Chapter 4
Cells of the Vascular System
Omaida Velazquez, Bo Wang
The effort to define the cell types in the vascular system is an ongoing process. The dynamic nature of cells makes this a challenging task. Cells of the vascular system constantly change their properties even during the postnatal period. Certain cell types with high plasticity undergo significant phenotypic and functional change in response to external stimuli. Currently, we have been able to define five main cell types that reside in the vascular wall. These are endothelial cells, vascular smooth muscle cells, pericytes, fibroblasts, and vessel-residing stem cells/progenitor cells. Nonnative cell types such as macrophages, neutrophils, and circulating stem cells can also be found in the blood vessel wall. Their migration into the vessel wall from the blood or distal sites is usually a response to different pathophysiologic conditions.
Traditionally, different cell types are defined by means of microscopy and differentiated from one another on the basis of their morphology and location. Modern technology relies more on molecular biological markers, which allow us to appreciate the dynamic nature of defining a certain cell type. Cells of the same type express different cell markers during different stages to reflect the ongoing changes of the intracellular processes. On the other hand, there can be a vast overlap among different cell types as to the cell markers they express, making it more difficult to differentiate them. Despite our confusion and the limits of technology, there is a fast-growing body of knowledge about the molecular pathways by which different cell types interact with one another as well as the signaling involved in inducing certain changes in cell properties. In this chapter, we discuss the five native cell types found in the blood vessels, their interactions with one another, and the roles they play in some pathophysiologic processes.
Endothelial Cells
Endothelial cells (ECs) are the innermost cellular component of the vessels. They form a cellular layer called endothelium that extends continuously throughout the entire vascular system. Throughout the endothelium layer, ECs display vast structural and functional heterogeneity depending on the organ they reside in and the specific function related to that organ.
The endothelial cells are supported by a layer of acellular components called basal lamina. The basal lamina is a specially organized extracellular matrix. The nature of the basal lamina and its components are complex and dynamic in nature. It borders the basolateral surface of the endothelium, providing attachment and support as well as regulating behaviors of the ECs such as migration and cell division. The interaction between the endothelial cells and their acellular environment is highly intricate. In vitro studies have shown that cultures of ECs on different acellular components, including collagens, fibronectins, and reconstructed basement membranes, show different rates of production of gap junctions, actin messenger RNA, and protein, cell proliferation, and membrane specialization.1–3
Endothelial Cells as a Transporting System and Selective Barrier
The endothelial cells are considered the most active part of the vascular system. They come in direct contact with the blood and sense the signals that are delivered through blood flow. These signals can be physical or chemical. In response to these signals, the cells carry out a complex intracellular and intercellular downstream process. Such a response is crucial in processes such as cardiovascular homeostasis, vasomotor regulation, blood cell trafficking, hemostatic balance, permeability, proliferation, survival, and innate and adaptive immunity.4
Endothelial permeability serves an important role in regulating the substance exchange between luminal vessel and extraluminal environment. There are several ways signals or substances can be exchanged—paracellular transport, endocytosis, transcytosis, and fenestration (Fig. 4-1).
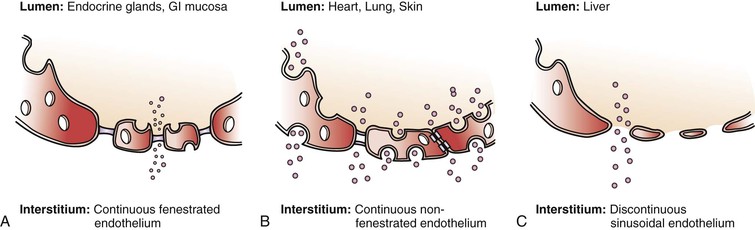
Figure 4-1 Different types of endothelium and permeability. A, Continuous fenestrated endothelium is usually found in secretory tissue, such as endocrine glands, renal glomerulus, and gastrointestinal (GI) mucosa. This type of cell has large fenestrations that are covered by a selective diaphragm, which allow certain particles to pass by fairly easily. Continuous fenestrated endothelium demonstrates greater permeability to water and small solutes than to albumin and larger macromolecules. The diaphragms of the fenestrae act as molecular filters. B, Continuous nonfenestrated endothelium is the most selective type of endothelium. It represents a tight barrier between the lumen and interstitial tissue. It is commonly found in tissue such as heart, skin, and lung. Substances can pass through the endothelium through either formed caveolae or special intracellular channels. In continuous nonfenestrated endothelium, water and small solutes pass between endothelial cells, whereas larger solutes pass through such cells either via transendothelial channels or by transcytosis, the latter process being mediated primarily by caveolae (see Fig. 4-2). C, Discontinuous, or sinusoidal, endothelium can be found inside the liver. Discontinuous endothelium is characterized by fenestrae (without diaphragms), gaps, and poorly organized basement membrane. (Adapted from Boron WF, et al: Medical physiology, Philadelphia, 2002, WB Saunders.)
Paracellular Transport
Molecules can move from blood to extraluminal environment through the paracellular (between cells) space between endothelial cells. This space is regulated by certain types of junctions. Two types of junctions are recognized at this space, tight junctions and adherent junctions.
Adherent junction, or zona adherens, connects the ECs on their sides to form a continuous cellular layer. Tight junction, or zona occludens, forms barriers between the lumen and extraluminal environment by tightly sealing the gaps between adjacent cells. The density of distribution for tight junctions varies in different parts of the vascular system. In large arteries, the tight junctions are well formed to prevent the leaking of luminal content. Toward the distal circulation, the number of tight junction decreases drastically in order to accommodate the function of substance exchange between the vascular beds and the tissues they supply. The brain’s vascular system has particularly rigid tight junctions, which protect neural tissue from fluctuations in blood composition. Adherent junctions are less common and exist primarily in large arteries. Their principal role is to permit inter–endothelial cell communication via movement of ions, metabolites, and regulatory factors.5
Fenestrations
Substances can also directly pass in and out of the ECs through fenestrations, or pores, on the cells. This special pathway is especially developed in secretory organs such as exocrine and endocrine glands, gastric and intestinal mucosa, choroid plexus, glomeruli, and renal tubules. Fenestrae are transcellular pores approximately 70 nm in diameter that extend through the entire thickness of the endothelial layer.4 Fenestrations usually have a thin layer of diaphragm formed by glycoproteins across their opening. This diaphragm provides a selective filter based on size for the fenestrations. As a fenestration gets larger, the endothelium becomes discontinuous. This type of endothelium exists especially in sinusoidal vascular beds, most notably the liver. These fenestrations are about 100 to 200 nm in diameter and have no diaphragm. Their underlying basement membrane is poorly formed (see Fig. 4-1).
Endocytosis
Another important pathway for transduction of information from the luminal space is through endocytosis. ECs possess clathrin-coated pits, clathrin-coated vesicles, multivesicular bodies, and lysosomes, which represent the structural components of the endocytotic pathway.6 The endocytosis can happen either via receptor-dependent or receptor-independent mechanisms. The receptor-dependent pathway is responsible for uptake of low-density lipoprotein (LDL), transferrin, albumin, ceruloplasmin, and advanced glycosylation end products. Liver sinusoidal ECs demonstrate particularly high rates of clathrin-mediated endocytosis.4
Transcytosis
Transcytosis is a means of transcellular transfer of molecules across the endothelium. This process is mediated by caveolae and vesiculo-vascular organelles (VVOs). The number of caveolae is highest in continuous nonfenestrated endothelium, particularly in heart, lung, and skeletal muscle.7 When caveolae are bounded to the cell membrane, it opens toward the luminal or abluminal side. After picking up the target particles, it forms a free vesicle inside the cytoplasm. The particle is transported inside the vesicle and is released when it fuses with the cell membrane on the opposite side of the cell. The density of caveolae is far more prominent in capillaries than other parts of the vascular system, reflecting the highly active molecular trafficking in the microcirculation. VVOs are observed mainly in the venular endothelium. They are aggregations of smaller vesicles. VVOs, which contain caveoline-1, are thought to arise from the fusion of individual caveolae (Fig. 4-2).
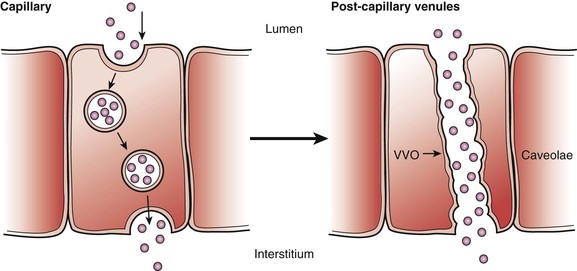
Figure 4-2 Transcytosis transportation of caveolae and vesiculo-vascular organelles (VVOs). Left, Caveolae are the major apparatus for transcytosis. They contain clathrin inside the vesicles that carrier protein for particles. When vesicles fuse with the luminal side of the cell, the clathrin are exposed to the luminal side to pick up molecules; afterwards, the vesicles are reformed, internalizing the molecules from the luminal side. Caveolae are particularly prevalent in capillaries of heart and skeletal muscle and rare in the blood-brain barrier. After being internalized, the molecules move to either lysosomes or endosomes or are released into the abluminal side of the cell. Right, The VVO is another way of transcytosis through endothelium. This form of transport is predominant on the postcapillary venules, which are much larger than caveolae. Many consider venules to be formed by the fusion of many smaller caveolae. Their number can greatly increase in response to inflammation, which result in leaky endothelium that causes loss of water and small molecules to the interstitium. (Adapted from Boron WF, et al: Medical physiology, Philadelphia, 2002, WB Saunders.)
Vasomotor Regulation
The endothelial cell is an important source of vasoactive agents. Each agent has its specific receptor on vascular smooth muscle cells. Binding on these receptors results in downstream molecular signaling, which eventually leads to a change in Ca2+ concentration. The end result is either vasoconstriction or vasodilation. Vasoactive agents can be divided into vasodilators and vasoconstrictors on the basis of their effects (Table 4-1).

Nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), and prostacyclin (prostaglandin I2 [PGI2]) are three vasodilators secreted by endothelial cells. The three vasodilators work in an integrated manner to maintain the health of the vasculature. Each mediator possesses the capacity to interact with components of the synthesis/activation processes for the other mediators.
The distribution of vasodilators in the circulating system varies in the body. NO and PGI2 responses are primarily found in conduit vessels, whereas EDHF predominates in resistance arteries.8,9 Moreover, it has been found that NO tonically inhibits EDHF responses. There is a compensatory increase in EDHF when NO is deficient or inhibited. In this way, the vasolidatory capacity of the vessel is maintained.10,11 PGI2 and NO interact in a similar manner, although this activity is often evident only after inhibition of nitric oxide synthase (NOS).12
Nitric Oxide
Nitric oxide, originally named endothelium-derived relaxing factor, is a potent vasodilator. It is produced by the enzyme NOS. Currently, three isoforms of oxide synthase have been identified on the basis of their location. Endothelial nitric oxide synthase (eNOS or NOS III) is found only in the endothelial cells. It is a constitutive form of NOS that is present all the time (Fig. 4-3).
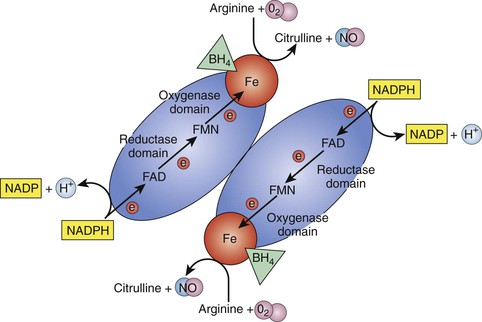
Figure 4-3 Dimer form of endothelial nitric oxide synthase (eNOS). For production of nitric oxide (NO), nitric oxide synthase (NOS) must exist in the dimer form. The monomer-formed molecule has two domains, the reductase domain and the oxygenase domain. Electrons (e−) are donated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) to the reductase domain and transferred through flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and Fe (iron) to the oxygenase domain. With the help of coenzyme tetrahydrobiopterin (BH4), arginine and O2 are catalyzed into NO and citrulline. H, Hydrogen; NADP, nicotinamide adenine dinucleotide phosphate.
The secretion of NO is under the control of several different mechanisms. Neuronal signal acetylcholine, inflammatory signal bradykinin, and increased shear stress can all increase the activity of eNOS. Other factors, such as substance P, thrombin, adenine nucleotides, and [Ca2+] (calcium ion concentration, can also affect eNOS. These molecules all affect the activity of eNOS through a Ca2+-and calmodulin (CaM)–dependent pathway. The molecular signals can trigger the entry of Ca2+, which binds to cytosolic CaM and then stimulates NOS. The activity of eNOS also requires cofactor tetrahydrobiopterin and NADPH (reduced nicotinamide adenine dinucleotide phosphate). Endothelial NOS catalyzes the formation of NO and L-citrulline from L-arginine. It is a lipophilic gas that can diffuse only a few cell lengths during its very short half-life. This feature limits the effect of NO on a local level.13
The receptor for NO inside the vascular smooth muscle cell (VSMC) is a soluble guanyl cyclase. Binding of NO activates the conversion from guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which in turn activates cGMP-dependent protein kinase (PKG). PKG phosphorylates myosin light chain kinase (MLCK), inhibiting its activity. This inhibition leads to a decrease in the activity of myosin light chain (MLC), whose activity depends on MLCK phosphorylation. The result is decreased interaction between myosin and actin, which leads to vasodilation.14,15
There are other pathways by which the NO can exert its effect on the VSMCs. In some blood vessels, cGMP can increase the level of cyclic adenosine monophosphate (cAMP) by preventing its degradation through inhibition of phosphodiesterase activity. An increase in cAMP results in muscle relaxation. NO may also induce relaxation by direct activation of Ca2+-dependent potassium (K+) channels in VSMCs.16 In addition, NO can function as a direct antagonist for the vasoconstrictors, including catecholamines, angiotensin II, and endothelin-1 (ET-1).17
In 1998, Furchgott Ignarro, and Murad shared the Nobel Prize for Physiology for their discovery of the NO signaling pathway. Today in medicine, NO donor drugs play an unparalleled role in the management of coronary artery disease and hypertensive emergency.
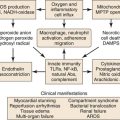
NO has commonly been regarded as a vessel-protective agent since its discovery; however, over the past decade, it has emerged as a fundamental signaling molecule that regulates virtually every critical cellular function and as a potent mediator of cellular damage in a wide range of conditions. We have introduced here the important functions of NO in protecting the vessel’s health, including vasodilation, antithrombosis, and anti-inflammation; however, the “double-edged sword” nature of NO is increasingly emphasized in recent literature.18,19 An important mediator for NO toxicity is peroxynitrite. NO is an antioxidant and a salvager of reactive oxygen species (ROS). The two substances, NO and ROS, form peroxynitrite when they meet. Peroxynitrite interacts with lipids, DNA, and proteins via either direct oxidative reactions or indirect radical-mediated mechanisms. These reactions trigger cellular responses ranging from subtle modulations of cell signaling to overwhelming oxidative injury, committing cells to necrosis or apoptosis. In vivo, peroxynitrite generation represents a crucial pathogenic mechanism in conditions such as stroke, myocardial infarction, chronic heart failure, diabetes, circulatory shock, chronic inflammatory diseases, cancer, and neurodegenerative disorders.18
NADPH oxidases such as superoxide anion are major sources of ROS in the vessel wall. The short-lived superoxide anion can rapidly form hydrogen peroxide. For the NOS enzyme to function normally, it must remain in a dimer form. Hydrogen peroxide can disrupt this dimer form.20 When NOS becomes uncoupled, it becomes a major producer of superoxide anion. To reverse back to dimer form, this process of reduction relies on an important cofactor, tetrahydrobiopterin (BH4). Under certain conditions, such as ischemic insult, the production of ROS overwhelms the NOS-BH4 reduction system.21
Growing evidence has shown that NO has an important role in modulating angiogenesis, especially in neovascularization developing in vivo in response to ischemia.22–24 This role is specifically restricted to eNOS-derived NO. NO derived from eNOS is involved in mobilizing endothelial progenitor cells (EPCs) to the site of injury.23,24 This function is impaired in diabetic patients. In such patients, eNOS is downregulated and wound healing potential is impaired as a result of decreased EPC mobilization from bone marrow. It has been found that this adverse condition can be reversed by hyperoxic conditions, providing evidence of hyperbaric O2 healing potential on a cell molecular level.24
It is believed that although sustained low concentrations of NO are mainly vasoprotective and excessive cytotoxic formation of NO (peroxynitrite) mainly results from inducible NOS (iNOS) and neuronal nitric oxide synthase (nNOS). White cell adherence is an early event in the development of atherosclerosis; therefore, NO may protect against the onset of atherogenesis. Furthermore, NO has been shown to inhibit DNA synthesis, mitogenesis, and proliferation of VSMCs. The inhibition of platelet aggregation and adhesion protects smooth muscle from exposure to platelet-derived growth factor(s). Therefore, NO also prevents a later step in atherogenesis, fibrous plaque formation. On the basis of the combination of those effects, endothelial NO probably represents the most important antiatherogenic defense principle in the vasculature. Many risk factors, such as diabetes and smoking, can lead to excess production of superoxide and diminished NO, thus damaging the endothelium.19
Endothelium-Derived Hyperpolarizing Factor NO, PGI2, and EDHF are the three native vasodilators that have been identified. Their effect on smooth muscle converges on causing hyperpolarization of the smooth muscle cell. It is believed that endothelium-dependent relaxation of vascular smooth muscle is a result of endothelial cell hyperpolarization.10–12,25–35
Endothelial cell hyperpolarization occurs as a result of influx of extracellular K+ through the opening of calcium-activated potassium channels in response to the rise in intracellular calcium. This endothelial hyperpolarization is transmitted to the smooth muscle through the gap junctions (ion channels) or the flow of K+ from the endothelial cell into the myoendothelial space. Smooth muscle hyperpolarization is then induced by the activation of rectifying potassium channels (KIR) or Na+/K+ adenosine triphosphatase (ATPase).25–29
EDHF is released by endothelial cells under the stimulation of acetylcholine, bradykinin, substance P, and increased shear stress. EDHF can cause hyperpolarization of VSMCs through three different pathways. First, it is able to diffuse across endothelial cells and activate calcium-activated potassium channels of VSMCs to promote K+ efflux. Second, EDHF can act in an autocrine manner to facilitate the activation of KCa channels on endothelial cells to cause endothelial hyperpolarization. Last, EDHF can enhance gap junction communication and increase the transmission of endothelial hyperpolarization to VSMCs25–30 (Fig. 4-4).
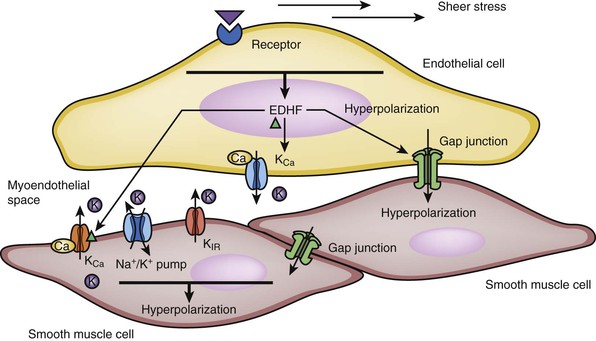
Figure 4-4 Hyperpolarization of smooth muscle cells induced by endothelium-derived hyperpolarizing factor (EDHF). EDHF causes smooth muscle cell relaxation by inducing hyperpolarization in the cell. The EDHF is released by stimulation of the acetylcholine, bradykinin, substance P receptor on the endothelial surface. Mechanical stress from blood flow can also lead to the release of EDHF. There are several mechanisms by which EDHF can cause hyperpolarization of smooth muscle cells. First, EDHF can diffuse through endothelial cells and bind the calcium-activated potassium channels of vascular smooth muscle cells (VSMCs), leading to potassium efflux. Second, endothelial cell hyperpolarization occurs as a result of efflux of extracellular K+ through the opening of calcium-activated potassium channels in response to the rise in intracellular calcium. This endothelial hyperpolarization can be transmitted to the smooth muscle cells through the gap junctions. This process in turn induces smooth muscle hyperpolarization via the activation of rectifying potassium channel (KIR) or Na+/K+ pump (adenosine triphosphatase [ATPase]). Third, pumping of K+ into the myoendothelial space can also directly lead to the hyperpolarization of VSMCs. KCa , calcium-sensitive potassium channel.
EDHF has been identified in coronary, peripheral, skin and venous vessels. It is defined as a group of molecules that carry out their vasodilatory function in the same manner rather than as a single type of molecule. This group of molecules includes hydrogen peroxide, epoxyeicosatrienoic acids (EEAs), anandamide, and C-natriuretic peptide.9
Prostacyclin
PGI2 is a product of arachidonic acid catalyzed by cyclooxygenase (COX). It is a potent vasodilator in arteries and vein, as well as an important platelet aggregation inhibitor. COX-1 is the constitutive form of the enzyme expressed in vascular endothelium that is thought to contribute to the maintenance of vascular homeostasis. COX-3, on the other hand, is an inducible form that is thought to be related to pathogenesis.31
PGI2 binds to specific receptors on smooth muscle cells. These are G protein–coupled adenyl cyclases that can increase cAMP levels, which in turn activate cAMP-dependent protein kinase (PKA). Like PKG, PKA phosphorylates MLCK, inhibiting MLCK activity, decreasing the interaction between myosin and actin, and leading to vasodilation.32 This mechanism of vasodilation is seen in NO-induced VSMC relaxation, as previously described. The difference is that NO activity depends on the activation of guanyl cyclase and cGMP rather than adenyl cyclase and cAMP.
Endothelin-1
ET-1 is the most potent vasoconstrictor. It is also the major isoform of endothelin found in humans. It is produced by ECs at a low physiologic level and inhibited by NO, PGI2, heparin, natriuretic peptides, and high levels of shear stress.33 It is converted from the pro-polypeptides by the membrane-bound ET-converting enzyme. ET-1 carries out its effects through two types of membrane–G protein–coupled receptors, ETA and ETB.34 The ET receptors can be found on VSMCs, adventitial fibroblasts, and endothelial cells. They are also expressed in kidney, liver, lung, and skin and are involved in a wide range of biological activities.
ET-1 binding to ETA receptors on VSMCs leads to cellular contraction. Abnormal vasoconstriction, vasospasm, and vascular hypertrophy all share the feature of abnormal ET receptor expression.35 In recent years, ET-1 has been identified as a major factor in endothelial dysfunction of various cardiovascular diseases, including coronary artery disease, peripheral artery disease, and stroke due to hypertension. Elevated blood and tissue levels of ET-1 have been identified in relation to the pathology of these diseases.36–39
At low concentrations, ET-1 can induces a paradoxical vasodilatation through activation of endothelial ETB receptors coupled with the release of NO, PGI2, and EDHF. Activation of ETB receptor–coupled G proteins results in an increase in phospholipase C (PLC) activity, generation of inositol 1,4,5-triphosphate (IP3) and diacylglycerol, and eventual release of Ca2+ from intracellular storage. This causes an increase in activity of the Ca2+/calmodulin-dependent NOS enzyme, which allows liberated NO to diffuse into the SMC layer, initiate production of cGMP, and develop the cellular relaxation response. ETA receptors are coupled to Gq/11, G12/13, and Gi heterotrimer G protein subunits, which link to inhibition of phospholipase C, RhoA–guanosine triphosphatase, and adenylyl cyclase, respectively.35
Endothelium-Derived Constricting Factor
Endothelium-derived constricting factor is a poorly defined term. Most literature uses it as a common name for the group of peptides that are produced by endothelium and can induce vasoconstriction.40 Endothelin-1 is the best-described member of the group. A few other peptides have been defined as members of the group. COX appears to be at least partially involved in their activities40,41 Superoxide anions and endoperoxides have been included as members of this group.41
Angiotensin II
Angiotensin II (Ang II) is a product of the renin-angiotensin-aldosterone axis. It is produced mainly in the lung and kidney endothelial cells as a systemic response to low blood osmolality and low pressure that is initiated in the kidney. It is produced by angiotensin-converting enzyme (ACE) in the endothelium as a product of angiotensin I. Angiotensin I is a product of angiotensinogen, which is produced in the liver. The process is catalyzed by the kidney-secreted enzyme renin. Angiotensin II is a potent vasoconstrictor for the vascular system. However, its special role in the regulation of renal perfusion has always been the main emphasis. Its effect on renal perfusion is mediated through the regulation of arteriole smooth muscle tone. Under high concentrations of angiotensin II, the efferent arterioles constrict more than the afferent arterioles, retaining the salt in circulation and increasing the colloid osmotic pressure in the blood. On the other hand, angiotensin II is also able to interact with specific receptors in the kidney to increase the uptake of Na+ and stimulate the thirst sensation in the hypothalamus. Finally, it stimulates the production of aldosterone in the adrenal cortex, which also promotes Na+ reabsorption in the collecting tubules and ducts.
The exact mechanism of how angiotensin II causes vascular constriction is still under study. It has been determined that the AT1B receptor, a subtype of angiotensin II type 1 (AT1) receptors, predominantly mediates contractions induced by angiotensin II.42 Binding of AT1 receptors leads to activation of the G protein Gq, which in turn activates phospholipase C, which hydrolyzes phosphatidylinositol-4,5-bisphosphate to generate inositol-1,4,5-trisphosphate and diacylglycerol. Inositol-1,4,5-trisphosphate activates the intracellular release of Ca2+, and extracellular Ca2+ can also enter the cell through Ca2+ channels located on the cell membranes. Ca2+-calmodulin–dependent MLCK can turn on MLC phosphorylation and vasoconstriction. Angiotensin II also activates phospholipase D, which converts phosphatidylcholine into choline and phosphatidic acid; the latter is converted to diacylglycerol rapidly. This pathway is thought to play a major role in the activation of protein kinase C in the sustained phase of angiotensin II–induced contraction.43
Hemostasis versus Antithrombosis/Fibrinolysis
The endothelial cells produce both anticoagulant factors and procoagulant factors. This makes them crucial in maintaining the balance between the two processes. The anticoagulant factors ECs produce include tissue factor pathway inhibitor (TFPI), heparin, thrombomodulin (TM), endothelial protein C receptor (EPCR), tissue-type plasminogen activator (t-PA), ecto-adenosine diphosphatase (ADPase), prostacyclin, and NO. The procoagulant, factors are plasminogen activator inhibitor-1 (PAI-1), von Willebrand factor (vWF), and protease activated receptors.4
Other than endothelium-derived factor, the coagulation cascade involves circulating platelet from bone marrow as well as clotting factors, fibrinogen, and the anticoagulants proteins C and S and antithrombin III, which are produced by the liver. These factors are been produced at a fairly constant rate and distributed evenly throughout the circulation. On the other hand, the production of endothelium-derived factors can vary among different vascular beds. This feature allows them to create a site-specific coagulating environment. For example, thrombomodulin is highly expressed in all vessel types and calibers in all organs, whereas EPCR is expressed predominantly in large arteries. Although von Willebrand factor is predominantly found on the venous side of the circulation, t-PA expression in the endothelium is restricted to arteries of the pulmonary system and central nervous system44–46 (Figs. 4-5 and 4-6).
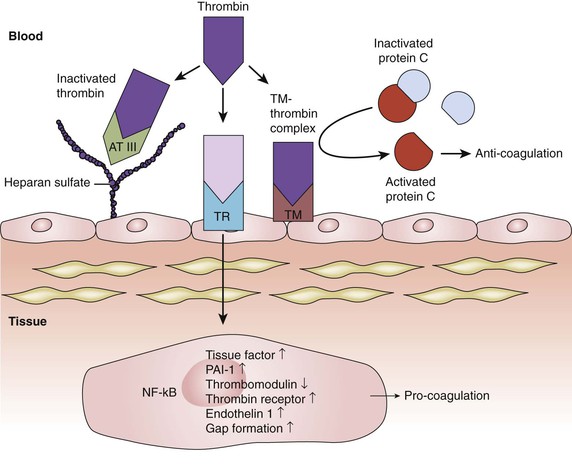
Figure 4-5 Inhibition of the thrombin pathway. Thrombin activation is a key step in the process of coagulation. Thrombin binding to a cell surface thrombin receptor (TR) can induce various pro-coagulation factors. Intact endothelium has various mechanisms to counteract coagulation. One of them is through the expression of heparan sulfate proteoglycans on endothelial cell surfaces. Heparan sulfate allows bound antithrombin III (AT III) to inhibit thrombin molecules generated by the coagulation cascade. Endothelial cells also synthesize and display thrombomodulin (TM). When thrombomodulin and thrombin complex (TM-thrombin complex), they work together to activate protein C, an enzyme that, when activated, destroys clotting factors and inhibits coagulation. NF-κB, Nuclear factor-κB; PAI-1, plasminogen activator inhibitor-1.
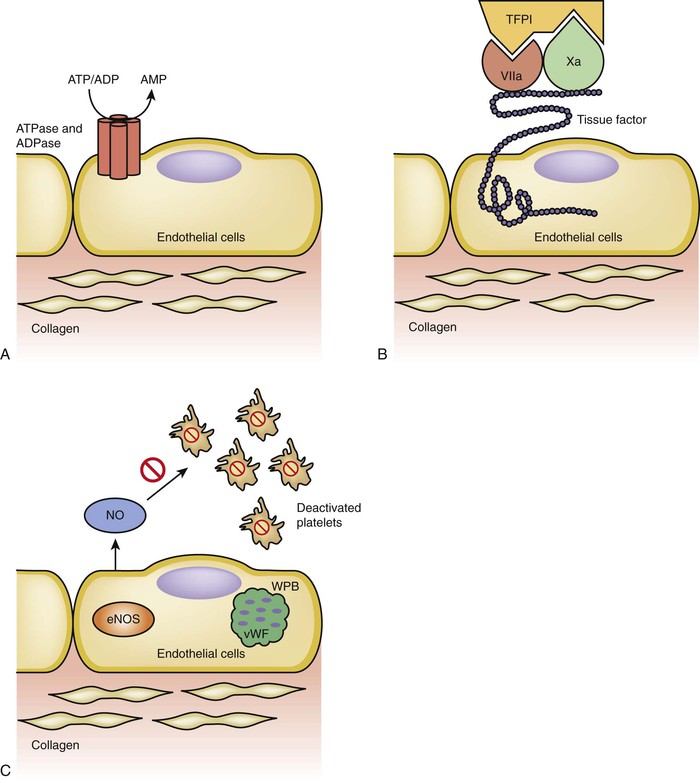
Figure 4-6 Anticoagulation pathways. A, Adenosine triphosphate (ATP) and adenosine diphosphate (ADP) are important factors in the process of platelet activation and thrombin release. Intact endothelial cells have ATP/ADPase that can convert ATP and ADP to adenosine monophosphate (AMP). B, Tissue factor pathway inhibitor (TFPI) is an inhibitor of the extrinsic pathway. Tissue factor exposure activation of factor VII, which in turn activates factor X, is a critical step in the extrinsic coagulation pathway. TFPI can bind to factors VII and X at the same time and deactivate the process of the extrinsic pathway. C, Platelet attachment to endothelium and activation are facilitated by von Willebrand factor (vWF). The endothelial cells can sequester vWF in Weibel-Palade bodies (WPBs) to prevent its exposure to platelet; furthermore, the constitutive form of nitric oxide synthase, extrinsic nitric oxide synthase (eNOS), also secretes nitric oxide (NO), which further inhibits platelet activation.
This site-specific distribution of clotting factors gives rise to site-specific thrombotic phenotypes. In the arterial system, platelet activation is based on the formation of thrombi, whereas in veins, fibrin is the main player in the formation of thrombi.46 Deficiency of specific factors is related to a thrombotic event that involves particular types of vessels or organs. For example, factor V Leiden is associated with increased risk of venous thrombosis but not of arterial myocardial infarction and stroke.47 Organ-specific thrombosis is observed in mice models with low levels of thrombomodulin, which increased fibrin deposition is found in the lung, heart, spleen, and liver but not in brain and kidney.48
The intact endothelium is an antithrombotic surface. It expresses heparin sulfate and chondroitin sulfate to prevent platelet binding. Endothelial cells also prevent the activation of platelets through the activity of ADPase, PGI2, and NO. The process of coagulation is initiated by the binding of factor VIIa with tissue factor complex. The binding is blocked by the tissue factor pathway inhibitor on the endothelial luminal surface. The action of thrombin, ATP, and ADP is a key component of the amplification of platelet aggregation signal. Heparin sulfate proteoglycans allows the binding of antithrombin III, which in turn inhibits thrombin molecule. ADPase blocks the platelet aggregation by metabolizing ADP to AMP and adenosine. The endothelium blocks coagulating factors from accessing the smooth muscle cells. Upon injury to the endothelium, the smooth muscle cells are exposed to circulating procoagulating factors, and thus the injured surface becomes an active site for neutrophil, monocyte, and platelet adhesion.49
NO, PGI2, and prostaglandin D2 are strong anticoagulative regulators able to interfere with platelet adhesion, activation, aggregation, secretion, and shape changes.50 PGI2 binds to specific receptors present on platelets that are linked to adenylate cyclase. Receptor activation leads to accumulation of intracellular cAMP in the platelet cytoplasm and downregulation of aggregation pathways.51 Thromboxane A2 (TxA2) and PGI2 usually plays each other’s counterparts in the process of coagulation. TxA2 is produced by platelet COX-1 and thromboxane synthase, and PGI2 is synthesized in vascular endothelial cells predominantly by the action of COX-2.51 PGI2 is continuously released into the circulation by the lungs to counter platelet aggregation from the release of TxA2. The PGI2/TxA2 ratio has been observed to be important; manipulation of this ratio with small doses of aspirin has beneficial effects similar to those of antithrombotic therapy.52 Endothelium-derived NO diffuses through the platelet membrane to bind its receptor. NO receptor is a soluble guanyl cyclase, like in the VSMCs. Binding to the receptor induces an elevation in cGMP and activation of cGMP-dependent kinases. This, in turn, inhibits intracellular Ca2+ increase, cytoskeletal rearrangements, integrin activation, and dense granule secretion, so the aggregation signal of platelet is blocked.53
Under normal conditions, the coagulation cascade is self-regulated. For example, NO production is in part triggered by the action of platelet-activating factor (PAF) on the endothelium.54 Although the importance of eNOS-produced NO has been confirmed, the contribution of other NOS species to platelet inhibition is poorly understood, and data on the subject are conflicting.50 The role of platelet-derived NO in the negative feedback system of platelet aggregation has been challenged, because independent studies have not demonstrated NOS messenger RNA and NOS activities in isolated platelets.55,56 The endothelial cells can respond to vasoactive agents, thrombin, or tumor necrosis factor-α (TNF-α) within minutes with the increased release of NO and prostacyclin. At the same time, the endothelial cells also recruit vesicles of pre-formed Weibel-Palade bodies to their plasma membranes. Weibel-Palade bodies are vesicles containing von Willebrand factor, P-selectin, and angiopoietin-S, which are involved in platelet binding, leukocyte recruitment, and inflammation modulation, respectively. Even though proteins C and S are not produced by endothelium, their activities occur in an endothelium-dependent fashion that involves multiple endothelially produced factors. Protein C and cofactor S can inactivate factors Va and VIIIa to turn off coagulation. The activation of protein C is facilitated by thrombomodulin and EPCR. EPCR binds protein C with high affinity to the surface of endothelial cells adjacent to the thrombomodulin-thrombin complex. Thrombomodulin transcription factor is enhanced in endothelial cells by arterial shear forces, which involve the transcription factor KLF-2.57 Arterial shear forces induce the transcription factor KLF2 and suppress inflammatory activation, a property that is lost in atherosclerosis-prone areas with disturbed blood flow.58–60
The key to the antithrombotic property of the endothelium is its integrity. If the endothelium is damaged or functions abnormally in any way, it will present a situation in which the disturbed balance favors thrombogenesis. Such disturbance has been recognized in clinical situations of traumatic vascular damage, diabetes, smoking, hypertension, and various other clinical diseases.
Leukocyte Trafficking
Migration of leukocytes from blood to peripheral tissue is crucial for the normal inflammatory process and also has important implications in pathologic conditions. The process, which is controlled by the endothelium, is a multistep cascade involving attachment, rolling, arrest, and transmigration. This process takes place almost exclusively in postcapillary venules.4
Leukocyte rolling is mediated by the interaction among endothelial E-selectin, P-selectin, and their respective leukocyte ligands. E-selectin is expressed in activated endothelium, and P-selectin is expressed constitutively. After the leukocytes are slowed down at the endothelial surface, firm adhesion is achieved by the interaction of endothelial intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM-1) with leukocyte integrins. Both adhesion molecules are induced by activation agonists of inflammatory signaling. Leukocytes can pass through the endothelium either paracellularly or transcellularly.61,62 This passage is facilitated by CD99 antigen, platelet endothelial cell adhesion molecule-1 (PECAM-1, also known as cluster of differentiation 31 [CD31]), and junctional adhesion molecules-1.63–65
Endothelial cells can be activated by endotoxin and inflammatory cytokines such as tumor necrosis factor-α αand interleukin-1. These factors all activate the classical nuclear factor-κB (NF-κB) pathway. Nuclear factor-κB induces the amplification of the genes required for inflammatory responses such as E-selectin, VCAM-1, ICAM-1, COX-2, PAI-1, and urokinase-type plasminogen activator (u-PA). NO can counteract this inflammatory activation. It can inhibit leukocyte adhesion to the vessel wall either by interfering with the ability of the leukocyte adhesion molecule CD11/CD18 (integrin) to form an adhesive bond with the endothelial cell surface or by suppressing CD11/CD18 expression on leukocytes.19 NO also plays a role in modulating or inhibit expression of VCAM, platelet endothelial cell adhesion molecule, and ICAM on the endothelial cell, thus inhibiting the adhesion and migration of leukocytes.58–60
The infiltration of leukocyte plays a major role in the development and progression of atherosclerosis. Activated leukocytes form a proinflammatory and prothrombotic condition, which generates oxygen radicals. Endothelial NOS produces NO, which can rapidly react with ROS to form peroxynitrite. This reduced availability of NO can impair endothelial function, reduce vasodilatation, and lead to proinflammatory and prothrombotic processes such as leukocyte adhesion and platelet aggregation.20
Recruitment of Bone Marrow–Derived Stem/Progenitor Cells
In addition to direct leukocyte adhesion, endothelial cells are involved in directing the homing of stem cells/progenitor cells. This involvement has important implications in both angiogenesis and vasculogenesis. Restoring blood flow to the site of injured tissue is a prerequisite to mounting a successful repair response. Bone marrow–derived EPCs play an important role in vasculogenesis in injured tissue. Stromal cell–derived factor-1α (SDF-1α) is the predominant chemokine that mediates this process. It has been found that under the stimulation of SDF-1α, endothelial cells and EPCs have the same upregulated expression of adhesion molecules. Through this mechanism, circulating EPCs are directed and homed at the injured endothelium and carry out their vasculogenesis function. The SDF-1α response to injury is downregulated in diabetic patients, contributing to the impaired wound healing ability of such patients.66 E-selectin is identified as the most important adhesion molecule that mediates this process of homing of EPCs. The use of SDF-1α engineered cell-based therapy has been shown to promote diabetic wound healing in mice by specifically upregulating E-selectin expression and increasing EPC homing.67 Furthermore, stem cells coated with E-selectin molecules showed an enhanced homing to injury tissue. This effect can overcome the impairment of injury tissue repair in diabetic individuals and restore their normal wound healing capability.67,68 The same process has been found to play a profound role in tumor angiogenesis and tumor growth in human melanoma. Suppression of E-selectin–mediated interaction between EPCs and endothelial cells and of EPC homing can serve as a novel target for inhibition of melanoma angiogenesis and tumor growth.69
Angiogenesis and Tumor
During embryonic development, angiogenesis contributes to organ growth and development, but during adulthood, most endothelial cells remain quiescent and angiogenesis occurs only in the cycling ovary and in the placenta during pregnancy. However, endothelial cells retain their ability to rapidly divide in response to physiologic stimulus of tissue repair and inflammation. The switch is maintained by groups of angiogenic and anti-angiogenic factors. Under normal conditions, the switch for angiogenesis is “Off.” There are various triggers to turn on this switch, which include metabolic stress, low PO2, low pH, hypoglycemia, mechanical stress, immune/inflammatory response, and genetic mutations (tumor).70 For example, when endothelial cells become hypoxic, they respond by upregulating hypoxia-inducible factor-1α (HIF-1α) and HIF-2α. They act as transcription factors for probably as many as 600 to 1800 genes, which include genes for metabolism cell survival, angiogenesis, and recruitment of leukocytes and progenitor cells.57
Angiogenesis involves multiple cell types, including endothelial cells, vascular smooth muscle cells, stromal cells, and parenchymal cells. The interactions among these cells occur through secreting factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and angiopoietins, as well as through cell extracellular matrix interaction. When the source for these factors becomes deregulated, as occurs in cancer, uncontrolled angiogenesis can ensue. This is the strategy that cancer cells use to harvest oxygen and nutrients for their expansion. Tumor vessels develop by sprouting or intussusception from preexisting vessels.70 The blood vessels that form within many tumors are often immature or defective, in that they show very poor investment with SMCs or pericytes and are greatly enlarged and extremely leaky. These so-called giant capillaries have a high propensity for tumor cell shedding and possible metastasis because they are readily penetrated by tumor cells.71,72 Circulating endothelial precursors, shed from the vessel wall or mobilized from the bone marrow, can also contribute to tumor angiogenesis.70 An important concept in tumor angiogenesis is that tumor blood vessels contain genetically normal and stable ECs, unlike tumor cells, which typically display genetic instability.73 ECs play mainly a responding and supporting role rather than a causal role in the process.
The structural abnormalities of the endothelium contribute to the spatial and temporal heterogeneity in tumor blood flow. Areas inside the tumor suffer from the pressure generated by proliferating cancer cells, which compress blood vessels and lymphatics. This creates zones of hypoxic and acidotic micro-environment inside the tumor. The cancer cells in this environment have adapted to this condition. Chemotherapy and radiation therapy demand normal blood flow and high oxygen to maximize their effects. However, this requirement is particularly challenging in these hypoxic and low–blood flow niches. A new concept that has emerged in the field of cancer therapy proposes that temporary normalization of the vessel structure is important to facilitate the delivery of chemotherapeutic agents and irradiation to these niches of hypoxic and acidotic cancer cells. Normalization can be achieved by providing anti-angiogenic agents to inhibit abnormal vessel growth. This concept emphasized and explained the importance of combining anti-angiogenic therapy and chemoradiation therapy in modern solid tumor therapy.74
Smooth Muscle Cells
Smooth muscle cells are the cellular component of the tunica media. They are arranged along with elastin and collagen in a highly organized fashion in order to withstand the continuous shear stress and transmural pressure exerted by the constant blood flow. The cells are surrounded by components of the extracellular matrix (ECM). The ECM is important in supporting the smooth muscle cell function. The SMCs are anchored by a basement membrane composed mainly of laminin, collagen type IV, heparan sulfate proteoglycan, entactin/nidogen, and fibronectins, which prevent SMCs from proliferating and migrating. This anchoring keeps SMCs in a contractile state as opposed to the synthetic state.75
Vascular Resistance
Functionally speaking, the arteries can be classified into muscular/resistance arteries and elastic arteries. Elastic arteries are the aorta and its large branches. They are large conductive vessels that have a thick collagen component in order to withstand the pressure exerted by blood flow. Small arteries and arterioles are considered resistance arteries. They contain a much thicker smooth muscle layer than connective tissue layer (collagen). The volume fraction of SMCs in the tunica media of small arteries is 70% to 85%. They are able to clamp down when stimulated, in order to shunt blood. Small arteries are defined as having diameters smaller than 500 µm. They provide about half of the total resistance in the circulation, and arterioles in the other half.76 This action is under the control of adrenergic nerves.76 An extreme example of this dominant muscular distribution can be seen in Raynaud’s phenomenon. Collagen in the vessel wall plays a role in maintaining the shape of the vessel. In this disease, the supportive collagen elements have regressed, complete vessel closure occurs because of the loss of the opposing force for smooth muscle constriction under cold conditions. This results in impaired distal circulation.13
In VSMCs, [Ca2+] elicits contraction by activating calmodulin. The Ca2+-calmodulin complex activates MLCK, which in turn phosphorylates the regulatory MLCK, which can then interact with actin to produce contraction. Phosphorylation of MLCK by PKA or PKG inactivates the enzyme, preventing contraction (Fig. 4-7). Smooth muscle cells can function as a syncytium when they are coupled with gap junctions. Although different neural and humoral agents act on smooth muscle cells through different membrane proteins and transduction pathways, their effects converge on regulating the activity of MLCK13 (Table 4-2).
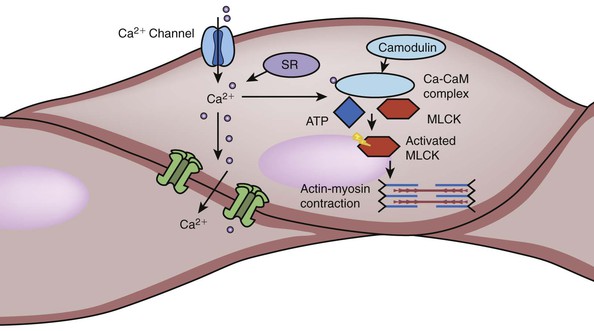
Figure 4-7 Activation of smooth muscle cells. The process of smooth muscle contraction starts with the increase in intracellular calcium. There are various mechanism that can lead to the influx of Ca2+ from extracellular environment as well as release of Ca2+ from sarcoplasmic reticulum (SR). When Ca2+ binds to calmodulin (CaM), the complex activates myosin light chain kinase (MLCK). MLCK is an enzyme that can phosphorylate myosin light chain using adenosine triphosphate (ATP). Phosphorylated myosin light chain can use this energy to change its conformation and interaction with actin filaments, thus leading to muscle contraction. Factors that affect smooth muscle contraction can achieve their effect by modifying the behavior of membrane Ca2+ channel, SR, calmodulin, and MLCK (see Table 4-2). There are also numerous gap junction or ion channels between bundled smooth muscle cells. When one muscle is activated, the signals of excitation can be transduced through this junction to cause neighboring cell contraction.
Table 4-2
Vasoconstriction and Vasodilation Pathways and Mechanisms
| Vasoconstrictive Pathway | Mechanism |
| Voltage-gated Ca2+ channels | Depolarization → voltage-gated Ca2+ channels open → Ca2+ entry → ↑[Ca2+] |
| Voltage-gated Na+ channels | Depolarization → voltage-gated Na+ channels open → more depolarization → voltage-gated Ca2+ channels open → Ca2+ entry → ↑[Ca2+] |
| Stretch-activated channels | Stretch → channels open → depolarization → voltage-gated Ca2+ channels open → Ca2+ entry → ↑[Ca2+] |
| Receptor-operated Ca2+ channels | Agonist (e.g., adenosine) → Receptor-operated Ca2+ channels open → Ca2+ entry → ↑[Ca2+] |
| Adrenergic receptor α1 | Agonist (e.g., norepinephrine) → Adrenoreceptor α1 → activation of Gαq → ↑PLC activity → ↑[IP3] and [DAG] → IP3 receptor in SR opens Ca2+ channels ↑[Ca2+] |
| Muscarinic receptor M2 | Agonist (e.g., acetylcholine) → Muscarinic receptor M2 → Adrenoreceptor α1 activation of Gαi → ↓AC activity → ↓[cAMP] → ↓PKA activity → ↓phosphorylation of MLCK → ↑MLCK activity → ↑phosphorylation of MLC |
| Endothelin receptor ETA | ET → Endothelin receptor ETA → ↑[IP3] and [DAG] → IP3 receptor in SR opens Ca2+ channels → ↑[Ca2+] |
| Purinergic receptor | ATP → P2X receptor (ligand-gated Ca2+ channel, an example of an ROC) → Ca2+ entry → ↑[Ca2+] |
| Na-Ca exchanger | Depolarization → voltage-gated Na+ channels open → Na+ entry → ↑[Na+] → slow Na-Ca exchange → ↑[Ca2+] |
| Voltage-gated K+ channels | Depolarization → voltage-gated K+ channels open → hyperpolarization → voltage-gated Ca2+ channels close → ↓[Ca2+] |
| Ca2+-dependent K+ channels | Hyperpolarization → voltage-gated Ca2+ channels close → ↓ [Ca2+] |
| ATP-sensitive K+ channels | ↓[ATP] → KATP channels open → hyperpolarization → Ca2+ channels close → ↓[Ca2+] |
| Adrenoreceptor β2 | Agonist (e.g., epinephrine) → adrenoreceptor β2 → activation of Gαs → ↑AC activity → ↑[cAMP] → ↑PKA activity → phosphorylation of MLCK → ↑MLCK activity → ↓phosphorylation of MLC |
| VIP receptor | Vasoactive peptide → VIP receptor → activation of Gαs → ↑ AC activity → ↑[cAMP] → ↑PKA activity → phosphorylation of MLCK → ↑MLCK activity → ↓phosphorylation of MLC |
| Both Ca2+-dependent and voltage-gated K+ channels open → hyperpolarization → Ca2+ channels close → ↓[Ca2+] | |
| Prostacyclin (PGI2) | PGI2 → ↑AC activity → ↑[cAMP] → ↑PKA activity → phosphorylation of MLCK → ↑MLCK activity → ↓phosphorylation of MLC |
| Histamine receptor H2 | Histamine → H2 receptor → activation of Gαs → ↑AC activity → ↑[cAMP] → ↑PKA activity → phosphorylation of MLCK → ↑MLCK activity → ↓phosphorylation of MLC |
| Purinergic receptor | Adenosine → A1, A2A, and A2B adenosine receptors → KATP → hyperpolarization → Ca2+ channels close → ↓[Ca2+] |
| ATP → purinergic P2Y receptors → activation of Gαq/11 → ↑PLC activity → ↑[Ca2+] → ↑NOS → NO release | |
| SR Ca2+ pump (SERCA) | Activation of Ca2+ pump (Ca-ATPase) in the SR → ↓[Ca2+] |
| Na-K pump | Activation of Na-K pump in plasma membrane → ↓[ Na+] → activation of Na-Ca exchange → ↓[Ca2+] |
AC, Adenylyl cyclase; ATP, adenosine triphosphate; Ca2+, calcium ion; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; ET, endothelin; Gαi, Gαq, Gαq11, Gαs, G protein subunits; IP3, inositol 1,4,5-triphosphate; KATP, ATP-sensitive potassium channel; MLC, myosin light chain; MLCK, MLC kinase; Na+, sodium ion; NO, nitric oxide; NOS, nitric oxide synthase; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; PLC, phospholipase C; ROC, receptor-operated channel; SERCA, sarcoplasmic/endoplasmic reticulum calcium-transporting ATPase; SR, sarcoplasmic reticulum; VIP, vasoactive intestinal polypeptide; →, leads to; ↑, increased/increase in; ↓, decreased/decrease in.
Original table from Boron WF, Boulpaep EL: Medical physiology: a cellular and molecular approach, Philadelphia, 2012, Saunders/Elsevier.
Vascular Smooth Muscle Differentiation
Vascular smooth muscle cells in adults are highly specialized cells whose primary function is regulation of blood vessel diameter through either contraction or relaxation. By means of this mechanism, animals can regulate blood pressure and blood flow distribution in the body. Under normal conditions, these cells exhibit very slow proliferation and synthetic activity. They have unique contractile proteins, ion channels, receptors, and signaling molecules that carry out the contractile function. However, vascular smooth muscle cells retain a high level of plasticity and can undergo remarkable phenotype change on the basis of environmental cues.
Earlier attempts have identified two main phenotypes for VSMCs, contractile and synthetic. Most mature VSMCs exhibit the contractile phenotype, a low-proliferative, nonmigratory, low-synthetic state. The cells are virtually completely committed to carrying out the contractile function. Synthetic phenotype has mainly been seen during vascular development and injury repair. During these processes, a group of VSMCs exhibits high rates of proliferation, migration, and production of ECM components, such as collagen, elastin, and proteoglycans, that make up a major portion of the blood vessel wall while at the same time acquiring contractile capabilities. During this stage, abundant gap junctions are formed between VSMCs and endothelial cells. The communication between endothelial cells and VSMCs and pericytes is critical for vascular maturation and vessel remodeling.77 Further studies have also provided evidence that circulating progenitor cells can contribute to neointima formation and repair following vascular injury.78 However, the extent to which these cells are involved in repair, as well as their destiny of forming mature VSMCs, has yet to be further proven.
Researchers believe that a phenomenon called “phenotype switching” is responsible for the synthetic phenotype; this switching is a process whereby contractile or synthetic VSMCs undergo major functional and morphologic changes in response to environmental signals, becoming the other phenotype. However, it is more important to be able to appreciate that these changes can happen at a much more subtle level and in vast varieties in order to respond to specific environmental changes. It is now recognized that the phenotypes of VSMCs cover a wide spectrum rather than two well-defined states. A new term was coined to describe this phenomenon on a more subtle level, “phenotype modulation.” This term includes modification in gene expression patterns, signaling mechanisms, contractility, and a full range of alterations in functional and structural properties.79
The major challenge in understanding the process of phenotype switch is the identification of the cell-specific markers for VSMCs. These markers usually are related to a specific state and function carried out by the cell. Because of the diverse functions of the SMCs and because most SMC markers are neither specific nor sensitive, to defining cells on the basis of these markers is particularly challenging. Furthermore, identifying the factors that induce phenotype modulation and the genes responsible for carrying out the changes in the cellular process will be the key to understanding the process. Factors that have so far been found to participate in phenotype switching include PDGF, tumor growth factor-β TGF-β, nitric oxide, ROS, and matrix metalloproteinases (MMPs).79
Certain disease models were able to provide great insight into this process. Because of this high plasticity, VSMCs are also predisposed to be affected by abnormal environmental changes causing adverse phenotypic changes that can result in the development of various pathologies such as hypertension and atherosclerosis.
Systemic hypertension is a popular cardiovascular disease with a complex etiology that differs from individual to individual. A common feature in most cases of hypertension is the increase in peripheral resistance as a result of greater vascular tone or smooth muscle cell contraction. This process of vascular remodeling believed to involve VSMC phenotypic switching.80,81 The change in contractility can be attributed to many factors, including changes in the intracellular calcium handling mechanism82 and alteration in the membrane potential.83 The VSMC phenotype undergoes profound changes, including hypertrophy, hyperplasia, increased synthesis of matrix materials, reorganization of cell-cell and cell-matrix contacts, and apoptosis associated with vessel rarefaction.82,84
VSMC phenotypic switching is also believed to play a crucial role in atherosclerosis. The role of VSMCs in atherosclerosis appears to vary, depending on the stage of the disease. VSMCs seem to be involved in atherosclerotic lesion development and progression,85,86 but at the same time, also to have a role in stabilizing the fibrous cap of the plaque. At the end stage of the disease of plaque rupture, VSMCs contribute to the destabilization of the plaque through activation of protease cascades.87,88
VSMCs exhibit a wide range of phenotypes depending on either the stage of lesion development or the location of the VSMCs within a lesion. Regulation of the transitions between phenotypes is a complex process under the influence of numerous factors. Most of the factors’ effects have been studied under in vitro conditions. Until today, little has been known about the in vivo process that regulates the phenotypic modulation of VSMCs. PDGF has been found to downregulate smooth muscle–selective markers in culture and to stimulate SMC proliferation and migration in arterial injury. TGF-β has been shown to promote SMC differentiation in cell culture and upregulation of smooth muscle–selective markers. Such function of TGF-β activity is thought to play a role in SMC matrix production and fibrotic stabilization of the developing atherosclerotic plaque. Matrix metalloproteinases are produced by SMCs and macrophages to actively modify the matrix in which SMCs reside and actively contribute to further phenotypic switching of the SMCs. Matrix production by SMCs and modification by MMPs are major factors contributing to the stability of plaque.85–88
Pericytes
Pericytes are supporting cells embedded within the vascular basement membrane surrounding the endothelial cells. They exist only in areas of microcirculation like capillaries, postcapillary venules, and terminal arterioles. One generally adopted view is that pericytes belong to the same lineage and category of cells as VSMCs and fibroblasts; however, no single molecular marker is known that can clearly distinguish pericytes from VSMCs or other mesenchymal cells. Because there is still confusion about the identity, ontogeny, and progeny of pericytes, they are currently defined with a mixture of criteria including location, morphology, and gene/protein expression pattern.89
Research evidence has shown that it is likely that pericytes in different parts of the body are from different embryonic origins. The majority of pericytes in the head region and thymus are derived from neural crest.90–93 The pericytes in gut, lung, and liver originate from mesothelium.94–96 VSMCs demonstrate similar various origins, their four developmental sources being secondary heart field, neural crest, somites, and splanchnic mesoderm.89
Mature pericytes are enveloped in a basement membrane that is continuous with the endothelial basement membrane. This membrane is not well defined within embryonic tissue and active angiogenesis. Here, the perivascular mesenchymal cells, VSMCs, and pericytes exist in their immature forms and are hard to distinguish from one another.89 This is no surprise, given the suggestion of a common lineage among these cell types. They have many common markers and are equipped with the ability to migrate and differentiate. It is likely that all three types of cells have the ability to develop into new pericytes in vivo when given the right signals.
Pericyte–Endothelial Cell Interaction
Pericyte density varies among different organs and vascular beds. The central nervous system vasculature has the highest density of pericytes, with a pericyte/EC ratio of 1 : 1. This ratio is merely 1 : 100 in skeletal muscle. Pericytes encircle the circumferences of the vessels with the extension of their cytoplasmic processes. The pericyte-EC interface is normally separated by basement membrane. Their communication is established through the abundant number of holes through the basement membrane. The pericytes extend their cytoplasmic fingers through the holes and insert into endothelial invaginations on the other side of the basement membrane. These peg-and-socket formations are the basic form of pericyte-EC communication. Other forms of communication, such as adhesion plaques, gap junction, and caveolae, are also important.97 It is believed that all the forms of communication support pericytes’ role of regulating capillary barriers, endothelial proliferation, and capillary diameter.89 In vitro studies have also shown that pericyte-EC interaction is important in regulating the basement membrane assembly98,99 (Fig. 4-8).
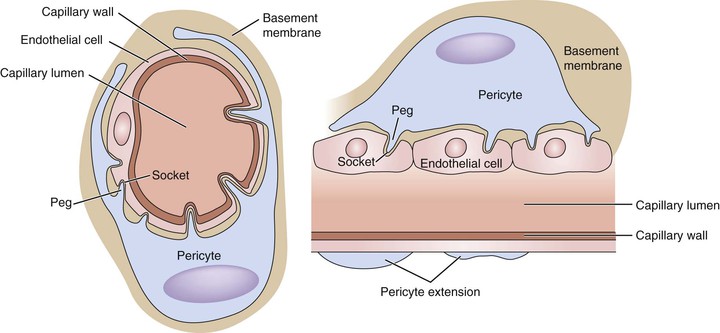
Figure 4-8 Pericyte-capillary interaction. The pericytes support the endothelial cell by extending their cytoplasmic processes, encircling the small vessels/capillaries. The endothelial cells and surrounding pericytes are separated by a layer of basement membrane. The interaction between the two types of cells is achieved in a “peg-and-socket” fashion. The basement membrane has numerous small holes. Through these holes, pericytes extend their cytoplasmic processes into the invaginations on the surfaces of endothelial cells. Important signals that affect various cellular processes are transduced through these bridges. One pericyte usually extends the length of several endothelial cells, but this feature very much varies from tissue to tissue. The central nervous system has the highest density of pericytes to endothelial cells, in a ratio close to 1 : 1.
Endothelial cell–pericyte interaction plays an important role in physiologic and pathologic functions. The two cell types interact through various signaling pathways. The PDGF-B/PDGF receptor-β (PDGFR-β) pathway is one of the best studied. PDGF-B is released from angiogenic endothelia cells and binds to PDGFR-β expressed on the surfaces of developing pericytes. This signal is responsible for recruiting pericytes to angiogenic sprouts.100 The same signal also plays a role in the proliferation and differentiation of aortic and venous VSMCs.89 Once secreted, PDGF-B is bound to the extracellular matrix, most likely the heparan sulfate proteoglycans. This binding plays a major role both in embryonic development and in postnatal angiogenesis. It is likely that other signaling pathways are working in parallel with the PDGF-B/PDGFR-β pathway, because different tissues seem to respond differently with the PDGF-B knockout in mice.101,102
TGF-β has been implicated in the induction of pericyte differentiation from mesenchymal stem cells (MSC) as well as pericyte proliferation. It is also involved in endothelial cell proliferation and differentiation. Both cell types secrete TGF-β as well as express TGF-β receptor, and the activation of cell differentiation and proliferation seems to require the collaboration between the two cell types.103 Two distinct type I TGF-β receptors, Alk-1 and Alk-5, are both expressed in endothelial cells and pericytes. However, the two receptors trigger different cellular effects. Activation of Alk-5 in mesenchymal cells promotes mitotic and migratory quiescence and differentiation into SMCs. Activation of Alk-1 promotes cell proliferation and migration and opposes SMC differentiation.103–105 Overall, Alk-5 seems to promote vessel maturation and Alk-1 to have the opposite effect.
The interaction between endothelial cells and pericyte is bidirectional. This feature is reflected in the angiopoietin/Tie-2 pathway. Angiopoietin-1 is expressed by pericytes and perivascular mesenchymal cells.106 The receptor for Ang-1, Tie-2, is predominantly expressed on endothelial cells.107 This pathway is proposed to play a role in maintaining endothelial cell maturation and stability and reducing vascular leakage.108–110 In humans, mutation of the TIE2 gene leads to venous malformation, in which focal loss of venous pericytes is observed.111 Angiopoietin-1 and Tie-2 are not involved directly in pericyte recruitment, but they do have important roles in blood vessel formation and stability. Overexpression of angiopoietin-1 leads to increased vascular branching and higher-order organization in immature vessels.112
Other pathways have also been determined to participate in the recruitment in pericytes. These pathways may work in an organ-specific fashion or in a compensatory fashion when the PDGF-B/PDGFR-β pathway is downregulated. The heparin-binding epidermal growth factor (HB-EGF)/EGF receptor (EGFR) pathway works in this way. This pathway has been found to be essential to cardiovascular development113,114 and also to protect against the loss of pericytes from intestinal vessels following mesenteric artery occlusion.115 Stromal cell–derived factor-1α/chemokine (C-X-C motif) receptor-4 (CXCR4) pathway also works in pericyte recruitment. SDF-1α promotes pericyte migration. It also cross-talks with the PDGF-B/ PDGFR-β pathway, because SDF-1α expression is stimulated by PDFG-B. In addition, Sonic hedgehog, notch, and Ephrin-Eph signaling pathways play major roles in pericyte and endothelial cell communication.
Blood-Brain Barrier
Brain vessels possess the special quality of been extremely selective for blood content, called the blood-brain barrier. Pericytes play a critical role in the maturation and maintenance of the blood-brain barrier.116,117 The brain vessels appear to have specific markers.118,119 Central nervous system vasculature also has the highest pericyte coverage among all analyzed organs. The traditional view of blood-brain barrier has focused on the endothelial junctions. However, later studies show that the role of pericytes in the brain vasculature can regulate the permeability of endothelial cell junctions. The endothelial cells here normally have a very low rate of transcytosis. Pericyte deficiency leads to an increase in brain vessel permeability, the extent of which correlates directly with the density of brain pericytes. The increase in permeability is a result of upregulated endothelial transcytosis. Several genes that are considered to play roles in the regulation of endothelial permeability are dysregulated in pericyte-deficient blood vessels. They include vascular endothelial growth factor-α, angiotensin-2, and adrenomedullin.116,117
Blood Flow Regulation
Very early on, pericytes were found to have contractile features like VSMCs, so the first function suggested for pericytes was to cause capillary constriction. However, the physiologic significance of pericyte contractility, and even the validity of this contractile property of pericytes, are still being challenged and are currently under investigation. Pericyte-mediated capillary constriction in response to vasoactive substances and neurotransmitters has been observed in vivo and as well as in organ cultures.120,121 Studies have also demonstrated that combined with the vasoconstriction at the arteriole level, pericyte-mediated capillary constriction is observed under certain conditions, such as oxidative stress.122 It has been suggested that this constriction may play a protective role during anoxic insults to the brain.
Pericyte Plasticity
The relationship between MSCs and pericytes is intimate but still poorly defined. Cultured pericytes have shown to differentiate in vitro into osteoblasts, adipocytes, chondrocytes, VSMCs, and skeletal muscles.123–126 These are defining features of MSCs. At the same time, MSCs were found to reside in a perivascular niche very close to pericytes.127 Research has now raised the concept that MSCs can come from pericytes, as well as white adipocyte progenitors, muscle stem cells, and even neural stem cells.128–134 Studies have been compared cell markers, but the results are usually limited by their lack of sensitivity and specificity. Despite the problem with defining pericytes, it is clear that they have a huge potential for plasticity. Perhaps it is exactly this high plasticity potential that has made defining pericytes with specific markers so difficult.
Fibrosis Generation
Pericytes’ plasticity may also be reflected in their relationships with fibroblasts and myofibroblasts. Later studies have pointed to pericytes as myofibroblast precursors in the pathologic process of fibrosis in liver and kidneys and of systemic sclerosis.135–137 One of the critical steps in fibrosis progression is the activation of myofibroblast progenitors. Myofibroblasts are fibroblast-like cells that deposit pathologic extracellular matrix and are directly responsible for the extent of fibrosis. Traditionally, myofibroblasts were considered activated fibroblasts. Currently there are several hypotheses about the cellular origins of myofibroblasts, including resident fibroblasts, fibrocytes, epithelial cells, bone marrow–derived cells, endothelial cells, and pericytes. The origin of myofibroblasts is far from being elucidated, but accumulating evidence indicates that pericytes may constitute an important source.
Pericytes in Tumor
The interaction between pericytes and endothelial cells plays an important role in angiogenesis, as previously mentioned. Therefore, it is not hard to infer that pericytes may also play a role in cancer biology. Pericytes are an obligatory component of the tumor stroma. The tumor stroma is the tumor’s microenvironment, which constitutes the extracellular matrix components as well as cell types such as fibroblasts, myofibroblasts, endothelial cells, and pericytes. The tumor stroma is an essential component of tumor growth, invasiveness, and metastasis.138,139
Pericytes are a ubiquitous part of the tumor microenvironment.140 As in physiologic angiogenesis, pericytes are recruited into tumor blood vessels. PDGF-B/PDGFR-β signaling, heparin-binding epidermal growth factor, and SDF-1α are found to play a part in recruiting pericytes to tumor vasculature sharing same mechanisms that were found in developmental angiogensis.141–143 However, the pericyte investment of tumor blood vessels is clearly abnormal. The extent of pericyte coverage on tumor vessels lower than on normal vessels, and pericyte–endothelial cell peg-and-socket communication is much looser, with cytoplasmic processes that penetrate deep in the tumor parenchyma.144 The exact cause of the abnormal pericyte behavior in tumor is still unknown. The potential of pericytes as a target for therapeutic intervention in cancer is currently under investigation.
Fibroblasts and Myofibroblasts
Fibroblast is the main cell type in the adventitia of the vessel wall. Adventitial fibroblasts are thought to arise locally from the mesenchyme, except in the coronary artery, in which a common proepicardial origin has been demonstrated for the medial SMCs and adventitial fibroblasts.145 Traditionally, the adventitia has been considered an essential supportive tissue, and the fibroblasts to serve mainly in a secretory function for the production of collagen and proteoglycans. This view has changed dramatically, because it has now been shown that adventitia and fibroblasts not only participate in structural support but also play principle roles in injury and inflammatory response, vessel remodeling, and various pathologic processes.
Fibroblasts are morphologically characterized as adherent, flat, spindle-shaped cells with flat, oval nuclei. They lack CD34, cytokeratin, and CD45 and express vimentin. They display a heterogeneous phenotype, so it is challenging to establish a definite description. The fibroblast population in one organism is made up of various subsets of cells, each with distinct protein expression profiles and differing functions.146 This feature may arise from their heterogeneous origins. Fibroblasts are generally thought to be of mesenchymal or neural crest origin and can arise from other cell types postnatally.
Fibroblasts in Tissue Injury and Remodeling
The adventitia has been indicated as the principle “injury-sensing tissue” within the artery. It is able to respond to different stimuli in an outside-in manner by originating and coordinating changes that progress toward the intima and finally lead to vessel remodeling.147 Adventitial fibroblasts play a pivotal role in this remodeling process, owing to their plasticity.148 When stimulated, fibroblasts undergo phenotypical change to myofibroblasts. Myofibroblasts are specialized cells with contractile as well as migratory properties. This was initially observed at the site following angioplasty and stent placement. Adventitial fibroblast cells underwent proliferation to a greater extent than the medial cells 2 to 3 days after angioplasty, and many adventitial cells that migrated to the neointima were later proven to be myofibroblasts. This process is called vessel neointimal hyperplasia.149,150
The myofibroblast has pivotal function in tissue repair and remodeling. This function contributes to various pathologic conditions, such as hypertrophic scars, fibromatoses, systemic sclerosis, organ fibrosis, and stroma reaction to epithelial tumors.145 Although resident fibroblasts are the main progenitors for myofibroblasts, several other possible sources for them have been identified, including SMCs, pericytes, mesenchymal stem cells, endothelial cells, and bone marrow–derived fibrocytes.151
As injury occurs, cytokines secreted by inflammatory cells activate fibroblasts in the arterial adventitia. The activated fibroblasts start to synthesize ECM components and organize into a mechanically supportive structure, which can produce a significant contractile force.151,152 The activated fibroblast, called a proto-myofibroblast, is characterized by the development of contractile bundles composed of β- and γ-cytoplasmic actins. This increased mechanical force within the ECM can further induce changes in proto-myofibroblasts, characterized by the expression of α-smooth muscle actin (α-SMA). Transforming growth factor-β1 can induce the same process in fibroblasts. The contractile force formed by myofibroblasts is a long-lasting and isometric tension resulting in a steady, permanent contraction. The same mechanism is observed in normal wound granulation tissue.153 When tissue repair is finished, myofibroblasts undergo massive apoptosis. Lack of this apoptosis can lead to conditions such as fibrosis and fibromatoses.152
To understand the role of fibroblast in vessel remodeling, one must first understand the concept of vessel remodeling. Many pathologic conditions can induce noxious stimuli to the structure and functions of the vessel wall. These can be commonly seen in hypertension as well as various types of vascular injury such as atherosclerosis and vessel overdistention due to balloon dilatation.154–157 In attempt to adapt to these stimuli, the vessels undergo changes known as remodeling. Remodeling is divided into two types, positive or adaptive remodeling and negative or constrictive remodeling.145
Positive remodeling principally involves inflammation-induced protease activity. It was originally described in the early stages of coronary atherosclerosis, in which vessel enlargement occurs in the presence of an increased burden of obstructive plaque in order to maintain a constant flow. It consists of an outward expansion of the vessel wall, which partly leads to a loss in collagen and reduction of smooth muscle component that results in medial and adventitial thinning. The result is a permanent change in the lumen size and relative composition of the vessel wall.158 If this reaction escapes self-limiting control, remodeling can lead to a maladaptive response, resulting in a reduction in vessel lumen size called negative remodeling.159 Negative remodeling is mediated mostly by a rapid proliferation of adventitial fibroblasts and the switch from adventitial fibroblast to myofibroblast. The result is the formation of a thickened adventitia rich in myofibroblasts and collagen fibers. Negative remodeling often correlates with high-grade stenosis associated with stable cardiovascular disease.160
Besides the atherosclerotic arteries and restenosis of vessels, adventitial myofibroblasts are observed to be involved in systemic and pulmonary hypertension, vein graft remodeling, coronary transplant vasculopathy, and inflammatory abdominal aortic aneurysms.161–164
Fibroblasts and Vascular Inflammation
Endothelial activation and leukocyte extravasation are key events in vascular inflammation. There is a growing appreciation of stromal fibroblasts’ role in production of cytokines, growth factors, and proteases and in maintaining acute and chronic inflammatory conditions. These fibroblasts are able to modulate endothelial cell functions in a paracrine manner, including pro-inflammatory activation and promotion of angiogenesis as well as promotion of leukocyte infiltration into tissues.
Traditionally, vascular inflammation has been considered an inside-out response, beginning with endothelial activation followed by leukocyte attachment and migration out of the vessels. Increase evidence points to a signaling pathway that occurs from outside in and is mediated by stromal fibroblasts. Fibroblasts when stimulated can produce abundant amount of signaling factors to stimulate endothelial cell activation and they become chemotactic to leukocytes; this in turn amplifies the proinflammatory effect of the vessel.165–167 Activated fibroblasts found in inflamed tissue produce cytokines, growth factors, and proteases in pathologic conditions such as tissue repair, fibrosis, pathologic organ remodeling, and cancer.168–170 Fibroblasts have been suggested to function as sentinel cells that are capable of proinflammatory activation, including management of cytokine secretion and leukocyte infiltration. The transcription of many cytokines and growth factor is regulated by the proinflammatory gatekeeper nuclear factor κB.171
The first response in vascular inflammation consists mainly of endothelial cells and leukocytes, but stromal fibroblasts are capable of inducing and prolonging inflammation, including managing the switch between acute and persistent inflammation.167 Prolonged inflammation can be harmful, because this powerful defense and reconstruction mechanism can also destroy healthy tissue. Fibroblasts are implicated in many chronic inflammatory conditions, such as venous ulcers,172 interstitial fibrosis of the lung and kidney,173,174 hypertrophic scar,175 and granulomas.172,176
Stem/Progenitor Cells
Today, multipotent adult stem/progenitor cells have been identified in almost all human organs. This development has huge implications for regenerative and therapeutic medicine. The potential to harvest, expand, and apply these different cell lines has been extensively investigated. Research has identified the blood vessel wall as a source of stem/progenitor cells. Together with bone marrow–derived endothelial progenitor cells, stem/progenitor cells are responsible for postembryonic neovascularization and angiogenesis. Currently, at least three distinct subpopulations of stem cells have been identified in the vessel wall. They are myogenic endothelial cells, pericytes, and adventitial cells, which reside in the intima, peri-intima, and adventitia, respectively, together called human blood vessel–derived stem cells (hBVSC).177–181 Although most of the studies have been limited to in vitro and animal research, the results have been promising.
Myogenic endothelial cells coexpress both myogenic (CD56) and endothelial (CD34 and CD144) cell markers. Such cells display developmental capacities similar to those of MSCs in vitro, including myogenesis, osteogenesis, chondrogenesis, and adipogenesis. They also express classic MSC markers. They are primarily located in the intimal compartment of the blood vessels. The therapeutic potential of myogenic endothelial cells has been successfully proven for skeletal muscle repair/regeneration and cardiac repair in animal models.182
We have talked extensively about pericytes and their physiologic and pathologic roles in previous section, as well as a possible origin for MSCs. Pericytes are the first type of stem cell found in the human blood vessel.183 With their inherent functions in vascular physiology, pericytes seem to be the ideal donor cell population for cardiovascular repair.184 Moreover, a human pericyte–based small-diameter vascular graft has been successfully engineered with high patency after long-term transplantation in an animal model.185
The emerging concept is that the adventitia is not merely a structure support but rather a dynamic reservoir of stem cells that participate in vascular remodeling and regeneration of surrounding tissues. The potential application of adventitial cells in the clinical setting has thus far been focused on cardiovascular repair and regeneration. It has been demonstrated that adventitial cells interact with endothelial cells and promote the formation and stabilization of the capillary-like structure.178 These progenitor cells were first found within the adventitia of the aortic root of mice. They were found to express a number of markers characteristic of progenitor cells. Under normal conditions, they are exclusively found in the adventitia, not the media or intima. Their location is maintained by a layer of Sonic hedgehog protein located between the media and the adventitia. Sonic hedgehog protein is known to be important in the retention of other progenitor cell populations in other tissues but prevents them from migrating freely.186–188 However, during injury and pathologic development, adventitial cells can migrate into the intima, and they have been shown to participate in the development of neointimal hyperplasia.189
Selected Key References
Aird WC. Phenotypic heterogeneity of the endothelium. I: structure, function, and mechanisms. Circ Res. 2007;100:158–173.
Provides comprehensive information about endothelial cells, including the vast heterogeneity in their phenotypes and functions. It emphasizes a new important concept that the endothelium in different parts of the body has special properties allowing it to carry out organ-specific functions.
Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
An important review on the current understanding of pericytes, their role in pathologic conditions, and their plastic and dynamic nature as well as their potential in regenerative medicine.
Boron WF, Boulpaep EL. Medical physiology: a cellular and molecular approach. Saunders/Elsevier: Philadelphia; 2012.
Textbook with a lot of fundamental information on the physiology of the vascular system and classic pathway, which have been well studied.
Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714.
Summarizes our current understanding of the function of nitric oxide and endothelial nitric oxide synthase. Contains the important concept of the “double-edged-sword” nature of nitric oxide in its protective and destructive properties.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
Explains an emerging concept in cancer therapy: To maximize the potential of chemoradiation therapy, normalization of tumor vasculature is more important than its destruction. This concept provides new understanding in antiangiogenic therapy and its role in combination with chemoradiation therapy.
Owens K, Meena S, Kumar MS, Brian R, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
Offers evidence for the new concept of the dynamic nature of the smooth muscle cells in the vascular wall. The vascular smooth muscle cells not only provide resistance and control the blood flow but also are capable of responding to a variety of stimulus and undergoing phenotypic and functional changes in order to adapt to the environment.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Merwin J, et al. Transforming growth factor β1 modulates extracellular matrix organization and cell-cell junctional complex formation during in vitro angiogenesis. J Cell Physiol. 1990;142:117–128.
2. Kocher O, et al. Modulation of actin mRNAs in cultured capillary endothelial and aortic endothelial and smooth muscle cells by matrix components and TGF-β1. In Vitro. 1989;25:424–434.
3. Grand D, et al. Intracellular mechanisms involved in basement membrane–induced blood vessel differentiation in vitro. In Vitro Cell Dev Biol. 1991;27A:327–336.
4. William C. Aird phenotypic heterogeneity of the endothelium. I: structure, function, and mechanisms. Circ Res. 2007;100:158–173.
5. Levy B, Tedgui A. Biology of the arterial wall. Kluwer Academic: Norwell, MA; 1999.
6. Muro S, et al. Endothelial endocytic pathways: gates for vascular drug delivery. Curr Vasc Pharmacol. 2004;2:281–299.
7. Bendayan M. Morphological and cytochemical aspects of capillary permeability. Microsc Res Tech. 2002;57:327–349.
8. Shimokawa H, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711.
9. Bellien J, et al. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam Clin Pharmacol. 2008;22:363–377.
10. Nishikawa Y, et al. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am J Physiol. 2000;279:H459–H465.
11. Bauersachs J, et al. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347.
12. Wu Y, et al. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2456–H2461.
13. Boron WF, et al. Medical physiology: a cellular and molecular approach. Saunders/Elsevier: Philadelphia; 2012.
14. Ignarro L. Haem-dependent activation of guanylate cyclase and cyclic GMP formation by endogenous nitric oxide: a unique transduction mechanism for transcellular signaling. Pharmacol Toxicol. 1990;67:1–7.
15. Hobbs A. Soluble guanylate cyclase: the forgotten sibling. Trends Pharmacol Sci. 1997;18:484–490.
16. Bolotina V, et al. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853.
17. Boulanger C, et al. Release of endothelin from the porcine aorta: inhibition of endothelium derived nitric oxide. J Clin Invest. 1990;85:587–590.
18. Pacher P, et al. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424.
19. Förstermann U, et al. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714.
20. Muller G, et al. Nitric oxide, NAD(P)H oxidase, and atherosclerosis. AntioxRedox Signal. 2009;11:1711–1713.
21. Landmesser U, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209.
22. Murohara T. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567–2578.
23. Park B. eNOS affects both early and late collateral arterial adaptation and blood flow recovery after induction of hindlimb ischemia in mice. J Vasc Surg. 2010;51:165.
24. Katherine A, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1α. J Clin Invest. 2007;117:1249–1259.
25. MacGuire JJ, et al. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s). Can J Physiol Pharmacol. 2001;79:443–470.
26. Busse R, et al. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2000;23:374–380.
27. Feletou M, et al. EDHF: the complete story. CRC Taylor and Francis: Boca Raton; 2006.
28. Feletou M, et al. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol. 2006;26:1215–1225.
29. Fleming I. Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol Res. 2004;49:525–533.
30. Fleming I, et al. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:2612–2618.
31. Villar IC, et al. Novel aspects of endothelium-dependent regulation of vascular tone. Kidney Int. 2006;70:840–853.
32. Mitchell JA, et al. COX isoforms in the cardiovascular system: understanding the activities of non-steroidal anti-inflammatory drugs. Nat Rev Drug Discov. 2006;5:75–86.
33. Vallance P, Webb D. Vascular endothelium in human physiology and pathophysiology. Harwood Academic Publishers: Amsterdam; 2000.
34. Abraham D, et al. Endothelin—role in vascular disease. Rheumatology (Oxford). 2008;47(Suppl 5):v23–v24.
35. Abraham D, et al. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9(Suppl 2):S2.
36. Feldstein C, et al. Role of endothelins in hypertension. Am J Ther. 2007;14:147–153.
37. Iglarz M, et al. Mechanisms of ET-1-induced endothelial dysfunction. J Cardiovasc Pharmacol. 2007;50:621–628.
38. Mangiafico RA, et al. Plasma endothelin-1 release in normal and varicose saphenous veins. Angiology. 1997;48:769–774.
39. McCarron RM, et al. Endothelial-mediated regulation of cerebral microcirculation. J Physiol Pharmacol. 2006;57(Suppl 11):133–144.
40. Rapoport RM, et al. Effects of EDCF and endothelin on aorta phosphatidylinositol hydrolysis and contraction in rat. Am J Physiol Cell Physiol. 1990;258:C122–C131.
41. Vanhoutte PM. Role of calcium and endothelium in hypertension, cardiovascular disease, and subsequent vascular events. J Cardiovasc Pharmacol. 1992;19(Suppl 3):S6–S10.
42. Zhou Y, et al. AT1b receptor predominantly mediates contractions in major mouse blood vessels. Circ Res. 2003;93:1089–1094.
43. Lasségue B, et al. Phosphatidylcholine is a major source of phosphatidic acid and diacylglycerol in angiotensin II-stimulated vascular smooth-muscle cells. Biochem J. 1993;292:509–517.
44. Laszik Z, et al. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–3640.
45. Ishii H, et al. Thrombomodulin, an endothelial anticoagulant protein, is absent from the human brain. Blood. 1986;67:362–365.
46. Aird WC. Vascular bed-specific hemostasis: role of endothelium in sepsis pathogenesis. Crit Care Med. 2001;29:S28–S35.
47. Juul K, et al. Factor V Leiden: The Copenhagen City Heart Study and 2 meta-analyses. Blood. 2002;100:3–10.
48. Weiler-Guettler H, et al. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101:1983–1991.
49. A randomized trial of aspirin and sulfinpyrazone in threatened stroke. The Canadian Cooperative Study Group. N Engl J Med. 1978;299:53–59.
50. Broos K, et al. Platelets at work in primary hemostasis. Blood Rev. 2011;25:155–167.
51. Vallance P, Webb D. Vascular endothelium in human physiology and pathophysiology. Harwood Academic Publishers: Amsterdam, Netherlands; 2000.
52. Giles TD, et al. Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens (Greenwich). 2012;14:198–205.
53. Walter U, et al. cGMP and cGMP-dependent protein kinase in platelets and blood cells. Handb Exp Pharmacol. 2009;5:33–48.
55. Gambaryan S, et al. NO-synthase-/NO-independent regulation of human and murine platelet soluble guanylyl cyclase activity. J Thromb Haemost. 2008;6:1376–1384.
56. Tymvios C, et al. Platelet aggregation responses are critically regulated in vivo by endogenous nitric oxide but not by endothelial nitric oxide synthase. Br J Pharmacol. 2009;158:1735–1742.
57. van Hinsbergh VWM. Endothelium—role in regulation of coagulation and inflammation. Semin Immunopathol. 2012;34(1):93–106.
58. Carreau A, et al. Nitric oxide modulates the expression of endothelial cell adhesion molecules involved in angiogenesis and leukocyte recruitment. Exp Cell Res. 2011;317:29–41.
59. Dal Secco D, et al. Nitric oxide inhibits neutrophil migration by a mechanism dependent on ICAM-1: role of soluble guanylate cyclase. Nitric Oxide. 2006;15:77–86.
60. Sun Y, et al. Nitric oxide inhibits T cell adhesion and migration by down-regulation of beta1-integrin expression in immunologically liver-injured mice. Int Immunopharmacol. 2006;6:616–626.
61. Feng D, et al. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med. 1998;187:903–915.
62. Carman CV, et al. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388.
63. Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036.
64. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314.
65. Aurrand-Lions M, et al. The last molecular fortress in leukocyte trans-endothelial migration. Nat Immunol. 2002;3:116–118.
66. Gallagher KA, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259.
67. Liu ZJ, et al. Identification of E-selectin as a novel target for the regulation of postnatal neovascularization: implications for diabetic wound healing. Ann Surg. 2010;252:625–634.
68. Castilla DM, et al. A novel autologous cell-based therapy to promote diabetic wound healing. Ann Surg. 2012;256:560–572.
69. Liu ZJ, et al. Inhibition of tumor angiogenesis and melanoma growth by targeting vascular E-selectin. Ann Surg. 2011;254(3):450–456.
70. Carmeliet P, et al. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257.
71. Eberhard A, et al. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–1393.
72. Morikawa S, et al. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160:985–1000.
73. Liu J, et al. The origins of vascularization in tumors. Front Biosci. 2012;17:2559–2565.
74. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
75. Thyberg J. Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int Rev Cytol. 1996;169:183–265.
76. Mulvany M, et al. Structure and function of small artery. Physiol Rev. 1990;70:922–946.
77. Hungerford JE, et al. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res. 1999;36:2–27.
78. Han CI, et al. Circulating bone marrow cells can contribute to neointimal formation. J Vasc Res. 2001;38:113–119.
79. Owens GK, et al. Wamhoff: Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
80. Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504.
81. Mulvany MJ, et al. Vascular remodeling. Hypertension. 1996;28:505–506.
82. Hermsmeyer K. Calcium channel function in hypertension. J Hum Hypertens. 1993;7:173–176.
83. Wellman GC, et al. Membrane depolarization, elevated Ca2+ entry, and gene expression in cerebral arteries of hypertensive rats. Am J Physiol Heart Circ Physiol. 2001;281:H2559–H2567.
84. Intengan HD, et al. Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension. 2000;36:312–318.
85. Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517.
86. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. N Engl J Med. 1993;362:801–809.
87. Galis ZS, et al. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262.
88. Galis ZS, et al. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions in human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503.
89. Armulik A, et al. Dev Cell. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;16(21):193–215.
90. Müller SM, et al. Neural crest origin of perivascular mesenchyme in the adult thymus. J Immunol. 2008;180:5344–5351.
91. Foster K, et al. Contribution of neural crest-derived cells in the embryonic and adult thymus. J Immunol. 2008;180:3183–3189.
92. Korn J, et al. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J Comp Neurol. 2002;442:78–88.
93. Etchevers HC, et al. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development. 2001;128:1059–1068.
94. Asahina K, et al. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011;53:983–995.
95. Que J, et al. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc Natl Acad Sci USA. 2008;105:16626–16630.
96. Wilm B, et al. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development. 2005;132:5317–5328.
97. Díaz-Flores L, et al. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24:909–969.
98. Stratman AN, et al. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101.
99. Stratman AN, et al. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood. 2010;116:4720–4730.
100. Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23.
101. Hellström M, et al. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055.
102. Lindahl P, et al. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245.
103. Chen S, et al. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF-beta. Nucleic Acids Res. 2003;31:1302–1310.
104. Goumans MJ, et al. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–1753.
105. Ota T, et al. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J Cell Physiol. 2002;193:299–318.
106. Sundberg C, et al. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest. 2002;82:387–401.
108. Falcón BL, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175:2159–2170.
109. Gaengel K, et al. Endothelial mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638.
110. Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514.
111. Vikkula M, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190.
112. Thurston G, et al. Angiopoietin causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132:3317–3326.
113. Iwamoto R, et al. Heparin binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA. 2003;100:3221–3226.
114. Nanba D, et al. Loss of HB-EGF in smooth muscle or endothelial cell lineages causes heart malformation. Biochem Biophys Res Commun. 2006;350:315–321.
115. Yu X, et al. Heparin-binding EGF-like growth factor protects pericytes from injury. J Surg Res. 2012;172:165–176.
116. Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561.
117. Daneman R, et al. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566.
118. Bondjers C, et al. Microarray analysis of blood microvessels from PDGF-B and PDGF-R beta mutant mice identifies novel markers for brain pericytes. FASEB J. 2006;20:1703–1705.
119. Heglind M, et al. Lack of the central nervous system- and neural crest-expressed forkhead gene Foxs1 affects motor function and body weight. Mol Cell Biol. 2005;25:5616–5625.
120. Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243.
121. Hamilton NB, et al. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics. 2010;21:2.
122. Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037.
123. Collett G, et al. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ Res. 2003;92:1123–1129.
124. Dellavalle A, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–267.
125. Doherty MJ, et al. Vascular pericytes express osteogenic potential in vitro and in vivo. J Bone Miner Res. 1998;13:828–838.
126. Farrington-Rock C, et al. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation. 2004;110:2226–2232.
127. Shi S, et al. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Miner Res. 2003;18:696–704.
128. Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313.
129. Davidoff MS, et al. Progenitor cells of the testosterone-producing Leydig cells revealed. J Cell Biol. 2004;167:935–944.
130. Feng J, et al. Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proc Natl Acad Sci USA. 2011;108:6503–6508.
131. Olson LE, et al. PDGFRb signaling regulates mural cell plasticity and inhibits fat development. Dev Cell. 2011;20:815–826.
132. Tang W, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–586.
133. Dellavalle A, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–267.
134. Dore-Duffy P, et al. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab. 2006;26:613–624.
135. Fabris L, et al. Epithelial-mesenchymal interactions in biliary diseases. Semin Liver Dis. 2011;31:11–32.
136. Schrimpf C, et al. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20:297–305.
137. Mahoney WM Jr, et al. A unifying hypothesis for scleroderma: identifying a target cell for scleroderma. Curr Rheumatol Rep. 2011;13:28–36.
138. Polyak K, et al. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38.
139. Joyce JA, et al. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252.
140. Hanahan D, et al. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
141. Abramsson A, et al. Analysis of mural cell recruitment to tumor vessels. Circulation. 2002;105:112–117.
142. Nolan-Stevaux O, et al. Differential contribution to neuroendocrine tumorigenesis of parallel EGFR signaling in cancer cells and pericytes. Genes Cancer. 2010;1:125–141.
143. Song N, et al. Overexpression of platelet-derived growth factor-BB increases tumor pericyte content via stromal-derived factor-1alpha/CXCR4 axis. Cancer Res. 2009;69:6057–6064.
144. Morikawa S, et al. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160:985–1000.
145. Coen M, et al. Myofibroblast-mediated adventitial remodeling: an underestimated player in arterial pathology. Arterioscler Thromb Vasc Biol. 2011;31:2391–2396.
146. Chang HY, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–12882.
147. Stenmark KR, et al. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda). 2006;21:134–145.
148. Patel S, et al. Characteristics of coronary smooth muscle cells and adventitial fibroblasts. Circulation. 2000;101:524–532.
149. Scott NA, et al. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation. 1996;93:2178–2187.
150. Shi Y, et al. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94:1655–1664.
151. Hinz B, et al. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816.
152. Tomasek JJ, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363.
153. Follonier Castella L, et al. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316:2390–2401.
154. Mulvany MJ, et al. Vascular remodeling. Hypertension. 1996;28:505–506.
155. Glagov S, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375.
156. Gruntzig A. Transluminal dilatation of coronary-artery stenosis. Lancet. 1978;1:263.
157. De Meyer GRY, et al. Possible mechanisms of collar-induced intimal thickening. Arterioscler Thromb Vasc Biol. 1997;17:1924–1930.
158. Pasterkamp G, et al. Relation of arterial geometry to luminal narrowing and histologic markers for plaque vulnerability: the remodeling paradox. J Am Coll Cardiol. 1998;32:655–662.
159. Zalewski A, et al. Vascular myofibroblasts: lessons from coronary repair and remodeling. Arterioscler Thromb Vasc Biol. 1997;17:417–422.
160. Shi Y, et al. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93:340–348.
161. Herrmann J, et al. Differential effect of experimental hypertension and hypercholesterolemia on adventitial remodeling. Arterioscler Thromb Vasc Biol. 2005;25:447–453.
162. Shi Y, et al. Remodeling of autologous saphenous vein grafts: the role of perivascular myofibroblasts. Circulation. 1997;95:2684–2693.
163. Subramanian SV, et al. Reprogramming of vascular smooth muscle alpha-actin gene expression as an early indicator of dysfunctional remodeling following heart transplant. Cardiovasc Res. 2002;54:539–548.
165. Maiellaro K, et al. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648.
166. McGettrick HM, et al. Fibroblasts from different sites may promote or inhibit recruitment of flowing lymphocytes by endothelial cells. Eur J Immunol. 2009;39:113–125.
167. Buckley CD, et al. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204.
168. Orimo A, et al. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–1601.
169. Powell DW, et al. Myofibroblasts. I: paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9.
170. Smith RS, et al. Fibroblasts as sentinel cells: synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317–322.
171. Newton R, et al. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem Biophys Res Commun. 1997;237:28–32.
172. Harding KG, et al. Wound chronicity and fibroblast senescence—implications for treatment. Int Wound J. 2005;2:364–368.
173. Mornex JF, et al. From granuloma to fibrosis in interstitial lung diseases: molecular and cellular interactions. Eur Respir J. 1994;7:779–785.
174. Gonzalez-Cuadrado S, et al. Expression of leucocyte chemoattractants by interstitial renal fibroblasts: up-regulation by drugs associated with interstitial fibrosis. Clin Exp Immunol. 1996;106:518–522.
175. Desmouliere A, et al. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12.
176. Powell DW, et al. Myofibroblasts. I: paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9.
177. Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313.
178. Campagnolo P, et al. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation. 2010;121:1735–1745.
179. Zheng B, et al. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nature Biotechnology. 2007;25:1025–1034.
180. Dellavalle A, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nature Cell Biology. 2007;9:255–267.
181. Corselli M, et al. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells and Development. 2012;21:1299–1308.
182. Zheng B, et al. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nature Biotechnology. 2007;25:1025–1034.
183. Schor AM, et al. Pericytes derived from the retinal microvasculature undergo calcification in vitro. J Cell Sci. 1990;97:449–461.
184. Chen CW, et al. Human blood-vessel-derived stem cells for tissue repair and regeneration. J Biomed Biotechnol. 2012;2012:597439.
185. He W, et al. Pericyte-based human tissue engineered vascular grafts. Biomaterials. 2010;31:8235–8244.
186. Adolphe C, et al. An in vivo comparative study of sonic, desert and Indian hedgehog reveals that hedgehog pathway activity regulates epidermal stem cell homeostasis. Development. 2004;131:5009–5019.
187. Ahn S, et al. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897.
188. Uhmann A, et al. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood. 2007;110:1814–1823.
189. Tigerstedt NM, et al. Vascular cell kinetics in response to intimal injury ex vivo. J Vasc Res. 2010;47:35–44.