CHAPTER 30 Cellular Adhesion
All cells interact with molecules in their environment, in many cases relying on cell surface adhesion proteins to bind these molecules. Multicellular organisms are particularly dependent on adhesion of cells to each other and the extracellular matrix (ECM). During development, carefully regulated genetic programs specify cell-cell and cell-matrix interactions that determine the architecture of each tissue and organ. Some adhesive interactions are stable. Muscle cells must adhere firmly to each other and to the connective tissue of tendons to transmit force to the skeleton (see Chapter 39). Skin cells must also bind tightly to each other and the underlying connective tissue to resist abrasion (see Fig. 35-6). On the other hand, many cellular interactions are transient and delicate. At sites of inflammation, leukocytes bind transiently to endothelial cells lining small blood vessels and then use transient interactions with the ECM to migrate through connective tissue (look ahead to Figs. 30-13 and 30-14).
Cells use a relatively small repertoire of adhesion mechanisms to interact with matrix molecules and each other. This conceptual breakthrough came when comparisons of amino acid sequences showed that most adhesion proteins fall into five large families (Fig. 30-1). Within each of these distinctive families, ancestral genes duplicated and diverged during evolution, giving rise to adhesion proteins with the many different specificities that are required for embryonic development, maintenance of organ structure, and migrations of cells of our defense systems. Common properties within each family allowed the appreciation of general mechanisms to emerge from characterizing a few examples. Several important adhesion proteins fall outside the five major families, and additional families may emerge from continued research.
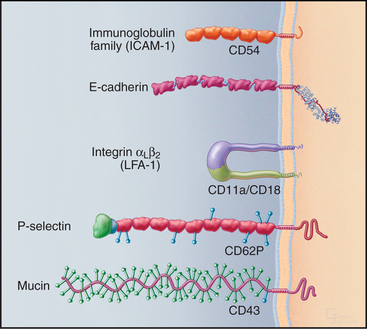
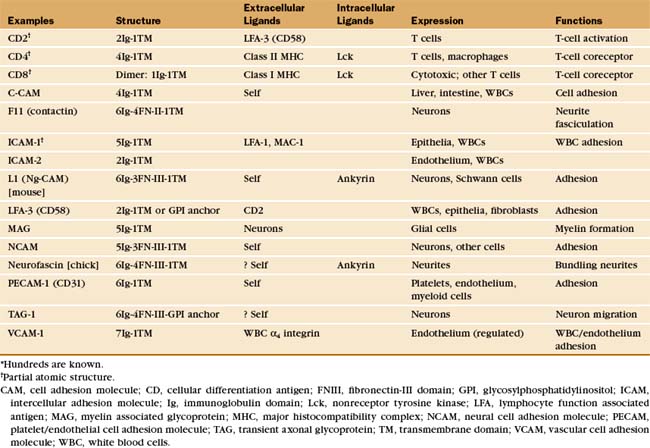
Many adhesion proteins were named before they were classified into families. Tables 30-1 through 30-5 are designed to help the reader with the nomenclature. Many adhesion proteins are named “CD” followed by a number. This stands for “clusters of differentiation,” a term that is used to classify cell surface antigens recognized by monoclonal antibodies, independent of any knowledge about the structure or function of the antigen. Hence, members of the four major families of adhesion proteins have CD numbers.
This chapter first highlights some general features of adhesion proteins and then introduces four major families: immunoglobulin–cell adhesion molecules (Ig-CAMs), cadherins, integrins, and selectins. While learning about each family, the reader should not lose track of an important point: These receptors rarely act alone. Rather, they usually function as parts of multicomponent systems. Two examples at the end of the chapter illustrate the cooperation that is required for leukocytes to respond to inflammation and for platelets to repair damage to blood vessels. Chapter 31 on intercellular junctions, Chapter 32 on specialized connective tissues, and Chapter 38 on cellular motility provide more examples of cellular adhesion.
General Principles of Cellular Adhesion
Second Principle of Adhesion
Many adhesion proteins bind one main ligand, and many ligands bind a single type of receptor (refer to Tables 30-1 through 30-5). If this one-to-one pairing were the rule, adhesion would be simple indeed. However, many exceptions exist, particularly in the integrin family of receptors (Table 30-3). These receptors generally bind more than one ligand, and some ligands, such as fibronectin, bind more than one integrin. One can generalize about the ligands for the several families of cell adhesion molecules:
Third Principle of Adhesion
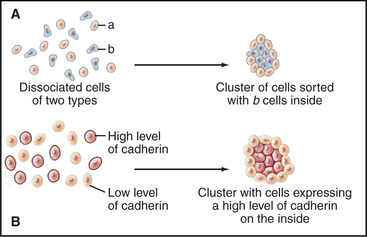
Cells modulate adhesion by controlling the surface density, state of aggregation, and state of activation of their adhesion receptors. Surface density reflects not only the level of synthesis but also the partitioning of adhesion molecules between the plasma membrane and intracellular storage compartments. For example, endothelial cells express P-selectin constitutively but store it internally in membranes of cytoplasmic vesicles. When inflammatory cytokines activate endothelial cells, these vesicles fuse with the plasma membrane, exposing P-selectin on the cell surface, where it binds white blood cells (Fig. 30-13). The importance of surface density is illustrated by an experiment in which cells that express different levels of the same cadherin are mixed together. Over time, they sort out from each other, the more adherent cells forming a cluster surrounded by the less adherent cells (Fig. 30-2). Such differential expression of cadherin determines the position of the oocyte in Drosophila egg follicles. Intracellular signals control the extracellular binding activity of integrins and cadherins. A variety of extracellular stimuli activate intracellular signaling pathways in lymphocytes, platelets, and other cells, which enhance or inhibit the ligand-binding activity of integrins already located on the cell surface. Integrin activation also regulates cellular interactions during development.
Fourth Principle of Adhesion
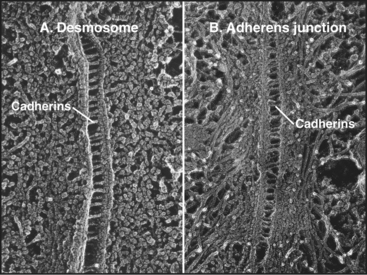
The rates of ligand binding and dissociation are important determinants of cellular adhesion. Many cell surface adhesion proteins (including members of the Ig-CAM, cadherin, integrin, and selectin families) bind their ligands weakly in comparison with other specific macromolecular interactions, such as the interaction of antigens and antibodies, hormones and receptors, or transcription factors and DNA. The measured dissociation equilibrium constants for these adhesion receptors are in the range of 1 to 100 mM, reflecting high rate constants (>1 s−1) for dissociation of ligand. In some cases, this makes good biological sense. Rapidly reversible interactions allow white blood cells to roll along the endothelium of blood vessels (Fig. 30-13). Transient adhesion also allows fibroblasts to migrate through connective tissue. On the other hand, the interactions of cells in epithelia and muscle appear to be more stable, perhaps owing to multiple weak interactions between clustered adhesion proteins cooperating to stabilize adherens junctions and desmosomes (Fig. 31-7). The combined strength of these bonds is said to increase the “avidity” of the interaction.
Identification and Characterization of Adhesion Receptors
Insights about the functions of adhesion receptors have usually come in several steps. Localization of a protein on specific cells frequently provides the first clues. Typically, the expression of each protein is restricted to a subset of cells or to a specific time during embryonic development or both. Next, investigators use specific antibodies to test for the participation of the adhesion protein in cellular interactions in vitro or in tissues. Blistering skin diseases called pemphigus illustrate the serious consequences when pathological autoantibodies disrupt adhesion between skin cells expressing the antigen (see the sections “Desmosomes” and “Adhesion to the Extracellular Matrix” in Chapter 31). Both human genetic diseases and experimental genetic knockouts in mice and other organisms produce defects caused by the absence of adhesion proteins. In leukocyte adhesion deficiency, white blood cells lack the b2 integrin that is required to bind the endothelial cells that line blood vessels. These defective white blood cells fail to bind to blood vessel walls or to migrate into connective tissue at sites of infection. Similarly, patients with a bleeding disorder called Bernard-Soulier syndrome lack one of the adhesion receptors for von Willebrand factor, a protein that promotes platelet aggregation. Loss of cadherins contributes to the spread of some cancer cells.
Immunoglobulin Family of Cell Adhesion Molecules
The Ig-CAM family contains hundreds of adhesion proteins, each with one to seven extracellular domains, similar to immunoglobulin domains, anchored to the plasma membrane by a single transmembrane helix (Fig. 30-3 and Table 30-1). Crystal structures established the antibody-like fold of the extracellular domains of several Ig-CAMS. These compact Ig domains consist of 90 to 115 residues folded into seven to nine β-strands in two sheets, usually stabilized by an intramolecular disulfide bond. The N- and C-termini are at opposite ends of these domains, allowing the formation of linear arrays of immunoglobulin domains.
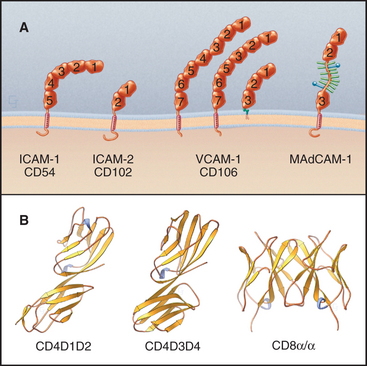
Some Ig-CAMs consist of a single polypeptide, but others are multimeric, with two (CD8) or four (see Fig. 27-8 for the T-cell receptor) subunits. Some nervous system Ig-CAMs have three or four fibronectin III (FN-III) domains between the immunoglobulin domains and the membrane anchor. The C-terminal cytoplasmic tails of these receptors vary in sequence and binding sites. The cytoplasmic domains of the lymphocyte accessory receptors CD4 and CD8 bind protein tyrosine kinases required for cellular activation (see Fig. 27-8). The cytoplasmic domains of neuronal Ig-CAMs bind PDZ domain proteins or membrane skeleton (see Fig. 7-10).
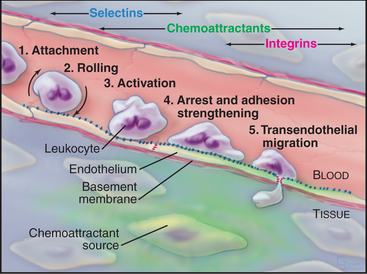
Differentiated metazoan cells express Ig-CAMs selectively, especially during embryonic development, when they may contribute to the specificity of cellular interactions required to form the organs. Neurons and glial cells express specific Ig-CAMs that guide the growth of neurites, mediate synapse formation and promote the formation of myelin sheaths. In adults, interaction of endothelial cell ICAM-1 with a white blood cell integrin is essential for adhesion and movement of the leukocytes into the connective tissue at sites of inflammation (Fig. 30-13).
Like other cell adhesion proteins, Ig-CAMs participate in signaling processes. Best understood are interactions of lymphocytes with antigen-presenting cells during immune responses. Ig-CAMs reinforce the interaction of antigen-specific T-cell receptors with major histocompatibility complex molecules carrying appropriate antigens on other cells (see Fig. 27-8). Although individual interactions are weak, the combination of specific (T-cell receptor) and nonspecific (CD2 and CD4) interactions with the target cell is sufficient to initiate signaling.
Cadherin Family of Adhesion Receptors
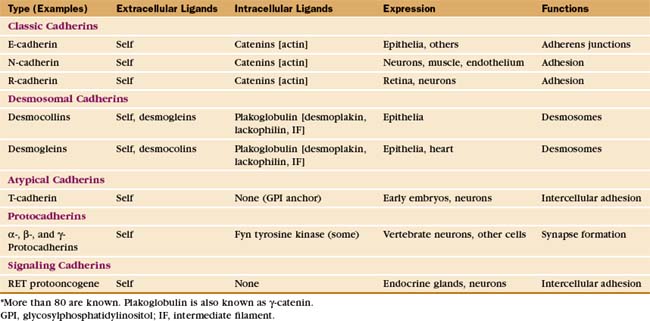
The complex architecture of organs in vertebrates depends on Ca2+-dependent associations between the cells mediated by more than 80 cadherins (Table 30-2). Their name derives from “calcium-dependent adhesion” protein. Genes for cadherin domains appeared in unicellular precursors of sponges, an early step toward the evolution of metazoan organisms.
Cadherins generally interact with like cadherins on the surfaces of other cells in a calcium-dependent fashion, but research is uncovering a growing list of examples of heterophilic interactions. Homophilic interactions of cadherins link epithelial and muscle cells to their neighbors, especially at specialized adhesive junctions called adherens junctions and desmosomes (Fig. 30-4; also see Fig. 31-7). The cytoplasmic domains of cadherins interact with actin filaments or intermediate filaments to reinforce these junctions and maintain the physical integrity of tissues. Contacts mediated by cadherins also influence cellular growth and migration, including suppression of growth and invasion of tu-mors, as well as formation of synapses in the nervous system.
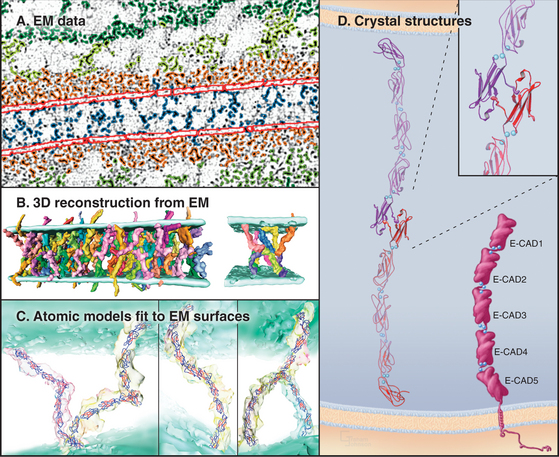
The structural hallmark of the cadherin family is the CAD domain (Figs. 30-5 and 30-6). CAD domains consist of about 110 residues folded into a sandwich of seven β-strands. This fold is similar to immunoglobulin and FN-III domains, but the limited sequence homology suggests independent origins and convergent evolution. N- and C-termini are on opposite ends of CAD domains. Ca2+ bound to three sites between adjacent CAD domains links them together into rigid rods. Without Ca2+, the domains rotate freely around their linker peptides.
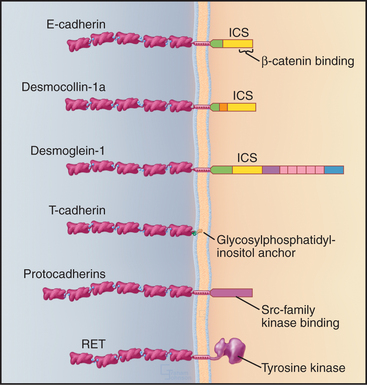
Many cadherins have five extracellular CAD domains. A single α-helix links classic cadherins and desmosomal cadherins to the plasma membrane, but T-cadherin has a glycosylphosphatidylinositol (GPI) anchor (see Fig. 7-9). Cytoplasmic domains vary in size, sequence, and binding sites for associated proteins. The proto-oncogene RET is a cadherin with a cytoplasmic tyrosine kinase domain.
Crystals of cadherins revealed how N-terminal CAD1 domains interact (Fig. 30-6A). A flexible strand located at the N-terminus of each CAD1 domain interacts with the CAD1 domain of its partner. A conserved tryptophan fits into a hydrophobic pocket of the partner CAD1 domain, forming the reciprocal interactions that link the partners together head to head. In crystals of C-cadherin, the CAD1 domains are antiparallel, suitable for a “trans-interaction” with a partner on another cell. In crystals of N-cadherin, this same exchange of N-terminal strands links parallel cadherins suitable for a “cis-interaction” with a cadherin on the same membrane. Three-dimensional reconstructions of electron micrographs of desmosomes show trans- and cis-interactions (Fig. 30-6B). Cadherins are synthesized with a small domain before the interaction strand, which must be removed by proteolysis to allow binding to another cadherin.
Cytoplasmic associations of cadherins with the cytoskeleton and adapter proteins contribute to adhesion by stabilizing the physical links between cells (Fig. 30-6C). The cytoplasmic tails of classic cadherins bind along the entire length of the adapter protein β-catenin (catenin is “link” in Greek), a long, twisted coil of 36 short α-helices (see Fig. 7-9F). α-Catenin binds both β-catenin and actin filaments, but these interactions appear to be mutually exclusive, so other proteins must help to link cadherins to actin. The more complicated cytoplasmic domains of desmosomal cadherins (desmocollins and desmogleins) interact with γ-catenin (a relative of β-catenin called plakoglobin) and desmoplakin. Desmoplakin links these cadherins to keratin intermediate filaments (see Fig. 31-7). The tails of some cadherins interact with formins, proteins that nucleate and elongate actin filaments (see Fig. 33-12).
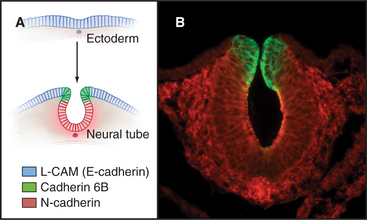
Differential expression and regulation of cadherins help to guide organ formation during embryonic development (Fig. 30-7). Cells with matching cadherins bind together and exclude cells that do not share those cadherins (or other appropriate adhesion receptors), although the mechanism is more complicated than differential affinities of cadherins for each other. For example, cadherins can be activated or inactivated from inside the cell by signaling pathways that are responsive to growth factors or other adhesion proteins. In other situations, Ig-CAMs facilitate the assembly of cadherins in adhesive junctions. In addition to helping with the mechanical sorting of embryonic cells, cadherins produce signals that influence cellular proliferation and differentiation. All cells of early embryos express several different cadherins, but as soon as the embryo forms three germ layers, the ectoderm on the outside surface expresses E-cadherin. In its absence, embryos die. Subsequently, when ectoderm folds inward to form the neural tube, the cells switch to expressing N-cadherin. Later in development, cells in specialized organs typically express characteristic cadherins, such as those in osteoblasts (OB-cadherin), kidney (K-cadherin), and muscle (M-cadherin). A giant-sized cadherin appears to form links between sensory stereocilia on the hair cells in the inner ear. These tip links pull open ion channels when the stereocilia move in response to sound waves.
Cadherins and catenins also participate in transduction of extracellular signals that control cell proliferation, migration, and differentiation. Cadherins contribute to the signal for “contact inhibition” of growth and motility produced when epithelial cells interact. Within a few seconds after epithelial cells contact each other, adherens junctions form. Signals originating from cadherins suppress proliferation of normal cells and inhibit the spread of cancer cells that arise due to somatic mutations. Loss of E-cadherin can contribute to the transition from benign to invasive malignant tumors. Expression of E-cadherin can correct this adhesion defect in tissue culture cells. Genetic defects in E-cadherin predispose people to stomach cancer. The oncogenic tyrosine kinase Src (see Box 27-1) phosphorylates both E-cadherin and β-catenin. This is associated with loss of adhesion of epithelial cells, suggesting one way in which transformation might alter cellular adhesion.
The RET proto-oncogene signals through its cytoplasmic tyrosine kinase domain (Fig. 30-5). Point mutations in the segment between the CAD domains and the plasma membrane or tyrosine kinase of RET cause dominantly inherited cancers of endocrine glands. These mutations cause constitutive dimerization of the receptor or activation of the tyrosine kinase or both, leading to neoplastic transformation. On the other hand, mutations that disable RET cause Hirschsprung’s disease. Autonomic nerves in the wall of the intestines fail to develop, causing severe dysfunction.
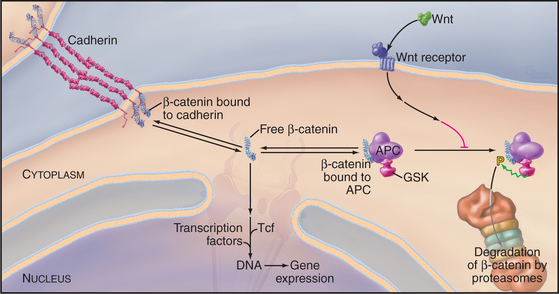
β-Catenin participates in a signal transduction pathway that regulates gene expression during embryonic development (Fig. 30-8). The pathway was discovered in Drosophila as part of the mechanism that determines the polarity of segments in early embryos. Vertebrates have a similar pathway. Most β-catenin is bound to cadherins, but a second pool exchanges between a cytoplasmic protein complex and the nucleus. Nuclear β-catenin recruits transcription factors to regulate the expression of genes that regulate cellular proliferation and tissue differentiation. In resting cells, cytoplasmic β-catenin turns over rapidly, so little enters the nucleus. Degradation is controlled by a cytoplasmic complex that includes the product of the APC gene (defective in patients with familial adenomatous polyposis coli, giving rise to multiple precancerous polyps in the large intestine) and glycogen synthase kinase (GSK), a protein kinase that phosphorylates β-catenin. Phosphorylated β-catenin is ubiquinated and degraded by proteasomes. Loss of APC or mutations in the phosphorylation site on β-catenin result in excess free β-catenin that enters the nucleus and stimulates proliferation. A family of extracellular signaling proteins (19 in humans) called Wnts (from the original Drosophila gene Wingless and the mouse proto-oncogene Int-1) activate the β-catenin gene expression pathway. Wnts bind to a large extracellular domain of seven-helix receptors and another class of receptors in the plasma membrane. Several steps downstream in an incompletely characterized pathway, the Wnt signal inhibits GSK. Inhibition of GSK stops β-catenin proteolysis and raises the concentration of β-catenin that is free to enter the nucleus. Stem cell proliferation is one of many developmental events influenced by Wnt signaling and adhesion by cadherins (see Box 41-1).
Integrin Family of Adhesion Receptors
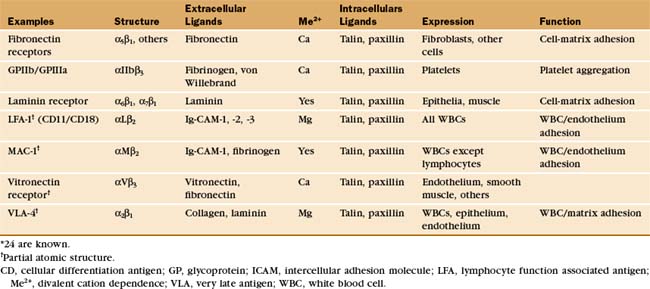
Integrins are the main cellular receptors for the ECM (Table 30-3). Certain integrins bind adhesion molecules on other cells or protein growth factors. These interactions generate signals that control cell growth and structure. Fibroblasts and white blood cells use integrins to adhere to fibronectin and collagen as they move through the ECM. Integrins bind epithelial and muscle cells to laminin in the basal lamina, providing the physical attachments necessary to transmit internal forces to the matrix and to resist external forces. When defects in small blood vessels need repair, integrins allow platelets to adhere to basement membrane collagen and to each other via plasma fibrinogen. Mouse sperm bind integrins on the egg membrane during fertilization. Other integrins cooperate with adhesion receptors of the Ig-CAM, mucin, and selectin families to facilitate the adhesion of white blood cells to endothelial cells at sites of inflammation. Some cells supplement integrins with structurally distinct matrix adhesion proteins, such as muscle dystroglycans and platelet GPIb-IX-V. Together, these interactions are essential for tissue development and integrity in multicellular organisms. Genetic losses of integrin function result in several human diseases.
Integrins tend to be more promiscuous than most adhesion receptors, as some bind to several protein ligands, and many matrix molecules bind to more than one integrin. For example, fibronectin binds to at least nine different integrins, and both laminin and von Willebrand’s factor bind at least five different integrins. This promiscuity may reflect common motifs. About one third of matrix ligands for integrins involve the sequence motif arginine-glycine-aspartic acid (RGD) or other simple sequences in otherwise unrelated proteins. Multiple integrins with overlapping ligand-binding activity provide cells with diverse pathways to activate different signaling pathways.
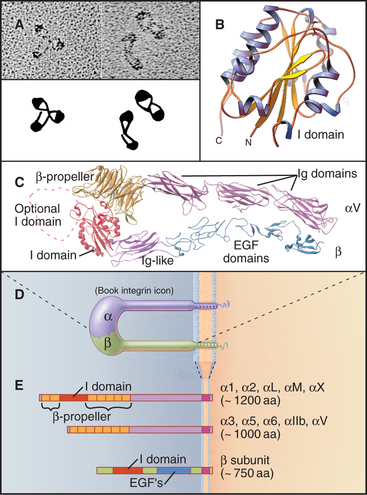
Integrins are heterodimers of two transmembrane polypeptides called α- and β-chains, which both contribute to ligand-binding specificity (Fig. 30-9). Vertebrate cells use a combinatorial strategy to establish their integrin repertoire by selectively expressing a subset of 18 different α-chains and 8 β-chains. These chains combine to form at least 24 different kinds of dimers, each with different ligand-binding specificity. Alternative mRNA splicing (see Fig. 16-6) also adds to the diversity of integrin isoforms.
With the exception of red blood cells, integrins are present in the plasma membranes of most animal cells, including sponges and corals from phyla that branched early in evolution (see Fig. 2-9). Many vertebrate cells express b1 and b3 integrins for adhesion to the ECM. Only white blood cells express b2 integrins, which they use to bind endothelial cells lining the walls of blood vessels. Only platelets express aIIb integrins, important receptors for soluble adhesive ligands in plasma, such as fibrinogen.
The ligand-binding domains of the α- and β-chains form a globular head connected to the plasma membrane by 16-nm legs (Fig. 30-9). All integrin β-chains and a subset of integrin α-chains have an I-domain (inserted domain) with a bound divalent cation that interacts with acidic residues of ligands. All α-chains have an N-terminal β-propeller domain similar to a Gb subunit of a trimeric G protein (see Fig. 25-9). Interaction of the α-chain propeller domain with the β-chain I-domain holds the integrin dimer together, remark-ably like the interaction between Ga and Gb subunits of trimeric G-proteins. Ligands bind to both the I-domains and the β-propeller. Single transmembrane segments anchor both integrin chains to the cell. Short (a ≥ 77 residues; b = 40 to 60 residues, except b4 = 1000 residues) C-terminal cytoplasmic tails contribute to efficient heterodimer assembly. Tail se-quences of homologous chains are conserved between species, and several have important roles in signal trans-duction.
Both α- and β-chains participate in binding at least two sites on ligands. Integrin a5b1 binds two sites on fibronectin: an RGD sequence on a surface loop of FN-III domain 10 and a secondary site on the adjacent FN-III domain 9 (see Fig. 29-15). Neither site is sufficient for binding, so simple RGD peptides can dissociate fibronectin. Integrin binding sites of some ligands are on separate polypeptide chains. The RDD binding site for integrin a1b1 is on three different polypeptide chains of the type IV collagen triple helix.
Ligands on both sides of the plasma membrane influence the conformations of integrins (Fig. 30-10). The open state has the highest affinity for extracellular ligands, with the head held above the membrane by extended, widely placed legs. The closed state has a low affinity for extracellular ligands, with the head bent over on closely spaced legs. Binding of extracellular ligands stabilizes the open state, and the wide spacing of the cytoplasmic domains presumably influences the activities of signal transduction proteins associated with the cytoplasmic domains. Operating in the opposite direction, “inside-out signals” can influence the affinity of integrins for extracellular ligands by favoring the open state.
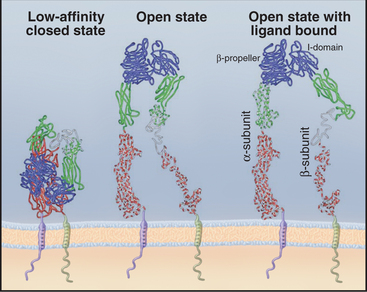
Even in the open state, integrins generally have a low affinity for extracellular ligands. For example, the micromolar Kd for integrin a5b1 binding fibronectin results in rapid association and dissociation, allowing cells to adjust their grip on fibronectin in the matrix as they move through connective tissue. Nonadhesive RGD proteins, such as tenascin (see Fig. 29-17), may modulate these interactions by competing with fibronectin and other ligands for binding integrins.
Cytoplasmic tails of integrins interact directly or indirectly with a remarkable variety of signaling and structural proteins (Fig. 30-11). These interactions are best understood at focal contacts, specialized sites where integrins cluster together to transduce transmembrane signals and link actin filaments to the ECM. The adapter proteins talin and vinculin link the cytoplasmic domains of b integrins to actin filaments at the ends of stress fibers. Talin transmits signals that activate integrins from the cytoplasm. Paxillin links integrins to signaling proteins, forming a scaffold for Src family tyrosine kinases (see Fig. 25-3) and focal adhesion kinase (a novel tyrosine kinase lacking SH2 and SH3 domains).
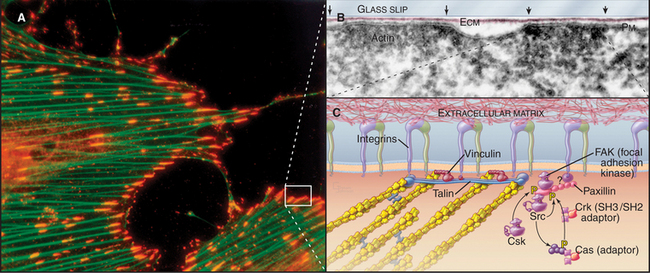
As in other signaling systems (see Chapters 24 and 27), conformational changes (Fig. 30-10) or physical aggregation of integrins may activate focal adhesion kinase and other associated kinases by bringing them close enough together to transphosphorylate each other. Aggregation of integrins by multivalent extracellular ligands or force on the integrins promotes interaction of integrin cytoplasmic domains with the dimeric protein talin. Talin in turn interacts with actin filaments, as well as with multiple signal transduction proteins (Fig. 30-11C). Ligand binding to integrins also activates Rho-family GTPases. Focal adhesion kinase has a central role in transducing these signals. Mouse mutants that lack focal adhesion kinase die during development, but surprisingly, their cells assemble focal contacts with high levels of tyrosine-phosphorylated proteins.
Integrins also participate in the decision of cells to undergo apoptosis, programmed cell death (see Chapter 46). Normal epithelial cells require anchorage to the basal lamina by b4 integrins to grow and divide. When forced to live in suspension or when dissociated from the matrix by RGD peptides, these cells arrest in the G1 phase of the cell cycle (see Chapter 41) and eventually undergo apoptosis. Anchorage by other adhesion proteins will not substitute for integrins. Loss of contact with the basal lamina may contribute to the terminal differentiation and death of cells in the upper levels of stratified epithelia, such as skin (see Figs. 35-6 and 40-1). Epithelial cancers typically lose this integrin-mediated, anchorage dependence for growth, one of the normal limitations on uncontrolled proliferation in inappropriate locations.
Experiments with neutralizing antibodies and competitive peptides provided initial clues about the functions of integrins, but genetic diseases and experimental gene disruptions provide more definitive answers. For example, RGD peptides and integrin antibodies inhibit cell migration and embryonic development by competing with fibronectin. Like null mutations in fibronectin (see Fig. 29-15), homozygous disruption of the integrin a4 or a5 genes is lethal during development. Cells that lack these integrins can form focal contacts in vitro, but fibronectin receptors using other a subunits cannot substitute for a5 in vivo. Dysfunction of b2 integrins is not lethal, but patients are highly susceptible to infections, owing to defects in the emigration of white blood cells from the blood at sites of infection (Fig. 30-13).
Selectin Family of Adhesion Receptors
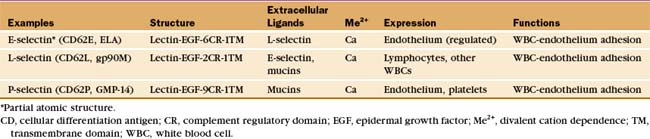
White blood cells and platelets use selectins to interact with vascular endothelial cells. In lymph nodes or at sites of inflammation, selectins snare circulating white blood cells, allowing them to roll over the surface of endothelial cells and eventually to exit the blood (Fig. 30-13). Selectins (Table 30-4) contribute to adhesion in other systems, including the initial binding of early mammalian embryos to the wall of the mother’s uterus.
The defining feature of selectins is a calcium-dependent lectin domain (Fig. 30-12) that binds O-linked sulfated oligosaccharides containing sialic acid and fucose. The lectin domain sits at the end of a rod-shaped projection that is anchored to the plasma membrane by a single transmembrane sequence.
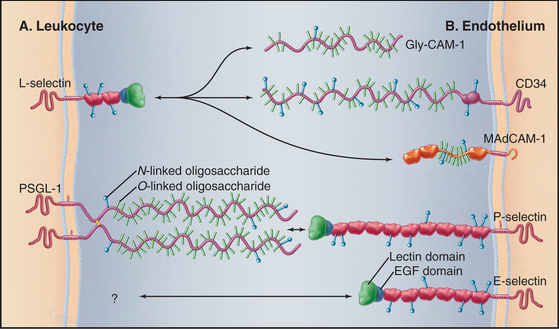
Bonds between selectins and their mucin ligands have high tensile strength (withstanding forces over 100 pN) but form and dissociate rapidly, on a second time scale. Low forces on these bonds prolong their lifetimes modestly, whereas high forces promote dissociation. Consequently, few selectin-mucin bonds are required to tether white blood cells to the endothelium, whereas the brief lifetime of the bonds allows blood flow to propel the cells with a rolling motion over the surface of the endothelium (Fig. 30-13).
Inflammatory mediators regulate selectins in several different ways. Activation of endothelial cells with histamine or platelets with thrombin causes vesicles storing P-selectin to fuse with the plasma membrane, exposing the selectin on the cell surface. Various inflammatory agents stimulate endothelial cells to synthesize E-selectin and P-selectin. Activation of white blood cells increases the affinity of L-selectin for mucins and later leads to its proteolytic release from the cell surface. Furthermore, selectin binding to mucins initiates intracellular signals that result in Ca2+ release inside the cell.
Other Adhesion Receptors
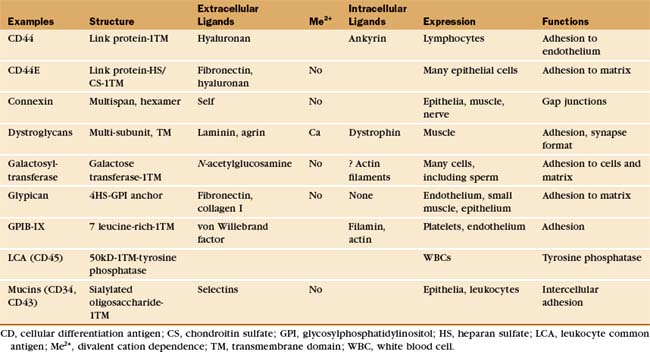
Table 30-5 lists a variety of adhesion receptors that fall outside the four main families. See Chapter 25 for CD45 and Chapter 31 for connexins.
Mucins
The extracellular segments of mucins are rich in serine and threonine, which are heavily modified with acidic oligosaccharide chains (Fig. 30-12). Because of their strong negative charge, these proteins extend like rods up to 50 nm from the cell surface. Mucins on endothelial cells or white blood cells interact with complementary selectins on the other cell type. Endothelial mucin CD34 interacts with white blood cell L-selectin, whereas endothelial P-selectin interacts with white blood cell PSGL-1 mucin. This interaction depends on anchoring of the cytoplasmic domain of PSGL-1 to the actin cytoskeleton. Other mucins are displayed on the surface of or secreted by epithelia lining the respiratory and gastrointestinal tracks.
Galactosyltransferase
One enzyme, galactosyltransferase, is also an adhesion receptor. This enzyme is usually considered in another context: protein glycosylation in the Golgi apparatus (see Chapter 21). However, the messenger RNA (mRNA) for galactosyltransferase has two alternative initiation sites, one of which adds 13 amino acids to the cytoplasmic, N-terminus of this transmembrane protein. The longer enzyme moves to the cell surface rather than being retained in the Golgi apparatus. On the cell surface, the enzyme can bind oligosaccharides that terminate in N-acetylglucosamine. These ligands are found on both cell surface and matrix proteins. The complex of transferase and ligand oligosaccharide is stable, because the galactose-nucleotide substrate added to the oligosaccharide in the Golgi apparatus is not available outside the cell to complete the reaction. During fertilization, a surface galactosyltransferase mediates the initial contact of mouse sperm with the matrix surrounding the egg (called the zona pellucida). This association induces secretion of the contents of the sperm acrosomal vesicle, including an enzyme that destroys the transferase binding site on the ma-trix so that the sperm can proceed through the zona to fuse with the egg. The enzyme is present on the surface of many cells that migrate during embryogenesis and may contribute to their interactions with the matrix.
Adhesion Receptors with Leucine-Rich Repeats (GPIb-IX-V)
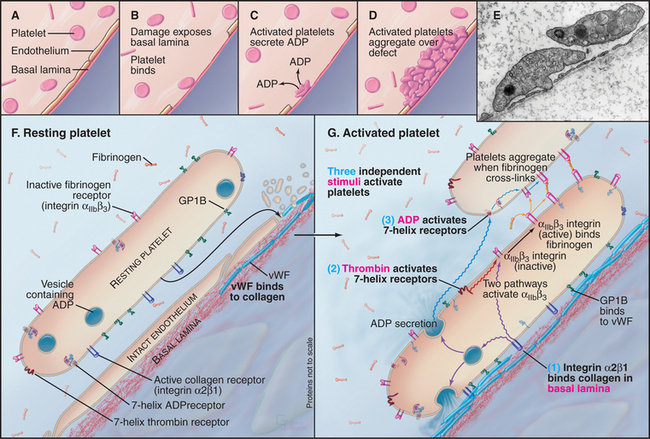
The platelet receptor for the adhesive glycoprotein called von Willebrand factor (Fig. 30-14) is a disulfide-bonded complex of four transmembrane polypeptides: GPIba GPIbb, GPIX, and GPV. Leucine-rich repeats at the end of a long stalk bind von Willebrand factor (see Fig. 24-12 for another example of receptors with leucine-rich repeats). Platelets bind to von Willebrand factor to initiate the repair of damaged blood vessels. This interaction also generates an intracellular signal that enhances affinity of integrin aIIbb3 for fibrinogen and reorganizes the cytoskeleton.
Dystroglycan/Sarcoglycan Complex
In muscles, a complex of transmembrane glycoproteins links a network of dystrophin and actin filaments on the inside of the plasma membrane to two proteins of the extracellular basal lamina, a2 laminin and agrin (see Fig. 39-9 and Table 39-2). These protein associations stabilize the muscle plasma membrane from inside and outside. This muscle membrane skeleton resembles in concept and function the actin-spectrin network of red blood cells (see Fig. 7-10). Genetic defects or deficiencies in dystrophin, transmembrane linker proteins of the dystroglycan/sarcoglycan complex, or a2 laminin cause muscular dystrophy in humans, most likely owing to the mechanical instability of the membrane, leading to cellular damage and eventual atrophy of the muscle. Chapter 39 provides details on their role in muscle function and disease. In other tissues, nonmuscle cells express many of these proteins (or their homologs), where they may contribute to adhesion to the ECM. Some pathogens use the dystroglycan complex to bind their cellular targets. Arenavirus, the cause of Lassa fever, binds directly to α-dystroglycan, and the leprosy bacterium binds laminin-2.
Examples of Dynamic Adhesion
Cellular Adhesion between Leukocytes and Endothelial Cells in Response to Inflammation
Movement of white blood cells from blood to sites of inflammation in connective tissue illustrates how cells integrate the activities of selectins, mucins, integrins, Ig-CAMs, and chemoattractant receptors. Infection or inflammation in connective tissue attracts lymphocytes as well as neutrophils and monocytes, the main phagocytes circulating in blood (see Fig. 28-7).
In the absence of inflammation, neutrophils flow rapidly over the surface of endothelial cells but do not bind to them because the appropriate pairs of adhesion molecules are not exposed or activated or both. Infection or other inflammation in nearby tissues causes neutrophils to bind to the vascular endothelium and to move out of the blood into the tissue. Neutrophils adhere to the endothelium in three sequential but overlapping steps (Fig. 30-13):
A similar mechanism and a partially overlapping set of receptors attract blood monocytes and eosinophils to sites of inflammation. Once they are in connective tissue, interactions of monocyte integrins with matrix molecules trigger the expression of genes required for differentiation into macrophages (see Chapter 28).
Lymphocytes (see Fig. 28-9) patrol the body, circulating from the blood through organs to lymphoid tissues and through the lymphatic circulation back to the blood. This “recirculation” requires lymphocytes to recognize endothelial cells in organs and specific lymphoid tissues where they exit from the blood. Lymphocytes use L-selectin, three different mucin-like proteins, and a4b2 integrins to bind to these target endothelial cells. Lymphocytes from mice that lack L-selectin do not roll on endothelial cells or accumulate in lymph nodes. Antibodies that block a4 integrins mitigate inflammation in the autoimmune disease multiple sclerosis by interfering with movement of lymphocytes into the brain, although side effects have limited their widespread use.
Platelet Activation and Adhesion
Platelets aggregate at sites where damage to vascular endothelial cells exposes the underlying basal lamina (Fig. 30-14). This process requires the coordinated activity of a variety of receptors, including integrins, leucine-rich repeat adhesion proteins, and seven-helix receptors. These reactions prevent bleeding and bruising, but inappropriate activation of platelets produces clots in blood vessels, causing heart attacks and strokes. To understand the good effects and avert the bad, investigators have studied platelet activation and adhesion in great detail.
Damage to the endothelium usually initiates platelet activation by exposing platelets to von Willebrand factor and collagen in the basal lamina. Under conditions of high shear, GPIb-IX-V interacts strongly with von Willebrand factor bound to basal lamina collagen. This interaction transiently tethers platelets to the basal lamina and favors binding of integrin a2b1 to collagen. Exposure to soluble agonists such as ADP or thrombin also activates platelets and promotes their aggregation. Within seconds of activation, platelet aIIbb3 integrins convert to a high-affinity state (Kd < mM) and bind tightly to fibrinogen. Dimeric fibrinogen links platelets into aggregates.
Agonists activate platelet aIIbb3 integrins through three different pathways:
Platelet aggregation is disadvantageous in the normal circulation, so several mechanisms actively inhibit platelet activation. Endothelial cells produce both nitric oxide and an eicosanoid, prostacyclin (PGI2), which inhibit platelet activation (see Fig. 26-9). Nitric oxide acts through cyclic guanosine monophosphate (cGMP), and prostacyclin acts through cyclic adenosine monophosphate (cAMP; see Fig. 26-1). Drugs that inhibit aIIbb3 are being tested to treat heart attacks.
Arnaout MA, Mahalingam B, Xiong J-P. Integrin structure, allostery and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381-410.
Brown EJ, Fraizer WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130-135.
Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility and signaling. Annu Rev Cell Dev Biol. 1996;12:463-519.
Campbell ID, Ginsberg MH. The talin-tail interaction places integrin activation on FERM ground. Trends Biochem Sci. 2004;29:429-435.
Critchley DR. Focal adhesions: The cytoskeletal connection. Curr Opin Cell Biol. 2000;12:133-139.
DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003;15:572-582.
Frank M, Kemler R. Protocadherins. Curr Opin Cell Biol. 2002;14:557-562.
Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev MolCell Biol. 2005;6:622-634.
Harwood A, Coates JC. A prehistory of cell adhesion. Curr Opin Cell Biol. 2004;16:470-476.
Henry MD, Campbell KP. Dystroglycan: An extracellular matrix receptor linked to the cytoskeleton. Curr Opin Cell Biol. 1996;8:625-631.
Kalderon D. Similarities between the Hedgehog and Wnt signaling pathways. Trends Cell Biol. 2002;12:523-531.
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810.
McEver RP. Selectins: Lectins that initiate cell adhesion under flow. Curr Opin Cell Biol. 2002;14:581-586.
Mould AP, Humphries MJ. Regulation of integrin function through conformational complexity: Not simply a knee-jerk reaction? Curr Opin Cell Biol. 2004;16:544-551.
Nayal A, Webb DJ, Horwitz AF. Talin: An emerging focal point of adhesion dynamics. Curr Opin Cell Biol. 2004;16:94-98.
Nelson WJ, Nusse R. Convergence of Wnt, b-catenin and cadherin pathways. Science. 2004;303:1483-1487.
Patel SD, Chen CP, Bahna F, et al. Cadherin-mediated cell-cell adhesion: Sticking together as a family. Curr Opin Struct Biol. 2003;13:690-698.
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-850.
Rosen SD. Ligands for L-selectin: Homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129-156.
Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697-715.
Seto ES, Bellen HJ. The ins and outs of Wingless signaling. Trends Cell Biol. 2004;14:45-53.
Shapiro L, Colman DR. The diversity of cadherins and implications for a synaptic adhesive code in the CNS. Neuron. 1999;23:427-430.
Springer TA. Traffic signals for lymphocyte and leukocyte emigration: The multi-step paradigm. Cell. 1994;76:301-314.
Turner CE. Paxillin and focal adhesion signaling. Nat Cell Biol. 2000;2:E231-E236.
van der Merwe PA, Barclay AN. Transient intercellular adhesion: The importance of weak protein-protein interactions. Trends Cell Biol. 1994;19:354-358.
Vestweber D. Regulation of endothelial cell contacts during leukocyte extravasation. Curr Opin Cell Biol. 2002;14:587-593.
Webb DJ, Brown CM, Horwitz AF. Illuminating adhesion complexes in migrating cells: Moving toward a bright future. Curr Opin Cell Biol. 2003;15:614-620.
Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207-235.
Xiao T, Takagi J, Coller BS, et al. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59-67.