[level-membership-for-neurology-category]
Chapter 64 Channelopathies
Episodic and Electrical Disorders of the Nervous System

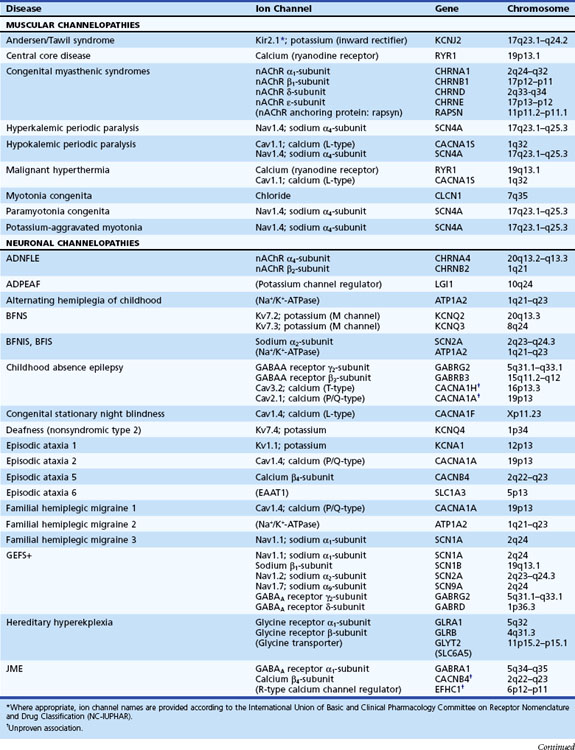
Channelopathies are disorders caused by ion channel dysfunction. Because of the great diversity of ion channel proteins and their expression in different tissues, channelopathies comprise a wide variety of clinical diseases (Table 64.1), the discovery of which helps elucidate how ion channels function in both illness and health. The periodic paralyses—the first group of ion channel disorders characterized at a molecular level—defined the field of channelopathies, which now encompasses diseases not only in muscle but also in the kidney (Bartter syndrome), epithelium (cystic fibrosis), and heart (long QT syndrome) as well as neurons. Because muscles and neurons are electrical organs, it is not surprising that most channelopathies are associated with neurological disease. Despite significant heterogeneity, a pervasive feature of neurological channelopathies is a paroxysmal phenotype. After a brief introduction to ion channels, this chapter describes disorders caused by congenital and acquired dysfunction of ion channels expressed in skeletal muscle and neurons.
Ion Channels
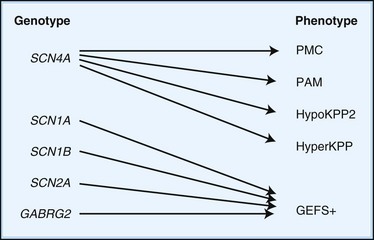
Depending on the location within the channel, mutations could alter voltage-dependent activation, ion selectivity, or time and voltage dependence of inactivation. Thus, two different mutations within the same gene can result in dramatically different physiological defects. For example, a mutation that prevents or slows inactivation could lead to a persistent ionic current. Conversely, a mutation elsewhere in the same gene that prevents activation will decrease ionic current. Phenotypic heterogeneity describes how different mutations in a single gene cause distinct phenotypes. For instance, mutations in the skeletal muscle voltage-dependent sodium channel can result in hyperkalemic periodic paralysis, hypokalemic periodic paralysis, potassium-aggravated myotonia, or paramyotonia congenita (see Table 64.1 and Fig. 64.1). In contrast, genetic heterogeneity occurs when a consistent clinical syndrome results from a variety of underlying mutations.
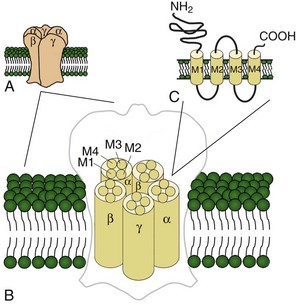
Voltage-gated potassium channels (VGKC) consist of four homologous α-subunits that combine to create a complete channel. Like the other channels described later, humans possess many distinct VGKC genes, and the resulting channels exhibit specialized properties and display rich tissue-type and cellular-compartment specificity. Each α-subunit contains six transmembrane segments (S1 to S6) linked by extracellular and intracellular loops (Fig. 64.2). The S5-S6 loop penetrates deep into the central part of the channel and lines the pore. The S4 segment contains positively charged amino acids and acts as the voltage sensor. These channels serve many functions, most notably to establish the resting membrane potential and to repolarize cells following an action potential. A unique class of potassium channel, the inwardly rectifying potassium channel, is homologous to the S5 to S6 segment of the VGKC. Because the voltage-sensing S4 domain is absent, voltage dependence results from a voltage-dependent blockade by magnesium and polyamines.
Voltage-gated sodium and calcium channels are highly homologous and share homology with VGKCs, from which they evolved. The α-subunits contain four highly homologous domains in tandem within a single transcript (DI–DIV) (Fig. 64.3). Each domain resembles a VGKC α-subunit, with six transmembrane segments as described earlier. Sodium and calcium channels differ in several regards, despite their many similarities. The amino acid sequence forming the selectivity filter and the modulatory auxiliary subunits are different. The sodium channel is composed of an α- and a β-subunit, and the calcium channel is composed of a pore-forming α1-subunit, an intracellular β-subunit, a membrane-spanning γ-subunit, and a membrane-anchoring α2δ-subunit. Sodium channels mediate fast depolarization and underlie the action potential, whereas voltage-gated calcium channels (VGCCs) mediate neurotransmitter release and allow the calcium influx that leads to second messenger effects.
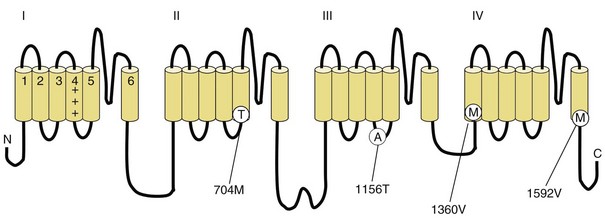
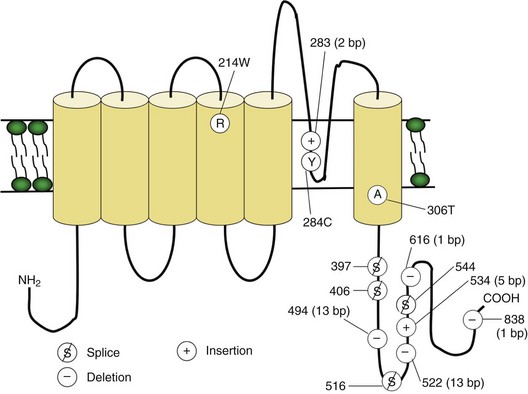
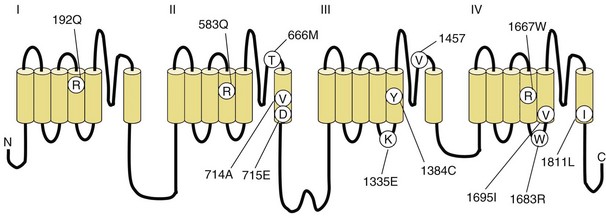
Fig. 64.3 Diagram of the voltage-gated sodium channel and the four most common mutations causing hyperkalemic periodic paralysis. The α-subunit consists of four highly homologous domains (I to IV), each containing six transmembrane segments (S1 to S6). When inserted into the membrane, the four domains fold so as to encircle a central ion-selective pore lined by the S5/S6 loop. Analogous to the potassium channel (see Fig. 64.2), the S4 segments contain positively charged residues conferring voltage dependence on the channel. Auxiliary β-subunits are not shown.
Ligand-gated ion channels activate on binding with their respective agonists. Among the diverse array of ligand-gated channels, GABAA, glycine, and nicotinic acetylcholine receptors (nAChRs) possess the only known disease-causing mutations. Although distinguished by their ligand binding and ion permeability, channels gated by GABA, glycine, and acetylcholine share several structural similarities. Five intrinsic membrane subunits assemble to form hetero- or homopentamers. Each subunit contains four transmembrane domains (M1 to M4), the second of which lines the pore and determines ionic selectivity (Fig. 64.4). Subunits contributing to nAChRs at the neuromuscular junction differ from those expressed in the central nervous system, explaining why mutation of one gene may cause seizures without affecting neuromuscular transmission, or vice versa. Binding of acetylcholine opens the channel, which conducts monovalent cations (Na+ and K+) with little or no selectivity, and some are additionally permeable to calcium. Channel activation results in membrane depolarization and excitation of the postsynaptic neuron or muscle fiber.
Genetic Disorders of Muscular Ion Channels
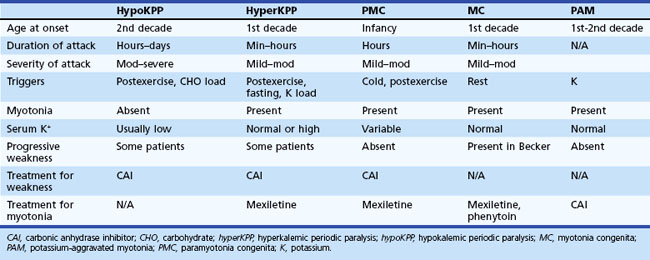
The periodic paralyses and nondystrophic myotonias encompass several skeletal muscle disorders inherited as autosomal dominant traits. These disorders include hypokalemic periodic paralysis (hypoKPP), hyperkalemic periodic paralysis (hyperKPP), paramyotonia congenita, myotonia congenita, potassium-aggravated myotonia, and Andersen-Tawil syndrome. General features include episodic weakness or stiffness, interictal return to an asymptomatic state, and responsiveness to carbonic anhydrase inhibition (see Table 64.3).
Hypokalemic Periodic Paralysis
Pathophysiology
In up to 70% of cases, the responsible mutation has been linked to a gene encoding a VGCC on chromosome 1q (Venance et al., 2006). The gene, CACNA1S, encodes the α1-subunit of the dihydropyridine-sensitive L-type VGCC found in skeletal muscle. This channel functions as the voltage sensor of the ryanodine receptor and plays an important role in excitation-contraction coupling in skeletal muscle. Four mutations in the S4 segments are responsible for voltage sensitivity. Two mutations, one involving arginine-to-histidine substitutions within the highly conserved S4 segments of DII and DIV (Arg-528-His and Arg-1239-His) account for most cases. The others involve arginine-to-glycine substitutions at the same locations.
Some 10% to 20% of families with hypoKPP have mutations in the gene encoding the α-subunit of the skeletal muscle voltage-gated sodium channel (SCN4A) on chromosome 17q. This is the same channel implicated in hyperKPP and other disorders described later (see Fig. 64.1). Evidence suggests that this sodium channel–associated syndrome is phenotypically different from the more common CACNA1S form. A proposed separate clinical entity, hypoKPP2, may be distinguishable from hypoKPP associated with CACNA1S by the presence of myalgias following paralytic attacks, and the presence of tubular aggregates instead of vacuoles in the muscle biopsy. In some patients, acetazolamide worsens symptoms (Bendahhou et al., 2001; Sternberg et al., 2001). In a large retrospective series, hypoKPP2 was associated with an older age of onset and shorter duration of attacks than classical hypoKPP (Miller et al., 2004).
Whether involving SCN4A or CACNA1S, virtually all mutations causing hypoKPP involve an S4 voltage-sensor domain. In the case of the sodium channel, these mutations allow a leak current to pass through the “gating pore” at resting membrane potentials, bypassing the central channel pore and leading to inappropriate muscle fiber depolarization and consequent channel inactivation and action potential failure (Sokolov et al., 2007). Speculation exists that this phenomenon may also occur in mutated VGCCs.
Diagnosis
An accurate medical history is essential for the diagnosis because observation of attacks is unusual, and patients are often normal between attacks. Characteristic features of hypoKPP that distinguish it from hyperKPP are that paralytic attacks are less frequent, longer lasting, precipitated by a carbohydrate load, and often begin during sleep (Table 64.2). Potassium concentrations are usually low during an attack, although concentrations less than 2 mM should suggest a secondary form of periodic paralysis. Electrocardiogram (ECG) changes such as increased PR and QT intervals, T-wave flattening, and prominent U waves suggest an underlying hypokalemia. Provocative testing can be dangerous and is not routine. Test performance requires a hospitalized setting with continuous cardiac monitoring and should be performed only in patients without cardiac or renal disease. After giving an oral glucose load (2-5 mg/kg up to a maximum of 100 g) without or, less commonly, with subcutaneous insulin (0.1 U/kg), perform serial examinations of strength while monitoring serum glucose and potassium concentrations. Other diagnostic tests include electromyography (EMG), which may show decreased compound muscle action potential amplitudes during attacks compared with interictal values. Muscle histology reveals nonspecific myopathic changes of tubular aggregates or vacuoles within fibers. Genetic testing should render muscle biopsy and provocative testing obsolete for diagnosis.
Thyrotoxic periodic paralysis may be clinically indistinguishable from hypoKPP, except that it is not familial and serum potassium levels are often lower than in familial hypoKPP (<2.5). Some cases may be associated with a mutation in KCNJ18, the gene encoding a novel inwardly rectifying potassium channel (Ryan et al., 2010). All patients with hypoKPP require screening for hyperthyroidism, as the treatment—correction of the thyroid disorder—differs from that outlined later for idiopathic hypoKPP. Exclude other secondary forms of hypokalemic paralysis when serum potassium concentrations remain low between attacks. Renal, adrenal, and gastrointestinal causes of persistent hypokalemia are common, and thiazide diuretic use or licorice (glycyrrhizic acid) intoxication are considerations.
Treatment
An effective holistic approach to treatment includes lifestyle modifications and acute and chronic pharmacological intervention. Dietary modification to avoid high carbohydrate loads and refraining from excessive exertion helps prevent attacks. Oral potassium (5-10 g load) reverses paralysis during an acute attack. Prophylactic use of acetazolamide (Table 64.3) decreases the frequency and severity of attacks. Dichlorphenamide is another carbonic anhydrase inhibitor that effectively prevents attacks, as demonstrated in a randomized clinical trial (Tawil et al., 2000) where the average dose was 100 mg daily. More potent than acetazolamide, dichlorphenamide may be useful when efficacy of the former begins to fail. Many believe, without supporting evidence, that reducing the frequency of paralytic attacks provides protection against the development of myopathy. As insight into molecular pathophysiology expands, new treatment possibilities—and a new understanding of old treatments—seem likely (Matthews and Hanna, 2010).
| Use | Prophylactic agent for some channelopathies (see text) |
| Mechanism | Inhibits carbonic anhydrase |
| Dosing | Adults: start 125 mg daily, titrating as needed up to a maximum daily dose of 1000-1500 mg, divided bid–qid. An extended release formulation is available. Children: consult a pharmacist. |
| Side effects | Taste changes (especially for carbonated drinks), fatigue, paresthesias, metabolic acidosis, blurred vision, myelosuppression, nephrolithiasis, etc. (Increased dietary citrate might be recommended to compensate for decreased urinary citrate observed during acetazolamide therapy.) |
| Monitoring | Check electrolytes, BUN, creatinine, and CBC at baseline and periodically throughout therapy. |
| Metabolism | None; excreted unchanged by kidneys |
Please note that this table is for brief informational purposes only. Prescribing physicians should consult a pharmacist or an appropriate reference for complete and updated information.
bid, Twice daily; BUN, blood urea nitrogen; CBC, complete blood cell count; qid, 4 times daily.
Hyperkalemic Periodic Paralysis
Clinical
Characteristic of this disorder is episodic weakness precipitated by hyperkalemia. Although the weakness is generally milder than in hypoKPP, it can be sufficiently severe in hyperKPP to cause flaccid quadriparesis. As in hypoKPP, respiratory and ocular muscles are unaffected and consciousness is preserved. Frequency of attacks varies from several per day to several per year. Attacks are usually brief, lasting 15 to 60 minutes, but may last up to days. Unlike hypoKPP, myotonia is present between attacks. Onset is usually in infancy or childhood, and characteristic attacks occur by adolescence. Triggers include rest after vigorous exercise, foods high in potassium, stress, and fatigue. Despite its name, hyperKPP is often associated with a normal serum potassium concentration during an attack (see following Diagnosis section). Most patients experience a subacute onset, and some describe paresthesias or a sensation of muscle tension prior to attacks. In these situations, mild exercise may abort or lessen the severity of the attack. Mild weakness may persist afterward, and the later development of a progressive myopathy is common.
Pathophysiology
HyperKPP is as an autosomal dominant disorder, with some sporadic cases. The disorder links to SCN4A, the same gene responsible for a minority of hypoKPP cases. Among several identified missense mutations, four account for about two-thirds of cases (see Fig. 64.1). Functional expression of naturally occurring mutations demonstrated a decrease in the voltage threshold of channel activation or abnormally prolonged channel opening or both (Bendahhou et al., 2002; Hayward et al., 1999), effectively increasing the depolarizing inward current. If sustained long enough, this would lead to inactivation of the sodium channels, transitory cellular inexcitability, and weakness.
Diagnosis
Despite advances in defining the underlying genetic mutations, a thorough medical and family history and physical examination remain the best diagnostic tools. Furthermore, genetic testing is not generally available, and the high number of causative sodium channel mutations makes widespread use of genetic testing unlikely in the near future. Serum potassium is normal between attacks and even during many attacks. Unlike hypoKPP, potassium administration may precipitate an attack. In the absence of provocative testing, the basis for diagnosis is the clinical presentation. Myotonia is present in many patients between attacks, either spontaneously or after muscle percussion, and failure to produce myotonia discriminates hypoKPP from hyperKPP (see Table 64.2). Take care not to confuse subjective muscle stiffness with objective changes. Peaked T waves on ECG suggest hyperkalemia and are an aid to diagnosis. As in hypoKPP, serum creatine kinase concentrations may be normal or elevated. Electrodiagnostic studies are useful for demonstrating subclinical myotonic discharges, not seen in hypoKPP. Nonspecific findings such as fibrillation potentials and small polyphasic motor unit potentials occur during late stages of disease.
Treatment
The goal of therapy is to abort the acute attacks and prevent future attacks. Acute attacks are often sufficiently brief and mild so as not to require acute intervention. In more severe attacks, aim treatment at lowering extracellular potassium levels. Mild exercise or eating a high sugar load (juice or a candy bar) may suffice, as insulin drives extracellular potassium into cells. Thiazide diuretics and inhaled β-adrenergic agonists are similarly helpful, and intravenous calcium gluconate may be useful when weakness is very severe. To prevent attacks, a diet low in potassium and high in carbohydrates may obviate the need for prophylactic drug therapy. Oral dichlorphenamide was useful for prophylaxis in one randomized controlled trial (Tawil et al., 2000). Acetazolamide (see Table 64.3) and thiazide diuretics are useful as well. Successful prophylaxis may decrease the later onset of myopathy, although direct proof of this hypothesis is lacking. Finally, myotonic symptoms are troublesome in some patients; based on the underlying pathophysiology, sodium channel blockers would seem an effective therapy, and mexiletine is commonly used for this purpose.
Paramyotonia Congenita
Treatment
Symptoms are generally mild and infrequent. Direct treatment, when required, at either myotonia or weakness or both. Sodium channel blockers such as mexiletine are sometimes effective in reducing the frequency and severity of myotonia. Patients with weakness often respond to agents used to treat hyperKPP (e.g., thiazides, acetazolamide). A single case report suggests the possible use of pyridostigmine (Khadilkar et al., 2010). Cold avoidance reduces the frequency of attacks.
Myotonia Congenita
Clinical
Inheritance of myotonia congenita (MC) is either as an autosomal dominant (Thomsen disease) or recessive (Becker myotonia) trait. The main feature is myotonia or delayed muscle relaxation after contraction. Forceful movement abruptly initiated after several minutes of rest causes the most pronounced myotonic stiffness. The myotonia of MC displays a warm-up phenomenon in which the myotonia decreases or vanishes completely when repeating the same movement several times, in contrast to the myotonia seen in patients with PMC. The onset of Thomsen myotonia is often within the first decade, whereas the onset of Becker myotonia is generally at 10 to 14 years of age. Although myotonia can affect all skeletal muscles, it is especially prominent in the legs, where it is occasionally severe enough to impede a patient’s ability to walk or run. In rare cases, sudden noise causes sufficient generalized stiffness to make the patient fall to the ground and remain rigid for several seconds. The recessive and dominant forms share many similarities, but some clinical features help distinguish the two. In general, patients with recessive disease experience transitory bouts of weakness after periods of disuse and may develop progressive myopathy (Kornblum et al., 2010); in addition, muscle hypertrophy and disease severity are greater than in the dominant form. Becker myotonia is more common than Thomsen disease.
Pathophysiology
Electrical instability of the sarcolemma leads to muscle stiffness by causing repetitive electric discharges of affected muscle fibers. Early in vivo studies in myotonic goats revealed greatly diminished sarcolemmal chloride conductance in affected muscle fibers. This causes a depolarization of the sarcolemmal membrane and muscle hyperexcitability. Genetic linkage analysis for both recessive and dominant forms of MC pointed to a locus on chromosome 7q, where the responsible gene, CLCN1, encodes the major skeletal muscle chloride channel. More than 70 mutations have been identified within CLCN1, and interestingly, some of these mutations are recognized to cause both dominant and recessive forms (Zhang et al., 2000). Examination of the functional effects of several myotonia-causing CLCN1 mutations in heterologous expression systems reveal effects on channel gating, usually resulting in a decreased chloride conductance (Zhang et al., 2000).
Potassium-Aggravated Myotonia
Pathophysiology
PAM links to chromosome 17q, where mutations in the SCN4A gene cause the disease. This sodium channel is the same one implicated in hyperKPP, PMC, and the sodium channel subtype of hypoKPP (see earlier discussion and Fig. 64.1). Functional expression studies reveal that the disease-causing mutations lead to a large persistent sodium current secondary to an increased rate of recovery from inactivation and an increased frequency of late channel openings (Wu et al., 2001). The cause of myotonia is this enhanced inward current, which leads to prolonged depolarization and subsequent membrane hyperexcitability.
Andersen-Tawil Syndrome
Clinical
Andersen-Tawil syndrome (ATS) is a rare autosomal dominant disorder characterized by the triad of periodic paralysis, cardiac arrhythmias, and dysmorphic features (including hypertelorism, micrognathia, low-set ears, high-arched or cleft palate, short stature, and clinodactyly) (Yoon et al., 2006a). The periodic paralysis, often triggered by rest after exercise, prolonged rest, and stress, is often the presenting symptom and can be hypo-, normo-, or hyperkalemic. The cardiac phenotype, often discovered later, includes prolonged QT intervals, but bidirectional ventricular tachycardia is common. Despite the known association of cardiac arrhythmias in rare periodic paralysis patients, ATS was only recognized as a separate entity in 1971. In families segregating an ATS allele, the phenotypic expressivity can vary greatly. Patients can manifest one, two, or three features of the triad, and the severity of any one feature can be extremely variable. Rare individuals are asymptomatic disease-gene carriers. Finally, ATS patients may exhibit neurocognitive deficits in executive function and abstract reasoning (Yoon et al., 2006b).
Pathophysiology
Mutations in the KCNJ2 gene on chromosome 17q account for approximately two-thirds of ATS probands. KCNJ2 encodes a widely expressed inwardly rectifying potassium channel (Plaster et al., 2001). Interestingly, among all identified probands, about 50% have an autosomal dominant disorder, and identification of sporadic cases with de novo mutations is common. The mechanisms of channel dysfunction are heterogeneous, including impaired phospholipid binding, pore function, or protein trafficking. Because VGKCs are tetrameric complexes, many (if not all) of the mutations are dominant negative.
Diagnosis
Previous studies that took into account the variable penetrance of ATS classified individuals as affected if two of three criteria were met: paroxysmal weakness, prolonged QT interval with or without ventricular dysrhythmias, or characteristic dysmorphic features (Yoon et al., 2006a). ATS should be included in the differential diagnosis of any individual with documented long-QT syndrome, even in the absence of periodic paralysis or dysmorphism. Some family members of patients with the full clinical triad show only prolonged QT intervals. Similarly, perform ECG on all patients with suspected periodic paralysis for careful measurement of the QT interval.
Treatment
Treatment of the prolonged QT interval depends on the severity of the underlying arrhythmias. The use of beta-blockers is a mainstay of treatment in long QT. Clinical experience shows that patients with ATS tolerate these agents well. In the presence of syncope due to sustained ventricular tachycardia, the placement of an implantable defibrillator is useful. Some evidence suggests that flecainide may be effective in the treatment of severe ATS-associated ventricular arrhythmias (Bökenkamp et al., 2007; Pellizzón et al., 2008).
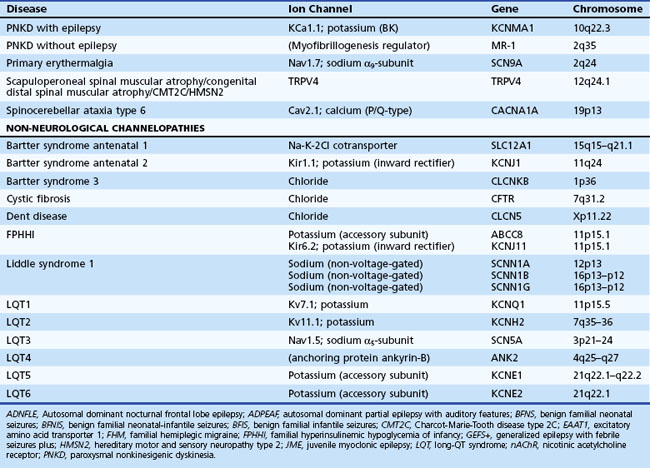
Genetic Disorders of Neuronal Ion Channels
Familial Hemiplegic Migraine
Clinical
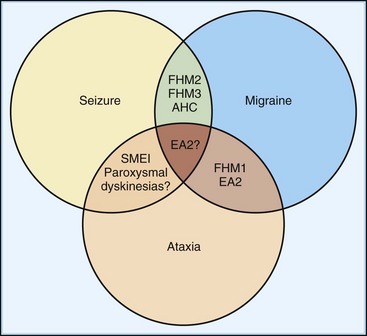
Familial hemiplegic migraine (FHM) is a rare autosomal dominant subtype of migraine characterized by lateralized motor weakness of variable intensity—hemiparesis to hemiplegia. Three subtypes, genetically defined (see later discussion), are distinguishable by clinical characteristics. FHM type 1 (FHM1) accounts for about 50% to 75% of families. In addition to weakness, auras always involve additional symptoms including sensory, visual, and language disturbances (Ducros et al., 2001). Severe aura attacks may last days or weeks and may involve fever, meningismus, and impaired consciousness ranging from confusion to coma. Recovery between attacks is typically complete, although 30% to 50% of patients with FHM1 exhibit permanent progressive cerebellar signs that include nystagmus, gait or limb ataxia, or dysarthria. These manifestations of FHM1 overlap with episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (Fig. 64.5), all caused by mutations in the same gene (see following discussion). By contrast, the characteristic features of FHM2 are an absence of cerebellar signs and a tendency for lower penetrance than in FHM1. Ataxia does not occur in FHM3, a rarer and more recently defined entity. Importantly, patients with FHM exhibit a spectrum of disease expression, and subtype classification is likely to evolve with our knowledge of underlying genetics.
Pathophysiology
Familial hemiplegic migraine is a genetically heterogeneous condition that links to three loci on chromosomes 1q, 2q, and 19p; other loci are possible (Ducros et al., 1997). FHM1 includes the 50% to 75% of families that show genetic linkage to chromosome 19p13, where missense mutations in the CACNA1A gene cause the disease (Ophoff et al., 1996). This gene encodes the pore-forming α1A-subunit of the neuronal P/Q-type VGCC, which is distributed widely throughout the brain. It is present at motor nerve terminals and the neuromuscular junction and is the principal calcium channel expressed by cerebellar Purkinje and granular neurons. These channels play an integral role in the action potential–triggered presynaptic calcium influx at nerve terminals that triggers vesicular fusion. At least 17 different missense mutations and one nonsense mutation (Jen et al., 1999) are reported in CACNA1A (Fig. 64.6). These cause a variety of altered but not lost functions: changes in channel conductance, kinetics of inactivation, and current density (Hans et al., 1999; Kraus et al., 2000). Indeed, these mutations may confer gain of function, because many appear to cause a hyperpolarizing shift in the activation voltage, meaning that channels open and permit calcium influx at abnormally low membrane potentials. In one extreme case, the S218L mutation confers both the tendency to open near the resting potential of many neurons (−60 to −50 mV) and overall slowing and reduction in channel inactivation, thus causing marked increases in overall calcium influx. In keeping with this extreme biophysical profile, S218L produces a severe clinical phenotype of aura attacks triggered by minor head trauma and leading to deep coma and prolonged cerebral edema (Tottene et al., 2005). On the other hand, some loss-of-function mutations associated with episodic ataxia type 2 (EA2) may also cause migraine symptoms (Jen et al., 2004), suggesting that a direct correlation between presynaptic VGCC-mediated calcium entry and disease severity is too simplistic. Importantly, the mechanism linking altered channel biophysics to disease expression is controversial and defies a simple model.
As mentioned earlier, other mutations in CACNA1A cause different dominantly inherited neurological disorders, including EA2 and spinocerebellar ataxia type 6 (SCA6). Although most patients with FHM1 also suffer persistent cerebellar deficits, a few mutations in CACNA1A cause only hemiplegic migraine (Ducros et al., 2001), and one mutation appears to cause classical migraine with aura but no weakness (Serra et al., 2010). These observations suggest that a spectrum of disease exists from pure ataxia at one extreme to classical migraine at the other.
Patients with FHM2, representing 10% to 20% of all FHM sufferers, exhibit mutations in the ATP1A2 gene encoding the α2-subunit of the Na+/K+-ATPase, a protein responsible for maintaining the membrane potential in neurons and other cells (De Fusco et al., 2003). This pump plays a critical role in establishing transmembrane ionic gradients and is in this way directly integral to the function of innumerable ion channels and other proteins. Many missense mutations have been identified, probably resulting in a loss of function but not loss of surface expression (De Fusco et al., 2003), possibly by a reduced affinity for potassium (Segall et al., 2004). Although an ionic mechanism is likely, the precise pathophysiological connection to hemiplegic aura or head pain remains obscure. Additional mutations in ATP1A2 may also cause a form of benign familial infantile seizures (see following section on Epilepsy) (Vanmolkot et al., 2003) and alternating hemiplegia of childhood (Swoboda et al., 2004). A single mutation was found to cause hemiplegic migraine with cerebellar findings (Spadaro et al., 2004), challenging the conception this association is specific for CACNA1A mutations.
FHM3 involves mutations in the SCN1A gene on chromosome 2q24 that encodes the neuronal voltage-gated sodium channel α1-subunit. To date, three missense mutations have been identified, and the two that have been characterized electrophysiologically each confer upon the channel more rapid recovery from fast inactivation and thus the potential for faster firing frequency and neuronal hyperexcitability (Castro et al., 2009; Dichgans et al, 2005). Other mutations in SCN1A cause some cases of severe myoclonic epilepsy of infancy and generalized epilepsy with febrile seizures plus (see following section on Epilepsy). Interestingly, mutations causing epilepsy occur nearby those causing FHM3, and one mutation—L263V—causes both clinical phenotypes (Castro et al., 2009). Epilepsy has also been reported in cases of FHM1 and FHM2, pointing to the intriguing possibility of a pathogenic link between seizure and migraine.
Diagnosis
Genetic testing is not widely available, so the diagnosis relies on a thorough history and physical examination. Family history is clearly helpful in demonstrating autosomal dominance, but it is important to note that typical migraine syndromes may also show a strong (if less regular) familial pattern. Certain clinical elements of FHM help distinguish it from classical migraine with aura. Although the aura symptoms may be similar to those in classical migraine, FHM patients are more likely to experience motor, speech, and sensory symptoms. Furthermore, the duration of the headache, as well as the duration of the visual and sensory components of the aura, are typically greater in FHM patients (Thomsen et al., 2002). Although patients with classical migraine often experience the aura in isolation from the headache, FHM patients experience an associated headache virtually all the time. Infrequently, FHM patients may experience bilateral weakness, whereas this is generally not the case in patients with classical migraine. Finally, although defined genetically, one might clinically suspect FHM based on an association with cerebellar abnormalities or seizures (see Fig. 64.5).
Treatment
Anecdotal evidence suggests that acetazolamide reduces the frequency of migraine attacks (Battistini et al., 1999; Jen et al., 2004). As suggested by functionally enhanced calcium currents in many cases of FHM, the calcium channel blocker, verapamil, aborted an attack when administered intravenously (Yu and Horowitz, 2001). To date, no controlled study has tested the efficacy of these or other agents in FHM. The guidelines for treating common migraine may be applied, except that triptans and ergotamine are not used, out of concern—possibly unwarranted—for stroke (Artto et al., 2007). Nimodipine is contraindicated because of the risk of worsening symptoms (Mjåset and Russell, 2008).
Familial Episodic Ataxias
The familial episodic ataxias (EAs) are rare, dominantly inherited diseases characterized by episodes of ataxia of early onset, often with completely normal cerebellar function between attacks (Jen et al., 2007). Of the seven syndromes now recognized (EA1-EA7), the two most common forms, EA1 and EA2, are best described.
Clinical
Patients with EA2 experience episodes of truncal ataxia lasting hours to days, precipitated by exertion and stress. Age of onset varies between childhood and young adulthood but is most frequently in the second decade. Vertigo, nausea, and vomiting are present in more than half of patients, and many exhibit spontaneous nystagmus that is not seen interictally. Between episodes, the patient returns to normal but frequently displays gaze-evoked nystagmus with features typical of rebound nystagmus (Jen et al., 2007). Less commonly, positional and, later in the disease course, spontaneous downbeat nystagmus occurs. Approximately half of EA2 patients report headaches that meet criteria for migraine. Since mutations causing EA2 are the same gene as in FHM1, this is expected.
Pathophysiology
The mutation underlying EA1 is on chromosome 12. At least 15 missense mutations have been described in the responsible gene, KCNA1, which encodes a delayed rectifier VGKC that opens with a delay after membrane depolarization, allowing K+ efflux and membrane repolarization (see Fig. 64.2). Coexpression of mutant and wild-type channels results in delayed outward potassium current and impaired membrane repolarization following an action potential (Zerr et al., 1998), a dominant-negative effect that could lead to increased neuronal excitability and neurotransmitter release. The delayed rectifier potassium channel is widely expressed in the nervous system, with highest levels in the cerebellum and myelinated axons of peripheral nerves. In the cerebellum, an imbalance between inhibition and excitation could result in brief episodic incoordination. Similarly, in the peripheral motor nerves, impaired repolarization could lead to repetitive neuronal activity and resultant myokymia. Suggesting that these two effects may be mechanistically distinct is a KCNA1 missense mutation that causes an episodic ataxia more akin to EA2 without myokymia (Lee et al., 2004a), and another missense mutation resulting in myokymia without ataxia (Rea et al., 2002).
Mutations in CACNA1A cause EA2 (Ophoff et al., 1996). Whereas FHM1 is associated with missense mutations in the same gene, the genetic alterations associated with EA2 seem more dramatic. Of the approximately 30 mutations described so far, most are truncating, a few missense, one insertional, and one due to expansion of a triplet CAG domain. Functional expression studies of some of these mutations reveal loss of voltage sensitivity or complete loss of channel function (Guida et al., 2001), suggesting that haploinsufficiency may underlie the phenotype. Some mutations may additionally interfere with protein folding and trafficking (Wan et al., 2005).
EA5 results from mutation of the VGCC β4-subunit gene CACNB4, also implicated in idiopathic generalized epilepsy (see following section on Epilepsy); it is clinically similar to EA2 except that seizures are an additional feature (Escayg et al., 2000a). Mutations in SLC1A3, the gene encoding the excitatory amino acid transporter EAAT1, cause EA6, an episodic ataxia associated with hemiplegia and seizures (Jen et al., 2005). The genes for EA3 (1q42), EA4, or EA7 (19q13) have not been identified.
Diagnosis
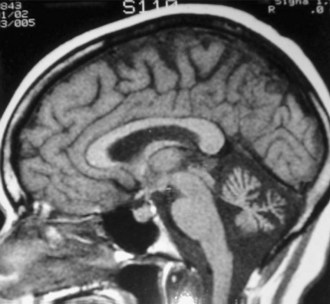
Family history is helpful in making the diagnosis, despite rare cases of de novo mutations. The probability of examining a patient during an attack is low, so careful examination for interictal signs are important. Although almost half of EA1 patients complain of diplopia, they do not have nystagmus, which helps distinguish them from EA2 patients. The brevity of attacks and the persistent interictal myokymia seen in EA1 also help distinguish this from EA2. EA2 patients may demonstrate interictal end-point tremor or impaired suppression of the vestibulo-ocular reflex. They tend to have subtle and slowly progressive interictal cerebellar signs, particularly gaze-evoked nystagmus. Although progressive ataxia often develops, it is rarely severe enough to prevent walking without assistance. Magnetic resonance imaging (MRI) may detect cerebellar atrophy, especially of the anterior vermis (Fig. 64.7), whereas EA1 patients have no cerebellar atrophy. Interestingly, there appears to be an increased incidence of epilepsy in both EA1 and EA2 patients, emphasizing the overlapping symptoms possibly due to shared mechanisms (see Fig. 64.5).
Treatment
Acetazolamide reduces the severity of attacks. Response to treatment varies among families, but generally, patients with EA2 have a greater response than those with EA1. Carbamazepine or 4-aminopyridine (Strupp et al., 2004) are beneficial in some patients. Because stress and strenuous exercise often exacerbate attacks, lifestyle modification can be quite effective.
Hereditary Hyperekplexia
Pathophysiology
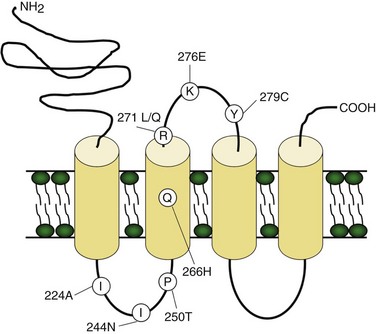
Pathogenic mutations involve the GLRA1 gene on chromosome 5q encoding the glycine receptor α1-subunit. The glycine receptor is a heteropentameric ligand-gated ion channel composed of three ligand-binding α1-subunits and two β-subunits, mediating fast inhibitory neurotransmission in the brainstem and spinal cord. Several dominant, recessive, and de novo GLRA1 missense point mutations account for familial hyperekplexia patients. Most mutations flank the M2 pore-forming domain (Fig. 64.8). The physiological consequence of mutations in the α1-subunit is decreased glycine sensitivity, impaired channel opening, and uncoupling of agonist binding from channel activation. These changes reduce glycinergic inhibition and increase neuronal excitability.
Mutations in the gene encoding the glycine receptor β-subunit (GLRB) also cause hyperekplexia. An example is a compound heterozygous patient with spontaneous hyperekplexia possessing a missense mutation on one allele and a splice-site mutation on the other (Rees et al., 2002). Electrophysiological studies showed reduced sensitivity of the mutant channel to agonist, suggesting impaired binding of glycine to the channel.
Finally, loss-of-function mutations in GLYT2 (also named SLC6A5), the gene encoding the neuronal glycine transporter 2 protein, have been identified in three families with hereditary hyperekplexia. Glycine transporters mediate synaptic reuptake of the neurotransmitter, and its genetic deletion reproduces hyperekplexia in mice (Eulenburg et al., 2006). Reduction of glycine transporter function is expected to prolong glycine neurotransmission; how this leads to hyperekplexia is unknown.
Hereditary Peripheral Nerve Disorders
Primary erythermalgia, also known as familial erythromelalgia or Weir Mitchell disease, is an autosomal dominant disorder characterized by burning pain and redness in the limbs in response to warmth or moderate exercise (Dib-Hajj et al., 2008; Yang et al., 2004). It is due to mutations in SCN9A, encoding a voltage-gated sodium channel selectively expressed in small-diameter dorsal root ganglion neurons (mostly nociceptors) and sympathetic ganglion neurons. These mutations enhance current through the channels by decreasing the voltage threshold for activation, among other effects. The discovery that expression of this sodium channel is narrowly restricted to a population of pain-mediating neurons, and that its enhanced function can result in pain, raises the exciting hypothesis that selective channel blockade may result in relief of chronic pain at least in primary erythermalgia and perhaps in other conditions. Supporting a more general role for this gene, some polymorphisms correlate with a lowered pain threshold in patients without primary erythermalgia (Reimann et al., 2010). Loss-of-function mutations in the same gene lead to an inherited insensitivity to pain. A selective antagonist of this channel does not yet exist.
Scapuloperoneal spinal muscular atrophy, congenital distal spinal muscular atrophy, and Charcot-Marie-Tooth disease type 2C (or hereditary motor sensory neuropathy type 2) are related disorders. Characteristic of all is autosomal dominant inheritance, muscle weakness and wasting, and other features that can include arthrogryposis, scoliosis, or vocal cord paralysis. These disorders result from mutations in TRPV4, the gene on chromosome 12q encoding the vanilloid transient receptor potential (TRP) protein, a peripheral nerve nonselective cation channel activated by many noxious stimuli such as heat, mechanical stress, osmotic pressure, and inflammatory cytokines (Auer-Grumbach et al., 2010; Deng et al., 2010; Landoure et al., 2010). Although the pathogenic mechanism remains unclear, all known mutations affect the ankyrin repeat region of the protein, where regulatory proteins, second messengers, and other TRPV4 channels normally bind.
Epilepsy
Familial Focal Epilepsies
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
ADNFLE was initially shown to be caused by a point mutation in the nAChR α4-subunit gene CHRNA4 on chromosome 20q (Steinlein et al., 1995). Four mutations in CHRNA4 were identified in several ethnic groups. Mutations have also been identified in CHRNB2 (Phillips et al., 2001) and CHRNA2 (Aridon et al., 2006), encoding the nAChR β2– and α2-subunits. The β2-subunit associates closely with the α4-subunit, and the α4– β2 combination is the dominant subtype of nAChR in the brain. Mutations in CHRNA4 and CHRNB2 involve M2, the second transmembrane domain and the part of the protein thought to line the channel pore. Some cases of ADNFLE link to chromosome 15q24, close to a cluster of three other nAChR subunits, although the responsible mutations have not been elucidated. Mutations result in increased sensitivity both to activation by acetylcholine and to block by carbamazepine. This latter in vitro observation bears clinical relevance because ADNFLE patients have a good therapeutic response to carbamazepine. Given the wide distribution of nAChRs within the brain, it remains unclear why the epilepsy is focal and why the seizures arise selectively in the frontal lobes.
Familial Temporal Lobe Epilepsies
Temporal lobe epilepsies exist in both sporadic and inherited forms, and the genetics of familial syndromes remains largely unknown (Andermann et al., 2005). The onset of familial lateral temporal lobe epilepsy (FLTLE), or autosomal dominant partial epilepsy with auditory features (ADPEAF), a benign syndrome distinguished from the more common mesial form by characteristic auditory auras, is in the second or third decade, and transmission is autosomal dominant with incomplete penetrance. Approximately 50% of cases involve mutations in the leucine-rich, glioma-inactivated gene 1 (LGI1) on chromosome 10q (Kalachikov et al., 2002; Morante-Redolat et al., 2002), whose product was shown to complex with presynaptic A-type VGKCs (Schulte et al., 2006). Normally, LGI1 appears to reduce channel inactivation, and its dysfunction results in faster inactivation kinetics and, likely, neuronal hyperexcitability.
Idiopathic Generalized Epilepsies
The idiopathic generalized epilepsies (IGEs) are among the most common seizure disorders, occurring at an overall frequency of 15% to 20% among cohorts of adults and children (Jallon and Latour, 2005). A strong genetic component to IGE transmission exists, but the pattern of inheritance varies between individual disorders. Some rare syndromes, including benign familial neonatal seizures and generalized epilepsy with febrile seizures plus, are monogenic autosomal dominant traits, now known to be due to mutations in ion channel genes. Other more common syndromes—juvenile myoclonic epilepsy (JME), childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), and epilepsy with grand mal seizures on awakening (EGMA)—exhibit a more complex pattern of inheritance. These disorders, which relate to each other by a continuum of clinical phenotypes and similar EEG findings (see Chapter 67) probably encompass a broad range of individual diseases. We now know that aberrant ion channels underlie at least some of these diseases.
Benign Familial Neonatal Seizures
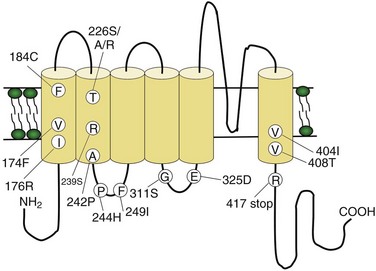
Two BFNS-causing genes have been identified, one on chromosome 20q and the other on chromosome 8q. The mutated genes both encode VGKCs: KCNQ2 and KCNQ3, respectively (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998). These channels activate by membrane depolarization and contribute to the repolarization of the action potential. Reports appear of missense amino acid deletion, splice-site frameshift mutations, and gene deletions; most involve the KCNQ2 gene (Fig. 64.9). Functional expression of mutant channels results in reduced potassium current, likely leading to impaired membrane repolarization and thus increased neuronal excitation. KCNQ2- and KCNQ3-encoded products combine to form a heteromeric channel underlying the M-current (Wang et al., 1998), a potassium conductance found widely in the central nervous system that plays a crucial role in the regulation of neuronal excitability. Coexpressing the mutant gene (KCNQ2 or KCNQ3) together with its wild-type allele and its wild-type partner results in mild (25%) reduction in M-current amplitude. Although it is unclear how the mutated channels either independently or in the form of the M-current lead to seizures, KCNQ2 channels are concentrated in the septum and hippocampus, areas key for control of rhythmic brain activity and neuronal synchronization, and both associated with the generation of epileptic seizures. Thus, even slight alterations in neuronal excitability through impaired KCNQ2/KCNQ3 channel-mediated repolarization could presumably lead to aberrant neuronal synchronization and seizures. In light of reports that lamotrigine and carbamazepine enhance neocortical potassium currents in vitro, they may be of particular use in BFNS patients.
Generalized Epilepsy with Febrile Seizures Plus
A high level of genetic heterogeneity in GEFS+ exists (see Fig. 64.1). Mutations in four voltage-gated sodium channel genes (SCN1B on chromosome 19q and SCN1A, SCN2A, and SCN9A on chromosome 2q) (Escayg et al., 2000b; Singh et al., 2009; Sugawara et al., 2001; Wallace et al., 1998) and two GABAA receptor genes (GABRG2 and GABRD) encoding the γ2– and δ-subunits (Baulac et al., 2001; Dibbens et al., 2004) cause GEFS+ (Fig. 64.10). Evidence exists for other mutated genes not yet identified. Functional analysis of several mutated sodium channels reveals slow inactivation, enhanced inward sodium current, and thus neuronal hyperexcitability (Lossin et al., 2002). Functional studies of the two GABRG2 mutations showed reduced channel conductance in one case and abolished benzodiazepine sensitivity in the other. The GABRD mutations reduce current amplitude, each leading in different ways to a decrease in synaptic inhibition and thus an increase in neuronal excitability. These functional observations evoke compelling molecular explanations for clinical disease and, like ADNFLE and BFNS, illustrate how heterogeneous genetic defects may converge on a single clinical phenotype.
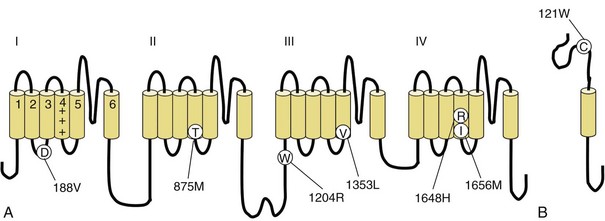
Fig. 64.10 Mutations in the voltage-gated Na+ channel causing generalized epilepsy with febrile seizures plus (GEFS+). The α-subunit is encoded by SCN1A (A), and the β1-subunit (B) is encoded by the SCN1B gene. Disease-causing mutations are shown. Mutations in other genes, including SCN2A and GABRG2, also cause GEFS+ (see Fig. 64.1).
SCN2A Mutation and Other Early Childhood Seizures
Other rare convulsive disorders of early childhood include benign familial infantile seizures and benign familial neonatal-infantile seizures, differentiated from BFNS by the age of onset. At least some cases involve mutations in the sodium channel α2-subunit gene, SCN2A, the same gene affected in some cases of GEFS+ (see preceding section). Two mutations are present in families with seizures beginning at 1 to 3 months of age and ending at around 4 months (“neonatal-infantile”) (Heron et al., 2002). Both mutations occur in cytoplasmic loops, inhibiting channel inactivation. A third mutation, also affecting a cytoplasmic loop, was identified in a family with similar seizures beginning at 4 to 12 months of age (“infantile”) (Striano et al., 2006). Of note, ATP1A2 mutations account for some cases of benign familial infantile seizures (see previous section on Familial Hemiplegic Migraine).
Juvenile Myoclonic Epilepsy
Juvenile myoclonic epilepsy accounts for 4% to 10% of all epilepsy (Jallon and Latour, 2005). Featuring myoclonic jerks, generalized tonic-clonic seizures, and absence spells, JME typically begins during adolescence (see Chapter 67). A genetic pattern is clear, but the mixed inheritance pattern suggests multiple heritable causes. One French-Canadian family suffers an autosomal dominant form of JME associated with a mutation in the GABAA receptor α1-subunit gene, GABRA1 (Cossette et al., 2002), that results in loss of function and possible neuronal hyperexcitability (Krampfl et al., 2005). Less well described is the possible role of the VGCC β4-subunit gene, CACNB4, in JME and episodic ataxia (see previous discussion) (Escayg et al., 2000a). GABRD variations may influence susceptibility to JME (Dibbens et al., 2004).
EFHC1 is a gene on chromosome 6p, and five missense mutations have been identified in 6 of 44 JME families in whom it was sequenced (Suzuki et al., 2004). Although the precise function of the EFHC1 protein is unknown, it binds specifically to R-type VGCCs, alters their function, and perhaps thereby influences cell death pathways when transfected into cells in vitro. Whether or not this model turns out to be correct, it illustrates that an inherited channelopathy may result not only from mutation of a channel gene itself, but also of genes whose products regulate the function of otherwise normal channels (Fig. 64.11).
Chromosome 15q has genetic loci implicated by linkage studies as possible contributors to some cases of JME. This area is known to contain, among other genes, the α7-subunit of the nicotinic acetylcholine receptor, CHRNA7 (Elmslie et al., 1997).
Similar to JME is familial adult myoclonic epilepsy (FAME) and autosomal dominant cortical myoclonus and epilepsy (ADCME), characterized by autosomal dominant inheritance, adult onset, varying degrees of myoclonus in the limbs, rare tonic-clonic seizures, and a benign course. These syndromes bear some similarity to JME except for the adult onset and the highly penetrant autosomal dominant transmission. Whereas the genes are not yet known, the FAME gene has been mapped to chromosome 8q24 (Plaster et al., 1999) and the ADCME gene to chromosome 2q11.1–q12.2 (Guerrini et al., 2001).
Childhood Absence Epilepsy
Childhood absence epilepsy, a syndrome less common than JME and typified by brief, frequent absence spells (see Chapter 67), exhibits a multigenic pattern of inheritance. Mutations in GABRB3, encoding the GABAA receptor β3-subunit, appear to cause CAE in some families (Tanaka et al., 2008; Urak et al., 2006). A mutation in the GABAA receptor γ2-subunit gene, GABRG2, on chromosome 5q, conferring loss of benzodiazepine-induced enhancement of GABA-induced currents in vitro, is present in a family with CAE and an increased incidence of febrile seizures (Wallace et al., 2001). Mutations in the T-type VGCC gene, CACNA1H, may contribute to some cases of CAE (Chen et al., 2003) and influence susceptibility to other IGEs (Heron et al., 2007). Mutations in other genes—CACNA1A, GABRA1, and a locus on chromosome 8q24—may also play a role.
Epilepsy and Paroxysmal Dyskinesia
A recognized association between IGE and paroxysmal dyskinesia exists. Characterizing this group of episodic movement disorders are recurrent attacks of dystonia, chorea, athetosis, or ballismus. In a family with an autosomal dominant pattern of generalized epilepsy (mostly absence attacks) and paroxysmal nonkinesigenic dyskinesia (PNKD, a type of paroxysmal dyskinesia in which alcohol, coffee, stress, and fatigue trigger attacks), a mutation of KCNMA1 on chromosome 10q was identified (Du et al., 2005). This gene encodes the α-subunit of the BK channel, a potassium channel that normally activates with both membrane depolarization and a rise in intracellular calcium; the mutation heightens calcium sensitivity, enhancing channel activity. Ethanol may also activate the BK channel (Davies et al., 2003), suggesting a mechanism whereby alcohol consumption in synergy with the mutation may precipitate a dyskinetic attack (Du et al., 2005). That a single ion channel mutation can cause both paroxysmal dyskinesia and epilepsy implies a pathophysiological connection between the basal ganglia and the cortex. PNKD without epilepsy is linked to mutation of the myofibrillogenesis regulator gene, MR1 (Lee et al., 2004b); the mechanism, and whether it involves an ion channel, is unknown.
Theoretical Considerations
The phenotypic and EEG characteristics of various IGEs overlap to a considerable extent, implying that the boundaries separating some of these disorders may be blurred. Moreover, the relationships between molecular defects and clinical expression are irregular. Mutations in some genes (e.g., SCN2A, GABRG2) lead to heterogeneous clinical syndromes or genotype-phenotype divergence. Genotype-phenotype convergence is typified by ADNFLE, BFNS, and GEFS+, each of which may result from myriad underlying genetic roots (see Fig. 64.1). These considerations challenge existing definitions of disease, which will likely shift as knowledge advances. Cheaper and more widely available genetic tests will eventually free clinicians from the ambiguities of syndromic classification, and the elucidation of the molecular basis of familial epilepsy syndromes will eventually lead to tailored pharmacological treatments.
Autoimmune Channelopathies
Myasthenia Gravis
The best characterized autoimmune disease, myasthenia gravis, results in most cases from circulating antibodies directed against nAChRs. Polyclonal immunoglobulin G (IgG) reacts against variable extracellular nAChR epitopes at the neuromuscular junction, with the main immunogenic region located on each of two α-subunits of the heteropentameric receptor complex (Tzartos et al., 1998). Antibodies produce disease by activating complement, causing lysis of the muscle membrane, and by cross-linking AChRs, leading to accelerated protein degradation. Understanding of the cause of autoantibody production is poor, but it probably results from multiple disease processes, sometimes involving thymoma or thymic hyperplasia (see Chapter 78).
Lambert-Eaton Myasthenic Syndrome
Like myasthenia gravis, Lambert-Eaton myasthenic syndrome (LEMS) produces weakness by obstructing neuromuscular transmission. However, its clinical manifestations are distinct because its pathology localizes to the presynaptic rather than postsynaptic membrane. There, autoantibodies directed against P/Q-type VGCCs impair presynaptic calcium influx, thereby reducing action potential–triggered vesicle release. These autoantibodies are not specific to the neuromuscular junction, and patients also exhibit autonomic dysfunction. However, the antibodies probably do not cross the blood-brain barrier to a sufficient extent to cause diseases mimicking CACNA1A mutants—FHM1, EA2, SCA6, or seizures—although some patients may exhibit ataxia (see following section on Paraneoplastic Cerebellar Degeneration). Conversely, weakness is not a typical feature in patients with loss-of-function CACNA1A mutations, although EMG studies reveal expected neuromuscular abnormalities (Jen et al., 2001). Small-cell lung cancer cells express VGCCs, likely triggering immunogenesis in the large proportion of patients in whom LEMS turns out to be a paraneoplastic phenomenon. As in myasthenia gravis, polyclonal antibodies may be detectable in LEMS. The extracellular S5-S6 linker regions of the α-subunit are the proposed pathogenic epitopes (Takamori, 2004).
Acquired Neuromyotonia (Isaacs Syndrome)
Isaacs syndrome presents with painful muscle cramps, slow muscle relaxation after contraction, and hyperhidrosis. Characteristic features of acquired neuromyotonia are myokymic and neuromyotonic discharges on EMG, reflecting spontaneous muscle activity driven by abnormal peripheral nerve firing. The syndrome may occur in association with thymoma. The autoimmune target is the VGKC found along peripheral motor axons and responsible for repolarization after action potential firing (Shillito et al., 1995). This is the same channel affected in EA1, and the mechanism of interictal myokymia is identical: reduced potassium efflux from axons and nerve terminals lead to higher membrane potentials, inappropriate action potential firing, and acetylcholine release. This same phenomenon probably also affects autonomic fibers, explaining why excessive sweating, salivation, and lacrimation are often observed. The action of these antibodies is complement-independent and appears to involve cross-linking of channels (Tomimitsu et al., 2004).
VGKC antibodies probably also cause Morvan syndrome, which encompasses all the clinical features of acquired neuromyotonia plus a fluctuating delirium (Barber et al., 2000; Lee et al., 1998). Supporting a role for circulating autoantibodies is the effectiveness of plasma exchange (Liguori et al., 2001). VGKC antibodies were present in a series of patients suffering from an encephalopathy indistinguishable from limbic encephalitis, with temporal lobe-onset seizures and behavioral and cognitive disturbances, but no neuromuscular abnormality (Thieben et al., 2004). Thus, a peripheral-to-central spectrum of disease may exist, with acquired neuromyotonia on the one extreme and VGKC antibody-associated limbic encephalitis on the other.
Paraneoplastic Cerebellar Degeneration
Paraneoplastic cerebellar degeneration (PCD), presenting as a rapidly progressive ataxic syndrome, most commonly occurs in cases of breast, ovarian, and lung malignancies. PCD probably represents a spectrum of diseases associated with distinct autoimmune targets. Among these may be the anti-P/Q-type VGCC antibodies found in small-cell cancer. Although these antibodies more typically cause peripheral disease (LEMS), they are found less commonly in the CSF of patients with PCD (Graus et al., 2002). Purkinje cell loss is the pathological hallmark of PCD; these cells express high levels of P/Q-type VGCCs, and it seems likely that antibodies directed against these channels are pathogenic. PCD associated with small-cell lung cancer may occur with or without neuromuscular dysfunction, and the factors predisposing to central versus peripheral action of the paraneoplastic antibodies are unknown. Finally, despite the fact that the same molecule may be the target of pathology in PCD and CACNA1A-associated diseases (FHM1, EA2, SCA6), PCD is not an episodic disorder but rather characterized by rapid deterioration, perhaps due to target cell destruction.
Limbic Encephalitis
The term limbic encephalitis encompasses an array of autoimmune disorders associated with psychiatric symptoms, cognitive dysfunction, and seizures, often in the context of an underlying malignancy. Some of these disorders result from an autoantibody directed against a brain ion channel. VGKC antibody-associated limbic encephalitis, mentioned earlier, is one example. Another example is anti–N-methyl-d-aspartate (NMDA) receptor limbic encephalitis (Dalmau et al., 2008). This syndrome, which occurs mainly in women, causes psychiatric symptoms, seizures, delirium, and other neurological symptoms. Like PCD, anti-NMDA receptor encephalitis strongly suggests an underlying malignancy, typically ovarian teratoma. NMDA receptors represent one of three classes of glutamate-gated ion channels in the brain; α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors represent another class, and antibodies against those receptors may also cause limbic encephalitis (Lai et al., 2009). Antibodies against GABAB receptors were isolated in a series of patients with limbic encephalitis and prominent seizures (Lancaster et al., 2010). While not an ion channel, this G protein–coupled GABA receptor regulates ion channels and synaptic transmission. As for PCD, acquired neuromyotonia, and LEMS, limbic encephalitis should be treated with plasma exchange, steroids, or other immunosuppressant therapies in parallel with an aggressive search for an underlying neoplasm.
Summary
Advances in molecular biology and electrophysiology allowed the characterization of a new group of disorders termed the channelopathies. Understanding the underlying pathophysiology of these diseases has not only expanded our knowledge of basic ion channel physiology but, more importantly, has also provided insight into mechanisms of common neurological disorders such as epilepsy and migraine. The channelopathies, a seemingly heterogeneous group of diseases, share striking similarities. Most have intermittent symptoms despite the invariant presence of the mutation, with interictal return to a normal state. Exacerbating factors such as stress, exertion, and fatigue are common to many of the channelopathies. Response to treatment with carbonic anhydrase inhibitors (acetazolamide) is an almost universal feature among the genetic syndromes (see Table 64.3), leading some clinicians to use acetazolamide responsiveness as diagnostic litmus test for the channelopathies. The mechanism by which acetazolamide prevents and ameliorates attacks is not completely understood, although recent evidence suggests activation of potassium channels may play a role.
These similarities suggest a common underlying pathophysiological basis shared among the channelopathies. In fact, many of the channelopathies, such as EA2 and FHM, share several characteristics. This chapter has attempted to provide a clinical approach to the recognition, diagnosis, and treatment of the channelopathies, for which widespread genetic testing is typically not available. Furthermore, knowing where the genetic mutation is located does not necessarily predict the clinical phenotype. Diagnostic confusion arises from phenotypic variability within a given syndrome and phenotypic similarities among different syndromes, as well as the realization that mutations in the same gene can produce seemingly unrelated phenotypes (see Fig. 64.1). This underscores the importance of careful clinical assessment. The clinician’s most useful diagnostic tool remains a detailed history and physical examination.
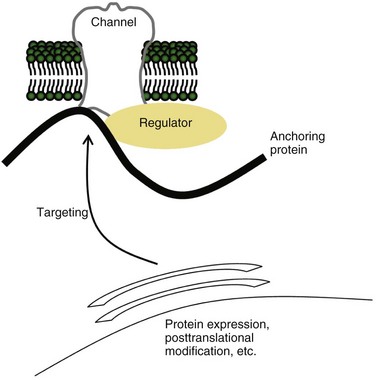
Finally, disruption of ion channel function may occur in more complex ways than simply by mutation in an ion channel gene or by an autoantibody to the channel protein itself. Our appreciation of the complex association of ion channels with other proteins that target, anchor, regulate, or otherwise influence their behavior is growing (see Fig. 64.11). Examples include EFHC1 and LGI1, discussed earlier in the chapter, which encode ion channel–regulating proteins and cause ion channel dysfunction when mutated. Mutation in ankyrin-B, an anchoring protein that co-localizes a diverse range of ion channels and other membrane proteins, causes long-QT syndrome type 4 (Mohler et al., 2003). Mutations in similar anchoring proteins are likely to be discovered in neurological disease; perhaps Schwartz-Jampel syndrome is an example. This rare autosomal recessive syndrome characterized by myotonia and chondrodysplasia results from mutations in HSPG2, the gene encoding perlecan, the heparan sulfate proteoglycan enriched in basement membranes and cartilage (Stum et al., 2006); the pathophysiological link between perlecan and myotonia has yet to be established. The spectrum of episodic and electrical disorders of the nervous system will continue to grow.
Andermann F., Kobayashi E., Andermann E. Genetic focal epilepsies: state of the art and paths to the future. Epilepsia. 2005;46:61-67.
Aridon P., Marini C., Di Resta C., et al. Increased sensitivity of the neuronal nicotinic receptor [alpha]2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79:342-350.
Artto V., Nissilä M., Wessman M., et al. Treatment of hemiplegic migraine with triptans. Eur J Neurol. 2007;14:1053-1056.
Auer-Grumbach M., Olschewski A., Papic L., et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42:160-164.
Barber P.A., Anderson N.E., Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology. 2000;54:771-772.
Battistini S., Stenirri S., Piatti M., et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. 1999;53:38-43.
Baulac S., Huberfeld G., Gourfinkel-An I., et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46-48.
Bendahhou S., Cummins T.R., Griggs R.C., et al. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol. 2001;50:417-420.
Bendahhou S., Cummins T.R., Kula R.W., et al. Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology. 2002;58:1266-1272.
Biervert C., Schroeder B.C., Kubisch C., et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403-406.
Bökenkamp R., Wilde A.A., Schalij M.J., et al. Flecainide for recurrent malignant ventricular arrhythmias in two siblings with Andersen-Tawil syndrome. Heart Rhythm. 2007;4:508-511.
Castro M.-J., Stam A., Lemos C., et al. First mutation in the voltage-gated NaV1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia. 2009;29:308-313.
Charlier C., Singh N.A., Ryan S.G., et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family [see comments]. Nat Genet. 1998;18:53-55.
Chen Y., Lu J., Pan H., et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239-243.
Cossette P., Liu L., Brisebois K., et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184-189.
Dalmau J., Gleichman A.J., Hughes E.G., et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091-1098.
Davies A.G., Pierce-Shimomura J.T., Kim H., et al. A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell. 2003;115:655-666.
De Fusco M., Marconi R., Silvestri L., et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump [alpha]2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192-196.
Deng H.-X., Klein C.J., Yan J., et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42:165-169.
Dib-Hajj S.D., Yang Y., Waxman S.G., et al. Chapter 4: genetics and molecular pathophysiology of Nav1.7-related pain syndromes. In: Advances in Genetics. Academic Press; 2008:85-110.
Dibbens L.M., Feng H.-J., Richards M.C., et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13:1315-1319.
Dichgans M., Freilinger T., Eckstein G., et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371-377.
Du W., Bautista J.F., Yang H., et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733-738.
Ducros A., Denier C., Joutel A., et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17-24.
Ducros A., Joutel A., Vahedi K., et al. Mapping of a second locus for familial hemiplegic migraine to 1q21-q23 and evidence of further heterogeneity. Ann Neurol. 1997;42:885-890.
Elmslie F.V., Rees M., Williamson M.P., et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997;6:1329-1334.
Escayg A., De Waard M., Lee D.D., et al. Coding and noncoding variation of the human calcium-channel beta4- subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531-1539.
Escayg A., MacDonald B.T., Meisler M.H., et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343-345.
Eulenburg V., Becker K., Gomeza J., et al. Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun. 2006;348:400-405.
Graus F., Lang B., Pozo-Rosich P., et al. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59:764-766.
Guerrini R., Bonanni P., Patrignani A., et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: a newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain. 2001;124:2459-2475.
Guida S., Trettel F., Pagnutti S., et al. Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2. Am J Hum Genet. 2001;68:759-764.
Hans M., Luvisetto S., Williams M.E., et al. Functional consequences of mutations in the human alpha 1A calcium channel subunit linked to familial hemiplegic migraine. J Neurosci. 1999;19:1610-1619.
Hayward L.J., Sandoval G.M., Cannon S.C. Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology. 1999;52:1447-1453.
Heron S.E., Crossland K.M., Andermann E., et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851-852.
Heron S.E., Khosravani H., Varela D., et al. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol. 2007;62:560-568.
Jallon P., Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46:10-14.
Jen J., Kim G.W., Baloh R.W. Clinical spectrum of episodic ataxia type 2. Neurology. 2004;62:17-22.
Jen J., Wan J., Graves M., et al. Loss-of-function EA2 mutations are associated with impaired neuromuscular transmission. Neurology. 2001;57:1843-1848.
Jen J., Yue Q., Nelson S.F., et al. A novel nonsense mutation in CACNA1A causes episodic ataxia and hemiplegia. Neurology. 1999;53:34-37.
Jen J.C., Graves T.D., Hess E.J., et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130:2484-2493.
Jen J.C., Wan J., Palos T.P., et al. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65:529-534.
Kalachikov S., Evgrafov O., Ross B., et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335-341.
Khadilkar S.V., Singh R.K., Mansukhani K.A., et al. Relief from episodic weakness with pyridostigmine in paramyotonia congenita: a family study. Muscle Nerve. 2010;41:133-137.
Kornblum C., Lutterbey G.G., Czermin B., et al. Whole-body high-field MRI shows no skeletal muscle degeneration in young patients with recessive myotonia congenita. Acta Neurol Scand. 2010;121:131-135.
Krampfl K., Maljevic S., Cossette P., et al. Molecular analysis of the A322D mutation in the GABA receptor alpha-subunit causing juvenile myoclonic epilepsy. Eur J Neurosci. 2005;22:10-20.
Kraus R.L., Sinnegger M.J., Koschak A., et al. Three new familial hemiplegic migraine mutants affect P/Q-type Ca2+ channel kinetics. J Biol Chem. 2000;275:9239-9243.
Lai M., Hughes E.G., Peng X., et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424-434.
Lancaster E., Lai M., Peng X., et al. Antibodies to the GABAB receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67-76.
Landoure G., Zdebik A.A., Martinez T.L., et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet. 2010;42:170-174.
Lee E.K., Maselli R.A., Ellis W.G., et al. Morvan’s fibrillary chorea: a paraneoplastic manifestation of thymoma. J Neurol Neurosurg Psychiatry. 1998;65:857-862.
Lee H., Wang H., Jen J.C., et al. A novel mutation in KCNA1 causes episodic ataxia without myokymia. Hum Mutat. 2004;24:536.
Lee H.Y., Xu Y., Huang Y., et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. 2004;13:3161-3170.
Liguori R., Vincent A., Clover L., et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001;124:2417-2426.
Lossin C., Wang D.W., Rhodes T.H., et al. Molecular basis of an inherited epilepsy. Neuron. 2002;34:877-884.
Matthews E., Hanna M.G. Muscle channelopathies: does the predicted channel gating pore offer new treatment options for hypokalaemic periodic paralysis? J Physiol. 2010.
Miller T.M., Dias da Silva M.R., Miller H.A., et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004;63:1647-1655.
Mjåset C., Russell M. Intravenous nimodipine worsening prolonged attack of familial hemiplegic migraine. J Headache Pain. 2008;9:381-384.
Mohler P.J., Schott J.J., Gramolini A.O., et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634-639.
Morante-Redolat J.M., Gorostidi-Pagola A., Piquer-Sirerol S., et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119-1128.
Ophoff R.A., Terwindt G.M., Vergouwe M.N., et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543-552.
Pellizzón O.A., Kalaizich L., Ptáçek L.J., et al. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008;19:95-97.
Phillips H.A., Favre I., Kirkpatrick M., et al. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225-231.
Plaster N.M., Tawil R., Tristani-Firouzi M., et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511-519.
Plaster N.M., Uyama E., Uchino M., et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology. 1999;53:1180-1183.
Rea R., Spauschus A., Eunson L.H., et al. Variable K+ channel subunit dysfunction in inherited mutations of KCNA1. J Physiol (Lond). 2002;538:5-23.
Rees M.I., Lewis T.M., Kwok J.B.J., et al. Hyperekplexia associated with compound heterozygote mutations in the β-subunit of the human inhibitory glycine receptor (GLRB). Hum Mol Genet. 2002;11:853-860.
Reimann F., Cox J.J., Belfer I., et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A. 2010;107:5148-5153.
Ryan D.P., da Silva M.R., Soong T.W., et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
Schulte U., Thumfart J.-O., Klocker N., et al. The epilepsy-linked lgi1 protein assembles into presynaptic kv1 channels and inhibits inactivation by Kv[beta]1. Neuron. 2006;49:697-706.
Segall L., Scanzano R., Kaunisto M.A., et al. Kinetic alterations due to a missense mutation in the Na,K-ATPase α2 subunit cause familial hemiplegic migraine type 2. J Biol Chem. 2004;279:43692-43696.
Serra S.A., Cuenca-Leon E., Llobet A., et al. A mutation in the first intracellular loop of CACNA1A prevents P/Q channel modulation by SNARE proteins and lowers exocytosis. Proc Natl Acad Sci U S A. 2010;107:1672-1677.
Shillito P., Molenaar P.C., Vincent A., et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol. 1995;38:714-722.
Singh N.A., Charlier C., Stauffer D., et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns [see comments]. Nat Genet. 1998;18:25-29.
Singh N.A., Pappas C., Dahle E.J., et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649.
Sokolov S., Scheuer T., Catterall W.A. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76-78.
Spadaro M., Ursu S., Lehmann-Horn F., et al. A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics. 2004;5:177-185.
Steinlein O.K., Mulley J.C., Propping P., et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201-203.
Sternberg D., Maisonobe T., Jurkat-Rott K., et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. 2001;124:1091-1099.
Striano P., Bordo L., Lispi M.L., et al. A novel SCN2A mutation in family with benign familial infantile seizures. Epilepsia. 2006;47:218-220.
Strupp M., Kalla R., Dichgans M., et al. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology. 2004;62:1623-1625.
Stum M., Davoine C.-S., Vicart S., et al. Spectrum of HSPG2 (perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum Mutat. 2006;9999:n/a.
Sugawara T., Tsurubuchi Y., Agarwala K.L., et al. A missense mutation of the Na+ channel alpha II subunit gene Nav1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384-6389.
Suzuki T., Delgado-Escueta A.V., Aguan K., et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842-849.
Swoboda K.J., Kanavakis E.K., Xaidara A., et al. Alternating hemiplegia of childhood or familial hemiplegic migraine?: a novel ATP1A2 mutation. Ann Neurol. 2004;55:884-887.
Takamori M. Lambert-Eaton myasthenic syndrome as an autoimmune calcium channelopathy. Biochem Biophys Res Commun. 2004;322:1347-1351.
Tanaka M., Olsen R.W., Medina M.T., et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249-1261.
Tawil R., McDermott M.P., Brown R.Jr, et al. Randomized trials of dichlorphenamide in the periodic paralyses. Working Group on Periodic Paralysis. Ann Neurol. 2000;47:46-53.
Thieben M.J., Lennon V.A., Boeve B.F., et al. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004;62:1177-1182.
Thomsen L.L., Eriksen M.K., Roemer S.F., et al. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125:1379-1391.
Tomimitsu H., Arimura K., Nagado T., et al. Mechanism of action of voltage-gated K+ channel antibodies in acquired neuromyotonia. Ann Neurol. 2004;56:440-444.
Tottene A., Pivotto F., Fellin T., et al. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678-17686.
Tzartos S.J., Barkas T., Cung M.T., et al. Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev. 1998;163:89-120.
Urak L., Feucht M., Fathi N., et al. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet. 2006;15:2533-2541.
Vanmolkot K.R.J., Kors E.E., Hottenga J.J., et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54:360-366.
Venance S.L., Cannon S.C., Fialho D., et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006;129:8-17.
Wallace R.H., Marini C., Petrou S., et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49-52.
Wallace R.H., Wang D.W., Singh R., et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366-370.
Wan J., Khanna R., Sandusky M., et al. CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics. Neurology. 2005;64:2090-2097.
Wang H.-S., Pan Z., Shi W., et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890-1893.
Wu F.F., Takahashi M.P., Pegoraro E., et al. A new mutation in a family with cold-aggravated myotonia disrupts Na(+) channel inactivation. Neurology. 2001;56:878-884.
Yang Y., Wang Y., Li S., et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41:171-174.
Yoon G., Oberoi S., Tristani-Firouzi M., et al. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. 2006;140A:312-321.
Yoon G., Quitania L., Kramer J.H., et al. Andersen-Tawil syndrome: definition of a neurocognitive phenotype. Neurology. 2006;66:1703-1710.
Yu W., Horowitz S.H. Familial hemiplegic migraine and its abortive therapy with intravenous verapamil. Neurology. 2001;57:1732-1733.
Zerr P., Adelman J.P., Maylie J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J Neurosci. 1998;18:2842-2848.
Zhang J., Bendahhou S., Sanguinetti M.C., et al. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology. 2000;54:937-942.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 64 Channelopathies
Episodic and Electrical Disorders of the Nervous System
Channelopathies are disorders caused by ion channel dysfunction. Because of the great diversity of ion channel proteins and their expression in different tissues, channelopathies comprise a wide variety of clinical diseases (Table 64.1), the discovery of which helps elucidate how ion channels function in both illness and health. The periodic paralyses—the first group of ion channel disorders characterized at a molecular level—defined the field of channelopathies, which now encompasses diseases not only in muscle but also in the kidney (Bartter syndrome), epithelium (cystic fibrosis), and heart (long QT syndrome) as well as neurons. Because muscles and neurons are electrical organs, it is not surprising that most channelopathies are associated with neurological disease. Despite significant heterogeneity, a pervasive feature of neurological channelopathies is a paroxysmal phenotype. After a brief introduction to ion channels, this chapter describes disorders caused by congenital and acquired dysfunction of ion channels expressed in skeletal muscle and neurons.
Ion Channels
Depending on the location within the channel, mutations could alter voltage-dependent activation, ion selectivity, or time and voltage dependence of inactivation. Thus, two different mutations within the same gene can result in dramatically different physiological defects. For example, a mutation that prevents or slows inactivation could lead to a persistent ionic current. Conversely, a mutation elsewhere in the same gene that prevents activation will decrease ionic current. Phenotypic heterogeneity describes how different mutations in a single gene cause distinct phenotypes. For instance, mutations in the skeletal muscle voltage-dependent sodium channel can result in hyperkalemic periodic paralysis, hypokalemic periodic paralysis, potassium-aggravated myotonia, or paramyotonia congenita (see Table 64.1 and Fig. 64.1). In contrast, genetic heterogeneity occurs when a consistent clinical syndrome results from a variety of underlying mutations.
Voltage-gated potassium channels (VGKC) consist of four homologous α-subunits that combine to create a complete channel. Like the other channels described later, humans possess many distinct VGKC genes, and the resulting channels exhibit specialized properties and display rich tissue-type and cellular-compartment specificity. Each α-subunit contains six transmembrane segments (S1 to S6) linked by extracellular and intracellular loops (Fig. 64.2). The S5-S6 loop penetrates deep into the central part of the channel and lines the pore. The S4 segment contains positively charged amino acids and acts as the voltage sensor. These channels serve many functions, most notably to establish the resting membrane potential and to repolarize cells following an action potential. A unique class of potassium channel, the inwardly rectifying potassium channel, is homologous to the S5 to S6 segment of the VGKC. Because the voltage-sensing S4 domain is absent, voltage dependence results from a voltage-dependent blockade by magnesium and polyamines.
Voltage-gated sodium and calcium channels are highly homologous and share homology with VGKCs, from which they evolved. The α-subunits contain four highly homologous domains in tandem within a single transcript (DI–DIV) (Fig. 64.3). Each domain resembles a VGKC α-subunit, with six transmembrane segments as described earlier. Sodium and calcium channels differ in several regards, despite their many similarities. The amino acid sequence forming the selectivity filter and the modulatory auxiliary subunits are different. The sodium channel is composed of an α- and a β-subunit, and the calcium channel is composed of a pore-forming α1-subunit, an intracellular β-subunit, a membrane-spanning γ-subunit, and a membrane-anchoring α2δ-subunit. Sodium channels mediate fast depolarization and underlie the action potential, whereas voltage-gated calcium channels (VGCCs) mediate neurotransmitter release and allow the calcium influx that leads to second messenger effects.
Fig. 64.3 Diagram of the voltage-gated sodium channel and the four most common mutations causing hyperkalemic periodic paralysis. The α-subunit consists of four highly homologous domains (I to IV), each containing six transmembrane segments (S1 to S6). When inserted into the membrane, the four domains fold so as to encircle a central ion-selective pore lined by the S5/S6 loop. Analogous to the potassium channel (see Fig. 64.2), the S4 segments contain positively charged residues conferring voltage dependence on the channel. Auxiliary β-subunits are not shown.
Ligand-gated ion channels activate on binding with their respective agonists. Among the diverse array of ligand-gated channels, GABAA, glycine, and nicotinic acetylcholine receptors (nAChRs) possess the only known disease-causing mutations. Although distinguished by their ligand binding and ion permeability, channels gated by GABA, glycine, and acetylcholine share several structural similarities. Five intrinsic membrane subunits assemble to form hetero- or homopentamers. Each subunit contains four transmembrane domains (M1 to M4), the second of which lines the pore and determines ionic selectivity (Fig. 64.4). Subunits contributing to nAChRs at the neuromuscular junction differ from those expressed in the central nervous system, explaining why mutation of one gene may cause seizures without affecting neuromuscular transmission, or vice versa. Binding of acetylcholine opens the channel, which conducts monovalent cations (Na+ and K+) with little or no selectivity, and some are additionally permeable to calcium. Channel activation results in membrane depolarization and excitation of the postsynaptic neuron or muscle fiber.
Genetic Disorders of Muscular Ion Channels
The periodic paralyses and nondystrophic myotonias encompass several skeletal muscle disorders inherited as autosomal dominant traits. These disorders include hypokalemic periodic paralysis (hypoKPP), hyperkalemic periodic paralysis (hyperKPP), paramyotonia congenita, myotonia congenita, potassium-aggravated myotonia, and Andersen-Tawil syndrome. General features include episodic weakness or stiffness, interictal return to an asymptomatic state, and responsiveness to carbonic anhydrase inhibition (see Table 64.3).
Hypokalemic Periodic Paralysis
Pathophysiology
In up to 70% of cases, the responsible mutation has been linked to a gene encoding a VGCC on chromosome 1q (Venance et al., 2006). The gene, CACNA1S, encodes the α1-subunit of the dihydropyridine-sensitive L-type VGCC found in skeletal muscle. This channel functions as the voltage sensor of the ryanodine receptor and plays an important role in excitation-contraction coupling in skeletal muscle. Four mutations in the S4 segments are responsible for voltage sensitivity. Two mutations, one involving arginine-to-histidine substitutions within the highly conserved S4 segments of DII and DIV (Arg-528-His and Arg-1239-His) account for most cases. The others involve arginine-to-glycine substitutions at the same locations.
Some 10% to 20% of families with hypoKPP have mutations in the gene encoding the α-subunit of the skeletal muscle voltage-gated sodium channel (SCN4A) on chromosome 17q. This is the same channel implicated in hyperKPP and other disorders described later (see Fig. 64.1). Evidence suggests that this sodium channel–associated syndrome is phenotypically different from the more common CACNA1S form. A proposed separate clinical entity, hypoKPP2, may be distinguishable from hypoKPP associated with CACNA1S by the presence of myalgias following paralytic attacks, and the presence of tubular aggregates instead of vacuoles in the muscle biopsy. In some patients, acetazolamide worsens symptoms (Bendahhou et al., 2001; Sternberg et al., 2001). In a large retrospective series, hypoKPP2 was associated with an older age of onset and shorter duration of attacks than classical hypoKPP (Miller et al., 2004).
Whether involving SCN4A or CACNA1S, virtually all mutations causing hypoKPP involve an S4 voltage-sensor domain. In the case of the sodium channel, these mutations allow a leak current to pass through the “gating pore” at resting membrane potentials, bypassing the central channel pore and leading to inappropriate muscle fiber depolarization and consequent channel inactivation and action potential failure (Sokolov et al., 2007). Speculation exists that this phenomenon may also occur in mutated VGCCs.
Diagnosis
An accurate medical history is essential for the diagnosis because observation of attacks is unusual, and patients are often normal between attacks. Characteristic features of hypoKPP that distinguish it from hyperKPP are that paralytic attacks are less frequent, longer lasting, precipitated by a carbohydrate load, and often begin during sleep (Table 64.2). Potassium concentrations are usually low during an attack, although concentrations less than 2 mM should suggest a secondary form of periodic paralysis. Electrocardiogram (ECG) changes such as increased PR and QT intervals, T-wave flattening, and prominent U waves suggest an underlying hypokalemia. Provocative testing can be dangerous and is not routine. Test performance requires a hospitalized setting with continuous cardiac monitoring and should be performed only in patients without cardiac or renal disease. After giving an oral glucose load (2-5 mg/kg up to a maximum of 100 g) without or, less commonly, with subcutaneous insulin (0.1 U/kg), perform serial examinations of strength while monitoring serum glucose and potassium concentrations. Other diagnostic tests include electromyography (EMG), which may show decreased compound muscle action potential amplitudes during attacks compared with interictal values. Muscle histology reveals nonspecific myopathic changes of tubular aggregates or vacuoles within fibers. Genetic testing should render muscle biopsy and provocative testing obsolete for diagnosis.
Thyrotoxic periodic paralysis may be clinically indistinguishable from hypoKPP, except that it is not familial and serum potassium levels are often lower than in familial hypoKPP (<2.5). Some cases may be associated with a mutation in KCNJ18, the gene encoding a novel inwardly rectifying potassium channel (Ryan et al., 2010). All patients with hypoKPP require screening for hyperthyroidism, as the treatment—correction of the thyroid disorder—differs from that outlined later for idiopathic hypoKPP. Exclude other secondary forms of hypokalemic paralysis when serum potassium concentrations remain low between attacks. Renal, adrenal, and gastrointestinal causes of persistent hypokalemia are common, and thiazide diuretic use or licorice (glycyrrhizic acid) intoxication are considerations.
Treatment
An effective holistic approach to treatment includes lifestyle modifications and acute and chronic pharmacological intervention. Dietary modification to avoid high carbohydrate loads and refraining from excessive exertion helps prevent attacks. Oral potassium (5-10 g load) reverses paralysis during an acute attack. Prophylactic use of acetazolamide (Table 64.3) decreases the frequency and severity of attacks. Dichlorphenamide is another carbonic anhydrase inhibitor that effectively prevents attacks, as demonstrated in a randomized clinical trial (Tawil et al., 2000) where the average dose was 100 mg daily. More potent than acetazolamide, dichlorphenamide may be useful when efficacy of the former begins to fail. Many believe, without supporting evidence, that reducing the frequency of paralytic attacks provides protection against the development of myopathy. As insight into molecular pathophysiology expands, new treatment possibilities—and a new understanding of old treatments—seem likely (Matthews and Hanna, 2010).
| Use | Prophylactic agent for some channelopathies (see text) |
| Mechanism | Inhibits carbonic anhydrase |
| Dosing | Adults: start 125 mg daily, titrating as needed up to a maximum daily dose of 1000-1500 mg, divided bid–qid. An extended release formulation is available. Children: consult a pharmacist. |
| Side effects | Taste changes (especially for carbonated drinks), fatigue, paresthesias, metabolic acidosis, blurred vision, myelosuppression, nephrolithiasis, etc. (Increased dietary citrate might be recommended to compensate for decreased urinary citrate observed during acetazolamide therapy.) |
| Monitoring | Check electrolytes, BUN, creatinine, and CBC at baseline and periodically throughout therapy. |
| Metabolism | None; excreted unchanged by kidneys |
Please note that this table is for brief informational purposes only. Prescribing physicians should consult a pharmacist or an appropriate reference for complete and updated information.
bid, Twice daily; BUN, blood urea nitrogen; CBC, complete blood cell count; qid, 4 times daily.
Hyperkalemic Periodic Paralysis
Clinical
Characteristic of this disorder is episodic weakness precipitated by hyperkalemia. Although the weakness is generally milder than in hypoKPP, it can be sufficiently severe in hyperKPP to cause flaccid quadriparesis. As in hypoKPP, respiratory and ocular muscles are unaffected and consciousness is preserved. Frequency of attacks varies from several per day to several per year. Attacks are usually brief, lasting 15 to 60 minutes, but may last up to days. Unlike hypoKPP, myotonia is present between attacks. Onset is usually in infancy or childhood, and characteristic attacks occur by adolescence. Triggers include rest after vigorous exercise, foods high in potassium, stress, and fatigue. Despite its name, hyperKPP is often associated with a normal serum potassium concentration during an attack (see following Diagnosis section). Most patients experience a subacute onset, and some describe paresthesias or a sensation of muscle tension prior to attacks. In these situations, mild exercise may abort or lessen the severity of the attack. Mild weakness may persist afterward, and the later development of a progressive myopathy is common.
Pathophysiology
HyperKPP is as an autosomal dominant disorder, with some sporadic cases. The disorder links to SCN4A, the same gene responsible for a minority of hypoKPP cases. Among several identified missense mutations, four account for about two-thirds of cases (see Fig. 64.1). Functional expression of naturally occurring mutations demonstrated a decrease in the voltage threshold of channel activation or abnormally prolonged channel opening or both (Bendahhou et al., 2002; Hayward et al., 1999), effectively increasing the depolarizing inward current. If sustained long enough, this would lead to inactivation of the sodium channels, transitory cellular inexcitability, and weakness.
Diagnosis
Despite advances in defining the underlying genetic mutations, a thorough medical and family history and physical examination remain the best diagnostic tools. Furthermore, genetic testing is not generally available, and the high number of causative sodium channel mutations makes widespread use of genetic testing unlikely in the near future. Serum potassium is normal between attacks and even during many attacks. Unlike hypoKPP, potassium administration may precipitate an attack. In the absence of provocative testing, the basis for diagnosis is the clinical presentation. Myotonia is present in many patients between attacks, either spontaneously or after muscle percussion, and failure to produce myotonia discriminates hypoKPP from hyperKPP (see Table 64.2). Take care not to confuse subjective muscle stiffness with objective changes. Peaked T waves on ECG suggest hyperkalemia and are an aid to diagnosis. As in hypoKPP, serum creatine kinase concentrations may be normal or elevated. Electrodiagnostic studies are useful for demonstrating subclinical myotonic discharges, not seen in hypoKPP. Nonspecific findings such as fibrillation potentials and small polyphasic motor unit potentials occur during late stages of disease.
Treatment
The goal of therapy is to abort the acute attacks and prevent future attacks. Acute attacks are often sufficiently brief and mild so as not to require acute intervention. In more severe attacks, aim treatment at lowering extracellular potassium levels. Mild exercise or eating a high sugar load (juice or a candy bar) may suffice, as insulin drives extracellular potassium into cells. Thiazide diuretics and inhaled β-adrenergic agonists are similarly helpful, and intravenous calcium gluconate may be useful when weakness is very severe. To prevent attacks, a diet low in potassium and high in carbohydrates may obviate the need for prophylactic drug therapy. Oral dichlorphenamide was useful for prophylaxis in one randomized controlled trial (Tawil et al., 2000). Acetazolamide (see Table 64.3) and thiazide diuretics are useful as well. Successful prophylaxis may decrease the later onset of myopathy, although direct proof of this hypothesis is lacking. Finally, myotonic symptoms are troublesome in some patients; based on the underlying pathophysiology, sodium channel blockers would seem an effective therapy, and mexiletine is commonly used for this purpose.
Paramyotonia Congenita
Treatment
Symptoms are generally mild and infrequent. Direct treatment, when required, at either myotonia or weakness or both. Sodium channel blockers such as mexiletine are sometimes effective in reducing the frequency and severity of myotonia. Patients with weakness often respond to agents used to treat hyperKPP (e.g., thiazides, acetazolamide). A single case report suggests the possible use of pyridostigmine (Khadilkar et al., 2010). Cold avoidance reduces the frequency of attacks.
Myotonia Congenita
Clinical
Inheritance of myotonia congenita (MC) is either as an autosomal dominant (Thomsen disease) or recessive (Becker myotonia) trait. The main feature is myotonia or delayed muscle relaxation after contraction. Forceful movement abruptly initiated after several minutes of rest causes the most pronounced myotonic stiffness. The myotonia of MC displays a warm-up phenomenon in which the myotonia decreases or vanishes completely when repeating the same movement several times, in contrast to the myotonia seen in patients with PMC. The onset of Thomsen myotonia is often within the first decade, whereas the onset of Becker myotonia is generally at 10 to 14 years of age. Although myotonia can affect all skeletal muscles, it is especially prominent in the legs, where it is occasionally severe enough to impede a patient’s ability to walk or run. In rare cases, sudden noise causes sufficient generalized stiffness to make the patient fall to the ground and remain rigid for several seconds. The recessive and dominant forms share many similarities, but some clinical features help distinguish the two. In general, patients with recessive disease experience transitory bouts of weakness after periods of disuse and may develop progressive myopathy (Kornblum et al., 2010); in addition, muscle hypertrophy and disease severity are greater than in the dominant form. Becker myotonia is more common than Thomsen disease.
Pathophysiology
Electrical instability of the sarcolemma leads to muscle stiffness by causing repetitive electric discharges of affected muscle fibers. Early in vivo studies in myotonic goats revealed greatly diminished sarcolemmal chloride conductance in affected muscle fibers. This causes a depolarization of the sarcolemmal membrane and muscle hyperexcitability. Genetic linkage analysis for both recessive and dominant forms of MC pointed to a locus on chromosome 7q, where the responsible gene, CLCN1, encodes the major skeletal muscle chloride channel. More than 70 mutations have been identified within CLCN1, and interestingly, some of these mutations are recognized to cause both dominant and recessive forms (Zhang et al., 2000). Examination of the functional effects of several myotonia-causing CLCN1 mutations in heterologous expression systems reveal effects on channel gating, usually resulting in a decreased chloride conductance (Zhang et al., 2000).
Potassium-Aggravated Myotonia
Pathophysiology
PAM links to chromosome 17q, where mutations in the SCN4A gene cause the disease. This sodium channel is the same one implicated in hyperKPP, PMC, and the sodium channel subtype of hypoKPP (see earlier discussion and Fig. 64.1). Functional expression studies reveal that the disease-causing mutations lead to a large persistent sodium current secondary to an increased rate of recovery from inactivation and an increased frequency of late channel openings (Wu et al., 2001). The cause of myotonia is this enhanced inward current, which leads to prolonged depolarization and subsequent membrane hyperexcitability.
Andersen-Tawil Syndrome
Clinical
Andersen-Tawil syndrome (ATS) is a rare autosomal dominant disorder characterized by the triad of periodic paralysis, cardiac arrhythmias, and dysmorphic features (including hypertelorism, micrognathia, low-set ears, high-arched or cleft palate, short stature, and clinodactyly) (Yoon et al., 2006a). The periodic paralysis, often triggered by rest after exercise, prolonged rest, and stress, is often the presenting symptom and can be hypo-, normo-, or hyperkalemic. The cardiac phenotype, often discovered later, includes prolonged QT intervals, but bidirectional ventricular tachycardia is common. Despite the known association of cardiac arrhythmias in rare periodic paralysis patients, ATS was only recognized as a separate entity in 1971. In families segregating an ATS allele, the phenotypic expressivity can vary greatly. Patients can manifest one, two, or three features of the triad, and the severity of any one feature can be extremely variable. Rare individuals are asymptomatic disease-gene carriers. Finally, ATS patients may exhibit neurocognitive deficits in executive function and abstract reasoning (Yoon et al., 2006b).
Pathophysiology
Mutations in the KCNJ2 gene on chromosome 17q account for approximately two-thirds of ATS probands. KCNJ2 encodes a widely expressed inwardly rectifying potassium channel (Plaster et al., 2001). Interestingly, among all identified probands, about 50% have an autosomal dominant disorder, and identification of sporadic cases with de novo mutations is common. The mechanisms of channel dysfunction are heterogeneous, including impaired phospholipid binding, pore function, or protein trafficking. Because VGKCs are tetrameric complexes, many (if not all) of the mutations are dominant negative.
Diagnosis
Previous studies that took into account the variable penetrance of ATS classified individuals as affected if two of three criteria were met: paroxysmal weakness, prolonged QT interval with or without ventricular dysrhythmias, or characteristic dysmorphic features (Yoon et al., 2006a). ATS should be included in the differential diagnosis of any individual with documented long-QT syndrome, even in the absence of periodic paralysis or dysmorphism. Some family members of patients with the full clinical triad show only prolonged QT intervals. Similarly, perform ECG on all patients with suspected periodic paralysis for careful measurement of the QT interval.
[/not-level-membership-for-neurology-category]
