Cellular mechanisms of neurological disease
The nervous system is subject to the full range of pathological processes found in other organs, together with a number of unique degenerative and demyelinating diseases. The basic pathological processes underlying these disorders will be discussed in this chapter (including inflammation, gliosis and neuronal cell death) before moving on to specific examples of neurological disorders in the chapters that follow. Demyelination is discussed separately in Chapter 14, in the context of multiple sclerosis.
Neuronal injury and death
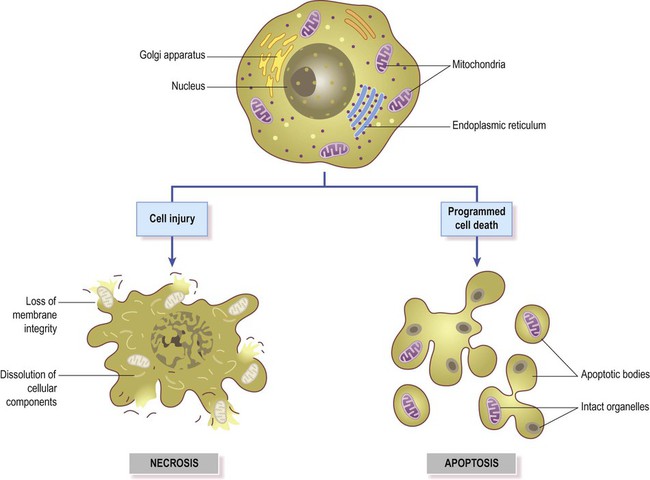
Nerve cells have a limited capacity to withstand pathological stimuli. Cell death occurs when the neuron reaches a ‘point of no return’ following irreversible damage to the plasma membrane, nuclear DNA or mitochondria. Since neurons are post-mitotic cells (meaning that they are unable to divide) they cannot usually be replaced in most parts of the brain and spinal cord. Exceptions include the hippocampus and olfactory bulb (where neurons can be replenished from a pool of stem cells). The two main forms of cell death are illustrated in Figure 8.1 and discussed below.
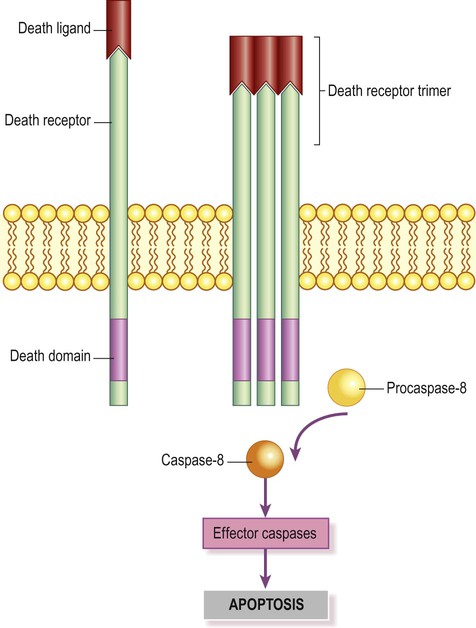
Apoptosis


Intrinsic pathway (Fig. 8.3)
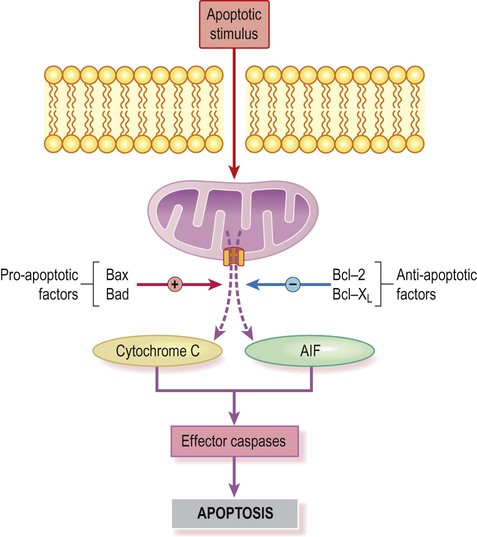
A key event is formation of the permeability transition pore (shown here in orange) in the mitochondrial membrane (coloured purple). This allows apoptosis-inducing factors to be released into the cytoplasm.
Disposal of the cell
Once a cell is committed to programmed cell death, its DNA and cytoskeleton are dismantled in an orderly manner. This is an active process that expends energy. A key step is activation of the enzyme caspase-activated DNAse (CAD) by effector caspases, which breaks down the DNA into nucleosomal units. Organelles are packaged into membrane-bound apoptotic bodies (see Fig. 8.1) which contain viable mitochondria. These structures express cell-surface markers that trigger their internalization by neighbouring cells. An example is the membrane constituent phosphatidylserine, which translocates from the inner to the outer leaflet of the plasma membrane. Phagocytes recognize and bind these molecules and internalize the apoptotic bodies for degradation. The entire process is carefully orchestrated and, in contrast to necrosis, there is no inflammatory reaction.





Axonal damage
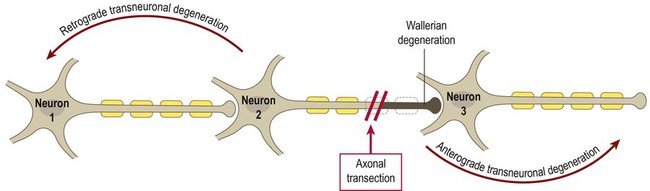
Axonal regrowth
Axons are able to regenerate following peripheral nerve damage and may re-establish connections with muscle fibres or glands. The distal tips of the severed axons form growth cones (Fig. 8.5) which ‘crawl’ along residual Schwann cell basement membranes to reinnervate target structures. This process occurs after peripheral nerve injury (see Clinical Box 8.1) but does not seem to be possible in the brain and spinal cord.
 Clinical Box 8.1:
Clinical Box 8.1: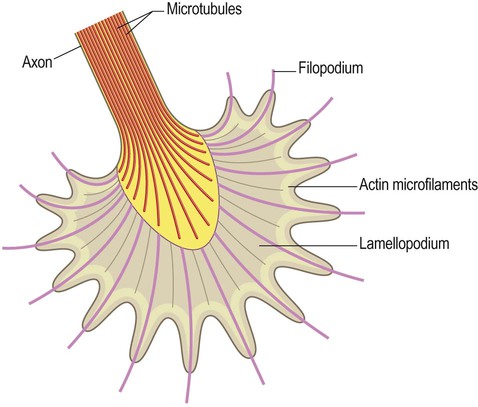




Cell death mechanisms
Excitotoxicity
Excessive stimulation by excitatory neurotransmitters (such as glutamate) can cause neuronal cell death in a process known as excitotoxicity. Intense glutamatergic stimulation leads to prolonged neuronal depolarization, lifting the magnesium blockade of NMDA (N-methyl D-aspartate) receptors. In this situation, free calcium ions are able to flood the neuronal cytoplasm via liberated NMDA receptors, as well as via calcium-permeable AMPA (alpha-amino, 3-hydroxy-4-isoxasole-propionic acid) receptors and voltage-gated calcium channels (see Ch. 7). This leads to further depolarization, with additional glutamate release, generating a vicious cycle. In addition to acute excitotoxic injury, there is evidence that low-grade excitotoxicity may cause chronic neuronal damage in some disorders (e.g. motor neuron disease; see Ch. 4, Clinical Box 4.9). Accumulation of intracellular free calcium is an important final common event in excitotoxicity neuronal cell death.




Oxidative stress



Mitochondria continuously produce superoxide anion which is catabolized by superoxide dismutase to form hydrogen peroxide (H2O2). This is also a reactive oxygen species and is degraded by catalase. Excessive generation of free radicals or reduced capacity of the normal scavenging mechanisms leads to oxidative stress, with abnormal cross-linkages forming between nucleic acids, lipids, carbohydrates and proteins as free radicals react with them indiscriminately (Fig. 8.6). Oxidative stress may be exacerbated by age-related mitochondrial abnormalities due to mutations in mitochondrial DNA that accrue during the lifetime of an individual.
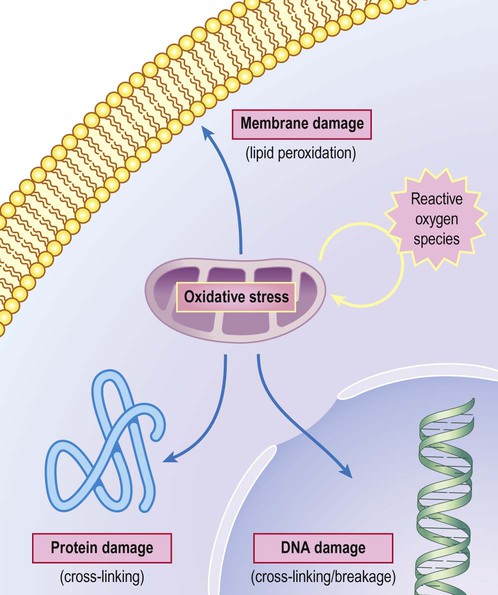
Reactive oxygen (and nitrogen) species damage key cellular components such a proteins, cell membrane and DNA, leading to cell death.





Inflammation and gliosis
Reactive gliosis
Astrocytosis (Fig. 8.7)
Following brain injury, nearby astrocytes enlarge, multiply and increase their expression of glial fibrillary acid protein (GFAP). Proliferation of astrocytes may be sufficient to fill in a small tissue defect, but larger areas of damage (e.g. following a major stroke, see Ch. 10) are transformed into a fluid-filled cystic cavity lined by a glial scar. Astrocytes secrete (i) cytokines that recruit inflammatory cells from the blood and (ii) various trophic factors, including:

Photomicrograph showing the spider-like appearance of reactive astrocytes using immunohistochemistry (antibody labelling) for glial fibrillary acidic protein (GFAP).



These are chemical mediators that promote neuronal survival and axon sprouting. They are released into the extracellular fluid, but can also be delivered directly to the neuronal cytoplasm via intercellular gap junctions (see Ch. 7).
Microgliosis (Fig. 8.8)
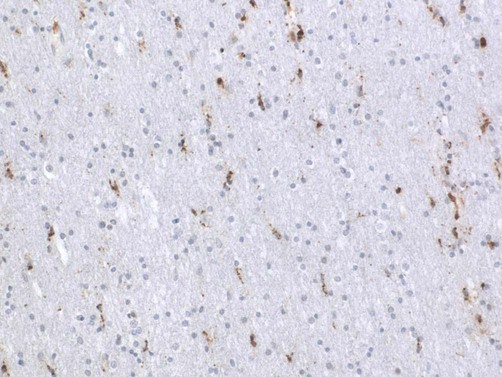
Photomicrograph of reactive microglial cells using immunohistochemistry for the macrophage marker CD68.
Activated microglia are immunocompetent cells that express MHC class II (major histocompatibility) proteins and are antigen-presenting cells. They may therefore contribute to T-cell-mediated immune responses and have been implicated in the inflammatory demyelinating disease multiple sclerosis (Ch. 14). Microglial activation is also a component of most neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease (Chs 12 and 13).
Acute and chronic inflammation
A number of terms are used to describe the site and distribution of acute and chronic inflammation in the nervous system. Meningitis is inflammation of the meninges (protective coverings of the brain). The term pachymeningitis is used if the dura is predominantly affected or leptomeningitis if inflammation is centred on the arachnoid, pia and subarachnoid space (see Ch. 1). The features of acute bacterial meningitis are discussed in Clinical Box 8.2.
 Clinical Box 8.2: Acute bacterial meningitis
Clinical Box 8.2: Acute bacterial meningitis
The main clinical features of meningitis are fever, headache and drowsiness, together with nausea, vomiting and photophobia (sensitivity to light). Clinical examination may show neck stiffness and evidence of raised intracranial pressure including papilloedema (swelling of the optic discs; see Ch. 5, Fig. 5.16). Unlike viral meningitis, a self-limiting illness from which most patients make a complete recovery, the mortality in bacterial meningitis (Fig. 8.9) is around 10% and a significant proportion of survivors are left with neurological deficits, cranial nerve palsies, hydrocephalus or epilepsy.
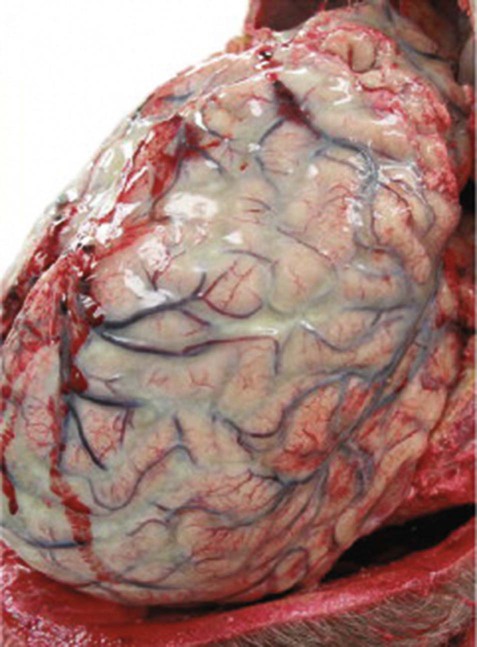
Post-mortem photograph showing thick green-yellow purulent exudate filling the subarachnoid space over the surface of the cerebral hemispheres. From Kleinschmidt-DeMasters, BK and Tyler, KL: Practical Surgical Neuropathology: A Diagnostic Approach (Churchill Livingstone 2010) with permission.
Inflammation in the CNS



The term myelitis indicates inflammation of the spinal cord, such as the inflammatory spinal cord disease poliomyelitis (caused by infection with a poliovirus); whereas combined inflammation of the brain and spinal cord is referred to as encephalomyelitis. It should be emphasized that these terms are descriptive and do not indicate the underlying cause of the inflammation.




Neurodegeneration
The neurodegenerative diseases are a heterogeneous group of progressive, incurable neurological disorders that are more common in later life. They often present with dementia (e.g. Alzheimer’s disease; Ch. 12) or as a movement disorder (e.g. Parkinson’s disease; Ch. 13). Most cases are classified as sporadic or idiopathic, meaning that the cause is not known. Inherited forms often exist, but they are less common and tend to present at an earlier age.
General features
In most neurodegenerative diseases there is a selective loss of certain populations of nerve cells, associated with deposits of an abnormal protein or peptide in neurons and/or glia. These disorders are therefore referred to as proteinopathies (Fig. 8.10). In cases where inherited (familial) forms of a disease have been identified, the mutation often affects the protein itself or an enzyme involved in its processing.
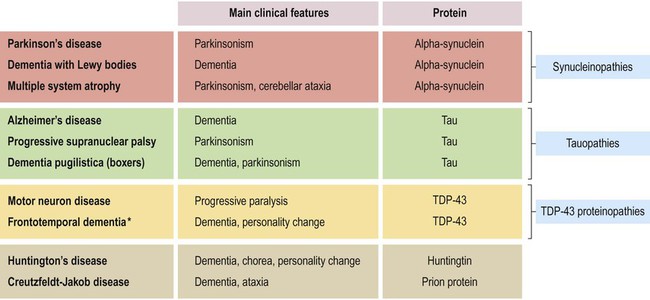
A number of common and/or important neurodegenerative disorders are shown, together with the main protein that accumulates in neurons or glial cells. *[NB: approximately 50% of frontotemporal dementias are TDP43-proteinopathies; in other cases the pathological inclusions are composed of tau (40%) or other proteins (10%)].
Protein folding and misfolding
Normal protein folding
Nuclear DNA encodes the primary structure of proteins, consisting of the basic amino acid sequence. Attainment of the correct three-dimensional conformation requires protein folding (Fig. 8.11). This transforms the linear amino acid sequence into more complex spatial arrangements with alpha-helices and beta-pleated sheets that make up the secondary structure. Further folding gives rise to a three-dimensional globular protein with a particular tertiary structure. Association with other proteins may occur, to form a multi-protein complex with its own quaternary structure. Protein folding relies on physical and chemical properties of the constituent amino acid residues (i.e. attraction and repulsion by hydrogen bonds, electrostatic forces and hydrophobic interactions).
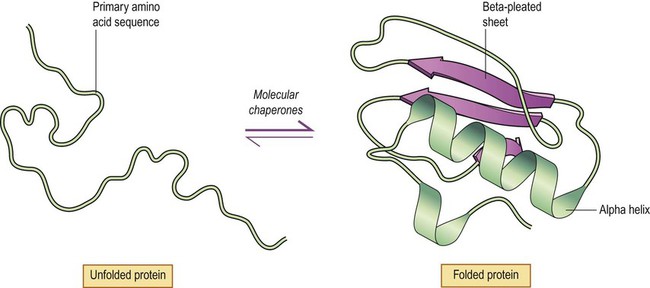
The amino acid sequence of an unfolded protein is encoded by the nuclear DNA (primary structure). This leads to spontaneous formation of alpha helices and beta-pleated sheets (secondary structure) and further folding to produce a globular protein (tertiary structure). The process of protein folding is promoted and accelerated by molecular chaperone proteins.
Disposal of abnormal proteins
Abnormal proteins are earmarked for destruction by tagging them with the 8.5 kDa protein ubiquitin (Fig. 8.12). This is attached in a series of enzyme-catalysed steps involving activating (E1), conjugating (E2) and ligating (E3) enzymes. These add a polyubiquitin chain that is composed of multiple ubiquitin monomers. Once ubiquitinated, the abnormal protein is targeted to the proteasome. This is a large multi-subunit protein complex that digests proteins into peptide fragments and amino acids in an active (ATP-dependent) process.
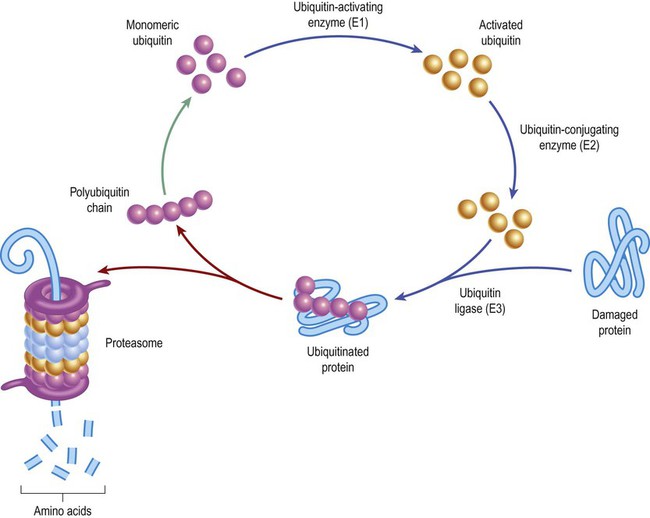
Damaged or incorrectly folded proteins are tagged with ubiquitin and earmarked for degradation (to amino acids) by the proteasome. Digestion within the proteasome is an ATP-dependent process that consumes cellular energy. Conversion of monomeric ubiquitin to its active form (see upper part of figure) is also an ATP-dependent step.




Protein aggregation
Aggregation of abnormal proteins in neurons (or glial cells) gives rise to structures called inclusion bodies. These are difficult to identify by routine light microscopy and are better visualized by silver staining or immunohistochemistry (antibody-labelling of specific proteins). Intracellular inclusions are often cytoplasmic (e.g. in Alzheimer’s and Parkinson’s diseases; Fig. 8.13) but in some cases are found within the nucleus. Abnormal proteins may also accumulate in the extracellular compartment (between nerve cells).
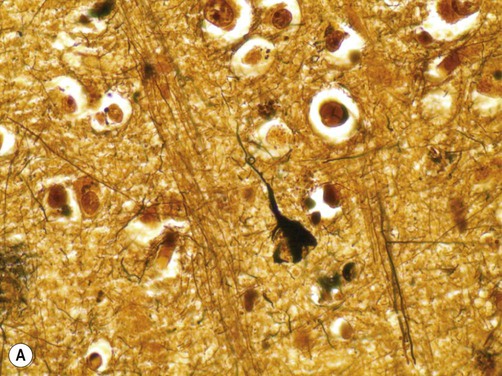
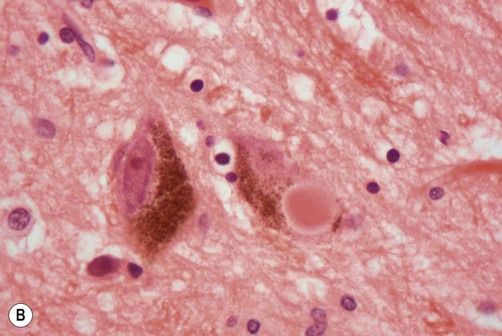
(A) A neurofibrillary tangle (composed of abnormally phosphorylated tau protein) in the cytoplasm of a cortical pyramidal neuron in a patient with Alzheimer’s disease [demonstrated using the modified Bielschowsky silver stain]. From Prayson, R: Neuropathology 1e (Churchill Livingstone 2005) with permission; (B) Micrograph showing two neurons in the substantia nigra in a patient with Parkinson’s disease. The neuron on the left has a large nucleus and prominent nucleolus with abundant brown (neuromelanin) pigment in the cytoplasm. The nucleus of the neuron on the right is not fully seen in this section, but the cytoplasm contains a bright pink Lewy body which is surrounded by a characteristic pale halo [Routine haematoxylin and eosin (H&E)-stained section]. Courtesy of Professor Steve Gentleman.
Amyloid
All forms of amyloid take up certain tissue stains such as Congo red and thioflavin S. Due to the regular, crystalline arrangement of the amyloid fibrils, the deposits also have the ability to rotate the plane of polarized light, termed birefringence. As a result, amyloid deposits stained with Congo red have a characteristic apple green colour when viewed under polarized light (Fig. 8.14).
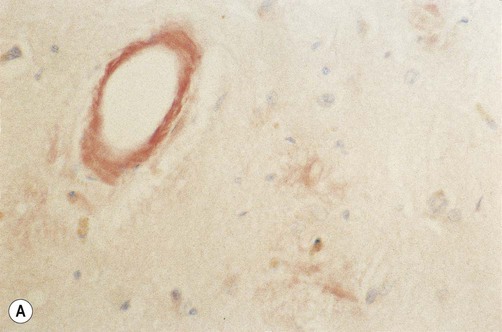
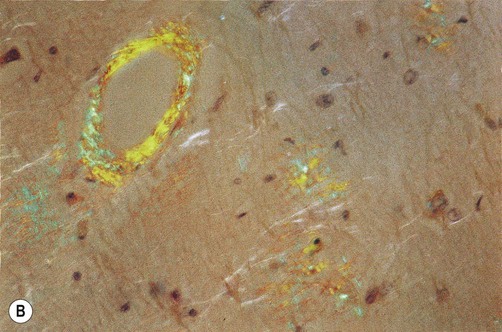
(A) Photomicrograph showing deposition of amyloid in a blood vessel in a case of cerebral amyloid angiopathy [stained with Congo red]; (B) Fluorescence microscopy showing the characteristic apple-green birefringence of amyloid [Congo red stain]. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
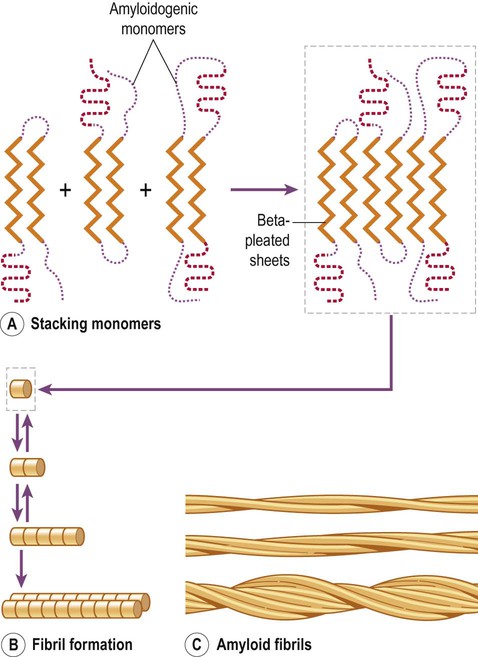
Amyloid fibril formation
The process of amyloid formation is illustrated in Figure 8.15. Fibrillogenesis is the process by which a peptide forms insoluble aggregates of amyloid. It involves three sequential steps. A peptide with a beta-pleated sheet structure must first be produced in sufficient quantities. The second step is nucleation, which starts the process of fibril formation. It requires a supportive microenvironment with a sufficiently high protein concentration, together with various permissive factors including appropriate acidity (pH), temperature or the presence of certain metallic ions. Finally, the phase of fibril growth involves the sequential addition of monomeric peptide units (each with a beta-sheet structure) to form an extending chain. This leads to the gradual assembly of oligomeric species (or protofibrils) which associate to form mature amyloid fibrils.





Prion diseases
General characteristics
Prion diseases have been described in a number of animals including sheep (scrapie) and cattle (bovine spongiform encephalopathy or BSE) in which they cause a rapid and devastating neurological decline that quickly ends in death. In humans, the classical form of prion disease is Creutzfeldt–Jakob disease or CJD, a sporadic disorder of later life characterized by an aggressive and rapidly fatal dementia (Clinical Box 8.3). There are a few very rare genetic forms of prion disease including fatal familial insomnia (FFI) and Gerstmann–Sträussler–Scheinker syndrome (GSS) that are all inherited in an autosomal dominant manner. Each of these has distinguishing clinical and pathological features, but all are incurable and ultimately fatal.
 Clinical Box 8.3: Sporadic CJD
Clinical Box 8.3: Sporadic CJD
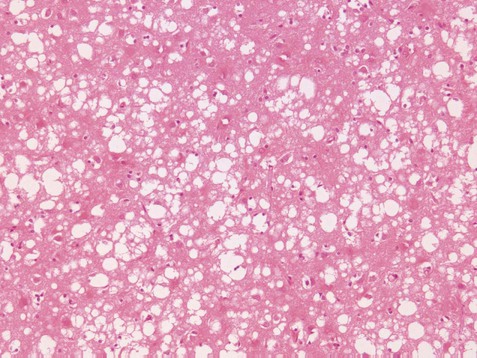
Creutzfeldt–Jakob disease is a sporadic neurodegenerative disease which affects one in a million people worldwide per year, most of whom are over the age of 65. It is characterized by a very aggressive dementia that leads to death in a matter of weeks. This is often accompanied by abnormal movements including electric-shock-like myoclonic jerks. The EEG shows characteristic periodic complexes in 60–80% of cases, consisting of biphasic or triphasic ‘sharp waves’ at a frequency of 1–2 Hz. The cerebrospinal fluid may contain increased levels of certain proteins (e.g. 14-3-3 and S100b). Examination of the brain after death shows widespread spongiform degeneration of the cerebral cortex (Fig. 8.16), accompanied by neuronal loss, gliosis and deposition of abnormal prion protein.
Infectivity
In addition to sporadic and inherited forms, prion diseases have the unique property (among degenerative diseases) of infectivity. They are therefore also referred to as transmissible spongiform encephalopathies (TSEs). In the 1990s in the UK and Europe, bovine spongiform encephalopathy entered the human food chain via contaminated beef and led to a new variant of CJD (Clinical Box 8.4). Prion diseases have also been transmitted iatrogenically (Greek: iatros, doctor) by blood transfusions and growth hormone supplements (obtained from human donors) and various neurosurgical procedures (via contaminated instruments or dural grafts).
 Clinical Box 8.4: New variant CJD
Clinical Box 8.4: New variant CJD
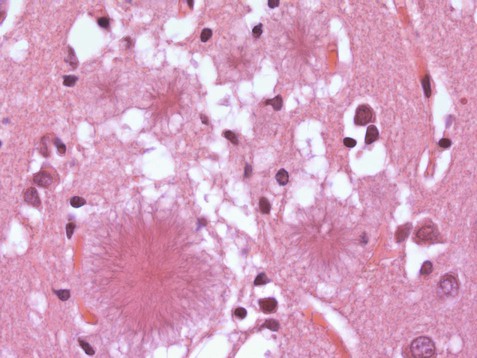
The new variant of CJD was first identified by its microscopic appearance. This differs from that of classical CJD and includes characteristic florid plaques, named for their resemblance to flowers (Fig. 8.17). The new variant affects younger people (usually below the age of 30) and has a longer duration, typically a year or more. Psychiatric features and cerebellar ataxia are also common and myoclonic jerks are absent. MRI scanning characteristically shows T2-hyperintensity in the posterior thalamus (the pulvinar sign). As with classical CJD, the condition is incurable and fatal, but it is much rarer, with fewer than 180 cases reported in the UK (representing 80% of the worldwide total).




Prion protein
The infective agent in prion disease is unique since it is non-cellular, has no DNA or RNA and appears to be composed entirely of protein (the ‘protein only hypothesis’). It is an abnormally folded form of cellular prion protein (or PrPc) which is present in many different tissue types. The pathogenic form has the same primary amino acid structure, but a different secondary structure that is rich in beta-pleated sheets rather than alpha-helices and is able to form amyloid deposits. This infective form is designated PrPSc (scrapie variant). The classical model of normal and abnormal prion protein structure is illustrated in Figure 8.18, but the actual three-dimensional structure is not known with certainty.
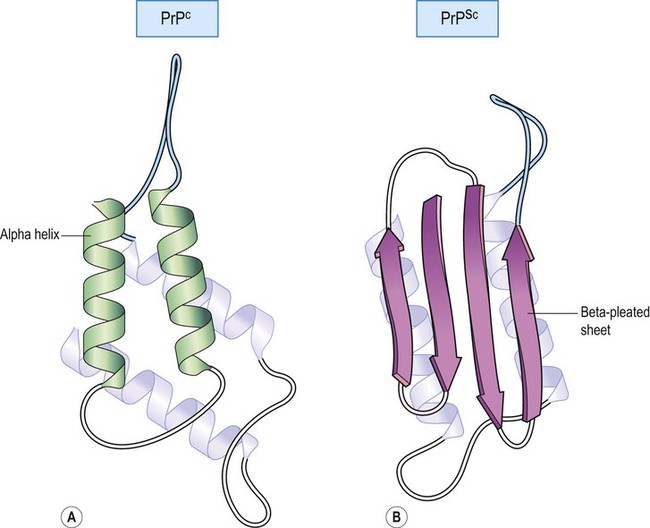
(A) Normal prion protein, with a secondary structure that is rich in alpha helices; (B) Abnormal prion protein has an identical amino acid sequence but is abnormally folded; abnormal prion protein is rich in beta-pleated sheets.
Codon 129



All pathologically confirmed cases of variant CJD have occurred in people who are homozygous for methionine at codon 129 (MM). This has therefore been referred to as the susceptibility genotype, which implies that the other forms confer resistance to infection. In keeping with this idea, mice that are homozygous for valine (VV) are virtually immune to inoculation with abnormal prion protein whereas heterozygotes (MV) show intermediate susceptibility. However, since the incubation period for prion diseases is sometimes measured in decades, the possibility remains that new cases of variant CJD will eventually emerge in people who do not have the susceptibility genotype.
Conversion of PrPc to PrPSc
Conversion models
There are two main models that attempt to describe how cellular prion protein may be converted to the abnormal form. The first is the template-directed refolding (heterodimer) model (Fig. 8.19). This suggests that abnormal prion protein is able to recruit the cellular form and form a heterodimer with it, acting as a template to catalyse its conversion to the scrapie form. The converted prion can then recruit more cellular protein or polymerise to form amyloid fibrils.
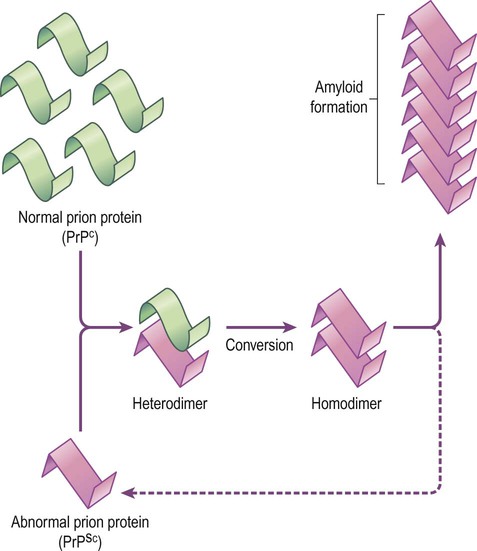
One possible mechanism of prion formation [illustrated here] is called template-directed refolding. The abnormal prion protein recruits normal (cellular) prion protein and induces a conformation change, converting it to abnormal prion protein. The abnormal protein is rich in beta pleated sheets which can stack to form amyloid.




