Replicative Senescence and the Hayflick Limit
Senescence and Viral Oncoproteins
Premature Senescence
Senescence in Vivo
Convergence of Senescence Stimuli on Two Major Pathways
p16INK4A/RB Pathway
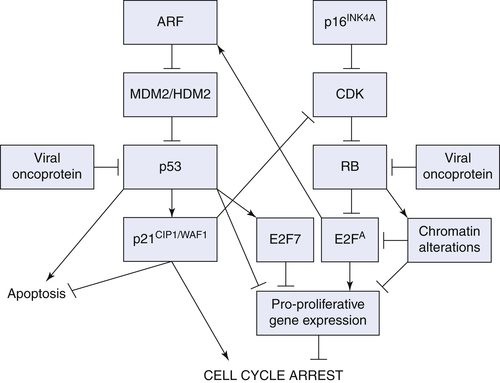
ARF/p53/p21CIP1/WAF1 Pathway
The Senescence-Associated Secretory Phenotype
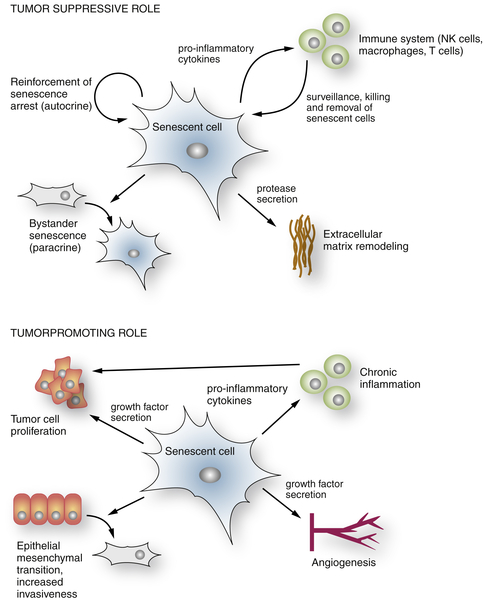
Senescence and Noncancer Disease States
Conclusions and Perspectives
1. The serial cultivation of human diploid cell strains . Exp Cell Res . 1961 ; 25 : 585 – 621 .
2. The limited in vitro lifetime of human diploid cell strains . Exp Cell Res . 1965 ; 37 : 614 – 636 .
3. Variation in the life-span of clones derived from human diploid cell strains . J Cell Biol . 1974 ; 62 : 48 – 53 .
4. Telomeres shorten during ageing of human fibroblasts . Nature . 1990 ; 345 : 458 – 460 .
5. Telomeres and their control . Annu Rev Genet . 2000 ; 34 : 331 – 358 .
6. Senescence induced by altered telomere state, not telomere loss . Science . 2002 ; 295 : 2446 – 2449 .
7. A DNA damage checkpoint response in telomere-initiated senescence . Nature . 2003 ; 426 : 194 – 198 .
8. Defining the molecular mechanisms of human cell immortalization . Biochim Biophys Acta . 1991 ; 1072 : 1 – 7 .
9. Telomeres and telomerase in normal and cancer stem cells . FEBS Lett . 2010 ; 584 : 3819 – 3825 .
10. Telomerases . Annu Rev Biochem . 1992 ; 61 : 113 – 129 .
11. Extension of life-span by introduction of telomerase into normal human cells . Science . 1998 ; 279 : 349 – 352 .
12. Telomerase is not an oncogene . Oncogene . 2002 ; 21 : 494 – 502 .
13. Specific association of human telomerase activity with immortal cells and cancer . Science . 1994 ; 266 : 2011 – 2015 .
14. Creation of human tumour cells with defined genetic elements . Nature . 1999 ; 400 : 464 – 468 .
15. High frequency of SV40 transformation of mouse cell line 3T3 . Virology . 1966 ; 28 : 756 – 759 .
16. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture . Nature . 1983 ; 304 : 602 – 606 .
17. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes . Nature . 1983 ; 304 : 596 – 602 .
18. Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen . Exp Cell Res . 1989 ; 184 : 109 – 118 .
19. A role for both RB and p53 in the regulation of human cellular senescence . Exp Cell Res . 1991 ; 196 : 33 – 39 .
20. siRNA and shRNA as anticancer agents in a cervical cancer model . Methods Mol Biol . 2008 ; 442 : 159 – 172 .
21. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells . J Virol . 2003 ; 77 : 6066 – 6069 .
22. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells . J Virol . 2004 ; 78 : 4063 – 4073 .
23. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence . J Virol . 2007 ; 81 : 2102 – 2116 .
24. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts . Cancer Res . 1999 ; 59 : 1539 – 1543 .
25. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a . Cell . 1997 ; 88 : 593 – 602 .
26. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling . Genes Dev . 1998 ; 12 : 3008 – 3019 .
27. Senescence of human fibroblasts induced by oncogenic Raf . Genes Dev . 1998 ; 12 : 2997 – 3007 .
28. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence . Genes Dev . 2007 ; 21 : 43 – 48 .
29. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion . Nat Cell Biol . 2009 ; 11 : 973 – 979 .
30. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer . Nat Cell Biol . 2011 ; 13 : 292 – 302 .
31. A negative feedback signaling network underlies oncogene-induced senescence . Cancer Cell . 2006 ; 10 : 459 – 472 .
32. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis . Nat Cell Biol . 2007 ; 9 : 493 – 505 .
33. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects . Cancer Cell . 2004 ; 5 : 375 – 387 .
34. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy . Cell . 2002 ; 109 : 335 – 346 .
35. DNA damage is able to induce senescence in tumor cells in vitro and in vivo . Cancer Res . 2002 ; 62 : 1876 – 1883 .
36. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers . Cancer Res . 2005 ; 65 : 2795 – 2803 .
37. Tumour biology: senescence in premalignant tumours . Nature . 2005 ; 436 : 642 .
38. Oncogene-induced senescence as an initial barrier in lymphoma development . Nature . 2005 ; 436 : 660 – 665 .
39. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland . Mol Cell Biol . 2005 ; 25 : 2660 – 2672 .
40. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis . Nature . 2005 ; 436 : 725 – 730 .
41. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression . Cancer Cell . 2008 ; 14 : 146 – 155 .
42. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis . Cancer Cell . 2010 ; 18 : 135 – 146 .
43. BRAFE600-associated senescence-like cell cycle arrest of human naevi . Nature . 2005 ; 436 : 720 – 724 .
44. High frequency of BRAF mutations in nevi . Nat Genet . 2003 ; 33 : 19 – 20 .
45. BRAF and NRAS mutations in melanoma and melanocytic nevi . Melanoma Res . 2006 ; 16 : 267 – 273 .
46. Molecular markers of tumor progression in melanoma . Curr Genomics . 2009 ; 10 : 231 – 239 .
47. Cellular mechanisms of tumour suppression by the retinoblastoma gene . Nat Rev Cancer . 2008 ; 8 : 671 – 682 .
48. p14ARF links the tumour suppressors RB and p53 . Nature . 1998 ; 395 : 124 – 125 .
49. Cyclin D-dependent kinases, INK4 inhibitors and cancer . Biochim Biophys Acta . 2002 ; 1602 : 73 – 87 .
50. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis . Nature . 2001 ; 413 : 86 – 91 .
51. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF . Cell . 1997 ; 91 : 649 – 659 .
52. The retinoblastoma gene family: cousins with overlapping interests . Trends Genet . 1998 ; 14 : 223 – 229 .
53. Targeted disruption of p107: functional overlap between p107 and Rb . Genes Dev . 1996 ; 10 : 1621 – 1632 .
54. Shared role of the pRB-related p130 and p107 proteins in limb development . Genes Dev . 1996 ; 10 : 1633 – 1644 .
55. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization . Genes Dev . 2000 ; 14 : 3037 – 3050 .
56. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry . Nature . 2003 ; 424 : 223 – 228 .
57. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence . Cell . 2003 ; 113 : 703 – 716 .
58. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence . Cancer Cell . 2010 ; 17 : 376 – 387 .
59. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence . Semin Cancer Biol . 2011 ; 21 : 367 – 376 .
60. Molecular dissection of formation of senescence-associated heterochromatin foci . Mol Cell Biol . 2007 ; 27 : 2343 – 2358 .
61. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence . Proc Natl Acad Sci U S A . 2012 ; 109 : 8971 – 8976 .
62. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice . Oncogene . 1993 ; 8 : 2457 – 2467 .
63. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas . Nature . 2007 ; 445 : 656 – 660 .
64. Restoration of p53 function leads to tumour regression in vivo . Nature . 2007 ; 445 : 661 – 665 .
65. Modeling the therapeutic efficacy of p53 restoration in tumors . Cell . 2006 ; 127 : 1323 – 1334 .
66. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras . Oncogene . 1999 ; 18 : 4974 – 4982 .
67. PML is induced by oncogenic ras and promotes premature senescence . Genes Dev . 2000 ; 14 : 2015 – 2027 .
68. PML is a direct p53 target that modulates p53 effector functions . Mol Cell . 2004 ; 13 : 523 – 535 .
69. Regulation of E2Fs and senescence by PML nuclear bodies . Genes Dev . 2011 ; 25 : 41 – 50 .
70. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence . Genes Dev . 2012 ; 26 : 1546 – 1557 .
71. E2F7, a novel target, is upregulated by p53 and mediates DNA damage-dependent transcriptional repression . Genes Dev . 2012 ; 26 : 1533 – 1545 .
72. Senescence of activated stellate cells limits liver fibrosis . Cell . 2008 ; 134 : 657 – 667 .
73. Granule exocytosis mediates immune surveillance of senescent cells . Oncogene . 2013 ; 32(15) : 1971 – 1977 . doi: 10.1038/onc.2012.206 Epub 2012 Jul 2 .
74. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development . Nature . 2011 ; 479 : 547 – 551 .
75. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity . Genes Dev . 2011 ; 25 : 2125 – 2136 .
76. Chemokine signaling via the CXCR2 receptor reinforces senescence . Cell . 2008 ; 133 : 1006 – 1018 .
77. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network . Cell . 2008 ; 133 : 1019 – 1031 .
78. Opposing roles of NF-kappaB in anti-cancer treatment outcome unveiled by cross-species investigations . Genes Dev . 2011 ; 25 : 2137 – 2146 .
79. Senescent cells develop a PARP-1 and nuclear factor-κB-associated secretome (PNAS) . Genes Dev . 2011 ; 25 : 1245 – 1261 .
80. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7 . Cell . 2008 ; 132 : 363 – 374 .
81. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen . PLoS One . 2010 ; 5 : e9188 .
82. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence . J Biol Chem . 2006 ; 281 : 29568 – 29574 .
83. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion . Cancer Res . 2007 ; 67 : 3117 – 3126 .
84. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging . Proc Natl Acad Sci U S A . 2001 ; 98 : 12072 – 12077 .
85. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor . PLoS Biol . 2008 ; 6 : 2853 – 2868 .
86. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer . Cancer Cell . 2012 ; 21 : 793 – 806 .
87. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells . Oncogene . 2012 ; 31 : 3148 – 3163 .
88. Cancer and aging: a model for the cancer promoting effects of the aging stroma . Int J Biochem Cell Biol . 2002 ; 34 : 1401 – 1414 .
89. Role of telomere in endothelial dysfunction in atherosclerosis . Curr Opin Lipidol . 2002 ; 13 : 537 – 543 .
90. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging . Oncogene . 1997 ; 15 : 203 – 211 .
91. Cellular senescence in aging primates . Science . 2006 ; 311 : 1257 .
92. Accumulation of senescent cells in mitotic tissue of aging primates . Mech Ageing Dev . 2007 ; 128 : 36 – 44 .
93. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders . Nature . 2011 ; 479 : 232 – 236 .
94. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence . Mol Cell . 2009 ; 36 : 2 – 14 .
95. Inflammatory networks during cellular senescence: causes and consequences . Trends Mol Med . 2010 ; 16 : 238 – 246 .
96. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing . Nat Cell Biol . 2010 ; 12 : 676 – 685 .