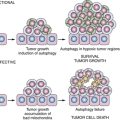
mTORC1 and Autophagy
mTORC1 and p53
Beyond Cell Growth: Does mTOR Regulate Organ and Organism Growth?
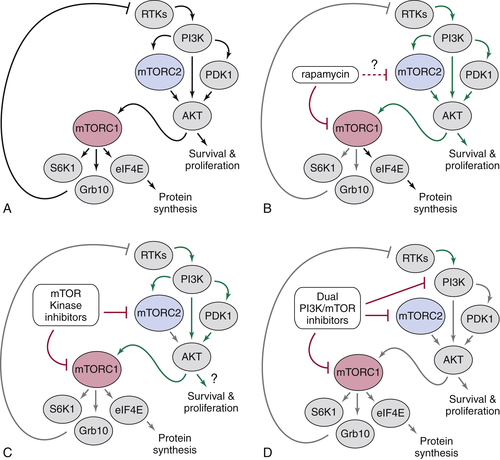
The mTORC2 Pathway
Controlling Body Size
Targeting mTOR Signaling as a Treatment for Cancer and Other Human Diseases of Cell Growth
1. Cell size control . Encyclopedia of Life Sciences . New York, NY : John Wiley & Sons, Ltd ; 2005 .
2. mTOR signaling in growth control and disease . Cell . 2012 ; 149 : 274 – 293 .
3. Molecular mechanisms of mTOR-mediated translational control . Nat Rev Mol Cell Biol . 2009 ; 10 : 307 – 318 .
4. mTOR mediated anti-cancer drug discovery . Drug Discov Today Ther Strateg . 2009 ; 6 : 47 – 55 .
5. Rapamycin passes the torch: a new generation of mTOR inhibitors . Nat Rev Drug Discov . 2011 ; 10 : 868 – 880 .
6. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling . Science . 2011 ; 332 : 1317 – 1322 .
7. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling . Science . 2011 ; 332 : 1322 – 1326 .
8. The multifaceted role of mTORC1 in the control of lipid metabolism . EMBO Rep . 2013 ; 14 ( 3 ) : 242 – 251 .
9. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1 . Science . 2013 ; 339 : 1323 – 1328 .
10. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis . Science . 2013 ; 339 : 1320 – 1323 .
11. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase . Science . 2011 ; 334 : 678 – 683 .
12. Nutrient sensing, metabolism, and cell growth control . Mol Cell . 2013 ; 49 : 379 – 387 .
13. AKT/PKB signaling: navigating downstream . Cell . 2007 ; 129 : 1261 – 1274 .
14. Amino acid signalling upstream of mTOR . Nat Rev Mol Cell Biol . 2013 ; 14 : 133 – 139 .
15. Amino acids and mTORC1: from lysosomes to disease . Trends Mol Med . 2012 ; 18 : 524 – 533 .
16. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival . Nature . 2013 ; 493 : 679 – 683 .
17. Defining the role of mTOR in cancer . Cancer Cell . 2007 ; 12 : 9 – 22 .
18. Current development of mTOR inhibitors as anticancer agents . Nat Rev Drug Discov . 2006 ; 5 : 671 – 688 .
19. Targeting PI3K signalling in cancer: opportunities, challenges and limitations . Nat Rev Cancer . 2009 ; 9 : 550 – 562 .
20. Targeting the phosphoinositide 3-kinase pathway in cancer . Nat Rev Drug Discov . 2009 ; 8 : 627 – 644 .
21. Deconvoluting the context-dependent role for autophagy in cancer . Nat Rev Cancer . 2012 ; 12 : 401 – 410 .
22. The role of autophagy during the early neonatal starvation period . Nature . 2004 ; 432 : 1032 – 1036 .
23. Mammalian autophagy: core molecular machinery and signaling regulation . Curr Opin Cell Biol . 2009 ; 22 : 124 – 131 .
24. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism . Nat Cell Biol . 2011 ; 13 : 1016 – 1023 .
25. AMPK and mTOR in cellular energy homeostasis and drug targets . Annu Rev Pharmacol Toxicol . 2012 ; 52 : 381 – 400 .
26. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res . 2006 ; 66 : 2885 – 2888 .
27. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways . Genes Dev . 2006 ; 20 : 267 – 275 .
28. p53 regulates mitochondrial respiration . Science . 2006 ; 312 : 1650 – 1653 .
29. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy . Oncogene . 2010 ; 29 : 3733 – 3744 .
30. mTOR complex 2 signaling and functions . Cell Cycle . 2011 ; 10 : 2305 – 2316 .
31. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice . Cancer Cell . 2009 ; 15 : 148 – 159 .
32. The nuts and bolts of AGC protein kinases . Nat Rev Mol Cell Biol . 2010 ; 11 : 9 – 22 .
33. A nutrient sensor mechanism controls Drosophila growth . Cell . 2003 ; 114 : 739 – 749 .
34. Growing roles for the mTOR pathway . Curr Opin Cell Biol . 2005 ; 17 : 596 – 603 .
35. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14 . Nat Med . 2007 ; 13 : 38 – 45 .
36. The pharmacology of mTOR inhibition . Sci Signal . 2009 ; 2 : pe24 .
37. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1 . J Biol Chem . 2009 ; 284 : 8023 – 8032 .
38. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB . Mol Cell . 2006 ; 22 : 159 – 168 .
39. An expanding role for mTOR in cancer . Trends Mol Med . 2005 ; 11 : 353 – 361 .
40. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease . Oncogene . 2005 ; 24 : 7475 – 7481 .
41. TOR signaling in growth and metabolism . Cell . 2006 ; 124 : 471 – 484 .
42. Ras, PI(3)K and mTOR signalling controls tumour cell growth . Nature . 2006 ; 441 : 424 – 430 .