.18,19 Timing of decompression before bilateral vocal cord paralysis may predict a better outcome.63 Pollack and colleagues64 have described favorable outcomes for patients undergoing early surgical intervention, that is, occipital craniectomy and cervical laminectomy for neurogenic dysphagia.
Indications for surgical intervention are based on clinical symptoms. The presence of a significant syrinx or one that has progressively enlarged should be considered for therapy. Symptoms include inspiratory stridor at rest or progressive by history, aspiration pneumonia due to palatal dysfunction or gastroesophageal reflux, central apnea with or without cyanosis, especially during sleep, opisthotonus, functionally significant or progressive spasticity of the upper extremities, and functionally significant or progressive truncal or limb ataxia.23,24
Before considering surgical decompression, patients with symptomatic Chiari II malformation must have physiologic intracranial pressure. This assurance requires a functioning shunt. It has been our experience and that of others, that a properly functioning ventricular shunt can often obviate the need for decompression of hindbrain herniation. Many times, this can only be determined by surgical inspection of the shunt. Caldarelli and colleagues20 reported that of 11 symptomatic CIIM patients, 5 had resolution of their symptoms following shunt revision alone. Milhorat and associates65 found in a retrospective study of a small number of patients that improvement in the size of their syrinx was observed after only ventriculoperitoneal shunting or revision. Tomita and McLone66 concluded that shunt revision can reverse acute respiratory arrest. In contrast, lower cranial nerve findings may not improve after the shunt revision but rather only after posterior fossa decompression.
One important caveat is that stable ventricular size is not a reliable indicator of a functioning ventricular shunt. Iskandar and colleagues67 found that in 20% of myelodysplastic children with shunted hydrocephalus, CT studies were unchanged in the presence of shunt malfunction as demonstrated by surgical exploration following continued clinical decline. After shunt revision, all patients in this myelodysplasia group had resolution of preoperative symptoms (Figs. 179-10 and 179-11).
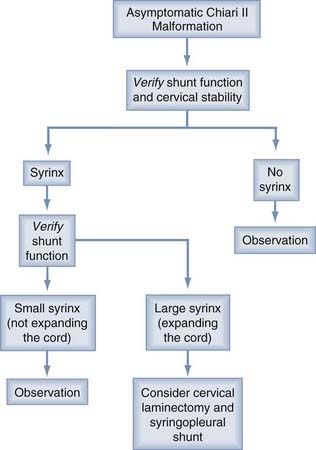
FIGURE 179-10 Flow diagram for the treatment of asymptomatic patients with the Chiari II malformation.
Complications
Posterior fossa decompression is relatively safe but not without complications. Direct vascular or neural injury, pseudomeningocele, CSF leaks, and meningitis are well recognized. Bleeding from dural venous lakes can be profuse at times. Less common complications include occipital-cervical instability, acute postoperative hydrocephalus secondary to infratentorial hygromas, and anterior brainstem compression from a retroflexed odontoid.63,68 A complication unique to posterior fossa craniectomies is cerebellar slump or ptosis, which results from extending a craniectomy so far laterally that the cerebellum herniates through the craniectomy defect. This can cause headaches (different from typical Chiari I headaches), obstruction of CSF flow with syrinx formation, and a variety of motor, sensory, and cranial nerve deficits. Cranioplasty to buttress the cerebellum into place is the most definitive treatment. Simple shunting is not adequate. We have not seen this complication in more than 400 patients by limiting the bony removal to the width of the spinal dura. Complications in our early series of 130 patients were minimal and included acute postoperative hydrocephalus in 2 patients, requiring temporary external ventricular drainage. Severe anterior brainstem compression from a retroflexed odontoid required a transoral odontoidectomy in 1 patient. Neither pseudomeningocele, meningitis, nor cerebellar slump was observed.6 In an attempt to more specifically categorize these complications in Chiari I patients and syringomyelia, Menezes69 reviewed a series of 35 children and identified a set of complications associated with surgery. These included excessive bleeding from venous lakes, failure to get into the fourth ventricle secondary to adhesions, persistent variation of blood pressure and heart rate, failure to awaken, respiratory compromise, and weakness. Although any of these can occur, many can be avoided by meticulous preparation and surgical execution and a thorough understanding of the pathology.
Results and Prognosis
Chiari I Malformation
With the natural history known incompletely, surgical data accumulated from a series of retrospective studies claiming to improve on the natural history should be looked at cautiously. Saez and associates70 attempted to classify patients into preoperative prognostic categories. The poorest prognosis was seen in patients with central cord signs; the best prognosis was found in patients with paroxysmal intracranial hypertension.
Chiari II Malformation
The natural history of these lesions can be disastrous. Therefore, the idea of surgical intervention early in a child with symptoms is widely accepted, although only a few studies exploring the role of decompression in Chiari II patients are available in the literature. Pollack and colleagues64 have prospectively treated neonates and young infants displaying symptomatic CIIMs with urgent laminectomies and limited suboccipital craniectomies. Ten of the 13 children recovered normal or almost normal neurological function postoperatively, whereas the other 3 exhibited bilateral vocal cord paralysis and severe central hypoventilation. The authors concluded that early recognition and treatment of brainstem compromise in Chiari II patients produces prompt and long-term clinical recovery.
Follow-Up
Chiari I Malformation
From our initial series of CIM, eight patients required a second posterior fossa exploration for syrinx persistence.6 Again, no attempt was made to manipulate the syrinx. The second surgery is more aggressive than the first. Unilateral tonsillar coagulation was performed for each patient. Stents were placed in four patients when free egress of CSF from the fourth ventricle was not achieved after lysis of subarachnoid adhesions and tonsil coagulation, or when no obvious pathology explaining syrinx persistence was identified. Syringes improved in four of six patients, and imaging studies are pending in the others. Interestingly, stents were not placed in the two patients who failed to improve. We stress that re-exploration of the posterior fossa is the best strategy for dealing with a recalcitrant syrinx.
Aboulezz AO, Sartor K, Geyer CA, et al. Position of cerebellar tonsils in the normal population and in patients with Chiari malformation: a quantitative approach with MR imaging. J Comput Assist Tomogr. 1985;9:1033-1036.
Badie B, Mendoza D, Batzdorf U. Posterior fossa volume and response to suboccipital decompression in patients with Chiari I malformation. Neurosurgery. 1995;37:214-218.
Curnes JT, Oakes WJ, Boyko OB. MR imaging of hindbrain deformity in Chiari II patients with and without symptoms of brainstem compression. AJNR Am J Neuroradiol. 1989;10:293-302.
Elster AD, Chen MY. Chiari I malformations: clinical and radiologic reappraisal. Radiology. 1992;183:347-353.
Grabb PA, Mapstone TB, Oakes WJ. Ventral brain stem compression in pediatric and young adult patients with Chiari I malformations. Neurosurgery. 1999;44:520-527.
Greenlee JD, Donavan KA, Hasan DM, et al. Chiari I malformation in the very young child: the spectrum of presentations and experience in 31 children under age 6 years. Pediatrics. 2002;110:1212-1219.
Heinz R, Curnes J, Friedman A, et al. Exophytic syrinx, an extreme form of syringomyelia: CT, myelographic, and MR imaging features. Radiology. 1992;183:243-246.
Iskandar B, McLaughlin C, Mapstone TB, et al. Pitfalls in the diagnosis of ventricular shunt dysfunction: radiology reports and ventricular size. Pediatrics. 1998;101:1031-1036.
Iskandar BJ, Hedlund GL, Grabb PA, et al. The resolution of syringohydromyelia without hindbrain herniation after posterior fossa decompression. J Neurosurg. 1998;89:212-216.
Marin-Padilla M, Marin-Padilla TM. Morphogenesis of experimentally induced Arnold-Chiari malformation. J Neurol Sci. 1981;50:29-55.
McLendon RE, Crain BJ, Oakes WJ, et al. Cerebral polygyria in the Chiari Type II (Arnold-Chiari) malformation. Clin Neuropathol. 1985;4:200-205.
McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
Meadows J, Kraut M, Guarnieri M, et al. Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000;92:920-926.
Menezes AH. Chiari I malformations and hydromyelia: complications. Pediatr Neurosurg. 1991;17:146-154.
Naidich TP, McLone DG, Fulling KH. The Chiari II malformation: Part IV. The hindbrain deformity. Neuroradiology. 1983;25:179-197.
Naidich TP, Pudlowski RM, Naidich JB, et al. Computed tomographic signs of the Chiari II malformation. Part I: Skull and dural partitions. Radiology. 1980;134:65-71.
Naidich TP, Pudlowski RM, Naidich JB. Computed tomographic signs of Chiari II malformation. II: Midbrain and cerebellum. Radiology. 1980;134:391-398.
Naidich TP, Pudlowski RM, Naidich JB. Computed tomographic signs of the Chiari II malformation. III: Ventricles and cisterns. Radiology. 1980;134:657-663.
Nohria V, Oakes WJ. Chiari I malformation: a review of 43 patients. Pediatr Neurosurg. 1990;16:222-227.
Oakes WJ. The Chiari malformations of the child. In: Menezes AH, Sonntag VKH, editors. Principles of Spinal Surgery. New York: McGraw-Hill; 1996:379-394.
Paul KS, Lye RH, Strang FA, et al. Arnold-Chiari malformation. Review of 71 cases. J Neurosurg. 1983;58:183-187.
Pollack IF, Kinnunen D, Albright AL. The effect of early craniocervical decompression on functional outcome in neonates and young infants with myelodysplasia and symptomatic Chiari II malformations: results from a prospective series. Neurosurgery. 1996;38:703-710.
Tubbs RS, McGirt MJ, Oakes WJ. Surgical experience in 130 pediatric patients with Chiari I malformations. J Neurosurg. 2003;99:291-296.
Tubbs RS. Oakes: Chiari Malformations. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: Saunders; 2004:3347-3361.
Williams B. Cerebrospinal fluid pressure-gradients in spina bifida cystica, with special reference to the Arnold-Chiari malformation and aqueductal stenosis. Dev Med Child Neurol Suppl. 1975;35:138-150.
1 Koehler PJ. Chiari’s description of cerebellar ectopy (1891). With a summary of Cleland’s and Arnold’s contributions and some early observations on neural-tube defects. J Neurosurg. 1991;75:823-826.
2 Tubbs RS. Oakes: Chiari malformations. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: Saunders; 2004:3347-3361.
3 Oakes WJ. The Chiari malformations of the child. In: Menezes AH, Sonntag VKH, editors. Principles of Spinal Surgery. New York: McGraw-Hill; 1996:379-394.
4 Oakes WJ. Chiari malformations, hydromyelia, syringomyelia. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1996:3593-3616.
5 Iskandar BJ, Hedlund GL, Grabb PA, et al. The resolution of syringohydromyelia without hindbrain herniation after posterior fossa decompression. J Neurosurg. 1998;89:212-216.
6 Tubbs RS, McGirt MJ, Oakes WJ. Surgical experience in 130 pediatric patients with Chiari I malformations. J Neurosurg. 2003;99:291-296.
7 Tubbs RS, Iskandar BJ, Bartolucci AA, et al. A critical analysis of the Chiari 1.5 malformation. J Neurosurg. 2004;101:179-183.
8 Levy WJ, Mason L, Hahn JF. Chiari malformation presenting in adults: a surgical experience in 127 cases. Neurosurgery. 1983;12:377-390.
9 Paul KS, Lye RH, Strang FA, et al. Arnold-Chiari malformation. Review of 71 cases. J Neurosurg. 1983;58:183-187.
10 Nohria V, Oakes WJ. Chiari I malformation: a review of 43 patients. Pediatr Neurosurg. 1990;16:222-227.
11 Passo M, Shults WT, Talbot T, et al. Acquired esotropia. A manifestation of Chiari I malformation. J Clin Neuro ophthalmol. 1984;4:151-154.
12 Lewis AR, Kline LB, Sharpe JA. Acquired esotropia due to Arnold-Chiari malformation. J Neuro ophthalmol. 1996;16:49-54.
13 Bronstein AM, Miller DH, Rudge P, et al. Down beating nystagmus: magnetic resonance imaging and neuro-otological findings. J Neurol Sci. 1987;81:173-184.
14 Gingold SI, Winfield JA. Oscillopsia and primary cerebellar ectopia: case report and review of the literature. Neurosurgery. 1991;29:932-936.
15 Greenlee JD, Donavan KA, Hasan DM, et al. Chiari I malformation in the very young child: the spectrum of presentations and experience in 31 children under age 6 years. Pediatrics. 2002;110:1212-1219.
16 Muhonen MG, Menezes AH, Sawin PD, et al. Scoliosis in pediatric Chiari malformations without myelodysplasia. J Neurosurg. 1992;77:69-77.
17 Lewonowski K, King JD, Nelson MD. Routine use of magnetic resonance imaging in idiopathic scoliosis patients less than eleven years of age. Spine. 1992;17:S109-S116.
18 Nokes SR, Murtagh FR, Jones JD, et al. Childhood scoliosis: MR imaging. Radiology. 1987;164:791-797.
19 Isu T, Chono Y, Iwasaki Y, et al. Scoliosis associated with syringomyelia presenting in children. Childs Nerv Syst. 1992;8:97-100.
20 Caldarelli M, Ceddia A, Di Rocco C, et al. Chiari type II malformation: a rare neurologic emergency. J Pediatr Neurosci. 1987;3:191-205.
21 Wealthall SR, Whittaker GE, Greenwood N. The relationship of apnoea and stridor in spina bifida to other unexplained infant deaths. Dev Med Child Neurol. 1974;16:107-116.
22 Yamada H, Tanaka Y, Nakamura S. Laryngeal stridor associated with the Chiari II malformation. Childs Nerv Syst. 1985;1:312-318.
23 Cochrane DD, Adderley R, White CP, et al. Apnea in patients with myelomeningocele. Pediatr Neurosurg. 1990;16:232-239.
24 Worley G, Erwin CW, Schuster JM, et al. BAEPs in infants with myelomeningocele and later development of Chiari II malformation-related brainstem dysfunction. Dev Med Child Neurol. 1994;36:707-715.
25 Lennerstrand G, Gallo JE, Samuelsson L. Neuro-ophthalmological findings in relation to CNS lesions in patients with myelomeningocele. Dev Med Child Neurol. 1990;32:423-431.
26 Elster AD, Chen MY. Chiari I malformations: clinical and radiologic reappraisal. Radiology. 1992;183:347-353.
27 Meadows J, Kraut M, Guarnieri M, et al. Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000;92:920-926.
28 Aboulezz AO, Sartor K, Geyer CA, et al. Position of cerebellar tonsils in the normal population and in patients with Chiari malformation: a quantitative approach with MR imaging. J Comput Assist Tomogr. 1985;9:1033-1036.
29 Barkovich AJ, Wippold FJ, Sherman JL, et al. Significance of cerebellar tonsillar position on MR. AJNR Am J Neuroradiol. 1986;7:795-799.
30 Mikulis DJ, Diaz O, Egglin TK, et al. Variance of the position of the cerebellar tonsils with age: preliminary report. Radiology. 1992;183:725-728.
31 Curnes JT, Oakes WJ, Boyko OB. MR imaging of hindbrain deformity in Chiari II patients with and without symptoms of brainstem compression. AJNR Am J Neuroradiol. 1989;10:293-302.
32 Naidich TP, Pudlowski RM, Naidich JB. Computed tomographic signs of Chiari II malformation. II: Midbrain and cerebellum. Radiology. 1980;134:391-398.
33 Naidich TP, McLone DG, Fulling KH. The Chiari II malformation: Part IV. The hindbrain deformity. Neuroradiology. 1983;25:179-197.
34 McLendon RE, Crain BJ, Oakes WJ, et al. Cerebral polygyria in the Chiari Type II (Arnold-Chiari) malformation. Clin Neuropathol. 1985;4:200-205.
35 Naidich TP, Pudlowski RM, Naidich JB, et al. Computed tomographic signs of the Chiari II malformation. Part I: Skull and dural partitions. Radiology. 1980;134:65-71.
36 Schmitt HP. “Inverse Chiari type II syndrome” in untreated hydrocephalus and its relationship to typical Arnold-Chiari syndrome. Brain Dev. 1981;3:271-275.
37 Naidich TP, Pudlowski RM, Naidich JB. Computed tomographic signs of the Chiari II malformation. III: Ventricles and cisterns. Radiology. 1980;134:657-663.
38 Van den Hof MC, Nicolaides KH, Campbell J, et al. Evaluation of the lemon and banana signs in one hundred thirty fetuses with open spina bifida. Am J Obstet Gynecol. 1990;162:322-327.
39 Nicolaides KH, Campbell S, Gabbe SG, et al. Ultrasound screening for spina bifida: cranial and cerebellar signs. Lancet. 1986;2:72-74.
40 Yu HC, Deck MD. The clivus deformity of the Arnold-Chiari malformation. Radiology. 1971;101:613-615.
41 Heinz R, Curnes J, Friedman A, et al. Exophytic syrinx, an extreme form of syringomyelia: CT, myelographic, and MR imaging features. Radiology. 1992;183:243-246.
42 Samuelsson L, Bergstrom K, Thuomas KA, et al. MR imaging of syringomyelia and Chiari malformations in myelomeningocele patients with scoliosis. AJNR Am J Neuroradiol. 1987;8:539-546.
43 Bhadelia RA, Bogdan AR, Wolpert SM, et al. Cerebrospinal fluid flow waveforms: analysis in patients with Chiari I malformation by means of gated phase-contrast MR imaging velocity measurements. Radiology. 1995;196:195-202.
44 Armonda RA, Citrin CM, Foley KT, et al. Quantitative cine-mode magnetic resonance imaging of Chiari I malformations: an analysis of cerebrospinal fluid dynamics. Neurosurgery. 1994;35:214-223.
45 Tachibana S, Iida H, Yada K. Significance of positive Queckenstedt test in patients with syringomyelia associated with Arnold-Chiari malformations. J Neurosurg. 1992;76:67-71.
46 Stone JL, Bouffard A, Morris R, et al. Clinical and electrophysiologic recovery in Arnold-Chiari malformation. Surg Neurol. 1983;20:313-317.
47 Holliday PO, Pillsbury D, Kelly DL, et al. Brain stem auditory evoked potentials in Arnold-Chiari malformation: possible prognostic value and changes with surgical decompression. Neurosurgery. 1985;16:48-53.
48 Morioka T, Kurita-Tashima S, Fujii K, et al. Somatosensory and spinal evoked potentials in patients with cervical syringomyelia. Neurosurgery. 1992;30:218-222.
49 McLone DG, Knepper PA. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
50 Nishikawa M, Sakamoto H, Hakuba A, et al. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40-47.
51 Sathi S, Stieg PE. “Acquired” Chiari I malformation after multiple lumbar punctures: case report. Neurosurgery. 1993;32:306-309.
52 Chumas PD, Drake JM, Del Bigio MR. Death from chronic tonsillar herniation in a patient with lumboperitoneal shunt and Crouzon’s disease. Br J Neurosurg. 1992;6:595-599.
53 Badie B, Mendoza D, Batzdorf U. Posterior fossa volume and response to suboccipital decompression in patients with Chiari I malformation. Neurosurgery. 1995;37:214-218.
54 Marin-Padilla M, Marin-Padilla TM. Morphogenesis of experimentally induced Arnold-Chiari malformation. J Neurol Sci. 1981;50:29-55.
55 Tubbs RS, Webb D, Abdullatif H, et al. Posterior cranial fossa volume in patients with rickets: insights into the increased occurrence of Chiari I malformation in metabolic bone disease. Neurosurgery. 2004;55:380-383.
56 Gardner WJ. Hydrodynamic mechanism of syringomyelia: its relationship to myelocele. J Neurol Neurosurg Psychiatr. 1965;28:247-259.
57 Pillay PK, Awad IA, Hahn JF. Gardner’s hydrodynamic theory of syringomyelia revisited. Cleve Clin J Med. 1991;59:373-380.
58 Sehgal AD. Chiari I and syringomyelia [letter; comment]. J Neurosurg. 1994;81:811-813.
59 Williams B. Cerebrospinal fluid pressure-gradients in spina bifida cystica, with special reference to the Arnold-Chiari malformation and aqueductal stenosis. Dev Med Child Neurol Suppl. 1975;35:138-150.
60 Oldfield EH, Muraszko K, Shawker TH, et al. Pathophysiology of syringomyelia associated with Chiari I malformation of the cerebellar tonsils. Implications for diagnosis and treatment. J Neurosurg. 1994;80:3-15.
61 Schijman E, Steinbok P. International survey on the management of Chiari I malformation and syringomyelia. Childs Nerv Syst. 2004;20:341-348.
62 Williams B. A blast against grafts: on the closing and grafting of the posterior fossa dura. Br J Neurosurg. 1994;8:275-278.
63 Grabb PA, Mapstone TB, Oakes WJ. Ventral brain stem compression in pediatric and young adult patients with Chiari I malformations. Neurosurgery. 1999;44:520-527.
64 Pollack IF, Kinnunen D, Albright AL. The effect of early craniocervical decompression on functional outcome in neonates and young infants with myelodysplasia and symptomatic Chiari II malformations: results from a prospective series. Neurosurgery. 1996;38:703-710.
65 Milhorat TH, Johnson WD, Miller JI, et al. Surgical treatment of syringomyelia based on magnetic resonance imaging criteria. Neurosurgery. 1992;31:231-244.
66 Tomita T, McLone DG. Acute respiratory arrest. A complication of malformation of the shunt in children with myelomeningocele and Arnold-Chiari malformation. Am J Dis Child. 1983;137:142-144.
67 Iskandar B, McLaughlin C, Mapstone TB, et al. Pitfalls in the diagnosis of ventricular shunt dysfunction: radiology reports and ventricular size. Pediatrics. 1998;101:1031-1036.
68 Elton S, Tubbs RS, Wellons JC3rd. Acute hydrocephalus following a Chiari I decompression. Pediatr Neurosurg. 2002;36:101-104.
69 Menezes AH. Chiari I malformations and hydromyelia: complications. Pediatr Neurosurg. 1991;17:146-154.
70 Saez RJ, Onofrio BM, Yanagihara T. Experience with Arnold-Chiari malformation, 1960 to 1970. J Neurosurg. 1976;45:416-422.






