[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Unlike tumor cells with PTEN loss, tumor cells that have lost TSC function are generally less belligerent. It is now accepted that this is due to negative feedback loops that suppress the PI3K-AKT pathway when mTORC1 activity is high.
2,6,7 For example, the mTORC1 substrate S6K1 can directly phosphorylate and inactivate the IRS1 and IRS2 proteins, two mediators of PI3K activation, which decreases PI3K-AKT activity. A second feedback mechanism occurs through the direct mTORC1 substrate Grb10, which is activated by mTORC1 and functions to inhibit both the insulin/IGF receptors and IRS proteins. Negative feedback mechanisms would normally keep cell growth and nutrient uptake in balance, but in the absence of TSC, negative feedback squelches PI3K and AKT activity, which is important for promoting cell survival and metastasis. This may be why tuberous sclerosis (discussed later), which is caused by mutations in TSC, generally does not develop into metastatic cancer. Because PTEN and PI3K are downstream of the insulin/IGF receptors and IRS proteins, cancers driven by loss of PTEN or PI3K activation are uncoupled from these particular mTORC1-driven negative feedback loops, explaining why they are typically associated with more aggressive cancer.
mTORC1 and Autophagy
Macroautophagy, or simply autophagy, is a catabolic process induced on nutrient limitation or stress in which autophagosomes form and fuse with the lysosome to create an autophagolysosome. The autophagolysosome is the final destination for proteins and organelles that are broken down into their basic components to be recycled as sources of cellular energy and metabolites during starvation. Autophagy has an important role in cell survival during times of energy crisis. Under normal growth conditions, autophagy also salvages and recycles cellular junk such as excess or damaged organelles. Newborn mice require autophagy to survive a brief starvation period immediately after birth, and defects in autophagy are associated with cancer, degenerative disorders, and aging.
21–23 A connection between mTORC1 signaling at autophagy first became apparent in yeast and mammalian cells in studies where rapamycin was found to induce autophagy. Genetic inactivation of TOR in
Drosophila also induces autophagy and confirmed that mTORC1 not only positively controls anabolic processes, but suppresses catabolic processes opposing growth.
Recent studies have revealed that the regulation of autophagy by mTORC1 is at least partly direct and occurs in coordination with AMPK.
24,25 More than 30 autophagy
(ATG) genes have been identified that control autophagy. In nutrient-rich conditions, mTORC1 associates with and directly phosphorylates ULK1/ATG1, the kinase subunit of a complex that is required for autophagy induction. Phosphorylation of ULK1 by mTORC1 inhibits its activity and reduces autophagic vesicle formation. In nutrient-deprived cells, AMPK not only inhibits mTORC1 by phosphorylating TSC2 and Raptor (as described earlier), but also directly activates ULK1 kinase activity. When mTORC1 activity decreases, ULK1 is dephosphorylated, allowing AMPK to now interact with, phosphorylate, and activate ULK1 to induce autophagy. This dual regulatory circuit emphasizes the tight balance between anabolic and catabolic processes.
Interestingly, prolonged autophagy, which delivers its cargo to the lysosome, eventually reactivates mTORC1. Thus, in nutrient-starved cells, autophagy may also be required to keep mTORC1 on in a minimally activated state to promote the synthesis of certain macromolecules essential for survival. The reactivation of mTORC1 also promotes the formation of nascent lysosomes that bud from the autophagosome. In fact, recent studies further suggest that mTORC1 may normally regulate lysosome formation through its ability to control that nuclear localization (and thus activity) of a transcription factor called TFEB. TFEB regulates the expression of many genes important for lysosome formation. Thus, the connections among nutrient and energy sensing, mTORC1 growth control, and protein/organelle turnover by autophagy and the lysosome are complex, and it will be interesting to determine if and how defects in these connections promote cancer.
Although the connections between mTORC1 and autophagy are being worked out, the strong connections between mTORC1 and cancer suggest that autophagy may have a role in tumorigenesis, and evidence supports this hypothesis.
21 For example, mice deficient for a pro-autophagy gene, called
beclin-1, have an increased frequency of spontaneous tumor formation, suggesting that
beclin-1 is a tumor suppressor gene. How could inactivation of autophagy promote tumorigenesis? It is difficult to speculate at this time because little is known about Beclin 1, but perhaps some transformed cells rely on autophagy to remove damaged organelles or for complete self-disposal. In this model, mutations that prevent autophagy would promote cell survival. Interestingly, the Bcl-2 pro-survival oncoprotein, which has long been thought to inhibit apoptosis, binds and inhibits Beclin 1–dependent autophagy.
26 This suggests that the oncogenic activity of Bcl-2 might be linked to inhibiting nonapoptotic autophagic cell death. Contrary to having tumor suppressor capacity, autophagy could promote tumorigenesis in some cases. When cancer cells experience nutrient limitation, autophagy may provide the rations necessary to sustain the most essential bioenergetic cellular process. This may allow cancer cells to survive until angiogenesis is initiated or other deleterious mutations occur. Understanding the role of autophagy in different cancer cells will be important to designing mTOR-focused treatment strategies.
mTORC1 and p53
Mutation of the p53 tumor suppressor is one of the most common genetic abnormalities associated with human cancer. DNA or spindle damage, telomere shortening, various stresses, or oncogenic mutations will usually initiate a p53-dependent checkpoint that arrests cells in G
1 or trigger apoptosis (depending on the cell type and signals present). This has led to speculation that such a checkpoint mechanism involving p53 might communicate with the mTORC1 pathway to discourage growth when conditions are unfavorable. Early reports from work with cultured cells suggest that AMPK and p53 may communicate in some type of checkpoint mechanism to restrict growth in low-glucose conditions or when DNA is damaged, although it is unclear from this early work if and how directly mTORC1 regulation might be connected.
27 Mounting evidence also indicates that a communication link exists between p53 and the regulation of mitochondrial respiration, particularly in mediating a cellular switch to preferentially deriving energy from glycolysis rather than aerobic respiration—a hallmark characteristic of cancer cells.
28 It is well known that p53 and
the mitochondria, and to some extent mTOR, have roles in apoptosis. Because signals from the mitochondria are also thought to control mTORC1’s ability to mediate growth, it seems likely that a complicated signaling circuitry involving both the p53- and mTOR-dependent pathway exists.
Beyond Cell Growth: Does mTOR Regulate Organ and Organism Growth?
The mTORC2 Pathway
The role of TOR in building cell mass is well documented and conserved in all eukaryotic organisms examined. However, extending a role for mTOR in controlling organ and organism growth is more complicated, because most tissues grow by mechanisms involving the collective coordination of cell growth, cell division, and cell death pathways. The finding that mTOR exists in a second complex called mTORC2 does suggest that mTOR may have broad influence over cell growth, cell division, cell death, metabolism, and many other processes essential for building tissues and organisms. Many of these additional targets of mTOR activity, although not completely worked out yet, are likely to be important in cancer progression, and thus emerging therapeutic strategies are now aiming to more broadly target mTOR signaling by inhibiting both mTORC1 and mTORC2.
The second mTOR complex also contains mLST8 and Deptor, but instead of Raptor and PRAS40, mTORC2 contains the Rictor, mSin1, and PROTOR proteins (see
Figure 12-1).
2 Understanding the function of mTORC2 initially eluded researchers because the complex is insensitive to acute rapamycin treatment, and thus rapamycin could not be used to probe for mTORC2 substrates. However, advances in RNA-interference technology allowed the specific depletion of Rictor from cells, leading to the discovery that mTOR, when outfitted with mTORC2-specific regulatory proteins, phosphorylates and activates the AKT kinase. Current models suggest that on recruitment to the membrane after growth factor stimulation, AKT is phosphorylated by mTORC2 in conjunction with PDK1, and this co-regulation is presumed necessary for full AKT activation. How mTORC2 activity itself is controlled remains to be worked out, but initial experiments indicate that growth factors also modulate mTORC2 activity by an unknown mechanism.
AKT activity influences cell growth, cell proliferation, cell survival, metabolism, and many other processes.
13 Thus, by coupling mTOR activity with proliferation and survival, the discovery of mTORC2, together with the well-known role of mTORC1 in controlling cell size, strengthens the argument that mTOR growth regulation extends beyond cell autonomous growth to organ and organism growth. Moreover, because of the widespread role of PI3K-AKT signaling in cancer (see
Table 12-1), the finding that mTORC2 phosphorylates and activates AKT is a compelling additional link between mTOR and cancer. In fact, a number of studies now suggest that selectively targeting mTORC2 could also be a promising therapeutic strategy.
29,30 What is interesting about potentially targeting mTORC2 is that in many cases, loss of mTORC2 function (by genetic approaches) is less toxic to normal cells than targeting mTORC1 function. In contrast, mTORC2 activity is required for transformation, particularly when transformation is driven by loss of PTEN.
31 Thus, mTORC2 might be more essential to certain cancer cells than to many normal cells. The challenge of selectively targeting mTORC2 is that beyond the mTOR catalytic domain, the understanding of mTORC2 structure and regulation is only vague, and thus it is currently difficult to predict what type of targeting strategy might best achieve this goal.
In retrospect, it is not surprising that mTOR can also phosphorylate AKT, because the mTORC2 phosphorylation site on AKT (S473) is structurally similar to the mTORC1 site on S6K1 (T389), both of which are present in carboxy-terminal hydrophobic motifs. In fact, both S6K and AKT belong to a larger family of structurally related kinases called the AGC kinases.
32 The discovery of the mTORC2-AKT signaling module also sets up the peculiar situation whereby mTOR can regulate itself in a manner dependent on what proteins it interacts with. For instance, mTORC2-mediated phosphorylation of AKT is proposed to promote subsequent phosphorylation and inactivation of TSC2 and PRAS40 (as described earlier), which would place mTORC2 upstream of mTORC1 regulation. The extent to which normal and cancer cells rely on such regulation is under investigation. How other AKT substrates are regulated by mTORC2 is also largely unclear. In addition to AKT, mTORC2 can directly phosphorylate two additional AGC kinases called SGK and PKC. SGK is most similar to AKT, except that it lacks the membrane-targeting PH domain that brings AKT to its activation site at the cell membrane. SGK also has some overlapping functions with AKT, but currently the role of mTORC2-mediated SGK and PKC regulation in cancer is unknown.
Controlling Body Size
It is a remarkable feat for mammals to coordinate the development of their organs to appropriate sizes that are proportional to overall body size. The endocrine system is responsible for conducting this massively orchestrated systemic growth. The major hormone responsible for controlling postnatal growth is growth hormone (GH). GH is synthesized in the pituitary gland. After its release into the blood, circulating GH stimulates the release of insulin-like
growth factors (IGFs) from the liver. IGFs subsequently stimulate growth of bone and muscle, which are the two organs most relevant to determining body size.
The findings (1) that mTOR regulates cell growth, proliferation, and survival and (2) that both mTORC1 and mTORC2 are downstream of insulin/IGF signaling raise the question of whether mTOR is a master regulator of body size. The role of mTOR as a nutrient-sensitive regulator of eukaryotic cell growth (i.e., as part of mTORC1) is an ancient function conserved from yeast to humans. During the evolution of multicellular organisms, growth factor signaling was grafted onto the mTORC1 pathway, probably to modulate autonomous cell growth in conjunction with systemic nutrient availability and signals from other cells and tissues. The discovery that mTOR (as part of mTORC2) regulates AKT further suggests that additional pathways independent of mTORC1 may contribute to the regulation of cell growth, proliferation, survival, and metabolism. Many mTORC2 components are also conserved in yeast—although the protein sequences and downstream functions are more diverged compared to mTORC1, suggesting the cellular functions of mTORC2 may have also diverged. Because of mTOR’s remarkable ability to integrate numerous signals and control many aspects of cell growth, it seems poised to be a master regulator of organ and body size control. Studying this in mammals has been difficult, however, because mice null for mTOR and the essential mTORC1 and mTORC2 regulatory subunits are embryonic lethal. In
Drosophila, the fat body (which may be equivalent to the vertebrate liver) functions as a nutrient-sensing organ that can control body growth through a TOR-dependent mechanism.
33 Moreover, mice and flies deficient for S6K1 are viable but reduced in size because of smaller cells, implying a link between mTORC1 signaling and body size control.
34 Thus, limited evidence in model organisms suggests that mTOR could influence body size by systemic as well as cell-autonomous mechanisms. Interestingly, in humans a rare mutation in the p14/LAMTOR2 subunit of the Ragulator delays growth and causes immunosuppression and hypopigmentation.
35 As novel upstream regulators and substrates of mTOR are unveiled, it will be interesting to determine if genetic variation in components of the mTOR network has a role in determining organ and organism size.
Targeting mTOR Signaling as a Treatment for Cancer and Other Human Diseases of Cell Growth
With a role for mTOR signaling in cancer now firmly established, developing molecules that inhibit mTOR for therapeutic purposes is a significant focus of the pharmaceutical industry. As mentioned earlier, rapamycin (also known as
sirolimus) is used clinically for immunosuppression (because of its ability to suppress T-cell proliferation), to prevent restenosis (by preventing the growth of vascular smooth muscle cells), and to treat certain cancers.
5,17,36 Analogs of rapamycin have also been developed for cancer therapy (CCI779/temsirolimus, AP23573, and RAD001/everolimus—collectively called rapalogues). Unfortunately, rapamycin as a broad anticancer drug has had limited success as a single-agent therapeutic, even against cancers with elevated PI3K-AKT signaling. The response to rapamycin is highly variable depending on the cancer, and only a few cancers, including mantle-cell lymphoma, renal-cell carcinoma, and endometrial cancers, have shown consistent positive responses. Temsirolimus was approved in 2007 to treat renal-cell carcinoma. Several trials are ongoing, although there is no clear biomarker predictive of rapamycin sensitivity.
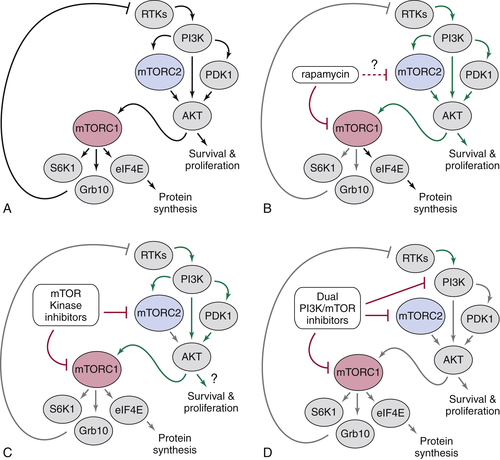
A number of reasons have been proposed to explain rapamycin’s general ineffectiveness as a cancer drug. One possibility (discussed earlier) is that inhibiting mTORC1 relieves strong negative feedback loops that function to suppress receptor tyrosine kinase (RTK) signaling and downstream PI3K-AKT activation (
Figure 12-2 ). Therefore, treating cells with rapamycin boosts AKT signaling, which in turn could drive AKT-dependent cell proliferation and survival pathways—clearly an undesirable response. The MAPK-ERK signaling pathway is also triggered by mTORC1-S6K1 inhibition. Using rapamycin in combination with PI3K or MAPK pathway inhibitors might prove to be a better strategy. Second, it is now clear that rapamycin only partially inhibits mTORC1. For example, although rapamycin results in rapid dephosphorylation and sustained inactivation of S6K, phosphorylation of 4E-BP1 is only partially and transiently decreased.
37 Therefore, translation is largely insensitive to rapamycin. As more mTORC1 substrates are being discovered, it will be interesting to determine why substrates vary in their sensitivity to rapamycin.
It is often stated that rapamycin specifically targets mTORC1, implying that it does not inhibit mTORC2. However, the actual situation is more complex.
2,38 In cultured cells, rapamycin acutely affects only mTORC1, but prolonged exposure disrupts mTORC2 integrity, likely by blocking the assembly of new mTORC2 complexes. In some cell types this results in decreased AKT phosphorylation, but often there is no apparent change or increased AKT phosphorylation. Notably, any mTORC2 complexes that remain intact following rapamycin treatment are still susceptible to losing feedback inhibition. This likely explains the cell-to-cell variability in AKT phosphorylation following prolonged rapamycin treatment. For example, even if most mTORC2 complexes are
disrupted by rapamycin, what remains intact is hyperactive, and consequently AKT phosphorylation can appear insensitive to rapamycin or increase. Exactly what determines whether AKT phosphorylation in a particular cancer cell will show increased, decreased, or no sensitivity to rapamycin remains a mystery. Nevertheless, inhibition of mTORC2 following chronic exposure to rapamycin could explain the drug’s effectiveness in certain cancers as well as some of its side effects. Dissecting the molecular mechanisms by which rapamycin functions and identifying biomarkers predictive of rapamycin sensitivity remains an important area of research.
Next-generation mTOR inhibitors are ATP competitive inhibitors (discussed earlier). Therefore, unlike the rapalogues (which are allosteric inhibitors), these drugs directly target mTOR kinase activity.
2,5 Consequently, the ATP competitive inhibitors more completely inhibit mTORC1 and also inhibit mTORC2. The mTOR ATP-competitive inhibitors are having a major impact in basic research, and several versions are being evaluated in clinical trials as anticancer drugs with the hope that they will outperform the rapalogues. Their tolerability remains to be seen, but early data are encouraging. Notably, losing feedback inhibition remains a challenge with these inhibitors because, despite the fact that they inhibit mTORC2 as well, the mTORC2 substrate AKT is also regulated by the PDK1 kinase, which increases activity following mTORC1 inhibition because the negative feedback is lost (see
Figure 12-2). A related class of inhibitors is called the
dual-specificity inhibitors, which target mTORC1, mTORC2, and PI3K. Dual inhibition is possible because the kinase domains of mTOR and PI3K are structurally similar, even though mTOR phosphorylates proteins and PI3K phosphorylates lipids. The dual-specificity inhibitors could have an advantage because by also inhibiting PI3K, they can mitigate the effects of losing the mTORC1-dependent negative feedback inhibition of PI3K-AKT signaling (see
Figure 12-2). Because many
research studies and clinical trials are ongoing with both ATP-competitive and dual-specificity mTOR inhibitors, it should soon become clear to what extent they will be useful clinically.
Figure 12-2 Model explaining how downstream signaling in cancer cells responds to currently available classes of mTOR pathway inhibitors (A) Simplified depiction of how the mTOR signaling circuitry is wired (described in the text). Note that PI3K is shown here activating mTORC2; however, exactly how growth factors activate mTORC2 is unknown. (B) Rapamycin (or the rapalogues) only partially inhibits mTORC1 and relieves strong mTORC1-dependent negative feedback inhibition of receptor tyrosine kinase (RTK)-PI3K signaling. Note that prolonged rapamycin treatment can inhibit mTORC2 in some cell types, but the therapeutic significance of this is unclear. (C) The mTOR kinase inhibitors are ATP-competitive inhibitors that target both complexes. However, these inhibitors also release PI3K from mTORC1-dependent feedback inhibition. (D) The dual-specificity inhibitors target both mTOR complexes and PI3K and thus may mitigate the consequences of losing feedback inhibition.
Beyond the obvious rationale for targeting mTOR in human cancer, a variety of tumor-prone syndromes are also linked to mutations that impinge on mTOR (see
Table 12-1).
18,39–42 These include tuberous sclerosis complex (caused by mutations in the
TSC1 or
TSC2 genes, a related disease called lymphangioleiomyomatosis or LAM (
TSC1 and
TSC2), Cowden disease
(PTEN), Bannayan-Riley-Ruvalcaba syndrome
(PTEN), Proteus and Proteus-like syndrome
(PTEN), Peutz-Jeghers syndrome (
LKB1—an upstream activator of AMPK), and neurofibromatosis (
NF1—a negative regulator of Ras-PI3K activity). Patients suffering from these syndromes are candidates for therapeutic intervention with mTOR inhibitors.
Tuberous sclerosis complex is characterized by benign tumors that can grow in the brain, kidney, heart, eyes, lungs, and skin and can be devastating and fatal depending on the degree of penetrance and location of the tumors. LAM usually affects women and results in abnormal growth of smooth muscle cells in the lungs that severely compromises respiration. The discovery that TSC negatively regulates mTORC1 propelled rapamycin into clinical trials to treat patients suffering from these diseases. Some features of tuberous sclerosis complex, such as the appearance of abnormally large astrocytomas in the brain, are reduced in size following rapamycin treatment, and everolimus is now FDA approved for treating this disease. Notably, the TSC complex may also have mTORC1-independent functions, and some studies have concluded that not all cellular phenotypes associated with losing TSC function are mTOR dependent. Although the rationale to use mTOR inhibitors to treat tuberous sclerosis complex and LAM is compelling, it will be important to consider the mTOR-independent roles of TSC when evaluating trial results.
Inactivation of PTEN can cause Cowden disease in addition to cancer. Cowden disease is characterized by hamartomas present in the skin, mucosa, gastrointestinal tract, bones, central nervous system, eyes, and genitourinary tract. Patients are at a high risk for breast and thyroid carcinoma. As described earlier, PTEN negatively regulates the PI3K-AKT signaling pathway, which can subsequently inactivate the TSC complex. Peutz-Jeghers syndrome is caused by inactivation of the LKB1 tumor suppressor and results in intestinal hamartomatous polyps and increased risk of intestinal cancer. LKB1 is a protein kinase that phosphorylates and activates AMPK. As discussed earlier, AMPK inactivates mTORC1 following energy depletion by promoting TSC function and by phosphorylating Raptor. Neurofibromatosis primarily results in tumors growing on nerve tissue but can also cause skin and bone abnormalities. The disease results from inactivation of NF1, a Ras-GTPase activating protein that, when absent, renders phosphorylation of TSC2 and S6K1 by AKT and mTORC1, respectively, resistant to growth factor (but not nutrient) withdrawal. Loss of NF1 leads to accumulation of Ras in the active GTP-bound state, suggesting that Ras, a well-known oncogene, also affects mTORC1 activity.
The links between signaling networks that control cell, organ, and organism growth through mTOR and the onset of human cancer suggest that therapeutic interventions targeting this pathway and its regulators could have promise. The potential for rapamycin and its analogs in treating cancer and other growth diseases captivated the pharmaceutical industry, and substantial investments in its development have led to extensive and ongoing clinical assessment. Despite years of research, rapamycin’s mechanism of action remains enigmatic, and it is now clear that rapamycin only partially inhibits mTORC1. Effective use of the drug as an anticancer agent will require a deeper understanding of why only subsets of cancer cells are sensitive and whether there are efficacious combination therapies that can be exploited. Discovering that mTOR exists in at least two distinct complexes with differing sensitivity to rapamycin provided a strong rationale for developing novel inhibitors that target both complexes. Preclinical and clinical trials should soon inform the community of how efficacious these drugs will be. Other new potential avenues for developing mTORC1 pathway inhibitors include targeting the amino acid– and glucose-sensing pathway that delivers mTORC1 to the lysosome. However, it is possible that mTORC1 is so essential for maintaining normal cellular homeostasis that inhibiting it systemically will be too toxic to normal cells. Selectively inhibiting mTORC2 is an alternative strategy, at least in PI3K-driven cancers, as this could be less toxic to normal cells and leave mTORC1-dependent negative feedback loops largely intact. This, however, may prove to be a daunting task, as there is currently no obvious mTORC2 functional domain (outside the mTOR kinase domain) or essential upstream kinase that can be targeted. Because unique interacting proteins define the mTOR complexes, potential exists for developing mTORC2-selective inhibitors that might disrupt complex integrity or localization. Obtaining greater knowledge of the structure and function of the individual mTORC2 subunits will be essential for this endeavor. The discovery that prominent cancer pathways impinge on mTOR signaling has provided many new potential avenues for drug development, and ongoing efforts to develop mTOR inhibitors will undoubtedly affect the care of cancer patients.
References
1. Guertin D.A. , Sabatini D.M. Cell size control . Encyclopedia of Life Sciences . New York, NY : John Wiley & Sons, Ltd ; 2005 .
2. Laplante M. , Sabatini D.M. mTOR signaling in growth control and disease . Cell . 2012 ; 149 : 274 – 293 .
3. Ma X.M. , Blenis J. Molecular mechanisms of mTOR-mediated translational control . Nat Rev Mol Cell Biol . 2009 ; 10 : 307 – 318 .
4. Liu Q. , Thoreen C. , Wang J. , Sabatini D. , Gray N.S. mTOR mediated anti-cancer drug discovery . Drug Discov Today Ther Strateg . 2009 ; 6 : 47 – 55 .
5. Benjamin D. , Colombi M. , Moroni C. , Hall M.N. Rapamycin passes the torch: a new generation of mTOR inhibitors . Nat Rev Drug Discov . 2011 ; 10 : 868 – 880 .
6. Hsu P.P. , Kang S.A. , Rameseder J. et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling . Science . 2011 ; 332 : 1317 – 1322 .
7. Yu Y. , Yoon S.-O. , Poulogiannis G. et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling . Science . 2011 ; 332 : 1322 – 1326 .
8. Ricoult S.J. , Manning B.D. The multifaceted role of mTORC1 in the control of lipid metabolism . EMBO Rep . 2013 ; 14 ( 3 ) : 242 – 251 .
9. Ben-Sahra I. , Howell J.J. , Asara J.M. , Manning B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1 . Science . 2013 ; 339 : 1323 – 1328 .
10. Robitaille A.M. , Christen S. , Shimobayashi M. et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis . Science . 2013 ; 339 : 1320 – 1323 .
11. Zoncu R. , Bar-Peled L. , Efeyan A. , Wang S. , Sancak Y. , Sabatini D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase . Science . 2011 ; 334 : 678 – 683 .
12. Yuan H.X. , Xiong Y. , Guan K.L. Nutrient sensing, metabolism, and cell growth control . Mol Cell . 2013 ; 49 : 379 – 387 .
13. Manning B.D. , Cantley L.C. AKT/PKB signaling: navigating downstream . Cell . 2007 ; 129 : 1261 – 1274 .
14. Jewell J.L. , Russell R.C. , Guan K.L. Amino acid signalling upstream of mTOR . Nat Rev Mol Cell Biol . 2013 ; 14 : 133 – 139 .
15. Efeyan A. , Zoncu R. , Sabatini D.M. Amino acids and mTORC1: from lysosomes to disease . Trends Mol Med . 2012 ; 18 : 524 – 533 .
16. Efeyan A. et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival . Nature . 2013 ; 493 : 679 – 683 .
17. Guertin D.A. , Sabatini D.M. Defining the role of mTOR in cancer . Cancer Cell . 2007 ; 12 : 9 – 22 .
18. Faivre S. , Kroemer G. , Raymond E. Current development of mTOR inhibitors as anticancer agents . Nat Rev Drug Discov . 2006 ; 5 : 671 – 688 .
19. Engelman J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations . Nat Rev Cancer . 2009 ; 9 : 550 – 562 .
20. Liu P. , Cheng H. , Roberts T.M. , Zhao J.J. Targeting the phosphoinositide 3-kinase pathway in cancer . Nat Rev Drug Discov . 2009 ; 8 : 627 – 644 .
21. White E. Deconvoluting the context-dependent role for autophagy in cancer . Nat Rev Cancer . 2012 ; 12 : 401 – 410 .
22. Kuma A. , Hatano M. , Matsui M. et al. The role of autophagy during the early neonatal starvation period . Nature . 2004 ; 432 : 1032 – 1036 .
23. Yang Z. , Klionsky D.J. Mammalian autophagy: core molecular machinery and signaling regulation . Curr Opin Cell Biol . 2009 ; 22 : 124 – 131 .
24. Mihaylova M.M. , Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism . Nat Cell Biol . 2011 ; 13 : 1016 – 1023 .
25. Inoki K. , Kim J. , Guan K.L. AMPK and mTOR in cellular energy homeostasis and drug targets . Annu Rev Pharmacol Toxicol . 2012 ; 52 : 381 – 400 .
26. Pattingre S. , Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res . 2006 ; 66 : 2885 – 2888 .
27. Levine A.J. , Feng Z. , Mak T.W. , You H. , Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways . Genes Dev . 2006 ; 20 : 267 – 275 .
28. Matoba S. , Kang J.G. , Patino W.D. et al. p53 regulates mitochondrial respiration . Science . 2006 ; 312 : 1650 – 1653 .
29. Sparks C.A. , Guertin D.A. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy . Oncogene . 2010 ; 29 : 3733 – 3744 .
30. Oh W.J. , Jacinto E. mTOR complex 2 signaling and functions . Cell Cycle . 2011 ; 10 : 2305 – 2316 .
31. Guertin D.A. , Stevens D.M. , Saitoh M. et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice . Cancer Cell . 2009 ; 15 : 148 – 159 .
32. Pearce L.R. , Komander D. , Alessi D.R. The nuts and bolts of AGC protein kinases . Nat Rev Mol Cell Biol . 2010 ; 11 : 9 – 22 .
33. Colombani J. , Raisin S. , Pantalacci S. , Radimerski T. , Montagne J. , Léopold P. A nutrient sensor mechanism controls Drosophila growth . Cell . 2003 ; 114 : 739 – 749 .
34. Sarbassov D. , Ali S.M. , Sabatini D.M. Growing roles for the mTOR pathway . Curr Opin Cell Biol . 2005 ; 17 : 596 – 603 .
35. Bohn G. , Allroth A. , Brandes G. et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14 . Nat Med . 2007 ; 13 : 38 – 45 .
36. Guertin D.A. , Sabatini D.M. The pharmacology of mTOR inhibition . Sci Signal . 2009 ; 2 : pe24 .
37. Thoreen C.C. , Kang S.A. , Chang J.W. et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1 . J Biol Chem . 2009 ; 284 : 8023 – 8032 .
38. Sarbassov D. , Ali S.M. , Sengupta S. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB . Mol Cell . 2006 ; 22 : 159 – 168 .
39. Guertin D.A. , Sabatini D.M. An expanding role for mTOR in cancer . Trends Mol Med . 2005 ; 11 : 353 – 361 .
40. Astrinidis A. , Henske E.P. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease . Oncogene . 2005 ; 24 : 7475 – 7481 .
41. Wullschleger S. , Loewith R. , Hall M.N. TOR signaling in growth and metabolism . Cell . 2006 ; 124 : 471 – 484 .
42. Shaw R.J. , Cantley L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth . Nature . 2006 ; 441 : 424 – 430 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Unlike tumor cells with PTEN loss, tumor cells that have lost TSC function are generally less belligerent. It is now accepted that this is due to negative feedback loops that suppress the PI3K-AKT pathway when mTORC1 activity is high.
2,6,7 For example, the mTORC1 substrate S6K1 can directly phosphorylate and inactivate the IRS1 and IRS2 proteins, two mediators of PI3K activation, which decreases PI3K-AKT activity. A second feedback mechanism occurs through the direct mTORC1 substrate Grb10, which is activated by mTORC1 and functions to inhibit both the insulin/IGF receptors and IRS proteins. Negative feedback mechanisms would normally keep cell growth and nutrient uptake in balance, but in the absence of TSC, negative feedback squelches PI3K and AKT activity, which is important for promoting cell survival and metastasis. This may be why tuberous sclerosis (discussed later), which is caused by mutations in TSC, generally does not develop into metastatic cancer. Because PTEN and PI3K are downstream of the insulin/IGF receptors and IRS proteins, cancers driven by loss of PTEN or PI3K activation are uncoupled from these particular mTORC1-driven negative feedback loops, explaining why they are typically associated with more aggressive cancer.
mTORC1 and Autophagy
Macroautophagy, or simply autophagy, is a catabolic process induced on nutrient limitation or stress in which autophagosomes form and fuse with the lysosome to create an autophagolysosome. The autophagolysosome is the final destination for proteins and organelles that are broken down into their basic components to be recycled as sources of cellular energy and metabolites during starvation. Autophagy has an important role in cell survival during times of energy crisis. Under normal growth conditions, autophagy also salvages and recycles cellular junk such as excess or damaged organelles. Newborn mice require autophagy to survive a brief starvation period immediately after birth, and defects in autophagy are associated with cancer, degenerative disorders, and aging.
21–23 A connection between mTORC1 signaling at autophagy first became apparent in yeast and mammalian cells in studies where rapamycin was found to induce autophagy. Genetic inactivation of TOR in
Drosophila also induces autophagy and confirmed that mTORC1 not only positively controls anabolic processes, but suppresses catabolic processes opposing growth.
Recent studies have revealed that the regulation of autophagy by mTORC1 is at least partly direct and occurs in coordination with AMPK.
24,25 More than 30 autophagy
(ATG) genes have been identified that control autophagy. In nutrient-rich conditions, mTORC1 associates with and directly phosphorylates ULK1/ATG1, the kinase subunit of a complex that is required for autophagy induction. Phosphorylation of ULK1 by mTORC1 inhibits its activity and reduces autophagic vesicle formation. In nutrient-deprived cells, AMPK not only inhibits mTORC1 by phosphorylating TSC2 and Raptor (as described earlier), but also directly activates ULK1 kinase activity. When mTORC1 activity decreases, ULK1 is dephosphorylated, allowing AMPK to now interact with, phosphorylate, and activate ULK1 to induce autophagy. This dual regulatory circuit emphasizes the tight balance between anabolic and catabolic processes.
Interestingly, prolonged autophagy, which delivers its cargo to the lysosome, eventually reactivates mTORC1. Thus, in nutrient-starved cells, autophagy may also be required to keep mTORC1 on in a minimally activated state to promote the synthesis of certain macromolecules essential for survival. The reactivation of mTORC1 also promotes the formation of nascent lysosomes that bud from the autophagosome. In fact, recent studies further suggest that mTORC1 may normally regulate lysosome formation through its ability to control that nuclear localization (and thus activity) of a transcription factor called TFEB. TFEB regulates the expression of many genes important for lysosome formation. Thus, the connections among nutrient and energy sensing, mTORC1 growth control, and protein/organelle turnover by autophagy and the lysosome are complex, and it will be interesting to determine if and how defects in these connections promote cancer.
Although the connections between mTORC1 and autophagy are being worked out, the strong connections between mTORC1 and cancer suggest that autophagy may have a role in tumorigenesis, and evidence supports this hypothesis.
21 For example, mice deficient for a pro-autophagy gene, called
beclin-1, have an increased frequency of spontaneous tumor formation, suggesting that
beclin-1 is a tumor suppressor gene. How could inactivation of autophagy promote tumorigenesis? It is difficult to speculate at this time because little is known about Beclin 1, but perhaps some transformed cells rely on autophagy to remove damaged organelles or for complete self-disposal. In this model, mutations that prevent autophagy would promote cell survival. Interestingly, the Bcl-2 pro-survival oncoprotein, which has long been thought to inhibit apoptosis, binds and inhibits Beclin 1–dependent autophagy.
26 This suggests that the oncogenic activity of Bcl-2 might be linked to inhibiting nonapoptotic autophagic cell death. Contrary to having tumor suppressor capacity, autophagy could promote tumorigenesis in some cases. When cancer cells experience nutrient limitation, autophagy may provide the rations necessary to sustain the most essential bioenergetic cellular process. This may allow cancer cells to survive until angiogenesis is initiated or other deleterious mutations occur. Understanding the role of autophagy in different cancer cells will be important to designing mTOR-focused treatment strategies.
mTORC1 and p53
Mutation of the p53 tumor suppressor is one of the most common genetic abnormalities associated with human cancer. DNA or spindle damage, telomere shortening, various stresses, or oncogenic mutations will usually initiate a p53-dependent checkpoint that arrests cells in G
1 or trigger apoptosis (depending on the cell type and signals present). This has led to speculation that such a checkpoint mechanism involving p53 might communicate with the mTORC1 pathway to discourage growth when conditions are unfavorable. Early reports from work with cultured cells suggest that AMPK and p53 may communicate in some type of checkpoint mechanism to restrict growth in low-glucose conditions or when DNA is damaged, although it is unclear from this early work if and how directly mTORC1 regulation might be connected.
27 Mounting evidence also indicates that a communication link exists between p53 and the regulation of mitochondrial respiration, particularly in mediating a cellular switch to preferentially deriving energy from glycolysis rather than aerobic respiration—a hallmark characteristic of cancer cells.
28 It is well known that p53 and
the mitochondria, and to some extent mTOR, have roles in apoptosis. Because signals from the mitochondria are also thought to control mTORC1’s ability to mediate growth, it seems likely that a complicated signaling circuitry involving both the p53- and mTOR-dependent pathway exists.
Beyond Cell Growth: Does mTOR Regulate Organ and Organism Growth?
The mTORC2 Pathway
The role of TOR in building cell mass is well documented and conserved in all eukaryotic organisms examined. However, extending a role for mTOR in controlling organ and organism growth is more complicated, because most tissues grow by mechanisms involving the collective coordination of cell growth, cell division, and cell death pathways. The finding that mTOR exists in a second complex called mTORC2 does suggest that mTOR may have broad influence over cell growth, cell division, cell death, metabolism, and many other processes essential for building tissues and organisms. Many of these additional targets of mTOR activity, although not completely worked out yet, are likely to be important in cancer progression, and thus emerging therapeutic strategies are now aiming to more broadly target mTOR signaling by inhibiting both mTORC1 and mTORC2.
The second mTOR complex also contains mLST8 and Deptor, but instead of Raptor and PRAS40, mTORC2 contains the Rictor, mSin1, and PROTOR proteins (see
Figure 12-1).
2 Understanding the function of mTORC2 initially eluded researchers because the complex is insensitive to acute rapamycin treatment, and thus rapamycin could not be used to probe for mTORC2 substrates. However, advances in RNA-interference technology allowed the specific depletion of Rictor from cells, leading to the discovery that mTOR, when outfitted with mTORC2-specific regulatory proteins, phosphorylates and activates the AKT kinase. Current models suggest that on recruitment to the membrane after growth factor stimulation, AKT is phosphorylated by mTORC2 in conjunction with PDK1, and this co-regulation is presumed necessary for full AKT activation. How mTORC2 activity itself is controlled remains to be worked out, but initial experiments indicate that growth factors also modulate mTORC2 activity by an unknown mechanism.
AKT activity influences cell growth, cell proliferation, cell survival, metabolism, and many other processes.
13 Thus, by coupling mTOR activity with proliferation and survival, the discovery of mTORC2, together with the well-known role of mTORC1 in controlling cell size, strengthens the argument that mTOR growth regulation extends beyond cell autonomous growth to organ and organism growth. Moreover, because of the widespread role of PI3K-AKT signaling in cancer (see
Table 12-1), the finding that mTORC2 phosphorylates and activates AKT is a compelling additional link between mTOR and cancer. In fact, a number of studies now suggest that selectively targeting mTORC2 could also be a promising therapeutic strategy.
29,30 What is interesting about potentially targeting mTORC2 is that in many cases, loss of mTORC2 function (by genetic approaches) is less toxic to normal cells than targeting mTORC1 function. In contrast, mTORC2 activity is required for transformation, particularly when transformation is driven by loss of PTEN.
31 Thus, mTORC2 might be more essential to certain cancer cells than to many normal cells. The challenge of selectively targeting mTORC2 is that beyond the mTOR catalytic domain, the understanding of mTORC2 structure and regulation is only vague, and thus it is currently difficult to predict what type of targeting strategy might best achieve this goal.
In retrospect, it is not surprising that mTOR can also phosphorylate AKT, because the mTORC2 phosphorylation site on AKT (S473) is structurally similar to the mTORC1 site on S6K1 (T389), both of which are present in carboxy-terminal hydrophobic motifs. In fact, both S6K and AKT belong to a larger family of structurally related kinases called the AGC kinases.
32 The discovery of the mTORC2-AKT signaling module also sets up the peculiar situation whereby mTOR can regulate itself in a manner dependent on what proteins it interacts with. For instance, mTORC2-mediated phosphorylation of AKT is proposed to promote subsequent phosphorylation and inactivation of TSC2 and PRAS40 (as described earlier), which would place mTORC2 upstream of mTORC1 regulation. The extent to which normal and cancer cells rely on such regulation is under investigation. How other AKT substrates are regulated by mTORC2 is also largely unclear. In addition to AKT, mTORC2 can directly phosphorylate two additional AGC kinases called SGK and PKC. SGK is most similar to AKT, except that it lacks the membrane-targeting PH domain that brings AKT to its activation site at the cell membrane. SGK also has some overlapping functions with AKT, but currently the role of mTORC2-mediated SGK and PKC regulation in cancer is unknown.
Controlling Body Size
It is a remarkable feat for mammals to coordinate the development of their organs to appropriate sizes that are proportional to overall body size. The endocrine system is responsible for conducting this massively orchestrated systemic growth. The major hormone responsible for controlling postnatal growth is growth hormone (GH). GH is synthesized in the pituitary gland. After its release into the blood, circulating GH stimulates the release of insulin-like
growth factors (IGFs) from the liver. IGFs subsequently stimulate growth of bone and muscle, which are the two organs most relevant to determining body size.
The findings (1) that mTOR regulates cell growth, proliferation, and survival and (2) that both mTORC1 and mTORC2 are downstream of insulin/IGF signaling raise the question of whether mTOR is a master regulator of body size. The role of mTOR as a nutrient-sensitive regulator of eukaryotic cell growth (i.e., as part of mTORC1) is an ancient function conserved from yeast to humans. During the evolution of multicellular organisms, growth factor signaling was grafted onto the mTORC1 pathway, probably to modulate autonomous cell growth in conjunction with systemic nutrient availability and signals from other cells and tissues. The discovery that mTOR (as part of mTORC2) regulates AKT further suggests that additional pathways independent of mTORC1 may contribute to the regulation of cell growth, proliferation, survival, and metabolism. Many mTORC2 components are also conserved in yeast—although the protein sequences and downstream functions are more diverged compared to mTORC1, suggesting the cellular functions of mTORC2 may have also diverged. Because of mTOR’s remarkable ability to integrate numerous signals and control many aspects of cell growth, it seems poised to be a master regulator of organ and body size control. Studying this in mammals has been difficult, however, because mice null for mTOR and the essential mTORC1 and mTORC2 regulatory subunits are embryonic lethal. In
Drosophila, the fat body (which may be equivalent to the vertebrate liver) functions as a nutrient-sensing organ that can control body growth through a TOR-dependent mechanism.
33 Moreover, mice and flies deficient for S6K1 are viable but reduced in size because of smaller cells, implying a link between mTORC1 signaling and body size control.
34 Thus, limited evidence in model organisms suggests that mTOR could influence body size by systemic as well as cell-autonomous mechanisms. Interestingly, in humans a rare mutation in the p14/LAMTOR2 subunit of the Ragulator delays growth and causes immunosuppression and hypopigmentation.
35 As novel upstream regulators and substrates of mTOR are unveiled, it will be interesting to determine if genetic variation in components of the mTOR network has a role in determining organ and organism size.
Targeting mTOR Signaling as a Treatment for Cancer and Other Human Diseases of Cell Growth
With a role for mTOR signaling in cancer now firmly established, developing molecules that inhibit mTOR for therapeutic purposes is a significant focus of the pharmaceutical industry. As mentioned earlier, rapamycin (also known as
sirolimus) is used clinically for immunosuppression (because of its ability to suppress T-cell proliferation), to prevent restenosis (by preventing the growth of vascular smooth muscle cells), and to treat certain cancers.
5,17,36 Analogs of rapamycin have also been developed for cancer therapy (CCI779/temsirolimus, AP23573, and RAD001/everolimus—collectively called rapalogues). Unfortunately, rapamycin as a broad anticancer drug has had limited success as a single-agent therapeutic, even against cancers with elevated PI3K-AKT signaling. The response to rapamycin is highly variable depending on the cancer, and only a few cancers, including mantle-cell lymphoma, renal-cell carcinoma, and endometrial cancers, have shown consistent positive responses. Temsirolimus was approved in 2007 to treat renal-cell carcinoma. Several trials are ongoing, although there is no clear biomarker predictive of rapamycin sensitivity.
A number of reasons have been proposed to explain rapamycin’s general ineffectiveness as a cancer drug. One possibility (discussed earlier) is that inhibiting mTORC1 relieves strong negative feedback loops that function to suppress receptor tyrosine kinase (RTK) signaling and downstream PI3K-AKT activation (
Figure 12-2 ). Therefore, treating cells with rapamycin boosts AKT signaling, which in turn could drive AKT-dependent cell proliferation and survival pathways—clearly an undesirable response. The MAPK-ERK signaling pathway is also triggered by mTORC1-S6K1 inhibition. Using rapamycin in combination with PI3K or MAPK pathway inhibitors might prove to be a better strategy. Second, it is now clear that rapamycin only partially inhibits mTORC1. For example, although rapamycin results in rapid dephosphorylation and sustained inactivation of S6K, phosphorylation of 4E-BP1 is only partially and transiently decreased.
37 Therefore, translation is largely insensitive to rapamycin. As more mTORC1 substrates are being discovered, it will be interesting to determine why substrates vary in their sensitivity to rapamycin.
It is often stated that rapamycin specifically targets mTORC1, implying that it does not inhibit mTORC2. However, the actual situation is more complex.
2,38 In cultured cells, rapamycin acutely affects only mTORC1, but prolonged exposure disrupts mTORC2 integrity, likely by blocking the assembly of new mTORC2 complexes. In some cell types this results in decreased AKT phosphorylation, but often there is no apparent change or increased AKT phosphorylation. Notably, any mTORC2 complexes that remain intact following rapamycin treatment are still susceptible to losing feedback inhibition. This likely explains the cell-to-cell variability in AKT phosphorylation following prolonged rapamycin treatment. For example, even if most mTORC2 complexes are
disrupted by rapamycin, what remains intact is hyperactive, and consequently AKT phosphorylation can appear insensitive to rapamycin or increase. Exactly what determines whether AKT phosphorylation in a particular cancer cell will show increased, decreased, or no sensitivity to rapamycin remains a mystery. Nevertheless, inhibition of mTORC2 following chronic exposure to rapamycin could explain the drug’s effectiveness in certain cancers as well as some of its side effects. Dissecting the molecular mechanisms by which rapamycin functions and identifying biomarkers predictive of rapamycin sensitivity remains an important area of research.
Next-generation mTOR inhibitors are ATP competitive inhibitors (discussed earlier). Therefore, unlike the rapalogues (which are allosteric inhibitors), these drugs directly target mTOR kinase activity.
2,5 Consequently, the ATP competitive inhibitors more completely inhibit mTORC1 and also inhibit mTORC2. The mTOR ATP-competitive inhibitors are having a major impact in basic research, and several versions are being evaluated in clinical trials as anticancer drugs with the hope that they will outperform the rapalogues. Their tolerability remains to be seen, but early data are encouraging. Notably, losing feedback inhibition remains a challenge with these inhibitors because, despite the fact that they inhibit mTORC2 as well, the mTORC2 substrate AKT is also regulated by the PDK1 kinase, which increases activity following mTORC1 inhibition because the negative feedback is lost (see
Figure 12-2). A related class of inhibitors is called the
dual-specificity inhibitors, which target mTORC1, mTORC2, and PI3K. Dual inhibition is possible because the kinase domains of mTOR and PI3K are structurally similar, even though mTOR phosphorylates proteins and PI3K phosphorylates lipids. The dual-specificity inhibitors could have an advantage because by also inhibiting PI3K, they can mitigate the effects of losing the mTORC1-dependent negative feedback inhibition of PI3K-AKT signaling (see
Figure 12-2). Because many
research studies and clinical trials are ongoing with both ATP-competitive and dual-specificity mTOR inhibitors, it should soon become clear to what extent they will be useful clinically.
Figure 12-2 Model explaining how downstream signaling in cancer cells responds to currently available classes of mTOR pathway inhibitors (A)
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading.
Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
The Molecular Basis of Cancer Expert Consult - Online