Chapter 45 Cerebral protection
The cerebral circulation is arguably the most important and most vulnerable in the body. Arrest of the circulation for only a few minutes can cause neuronal death. The concept of cerebral protection has been engaged across a broad spectrum of clinical scenarios. It has been incorporated into the prophylaxis, treatment and subsequent management of ischaemia and infarction and even into attempts to ameliorate postischaemic or anoxic damage following cardiorespiratory resuscitation. A complete review is beyond the scope of this chapter, but an understanding of the current approaches to cerebral protection is certainly helpful in the management of cerebral insults.
NORMAL BRAIN PHYSIOLOGY
COLLATERAL BLOOD SUPPLY
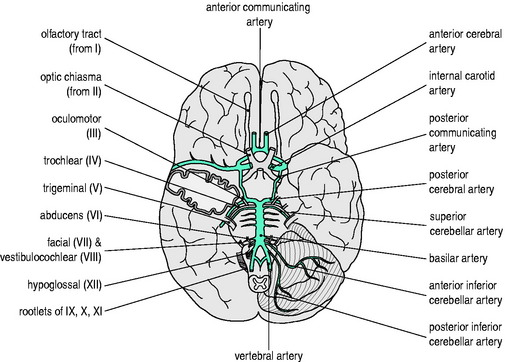
An elaborate vascular architecture is designed to ensure adequate CBF. The anterior cerebral circulation is provided by bilateral internal carotid arteries as they each divide into anterior and middle cerebral arteries. They provide approximately 70% of the cerebral circulation and supply the anterior cerebrum (frontal, parietal and temporal lobes) and anterior diencephalon (basal ganglia and hypothalamus). The remaining 30% of the cerebral circulation is provided posteriorly by the vertebrobasilar system which runs the length of the posterior fossa, supplying the brainstem, cerebellum and posterior portion of the cerebrum (occipital lobes) and diencephalon (thalamus). These two circulations are joined by communicating arteries, ultimately forming the circle of Willis (Figure 45.1). This joining of the anterior and posterior circulations at the base of the brain is the major component of cerebral vasculature in humans. Between these arterial distributions are watershed zones fed by leptomeningeal connections. In addition, persistent fetal arteries can infrequently provide collateral routes between the anterior and posterior arterial systems in the brain.
CEREBRAL BLOOD FLOW
CEREBRAL PERFUSION PRESSURE
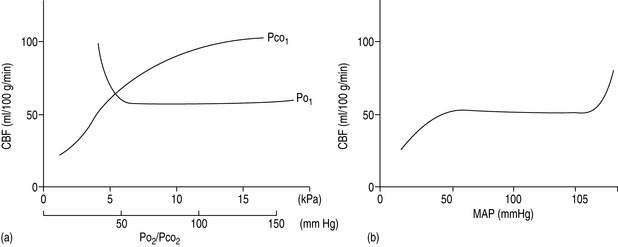
The cerebral vessels change diameter inversely with changing perfusion pressure: as CPP rises, the vessels constrict and as CPP falls the vessels dilate, such that blood flow is kept constant over a wide range of CPP (Figure 45.2a). This pressure autoregulation is thought to be controlled by local myogenic responses of the vessel wall to changes in intra-arterial pressure. At pressures above and below this range of 6.7–20 kPa (50–150 mmHg), cerebral perfusion becomes pressure-passive and increases or decreases in direct proportion to changes in CPP. The autoregulatory range varies with age, being shifted to the left in newborns and to the right in those with chronic hypertension. The latter is important to remember to avoid overtreating systolic blood pressure in such patients and thus incur the risk of cerebral ischaemia at the lower limits of autoregulation. Alternatively, cerebral perfusion above normal can be caused by acute hypertension overcoming the upper limits of autoregulation. This may lead to cerebral oedema secondary to increased hydrostatic pressures (hypertensive encephalopathy) and potentially lead to seizures or cerebral haemorrhage.
PaO2 AND PaCO2 EFFECTS
A second group of factors control CBF through an influence on the local metabolic milieu. Prominent in this mechanism are oxygen and carbon dioxide. Arterial content or partial pressure of oxygen in the normal or hyperoxic ranges causes very little change in CBF. Perhaps this represents a demand for another nutrient (i.e. glucose) or a need to remove waste products (i.e. carbon dioxide or metabolic acid). With the onset of hypoxaemia (PaO2 60 mmHg or 8 kPa), there is a prompt increase in CBF proportional to the decrease in blood oxygen content, in order to maintain oxygen delivery constant (Figure 45.2b).
There is also a direct relationship between CBF and PaCO2, such that cerebral perfusion increases with increasing PaCO2 (Figure 45.2b). This probably represents the need of the brain to maintain homeostatic pH by removing metabolic breakdown products more efficiently by increased blood flow. Unlike the response to oxygen, the CBF response to changes in PaCO2 is dramatic in the physiological range, such that for every 0.13 kPa (1.0 mmHg) change in PaCO2 there is a 1–2 ml/min per 100 g tissue change in CBF. Therefore, an increase in PaCO2 to 10.6 kPa (80 mmHg) will increase CBF to approximately 100 ml/min per 100 g and a decrease in PaCO2 to 2.7 kPa (20 mmHg) will decrease CBF to 25 ml/min per 100 g. Thus:
GLOBAL HYPOXIC/ISCHAEMIC INSULTS
These types of insult are caused by hypoxaemia and cardiovascular insufficiency or arrest, respectively. These are usually sudden, short and severe. If there is to be recovery, prompt return of oxygen delivery and spontaneous circulation are necessary. The recovery may be variable, depending on the severity and duration of the insult and the selective vulnerability of the cell types involved. Different mechanisms are responsible for reversible loss of cellular function and for irreversible cell death and there are also differences between the mechanisms that cause death of neurones, glia and endothelial cells. After 4–6 minutes of complete global ischaemia, there are signs of permanent histological damage in selective neuronal populations and the beginnings of neurological deficits in survivors. Outcome worsens significantly after 15 minutes of global ischaemia.1
BRAIN ISCHAEMIC AND INFARCTION PROCESSES
Changes in normal physiology begin to occur when blood flow is reduced.
EFFECTS OF ISCHAEMIA
This begins a cascade of events resulting in eventual cell death:
Other effects within the cell influence DNA and RNA production, hence inhibiting protein production. This may explain why cellular and clinical recovery is partial, even with restoration of ionic equilibrium and near-normal ATP levels after successful reperfusion. Necrosis is thought to occur in the core of the cerebral infarct following acute vascular occlusion, with further neurodegeneration occurring more slowly in the penumbra, by apoptosis or release of various immunological mediators.2
EXTRACELLULAR EFFECTS
Leukocytes are thought to be major contributors to reperfusion injury in that:
MANAGEMENT
In cases of head trauma, it is very important to prevent secondary brain injury. Reviews of intensive care practice have resulted in recommendations for treatment in this group of patients.3 These involve:
HYPERTENSION
Most patients have elevated blood pressure in the early phase after an acute ischaemic stroke. The mechanism and effects of this blood pressure elevation are not well understood and the benefits of intervention remain debated. Most experts would not advocate antihypertensive treatment in the acute phase after stroke, unless the blood pressure is particularly high. Currently hypertension (systolic blood pressure > 180 mmHg) should be treated if thrombolytics are to be administered, based on the trials involving these agents in which the blood pressure was lowered prior to treatment.4 Ongoing studies such as Blood Pressure in Acute Stroke Collaboration (BASC) should help define optimal management.
HYPERGLYCAEMIA
Hyperglycaemia has also been associated with an increased mortality and reduced functional outcome after stroke irrespective of whether the patient has a diabetic history or not.5 Proposed explanations for this are:
Hyperglycaemia may result in a hypercoaguable state, with decreased plasma fibrinolytic activity, which may delay reperfusion and increase haemorrhagic infarct conversion in tissue plasminogen activator-treated patients.6
Insulin, as well as having a glucose-lowering effect, has also been shown to be directly neuroprotective and this is the subject of a number of ongoing trials7 (Treatment of Hyperglycaemia in Ischaemic Stroke (THIS) trial, the Glucose Regulation in Acute Stroke Patients (GRASP) trial and the Glucose Insulin Stroke Trial UK (GIST-UK).
HYPERTHERMIA
An increased temperature increases cerebral metabolism, oxygen requirements, CBF and ICP. Hyperpyrexia following acute stroke adversely influences stroke severity, infarct size, functional outcome and mortality.8 A raised temperature should, therefore, be treated aggressively and any evidence of infection identified early and treated with appropriate antibiotics.
REVASCULARISATION
Ischaemic neuronal degeneration follows the onset of perfusion failure so that interventional therapy must begin immediately. Diagnostic imaging is required before using thrombolytic agents. For acute ischaemic stroke the National Institute of Neurological Disorders and Stroke (NINDS study) suggested efficacy if intravenous thrombolytic therapy is initiated within the first 3 hours of stroke symptoms (using recombinant tissue plasminogen activator).9
HAEMODILUTION
Current practice includes the use of hypervolaemic haemodilution and deliberate hypertension with augmentation of cardiac output using vasopressors and inotropic agents, particularly in the treatment of delayed ischaemic deficits after subarachnoid haemorrhage related to vasospasm. This therapy can be administered safely with intensive monitoring, but benefits are anecdotal, as it has not been tested with concurrent controls.10
Decreasing haematocrit and viscosity by haemodilution may improve flow and hence blood and oxygen delivery to areas with impaired arterial supply. Benefit in animal models of ischaemia has not been found in clinical trials of stroke. Trials of normovolaemic11 and hypervolaemic haemodilution12 have had complications from volume therapy in patients with associated heart disease.
Theoretical neuroprotective benefits of human haemoglobin-derived blood substitutes have not translated into clinical benefit, largely due to adverse events, and therefore clinical development of substitutes were discontinued.13
INTRACRANIAL PRESSURE
The relationship between ICP and intracranial volume is described by a non-linear pressure–volume curve. At lower intracranial volumes the ICP remains low and reasonably constant. Any increases in intracranial volume are compensated for by decreases in intracerebral blood or CSF volume. If greater intracranial volumes persist, this compensation is lost and ICP rises considerably, despite relatively small increases in intracerebral blood volume. Eventually at high levels of ICP cerebrovascular responses are lost, ICP equals MAP and CPP is very low.14
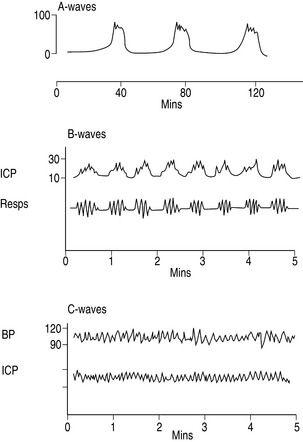
MONITORING OF ICP (Figure 45.3)
ICP-monitoring devices have been ranked by the Brain Trauma Foundation14 on their accuracy, stability and ability to drain CSF. Intraventricular and intraparenchymal catheters are seen as most favourable, followed by subdural, subarachnoid and epidural devices.
INTRAVENTRICULAR DEVICES
ICP is measured from an external pressure transducer attached to an intraventricular catheter inserted in one of the lateral ventricles using the external auditory meatus as the reference point. This device allows CSF sampling for culture or therapeutic drainage, the administration of antibotics and the ability to rezero following insertion. The main disadvantage is the high infection rate associated with their use.14,15 Infection rates increase significantly after 5 days of catheter insertion and with repeated use of the device’s flushing system. The use of antibiotic-impregnated catheters may lower the infection rate. Air, blood, tissue debris and ventricular collapse can all adversely affect measurement.
INTRAPARENCHYMAL DEVICES
ICP can be measured by either a miniature strain gauge pressure transducer mounted at the end of a thin catheter or by a fibreoptic cable which directs light to a miniature mirror at the end of the cable placed in brain tissue. These monitors are advantageous when lateral ventricles have collapsed secondary to raised ICP. This system is not dependent on fluid coupling for pressure transduction so waveform damping and artefacts are avoided. The infection rates are very low. Its main disadvantages include lack of recalibration in vivo, a tendency for baseline drift requiring transducer replacement, the inability to drain CSF and the ability to measure only localised pressure which may not be representative of the global ICP.15
REGULATION OF INTRACRANIAL PRESSURE
BRAIN COMPARTMENT
Reduction in the parenchymal compartment depends on removal of either free water or the lesion causing the raised ICP.
Free water must be moved across an intact blood–brain barrier. Mannitol increases plasma osmolarity and reduces brain oedema. In addition, it may have a beneficial effect on microcirculatory flow. It is also thought to have antioxidant effects, although these may not be clinically important. Hypertonic saline has reduced ICP in patients with haemorrhagic shock and traumatic brain injury when used for volume resuscitation (7.5%), and also for treatment of raised ICP refractory to mannitol (23.4%).16
Removal of tumour or blood clot, drainage of abscesses and extirpation of infarcted brain are all therapies aimed at improving compliance. Mounting evidence now suggests that there is a penumbra of functionally impaired but potentially reversible neuronal injury surrounding a haematoma. Indications for clot removal, despite numerous studies performed over the last four decades, has been controversial. The Surgical Trial in Intracerebral Haemorrhage (STICH) compared early surgical evacuation of haematoma with initial conservative treatment, but found no significant difference in outcomes between the two treatments.17
The rationale for removing part of the skull overlying the stroke in patients with or at risk of developing cerebral oedema and intracranial hypertension is simply to decompress the brain swelling and prevent herniation. Decompressive craniectomy has been assessed in experimental cerebral infarction and was effective in reducing death and neurological impairment whether performed 1 hour or 24 hours after induction of permanent middle cerebral artery occlusion.18 An uncontrolled trial comparing patients with hemicraniectomy with historical controls found that mortality rates were reduced from 80% to 35% in the surgical group.19 These non-randomised studies are clearly at risk of bias and properly controlled trials of craniectomy for malignant middle cerebral artery infarction are required.
BLOOD COMPARTMENT
Although a small component of intracranial volume, the blood compartment is the most compliant. Reduction in blood volume is useful in the treatment of raised ICP, especially in the acute setting. As explained above, hypoxia and hypercarbia can lead to hyperaemia and an increase in cerebral blood volume, potentially worsening ICP. Alternatively, induced hypocarbia leads to very rapid changes in blood flow and blood volume. Hyperventilation for raised ICP is controversial. Up until 10 years ago, patients were often aggressively hyperventilated to a PaCO2 of 25 mmHg in order to reduce ICP rapidly as a result of arteriolar vasoconstriction. Unfortunately, the resultant reduction in cerebral blood volume may be accompanied by a fall in global CBF, which may result in ischaemia and a worsened outcome. The Brain Trauma Foundation guidelines in the 1990s recommended that chronic hyperventilation (PaCO2 = 25 mmHg) should not be instituted in severe traumatic brain injury in patients with a normal ICP and that prophylactic hyperventilation to a PaCO2 of 35 mmHg should also be avoided in the first 24 hours after a severe traumatic brain injury, as it can reduce cerebral perfusion during a time of reduced CBF. Current studies suggest that ICP reduction, as a result of moderate hyperventilation within the first 24 hours and the resultant decrease in CBF, do not compromise cerebral metabolism.20 It is recommended that a secondary treatment be instituted as soon as possible to allow slow withdrawal of hyperventilation.21 If adaptation to hypocapnia has not occurred, hyperventilation can be reinstituted with the same effect. Prevention of seizures and hyperthermia lower the cerebral metabolic demand and reduce CBF and volume.
CEREBROSPINAL FLUID COMPARTMENT
CSF drainage can be used to reduce ICP but is only possible if there is a ventricular catheter in place. Care is taken regarding the route and rate of CSF drainage to avoid herniation of a mass lesion, either towards the other side of the brain or through the tentorium. In recent years there has been a move towards the use of less invasive methods to measure ICP. However, the Brain Trauma Foundation guidelines for the management of severe head injury provide some evidence supporting the increased use of CSF drainage for ICP control.22
HYPOTHERMIA
Hypothermia has been known to offer protection for years. In particular, drowning victims who were hypothermic have survived long periods of ischaemia. The mechanism of ICP reduction is unknown but may be due to a reduction in intracranial blood volume secondary to cerebral vasoconstriction or to alterations in metabolism. The protective effect of cooling appears to be much greater than that explained by changes in metabolism alone.
In contrast to preventing hyperthermia, the use of induced hypothermia is more complex. It has been used for protection extensively in coronary artery bypass surgery where hypothermia (28–30°C) is commonplace and deep hypothermia (< 20°C) has allowed prolonged circulatory arrest for surgery such as high thoracic and giant cerebral aneurysms. Animal work suggests that moderate, systemic hypothermia reduces the cerebral oedema and death after injury to the cerebral cortex. In several centres cooling has been used as therapy for severe head injuries. Unfortunately, a study which evaluated the efficacy of hypothermia in head injuries was halted after the enrolment of 392 patients because the treatment was ineffective.24 Cooling patients to 33°C within 8 hours after injury and maintaining hypothermia for 48 hours were not effective in improving the clinical outcome at 6 months and patients older than 45 years of age had a poorer outcome. This contrasted with two earlier studies,25,26 both of which demonstrated an improvement in outcome with cooling to 32°C.
In stroke patients there are no randomised controlled trials of hypothermia. Animal models suggest that hypothermia reduces ischaemic stroke lesion size.27 A small uncontrolled study of moderate hypothermia found reduced mortality from an expected value of 78% to 44% in patients with severe middle cerebral artery infarction.28 There are ongoing studies such as the Nordic Cooling Stroke Study (NOCSS).
In cardiac arrest the situation is changing. Although 50 years ago therapeutic hypothermia was used post cardiac arrest, it was abandoned because of the uncertain benefit. In the 1980s, animal studies indicated benefit with mild (32–35°C) rather than moderate or deep hypothermia. Two randomised clinical trials of hypothermia, after witnessed out-of-hospital cardiac arrest, demonstrated improved survival and neurological outcome after 12 or 24 hours of hypothermia at 33°C.29,30 The evidence from these two relatively small trials has led to recommendations as set out by the International Liaison Committee on Resuscitation:
It has also been suggested, currently without evidence, that hypothermia may be beneficial for other rhythms or for in-hospital cardiac arrest. It is not recommended in patients with severe cardiogenic shock, life-threatening arrhythmias, pregnant patients or those with a coagulopathy.31
The uptake of these recommendations is variable. Recently, in the UK, a survey suggested that only 26% of units currently undertook therapeutic hypothermia after cardiac arrest. The main reasons for not using therapeutic hypothermia appeared to be due to logistical or resource issues and a perceived lack of evidence or lack of consensus within individual intensive care unit teams.32
There is currently insufficient evidence to recommend therapeutic hypothermia in children resuscitated from cardiac arrest. Neonatal animal studies, however, have shown promising results with regard to neuroprotection offered by posthypoxic hypothermia, although it has been shown that the protective effects of hypothermia are lost without adequate sedation.33
ANAESTHETIC AGENTS
Barbiturates have shown convincing benefit, especially for focal ischaemia in many animal species. In one small clinical study of induced barbiturate coma during coronary bypass surgery, there was a reduction in focal deficits.34 Barbiturate-mediated neuroprotection was initially attributed to suppression of cerebral metabolic rate but more recently to redistribution of CBF to injured areas, the blockade of glutamate receptors and sodium channels, inhibition of free radical formation and potentiation of GABAergic activity.35 At present barbiturates are not commonly used either in coronary bypass surgery or in situations of global ischaemia. Barbiturates are now less commonly used in head-injured patients but can play a role in those patients who have intractable intracranial hypertension.
Propofol has been shown to be neuroprotective in vivo, in focal and global models of cerebral ischaemia. It decreases cerebral metabolic rate and hence CBF. It has been shown to have antioxidant properties, potentiate GABA-A-mediated inhibition of synaptic transmission and inhibit glutamate release. It delays neuronal death by being a free radical scavenger, preventing lipid peroxidation and modulating apoptosis-regulating proteins.35,36 Its side-effects include hypotension with a reducing CPP and hyperlipidaemia when an infusion of 200 μg/kg per min is used to produce burst suppression.37 This latter problem has been lessened by the introduction of a more concentrated formulation.
Inhalational agents have significant neuroprotective effects but the precise mechanism by which they reduce cerebral injury is unclear.36 It is possible that isoflurane may attenuate excitotoxicity by inhibiting glutamate release and its postsynaptic responses at both anaesthetic and EEG burst supression concentrations. The neuroprotection provided by volatile agents may also be attributable to their effect at GABA-A receptors and an ability to reduce the sympathetic vascular response to ischaemia. Certainly their effect on reducing cerebral metabolic rate is not sufficient to explain their neuroprotective properties.35,36 These potential benefits in adults are offset by recent studies which have suggested that in neonatal animal studies there may be disturbing increases in cerebral apoptosis, although this has yet to be confirmed. There is an increasing interest in xenon, which presently looks promising.
Nitrous oxide exhibits the neuroprotective and neurotoxic features of an NMDA antagonist. Studies, however, have shown that, when combined with an opioid, e.g. fentanyl, its neuroprotective effect during incomplete cerebral ischaemia is still inferior when compared to volatile agents.36
CALCIUM ANTAGONISTS
The influx of calcium from the extracellular space and from intracellular organelles has been implicated as the common mediator of cell death from a variety of causes. Calcium antagonists were among the first neuroprotective agents studied to prevent cerebral ischaemia. Despite the effects seen in animal models, human studies in both global and focal ischaemia have been disappointing. Two large trials of intravenous nimodipine in patients with acute ischaemic stroke were terminated early as neurological and functional outcome were significantly poorer in the nimodipine group.38,39 A close relationship was found between a reduction in diastolic and mean blood pressure in the group treated with nimodipine and an unfavourable neurological outcome. A review of 29 randomised acute stroke trials involving calcium antagonists concluded that the use of calcium antagonists could not be justified in patients with ischaemic stroke and that, although the published trials showed no overall effect on death and dependency, unpublished trials were associated with a statistically significant worse outcome.40 Nimodipine, however, has become the standard prophylactic treatment for cerebral vasospasm after subarachnoid haemorrhage, with a consequent decrease in cerebral infarction and better patient outcome.41 Benefits appear to be due to an effect on smaller penetrating vessels not seen by angiography, or a neuroprotective effect at the cellular level, rather than cerebral vasodilatation identifiable by angiography.
Nimodipine was shown to be neuroprotective in head-injured patients with traumatic subarachnoid haemorrhage.42 Regrettably, further studies to test the neuroprotective effect of nimodipine in severely head-injured patients with traumatic subarachnoid haemorrhage (including the multicentre study, HIT IV) failed to confirm the beneficial effects of nimodipine. A small group of patients with a Glasgow Coma Score < 9 did appear to be a have a better outcome in the nimodipine-treated group;43 however, a recent systematic review concluded that there was no beneficial effect on outcome.44 Its use therefore remains contentious.
STEROIDS
Glucocorticoids were used for over 30 years in the treatment of head injury despite the fact that randomised trials had failed to demonstrate their effectiveness reliably. They were thought to decrease cerebral oedema associated with breakdown of the blood–brain barrier (i.e. vasogenic oedema) and show improvement in central nervous system function with brain tumours and abscesses.45,46
The Corticosteroid Randomisation After Significant Head injury (CRASH) trial investigated the effects of a 48-hour infusion of methylprednisolone on death within 14 days or disability at 6 months in 10 008 adults with clinically significant head injury. The trial was stopped early, as at interim analysis, the steroid-treated subjects had significantly higher all-cause 2-week mortality (21.1% versus 17.9%, P = 0.0001). The 6-month mortality was also higher in steroid-treated subjects (25.7% versus 22.3%, P = 0.0001), with a trend toward increases in the combined endpoint of death or severe disability (38.1% versus 36.3%, P = 0.08). In neither report did the results differ by injury severity or time since injury.47 The cause for the increased mortality is unclear.
In subarachnoid and primary intracerebral haemorrhage, corticosteroids have also been commonly used. In 2005, a Cochrane Review concluded that there was no evidence to support the use of mineralocorticoids or glucocorticoids in subarachnoid haemorrhage or to support the use of glucocorticoids in primary intracerebral haemorrhage. Corticosteroid use may also be associated with adverse events.48
In acute spinal cord injury, high-dose methylprednisolone for 24 hours has been shown to offer a small but significant benefit, provided treatment begins within 8 hours of injury (National Spinal Cord Injury Study (NASCIS) II trial).49,50 This is still controversial. Adverse side-effects such as sepsis and poor wound healing were associated with the use of methylprednisolone, although some of these side-effects did not reach statistical significance. Currently steroid therapy in spinal cord injury is an unproven standard of care.51
EXPERIMENTAL THERAPY – THE FUTURE
Intracerebral haemorrhage is the least treatable form of stroke and is associated with a high morbidity and mortality. Recombinant activated factor VIIa (rFV11a) administered within 4 hours of onset and within 1 hour of a diagnostic CT scan has been shown in one trial to limit haematoma growth, reduce mortality and improve 90-day functional outcome. There was, however, a small increase in the frequency of thromboembolic events.58
1 Bedell S, Delbanco T, Cook E, et al. Survival after cardiopulmonary resuscitation in the hospital. N Engl J Med. 1983;309:569-576.
2 Dirnagi U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-397.
3 Maas AIR, Dearden M, Teassdale GM, et al. EBIC guidelines for management of severe head injury in adults. Acta Neurochir. 1997;139:286-294.
4 Brott T, Lu M, Kothari R, et al. Hypertension and its treatment in the NINDS rt-PA stroke trial. Stroke. 1998;29:1504-1509.
5 Capes SE, Hunt D, Malmberg K, et al. stress hyperglycaemia and prognosis of stroke in nondiabetic and diabetic patients; a systematic overview. Stroke. 2001;32:2426-2432.
6 Ribo M, Molina C, Montaner J, et al. Acute hyperglycaemia state is associated with lower t-PA-induced recanalisation rates in stroke patients. Stroke. 2005;36:1705-1709.
7 Strong AJ, Fairfield JE, Monteiro E, et al. Insulin protects cognitive function in experimental stroke. J Neurol Neurosurg Psychiatry. 1990;53:847-853.
8 Reith J, Jorgensen HS, Pedersen PM, et al. Body temperature in acute stroke: relation to stroke severity, infarct size, mortality and outcome. Lancet. 1996;347:422-425.
9 The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischaemic stroke. N Engl J Med. 1995;333:1581-1587.
10 Solomon R, Fink M, Lennihan L. Early aneurysm surgery and prophylactic hypervolemic, hypertensive therapy for the treatment of aneurysmal subarachnoid hemorrhage. Neurosurgery. 1988;23:699-704.
11 Italian Acute Stroke Study Group. Haemodilution in acute stroke: results of the Italian haemodilution trial. Lancet. 1988;1:318-321.
12 The Hemodilution in Stroke Study Group. Hypervolemic hemodilution treatment of acute stroke. Results of a randomised multicenter trial using pentastarch. Stroke. 1989;20:317-323.
13 Saxena R, Wijnhoud AD, Carton H, et al. Controlled safety study of a haemoglobin-based oxygen carrier, DCL Hb, in acute ischaemic stroke. Stroke. 1999;30:993-996.
14 Brain Trauma Foundation. Available online at www2.braintrauma.org/
15 Steiner LA, Andrews PDJ. Monitoring the injured brain: ICP and CBF. Br J Anaesth. 2006;97:26-38.
16 Qureshi AI, Suarez JI. Use of hypertonic saline solutions in treatment of cerebral oedema and intracranial hypertension. Crit Care Med. 2000;28:3301-3313.
17 Mendelow AD, Gregson BA, Fernandes HM, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387-397.
18 Forsting M, Reith W, Shabitz W-R, et al. Decompressive craniectomy for cerebral infarction. An experimental study in rats. Stroke. 1995;26:259-264.
19 Rieke K, Schwab S, Horn M, et al. Decompressive surgery in space occupying hemispheric infarction: results of an open, prospective trial. Crit Care Med. 1995;23:1576-1587.
20 Oertel M, Kelly DF, Lee JH, et al. Efficacy of hyperventilation, blood pressure elevation, and metabolic suppression therapy in controlling intracranial pressure after head injury. J Neursurg. 2002;97:1045-1053.
21 Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731-739.
22 Bullock MR, Povilshock JT. Indications for intracranial pressure monitoring. J Neurotrauma. 1996;13:667-669.
23 Ginsberg MD, Busto R. Combating hyperthermia in acute stroke: a significant clinical concern. Stroke. 1998;29:529-534.
24 Clifton GL, Miller ER, Choi SC, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344:556-563.
25 Clifton GL, Allen S, Barrodale P, et al. A phase II study of moderate hypothermia in severe brain injury. J Neurotrauma. 1993;10:263-271.
26 Marion DW, Obrist WD, Carlier PM, et al. The use of moderate therapeutic hypothermia for patients with severe head injuries: a preliminary report. J Neurosurg. 1993;79:354-362.
27 Baker J, Onesti T, Solomon R. Reduction by delayed hypothermia of cerebral infarction following middle cerebral artery occlusion in the rat: a time-course study. J Neurosurg. 1992;77:438-444.
28 Schwab S, Schwartz S, Spranger M, et al. Moderate hypothermia in the treatment of patients with severe middle cerebral artery infarction. Stroke. 1998;29:2461-2466.
29 The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurological outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
30 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
31 Nolan JP, Morley PT, Vanden Hoek TV, et al. Therapeutic hypothermia after cardiac arrest: an advisory statement by the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118-121.
32 Laver SR, Padkin A, Atalla A, et al. Therapeutic hypothermia after cardiac arrest: a survey of practice in intensive care units in the United Kingdom. Anaesthesia. 2006;61:873-877.
33 Tooley JR, Satas S, Porter H, et al. Head cooling with mild systemic hypothermia in anaesthetised piglets is neuroprotective. Ann Neurol. 2003;53:65-72.
34 Nussmeier NA, Arlund C, Slogoff S. Neuropsychiatric complications after cardiopulmonary bypass: cerebral protection by a barbiturate. Anesthesiology. 1986;64:165-170.
35 Kawagughi M, Furuya H, Patel PM. Neuroprotective effects of anaesthetic agents. J Anesth. 2005;19:150-156.
36 Sanders RD, Ma D, Maze M. Anaesthesia induced neuroprotection. Best Pract Res Clin Anaesthesiol. 2005;19:461-474.
37 Menon DK. Cerebral protection in severe brain injury: physiological determinants of outcome and their optimisation. Br Med Bull. 1999;55:226-258.
38 Wahlgren NG, MacMahon DG, De Keyser J, et al. Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke. Cerebrovasc Dis. 1994;4:20. –10
39 Bridgers S, Koch G, Munera C, et al. Intravenous nimodipine in acute stroke: interim analysis of randomised trial. Stroke. 1991;22:29.
40 Horn J, Limburg M. Calcium antagonists for ischaemic stroke: a systematic review. Stroke. 2001;32:570-576.
41 Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. Br Med J. 1989;298:636-642.
42 Harders A, Kakarieka A, Braakman R, et al. Traumatic subarachnoid haemorrhage and its treatment with nimodipine. J Neurosurg. 1996;85:82-85.
43 Clausen T, Bullock R. Medical treatment and neuroprotection in traumatic brain injury. Curr Pharm Design. 2001;7:1517-1532.
44 Vergouwen MD, Vermeulen M, Roos YB. Effect of nimodipine on outcome in patients with traumatic subarachnoid haemorrhage: a systematic review. Lancet Neurol. 2006;5:1029-1032.
45 Reulen H. Vasogenic brain oedema. New aspects in its formation, resolution and therapy. Br J Anaesth. 1976;48:741-752.
46 French LA, Galicich JH. The use of steroids for control of cerebral oedema. Clin Neurosurg. 1964;10:212-223.
47 CRASH trial collaborators. Final results of the MRC CRASH trial, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365:1957-1959.
48 Feigin V.L, Anderson N, Rinkel GJE, et al. Corticosteroids for aneurysmal subarachnoid haemorrhage and primary intracerebral haemorrhage. Cochrane Database of Systematic Reviews. 3, 2005. CD004583
49 Bracken M, Shepard M, Collins W, et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: one-year follow-up data. J Neurosurg. 1992;76:23-31.
50 Bracken MB, Shepard MJ, Collins W, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second national acute spinal cord injury study. N Engl J Med. 1990;322:1405-1411.
51 Hurlbert R. Strategies of a medical intervention in the management of acute spinal cord injury. Spine. 2006;31(suppl.):S16-21.
52 Sacco RL, DeRosa JT, Clarke Haley EJr, et al. Glycine antagonist in neuroprotection for patients with acute stroke. GAIN Americas: a randomised controlled trial. JAMA. 2001;285:1728-1729.
53 Meloni BP, Zhu H, Knuckey NW. Is magnesium neuroprotective following global and focal ischaemia? A review of published studies. Magnesium Res. 2006;19:123-137.
54 Saver JL, Kidwell C, Eckstein M, et al. Prehospital neuroprotective therapy for acute stroke: results of the Field Administration of Stroke Therapy–Magnesium (FAST-MAG) pilot trial. Stroke. 2004;35:106-108.
55 Wahlgren NG, for the CLASS study group. The Clomethiazole Acute Stroke Study (CLASS): results of a randomised controlled trial of clomethiazole versus placebo in 1360 acute stroke patients. J Stroke. 1999;230:21-28.
56 Diener HC. for the European and Australian Lubelozole Ischaemic Stroke Study Group. Multinational randomised controlled trial of lubelozole in acute ischaemic stroke. Cerebrovasc Dis. 1998;8:172-181.
57 Ginsberg MD. Adventures in the pathophysiology of brain ischaemia: penumbra, gene expression, neuroprotection. The 2002 Thomas Willis lecture. Stroke. 2003;34:214-223.
58 Mayer SA, Brun NC, Begtrup K, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2005;352:777-785.