21 Central Nervous System Tumors
Primary central nervous system (CNS) tumors accounted for an estimated 21,810 new cases diagnosed in the year 2008 in the United States,1 and for an estimated 13,810 deaths for the same year. The incidence of brain tumors has been slowly increasing at an average rate of 1.1% per year.2 However, the death rate has been decreasing over time, and the 5-year relative survival rate for primary brain tumors has improved from 24% during 1975 through 1977 to 35% during 1996 through 2003.1 There is an early peak of 2 to 3 per 100,000 in the annual age-specific incidence of primary brain tumors in children up to the age of 4 years. A decline occurs between 15 and 24 years, followed by a steady rise, reaching a plateau of 21 per 100,000 between 75 and 79 years of age.3 Several histopathologically different tumors arise in the brain, reflecting the diversity of phenotypically distinct cells within the CNS, that have a capacity for neoplastic transformation.4 Malignant gliomas are considered first in this chapter. Many of the principles of brain tumor management are discussed in this section. Tumors that are most common in children but that are also seen in adult patients are reviewed in Chapter 54 and are only briefly presented here. The chapter closes with a discussion on the management of metastatic brain tumors, which constitute the most common type of brain tumor encountered.
Malignant Glioma
Epidemiologic Statistics
Malignant gliomas make up 35% to 45% of primary brain tumors, and of these, nearly 85% are glioblastoma multiforme (GBM).4 The incidence of anaplastic astrocytoma (AA) peaks in children younger than 10 years of age and then remains constant in each subsequent decade of life. In contrast, GBM is uncommon before the age of 20 years, whereas the rate of occurrence increases dramatically after the age of 40.3 The incidence of malignant gliomas has increased at least twofold among the elderly during the previous two decades.5
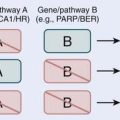
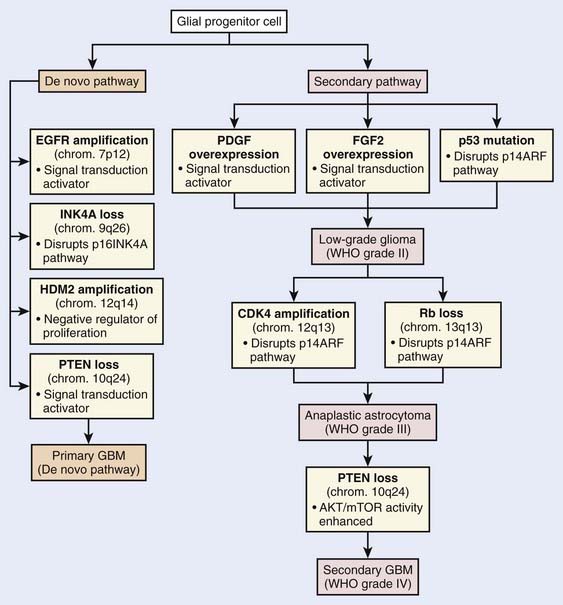
Current knowledge of the molecular biology and genetics of the astrocytic neoplasms has been well summarized.6 Two genetic pathways, a progression pathway and a de novo pathway, that lead to GBM development have been described (Fig. 21-1). Although primary and secondary GBMs are histologically indistinguishable, it is well accepted that distinct molecular aberrations and pathways lead to the formation of primary (de novo) versus secondary GBMs. Primary GBMs are thought to arise in a de novo manner, without any malignant precursor lesion. Amplification of the epidermal growth factor receptor (EGFR) is thought to be a critical event in the molecular cause of primary GBMs. Secondary GBMs, on the other hand, are thought to arise as a result of the malignant transformation of lower grade gliomas, with overexpression of platelet-derived growth factor, fibroblast growth factor–2, and cyclin-dependent kinase 4, as well as p53 mutations and loss of Rb playing major roles in such transformations. Loss of PTEN has been implicated in both pathways, although it is much more common in the pathogenesis of primary GBM.
Several hereditary syndromes are associated with an increased risk of brain tumors (Fig. 21-2). Neurofibromatosis-1 is the most common familial tumor syndrome associated with brain tumors.7 With the exception of Turcot syndrome, all are inherited in an autosomal dominant pattern. In addition to the association between hereditary syndromes and brain tumors, certain families of brain tumor patients have aggregations of brain tumors and extraneural malignancies. Germ-line p53 mutations have been frequently identified in glioma patients with multifocal lesions, a different second primary malignancy, and a family history of cancer.
Although some environmental factors have been linked with brain tumor development, they do not appear to be responsible for most brain tumors.8 Apart from a predisposing genetic syndrome that is present in less than 5% of patients with brain tumors, the only other firmly established cause is ionizing radiation. Radiation-induced gliomas have been reported, and 25% of such cases have arisen in children with acute lymphoblastic leukemia who received prophylactic cranial irradiation and chemotherapy.
Anatomy
The brain is divided into supratentorial and infratentorial compartments by the tentorium cerebelli. The supratentorial compartment includes the cerebral hemispheres and the sellar, pineal, and diencephalic regions, whereas the infratentorial compartment includes the midbrain, pons, medulla, and cerebellum. The cerebral hemispheres are connected by the corpus callosum and are divided into the frontal, parietal, occipital, and temporal lobes. The frontal lobes are concerned with behavior organization, planning and association, and speech; the parietal lobes with motor, sensory, and complex intellectual functions; the occipital lobes with vision; and the temporal lobes with behavior, memory, speech, emotion, and auditory-visual pathways.9 Most malignant gliomas arise in the cerebral hemispheres, and the lobar distribution is directly related to the amount of white matter present in each region.
The brain is housed within the bony calvarium, made up of the frontal, ethmoid, parietal, sphenoid, temporal, and occipital bones. The posterior margin of the lesser wings of the sphenoid marks the anterior aspect of the lateral cerebral (Sylvian) fissure, the junction of the frontal and temporal lobes of the brain. The base of the skull can be approximated on the surface of the patient by a line, called the orbitomeatal or Reid baseline, which extends from the infraorbital rim to the external auditory meatus.9
Pathologic Conditions
Neuropathologists have not agreed on a uniform classification system for brain tumors. However, the fourth edition of the World Health Organization (WHO) classification of tumors of the CNS, published in 2007, is based on the consensus of an international working group of pathologists and geneticists.10 It is the standard for the definition of brain tumors for clinical oncology and cancer research communities worldwide. According to this classification, CNS tumors are classified as follows: (1) tumors of neuroepithelial tissue, (2) germ cell tumors, (3) tumors of cranial and paraspinal nerves, (4) tumors of the sellar region, (5) tumors of the meninges, (6) lymphomas and hemopoietic neoplasms, and (7) metastatic tumors (Table 21-1).
Table 21-1 World Health Organization Classification of Primary Central Nervous System Tumors
| Major Classification of Tumors | WHO Grade |
|---|---|
| Astrocytic Tumors | |
| Pilocytic astrocytoma | I |
| Diffuse astrocytoma | II |
| Anaplastic astrocytomas | III |
| Glioblastoma | IV |
| Gliosarcoma | IV |
| Oligodendroglial Tumors | |
| Oligodendroglioma | II |
| Anaplastic oligodendroglioma | III |
| Oligoastrocytic Tumors | |
| Oligoastrocytoma | II |
| Anaplastic oligoastrocytoma | III |
| Ependymal Tumors | |
| Ependymoma | II |
| Anaplastic ependymoma | III |
| Choroid Plexus Tumors | |
| Choroid plexus papilloma | II |
| Atypical choroid plexus papilloma | III |
| Tumours of the pineal region | |
| Pineocytoma | I |
| Pineoblastoma | IV |
| Neuronal and Mixed Neuronalglial Tumors | |
| Anaplastic ganglioglioma | III |
| Tumors of the Pineal Region | |
| Pineocytoma | I |
| Pineoblastoma | IV |
| Embryonal Tumors | |
| Medulloblastoma | IV |
| CNS primitive neuroectodermal tumors (PNETs) | IV |
| Tumors of the Cranial and Paraspinal Nerves | |
| Schwannoma | I |
| Neurofibroma | I |
| Meningeal Tumors | |
| Meningeoma | I |
| Anaplastic meningioma | II |
| Hemangioblastoma | I |
| Tumors of the Sellar Region | |
| Craniopharyngioma | I |
| Pituicytoma | I |
Histologic grading is a means of predicting the biologic behavior of a neoplasm. In the clinical setting, tumor grade is a key factor influencing the choice of therapies, particularly determining the use of adjuvant radiation and specific chemotherapy protocols. The WHO classification of tumors of the nervous system includes a grading scheme that ranges across a wide variety of neoplasms rather than a strict histologic grading system.10 Grade I applies to lesions with low proliferative potential and the possibility of cure following surgical resection alone. Neoplasms designated grade II are generally infiltrative in nature and, despite low-level proliferative activity, often recur. Some type II tumors tend to progress to higher grades of malignancy, for example, low-grade diffuse astrocytomas that transform to AA and glioblastoma. Similar transformation occurs in oligodendroglioma and oligoastrocytoma. The designation WHO grade III is generally reserved for lesions with histologic evidence of malignancy, including nuclear atypia and brisk mitotic activity. The designation WHO grade IV is assigned to cytologically malignant, mitotically active, necrosis-prone neoplasms. Examples of grade IV neoplasms include glioblastoma, medulloblastoma, and lymphoma.
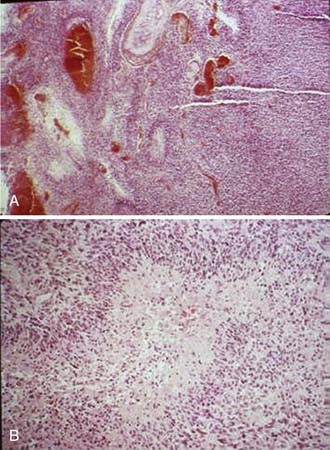
Astrocytic gliomas, which are the most common primary brain tumors, arise from astrocytes, the supporting cells of the brain and spinal cord. The cytoplasmic processes that extend from the astrocytes contain a characteristic filamentous protein, glial fibrillary acidic protein (GFAP), which provides an immunohistochemical marker for these tumors. The WHO defines diffusely infiltrative astrocytic tumors with cytologic atypia alone as grade II (diffuse astrocytoma), those also showing anaplasia and mitotic activity as grade III (AA), and tumors additionally showing microvascular proliferation or necrosis as WHO grade IV (Fig. 21-3). Necrosis may be of any type; perinecrotic palisading need not be present.
Clinical Presentation
The presenting symptoms and signs of brain tumors are divided into those associated with a mass effect and increased intracranial pressure and those that are focal. The most common presenting symptom is headache, and approximately 20% of patients with supratentorial tumors present with seizures. Alterations in personality, mood, mental capacity, and concentration are frequently seen early in the clinical course.4 The focal neurologic symptoms and signs observed with supratentorial brain tumors are summarized in Table 21-2.
| Anatomic Region | Symptoms or Signs or Both |
|---|---|
| Frontal lobe | Personality change |
| Slowing of contralateral hand movements | |
| Contralateral spastic hemiplegia | |
| Mood elevation | |
| Difficulty in adapting to new situations | |
| Loss of initiative | |
| Dysphagia + lip, tongue, and hand movements | |
| Apraxia (if dominant lobe involved) | |
| Bifrontal involvement | Bilateral hemiparesis |
| Spastic bulbar palsy | |
| Impairment of intellect | |
| Lability of mood | |
| Dementia | |
| Primitive grasp, suck, and snout reflex | |
| Temporal lobe | Impairment of recent memory |
| Homonymous quadrantanopsia | |
| Auditory hallucination | |
| Aggressive behaviour | |
| Non-dominant lobe | Minor perceptual problem |
| Spatial disorientation | |
| Dominant lobe | Dysnomia |
| Impaired perception of verbal command | |
| Fluent-Wernicke-like aphasia | |
| Parietal lobe | Mild hemiparesis |
| Mild to severe sensory loss | |
| Homonymous hemianopsia | |
| Visual inattention | |
| Non-dominant lobe | Perceptual abnormalities |
| Anosognosia | |
| Apraxia for self-dressing | |
| Dominant lobe | Alexia |
| Dysgraphia | |
| Other forms of apraxia | |
| Occipital lobe | Contralateral homonymous hemianopsia |
| Visual aberrations | |
| Bilateral occipital involvment | Cortical blindness |
| Thalamus or basal ganglia or both | Herniation syndrome |
| Contralateral sensory abnormalities | |
| Intermittent contralateral paresthesia | |
| Neuropathic pain syndrome | |
| Contralateral intention tremor | |
| Semiballistic-like movement disorder | |
| Hydrocephalus due to trapping of lateral horn of ventricle |
Routes of Spread
Malignant gliomas form as an expansile mass that conforms to the barriers of the cortical convolutions, deep nuclear structures, and adjacent myelinated nerve fibers.11 Microscopic examination typically shows a gradient of infiltrating tumor cells that decreases with the distance from the periphery of the mass. As they enlarge, malignant gliomas extend directly into adjacent lobes, infiltrate throughout the ipsilateral cerebral hemisphere, and disseminate along anatomically defined nerve fiber pathways. In some cases, individual cells, facilitated by the accompanying edema, may infiltrate for long distances from the main tumor mass. Multicentric gliomas are found in less than 5% of patients. Dissemination by seeding through the cerebrospinal fluid pathways occurs in approximately 10% of cases, but is usually a late event, often appearing at a time when its effects are inconsequential compared with those of the recurrent primary tumor mass. Metastases rarely arise outside the CNS.
Diagnostic Studies
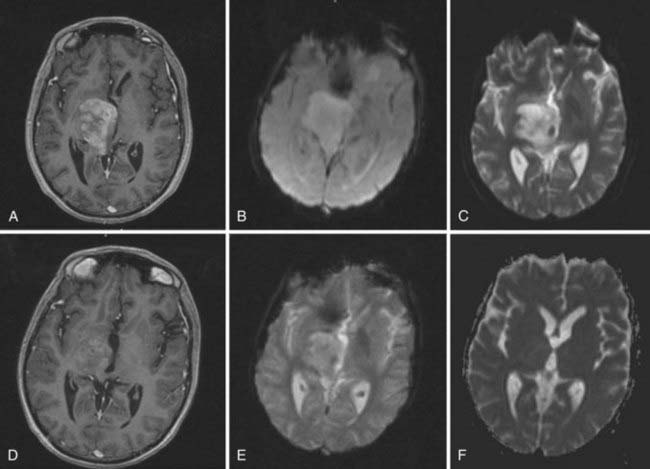
Magnetic resonance imaging (MRI) is the modality of choice for diagnosis and evaluation of intracranial neoplasms. It is the ideal modality for initial preoperative diagnosis, including tumor extent, treatment planning, and image-guided therapies because of its multiplanar capability, anatomic detail, and superior resolution.12,13 The ability to characterize tissue allows for improved assessment of fatty, hemorrhagic, cystic, necrotic, and vascular components, as well as mass effect and location. In malignant gliomas, on T1-weighted sequence enhanced with gadolinium contrast, the tumor appears as an irregular, ringlike configuration that may surround a central area of necrosis (Fig. 21-4). On T2-weighted fluid-attenuation image recovery (FLAIR) images, the changes that reflect edema extend beyond the boundaries of contrast enhancement that may harbor microscopic extension. In fact, tumor may even be found in normal-appearing brain beyond the T2-weighted abnormality.
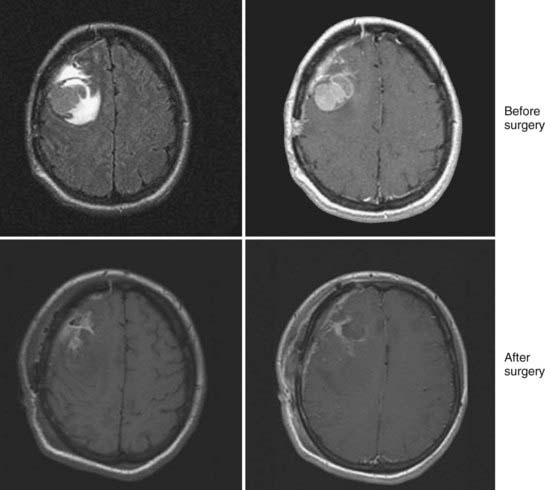
Imaging studies are performed within the first 48 hours after surgical resection to determine the presence of residual tumor and to provide a baseline for subsequent treatment. Enhancement resulting from surgical trauma may be indistinguishable from residual tumor even after a complete surgical resection. Postsurgical enhancement develops as early as the fifth postoperative day, peaks after 2 weeks, and may persist for months.14 Corticosteroids, which act to re-establish the blood-brain barrier, also have a profound effect on the area of enhancement, and diminution in the area of enhancement may be due to corticosteroids alone. To determine a radiographic response to therapy, patients should be on the same or a lower dose of corticosteroids than the dose at the time of the pretreatment scan.
Lacking with conventional MRI is an assessment of physiologic and functional information about the tumor. The gadolinium enhancement, as seen on T1-weighted MRI, reflects regions where there has been a breakdown of the blood-brain barrier. This may not be a reliable indicator of high-grade tumor because of the presence of nonenhancing tumor or contrast-enhancing necrosis.15 Similarly, T2-defined volume either overestimates or underestimates the microscopic or nonenhancing disease in a majority of patients. New MRI techniques, such as MR perfusion, MR diffusion, MR spectroscopy (MRS), and functional MRI (fMRI), can provide further physiologic characterization of the tumor related to hemodynamics, cellularity, and metabolism, respectively, and may be used to assess response to therapy.16
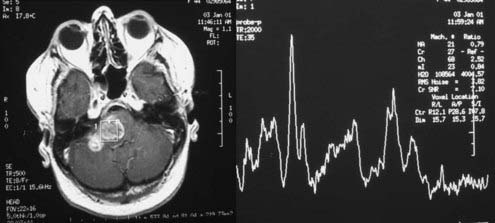
MRS is a noninvasive method to evaluate the malignancy of gliomas based on metabolite levels such as N-acetylaspartate (NAA), choline (Cho) compounds, total creatine (Cr), and lactate (Lac), or metabolite ratios such as Cho/NAA and Cho/Cr (Fig. 21-5). Spectroscopy also holds great promise in aiding target definition for radiotherapy treatment planning and evaluation of response to therapy and tumor recurrence.17 With the advantage of measuring tumor regional variations in abnormalities of metabolite levels, MRS has recently been used as an in vivo molecular imaging technique that assists in targeting and predicts response to radiation therapy for patients with brain gliomas.
In addition to MRS, brain fMRI has gained potential in clinical application recently as it can image the eloquent cortices, such as the motor cortex, the Broca area, the Wernicke area, and the visual cortex, which are difficult to identify on anatomic MRI. Incorporation of fMRI information into the radiotherapy treatment process can possibly allow a radiation oncologist to properly plan and deliver an adequate radiation dose while avoiding damage to the adjacent functional cortices in the treatment of gliomas.18
Dynamic MRI techniques can be used to measure important features of tumor vascularity in vivo, including the density and permeability of capillaries. Increased vessel numbers lead to higher regional cerebral (i.e., tumor) blood volume, which can be measured relative to healthy brain tissue by perfusion MRI techniques. Thus, relative cerebral blood volume provides analysis of capillary density in gliomas (Fig. 21-6). In adult astrocytomas, relative cerebral blood volumes correlate well with histopathologic grade and survival. A relative cerebral blood volume value of more than 1.75 is associated with the presence of high-grade tumor and with reduced survival in patients with gliomas.19
Prognostic Factors
A major contribution of the cooperative group brain tumor studies has been the identification of pretreatment characteristics that affect the outcome in patients with malignant gliomas. A nonparametric recursive partitioning technique was applied to an analysis of 1578 patients accrued to three successive Radiation Therapy Oncology Group (RTOG) trials.20 Age, histologic appearance, Karnofsky performance status (KPS), mental status, duration of symptoms, neurologic functional class, extent of surgery, and radiation dose were identified as significant partitioning covariates. As shown in Table 21-3, six patient classes were defined with median survival times ranging from 4.6 to 58.6 months and 2-year survival rates of 4% to 76%. These classes were used to define favorable (classes I and II, 12% of the patients evaluated), intermediate (classes III and IV, 43%), and poor (classes V and VI, 45%) prognosis subgroups. A subsequent reanalysis of the data in glioblastoma patients showed no statistical difference between class V and VI with a median survival time of 7.5 months.21 This information is important for interpreting correctly the results of studies comparing different treatment regimens and for assessing the potential of new therapeutic methodologies.
Table 21-3 Malignant Glioma Recursive Partitioning Analysis Classification
| Class | Definition |
|---|---|
| I | Age <50, anaplastic astrocytoma, and normal mental status |
| II | Age ≥50, KPS 70-100, anaplastic astrocytoma, and at least 3 months from time of first symptoms to initiation treatment |
| III | —Age <50, anaplastic astrocytoma and abnormal mental status |
| —Age <50, glioblastoma multiforme and KPS 90-100 | |
| IV | —Age <50, glioblastoma multiforme, KPS <90 |
| —Age ≥50, KPS 70-100, anaplastic astrocytoma and 3 months or less from time of first symptoms to start of treatment | |
| —Age >50, glioblastoma multiforme, surgical resection, and good neurologic function | |
| V | —Age ≥50, KPS 70-100, glioblastoma multiforme, either surgical resection and neurologic function that inhibits the ability to work or biopsy only followed by at least 54.4 Gy of RT |
| —Age ≥50, KPS <70, normal mental status | |
| VI | —Age ≥50, KPS <70, abnormal mental status |
| —Age ≥50, KPS 70-100, glioblastoma multiforme, biopsy only, receiving less than 54.4 Gy of RT |
Standard Therapeutic Approaches
Surgery
The combination of surgery, radiation therapy, and chemotherapy represents the standard approach to the treatment of malignant gliomas. Generally, surgery is performed through an open craniotomy. The goals of surgery are to provide a histologic diagnosis, to alleviate intracranial hypertension and focal neurologic deficits resulting from a mass effect, and to permit rapid corticosteroid dose tapering.22 Furthermore, a large tumor mass left in the brain can serve as a nidus for cerebral edema after radiotherapy. The influence of surgical resection in malignant gliomas has been controversial. The aim of palliation of symptoms was always clear, but the survival advantage was debated. However the evidence suggests that patients with more complete resections designed to minimize the volume of residual tumor live longer and have an improved functional status compared with those who undergo a biopsy or partial resection only. An analysis of an RTOG database of 645 patients with malignant gliomas revealed a median survival of 11.3 months with total resection, 10.4 months with subtotal resection, and 6.6 months with biopsy alone.23 However, tumor size was not found to be a predictor of survival. Unfortunately, there are no well-conducted randomized clinical trials in high-grade gliomas that have tested the extent of surgical resection. A poorly done study on 30 elderly patients with radiologically obvious malignant glioma randomized to biopsy alone versus resection showed a borderline survival benefit with resection.24 Advances in neurosurgery, including diagnostic ultrasound, lasers, ultrasonic tissue aspirators, cortical mapping, functional imaging, and computer-assisted stereotactic laser techniques, have improved the ability of neurosurgeons to radically remove intracranial tumors.22
A closed-needle biopsy, using computed tomography (CT)- or MRI-coupled stereotactic techniques, may be indicated in many clinical settings. A biopsy is preferred for tumors located in functionally important or inaccessible areas of the brain. In addition, surgical resection is not practical for patients with significant tumor infiltration across the midline and around the ventricular system, or for those with diffuse, nonfocal lesions.22
Radiation Therapy
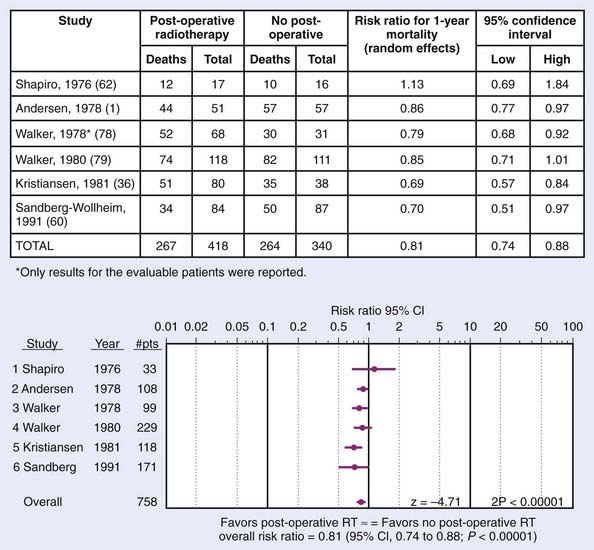
Randomized trials conducted by the Brain Tumor Cooperative Group (BTCG) and the Scandinavian Glioblastoma Study Group (SGSG) provided seminal evidence that external-beam irradiation favorably affects the outcome of malignant gliomas. BTCG trials 6901 and 7201 demonstrated a significant survival advantage for irradiated patients who received 50 to 60 Gy to the whole brain (in single daily fractions of 1.7-2.0 Gy, 5 days per week) either alone or with chemotherapy compared with those treated with either resection and supportive care only (P = 0.001) or with chemotherapy alone (P < 0.01).25,26 The median survival of patients receiving 60 Gy was 2.3 times longer than that observed for nonirradiated patients. Nearly 30% of irradiated patients in the SGSG trial maintained a full or partial working capacity, whereas the nonirradiated patients progressively deteriorated, and none regained their original performance level.27 Randomized trials of postoperative radiotherapy in gliomas is shown in Fig. 21-7. These studies were so convincing that virtually all patients with malignant gliomas receive adjuvant radiation therapy.
Chemotherapy
Until recently, the role of adjuvant chemotherapy in high-grade gliomas remained controversial. Historically, the nitrosoureas, especially carmustine (bis-chloroethyl-nitrosourea [BCNU]), was the most active single agent and no other drug or drug combination had been found to be more effective with the exception of the procarbazine, lomustine (CCNU), and vincristine (PCV) regimen.28 However, a retrospective analysis of RTOG data and a phase III trial conducted at the Medical Research Council (MRC) showed no benefit in survival with the PCV regimen. Similarly, no benefit of chemotherapeutic agents like tirapazamine, topotecan, paclitaxel (Taxol), interferon-β, and thalidomide were noted when used with standard radiation in RTOG phase II trials.28
Introduction of temozolomide has redefined the role of chemotherapy in gliomas and represents the new standard of care. It is a derivative of dacarbazine and an inactive prodrug that undergoes hydrolysis to active metabolite monomethyl triazeno imidazole carboximide when absorbed and results in methylation of guanine at the O6 and N7 positions at the deoxyribonucleic acid. It has several advantages over conventional chemotherapy agents. These include oral administration, rapid absorption, 100% bioavailability, ability to cross the blood-brain barrier, linear pharmacokinetics, and minimal delayed myelosuppression. A large, multicenter phase II trial done in recurrent gliomas refractory to PCV chemotherapy treated with temozolomide showed a 35% response rate (RR) and a 6-month progression-free survival (PFS) rate of 46%, and was subsequently Food and Drug Administration–approved for relapsed AA.29 Based on these responses, the European Organisation for Research and Treatment of Cancer (EORTC) conducted a phase III trial of 60 Gy of radiation with or without temozolomide at 75 mg/m2/day followed by six cycles of adjuvant therapy with temozolomide at 200 mg/m2 for 5 days per month in 543 patients.30 With a median follow-up of 28 months, the median overall survival improved from 12.1 to 14.6 months and the 2-year overall survival improved from 10.4% to 26.5% with the addition of temozolomide therapy.
Current focus of clinical development involves targeted therapies in glioma. Several agents have shown confirmed activity and are currently being investigated for integration in the initial management of malignant gliomas. These include antiangiogenic agents, tyrosine kinase blockers, inhibitors of Ras/MAPK pathways, and histone deacetylase inhibitors.31
Techniques of Radiotherapy
Most malignant gliomas are unifocal at the time of initial presentation, and after treatment the majority recur at or within 1 to 2 cm of their original location.32 Thus, limited treatment portals are used for malignant gliomas. Intracranial metastases that appear after partial brain irradiation do not affect the ultimate outcome, because they are nearly always accompanied by relapse at the primary tumor site. Whole-brain radiation therapy (WBRT) is commonly recommended for patients with multifocal tumors, but even for these lesions, relapses occur most frequently in the sites of known disease.32
The gross target volume (GTV) represents a three-dimensional (3-D) reconstruction of the location of the tumor, determined by merging data from contrast-enhanced CT scans and MRI studies. The integration of MRI into CT-based treatment planning provides complementary information for accurately defining the GTV. Little agreement exists as to the definition of the clinical target volume (CTV) or the planning target volume (PTV). A shrinking field approach is used in RTOG trials. The initial PTV (PTV1) encompasses the enhancing lesion (GTV) and edema (CTV) with a 2-cm margin. After 46 Gy of a conventionally fractionated treatment course, the PTV (PTV2) is reduced to include only the enhancing lesion with a 2.5 cm margin. Using the extent of edema to define the CTV has some limitations. The identification of peritumoral edema is subjective, and its volume may vary with corticosteroid dosage. An analysis of the patterns of failure using the RTOG-defined PTVs demonstrated that nearly all relapses occurred within the reduced PTV2 at the site of the enhancing tumor.33 Based on these data, we define the CTV by adding a margin of approximately 2.5 cm around the T1 contrast or a 1.5-cm margin around the FLAIR series. The PTV is determined by adding an additional 0.3- to 0.5-cm margin to the CTV to account for treatment uncertainties.
Intensity-modulated radiation therapy (IMRT) is an advanced form of 3D-CRT that uses inverse planning and computer-controlled radiation dose deposition. The advantage of precision delivery of radiation enables dose escalation and sparing of normal tissues. The brain IMRT treatment planning consists of immobilization using an Aquaplast face mask. A CT simulation with intravenous contrast is done with the acquisition of 1.5- to 3-mm thickness images. The previously obtained MRI data is superimposed on the CT images using the fusion program. GTV, CTV, and PTV are defined as in a 3D-CRT plan. All normal structures are outlined in three dimensions. User-defined constraints, including maximum and minimum dose constraints both for the PTV and the normal structures, are prescribed. As in 3D-CRT, noncoplanar beams are used. The plan consists of an intensity profile that is created for each beam. This is translated to leaf motion of the dynamic multileaf collimator using the inverse planning algorithm (Fig. 21-8). We have seen that the use of IMRT decreases the dose to the critical structures like optic nerves and brainstem when compared with the 3-D conformal plan.34 There is evidence that use of IMRT decreases the dose to the cochlea in children with medulloblastoma when used for the posterior fossa boost.
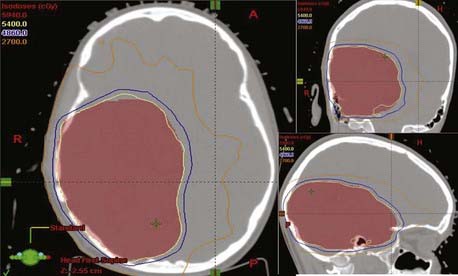
FIGURE 21-8 • Intensity-modulated radiation therapy treatment plan for a patient with high-grade glioma of right parietal region.
Special Issues With Radiation Management
Volume of Radiation
In the BTCG 8001 trial, patients were randomized to receive either 6000 cGy WBRT or 4300 cGy WBRT with 1720 cGy boost. No difference in survival or recurrence was noted.35 In a Japanese trial, patients were randomized to 4000 cGy WBRT with 1800 cGy boost or 5600 cGy local field radiation alone.36 Again, the 2-year survival rate was no different in this study (43% versus 39%), indicating that large-volume irradiation is unnecessary in high-grade gliomas.
Dose of Radiation
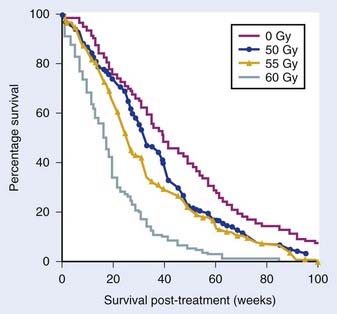
Radiation dose is another important consideration. In an MRC phase III trial, 444 patients were randomized to either 45 Gy in 20 fractions or 60 Gy in 30 fractions (1 : 2 randomization). A benefit of 60 Gy was seen in terms of 1-year survival (29% versus 39%) and the median survival (9 versus 11 months).37 In the RTOG phase III trial, patients received either 60 Gy WBRT or 60 Gy WBRT with a 10 Gy boost (Fig. 21-9). There was no benefit with the boost in terms of the median survival (9.3 versus 8.2 months).38 Researchers at the University of Michigan reported dose escalation to 80 Gy and 90 Gy with 3D-CRT and showed no difference in patterns of relapse or survival, indicating the absence of benefit with dose escalation beyond 60 Gy in malignant gliomas.39
Radiation Sensitizers
Multiple randomized trials conducted by the RTOG, the Brain Tumor Study Group (BTSG), and others have failed to show any survival benefit with misonidazole or other agents when compared with radiation therapy alone. Difluoromethylornithine, an inhibitor of polyamine synthesis, has not shown any benefit in a phase III clinical trial.40 Efaproxiral (RSR13), a synthetic allosteric modifier of hemoglobin, showed a median survival of 12.3 months when used along with 60-Gy radiation therapy in a phase II study, and is being presently tested in a phase III trial.41
Two halogenated pyrimidine analogues, bromodeoxyuridine (BUdR) and iododeoxyuridine, have been tested and in randomized trials have failed to show any benefit. Northern California Oncology Group data had shown a median survival of 252 weeks with BUdR with concomitant radiation and chemotherapy in AA compared with 82 weeks without BUdR.42 However, an RTOG trial of 60 Gy in 30 fractions combined with PCV chemotherapy with or without BUdR in 189 patients of AA had to be prematurely closed because of lack of benefit (1-year survival 68% versus 82%) and higher toxicity.43
Altered Fractionation
Again, multiple randomized trials conducted by RTOG, EORTC, and others have failed to show any survival benefit with hyperfractionation when compared with conventional radiation therapy alone.44 Similarly, several trials using different accelerated fractionation regimens have been reported; none have shown a survival benefit over conventional irradiation. An EORTC trial involving 60 Gy given in either conventional fractionation or with three fractions of 2 Gy given in a single day, 4 hours apart, in 340 patients showed no difference in survival or any increased toxicity.45 Doses of 64 and 70.4 Gy in 1.6-Gy, twice-daily fractions with BCNU tested in an RTOG trial showed no benefit.46 Shortening the treatment time has again failed to show any improvement.47 A Johns Hopkins trial of 30 Gy in 10 fractions followed by 2 weeks rest and then another 21 Gy in 7 fractions to reduced fields in 219 patients showed a survival similar to RTOG recursive partitioning analysis (RPA) groups I through VI patients.48
Brachytherapy
An earlier BTSG trial that randomized malignant glioma patients to 60 Gy radiation with or without iodine-125 (I-125) seed implant to 50 Gy showed a survival benefit with the addition of brachytherapy (median survival of 13 versus 16 months).49 Since the information was presented as an abstract only and never published, it remains difficult to establish that this approach clearly improves survival. However, a randomized trial done at Princess Margaret Hospital that randomized patients to 50-Gy external-beam radiation with or without temporary I-125 seed implant to 60 Gy did not show any survival benefit (median survival of 13.2 versus 13.8 months).50
Stereotactic Radiosurgery
Several retrospective trials have indicated that there is a possible survival advantage with the addition of a stereotactic radiosurgery (SRS) boost to high-grade gliomas. The RTOG conducted a randomized trial of conventional radiotherapy to 60 Gy and BCNU alone or preceded by a radiosurgery boost to 15 to 24 Gy in patients with GBM measuring 4 cm or less.51 However, the results were disappointing. The median survival (14 versus 13.7 months), 2-year survival (22% versus 18%), and 3-year survival (16% versus 8%), were similar with or without the boost. There was no improvement in any RPA class. No increased toxicities were seen with the addition of the SRS boost. Failures were still predominantly local (>90%).
Normal Tissue Reactions
Several adverse neurologic reactions may be observed in patients receiving cranial irradiation. They are classified into acute reactions, early-delayed reactions, and late-delayed injuries. With daily fractions of 1.8 to 2.0 Gy, the acute reaction most often presents as mild headache and nausea, beginning within a few hours after the first treatment and becoming progressively less severe with each succeeding fraction.52 The pathogenesis is thought to be increased cerebral edema caused by radiation-induced permeability changes in the blood-brain barrier. Corticosteroids may prevent or relieve most symptoms. Thus, if symptoms of increased intracranial pressure are present, patients undergoing cranial irradiation should be pretreated with corticosteroids (i.e., dexamethasone, 8-16 mg per 24 hours), administered for at least 48 to 72 hours before beginning treatment.
The early-delayed reaction (also called radiation encephalopathy) is characterized by transient, self-limited neurologic deterioration, somnolence, or focal encephalopathy.52 Early-delayed encephalopathy occurs in 15% to 40% of patients with primary or metastatic brain tumors. This reaction, which begins within 1 to 12 weeks after the completion of radiation therapy, usually peaks by 8 weeks after treatment and resolves spontaneously within the subsequent 4 months. Patients complain of headache, lethargy, and an exacerbation of their neurologic symptoms. At times, corticosteroid therapy and intensive medical support may be required. Declines in long-term memory within 1 to 2 months after irradiation followed by recovery 4 to 8 months later have also been observed.
Approximately 3% to 9% of patients irradiated for brain tumors develop clinically detectable focal radiation necrosis.53 Radiation necrosis may appear as early as 3 months after treatment, but usually develops within 1 to 2 years after treatment is completed. The breakdown of white matter may induce marked edema and mass effect. The symptoms of focal necrosis frequently recapitulate those of the tumor, leading the clinician to suspect recurrence. MRI may show a contrast-enhancing mass with extensive white matter alterations on T2-weighted images. Histopathologic findings are generally limited to the white matter and include focal coagulation necrosis and demyelination. Endothelial cell atypia and a unique form of fibrinoid necrosis of small arterial vessels are characteristic features that suggest the underlying pathophysiology.
Radiation necrosis is uncommon at doses less than 60 Gy with conventionally fractionated irradiation. The probability of necrosis increases with larger daily fraction sizes (2.2-2.5 Gy).37 For patients treated with hyperfractionated irradiation (twice-daily fractions of 1.2 Gy), the incidence of necrosis increased from 4.6% for 64.8 Gy to 19.2% for 81.6 Gy. Individual host factors, including the use of chemotherapy, may alter sensitivity and increase the risk of injury.
Corticosteroids may improve or stabilize the neurologic symptoms associated with the effects of radiation injury. Surgical resection is frequently beneficial to patients with favorably situated focal necrotic lesions who deteriorate neurologically and become steroid-dependent. There is no evidence that anticoagulation may lead to clinical improvement when surgery is not feasible. However, recent reports suggest a role for bevacizumab in the management of radiation-induced brain necrosis.54
Diffuse white matter injury develops in at least 40% of patients irradiated for intracranial neoplasms.52 The T2-weighted MRI images reveal hyperintensity of the periventricular white matter. Cerebral cortical atrophy occurs in 17% to 50% of patients treated for brain tumors. Enlarged cerebral sulci and ventricular dilatation are seen on neuroimaging studies. The abnormalities that accompany white matter change and cerebral atrophy are discernible within the first year after irradiation and persist or progress thereafter. They tend to be more severe with larger treatment volumes, higher doses, older age, longer intervals after irradiation, and chemotherapy. Clinical features range from mild lassitude or personality change to incapacitating dementia. Some patients with cerebral atrophy may also develop gait abnormalities and urinary incontinence suggestive of the syndrome of normal pressure hydrocephalus. Many patients with radiographic changes have no symptoms, but in those who do, the degree of impairment correlates approximately with the severity of the MRI appearance.
Decreased levels of intellectual function have been observed in adults after cranial irradiation. Certain cognitive functions, such as memory, may be more susceptible to decline than others.52 Impairment is most pronounced in those who have had chemotherapy and WBRT. Intellectual decline is first discernible within 4 to 6 months after treatment and becomes more pronounced by 2 to 3 years of follow-up. Memory loss may prevent patients from returning to their premorbid occupation. However, an analysis55 found that 60% of long-term survivors irradiated for gliomas were employed at occupations comparable to those they had held before treatment. Patients treated with partial brain irradiation tended to have a higher KPS, superior memory function, and a better employment history compared with those who received WBRT.
Neuropsychologic testing of patients treated for a variety of brain tumors who failed to retain their premorbid social or occupational levels of function demonstrated that newly learned tasks requiring attention and immediate problem-solving ability were performed poorly. In contrast, tests that evaluated long-term memory and overlearned material were generally consistent with premorbid levels.53 IQ testing alone is a less sensitive indicator of changes in cognitive function in adults. Prompt neuropsychologic intervention when necessary and early return to work after treatment may lead to improvement in or recovery of cognitive function.
Outcome
The median survival times using conventional radiation therapy alone or with chemotherapy for patients with GBM is 10 to 14 months, whereas the 2-year survival rate is only 10% to 25%.30 The median survival for patients with AA is 36 months, and the 3-year survival rate is approximately 50%.
Low-Grade Astrocytoma
Incidence
Low-grade astrocytomas make up 5% to 15% of adult primary brain tumors and 67% of low-grade gliomas, the remainder of low-grade gliomas being mixed oligoastrocytomas (19%) and oligodendrogliomas (13%).4 Unlike the incidence of their malignant counterparts, the incidence of low-grade astrocytomas decreases with increasing age. They are most common between the ages of 20 and 40 years and rarely occur after the age of 50.
Anatomy
Low-grade astrocytomas in adults typically involve the cerebral hemispheres. Among 995 cases of supratentorial astrocytomas culled from the literature, 42% were located in the frontal lobes, 37% in the temporal, 5% in the parietal, and 10% in more than one lobe, whereas 6% presented at other sites.4
Pathologic Conditions
Astrocytomas are well-differentiated tumors that display increased cellularity compared with normal brain tissue and have mild to moderate nuclear pleomorphism (see Fig. 21-3). Microcysts are frequently present, a feature that distinguishes astrocytomas from reactive gliosis.54 The majority of low-grade astrocytomas are classified as grade II in the WHO classification. Juvenile pilocytic astrocytomas are classified as grade 1 in the WHO system (see Table 21-1).
The various astrocytoma subtypes, including the fibrillary, protoplastic, gemistocytic, and pilocytic forms, are distinguished by their intracytoplasmic fibrillary processes, demonstrated by staining for GFAP (Fig. 21-10). Fibrillary astrocytomas, the most common subtype, and protoplasmic astrocytomas have been referred to as “ordinary” astrocytomas and share a similar prognosis. Over time, at least 50% of these tumors transform into more anaplastic lesions. Gemistocytic astrocytomas are composed of large, plump astrocytes with abundant eosinophilic cytoplasm. Because gemistocytes commonly transform into highly anaplastic cells, they behave in an aggressive fashion and should be treated as anaplastic gliomas.
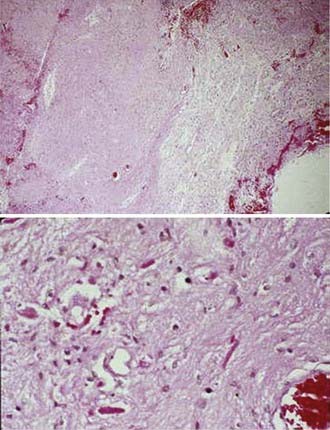
Pilocytic astrocytomas typically occur in the first two decades of life but also arise in adults. They are composed of fusiform cells with unusually long, wavy processes called Rosenthal fibers. Mitosis is rarely seen. Endothelial proliferation may be present, but does not signify malignancy.56 Pilocytic astrocytomas have a long natural history and rarely dedifferentiate into tumors with more malignant histologic appearances.
Although low-grade astrocytomas are often similar in their histologic appearance, their biologic behavior may vary considerably. In one study, a Ki-67 labeling index (LI) of more than 10% was associated with higher histologic grade and poorer survival, and this value was more significant than histologic grading.57 These data suggest that patients with a high LI may be considered for more aggressive treatment programs.
Clinical Presentation
About two thirds of adult patients with low-grade astrocytomas present with seizures but are otherwise neurologically intact.4 Others exhibit a slowly progressive neurologic syndrome consisting of headache, vomiting, motor deficit, visual or sensory loss, language disturbance, or personality change. Symptoms may be present for months or years before a diagnosis is made. Seizures are associated with a better survival, whereas the presence of functional deficits predicts a poorer outcome.
Diagnostic Studies
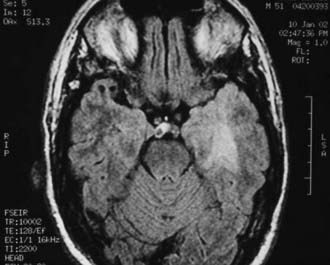
Pilocytic astrocytomas appear on imaging as discrete, enhancing lesions. The classic appearance is a large cyst with an enhancing mural nodule. Ordinary astrocytomas appear as diffuse, poorly defined, low-density, nonenhancing lesions.16 Approximately 40% of ordinary astrocytomas enhance on imaging, and calcification is found in 10% of cases.12,16 MRI typically shows low signal intensity changes on T1-weighted images, high signal intensity on T2, and an absence of enhancement (Fig. 21-11).
Prognostic Factors and Grouping
Bauman and associates have proposed a grouping system for predicting the survival for patients with low-grade gliomas using RPA based on a database of 401 patients.58 Age, KPS, and presence or absence of contrast enhancement were used as prognostic indicators. Group I (KPS <70 and age >40) had the worst median survival, at 12 months. Group II (KPS >70, age >40 and contrast enhancement present) had a median survival of 46 months. Group III (KPS >70, age >40 and contrast enhancement absent or KPS <70 and age <40) had a median survival of 87 months. Group IV (KPS >70, age 18-40) had the best median survival of 128 months.
Standard Therapeutic Approaches
Surgery
The goals of surgery are to establish a tissue diagnosis, to remove as much tumor as possible without increasing the neurologic deficit, and to remove an epileptogenic focus if present.59 Pilocytic astrocytomas are relatively well circumscribed, and 60% to 80% are amenable to total removal.60 Long-term survival approaches 100% after complete surgical resection. After partial resection, survival rates range from 80% to 90% at 5 years, 70% to 80% at 10 years, and 50% to 60% at 20 years. The 10- to 20-year survival after biopsy alone varies from 40% to 50%.
Resection of the more common diffuse astrocytomas is limited by the lack of clear demarcation between the infiltrating tumor and normal brain tissue. Newer surgical techniques make the attempted resection safer, more complete, and more likely to control seizures. Most series show an improvement in time to progression and survival with more extensive surgery.59 In one series, 80% of adult patients with completely resected tumors survived 5 years, compared with 50% for subtotal resection and 45% for biopsy. Other studies, however, have not shown a correlation between the extent of resection and prognosis.
The earlier diagnosis of patients with low-grade astrocytomas has raised questions regarding the timing of therapeutic intervention. There is general agreement that large symptomatic and progressive tumors should be treated at diagnosis, and a complete surgical resection should be attempted. A diagnostic biopsy should be performed on patients with deep or unresectable symptomatic tumors. Patients with small, asymptomatic (other than medically controlled seizures), apparently indolent tumors may be considered for close observation. Surgical intervention is offered should the tumor change its radiographic appearance or cause new symptoms or medically uncontrollable seizures.59
Alternatively, more aggressive local treatment in the form of surgery alone or surgery and postoperative irradiation can be offered. Arguments for performing immediate surgery are to confirm the diagnosis and to identify patients with nonenhancing anaplastic tumors. Complete resection of smaller tumors may improve survival,60 obviate the need for irradiation, and decrease the risk of malignant transformation, the most common cause of death from low-grade astrocytomas.
Radiation Therapy
Patient selection for and the timing and dose of postoperative irradiation are controversial issues. Postoperative irradiation is not indicated for completely resected pilocytic astrocytomas.60 There is insufficient data relative to the role of radiation therapy for incompletely resected pilocytic tumors. Therefore, after subtotal resection or biopsy, close follow-up and deferring treatment is generally recommended until there is evidence of disease progression.
Opinions differ regarding the need for postoperative irradiation for completely resected ordinary astrocytomas. The 5-year recurrence-free survival rates of patients with supratentorial astrocytomas or mixed oligoastrocytomas who undergo total or radical subtotal tumor resection range from 52% to 95%.59 The outcome in adult patients after total resection has been found in some series to be similar to that of patients undergoing less extensive surgery. Thus, in adults, postoperative irradiation has been recommended after complete resection by some, whereas others suggest that radiotherapy be withheld until recurrence.
Most retrospective reviews suggest that postoperative irradiation is beneficial for incompletely resected nonpilocytic astrocytomas. A major question concerns whether radiotherapy should be given immediately after surgery or be delayed until recurrence or progression. Patients with intractable seizures or with large, progressive, symptomatic, unresectable, or incompletely resected tumors should be considered for radiotherapy. Immediate postoperative irradiation has been recommended for patients older than 40 years who appear to benefit most from this treatment approach.61,62 Postoperative irradiation is commonly deferred in patients with well-controlled seizures who present with otherwise asymptomatic tumors. Proponents of this approach argue that with CT and MRI, the disease is diagnosed earlier in its natural history than in the past and that it is unclear whether early irradiation provides an outcome advantage over delayed irradiation, whether irradiation can delay or prevent tumor dedifferentiation, or whether it even alters the prognosis.
A randomized trial of early versus delayed radiotherapy in adult patients was conducted by the EORTC.63 In this trial, 311 patients with low-grade gliomas were randomized to no immediate therapy or 54-Gy postoperative radiation. In this study, biopsy alone was the surgical procedure in 40% of the patients; the histologic examination revealed oligodendroglioma in 25% of the patients. With a median follow-up of 5 years, the 5-year disease-free survival was better with immediate postoperative radiation (37% versus 44%). However, the 5-year overall survival was the same (63% versus 66%), indicating that deferring the postoperative therapy is an option for a selected group of patients.
The optimal dose of radiation remains unclear. In a randomized trial conducted by the EORTC involving 379 patients, no survival difference was observed when 45 Gy was compared with 59.4 Gy. With a median follow-up of 6 years, the 5-year disease free survival (47% versus 50%) and the 5-year overall survival were the same (58% versus 59%).64 In another combined North Central Cancer Treatment Group, RTOG, and ECOG trial, patients were randomized to receive either 50.4 Gy in 28 fractions or 64.8 Gy in 36 fractions. With a median follow-up of 6.3 years, the 5-year disease-free survival (44% versus 40%) and the 5-year overall survival were again the same (72% versus 64%), indicating that lower doses of radiation therapy are probably as effective as higher doses of radiation for low-grade gliomas.65
Chemotherapy
The role of chemotherapy is controversial in adult low-grade astrocytomas. In a Southwest Oncology Group trial, patients with incompletely excised tumors were randomly assigned to receive radiotherapy alone or with CCNU. The median survival of all patients was 4.45 years, with no difference between the two treatment arms.62 There was an RTOG trial that randomized high-risk, low-grade glioma patients to postoperative radiation to 54 Gy with or without PCV chemotherapy for six cycles. In an initial report, although the 5-year PFS was improved with the addition of chemotherapy (39% versus 61%), the overall survival was unchanged.65 Recently, temozolomide has shown its effectiveness in the initial treatment of low-grade glioma. In the largest reported retrospective analysis of 149 patients, Kaloshi and colleagues reported a 15% partial response (PR) and 37% stable disease with temozolomide as the initial therapy. The median PFS was 2.8 years and the 3-year overall survival was 70%.66 Based on this data, EORTC is conducting an ongoing trial in which patients are randomized to either radiotherapy or temozolomide at the time of initial diagnosis.
Outcome
The outcome of patients diagnosed and treated in the CT and MRI era is notably better than that reported in the past, when the conditions of diagnosis were different than they are now. Median survival times in recent series range from 7 to 12 years as compared with 5 years or less in older series, raising concerns over the value of the older literature in making treatment decisions today.65 The improved outcome appears to be related to the early diagnosis of neurologically intact patients who exhibit only seizures at the time of diagnosis.
The initial results of a prospective phase II randomized trial from RTOG (9802), which followed totally resected young patients and randomized unfavorable low-grade gliomas to radiation alone or with PCV chemotherapy showed that the 5-year PFS was poor in all the groups ranging from 39% to 61%. Only half of the favorable patients were disease free at 5 years.67 Addition of PCV chemotherapy resulted in a statistically insignificant improvement in PFS with no overall survival benefit. A subsequent RTOG trial (0424) that gave daily temozolomide with concurrent radiotherapy (54 Gy/30 fractions/6 weeks) followed by temozolomide for 12 cycles in high-risk low-grade gliomas has met its accrual target.
Oligodendroglioma
Incidence
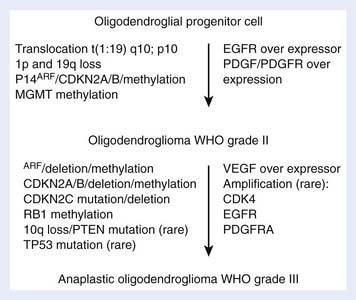
Oligodendrogliomas, which compose less than 5% of adult primary brain tumors, occur most commonly between the ages of 25 and 49 years.4 Losses of genetic information from chromosomes 1p (75%) and 19q (81%) are commonly seen in oligodendroglioma specimens, whereas losses on 17p (19%) and p53 gene mutations are notably less frequent, suggesting that early events in their oncogenesis are distinct from those associated with astrocytic tumors. The genetic pathway in the development of oligodendrogliomas is shown in Fig. 21-12. Anaplastic oligodendrogliomas show additional allelic losses involving chromosomes 9p and 10q and in some cases EGF-R gene amplification in a pattern similar to that of AAs, indicating a common progression pathway.68
Anatomy
More than 80% of oligodendrogliomas arise in the white matter of the cerebral hemispheres, most commonly in the frontal, temporal, and parietal lobes.4 Approximately 15% are found in the third or lateral ventricles, and the remainder arise in the posterior fossa.
Pathologic Conditions
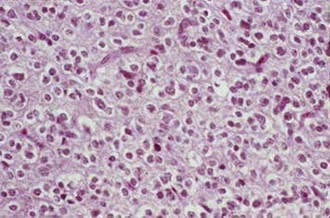
Oligodendrogliomas are composed of small, uniform cells with round central nuclei and distinct cytoplasmic borders. Formalin fixation causes a perinuclear halo that produces a “fried egg” or “honeycomb” appearance. The cells lack fibrillary cytoplasmic processes. A rich, vascular network, called “chicken-wire” vessels, divides the tumor into discrete lobules (Fig. 21-13). Anaplastic oligodendrogliomas, which represent only 3% to 5% of malignant gliomas,4 may evolve from low-grade oligodendrogliomas or arise de novo. They have recognizable oligodendroglial components, but also exhibit features of anaplasia, including nuclear pleomorphism, vascular endothelial proliferation, mitoses, and necrosis.68
A two-tiered system, low-grade and anaplastic, is used to grade oligodendrogliomas. In the new WHO classification (see Table 21-1), low-grade lesions are labeled grade II and anaplastic lesions are labeled as grade III.68 Patients with grade II tumors have a median survival of 9.8 years and 5- and 10-year survival rates of 73% and 49%, respectively, whereas those with grade III tumors have a median survival of 4.6 years and 5- and 10-year survival rates of 45% and 26%.65
The RTOG 9402 trial confirmed the prognostic significance of 1p/19q deletion and showed that only PFS was prolonged in patients who received neoadjuvant chemotherapy and in association with 1p/19q deletion. A subsequent tumor histopathologic review separated 115 tumors deemed to be classic for oligodendroglioma (CFO) from 132 lacking classic features of oligodendroglioma (NCFO) and evaluated the relationship of histopathologic condition and 1p/19q status to treatment and outcome.69 This study showed that that overall survival of patients with CFO was significantly longer than for patients with NCFO and was not affected by necrosis. Also, the classic oligodendroglial morphology was highly associated with 1p/19q deletion, present in 80% of CFO and in only 13% of NCFO. On multivariate analysis, both classic oligodendroglial morphology and 1p/19q deletion remained significantly associated with improved PFS and overall survival, suggesting that these features may in the future be predictive of therapeutic response and survival.69
Many oligodendrogliomas are admixed with astrocytoma or ependymoma components. The presence of up to 50% astroglial component is accepted to make the diagnosis of mixed oligoastrocytoma in the new WHO classification.66 The median survival for patients with low-grade mixed oligoastrocytomas is 7 years and the 5- and 10-year survival rates are 63% and 33%, respectively.70 Most oligoastrocytomas and 50% to 75% of oligodendrogliomas recur as AAs or GBM.
Clinical Presentation
Oligodendrogliomas present in a fashion similar to that of hemispheral astrocytomas. However, two features distinguish them from astrocytomas: the antecedent history, averaging 7 to 8 years, tends to be longer, and seizures are more common, occurring in 70% to 90% of patients by the time of diagnosis.4 Headache, altered mental status, papilledema, and focal neurologic deficits are also common at presentation.
Patterns of Spread
Oligodendrogliomas grow in a diffuse infiltrative pattern. They may invade the cerebral cortex or expand centrally into the deep midline structures. Ventricular extension accounts for a 5% to 10% incidence of spread through the cerebrospinal fluid pathways. Metastases may occur to the spinal cord and rarely to extracranial sites.4
Diagnostic Studies
A provisional diagnosis may be made by CT or MRI. Oligodendrogliomas are typically hypodense or isodense, poorly defined, and nonenhancing on CT. Calcification is present in 60% of cases, and peritumoral edema is minimal. They are of low signal intensity on T1-weighted MRI and hyperintense on T2-weighted studies except for regions of signal void that correspond to fragments of calcium (see Fig. 21-10).12 Some aggressive low-grade oligodendrogliomas enhance on CT and MRI.12 Anaplastic oligodendrogliomas and mixed gliomas more often enhance and may contain hemorrhagic and necrotic areas. Although these tumors do rarely spread through the cerebrospinal fluid, cytologic and spinal MRI studies are not obtained unless clinically warranted.
Standard Therapeutic Approaches
Surgery
Surgery is required for histologic confirmation. The principles of surgical resection are similar to those for cerebral astrocytomas, with gross total removal being the goal when this is consistent with good neurologic outcome. The margins of oligodendrogliomas appear to be more distinct than those of astrocytomas, but generally they are infiltrative, and surgical cure is unlikely. The extent of resection and postoperative tumor volume59 have been shown to affect survival in some series, but not in others. As in the case of low-grade astrocytomas, some small tumors that are asymptomatic except for controlled seizures can be observed, delaying surgical intervention until there is disease progression. However, if feasible, large, symptomatic, or progressive tumors should be resected.
Radiation Therapy
Conclusions regarding the value of radiotherapy are contradictory, and the lack of randomized trials precludes the statement of firm recommendations. The infrequent occurrence of oligodendrogliomas and their variable and often long natural history make it difficult to evaluate the effect of radiotherapy on these tumors. Certain histologic features affect the prognosis,71 and it is likely that many retrospective studies contain patients with both differentiated and anaplastic oligodendrogliomas.
Patients with completely resected low-grade oligodendrogliomas can be observed, deferring radiotherapy until the time of recurrence. Large, symptomatic, unresectable, or incompletely resected tumors should receive postoperative irradiation.61 On the other hand, certain small tumors that are asymptomatic except for controlled seizures may be observed after subtotal resection, delaying radiotherapeutic intervention until there is tumor progression. Patients with pure or mixed anaplastic oligodendrogliomas should routinely receive postoperative irradiation.
Chemotherapy
Chemotherapy is generally withheld in low-grade oligodendrogliomas until the time of disease progression. Anaplastic oligodendrogliomas are chemosensitive tumors. PCV regimen has produced RRs of 75% or more in newly diagnosed anaplastic oligodendrogliomas.72
To test the efficacy of chemotherapy in newly diagnosed pure and mixed anaplastic oligodendrogliomas, the RTOG conducted a trial comparing radiotherapy alone with neoadjuvant intensive PCV chemotherapy (four 6-week cycles) followed by radiotherapy. The median survival times were similar (4.9 years after PCV plus radiotherapy versus 4.7 years after radiotherapy alone). However, PFS time favored PCV plus radiotherapy (2.6 years versus 1.7 years for radiotherapy alone). Of these patients, 65% experienced grade 3 or 4 toxicity, and one patient died.72 In an EORTC trial, 368 patients were randomly assigned to either 59.4 Gy of radiotherapy in 33 fractions only or to the same radiotherapy followed by six cycles of standard PCV chemotherapy. Adjuvant PCV chemotherapy did not prolong overall survival (31 versus 40 months), but did increase PFS (23 versus 13.2 months).73 RTOG also has conducted another phase II trial of pre–radiation therapy with temozolomide chemotherapy for six cycles to delay radiation therapy (59.4 Gy) in partial responders or to even withhold it in complete responders in pure or mixed anaplastic oligodendroglioma patients. The study has met the accrual and results are awaited.
Radiation Therapy Techniques
Outcome
Although some authors have noted worse outcomes in mixed anaplastic tumors compared with pure anaplastic oligodendrogliomas, others have noted similar outcomes. Winger and colleagues74 reported median survival times of 1.1 years for mixed anaplastic oligoastrocytomas and 5.3 years for anaplastic oligodendrogliomas treated with radiotherapy and chemotherapy. In contrast, Shaw and associates75 found that patients with high-grade mixed oligoastrocytomas and high-grade oligodendrogliomas had similar outcomes with median survival times and 5- and 10-year survival rates of 4.3 years (45% and 23%, respectively) for high-grade oligoastrocytomas and 4.6 years (45% and 26%) for high-grade oligodendrogliomas.
Primary Central Nervous System Lymphoma
Epidemiologic Statistics
Until the 1980s, primary CNS lymphomas (PCNSLs) represented less than 2% of intracranial neoplasms. However, during the decade from 1985 through 1994, there was nearly a fivefold increase in incidence.2 Although this partly reflected the higher prevalence of acquired immunodeficiency syndrome (AIDS) during that time, the incidence was also noted within the apparently immunocompetent general population. However, a sharp decrease in incidence is now noted (with the peak in 1995) that mirrors the decrease in the AIDS rate and the introduction of more effective antiretroviral therapy.2 The peak incidence in human immunodeficiency virus (HIV)-negative patients is in the fifth to seventh decades of life with a 3 : 2 to 2 : 1 male/female ratio. In contrast, immunocompromised patients are frequently diagnosed in the third and fourth decades, and nearly all are male.76 The molecular genetics and pathogenesis of PCNSL have not been elucidated. Epstein-Barr virus (EBV) genome-protein expression is present in two thirds of HIV-related PCNSL, but in only 15% of immunocompetent patients, suggesting that EBV does not play an essential role in the pathogenesis of PCNSL in immunocompetent patients and that HIV-related and HIV-negative PCNSL may represent pathogenetically distinct entities.76
Anatomy and Routes of Spread
CNS lymphomas most frequently arise in the supratentorial paraventricular region of the brain, but they also occur in the cerebellum and brainstem. Rarely, they present only in the leptomeninges or spinal cord.77 Multifocal tumors are present at diagnosis in 25% to 50% of immunocompetent patients and in 60% to 80% of AIDS patients. CNS lymphomas tend to infiltrate extensively along the corpus callosum and other deep white matter tracts. They frequently traverse the ependyma to involve the ventricular surface or spread peripherally into the overlying leptomeninges. Cytologic examination of cerebrospinal fluid reveals malignant or suspicious cells in up to two thirds of immunocompetent patients and in nearly all AIDS patients. Ocular lymphoma is found in association with PCNSL in approximately 20% of patients at diagnosis.77 Lymphoma can also begin within the globe. Of patients presenting with ocular lymphoma, 50% to 80% will subsequently develop CNS lymphoma within a median of 9 months.
Pathologic Conditions
Histologic examination shows perivascular cuffs and sheets of lymphoid tumor cells infiltrating brain tissue with expansion of the Virchow-Robin spaces. The neoplastic cells are similar to those of non-Hodgkin lymphoma arising in extranodal sites. They are predominantly of B-cell origin, the majority being high-grade large cell or small noncleaved cell lymphomas, with large cell immunoblastic the most common subtype. Intermediate-grade lymphomas are less common, and low-grade subtypes and T-cell lymphomas are rare.78
Clinical Presentation
Presenting symptoms include focal neurologic deficits, seizures, neuropsychiatric disturbance, headache, lethargy, and confusion.4,76 Neck or back pain and symptoms or signs of myelopathy may indicate parenchymal spinal cord involvement, whereas blurred vision or floaters suggests ocular lymphoma.
Diagnostic Studies
The diagnostic evaluation begins with a cranial MRI. Single or multiple densely and uniformly contrast-enhancing lesions in the paraventricular regions, basal ganglia, thalamus, or corpus callosum are characteristic findings (Fig. 21-14). Irregular ring enhancement may be present on imaging studies in AIDS-related PCNSL, making it difficult to distinguish lymphoma from toxoplasmosis and other opportunistic infections.12
Cerebrospinal fluid cytologic examination and an ophthalmologic evaluation that includes a slit-lamp examination are also performed. A spinal MRI is obtained only if parenchymal spinal cord involvement is suspected.77 Because systemic involvement at the time of diagnosis (or relapse) is extremely rare, extensive testing for occult systemic lymphoma is not warranted. However, CNS metastases are common in AIDS patients with systemic non-Hodgkin lymphoma. Thus, a systemic staging evaluation is recommended for AIDS patients with a suspected PCNSL.
Standard Therapeutic Approaches
Surgery
The role of surgery is to establish a tissue diagnosis, and this is best obtained by stereotactic biopsy. Because of the multifocal nature of this tumor, extensive resections produce little survival benefit and increase the risk of postoperative deficits.77 Large single hemispheric lesions may be surgically reduced when intracranial pressure cannot be medically controlled.
Corticosteroids
PCNSLs respond dramatically to corticosteroid therapy. At least 90% of patients improve clinically, whereas 40% of lesions shrink considerably and 10% disappear on CT and MRI. Tumor biopsy after corticosteroid therapy may be problematic, due to the disappearance of a target for biopsy, and may yield nondiagnostic tissue. Thus, when the diagnosis of PCNSL is suspected, corticosteroids should be withheld, if possible, until a tissue diagnosis has been obtained.77
Radiation Therapy
Historically, WBRT with corticosteroids had remained the standard treatment for PCNSL. Focal radiation is not indicated for PCNSL because of the multicentric and infiltrating nature of the disease, but WBRT has never been compared with focal radiotherapy in a randomized controlled study. Shibamoto and colleagues have reported a retrospective analysis of patients who underwent focal radiotherapy with margins smaller than 4 cm or larger than 4 cm. In their study, patients with smaller margins had an out-of-field recurrence rate of 83% compared with 22% in the group with wider margins.79 Also, the median survival was longer in the group that received larger margins (28.5 months versus 15 months).
The RTOG examined the efficacy of high-dose radiotherapy in a prospective study (trial 8315).78 Patients with non-AIDS PCNSL were treated with WBRT to 40 Gy plus an additional 20 Gy boost to the tumor. The median survival from the start of radiotherapy was only 11.6 months, and the 1- and 2-year survival rates were 48% and 28%, respectively. Recurrence of disease in the brain occurred in 61% of patients, and most relapses occurred within the region that received 60 Gy, suggesting that there is no clear dose response over 40 Gy. For patients with primary leptomeningeal lymphoma, the craniospinal axis is generally treated to 36 Gy, and areas of grossly identifiable tumor are given an additional 9 to 14.4 Gy.
When ocular lymphoma is present at the time of diagnosis of cerebral lymphoma, the eyes are treated to 36 Gy concurrently with the brain. A similar dose is used if an ocular recurrence develops in a patient who has had previous cranial irradiation.77 Radiation therapy improves visual acuity in up to 75% of patients with symptomatic ocular lymphoma, unless extensive retinal damage has already occurred.77 The treatment of isolated ocular lymphoma is controversial. Although some advocate treatment with ocular radiotherapy and WBRT, most prefer administering only ocular radiotherapy because of the potential toxicity of WBRT.
Additional data regarding the dose of radiotherapy came from recent studies that have combined chemotherapy with WBRT. In two sequential studies, Bessell and colleagues80 studied the effect of a reduced dose of radiotherapy in patients with PCNSL after achieving a complete response (CR) with chemotherapy. After chemotherapy, patients from their initial study received 45 Gy WBRT. In the second study, the radiotherapy dose was reduced to 30.6 Gy in 26 patients who had a CR after chemotherapy. Among 25 patients younger than 60 years who achieved a CR after chemotherapy, the 3-year relapse risk was higher in the group that received the lower dose of radiotherapy (25% versus 83%). In addition, RT dose was also the only predictor of overall survival in this group (92% versus 60%). The RTOG study 93-10 was modified midway during the trial (because of the risk of neurotoxicity) so that patients who achieved CR after chemotherapy would receive 36 Gy hyperfractionated RT instead of 45 Gy standard WBRT. There was no difference in PFS, overall survival, or late neurotoxicity between the two groups.81
Chemotherapy
Several reports have documented improved survival when chemotherapy is added to radiation therapy.74,75,79 Lipophilic agents, such as methotrexate (MTX) and cytarabine, are used most frequently. High-dose, MTX-based regimens followed by WBRT has been shown in multiple phase II trials to increase survival (medians from 32-60 mo) compared with radiotherapy alone.74 Although high-dose MTX is an established chemotherapy agent for PCNSL, the optimal dose and the best combination regimen remain unclear. The RTOG 9310 protocol consisted of high-dose MTX (2.5 g/m2), PCV, procarbazine, and intrathecal (IT) MTX followed by 45 Gy WBRT and high-dose cytarabine. Of these patients, 58% achieved a CR and 36% had a PR after preirradiation chemotherapy. PFS was 24 months and overall survival was 37 months.80
Of concern in the PCNSL trials was the observation of dementia and ataxia among 27% of patients older than age 50 who achieved a CR. These findings were not seen in younger patients.77 In the RTOG 9310 study, there was no prospective neuropsychologic testing, and the 15% incidence of neurotoxicity reported was a minimum. This has led to the policy of deferring radiotherapy in selected older patients who respond to chemotherapy until the time of disease progression. Others are exploring regimens in which radiotherapy is withheld until recurrence to avoid radiation-induced neurotoxicity. EORTC reported a multicenter phase II study in patients older than 60 years treated with chemotherapy alone as initial treatment. The protocol consisted of high-dose MTX (1 g/m2), CCNU, procarbazine, methylprednisolone, IT MTX, and IT cytarabine. Only 42% achieved a CR. Overall median survival was 14 months, but this still compared favorably with a median survival of only 7 months after WBRT alone for older patients.82 Delayed neurotoxicity was reported in 12% of patients, indicating that aggressive chemotherapy itself could induce the late changes.
Outcome
Historically, survival times from the onset of symptoms to death with no treatment or with surgical resection alone ranged from 0.9 to 4.6 months. The median survival for WBRT alone varies from 12 to 18 months, and only 3% to 4% of patients survive for 5 years. Median survival times for treatment programs that include high-dose MTX–based chemotherapy range from 33 to 42 months. Age younger than 60 and good performance status are favorable prognostic indicators. Molecular markers are being studied as a prognostic factor for PCNSL.76
Uncommon Adult Primary Brain Tumors
Brainstem Glioma
Brainstem gliomas account for less than 2% of brain tumors in adults.4 In series of brainstem gliomas that include patients of all ages, adults constitute approximately one third of those reported. Radiation therapy alone by conventional fractionation to 54 Gy in symptomatic or large brainstem lesions remains the current standard of care. Overall, radiotherapy induces neurologic improvement, allows reduction or discontinuation of steroids, and is associated with radiologic response.83 The clinical response (85%) seems to be higher than the radiologic response (approximately 50%). No correlation has been shown between clinical and radiologic response. A survival benefit for radiologic responders has not been shown.
There are no prospective trials that have specifically addressed adult brainstem gliomas alone. As a result, the prognostic covariates identified in children may not necessarily be significant in the adult population. Shrieve and associates84 have reported 41 pediatric and 19 adult patients who received 66 to 78 Gy with a hyperfractionation schedule of 1.0 Gy twice daily. The median survival time and 1- and 2-year survival rates for children were 16 months and 63% and 32%, respectively, compared with 44 months and 68% and 53% for adults. The 2-year survival for patients with thalamic and midbrain tumors (40%) was similar to that of patients with tumors arising in the pons or medulla (57%, P = NS). Furthermore, the 2-year survival (54%) of patients with diffuse tumors was the same as that of patients with focal tumors (50%). In an update of that series, the median survival of 42 adult patients was 16 months, and the median time to progression was 11.4 months.85
So far no adjuvant treatment has shown any benefit over conventional radiation therapy alone for brainstem gliomas. The use of chemotherapy either before or after radiation has not shown any survival advantage.83 Concomitant chemotherapy and radiotherapy has been investigated in multiple pediatric trials and did not seem to confer a survival benefit.
Ependymoma
Ependymomas account for 2% of all intracranial tumors in adults.86 Although they are most common in children, approximately one third occur in adults. The incidence peaks at 5 years and again at 34 years of age. Between 30% and 40% of ependymomas arise in the supratentorial brain, and 40% to 60% of tumors in this location occur in adults. The remaining 60% to 70% of ependymomas arise in the infratentorial brain, and 25% of these occur in adults.
Although the correlation between survival and histopathologic grade is controversial, histopathologic grade is a useful predictor of outcome.87 Ependymomas are separated into low-grade and high-grade lesions (grades II and III in the new WHO classification). The 5-year survival for low-grade tumors ranges from 60% to 80%, whereas it varies from 10% to 47% for high-grade tumors. Supratentorial ependymomas generally have a poorer prognosis than their infratentorial counterparts because of a greater proportion of lesions that are high-grade87 and the tendency for larger volumes of residual disease to be present after surgical resection.
Many ependymomas cannot be completely excised because of their location and growth characteristics.87 Postoperative irradiation improves local tumor control and survival and is an accepted part of the standard treatment for these tumors. Although most ependymomas are slow-growing, circumscribed tumors that seldom spread, others are more aggressive and may disseminate throughout the cerebrospinal fluid pathways. The frequency of tumor seeding, the identification of patients who are at high risk for dissemination, and the volume of the CNS to treat have been controversial topics in the past. A literature review found that local relapse had the greatest effect on the development of spinal seeding, regardless of tumor grade or site.87 The incidence of spinal dissemination was 3.3% in locally controlled patients and 9.5% in those with uncontrolled primary tumors. The risk of seeding was not affected by prophylactic spinal irradiation.
The treatment of anaplastic ependymomas has remained controversial. Despite the apparent superiority of craniospinal irradiation in some series, the finding that spinal seeding is uncommon in the absence of local relapse, and the identification of similar patterns of failure in patients treated with local fields or to the craniospinal axis have led most investigators to question the routine use of craniospinal irradiation, WBRT, or even entire posterior fossa irradiation.87 Currently, at New York University we treat high-grade ependymomas with local fields using a PTV of 2 to 3 cm beyond the MRI-defined lesion to a dose of 59.4 Gy. The value of chemotherapy in adults with ependymomas and anaplastic ependymomas is not well defined.4
There are no prospective trials that have specifically addressed adult ependymoma. Metellus and colleagues have reported the results of the largest multi-institutional analysis of intracranial ependymomas in 152 adult patients diagnosed in 24 French neurosurgical university hospital centers from 1990 through 2004. The 5- and 10-year overall survival rates were 84.8% and 76.5%, respectively. The 5- and 10-year PFS rates were 63.5% and 52.8%, respectively. On multivariate analysis, overall survival rates were associated with histologic grade, extent of surgery, patient age, and KPS. The multivariate analysis also revealed that the risk of recurrence was associated with high histologic grade, incomplete resection and a KPS score of 80 or less. The effect of radiotherapy was found to be beneficial for incompletely resected low-grade ependymomas, and to a lesser extent for completely removed high-grade tumors.88
Medulloblastoma
Approximately 30% of patients with medulloblastoma are 16 years of age or older (median 22-28 years) when first diagnosed.89 In adults, medulloblastomas more frequently arise laterally in the cerebellar hemispheres or cerebellopontine angle (38%-67%) than in children (7%-9%). Thus, adults commonly present with lateral cerebellar dysfunction. The desmoplastic histopathologic variant of medulloblastoma is more common in adults (25%-47%) than in children (13%-18%). On microscopic examination, thick bundles of collagen are found to divide the tumor into follicular nests of anaplastic cells. These changes have been thought to result from extension of the tumor to the leptomeninges, which incites a proliferation of fibrous connective tissue. However, the finding of loss of heterozygosity at chromosome 9q with this histologic subtype suggests that desmoplastic medulloblastomas may be pathogenetically distinct from their classic counterparts.90 Approximately 30% of adults have disease outside the primary site,89 similar to the rates reported in the pediatric literature.
The role of chemotherapy in adult medulloblastomas is not well defined. Spreafico and colleagues retrospectively studied 26 consecutive adults treated for medulloblastoma using intensive chemotherapy pediatric protocols.91 With a median follow-up of 46 months, 5-year disease-free and overall survival rates were 65% and 73%, respectively. However, 87% of the patients developed grade IV hematologic toxicity. Prados and colleagues have found an improvement in survival and time to progression with adjuvant chemotherapy, and they recommend that chemotherapy be given to poor-risk patients.89
The prognostic factors for adult medulloblastomas include M stage, functional status at the time of radiotherapy, and extent of surgical resection.90 The T stage has less prognostic value in adults than in children,92 and tumor location and desmoplastic histologic appearance do not affect the prognosis. A recent histologic analysis of a large series of 181 medulloblastoma patients revealed a lower labeling index and higher apoptotic index in adults, indicating a biologic difference between the two age groups, thus potentially raising the question of less aggressive therapy for adult medulloblastoma.93
A retrospective review of literature suggests a 5-year survival rate range from 46% to 81%; 10-year rates vary from 38% to 55%, comparable to the rates reported in children.89,90 Brandes and colleagues have reported the results a multicentric phase II trial on 26 patients to study the value of prognostic factors and the outcome of medulloblastoma in adults. In low-risk patients, treatment consisted of 36 Gy to the craniospinal axis, supplemented by a local tumor dose of 18.8 Gy. In high-risk patients, two cycles of up-front chemotherapy were delivered before the same radiation therapy, followed by maintenance chemotherapy if metastatic disease was present. Over a 12-year period, 36 evaluable patients were enrolled. PFS at 5 years was higher in low-risk patients compared with the high-risk group: 76% versus 61%. Patients with M-negative disease showed a significantly better outcome than M-positive patients, with 75% showing PFS at 5 years versus 45%. They concluded that the overall PFS observed was comparable to that obtained in the pediatric series and suggested that a more effective therapy must be developed for high-risk patients.94
Brain Metastases
Epidemiologic Statistics
Metastases to the brain occur in as many as 30% of patients with systemic cancer and outnumber primary brain tumors by 8 : 1. In the United states, approximately 170,000 cancer patients are expected to develop brain metastases every year.95 Brain metastases exert a profound effect on the quality and length of survival, and despite the best current management, they represent the direct cause of death in one third to one half of affected patients.96 Although brain metastases can arise from any primary cancer, certain tumors such as melanoma and carcinomas of the lung (especially small cell and adenocarcinoma), breast, and kidney have a propensity to metastasize to the brain.95 Approximately one half of patients present with only a single lesion, and an additional 20% have only two. Most brain metastases, particularly those that arise from primary sites other than the lung, occur at a late stage when metastatic dissemination is present elsewhere in the body.
Anatomy
Brain metastases arise from hematogenous spread to the white matter of the watershed area of the brain at the junction of the gray and white matter. Most metastatic tumors distribute themselves between the supratentorial and infratentorial compartments in proportion to the relative weight and blood supply of these structures; that is, 85% in the cerebral hemispheres, 10% to 15% in the cerebellum, and 1% to 3% in the brainstem.97 Certain metastatic tumors, particularly small cell lung cancer and tumors arising in the prostate, uterus, or gastrointestinal tract, have a predilection to the cerebellum.
Most brain metastases grow as spherical, well-demarcated, solid masses that displace rather than destroy adjacent tissue. They may be surrounded by minimal to extensive edema. Some rapidly growing metastases undergo central necrosis or cystic change, and others, especially those from melanoma, choriocarcinoma, and testicular carcinoma, may be hemorrhagic. Carcinomatous meningitis is a clinical syndrome caused by widespread invasion of the cerebral cortex by small or microscopic foci of tumor that may or may not be seen on imaging studies.95
Pathologic Conditions
The microscopic appearance of a brain metastasis resembles the primary tumor from which it arises. Thus, in patients with an unknown primary cancer, extirpation of the metastasis with microscopic examination often provides an important clue as to the original site of the primary tumor.96
Clinical Presentation
The most common presenting signs and symptoms are summarized in Table 21-2. They usually begin insidiously and evolve over a period of days or a few weeks. The spread of edema through the white matter frequently determines the speed of onset and progression of signs and symptoms.95 Hemorrhage into a tumor may cause a more sudden onset or acute worsening of symptoms.
Diagnostic Studies
Gadolinium-enhanced MRI is the best diagnostic test for brain metastases.98 MRI can detect small lesions not seen on CT, particularly in the cerebellum and brainstem. The T2-weighted image reveals an area or areas of hyperintensity in the white matter, encompassing both the tumor and surrounding edema.
Although the clinical history combined with the results of MRI establish the diagnosis of brain metastasis with reasonable certainty, a definitive diagnosis of metastatic brain tumor cannot be made on scan results alone. Even a typical radiographically defined lesion may prove to be a primary tumor, an abscess, or another lesion.98 A known systemic cancer increases the likelihood of brain metastases, and multiple lesions make the diagnosis even more likely. However, if doubt exists, a biopsy is required to establish the correct diagnosis.
Grouping
Although no generally accepted grouping system exists for brain metastases, one was proposed by investigators from the RTOG based on a RPA of prognostic factors.99 Three groups were defined by KPS (<70 versus ≥70), age (<65 versus ≥65), status of the primary tumor (controlled versus uncontrolled), and extent of metastases (brain only versus brain and other sites). The definitions and corresponding survival rates for each group are shown in Table 21-4. The application of this scheme is important to ensure that new treatment techniques for brain metastases are tested in homogeneous patient groups.
Table 21-4 Staging System for Brain Metastases Based on Recursive Partitioning Analysis of Prognostic Factors
| Stage Characteristics | Median Survival | |
|---|---|---|
| I | KPS ≥70, primary controlled, age <65 y, metastasis to brain only | 7.1 |
| II | KPS ≥70, primary uncontrolled | 4.2 |
| KPS ≥70, primary uncontrolled, age ≥65 y | ||
| KPS ≥70, primary uncontrolled, age <65 y | ||
| KPS ≥70, primary uncontrolled, age <65 y, metastasis to Brain and other sites | ||
| III | KPS <70 | 2.3 |
Kamofsky performance status.
Data from Gasper L, Scott C, Rotman M, et al: Recursive partitioning analysis (RPA) of prognostic factors in three Radiation Therapy Oncology Group (RTOG) brain metastasis trials, Int J Radiat Oncol Biol Phys 37:745, 1997.
Standard Treatment Approaches
Because the majority of patients with metastatic brain lesions have or will soon develop widely disseminated disease, treatment is dictated by the need to achieve immediate short-term palliation and the desire for durable, symptom-free remission. With early diagnosis and vigorous treatment, many of the symptoms associated with brain metastases can be reversed, often returning the patients to a useful life, at least temporarily. The median survival of patients with symptomatic brain metastases is approximately 1 month without treatment and 2 months with corticosteroid administration. Survival is longer and the quality of life better if brain metastases are treated.95
Corticosteroids
Corticosteroids rapidly ameliorate many symptoms of brain metastasis and should be used at the onset for all symptomatic patients. Symptomatic but stable patients can begin with approximately 16 mg of dexamethasone (or equivalent) daily in two to four divided doses. Patients who are receiving WBRT should receive steroids for at least 48 hours before treatment. Steroid tapering may begin during week 2 of radiotherapy. For patients receiving 16 mg of dexamethasone, the drug should be tapered by 2 to 4 mg every fifth day. If the patient develops symptoms of either brain tumor or steroid withdrawal, the dosage is increased to the next level for 4 to 8 days before the taper is started again. If symptoms return after drug withdrawal, the full dexamethasone regimen is restarted.95 An alternative approach is to begin at 16 mg per day for 4 days followed by 8 mg for 4 days and 4 mg per day until the completion of treatment.
Surgery
Surgery is used to establish the diagnosis of metastatic brain disease when it is uncertain and as a treatment for single metastases. When the brain metastasis is resectable with a minimal risk of neurologic injury, surgical extirpation may provide local tumor control and immediate relief of neurologic signs and symptoms caused by a mass effect. Patients with a single accessible lesion, controlled or absent extracranial disease, a KPS of at least 70, age younger than 60, and a life expectancy of at least 2 months are most likely to benefit from surgery.100 Such patients live longer, have fewer recurrences of cancer in the brain, and enjoy a better quality of life compared with those treated by radiotherapy alone. After tumor removal, steroids can usually be discontinued, whereas it is more difficult to discontinue steroids after radiotherapy because residual tumor often remains as a nidus for continuing brain edema. However, only approximately 10% of patients in general are ideal candidates for surgical extirpation. Rates of complete resection vary from 70% to 79%, whereas the operative mortality is 1% to 2%, and 5% to 10% of patients develop worsening of neurologic deficits.100 The outcome after surgery varies with the primary tumor type. Renal cell, lung, and breast carcinomas carry a better prognosis (median survival 11-12 months), whereas melanoma and colorectal cancers have a worse outcome (6.5-8 months).
Although surgery is usually not the approach of choice for patients with multiple metastases, the presence of multiple brain metastases does not always constitute a contraindication to surgery. A patient with two accessible lesions that cannot be controlled by radiotherapy or chemotherapy (not lymphoma and leukemia, small cell lung cancer, or germ cell tumor) may be treated surgically.101 Furthermore, patients with a large metastatic lesion causing uncontrollable neurologic symptoms may be candidates for resection, even if multiple brain metastases are present, before irradiation of the smaller lesions.
Approximately 5% to 20% of patients with brain metastases present with an unknown primary tumor.102 Most metastases arise from lung or renal cell carcinoma and melanoma. When a patient without known cancer presents with brain metastases, a search for the primary tumor should ensue before treatment of the metastatic brain disease is begun. If no other lesion is found and if the brain lesion is single and surgically accessible, extirpation is indicated to establish the diagnosis and for therapy. If multiple lesions are present or if a single lesion is surgically inaccessible, a stereotactic needle biopsy is warranted.
Whole-Brain Radiation Therapy
Radiotherapy is the appropriate treatment for most patients with brain metastases, including those with multiple lesions and those with single metastases who are not candidates for surgery. The standard approach is to treat the whole brain to 30 to 37.5 Gy in 10 to 15 daily fractions of 2.5 to 3 Gy each over 2 weeks.103 The advantages of WBRT include simple clinical set-up, its ease of delivery on an outpatient basis, its low cost, its minimal disruption of quality of life, and its good palliation as a single modality. Depending on the symptom, the RR varies from 70% to 90%. Overall, neurologic function is improved in 50% of patients. Functional improvement depends on the degree of neurologic impairment at the time of treatment. Approximately two thirds of patients with severe neurologic dysfunction improve, whereas one third of those with moderate dysfunction improve to normal or near normal. The median time to improvement is 1 to 2 weeks in patients with severe dysfunction and 3 weeks for those with moderate impairment. The durability of response is problematic, as the median time to progression is only 2 to 3 months. Overall, 75% to 80% of remaining life is spent in an improved or stable neurologic state.103
RTOG has tried five schedules of fractionation varying from 2000 cGy in a week to 4000 cGy in 4 weeks in three randomized trials. All the regimens were similar in improvement in neurologic function (50%), duration of improvement (9-12 wk), time to progression (10 wk), survival (15-18 wk), or the quality of palliation.103 RTOG trials using accelerated fractionation (54.4 Gy in twice-daily 1.6-Gy fractions) or radiosensitizers (misonidazole/BrdU) did not improve the outcome (XX). Rapid dose schedules such as 10 Gy in one fraction or 15 Gy in two fractions have yielded a shorter duration of improvement, a lower RR, and earlier relapse. In addition, high-dose fractions often lead to severe acute side effects and in some cases cerebral herniation and death. A minority of patients, perhaps only 10% to 20%, who have been treated with 30 Gy in 10 fractions to the whole brain and who survive for longer than 1 year develop radiation-induced dementia. In a series by DeAngelis and colleagues,104 12 patients treated with WBRT to a dose of 25 to 39 Gy in 3 to 6 Gy per fraction, with or without surgery, developed late toxicity. All these patients had no evidence of disease by CT or at the time of surgery. All these patients had severe dementia, ataxia, and incontinence 5 to 36 months (median 14 months) after therapy. CT scan revealed evidence of cortical atrophy, ventricular dilatation, and hypodensity of white matter. Thus, for patients who have favorable characteristics and are expected to have a good prognosis (such as RTOG class I), either delaying WBRT by pursuing alternative options or using a smaller dose per fraction is recommended.
Most series report no survival differences by tumor type except for melanoma. Studies indicate that patients harboring metastases from breast and lung carcinoma are likely to respond to radiotherapy both clinically and by scan, and those who respond are unlikely to die of their metastatic brain tumor. Patients with melanoma and colon cancer are less likely to have a radiographic response even when there is a clinical response, and they are more likely to die of their metastatic brain tumor than of systemic disease.105 However, occasional responses by these patients to irradiation, documented by CT or MRI, make treatment of all patients desirable.
The outcome of three randomized trials comparing surgery and radiotherapy to WBRT alone is summarized in Fig. 21-15.106–110 In the study by Patchell and associates,100 the recurrence rate was 20% in the surgery group and 52% in the radiation-only group (P < 0.02), and the median time to recurrence was more than 59 weeks for combined treatment and 21 weeks for radiation alone (P < 0.0001). The median duration of functional independence, defined as KPS of 70 or more, was 38 weeks for combined treatment and 8 weeks for radiation alone (P < 0.005). Noordijk and colleagues found that the median survival of patients with inactive extracranial disease was 12 months for combined treatment and 7 months for radiotherapy alone (P = 0.02).111 In contrast, there was no benefit from combined treatment in patients with active extracranial disease or in those older than 60 years (median survival 5 months in both groups). However, Mintz and colleagues did not report a benefit in either survival or the quality of life with the addition of surgery in patients who received WBRT.110
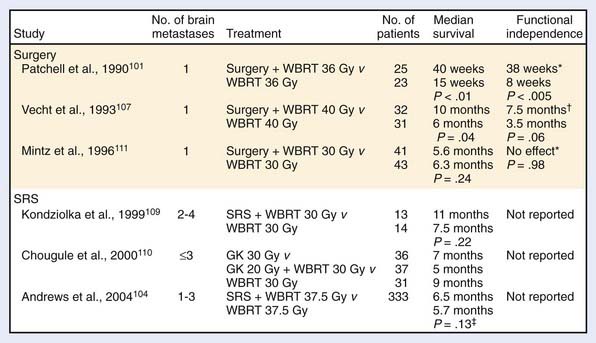
FIGURE 21-15 • Randomized studies of surgery/stereotactic radiosurgery versus whole-brain radiation therapy.
WBRT is generally recommended after surgical resection of metastatic tumors. There is good evidence that suggests that postoperative irradiation decreases relapse both at the site of surgical removal and elsewhere in the brain.100 Patchell and colleagues randomized 95 patients with solitary metastasis to surgery with or without 50.4 Gy WBRT. They noted a benefit of WBRT in terms of decreased recurrence at the site of original disease (10% versus 46%), recurrence at other sites in brain (14% versus 37%), recurrence anywhere in the brain (18% versus 70%), and death resulting from neurologic causes (14% versus 44%). However, the overall survival (48 weeks) and functionally independent survival (36 weeks) were the same in both the arms.100 Hence, a protracted radiotherapy course could be considered in favorable-prognosis patients because shorter treatment courses do not appear to prevent recurrence and may increase toxicity.
Stereotactic Radiosurgery
Several reports support the use of radiosurgery in the initial treatment of patients with one or two favorable brain metastases or in those who relapse after WBRT. Local control to complete radiologic response is noted in 80% to 90% of the cases, with a median survival of 9 months.112 These survival results are comparable to those observed for surgical resection. There has never been a randomized trial comparing SRS with surgical resection. A multi-institutional retrospective data of SRS for solitary metastasis that comes closest to addressing this issue was reported by Auchter and colleagues.113 A total of 122 patients with solitary brain metastasis were treated to a 37.5-Gy median dose WBRT with SRS boost to 15 to 24 Gy. With a 123-week follow-up, the local control was 86%. The infield recurrence rate was 14% and the intracranial recurrence outside SRS volume was 22%. The median overall and functionally independent survival rates were 56 and 44 weeks respectively, indicating that the SRS is at least equivalent if not superior to surgical resection.
There have been many randomized trials that have tried to address the benefit of SRS when given in addition to WBRT. Kondziolka and colleagues108 reported a small randomized trial of 27 patients with two to four metastases to WBRT with or without SRS. The median time to local failure was 6 versus 36 months, and the median survival was significantly better with the addition of SRS boost (7.5 versus 11 months).112 The local failure decreased from 100% to 8% at 1 year with SRS. Chougle and colleagues did not report survival benefit in any of the three arms treated with SRS, WBRT, and WBRT and SRS. However, this study is difficult to interpret because the arms were not balanced.109 In the RTOG 9508 trial, 333 patients with 1 to 3 metastases were randomized to 37.5-Gy WBRT with or without SRS boost.103 A survival benefit was noted in RPA class I, solitary metastasis, and non-small cell lung cancer patients. Local control at 1 year was 71% with WBRT compared with 82% with the addition of SRS boost. Improved KPS was also noted with the addition of SRS boost (27% versus 43%) at 6 months.
There is suggestion that treatment outcome may not be compromised by omitting immediate WBRT in patients treated with SRS for limited brain metastasis. A retrospective data by Sneed on 105 patients, of which 62 were treated with SRS alone, showed an overall survival of 11-month and 1-year PFS of 75%.114 Although the brain PFS was lower (28% versus 69%) with the omission of upfront WBRT, with salvage it was the same (62% versus 73%), indicating that it could be an option for patients with one to two metastases. A word of caution comes from Regine and colleagues, who note that 70% of patients were symptomatic at the time of recurrence and 60% had neurologic deficits, and recommend that adjuvant WBRT should be considered a mainstay of treatment in brain metastases.115
There are two randomized trials that have looked at stereotactic radiosurgery plus WBRT versus SRS alone for treatment of brain metastases. Aoyami and colleagues116 have reported the results of a Japanese randomized trial in 132 patients with one to four brain metastases. The primary end-point of this study was overall survival. The median survival time was similar in both arms (7.5 and 8.0 months). The 1-year brain tumor recurrence rate was 46.8% in the WBRT-with-SRS group, and 76.4% for the SRS-alone group. Salvage brain treatment was less frequently required in the WBRT-with-SRS group than with SRS alone. Death was attributed to neurologic causes in 22.8% of patients in the WBRT-with-SRS group and in 19.3% of those treated with SRS alone. However, there were no significant differences in systemic and neurologic functional preservation and toxic effects of radiation between the arms. The American College of Surgery Oncology Group is conducting a randomized phase III trial of radiosurgery with or without WBRT.
Chemotherapy
The role of chemotherapy has traditionally been minimal in the treatment of brain metastases. This has been the result of short survival and poor performance status associated with symptomatic brain metastases as well as the assumption that an intact blood-brain barrier would limit the delivery of large and hydrophilic drugs to the site of brain lesions. However, during the last few years, several phase II studies have illustrated the activity of chemotherapy in the treatment of brain metastases. Temozolomide has shown the maximum activity among the agents that have been studied. In a large phase II trial reported by Antonadou,117 patients were assigned to receive temozolomide (75 mg/m2) plus WBRT (10 fractions of 3 Gy each) or WBRT alone. In addition, patients in the combined-therapy group continued to receive temozolomide (200 mg/m2 days 1 through 5 every 4 weeks for six cycles) beginning 1 month after completing WBRT. The group receiving WBRT plus temozolomide experienced a significant improvement in RR compared with those receiving WBRT alone (53% versus 33%). However, no difference in neurologic response or overall survival was noted in the two arms. Use of experimental radiosensitizing agents like motexafin gadolinium or efaproxiral in phase III trials has not shown a benefit in overall survival.118 Inhibitors of the EGFR-associated tyrosine kinase like gefitinib (ZD1839) have shown responses in patients who had received previous WBRT, and is being explored in ongoing clinical trials.95
1 Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71-96.
2 Hoffman S, Propp JM, McCarthy BJ. Temporal trends in incidence of primary brain tumors in the United States, 1985–1999. Neuro Oncol. 2006;8:27-37.
3 Fisher JL, Schwartzbaum JA, Wrensch M, et al. Epidemiology of brain tumors. Neurol Clin. 2007;25:867-890.
4 Fine VH, Barker GFII, Markert MJ, et al. Neoplasms of the central nervous system. In: DeVita VTJr, Hellman S, Steven A, editors. Cancer: Principles and practice of oncology. 7th ed. Philadelphia: JB Lippincott; 2005:1834-1932.
5 Jemal A, Tiwari RC, Murray T, et al. Cancer statistics. CA Cancer J Clin. 2004;54:8-29.
6 Soni D, King JA, Kaye AH, et al. Genetics of glioblastoma multiforme: mitogenic signaling and cell cycle pathways converge. J Clin Neurosci. 2005;12:1-5.
7 Von Deimling A, Perry A. Neurofibromatosis type 1. In: Louis DN, Ohgaki H, Wiestler OD, et al, editors. WHO classification of tumors of the central nervous system. ed 4. Lyon: IARC; 2007:206.
8 Connelly JM, Malkin MG. Environmental risk factors for brain tumors. Curr Neurol Neurosci Rep. 2007;7:208-214.
9 Martin JH, Leonard ME, Radzyner HJ. Neuroanatomy: text and atlas, ed 3. McGraw-Hill; 2002.
10 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
11 Burger PC. Classification, grading and patterns of spread of malignant cerebral gliomas. Apuzzo. 1990. American Association of Neurological Surgeons. Park Ridge, Illinois. 3.
12 Henson JW, Gaviani P, Gonzalez RG. MRI in treatment of adult gliomas. Lancet Oncol. 2005;3:167-175.
13 Poussaint TY. Magnetic resonance imaging of pediatric brain tumors: state of the art. Top Magn Reson Imaging. 2001;12(6):411-433.
14 Albayrak B, Samdani AF, Black PM. Intraoperative magnetic resonance imaging in neurosurgery. Acta Neurochir (Wien). 2004;146:543-556.
15 Cha S. Update on brain tumor imaging: From anatomy to physiology. AJNR Am J Neuroradiol. 2006;3:475-487.
16 Poussaint TY, Rodriguez D. Advanced neuroimaging of pediatric brain tumors: MR diffusion, MR perfusion, and MR spectroscopy. Neuroimaging Clin N Am. 2006;16:169-192.
17 Narayana A, Chang J, Thakur S, et al. Use of MR spectroscopy and functional imaging in the treatment planning of gliomas. Br J Radiol. 2007;80:347-354.
18 Chang J, Kowalski A, Hou B, et al. Feasibility study of intensity-modulated radiotherapy (IMRT) treatment planning using brain functional MRI. Med Dosim. 2008;33:42-47.
19 Law M, Young RJ, Babb JS, et al. Gliomas: predicting time to progression or survival with cerebral blood volume measurements at dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging. Radiology. 2008;247(2):490-498.
20 Curran WJJr, Scott CB, Horton J, et al. Does extent of surgery influence outcome for astrocytoma with atypical or anaplastic foci (AAF)? A report from three Radiation Therapy Oncology Group (RTOG) clinical trials. Int J Radiat Oncol Biol Phys. 1993;26:239.
21 Shaw EG, Seiferheld W, Scott C, et al. Reexamining the Radiation Therapy Oncology Group recursive partitioning analysis for glioblastoma multiforme patients. Int J Radiat Oncol Biol Phys. 2003;57(suppl 2):S135-136.
22 Pang BC, Wan WH, Lee CK, et al. The role of surgery in high-grade glioma—is surgical resection justified? A review of the current knowledge. Ann Acad Med Singapore. 2007;36:358-363. Review
23 Simpson JR, Horton J, Scott C, et al. Influence on location and extent of surgical resection on survival of patients with glioblastoma multiforme: Results of three consecutive Radiation Therapy Oncology Group clinical trials. Int J Radiat Oncol Biol Phys. 1993;26:239-244.
24 Vuorinen V, Hinkka S, Färkkilä M, et al. Debulking or biopsy of malignant glioma in elderly people—a randomised study. Acta Neurochir (Wien). 2003;145:5-10.
25 Walker MD, Alexander EJr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. J Neurosurg. 1978;49:333.
26 Walker MD, Green SB, Byar DP, et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303:1323.
27 Kristiansen K, Hagen S, Kollevold T, et al. Combined modality therapy of operated astrocytomas grade III and IV. Confirmation of the value of postoperative irradiation and lack of potentiation of bleomycin on survival time: a prospective multicenter trial of the Scandinavian Glioblastoma Study Group. Cancer. 1981;47:649.
28 Reardon DA, Rich JN, Friedman HS, et al. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253-1265.
29 Yung WK, Albright RE, Olsen J, et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588.
30 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
31 Fine HA. Promising new therapies for malignant gliomas (Review). Cancer J. 2007;13:349-354.
32 Qi XS, Schultz CJ, Li XA. An estimation of radiobiologic parameters from clinical outcomes for radiation treatment planning of brain tumor. Int J Radiat Oncol Biol Phys. 2006;64:1570-1580.
33 Nelson DF, Curran WJJr, Scott C, et al. Hyperfractionated radiation therapy and bis-chlorethyl nitrosourea in the treatment of malignant glioma—possible advantage observed at 72 Gy on 1.2 Gy b.i.d. fractions: Report of the Radiation Therapy Oncology Group protocol 8302. Int J Radiat Oncol Biol Phys. 1993;25:193.
34 Narayana A, Yamada J, Berry S, et al. Intensity-modulated radiotherapy in high-grade gliomas: Clinical and dosimetric results. Int J Radiat Oncol Biol Phys. 2006;64:892-897.
35 Shapiro WR, Green SB, Burger PC, et al. Randomized trial of three chemotherapy regimens and two radiotherapy regimens and two radiotherapy regimens in postoperative treatment of malignant glioma, Brain Tumor Cooperative Group Trial 8001. J Neurosurg. 1989;71(1):1-9.
36 Bleehen NM, Stenning SP. A Medical Research Council trial of two radiotherapy doses in the treatment of grades 3 and 4 astrocytoma. The medical research council brain tumor Working Party. Br J Cancer. 1991;64:769.
37 Nelson DF, Diener-West M, Horton J, et al. Combined modality approach to treatment of malignant gliomas—Re-evaluation of RTOG 7401/ECOG 1374 with long-term follow-up: a joint study of the Radiation Therapy Oncology Group and the Eastern Cooperative Oncology Group. NCI Monogr. 1988;6:279.
38 Lee SW, Fraass BA, Marsh LH, et al. Patterns of failure following high-dose 3-D conformal radiotherapy for high-grade astrocytomas: a quantitative dosimetric study. Int J Radiat Oncol Biol Phys. 1999;43:79.
39 Deutsch M, Green SB, Strike TA, et al. Results of a randomized trial comparing BCNU plus radiotherapy, streptozotocin plus radiotherapy, BCNU plus hyperfractionated radiotherapy, and BCNU following misonidazole plus radiotherapy in the postoperative treatment of malignant glioma. Int J Radiat Oncol Biol Phys. 1989;16:1389.
40 Kleinberg L, Grossman SA, Carson K, et al. Survival of patients with newly diagnosed glioblastoma multiforme treated with RSR13 and radiotherapy: results of a phase II new approaches to brain tumor therapy CNS Consortium Safety and Efficacy Study. J Clin Oncol. 2002;20:3149.
41 Phillips TL, Levin VA, Ahn DK, et al. Evaluation of bromodeoxyuridine in glioblastoma multiforme: a Northern California Cancer Center phase II study. Int J Radiat Oncol Biol Phys. 1991;21:709.
42 Prados MD, Scott C, Sandler H, et al. A phase 3 randomized study of radiotherapy plus procarbazine, CCNU, and vincristine (PCV) with or without BUdR for the treatment of anaplastic astrocytoma: a preliminary report of RTOG 9404. Int J Radiat Oncol Biol Phys. 1999;45:1109.
43 Prados MD, Scott C, Curran WJJr, et al. Procarbazine, lomustine, and vincristine (PCV) chemotherapy for anaplastic astrocytoma: A retrospective review of radiation therapy oncology group protocols comparing survival with carmustine or PCV adjuvant chemotherapy. J Clin Oncol. 1999;11:3389-3395.
44 Nelson DF, Diener-West M, Weinstein AS, et al. A randomized comparison of misonidazole sensitized radiotherapy plus BCNU and radiotherapy plus BCNU for treatment of malignant glioma after surgery: final report of an RTOG study. Int J Radiat Oncol Biol Phys. 1986;12:1793.
45 Coughlin C, Scott C, Langer C, et al. Phase II, two-arm RTOG trial (94–11) of bischloroethyl-nitrosourea plus accelerated hyperfractionated radiotherapy (64.0 or 70.4 Gy) based on tumor volume (>20 or <20 cm[2], respectively) in the treatment of newly-diagnosed radiosurgery-ineligible glioblastoma multiforme patients. Int J Radiat Oncol Biol Phys. 2000;48:1351.
46 Brada M, Sharpe G, Rajan B, et al. Modifying radical radiotherapy in high grade gliomas: shortening the treatment time through acceleration. Int J Radiat Oncol Biol Phys. 1999;43:287.
47 Kleinberg L, Grossman SA, Piantadosi S, et al. The effects of sequential versus concurrent chemotherapy and radiotherapy on survival and toxicity in patients with newly diagnosed high-grade astrocytoma. Int J Radiat Oncol Biol Phys. 1999;44:535.
48 Green SB, Shapiro WR, Burger PC, et al. A randomized trial of interstitial radiotherapy (RT) for newly diagnosed malignant glioma: Brain Tumor Cooperative Group (BTCG) trial 8701 (Abstract). Proc Am Soc Clin Oncol. 1994;13:174.
49 Prados MD, Gutin PH, Phillips TL, et al. Highly anaplastic astrocytoma: a review of 357 patients treated between 1977 and 1989. Int J Radiat Oncol Biol Phys. 1992;23:3.
50 Laperriere NJ, Leung PM, McKenzie S, et al. Randomized study of brachytherapy in the initial management of patients with malignant astrocytoma. Int J Radiat Oncol Biol Phys. 1998;41:1005-1011.
51 Chang CH, Horton J, Schoenfeld D, et al. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer. 1983;52:997.
52 Shaw EG, Robbins ME. The management of radiation-induced brain injury. Cancer Treat Res. 2006;128:7-22.
53 Gutin PH. Treatment of radiation necrosis of the brain. In: Gutin PH, Leibel SA, Sheline GE, editors. Radiation injury to the nervous system. New York: Raven Press; 1991:271.
54 Gonzalez J, et al. Effect of bevacizumab on radiation necrosis of the brain. Int J Radiat Oncol Biol Phys. 2007;67:323-326.
55 Kleinberg L, Wallner K, Malkin MG. Good performance status of long-term disease-free survivors of intracranial gliomas. Int J Radiat Oncol Phys. 1993;26:129.
56 Scheithsuer WB, Hawkins C, Tihan T, et al. Pilocytic astrocytoma. In: Louis DN, Ohgaki H, Wiestler OD, et al, editors. WHO Classification of Tumors of the Central nervous system. ed 4. Lyon: IARC; 2007:14-24.
57 Calvar JA, Meli FJ, Romero C, et al. Characterization of brain tumors by MRS, DWI and Ki-67 labeling index. J Neurooncol. 2005;72:273-280.
58 Bauman G, Lote K, Larson D, et al. Pretreatment factors predict overall survival for patients with low-grade glioma: a recursive partitioning analysis. Int J Radiat Oncol Biol Phys. 1999;45:923.
59 Pouratian N, Asthagiri A, Jagannathan J, et al. Medscape. Surgery insight: the role of surgery in the management of low-grade gliomas (Review). Nat Clin Pract Neurol. 2007;3:628-639.
60 Wisoff JH, Abbott R, Epstein F. Surgical management of exophytic chiasmatic-hypothalamic tumors of childhood. J Neurosurg. 1990;73:661-667.
61 MacDonald DR. Low grade gliomas, mixed gliomas, and oligodendrogliomas. Semin Oncol. 1994;21:236.
62 Eyre HJ, Crowley JJ, Townsend JJ, et al. A randomized trial of radiotherapy versus radiotherapy plus CCNU for incompletely resected low-grade gliomas: a Southwest Oncology Group study. J Neurosurg. 1993;78:909.
63 Karim AB, Afra D, Cornu P. Randomized trial on the efficacy of radiotherapy for central low-grade glioma in the adult: EORTC Study 22845 with the MRC Study BR04: an interim analysis. Int J Radiat Oncol Biol Phys. 2002;52:316.
64 Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549-556.
65 Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: Initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2267-2276.
66 Kaloshi G, Benuaich-Amiel A, Diakite F, et al. Temozolomide for low grade gliomas: predictive impact of 1p/19q loss on response and outcome. Neurology. 2007;68:1831-1836.
67 Shaw E, Berkey B, Coons S, et al: Initial report of radiation therapy oncology group (RTOG) 9801: Prospective studies in adult low-grade glioma (LGG) [abstract], Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part I. Vol 24, No 18 (June 20 Suppl), 2006; 1500.
68 Von Deimling A, Reifenberger G, Kros MJ, et al. Oligodendroglial tumors. In: Louis DN, Ohgaki H, Wiestler OD, et al, editors. WHO classification of tumors of the central nervous system. ed 4. Lyon: IARC; 2007:54-67.
69 Giannini C, Burger PC, Berkey BA, et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 2008;18(3):360-369.
70 Brumer JM. Neuropathology of malignant gliomas. Semin Oncol. 1994;21:126.
71 Puduvalli VK, Hashmi M, McAllister LD, et al. Anaplastic oligodendrogliomas: prognostic factors for tumor recurrence and survival. Oncology. 2003;65:259-266.
72 Intergroup Radiation Therapy Oncology Group Trial 9402Cairncross G, Berkey B, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707-2714.
73 Van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715-2722.
74 Winger MJ, MacDonald DR, Cairncross JG. Supratentorial anaplastic gliomas in adults: the prognostic importance of extent of resection and prior low-grade glioma. J Neurosurg. 1989;71:487.
75 Shaw EG, Scheithauer BW, O’Fallon JR. Astrocytomas (A), oligoastrocytomas (OA), and oligodendrogliomas (O): a comparative survival study (Abstract). Neurology. 1992;42(suppl 3):342.
76 Iwamoto FM, DeAngelis LM. An update on primary central nervous system lymphoma. Hematol Oncol Clin North Am. 2006;20:1267-1285.
77 Deangelis LM, Iwamoto FM. An update on therapy of primary central nervous system lymphoma. Hematology Am Soc Hematol Educ Program. 2006:311-316.
78 Deckert M, Paulus W. Malignant lymphomas. In: Louis DN, Ohgaki H, Wiestler OD, et al, editors. WHO Classification of Tumors of the Central Nervous System. ed 4. Lyon: IARC; 2007:188-204.
79 Shibamoto Y, Hayabuchi N, Hiratsuka J, et al. Is whole-brain irradiation necessary for primary central nervous system lymphoma? Patterns of recurrence after partial-brain irradiation. Cancer. 2003;97:128-133.
80 Bessell EM, Lopez-Guillermo A, Villa S, et al. Importance of radiotherapy in the outcome of patients with primary CNS lymphoma: an analysis of the CHOD/BVAM regimen followed by two different radiotherapy treatments. J Clin Oncol. 2002;20:231-236.
81 Nelson DF, Martz KL, Bonner H, et al. Non-Hodgkin’s lymphoma of the brain: can high dose, large volume radiation therapy improve survival? Report on a prospective trial by the Radiation Therapy Oncology Group (RTOG): RTOG 8315. Int J Radiat Oncol Biol Phys. 1992;23:9-17.
82 Hoang-Xuan K, Taillandier L, Chinot O, et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: A multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21:2726-2731.
83 Hargrave D, Bartels U, Bouffet E. Diffuse brainstem glioma in children: Critical review of clinical trials. Lancet Oncol. 2006;7:241-248.
84 Shrieve DC, Wara WM, Edwards MS, et al. Hyperfractionated radiation therapy for gliomas of the brainstem in children and in adults. Int J Radiat Oncol Biol Phys. 1992;24:599-610.
85 Prados MD, Wara WM, Edwards MS, et al. The treatment of brain stem and thalamic gliomas with 78 Gy of hyperfractionated radiation therapy. Int J Radiat Oncol Biol Phys. 1995;32:85-91.
86 Chowdhary S, Green MR, Chamberlain M. Ependymomas. Curr Treat Options Neurol. 2006;8:309-318.
87 Figarella-Branger D, Metellus P, Barrié M, et al. [Intracranial ependymomas in adult patients. Diagnosis and histological prognostic factors]. Neurochirurgie. 2007;53:76-84.
88 Metellus P, Barrie M, Figarella-Branger D, et al. Multicentric French study on adult intracranial ependymomas: prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Brain. 2007;130:1338-1349.
89 Prados MD, Warnick RE, Wara WM, et al. Medulloblastoma in adults. Int J Radiat Oncol Biol Phys. 1994;32:1145.
90 Schofield D, West DC, Anthony DC, et al. Correlation of loss of heterozygosity at chromosome 9q with histological subtype in medulloblastomas. Am J Pathol. 1995;146:472.
91 Spreafico F, Massimino M, Gandola L, et al. Survival of adults treated for medulloblastoma using paediatric protocols. Eur J Cancer. 2005;41:1304-1310.
92 Frost PJ, Laperriere HJ, Wong CS, et al. Medulloblastoma in adults. Int J Radiat Oncol Biol Phys. 1995;32:951.
93 Sarkar C, Pramanik P, Karak AK, et al. Are childhood and adult medulloblastomas different? A comparative study of clinicopathological features, proliferation index and apoptotic index. J Neurooncol. 2002;59:49.
94 Brandes AA, Ermani M, Amista P, et al. The treatment of adults with medulloblastoma: a prospective study. Int J Radiat Oncol Biol Phys. 2003;57:755-761.
95 Langer CJ, Mehta MP. Current management of brain metastases, with a focus on systemic options. J Clin Oncol. 2005;23:6207-6219.
96 Posner JB. Neurologic complications of cancer. Philadelphia: FA Davis; 1995. pp. 37, 77, 311
97 Martin JH, Leonard ME, Radzyner HJ. Neuroanatomy: Text and Atlas, ed 3. McGraw-Hill; 2002.
98 Van der Molen AJ, Bellin MF. Extracellular gadolinium-based contrast media: differences in diagnostic efficacy. Eur J Radiol. 2008;66:168-174.
99 Gaspar L, Scott C, Rotman M, et al. Recursive portioning analysis (RPA) of prognostic factors in three Radiation Therapy Oncology Group (RTOG) brain metastases trials. Int J Radiat Oncol Biol Phys. 1997;37:745.
100 Patchell RA, Tibbs PA, Walsh JW, et al. A randomized trial of surgery in the treatment of single metastases to the brain. N Engl J Med. 1990;322:494.
101 Truong MT, St Clair EG, Donahue BR, et al. Results of surgical resection for progression of brain metastases previously treated by gamma knife radiosurgery. Neurosurgery. 2006;59:86-97.
102 Agazzi S, Pampallona S, Pica A, et al. The origin of brain metastases in patients with an undiagnosed primary tumour. Acta Neurochir (Wien). 2004;146:153-157.
103 Andrews DW, Scott CB, Sperduto PW, et al. Whole brain radiation therapy with or without stereotactic radiosurgery boost for patients with one to three brain metastases: phase III results of the RTOG 9508 randomised trial. Lancet. 2004;363:1665-1672.
104 DeAngelis LM, Delattre JY, Posner JB. Radiation-induced dementia in patients cure of brain metastases. Neurology. 1989;39:789.
105 Majer M, Samlowski WE. Management of metastatic melanoma patients with brain metastases (Review). Curr Oncol Rep. 2007;9:411-416.
106 Vecht CJ, Haaxma-Reiche H, Noordijk EM, et al. Treatment of single brain metastasis: radiotherapy alone or combined with neurosurgery? Ann Neurol. 1993;33:583-590.
107 Mehta MP, Rodrigus P, Terhaard CHJ, et al. Survival and neurologic outcomes in a randomized trial of motexafin gadolinium and whole-brain radiation therapy in brain metastases. J Clin Oncol. 2003;21:2529-2536.
108 Kondziolka D, Patel A, Lunsford LD, et al. Stereotactic radiosurgery plus whole brain radiotherapy versus radiotherapy alone for patients with multiple brain metastases. Int J Radiat Oncol Biol Phys. 1999;45:427-434.
109 Chougule PB, Burton-Williams M, Saris S, et al. Randomized treatment of brain metastasis with gamma knife radiosurgery, whole brain radiotherapy or both. Int J Radiat Oncol Biol Phys. 2000;48:114. (suppl; abstr 7)
110 Mintz AH, Kestle J, Rathbone MP, et al. A randomized trial to assess the efficacy of surgery in addition to radiotherapy in patients with a single cerebral metastasis. Cancer. 1996;78:1470-1476.
111 Noordijk EM, Vecht CJ, Haaxma-Reiche H. The choice of treatment of single brain metastasis should be based on extracranial tumor activity and age. Int J Radiat Oncol Biol Phys. 1994;29:711.
112 Hussain A, Brown PD, Stafford SL, et al. Stereotactic radiosurgery for brainstem metastases: survival, tumor control, and patient outcomes. Int J Radiat Oncol Biol Phys. 2007;67:521-524.
113 Auchter RM, Lamond JP, Alexander E, et al. A multiinstitutional outcome and prognostic factor analysis of radiosurgery for resectable single brain metastasis. Int J Radiat Oncol Biol Phys. 1996;35:27.
114 Flickinger JC, Kondziolka D, Lunsford LD, et al. A multi-institutional experience with stereotactic radiosurgery for solitary brain metastases. Int J Radiat Oncol Biol Phys. 1994;28:797.
115 Regine WF, Huhn JL, Patchell RA, et al. Risk of symptomatic brain tumor recurrence and neurologic deficit after radiosurgery alone in patients with newly diagnosed brain metastases: results and implications. Int J Radiat Oncol Biol Phys. 2002;52:333.
116 Aoyama H, Shirato H, Tago M, et al. Stereotactic radiosurgery plus whole-brain radiation therapy vs stereotactic radiosurgery alone for treatment of brain metastases: a randomized controlled trial. JAMA. 2006;295:2483-2491.
117 Antonadou D, Paraskevaidis M, Sarris G, et al. Phase II randomized trial of temozolomide and concurrent radiotherapy in patients with brain metastases. J Clin Oncol. 2002;20:3644-3650.
118 Chang JE, Robins HI, Mehta MP. Therapeutic advances in the treatment of brain metastases. Clin Adv Hematol Oncol. 2007;5:54-64.