Chapter 24 Cell Death, Apoptosis, and Autophagy in Retinal Injury
Modes of cell death
According to the Nomenclature Committee on Cell Death 2009, “Cell death can be classified according to its morphological appearance (which may be apoptotic, necrotic, autophagic or associated with mitosis), enzymological criteria (with and without the involvement of nucleases or of distinct classes of proteases, such as caspases, calpains, cathepsins and transglutaminases), functional aspects (programmed or accidental, physiological or pathological) or immunological characteristics (immunogenic or non-immunogenic).”1 A cell is normally considered dead once it has passed an irreversible phase in the death process.
Apoptosis
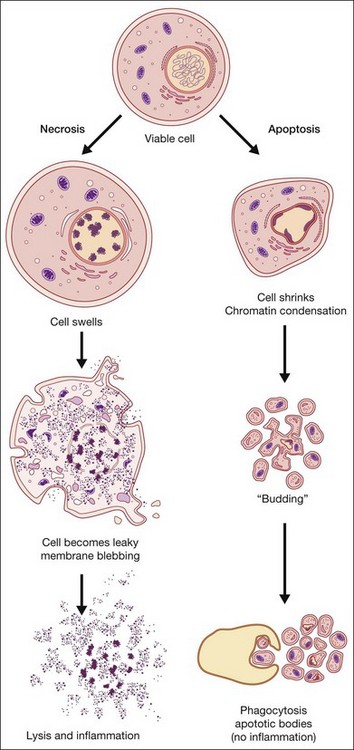
Apoptosis, or programmed cell death, has received extensive study due to its critical role in development, tissue homeostasis, and pathology.2,3 Furthermore, apoptosis does not elicit an inflammatory response, thus allowing “physiological” cell death to take place without pathological consequences. Morphological features of apoptosis include rounding up of the cell, reduction in cellular and nuclear volume (pyknosis), nuclear fragmentation, modification of cytoplasmic organelles, plasma membrane blebbing, and engulfment by neighboring cells (Fig. 24.1).1 Apoptosis can be initiated by a variety of stimuli through two distinct pathways.4 The extrinsic pathway is triggered after the interaction of death receptors present on the cell surface with their cognate ligands (e.g., Fas ligand, tumor necrosis factor (TNF)-α and TNF-related apoptosis-inducing ligand (TRAIL)) and which can start the downstream executioner caspase (cysteine aspartic acid proteases) cascade within seconds of ligand binding. By contrast, the intrinsic pathway is initiated by a multitude of intracellular triggers, collectively called “stress signals,” which can include oxidative damage, deoxyribonucleic acid (DNA) damage, loss of cell–cell contact, growth factor withdrawal, hypoxia, and endoplasmic reticulum stress that all target the mitochondria and induce the release of proapoptotic factors to the cytosol that activate caspases. Excessive or deficient apoptosis is involved in numerous disease states. Readers requiring more detail are directed to the work of Green and Reed.2,3
Necrosis
Necrosis has been defined as a type of uncontrolled cell death that can occur in response to infection, toxins, chemicals, injury, or lack of blood supply.2,5 Morphologically, necrosis is associated with cytoplasmic swelling (oncosis), rupture of the plasma membrane, swelling of cytoplasmic organelles, and moderate chromatin condensation (Fig. 24.1). The big pathophysiological difference between necrosis and apoptosis is inflammation. Necrosis culminates in the uncontrolled release of antigens which lead to activation of the immune system and inflammation whereas in apoptosis cell-bound bodies are formed which are phagocytosed by neighboring cells and there is an absence of inflammation. However, recent studies suggest that there is a molecular signaling network that can regulate the necrotic cell death pathway.6
Other
A number of other cell death pathways have been identified, of which autophagic cell death has gained some prominence. Increased autophagy (see below), such as that occurring under starvation, leads to self-destruction of intracellular organelles for provision of nutrients which, if starvation is not reversed, will culminate in self-destruction of cells and tissues. Autophagic cell death is morphologically defined as occurring in the absence of chromatin condensation, massive autophagic vacuolization, and little or no uptake by neighboring cells.1 Interestingly, autophagy is upregulated in a number of neurodegenerative diseases and thus may contribute to cell loss associated with these conditions. A number of atypical cell death modalities have also been identified and these are reviewed in Kroemer et al.1
Cross-talk between cell death pathways
Until recently, a requirement for gene expression was documented only for apoptotic and autophagic cell death. Interestingly, certain genes and their products, e.g., p53, Bcl-2 family proteins, and calpain, are important for both these modes of cell death.5–7 Recent work indicates that basal p53 activity suppresses autophagy, whereas the activation of p53 by some stimuli induces autophagy as well as apoptosis mediated by the Bcl-2 family proteins.6,7 Atg5 is essential for autophagy, but upon cleavage by calpains it has been reported that the truncated Atg5 associates with BclxL to promote cytochrome c release and caspase-dependent apoptosis.8 For many years, necrosis was regarded as the result of an accidental and uncontrolled process. However, accumulating evidence now suggests that necrotic cell death might also be mediated by a specific set of signal transduction pathways and degradative mechanisms. Similar to apoptosis, cell death with a necrotic appearance can contribute to embryonic development and adult tissue homeostasis. Some gene products, such as TNFR, CD95, TRAIL-R and RIP1, might trigger both apoptosis and necrosis, depending on interaction with other proteins.5 Moreover, there is cross-talk between these two cell death modalities. For example, inactivation of caspases might cause a shift from apoptosis to necrosis, or to a mixture of these two cell death modes.5 Thus cell death is not as easily defined as generally believed and there is considerable cross-talk between the different cell death mechanisms.
Autophagy and cell maintenance
Autophagy is essential for cellular housekeeping and homeostasis through the sequestration and transfer of intracellular components (e.g., protein aggregates, organelles) to lysosomes for degradation.9–11 In mammalian cells, three primary types of autophagy have been reported: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy (Fig. 24.2).12–16 These different types of autophagy are designed to nonselectively or selectively degrade different substrates and are regulated by different signaling pathways.
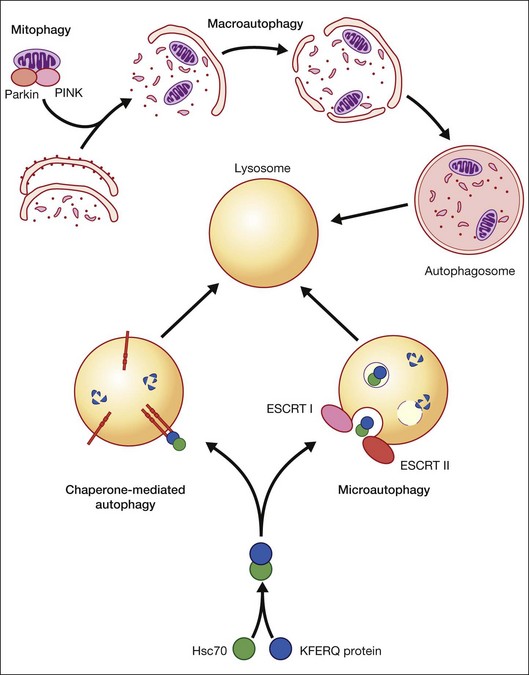
Macroautophagy is the best-characterized autophagy pathway that generally deals with larger substrates such as protein aggregates, intracellular pathogens, and dysfunctional organelles such as mitochondria (Fig. 24.2).9–11 The process of macroautophagy involves over 30 autophagy-related proteins (Atg) which regulate different stages of the autophagic response (Fig. 24.3). Macroautophagy is initiated by the sequestration of the cytosolic substrate into double membrane-bound phagophores which predominantly originate from the rough endoplasmic reticulum with possible contributions from the plasma membrane or mitochondria.17–20 The resulting autophagosome acquires endosomal and lysosomal proteins, ultimately maturing into a degradative autolysosome. The mTOR kinase complex is considered central to the signaling pathway of autophagy and can sense regulating conditions such as nutrient abundance, energy state, and growth factor levels.21,22 The PI3K-III complex consists of Vps34 and p150 and activators such as Beclin-1, Ambra1, ATG14, and UVRAG. This complex plays a crucial role in the induction of the autophagic process by generating PtdIns(3)P-rich membranes, which act as platforms for ATG protein recruitment and autophagosome nucleation.23 Antiapoptotic BH3 proteins such as BclxL and Bcl-2 bind to Beclin-1, having a negative impact on PI3K-III activity and autophagy. Elongation and completion of the phagophore are brought about by two ubiquitin-like conjugation systems, the Atg12-Atg5-Atg16 system and the Atg8-phosphatidylethanolamine (PE) system. Atg7 functions as an E1 enzyme in both systems, while Atg10 and Atg3 act as E2 enzymes for Atg12 and Atg8, respectively.24 The C-terminus of Atg8 is cleaved by Atg4 which primes the protein for conjugation to PE. The Atg12–Atg5–Atg16 complex recruits Atg8-PE to the elongating phagophore.25,26 At least eight different Atg8 orthologs belonging to two subfamilies (LC3 and GATE-16/GABARAP) occur in mammalian cells. LC3s are involved in elongation of the phagophore membrane whereas the GABARAP/GATE-16 subfamily is essential for a later stage in autophagosome maturation.27 The N-termini of LC3 and GATE-16 are required for autophagosome–lysosome fusion.28 Once in the lysosomes, substrates are degraded by the repertoire of lysosomal enzymes.29 It has been proposed that the autophagic elimination of mitochondria has its own specialized pathway.16 Critical to this process are the proteins PINK1, the E3 ubiquitin ligase, Parkin and possibly p62, a protein that binds to ubiquitin and LC3. PINK1 binds to uncoupled mitochondria which then facilitates the recruitment of Parkin which leads to ubiquitination of mitochondrial surface proteins. The ubiquitinated mitochondrion is then sequestered into the autophagosome through the likely actions of p62 and LC3.
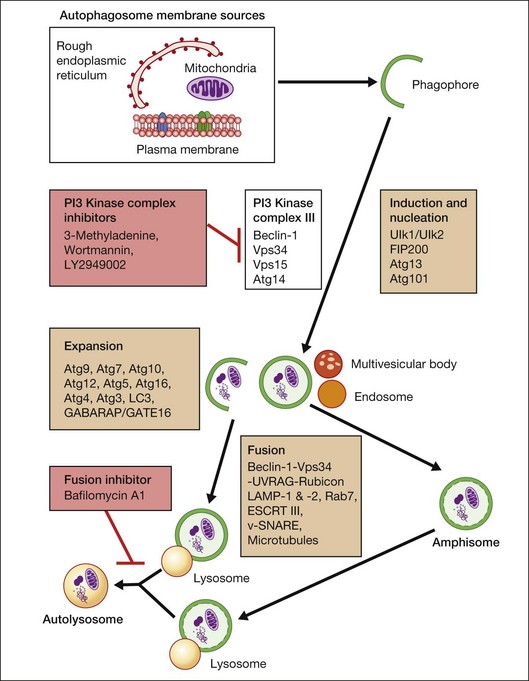
Autophagy can be activated by nutrient deprivation and environmental stress. For example, amino acid starvation and reactive oxygen species can stimulate autophagy.30,31 Recently, a distinction has been made between starvation- and stress-induced macroautophagy, also referred to as quality control autophagy. It has been observed that autophagic deficient cells tend to accumulate p62-rich aggregates, which in turn cause Nrf2 to be activated after separation from its interacting partner Keap1, which allows Nrf2 to mount an antioxidant response.32 In addition, histone deacetylase (HDAC)6 stands out as a key regulator in the autophagic response to oxidative damage, as HDAC6 is recruited to ubiquitinated autophagic substrates, where it stimulates autophagosome–lysosome fusion by promoting F-actin remodeling in a cortactin-dependent manner.33 However, HDAC6 and cortactin are dispensable for starvation-induced autophagy.
CMA differs from the other types of autophagy as it does not involve vesicle formation but, rather, a direct translocation of a specific set of soluble proteins across the lysosomal membrane for subsequent degradation (Fig. 24.2).34 CMA cargo substrates include enzymes, transcription factors, binding proteins, subunits of the proteasome and proteins involved in vesicular trafficking and contain a KFERQ-like motif which is recognized by the cytosolic chaperone, Hsc70. Binding of the chaperone/substrate to the cytosolic tail of lysosome-associated membrane protein type 2A (LAMP-2A) which spans the lysosomal membrane leads to translocation of the cargo across the membrane and into the lysosomal lumen for degradation.12 This pathway has been shown to be progressively ineffective with age, because of the age-related loss of LAMP-2A.35
Microautophagy involves internalization of cytosolic cargo (cytosolic proteins, glycogen, and ribosomes) through invaginations of the lysosomal membrane,36,37 which resembles the formation of multivesicular bodies (Fig. 24.2).13,34,38 Although the molecular mechanisms in mammalian cells are poorly understood, a recent study by Sahu et al. proposes that microautophagy relies on endosomal sorting complexes required for transport (ESCRT) I and III, which are necessary for the formation of the vesicles in which the cytosolic cargo is internalized.38 It appears that this pathway, like CMA, also involves Hsc70 interaction with a substrate containing a KFERQ-like motif and that mitophagy and CMA may share common upstream pathways.34,38
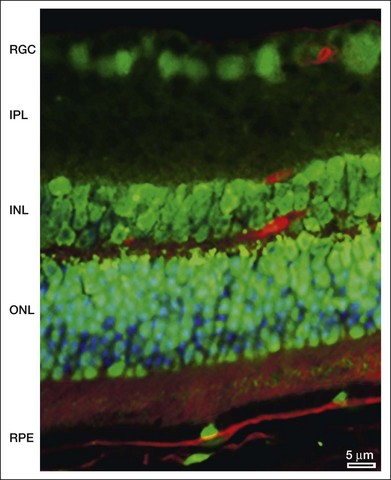
As will be discussed below, autophagy plays a critical role in maintaining retinal homeostasis as it is central, together with the proteosomal system, in the removal of damaged proteins and organelles in highly metabolic nondividing cells that exist in a pro-oxidative retinal environment. Autophagy proteins are strongly expressed in the retina (Fig. 24.4). However, problems occur when basal levels of autophagy become dysregulated as either a decrease or increase in autophagy flux will have significantly detrimental effects on cell function.39
AGE-related retinal cell loss
It is well recognized that the human retina undergoes numerous age-related changes which result in altered morphology, reduced function, and cell loss. Not surprisingly, this is associated with a significant reduction of retinal thickness as a function of age.40–42 Studies report that mean retinal thickness decreases by 0.53 µM/year40 and that, in the macula, retinal thickness and macular volume decrease by around 0.35 µM and 0.01 mm3/year.42 Changes in cell morphology include: nodular excrescences in rod outer segments43; accumulation of lipofuscin in photoreceptor inner segments and the retinal pigment epithelium (RPE)44–46; displacement of nuclei from the outer nuclear layer (ONL)47; and extension of ON-cone bipolar cell and horizontal cell processes into the ONL.48 Such evidence of retinal reorganization and plasticity has also been corroborated by animal studies.49,50 Reorganization of the dendrites could be an adaptive attempt to compensate for the lost circuitry due to photoreceptor loss and/or to make up for existing, yet dysfunctional, synapses.
There is an age-related decrease in the density of photoreceptor cells in the human retina, with rods appearing to be more vulnerable than cones.51,52 In the equatorial retina cones decrease uniformly at approximately 16 cells/mm2/year while the decrease in equatorial rods is greatest, 970 cells/mm2/year, between the second and fourth decades.52 By contrast, cone density remains relatively constant at the fovea up to the ninth decade.51–53 It therefore appears that rod photoreceptors are more vulnerable to loss during aging than cones and that photoreceptors in the fovea are less susceptible to attrition. Furthermore, compensatory adaptations have been reported following rod cell degeneration where the space vacated by dying rods is filled by enlarged rod inner segments from neighboring photoreceptors, resulting in similar rod coverage at all ages.51,54–56 Evidence that cones depend on survival factors secreted by rods may explain this differential vulnerability between rods and cones57–60 but remains a matter of intense debate.
The loss of photoreceptors appears to precede the loss of associated neural cells. The retinal nerve fiber layer thickness decreases dramatically with age40,61 and is associated with a significant loss of retinal ganglion cells (RGC) by as much as 150/mm2 over a period of 40+ years.41 RGC death at the equatorial regions follows a similar trend as the photoreceptors during aging, thus maintaining a constant photoreceptor-to-RGC ratio.52 Age-associated degeneration of the rod bipolar cells in the inner nuclear layer (INL) has been reported.62 Although rod cell death can initiate as early as the second decade of life, the bipolar cells start to degenerate only after the fourth decade and appreciably reduce by the ninth decade.52 So it seems to be a phenomenon secondary to rod cell loss. In a more comprehensive study done using multiphoton confocal microscopy to quantify neuron densities in the RGC layer, INL, and ONL, it was seen that the greatest neuronal loss occurs in the RGC layer and ONL in human aging retinas, whereas the INL is relatively preserved.63 It must also be remembered that RGCs are classified into a number of subtypes56 and so even a seemingly mild loss of RGC in the initial phases could imply the loss of a major subtype of RGC that could start affecting visual perception.
Despite numerous studies to determine age-related changes in RPE cell density, outcomes vary and are highly dependent on retinal location. Two studies have reported that RPE density decreases with age in the equatorial retina and is greatest in the periphery,52,64 with an estimated loss of 0.3% per year.64 By contrast, no significant age-related decrease in RPE cell density was observed at the foveal center, suggesting that, like foveal cones, the RPE cells in this region are more resistant to attrition than those outside the fovea.52 However, a further study reported that the macular region in aged eyes contained a significant number of apoptotic cells and these were greatest in the fovea.54 Given the disparity between studies on age-related changes in RPE cell density it is not surprising that there is no clear agreement to what extent the RPE-to-photoreceptor ratio changes with age, if at all.52,65
Evidence would suggest that age-related cell loss in the retina occurs by apoptosis since occasional apoptotic cells are observed in retinal sections plus the lack of any significant age-related inflammatory response which would occur via necrosis. The stimulus for age-related cell loss is also unclear but since the majority of cells affected are postmitotic or terminally differentiated, the accumulation of stochastic damage as occurs with aging in other tissues is plausible. In particular, oxidative damage is likely to play a significant role since the retina has high oxygen levels, is exposed to light, and has a number of highly metabolically active cell types, making it an ideal environment for the generation of reactive oxygen species.66 As already mentioned, the retina undergoes considerable remodeling throughout life to adapt to age-related cell loss. In addition, there is likely to be a basal level of limited cellular replacement through resident and bone marrow-derived stem or progenitor cells that have the capacity to differentiate into a number of different retinal cell types.67
Retinal damage: death and repair
Glaucoma and ganglion cell loss
Glaucoma is a heterogeneous group of diseases that leads to RGC death.68–70 Pathology is associated with “cupping” of the optic disc due to loss of ganglion cell axons. Death of RGCs in both postmortem specimens and experimental animal models takes place by apoptosis.71–73 Analysis of retinas from human donors suffering primary open-angle glaucoma demonstrated greater than 15 times more terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive apoptotic cells than the controls.74 Interestingly, apoptosis also accounts for the selective elimination of about 50% of excess RGCs during developmental organization of the visual pathway.75 Apoptosis of RGCs in glaucoma has generally been considered to occur as a result of mechanical damage due to an increase in intraocular pressure, but damage to RGCs can also occur in normal-pressure glaucoma.76 Other insults that have been reported to induce RGC apoptosis are neurotrophin deprivation, glial activation, ischemia, glutamate excitotoxicity, and oxidative stress.70 Confirmatory data that RGC apoptosis occurs by the intrinsic pathway that involves the mitochondria come from backcrossing DBA/2J mice that exhibit a spontaneous secondary glaucoma with Bax (one of several BH3 family death proteins) knockout mice that resulted in a mouse strain in which glaucomatous neurodegeneration was reduced.77
Despite the strong association between oxidative stress, mitochondrial damage, and RGC death, there have only been limited studies on the role of autophagy in RGC maintenance and glaucoma. Rodriguez-Muela and colleagues recently reported that autophagy promotes survival of RGCs after optic nerve axotomy in mice.78 Calpain-mediated cleavage of Beclin-1 and autophagy deregulation have been reported in a rat model of retinal ischemic injury that recapitulates features of glaucoma.79 Furthermore, blockade of autophagy increased cell death in cultured RGC, suggesting a prosurvival role of the autophagic process. By contrast, activation of autophagy in RGCs occurs after optic nerve transection and increased autophagy offers a protective role in cultured RGCs.80 Sternberg and colleagues demonstrated that autophagy provided a survival mechanism against caspase-dependent death of neonatal RGCs induced by axon damage.81
Diabetic retinopathy
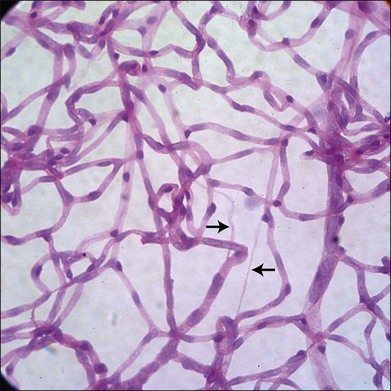
It has long been recognized that diabetes leads to a loss of pericytes and endothelial cells in the retinal vasculature (Fig. 24.5).82,83 Development of new techniques to study the vasculature allowed Cogan and colleagues to identify pericyte “ghosts” (intramural pockets in the vascular basement membrane lacking normal cell contents) as one of the earliest changes in DR.84 Pericyte loss is accompanied by loss of endothelial cells from the retinal vasculature, leading to “acellular” capillaries (intact vessel basement membrane devoid of cells lining the lumen). Cell death in these populations appears to occur predominantly via the intrinsic apoptotic pathway.85 These characteristic changes have long been considered a hallmark of DR. However, studies by Barber and others reveal that diabetes is also associated with increased loss of retinal neurons.86,87 STZ-diabetic rat retinas after 30 weeks of diabetes showed a 22% reduction in the thickness of the inner plexiform layer, a 10% reduction in RGCs, and a 14% reduction in the thickness of the INL.88 Clinical studies of diabetics using scanning laser polarimetry and optical coherence tomography have largely confirmed these findings in patients.89–91 It is also likely that other neuronal populations such as amacrine cells are also undergoing apoptosis in diabetes.92 The potential initiators of retinal cell apoptosis are many and include hyperglycemia, oxidative stress, reduced blood flow, ischemia, neuroinflammation and, specifically in the case of retinal neurons, glutamate excitotoxicity.85,86
There is surprisingly little information currently available on the potential role of autophagy in the pathogenesis of DR, even though there is extensive evidence that: (1) autophagy is dysregulated in other diabetic tissues93,94 and (2) damaged mitochondria are associated with the pathogenesis of DR.85 We have recently reported that autophagy flux is decreased in the retinal cells of diabetic rats compared to controls.95
The impact of cell death in the diabetic retina and the order in which different cell types die during the progression of DR are unclear. For example, pericyte dropout and acellular capillaries are observed in many diabetic animal models, yet they do not progress to the sight-threatening proliferative stage. Furthermore, the duration of diabetes in many patients may be 15 years or more before clinical abnormalities are observed in the retina, even though vascular and neuronal cell death will be occurring. A possible explanation for this chronic, rather than acute, attrition in the retina is a low level of cellular replacement from resident and bone marrow-derived stem or progenitor cells.67 Bone marrow-derived progenitor cells have the capacity to differentiate in a range of retinal vascular and neuronal cell types in response to retinal injury and in the case of the retinal vasculature can repopulate acellular capillaries in rodent models of DR.96,97 However, these bone marrow-derived progenitors are reduced and appear to be dysfunctional in diabetics and thus repair potential is attenuated.98,99
Macular degeneration
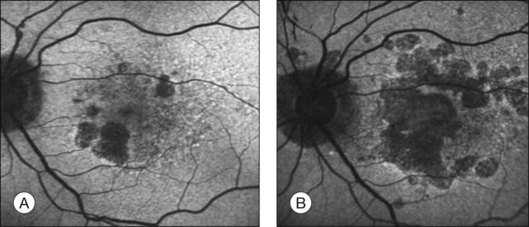
The challenge in studying retinal cell loss mechanisms in AMD is to be able to differentiate between the cell loss in the disease from that in the normal course of aging. However, reports concur that photoreceptor, RPE, and choroidal cell loss are accelerated in AMD and that this is regional and often focal. Clinically, areas of geographic atrophy can be observed within the retinal arcades in the eyes of patients with dry AMD and these lesions will increase in size with lengthening duration of AMD (Fig. 24.6). Areas of geographic atrophy show a continuous enlargement over time with a median growth rate of between 1 and 13 mm2/year and progression appears to be linear.100,101 The considerable variability between patients is unexplained but could reflect genetic susceptibility, diet, smoking, and light exposure. However, there is concordance of disease progression in bilateral geographic atrophy.102 Loss of RPE is normally associated with a reduction in the choroidal vasculature. Evidence of the degree of choroidal capillary loss and its relationship with RPE loss has been elegantly described by McLeod and colleagues.103 They observed a linear relationship between the loss of RPE and choriocapillaris in geographic atrophy. The vascular area was reduced by 50% in regions of complete RPE atrophy and there was extreme constriction of remaining viable capillaries. Adjacent to active choroidal neovascularization, choriocapillaris dropout was evident in the absence of RPE atrophy, resulting in a 50% decrease in vascular area. The authors concluded that the close association observed between degenerating RPE and choriocapillaris suggests that, at least in geographic atrophy, RPE atrophy occurs first, followed by choriocapillaris degeneration.
It has largely been considered that photoreceptor cell death occurs as a result of dysfunction or death of the underlying RPE. However, as discussed above in the aging section, there is significant rod photoreceptor loss as a function of age even in the presence of an apparently healthy RPE.51 Photoreceptor topography in both dry and wet AMD shows preferential loss of rods over cones.104 The total number of foveal cones in eyes with large drusen and basal deposits was similar to that of age-matched controls and the foveal cone mosaic appeared normal. By contrast, cones appeared large and misshapen and few rods remained in the parafovea and, by late-stage AMD, virtually all surviving photoreceptors in the macula were cones.104 In eyes from wet AMD donors, photoreceptors surviving on, or at the margins of, disciform scars were largely cones. Subsequent functional studies supported the histological evidence for preferential vulnerability of rods in aging and AMD.105 Although Jackson and colleagues concluded from these histological studies that photoreceptor loss is secondary to RPE dysfunction or death in AMD, it still remains possible that a photoreceptor abnormality could be the primary effect in AMD and that this leads to subsequent loss of the underlying RPE.
Despite the overwhelming evidence of retinal cell loss in AMD there are surprisingly few reports on the type of cell death, or the initiating insult(s), in human AMD tissue. Most studies have relied on cell culture assays and animal models with an AMD phenotype which indicate that cell death occurs primarily by apoptosis. The most detailed report is from Dunaief and colleagues who observed a statistically significant increase in TUNEL-positive apoptotic cells in the inner choroid, RPE, ONL, and INL of AMD donor eyes.106 This increase in the number of apoptotic cells was evident in 5 of 6 eyes with geographic atrophy and all eyes with exudative AMD. Interestingly, in eyes with drusen, only a few TUNEL-positive cells were observed in each nuclear layer except the RGC layer. From the same study TUNEL-positive photoreceptors are also evident over an area of disorganized RPE near an edge of atrophy. TUNEL-positive RPE cells were found most commonly near areas of atrophy and occurred more often in AMD eyes than in controls.106 While an interesting and informative study on cell death in AMD, the number of apoptotic cells seems high, especially in the neural retina, for a slowly progressive condition. By contrast, Xu et al. only observed apoptotic cells in the retina of 4 of 16 AMD donor eyes and the overall numbers of apoptotic cells appeared relatively low.107 Nordgaard and colleagues undertook proteomic analysis of the RPE from donor eyes at progressive stages of AMD.108 Several components of apoptotic signaling pathways (αA crystallin, VDAC1, HSP70, GST-π) demonstrated changes in expression early in AMD or changed linearly with AMD progression. Surprisingly, information on death mechanisms in the AMD is scarce but there is evidence from cell culture and animal models for both intrinsic and extrinsic apoptosis pathways. FAS-mediated apoptosis has been observed in retinal cells, which could explain the role of inflammation in AMD,109 and a multitude of studies have reported oxidative stress-induced apoptosis by the classic mitochondrial route.110–113 The recent finding of Alu RNA accumulation leading to RPE cytotoxicity in geographic atrophy and Dicer1 downregulation suggests a completely new mechanism of RPE degeneration.114 It should also be considered that cellular repair, cellular replacement, and damage control are critical in retinal homeostasis and a declining antioxidant system together with increased oxidative damage will play a major etiological role in AMD, and that once the threshold for damage is reached, multiple cellular processes for repair and regeneration will be impacted.115–118
There is increasing evidence that autophagy flux may be dysregulated in the RPE in AMD119,120 and is likely due to multiple factors that affect the initiation of autophagy and/or the fusion of autophagosomes with lysosomes. These will include lipofuscin accumulation, susceptibility to oxidative stress, mitochondrial damage, and lysosomal dysregulation, all of which have a strong association with AMD. To what extent changes in autophagy flux reflect alterations in the formation or elimination of autophagosomes and are a cause or consequence of AMD remains to be determined. Lipid peroxidation products reduce autophagy flux and increase lipofuscin accumulation in cultured RPE cells.121 An increase in lysosomal pH which is associated with the lipofuscin constituent A2E122 may impair autophagosome–lysosome fusion, as may the accumulation of lipofuscin granules within the lysosomal vacuome. It has been reported that drusen formation may reflect an increase in both mitochondrial damage and autophagy in the RPE.123 The researchers speculated that increased autophagy and the release of intracellular proteins via exosomes by the aged RPE may contribute to the formation of drusen. It is important to note that there is substantial cross-talk between autophagy and proteasomal degradation pathways which may also affect the status of the RPE.119
Retinal detachment
Retinal detachment resulting from full-thickness retinal breaks, subretinal exudation, and/or vitreoretinal traction is a common cause of photoreceptor loss.124 Cell death is highly dependent on the area and duration of the detachment. Analysis of tissue samples from patients with retinal detachment showed the presence of significant numbers of apoptotic cells by 24 hours, which peaked by 2 days and dropped to a low level by 7 days after detachment.125 These observations have been largely supported by experimental retinal detachment in cats and rats, both of which show a peak of apoptosis between 1 and 3 days postdetachment, which then declines.126,127 Some apoptotic cells were still evident at 28 days postdetachment. However, there is some debate whether apoptosis following retinal detachment occurs via the intrinsic or extrinsic pathways.124,128,129 Interestingly, retina RPE separation in rats causes a Fas-dependent activation of autophagy in injured photoreceptors and, if autophagy is inhibited, the time course and number of apoptotic cells are accelerated.130 Thus it appears that autophagy is activated to regulate the level of receptor apoptosis.
Retinal dystrophies
Retinal dystrophies encompass a heterogeneous group of inherited conditions, with more than 100 genes or loci identified so far.131 The most common subtype is retinitis pigmentosa (RP), which is characterized by progressive death of retinal rod and cone photoreceptors, and the disease proceeds toward reduction of peripheral field with tunnel vision and finally loss of sight.131,132 One of the major impediments in the comprehensive identification of the degenerative mechanisms of retinal dystrophies is the heterogeneity of the disease and involvement of multiple causal genes in the pathogenesis of the disease.133 The mode of rod cell death in several animal models of RP suggests death by apoptosis, which is in agreement with findings in RP retinas from donor eyes134 (reviewed by Travis135). Cone cells usually die as a secondary response to rod cell death, possibly because they depend on rod-secreted neurotrophic factors for survival.57,60
Studies of apoptotic mechanisms in the photoreceptors of RP models imply the involvement of caspase-dependent as well as independent pathways.136–138 DNA fragmentation was a regular feature encountered in the mouse models, indicating that photoreceptors in mouse models die of apoptosis. Administration of caspase-3 inhibitors inhibited photoreceptor apoptosis in the tubby mouse (Usher syndrome model).139 Caspase-independent modes of apoptosis may involve calpain and calcium ion excess in RP.140 In recent years it has been suggested that, although the primary cell death mode will be apoptosis, other modes of cell removal could be involved, including autophagy and complement-mediated lysis.141 Investigation for different cell death pathways in three independent mouse models of photoreceptor degeneration – the rd/rd mouse, the rds/rds mouse and the light-damage model in albino mice – shows that, apart from apoptotic cell death, several oxidative stress markers as well as elements of the autophagic and complement pathways are upregulated. While the induction of oxidative stress response genes is early, the induction of autophagy was only seen in damaged retinas when compared to controls, which made the authors conclude that autophagy specifically removes damaged photoreceptors from the retina.141 However, the data may also be interpreted as an attempt in the damaged retina to salvage the photoreceptors from initial stress which, when overwhelming, gives rise to autophagic death. Finally, the evidence of upregulation of high-mobility group box 1 (HMGB1) protein in human eyes with retinal detachment suggests that necrosis could also be a mode of photoreceptor death.142 It could well be this mode of cell death that accounts for some of the caspase-independent photoreceptor death pathways that have hitherto been thought to be apoptosis. However the relevance of necrosis in early stages of RP still needs to be investigated. Nevertheless, the heterogeneity of RP and similar retinal dystrophies necessitates the understanding of the earliest mechanisms of disease inception such that customized treatments may be catered according to the nature of disease pathology in a patient. Attempts to block cell death by one strategy may prove to be futile as the protective effect may only be successful for a short duration, after which the cell might proceed through another death mode.
Light damage
The retina is vulnerable to damage from ultraviolet radiation, visible light (400–700 nm), and infrared radiation.66,143,144 The extent and type of damage are highly dependent upon the wavelength, power, duration, area of coverage, cumulative exposure, and location (e.g., macular versus periphery). Light-induced retinal damage can occur via at least one of three mechanisms: (1) mechanical (high irradiances of short duration that cause sonic shock waves disrupting the tissue irreversibly); (2) photothermal (when incident energy is trapped, causing a rise of temperature 10°C or more); and (3) photochemical (shorter-wavelength visible light is absorbed and dissipated away, causing molecular alterations).66 Retinal photodamage is highly dependent on the presence of chromophores, of which the most obvious are the visual pigments, and in most species prolonged intense light exposure will lead to significant photoreceptor cell damage.145 However, other important chromophores include hemoglobin, melanin, lipofuscin, macular pigment, and flavins.66 Photochemical damage has been the most extensively studied form of light damage because it can cause retinal damage within the intensity range of ambient visible light.66,146 There are two well-defined types of retinal light damage. Class 1 damage has an action spectrum that is identical to the absorption spectrum of the visual pigment and the initial damage is in the photoreceptors.66,146,147 Class 2 damage has an action spectrum that peaks at shorter wavelengths, is generally confined to the RPE, and is often referred to as the blue light hazard.66,146,148
Most mechanistic studies on retinal photochemical damage have been undertaken in experimental animal models and all show that cell death occurs via apoptosis. Excess light has been shown to trigger apoptosis in rat retinas.149,150 Low-intensity light exposure for long durations as well as intense light exposure (3000 lux) for short durations up to 2 hours can cause severe photoreceptor degeneration by apoptosis followed by RPE degeneration.151,152 Both types of retinal photodamage, induced by exposure to either low- or high-intensity light, are dependent on the presence of, and regeneration of, rhodopsin in the retina.153–157 The extent of photodamage is correlated to the amount of rhodopsin present in the retina prior to the light damage,158,159 although direct evidence of rhodopsin causing damage is absent. Several rhodopsin intermediates, especially all-trans-retinal accumulation in the photoreceptor membrane due to decreased reduction by retinol dehydrogenase, have been implicated as mediators of photo-oxidation.160 Decrease of intracellular calcium levels during the phototransduction cycle has also been suggested to be an aspect of rhodopsin-mediated injury causing photoreceptor apoptosis.161 Light-induced photoreceptor apoptosis can either be caspase-mediated162–166 or caspase-independent.167 The most probable reason for difference of opinion in mechanisms like caspase involvement and signal transduction in photoreceptor death is that cellular response will vary according to the lighting conditions and, in reality, all of the discovered pathways could exist to decide on cell fate depending on the prevalence and type of light damage during an individual’s lifetime. Melanosomes and lipofuscin are considered the most significant fluorophores implicated in RPE damage. Irradiated lipofuscin granules or their constituents can generate high levels of reactive oxygen species.168–170 Several models of retinal degeneration in vitro and in vivo demonstrate the role of calcium in inducing cell death by activating degradative proteases such as calpain.171,172 Adaptor protein 1 (AP-1) signal transduction has also been implicated in bright light-induced photoreceptor death where there is an increase of AP-1 gene expression.173 Light can damage not only photoreceptor cells and RPE but also RGCs.174 Photosensitive RGCs contain melanopsin, suggesting that light damage could impact a specific subset of RGCs and contribute to the pathogenesis of glaucoma.175,176 The role of autophagy in retinal light damage has only received limited attention but Kunchithapautham and colleagues have shown that upregulation of autophagy can protect against light damage and oxidative stress in the retina.177,178
Therapeutic options
Neuroprotection
Neuroprotection offers a promising approach for preventing or slowing retinal cell loss. Neuroprotection is essentially defined as the use of therapeutic agents to prevent, hinder, and in some instances, reverse neuronal cell death whatever the primary injury.179–181 The strategies for neuroprotection vary considerably dependent upon the target cell, the nature of the factors initiating cell loss, and the stage of disease. Clearly, the best time for neuroprotection is early in the time course of the disease before any significant cell death has occurred. Three broad approaches have evolved: (1) blocking the pathways involved in the cellular damage (e.g., antioxidants); (2) inhibiting the cell death path directly (e.g., upregulation of Bcl-2 and inhibition of caspases); and (3) treatment with neurotrophic factors that suppress the intrinsic apoptosis pathway (e.g., brain-derived neurotrophic factor (BDNF), glial-derived neurotrophic factor (GDNF), pigment epithelial-derived factor (PEDF)). A summary of potential neuroprotective agents, their retinal cell targets, and their proposed mechanisms of action is shown in Table 24.1.
Table 24.1 A summary of potential neuroprotective agents, their retinal cell targets, and their proposed mechanisms of action
| Target cell | Mechanistic target | Therapeutic approaches |
|---|---|---|
| Retinal ganglion cells | Excitotoxicity | Memantine |
| Oxidative stress | Vitamin E | |
| Mitochondrial damage | Coenzyme Q10 | |
| Neurotrophin derivation | BDNF, gene therapy | |
| Photoreceptors | Apoptosis | Survival and growth factors (e.g., BDNF, GDNF) |
| RPE | Apoptosis | Survival and growth factors |
| Autophagy | Rapamycin | |
| Oxidative damage | Antioxidants, lutein, zeazanthin, zinc | |
| Mitochondrial damage | Antioxidants, resveratrol | |
| Lipofuscin system | Reducing the retinoid cycle Glutathione-S-transferase |
|
| Lysosomal system | Elevated pH (A2A adenosine receptor agonist) | |
| Bruch’s membrane | Subthreshold laser, MMP upregulation, microbial enzymes | |
| Choroid | Endothelial cell loss | Growth factors (e.g., VEGF) |
| Inflammation | Alternative complement pathway | Complement pathway inhibitors |
BDNF, brain-derived neurotrophic factor; GDNF, glial-derived neurotrophic factor; RPE, retinal pigment epithelium; MMP, matrix metalloproteinase; VEGF, vascular endothelial growth factor.
Light damage in rodents, which results in extensive photoreceptor and RPE cell loss, has been extensively studied in the search for effective neuroprotective agents.143 Light induction of photoreceptor apoptosis is dependent on the availability of 11-cis retinal for rhodopsin regeneration.182 Acute bright light-induced photoreceptor apoptosis involves activation of nitric oxide synthase (NOS) and generation of nitric oxide (NO), increased intracellular calcium, oxidative damage, and alterations in mitochondrial function.143,183 Inhibition of NOS activity by LNAME183,184 or application of the calcium channel blockers d-diltiazem, nilvadipine, or nicardipine167 protected photoreceptor cells following cell death in light-damaged retinas. Inhibition of the apoptotic cascade by reduction of the proapoptotic Bcl-2 family members Bax and Bak also protects the retina against light damage.185 The use of antioxidants (that are reactive oxygen species quenchers) like dimethylthiourea and N-phenyl-2-naphthylamine and the administration of exogenous neurotrophic factors such as BDNF, ciliary neurotrophic factor (CNTF), basic fibroblast growth factor (bFGF), erythropoietin (Epo), and PEDF have all been shown to be protective against light damage (reviewed by Wenzel et al.143). bFGF plays an important role in the endogenous defense against stress of retinal cells. Preconditioning with light186,187 or ischemic preconditioning188,189 increases the expression of bFGF and protects retinal cells against damaging doses of light or increased intraocular pressure. The neuroprotective effect could be enhanced by a combination of bFGF and PEDF.190 In contrast, no protection resulted against constant light exposure for 1 week when bFGF was expressed locally from a transgene delivered by adeno-associated virus vector191 or simian immunodeficiency virus vector.192 However, intravitreal injection of recombinant BDNF protein or its release from transgenic cell transplants193 protects the retina against 1–2 weeks of constant light exposure.
Inhibition of apoptotic pathways in a light-damaged neuron is relatively straightforward as the cell is otherwise healthy. However, in retinal cells harboring mutations, as is the case in retinal dystrophies, the cell has more than just the apoptotic cascade to address as the cause of the dystrophy and associated cell death will remain unless also treated. Photoreceptors are the target cells as these are the predominant cell type lost in the retinal dystrophies. Studies in animal models with inherited retinal diseases have used similar strategies to those described above for light damage. While considerable success has been achieved, this has not translated to clinical care and gene therapy to reverse the mutation, as in Leber’s congenital amaurosis, is perhaps a preferable option.194 Both PEDF and CNTF have shown considerable promise in slowing photoreceptor degeneration, whether provided by intraocular or gene transfer, in rd1, rd2, rds P216L, rhodopsin S334ter and P23H rodent models of inherited degeneration.195–200 CNTF, released by engineered cells, slowed retinal degeneration in the rcd1 dog model201 and, after intravitreal injection, in an autosomal dominant feline model of rod–cone dystrophy.202 Neurotrophin-3 can induce upregulation of bFGF, and thereby neuroprotection, by activation of TrkC in Müller cells.203 Viral delivery of a BDNF transgene delayed degeneration induced by the Q344ter mutation in rhodopsin204 and prolonged release from transgenic cell transplants into the eye slowed degeneration in RCS rats.205 Capsase-3 inhibition is protective in the rd1 degeneration by delaying cell death172,206 and preservation of retinal morphology.207 Caspase-3 inhibitors also provided protection in two other models of inherited retinal degeneration: the S334ter rat137 and the tubby mouse.139 Alternative approaches which have reduced photoreceptor loss and slowed the progression of retinal degeneration in animal models include: (1) overexpression of the X-linked inhibitor of apoptosis using gene therapy208,209; (2) sustained intravitreal delivery of fluocinolone acetonide, which suppresses microglial activation and inflammation210; (3) GDNF either given by subretinal injection or conjugated to nanoparticles211,212; and (4) neuroprotectin D1 (NPD1).110
Rodent and dog models of glaucoma have shown that agents that block glutamate excitoxicity, prevent mitochondrial dysfunction, reduce oxidative stress, or enhance neurotrophic factors all decrease or prevent the loss of RGCs (Table 24.1).213,214 The N-methyl-d-aspartate receptor antagonist was shown to be a highly effective neuroprotective agent in animal models of RGC death (reviewed by Cheung et al.213 and Danesh-Meyer214). However, the outcome of clinical trials, although showing a trend for improvement, was inconclusive. An alternative agent is brimonidone tartrate, which is a highly selective alpha2-adrenergic agonist that increases RGC survival in animal models.215 Coenzyme Q10, which is an essential cofactor of the electron transport chain, has been shown to have some neuroprotective effect on RGCs based on its antioxidant properties.216 Treatment with exogenous neurotrophins such as BNDF, nerve growth factor, and CNTF have all been shown to slow the loss of RGCs but not to inhibit RGC death in the long term. To improve outcomes, strategies that researchers have attempted include sustained release of neurotrophic factors via either gene therapy217 or encapsulated cell technology, which allows the intravitreal implantation of a chamber containing live cells programmed to release CNTF.218 More direct antiapoptotic strategies have included activation of the Bcl-2 pathways using cilostazol or 5-S-GAD which increases RGC survival in animal models.219,220
RPE cell loss is a hallmark feature of dry AMD. In vitro studies have identified a plethora of agents capable of either directly inhibiting apoptosis with, for example, caspase inhibitors, or indirectly by neutralizing the initiating factors leading to cell death. Macular carotenoids, NPD1, eEpo, resveratrol, and PEDF have all been shown to confer protection against oxidative stress-induced apoptosis.113,221–227 αB crystallin is apically secreted within exosomes by polarized human RPE and provides neuroprotection to adjacent cells.228 An alternative approach has been to upregulate antioxidants or molecular chaperones to cope with the increased oxidative stress.112,229 Neurotrophic protection of the RPE has been more difficult to model in vivo as there are no reliable models of geographic atrophy. However, the Age-Related Eye Disease Study demonstrated that oral supplementation with high levels of antioxidants and zinc can reduce the risk of disease progression to advanced AMD.230
The strategies described above are likely to be equally effective in other retinal conditions resulting in cell death. Imai and colleagues emphasize the importance of exogenous neurotrophic factors in reducing cell loss in DR231 while BDNF, bFGF, and inhibition of the FAS receptor impede apoptosis following retinal detachment.127,128
Modulating autophagy
As discussed earlier, autophagy is dysregulated in a number of retinal conditions. However, dependent on the type and stage of the disease, this dysregulation could reflect as decreased or increased autophagy. A similar trend is also observed in cancer and neurodegenerative diseases. That is, autophagy plays a role in tumor suppression or oncogenesis but can desensitize cells to cytotoxic agents.232 Thus, while enhancing autophagy could prevent tumor progression it could also reduce sensitivity to chemotherapeutic agents. Similarly, upregulation of autophagy can protect against Huntington’s disease in mouse models whereas enhancing autophagosome formation in Alzheimer’s disease can exacerbate the accumulation of Aβ.232 Therefore any therapy must ensure the correct balance of autophagy flux is attained for the targeted disease or cell type.
Many autophagy-related proteins and signaling molecules have been implicated in a number of events of autophagy, including signaling, sequestration, maturation, and degradation. The primary regulation of autophagy proceeds by mTor, GCN2, and PI3K-III signaling pathways. Autophagy is negatively regulated by mTor and Bcl-2/Bclxl and positively regulated by PI3K class III and GCN2/eIF2α. Compounds that inhibit mTor phosphorylation (rapamycin) or Bcl-2 interaction with Beclin-1 (ABT737) promote autophagy.233,234 PI3K-III can be activated by inhibiting the interactions between Bcl-2 and Beclin/Atg6 or by overexpressing Beclin-1 or Beclin-1-BD (Bcl-2 binding defective mutant) and suppression by Vps34 inhibitors, 3-methyladenine, or wortmannin.235,236 A number of mTOR-dependent and mTOR-independent agents have been identified that stimulate autophagy. Rapamycin is a well-established compound for inducing autophagy and attenuating neuronal cell death in a number of in vitro and in vivo experimental models. Other stimulators of autophagy include lithium and trehalose which enhance autophagy via an mTOR-independent mechanism.237,238 Unfortunately, despite their potency, rapamycin and lithium have significant side-effects which lessen enthusiasm for their clinical use in chronic neurodegenerative diseases.14,239,240 In an attempt to address this researchers are screening for small-molecule enhancers of rapamycin (SMERs) and small molecule inhibitors of rapamycin (SMIRs) with less cytotoxicity.241 The efficacy and specificity of compounds that activate autophagy in an mTOR-independent fashion have yet to be established.239,240 Gene therapy is also a possibility and overexpression of Atg7 can protect against anoxia/reoxygenation injury.242
Given the realization of the importance of autophagy in cancer and neurodegenerative diseases there has been an extensive effort to identify potential therapeutic regulators of autophagy. Those under development include HDAC inhibitors, mTOR inhibitors, BH3 domain mimetics, glycolytic inhibitors, inositol-lowering agents, and protein kinase inhibitors.241,243–245 Of particular interest in cancer has been the use of the antimalarial drug hydroxychloroquine (HCQ) which serves to inhibit autophagy by perturbing lysosomal function.243,244 HCQ is now being assessed in combination with a number of chemotherapy agents in phase I and II clinical trials.243,244 However, it has long been recognized that HCQ can cause ocular toxicity, with the most serious being an irreversible retinopathy. The dosage parameters associated with retinopathy are still uncertain but it has been suggested that, for doses of HCQ less than 6.5 mg/kg, the incidence of retinopathy is minimal.246 Nevertheless, some clinical trials are testing dosages that exceed these levels. Furthermore, shutting down lysosomal functions, whether it be autophagy or endocytosis, can dramatically alter cellular homeostasis and defense.
Proteins other than the autophagic pathway proteins have been implicated in autophagy. Caspases and calpains play key roles in cleavage and activation or inactivation of autophagy proteins (summarized by Kaminskyy and Zhivotovsky247). Cross-talk between the autophagy and apoptosis pathways is regulated by caspase cleavage of Beclin-1248 and also by p62/Sqstm1-Keap1 signaling.249 Similarly, modulation of the Sirt-1-Foxo pathway also modulates autophagy.250,251 Modulating reactive oxygen species levels with antioxidant compounds such as resveratrol and curcumin or overexpressing antioxidant enzymes can negatively modulate autophagy. Quenching reactive oxygen species will decrease mitochondrial damage and autophagy initiation. In addition, a lowering of reactive oxygen species will preserve the activity of lysosomal enzymes (reviewed by Scherz-Shouval and Elazar252). It is important to keep in mind that, when one type of autophagy is altered, the other types will also be affected. Chronic blockage of CMA promotes upregulation of macroautophagy,253 whereas acute blockage leads to macroautophagic dysregulation.254 Cells respond to blockage of macroautophagy by increasing CMA.254 Acute blockage of the proteasome upregulates macroautophagy,255 whereas chronic blockage leads to macroautophagic dysregulation.256 Some subunits of the proteasome are degraded by CMA,257 which may explain why blockage of CMA is associated with proteasome dysregulation.258 Interactions of microautophagy with other proteolytic systems remain undiscovered.
Cellular replacement
In order to maintain optimal tissue and organ function, dead cells need to be replaced. Amphibians and fish exhibit robust retinal regeneration which, unfortunately, is not retained in mammals. It has long been known that amphibians can regenerate a completely new and functional retina from the adjacent RPE, which restores vision.259,260 Retinal regeneration in fish is not from the RPE and instead occurs from an intrinsic progenitor that is derived from the Müller glia cells.261,262 Birds show some limited replacement of lost neurons which involves a mitotic subpopulation of Müller glia.263 Cellular replacement in the retina of mammals is very minimal, even in response to injury.260
A number of endogenous stem/progenitor populations have been reported but many, such as retinal stem cells in the ciliary margin zone, remain controversial.260 Studies in rodents have found evidence of progenitor gene expression in Müller glia after retinal damage but there is no definitive evidence that they produce differentiated and functional neurons. However, Ooto et al.264 did report that these “progenitor-like” Müller cells can express markers for bipolar cells and photoreceptors after injury. An alternative source of reparative cells could be derived from the bone marrow. Using chimeric mice transplanted with bone marrow cells expressing green fluorescent protein, it was shown that bone marrow-derived cells would home to the site of retinal injury and had the potential to differentiate into astrocytes, macrophages/microglia, endothelial cells, pericytes, and RPE.96,97 However, recruitment and integration occurred at a very low level. To overcome this, a recent study infected bone marrow-derived cells ex vivo with lentiviral vector expressing the RPE-specific gene RPE65 and injected these cells into the circulation of mice in which the RPE had been destroyed by sodium iodate.265 These transplanted cells homed to the neural retina Bruch’s membrane interface in large numbers and showed restoration of a functional RPE layer, with typical RPE phenotype, including coexpression of another RPE-specific marker, CRALBP, and photoreceptor outer-segment phagocytosis. Most importantly, retinal degeneration was prevented and visual function was restored to levels similar to those found in normal animals.265
An alternative approach, with greater clinical application, has been the transplantation of a variety of stem/progenitor cell types. Most focus has been on the RPE as it can be readily generated from embryonic stem cells (ESC) or induced pluripotent stem (iPS) cells.266–269 These cells have been successfully transplanted into a number of animal models of retinal degeneration and demonstrated recovery of vision, and ESC-derived RPE is now in phase I/II clinical trials. While a promising strategy, there remain concerns that: (1) ESC may be prone to teratoma formation270; (2) iPS cells contain protein-coding point mutations271; (3) success with RPE transplantation in humans has been modest272,273; and (4) transplantation is normally into a severely degenerated retina with late-stage disease.272,273 Despite these limitations, considerable progress has been made in the field of RPE replacement in the last decade. Repair of the neural retina is more complicated due to the need to form the requisite neural connections to perform vision. The most cited study is that of MacLaren and colleagues, who showed that donor cells can integrate into the adult or degenerating mouse retina provided the donor cells are derived from the developing retina at a time coincident with the peak of rod genesis.274 Importantly, these transplanted cells integrated and differentiated into functional rod photoreceptors that formed synaptic connections, and improved visual function in the host animals. Human ESC or iPS have been shown, under the right conditions, to be able to differentiate into rods and cones275–278 and when transplanted into the adult mouse retina can differentiate into photoreceptors and restore light responses in Crx-deficient mice.279 Since it is not the intention of this chapter to provide a detailed review of regenerative medicine for retinal repair, the reader is pointed to the following reviews and Chapter 35, Stem cells and cellular therapy.260,275,280–283
1 Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11.
2 Green DR. Means to an end: apoptosis and other cell death mechanisms. New York: Cold Spring Harbor Laboratory Press; 2011.
3 Green DR, Reed JC. Apoptosis: physiology and pathology. Cambridge: Cambridge University Press; 2011.
4 Pereira WO, Amarante-Mendes GP. Apoptosis: a programme of cell death or cell disposal? Scand J Immunol. 2011;73:401–407.
5 Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43.
6 Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323.
7 Zhivotovsky B, Orrenius S. Cell death mechanisms: cross-talk and role in disease. Exp Cell Res. 2010;316:1374–1383.
8 Yousefi S, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132.
9 Chen Y, Klionsky DJ. The regulation of autophagy – unanswered questions. J Cell Sci. 2011;124:161–170.
10 Hubbard VM, Valdor R, Macian F, et al. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology. 2011 Apr 3. [Epub ahead of print]
11 Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811.
12 Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–189.
13 Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy. 2011;7:673–682.
14 Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075.
15 Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131.
16 Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14.
17 Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667.
18 Ravikumar B, Moreau K, Jahreiss L, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757.
19 Ravikumar B, Moreau K, Rubinsztein DC. Plasma membrane helps autophagosomes grow. Autophagy. 2010;6:1184–1186.
20 Dunn WA, Jr. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1933.
21 Nobukuni T, Joaquin M, Roccio M, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243.
22 Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595.
23 Fimia GM, Di Bartolomeo S, Piacentini M, et al. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy. 2011;7:115–117.
24 Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. “Protein modifications: beyond the usual suspects” review series. EMBO Rep. 2008;9:859–864.
25 Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302.
26 Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–2100.
27 Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802.
28 Weidberg H, Shpilka T, Shvets E, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. 2011;20:444–454.
29 Saftig P. Physiology of the lysosome. In: Mehta A, Beck M, Sunder-Plassmann G. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis, 2006. Chapter 3
30 Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–564.
31 Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760.
32 Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223.
33 Lee JY, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980.
34 Shpilka T, Elazar Z. Shedding light on mammalian microautophagy. Dev Cell. 2011;20:1–2.
35 Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–31513.
36 Farre JC, Krick R, Subramani S, et al. Turnover of organelles by autophagy in yeast. Curr Opin Cell Biol. 2009;21:522–530.
37 Marzella L, Ahlberg J, Glaumann H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;36:219–234.
38 Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139.
39 Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549.
40 Alamouti B, Funk J. Retinal thickness decreases with age: an OCT study. Br J Ophthalmol. 2003;87:899–901.
41 Cavallotti C, Artico M, Pescosolido N, et al. Age-related changes in the human retina. Can J Ophthalmol. 2004;39:61–68.
42 Eriksson U, Alm A. Macular thickness decreases with age in normal eyes: a study on the macular thickness map protocol in the Stratus OCT. Br J Ophthalmol. 2009;93:1448–1452.
43 Marshall J, Grindle J, Ansell PL, et al. Convolution in human rods: an ageing process. Br J Ophthalmol. 1979;63:181–187.
44 Tucker GS. Refractile bodies in the inner segments of cones in the aging human retina. Invest Ophthalmol Vis Sci. 1986;27:708–715.
45 Iwasaki M, Inomata H. Lipofuscin granules in human photoreceptor cells. Invest Ophthalmol Vis Sci. 1988;29:671–679.
46 Boulton ME. Lipofuscin of the retinal pigment epithelium. In: Lois N, Forrester JV. Fundus autofluorescence. Philadelphia: Wolters Kluwer/Lipincott Williams & Wilkins, 2009.
47 Gartner S, Henkind P. Aging and degeneration of the human macula. 1. Outer nuclear layer and photoreceptors. Br J Ophthalmol. 1981;65:23–28.
48 Eliasieh K, Liets LC, Chalupa LM. Cellular reorganization in the human retina during normal aging. Invest Ophthalmol Vis Sci. 2007;48:2824–2830.
49 Mansergh F, Orton NC, Vessey JP, et al. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14:3035–3046.
50 Liets LC, Eliasieh K, van der List DA, et al. Dendrites of rod bipolar cells sprout in normal aging retina. Proc Natl Acad Sci U S A. 2006;103:12156–12160.
51 Curcio CA, Millican CL, Allen KA, et al. Aging of the human photoreceptor mosaic: evidence for selective vulnerability of rods in central retina. Invest Ophthalmol Vis Sci. 1993;34:3278–3296.
52 Gao H, Hollyfield JG. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1–17.
53 Curcio CA, Owsley C, Jackson GR. Spare the rods, save the cones in aging and age-related maculopathy. Invest Ophthalmol Vis Sci. 2000;41:2015–2018.
54 Sanyal S, Hawkins RK, Jansen HG, et al. Compensatory synaptic growth in the rod terminals as a sequel to partial photoreceptor cell loss in the retina of chimaeric mice. Development. 1992;114:797–803.
55 Jansen HG, Sanyal S. Synaptic plasticity in the rod terminals after partial photoreceptor cell loss in the heterozygous rds mutant mouse. J Comp Neurol. 1992;316:117–125.
56 Spear PD. Neural bases of visual deficits during aging. Vision Res. 1993;33:2589–2609.
57 Leveillard T, Mohand-Said S, Fintz AC, et al. The search for rod-dependent cone viability factors, secreted factors promoting cone viability. Novartis Found Symp. 2004;255:117–127. discussion 127–30, 177–8
58 Chalmel F, Leveillard T, Jaillard C, et al. Rod-derived cone viability factor-2 is a novel bifunctional-thioredoxin-like protein with therapeutic potential. BMC Mol Biol. 2007;8:74.
59 Fridlich R, Delalande F, Jaillard C, et al. The thioredoxin-like protein rod-derived cone viability factor (RdCVFL) interacts with TAU and inhibits its phosphorylation in the retina. Mol Cell Proteomics. 2009;8:1206–1218.
60 Yang Y, Mohand-Said S, Danan A, et al. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009;17:787–795.
61 Feuer WJ, Budenz DL, Anderson DR, et al. Topographic differences in the age-related changes in the retinal nerve fiber layer of normal eyes measured by Stratus optical coherence tomography. J Glaucoma. 2011;20:133–138.
62 Aggarwal P, Nag TC, Wadhwa S. Age-related decrease in rod bipolar cell density of the human retina: an immunohistochemical study. J Biosci. 2007;32:293–298.
63 Lei Y, Garrahan N, Hermann B, et al. Transretinal degeneration in ageing human retina: a multiphoton microscopy analysis. Br J Ophthalmol. 2011;95:727–730.
64 Panda-Jonas S, Jonas JB, Jakobczyk-Zmija M. Retinal pigment epithelial cell count, distribution, and correlations in normal human eyes. Am J Ophthalmol. 1996;121:181–189.
65 Dorey CK, Wu G, Ebenstein D, et al. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–1699.
66 Boulton M, Rozanowska M, Rozanowski B. Retinal photodamage. J Photochem Photobiol B. 2001;64:144–161.
67 Boulton M, Albon J. Stem cells in the eye. Int J Biochem Cell Biol. 2004;36:643–657.
68 Agarwal R, Gupta SK, Agarwal P, et al. Current concepts in the pathophysiology of glaucoma. Ind J Ophthalmol. 2009;57:257–266.
69 Kisiswa L, Dervan AG, Albon J, et al. Retinal ganglion cell death postponed: giving apoptosis a break? Ophthalmic Res. 2010;43:61–78.
70 Qu J, Wang D, Grosskreutz CL. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp Eye Res. 2010;91:48–53.
71 Pease ME, McKinnon SJ, Quigley HA, et al. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41:764–774.
72 Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999;18:39–57.
73 Quigley HA, Nickells RW, Kerrigan LA, et al. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 1995;36:774–786.
74 Kerrigan LA, Zack DJ, Quigley HA, et al. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997;115:1031–1035.
75 Levin LA. Mechanisms of optic neuropathy. Curr Opin Ophthalmol. 1997;8:9–15.
76 Baltmr A, Duggan J, Nizari S, et al. Neuroprotection in glaucoma – Is there a future role? Exp Eye Res. 2010;91:554–566.
77 Libby RT, Li Y, Savinova O, et al. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26.
78 Rodriguez-Muela N, Germain F, Marino G, et al. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2011;19:162–169.
79 Russo R, Berliocchi L, Adornetto A, et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144.
80 Kim SH, Munemasa Y, Kwong JM, et al. Activation of autophagy in retinal ganglion cells. J Neurosci Res. 2008;86:2943–2951.
81 Sternberg C, Benchimol M, Linden R. Caspase dependence of the death of neonatal retinal ganglion cells induced by axon damage and induction of autophagy as a survival mechanism. Braz J Med Biol Res. 2010;43:950–956.
82 Hammes HP, Lin J, Renner O, et al. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes. 2002;51:3107–3112.
83 Porta M. The importance of endothelial damage: Diabetic retinopathy. Adv Stud Med. 2005;5:S150–S158.
84 Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366–378.
85 Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7:1581–1587.
86 Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156–1163.
87 El-Remessy AB, Al-Shabrawey M, Khalifa Y, et al. Neuroprotective and blood–retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–244.
88 Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–791.
89 Lopes de Faria JM, Russ H, Costa VP. Retinal nerve fibre layer loss in patients with type 1 diabetes mellitus without retinopathy. Br J Ophthalmol. 2002;86:725–728.
90 van Dijk HW, Kok PH, Garvin M, et al. Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:3404–3409.
91 van Dijk HW, Verbraak FD, Kok PH, et al. Decreased retinal ganglion cell layer thickness in patients with type 1 diabetes. Invest Ophthalmol Vis Sci. 2010;51:3660–3665.
92 Gastinger MJ, Singh RS, Barber AJ. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci. 2006;47:3143–3150.
93 Chen ZF, Li YB, Han JY, et al. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy. 2011;7:12–16.
94 Gonzalez CD, Lee MS, Marchetti P, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2–11.
95 Qi X, Cai J, Dunn WR, et al. Circadian rhythmicity in the expression of autophagy proteins in normal and diabetic retinas. Invest Ophthalmol Vis Sci. 51, 2010. ARVO E-Abstract 5614
96 Chan-Ling T, Baxter L, Afzal A, et al. Hematopoietic stem cells provide repair functions after laser-induced Bruch’s membrane rupture model of choroidal neovascularization. Am J Pathol. 2006;168:1031–1044.
97 Grant MB, May WS, Caballero S, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612.
98 Busik JV, Tikhonenko M, Bhatwadekar A, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906.
99 Caballero S, Sengupta N, Afzal A, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967.
100 Holz FG, Bindewald-Wittich A, Fleckenstein M, et al. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143:463–472.
101 Schatz H, McDonald HR. Atrophic macular degeneration. Rate of spread of geographic atrophy and visual loss. Ophthalmology. 1989;96:1541–1551.
102 Fleckenstein M, Adrion C, Schmitz-Valckenberg S, et al. Concordance of disease progression in bilateral geographic atrophy due to AMD. Invest Ophthalmol Vis Sci. 2010;51:637–642.
103 McLeod DS, Grebe R, Bhutto I, et al. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:4982–4991.
104 Curcio CA, Medeiros NE, Millican CL. Photoreceptor loss in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:1236–1249.
105 Jackson GR, Owsley C, Curcio CA. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res Rev. 2002;1:381–396.
106 Dunaief JL, Dentchev T, Ying GS, et al. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442.
107 Xu GZ, Li WW, Tso MO. Apoptosis in human retinal degenerations. Trans Am Ophthalmol Soc. 1996;94:411–430. discussion 430–1
108 Nordgaard CL, Berg KM, Kapphahn RJ, et al. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822.
109 Jiang S, Wu MW, Sternberg P, et al. Fas mediates apoptosis and oxidant-induced cell death in cultured hRPE cells. Invest Ophthalmol Vis Sci. 2000;41:645–655.
110 Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–271.
111 Cai J, Nelson KC, Wu M, et al. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221.
112 Kasahara E, Lin LR, Ho YS, et al. SOD2 protects against oxidation-induced apoptosis in mouse retinal pigment epithelium: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:3426–3434.
113 Mansoor S, Gupta N, Patil AJ, et al. Inhibition of apoptosis in human retinal pigment epithelial cells treated with benzo(e)pyrene, a toxic component of cigarette smoke. Invest Ophthalmol Vis Sci. 2010;51:2601–2607.
114 Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330.
115 Shamsi FA, Boulton M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Invest Ophthalmol Vis Sci. 2001;42:3041–3046.
116 Jarrett SG, Boulton ME. Antioxidant up-regulation and increased nuclear DNA protection play key roles in adaptation to oxidative stress in epithelial cells. Free Radic Biol Med. 2005;38:1382–1391.
117 Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403.
118 Winkler BS, Boulton ME, Gottsch JD, et al. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32.
119 Kaarniranta K. Autophagy – hot topic in AMD. Acta Ophthalmol. 2010;88:387–388.
120 Mitter SK, Rao HV, Qi X, et al. Autophagy in the retina: A potential role in age-related macular degeneration. Adv Exp Med Biol. 2012;723:83–90.
121 Krohne TU, Stratmann NK, Kopitz J, et al. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp Eye Res. 2010;90:465–471.
122 Liu J, Lu W, Reigada D, et al. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4(–/–) mice: pharmacologic approaches and functional recovery. Invest Ophthalmol Vis Sci. 2008;49:772–780.
123 Wang AL, Lukas TJ, Yuan M, et al. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS ONE. 2009;4:e4160.
124 Yang L, Bula D, Arroyo JG, et al. Preventing retinal detachment-associated photoreceptor cell loss in Bax-deficient mice. Invest Ophthalmol Vis Sci. 2004;45:648–654.
125 Arroyo JG, Yang L, Bula D, et al. Photoreceptor apoptosis in human retinal detachment. Am J Ophthalmol. 2005;139:605–610.
126 Cook B, Lewis GP, Fisher SK, et al. Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci. 1995;36:990–996.
127 Hisatomi T, Sakamoto T, Goto Y, et al. Critical role of photoreceptor apoptosis in functional damage after retinal detachment. Curr Eye Res. 2002;24:161–172.
128 Besirli CG, Chinskey ND, Zheng QD, et al. Inhibition of retinal detachment-induced apoptosis in photoreceptors by a small peptide inhibitor of the fas receptor. Invest Ophthalmol Vis Sci. 2010;51:2177–2184.
129 Dong K, Sun X. Targeting death receptor induced apoptosis and necroptosis: A novel therapeutic strategy to prevent neuronal damage in retinal detachment. Med Hypotheses. 2011;77:144–146.
130 Besirli CG, Chinskey ND, Zheng QD, et al. Autophagy activation in the injured photoreceptor inhibits fas-mediated apoptosis. Invest Ophthalmol Vis Sci. 2011;52:4193–4199.
131 Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809.
132 Marigo V. Programmed cell death in retinal degeneration: targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle. 2007;6:652–655.
133 Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12:238–249.
134 Portera-Cailliau C, Sung CH, Nathans J, et al. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U S A. 1994;91:974–978.
135 Travis GH. Mechanisms of cell death in the inherited retinal degenerations. Am J Hum Genet. 1998;62:503–508.
136 Cottet S, Schorderet DF. Mechanisms of apoptosis in retinitis pigmentosa. Curr Mol Med. 2009;9:375–383.
137 Liu C, Li Y, Peng M, et al. Activation of caspase-3 in the retina of transgenic rats with the rhodopsin mutation s334ter during photoreceptor degeneration. J Neurosci. 1999;19:4778–4785.
138 Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605.
139 Bode C, Wolfrum U. Caspase-3 inhibitor reduces apototic photoreceptor cell death during inherited retinal degeneration in tubby mice. Mol Vis. 2003;9:144–150.
140 Doonan F, Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in mouse models of retinal degeneration. J Neurosci. 2003;23:5723–5731.
141 Lohr HR, Kuntchithapautham K, Sharma AK, et al. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006;83:380–389.
142 Arimura N, Ki-i Y, Hashiguchi T, et al. Intraocular expression and release of high-mobility group box 1 protein in retinal detachment. Lab Invest. 2009;89:278–289.
143 Wenzel A, Grimm C, Samardzija M, et al. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306.
144 Mainster MA, Boulton ME. Retinal toxicity. In: Albert DM, Jakobiec FA, Azar DT, et al. Principles and practice of ophthalmology. London: Elsevier; 2008:2195–2205.
145 Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134.
146 Wu J, Seregard S, Algvere PV. Photochemical damage of the retina. Surv Ophthalmol. 2006;51:461–481.
147 Noell WK, Walker VS, Kang BS, et al. Retinal damage by light in rats. Invest Ophthalmol. 1966;5:450–473.
148 Ham WT, Jr., Mueller HA, Ruffolo JJ, Jr., et al. Action spectrum for retinal injury from near-ultraviolet radiation in the aphakic monkey. Am J Ophthalmol. 1982;93:299–306.
149 Abler AS, Chang CJ, Ful J, et al. Photic injury triggers apoptosis of photoreceptor cells. Res Commun Mol Pathol Pharmacol. 1996;92:177–189.
150 Zhang SR, Li SH, Abler A, et al. Tissue transglutaminase in apoptosis of photoreceptor cells in rat retina. Invest Ophthalmol Vis Sci. 1996;37:1793–1799.
151 Hafezi F, Marti A, Munz K, et al. Light-induced apoptosis: differential timing in the retina and pigment epithelium. Exp Eye Res. 1997;64:963–970.
152 Hafezi F, Steinbach JP, Marti A, et al. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat Med. 1997;3:346–349.
153 Grimm C, Wenzel A, Hafezi F, et al. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat Genet. 2000;25:63–66.
154 Grimm C, Wenzel A, Williams T, et al. Rhodopsin-mediated blue-light damage to the rat retina: effect of photoreversal of bleaching. Invest Ophthalmol Vis Sci. 2001;42:497–505.
155 Keller C, Grimm C, Wenzel A, et al. Protective effect of halothane anesthesia on retinal light damage: inhibition of metabolic rhodopsin regeneration. Invest Ophthalmol Vis Sci. 2001;42:476–480.
156 Wenzel A, Reme CE, Williams TP, et al. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci. 2001;21:53–58.
157 Rozanowska M, Sarna T. Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem Photobiol. 2005;81:1305–1330.
158 Farber DB, Danciger JS, Organisciak DT. Levels of mRNA encoding proteins of the cGMP cascade as a function of light environment. Exp Eye Res. 1991;53:781–786.
159 Organisciak DT, Xie A, Wang HM, et al. Adaptive changes in visual cell transduction protein levels: effect of light. Exp Eye Res. 1991;53:773–779.
160 Kennedy MJ, Lee KA, Niemi GA, et al. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 2001;31:87–101.
161 Fain GL, Lisman JE. Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest Ophthalmol Vis Sci. 1999;40:2770–2772.
162 Wu J, Seregard S, Spangberg B, et al. Blue light induced apoptosis in rat retina. Eye (Lond). 1999;13:577–583.
163 Wu J, Gorman A, Zhou X, et al. Involvement of caspase-3 in photoreceptor cell apoptosis induced by in vivo blue light exposure. Invest Ophthalmol Vis Sci. 2002;43:3349–3354.
164 Westlund BS, Cai B, Zhou J, et al. Involvement of c-Abl, p53 and the MAP kinase JNK in the cell death program initiated in A2E-laden ARPE-19 cells by exposure to blue light. Apoptosis. 2009;14:31–41.
165 Tezel G, Kolker AE, Kass MA, et al. Parapapillary chorioretinal atrophy in patients with ocular hypertension. I. An evaluation as a predictive factor for the development of glaucomatous damage. Arch Ophthalmol. 1997;115:1503–1508.
166 Tezel G, Kolker AE, Wax MB, et al. Parapapillary chorioretinal atrophy in patients with ocular hypertension. II. An evaluation of progressive changes. Arch Ophthalmol. 1997;115:1509–1514.
167 Donovan M, Cotter TG. Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ. 2002;9:1220–1231.
168 Gaillard ER, Atherton SJ, Eldred G, et al. Photophysical studies on human retinal lipofuscin. Photochem Photobiol. 1995;61:448–453.
169 Rozanowska M, Jarvis-Evans J, Korytowski W, et al. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem. 1995;270:18825–18830.
170 Rozanowska M, Wessels J, Boulton M, et al. Blue light-induced singlet oxygen generation by retinal lipofuscin in non-polar media. Free Radic Biol Med. 1998;24:1107–1112.
171 Paquet-Durand F, Azadi S, Hauck SM, et al. Calpain is activated in degenerating photoreceptors in the rd1 mouse. J Neurochem. 2006;96:802–814.
172 Sharma AK, Rohrer B. Calcium-induced calpain mediates apoptosis via caspase-3 in a mouse photoreceptor cell line. J Biol Chem. 2004;279:35564–35572.
173 Hafezi F, Marti A, Grimm C, et al. Differential DNA binding activities of the transcription factors AP-1 and Oct-1 during light-induced apoptosis of photoreceptors. Vision Res. 1999;39:2511–2518.
174 Sang A, Cheng Y, Lu H, et al. Light-induced retinal ganglion cell damage in vivo involves Dexras1. Mol Vis. 2011;17:134–143.
175 Dacey DM, Liao HW, Peterson BB, et al. Melanopsin-expressing ganglion cells in primate retina signal colour and irradiance and project to the LGN. Nature. 2005;433:749–754.
176 Osborne NN, Li GY, Ji D, et al. Light affects mitochondria to cause apoptosis to cultured cells: possible relevance to ganglion cell death in certain optic neuropathies. J Neurochem. 2008;105:2013–2028.
177 Kunchithapautham K, Coughlin B, Lemasters JJ, et al. Differential effects of rapamycin on rods and cones during light-induced stress in albino mice. Invest Ophthalmol Vis Sci. 2011;52:2967–2975.
178 Kunchithapautham K, Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007;3:433–441.
179 Bahr M. Neuroprotection: models, mechanisms, and therapies. Weinheim, Germany: Wiley-VCH; 2004.
180 Faden AI, Stoica B. Neuroprotection: challenges and opportunities. Arch Neurol. 2007;64:794–800.
181 Whitcup SM. Clinical trials in neuroprotection. Prog Brain Res. 2008;173:323–335.
182 Ishizawa Y, Sharp R, Liebman PA, et al. Halothane binding to a G protein coupled receptor in retinal membranes by photoaffinity labeling. Biochemistry. 2000;39:8497–8502.
183 Donovan M, Carmody RJ, Cotter TG. Light-induced photoreceptor apoptosis in vivo requires neuronal nitric-oxide synthase and guanylate cyclase activity and is caspase-3-independent. J Biol Chem. 2001;276:23000–23008.
184 Goureau O, Jeanny JC, Becquet F, et al. Protection against light-induced retinal degeneration by an inhibitor of NO synthase. Neuroreport. 1993;5:233–236.
185 Hahn P, Lindsten T, Lyubarsky A, et al. Deficiency of Bax and Bak protects photoreceptors from light damage in vivo. Cell Death Differ. 2004;11:1192–1197.
186 Liu C, Peng M, Laties AM, et al. Preconditioning with bright light evokes a protective response against light damage in the rat retina. J Neurosci. 1998;18:1337–1344.
187 Li F, Cao W, Anderson RE. Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci. 2003;44:4968–4975.
188 Casson RJ, Chidlow G, Wood JP, et al. The effect of retinal ganglion cell injury on light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2004;45:685–693.
189 Casson RJ, Wood JP, Melena J, et al. The effect of ischemic preconditioning on light-induced photoreceptor injury. Invest Ophthalmol Vis Sci. 2003;44:1348–1354.
190 Cao W, Tombran-Tink J, Elias R, et al. In vivo protection of photoreceptors from light damage by pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2001;42:1646–1652.
191 Lau D, Flannery J. Viral-mediated FGF-2 treatment of the constant light damage model of photoreceptor degeneration. Doc Ophthalmol. 2003;106:89–98.
192 Spencer B, Agarwala S, Gentry L, et al. HSV-1 vector-delivered FGF2 to the retina is neuroprotective but does not preserve functional responses. Mol Ther. 2001;3:746–756.
193 Kano T, Abe T, Tomita H, et al. Protective effect against ischemia and light damage of iris pigment epithelial cells transfected with the BDNF gene. Invest Ophthalmol Vis Sci. 2002;43:3744–3753.
194 Bainbridge JW, Ali RR. Success in sight: The eyes have it! Ocular gene therapy trials for LCA look promising. Gene Ther. 2008;15:1191–1192.
195 Bok D, Yasumura D, Matthes MT, et al. Effects of adeno-associated virus-vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res. 2002;74:719–735.
196 Cayouette M, Smith SB, Becerra SP, et al. Pigment epithelium-derived factor delays the death of photoreceptors in mouse models of inherited retinal degenerations. Neurobiol Dis. 1999;6:523–532.
197 Huang SP, Lin PK, Liu JH, et al. Intraocular gene transfer of ciliary neurotrophic factor rescues photoreceptor degeneration in RCS rats. J Biomed Sci. 2004;11:37–48.
198 Liang FQ, Aleman TS, Dejneka NS, et al. Long-term protection of retinal structure but not function using RAAV.CNTF in animal models of retinitis pigmentosa. Mol Ther. 2001;4:461–472.
199 Schlichtenbrede FC, MacNeil A, Bainbridge JW, et al. Intraocular gene delivery of ciliary neurotrophic factor results in significant loss of retinal function in normal mice and in the Prph2Rd2/Rd2 model of retinal degeneration. Gene Ther. 2003;10:523–527.
200 Miyazaki M, Ikeda Y, Yonemitsu Y, et al. Simian lentiviral vector-mediated retinal gene transfer of pigment epithelium-derived factor protects retinal degeneration and electrical defect in Royal College of Surgeons rats. Gene Ther. 2003;10:1503–1511.
201 Tao W, Wen R, Goddard MB, et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43:3292–3298.
202 Chong NH, Alexander RA, Waters L, et al. Repeated injections of a ciliary neurotrophic factor analogue leading to long-term photoreceptor survival in hereditary retinal degeneration. Invest Ophthalmol Vis Sci. 1999;40:1298–1305.
203 Harada T, Harada C, Nakayama N, et al. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. 2000;26:533–541.
204 Okoye G, Zimmer J, Sung J, et al. Increased expression of brain-derived neurotrophic factor preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neurosci. 2003;23:4164–4172.
205 Lawrence JM, Keegan DJ, Muir EM, et al. Transplantation of Schwann cell line clones secreting GDNF or BDNF into the retinas of dystrophic Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2004;45:267–274.
206 Yoshizawa K, Kiuchi K, Nambu H, et al. Caspase-3 inhibitor transiently delays inherited retinal degeneration in C3H mice carrying the rd gene. Graefes Arch Clin Exp Ophthalmol. 2002;240:214–219.
207 Zeiss CJ, Neal J, Johnson EA. Caspase-3 in postnatal retinal development and degeneration. Invest Ophthalmol Vis Sci. 2004;45:964–970.
208 Leonard KC, Petrin D, Coupland SG, et al. XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS ONE. 2007;2:e314.
209 Renwick J, Narang MA, Coupland SG, et al. XIAP-mediated neuroprotection in retinal ischemia. Gene Ther. 2006;13:339–347.
210 Glybina IV, Kennedy A, Ashton P, et al. Photoreceptor neuroprotection in RCS rats via low-dose intravitreal sustained-delivery of fluocinolone acetonide. Invest Ophthalmol Vis Sci. 2009;50:4847–4857.
211 Delyfer MN, Simonutti M, Neveux N, et al. Does GDNF exert its neuroprotective effects on photoreceptors in the rd1 retina through the glial glutamate transporter GLAST? Mol Vis. 2005;11:677–687.
212 Read SP, Cashman SM, Kumar-Singh R. POD nanoparticles expressing GDNF provide structural and functional rescue of light-induced retinal degeneration in an adult mouse. Mol Ther. 2010;18:1917–1926.
213 Cheung W, Guo L, Cordeiro MF. Neuroprotection in glaucoma: drug-based approaches. Optom Vis Sci. 2008;85:406–416.
214 Danesh-Meyer HV. Neuroprotection in glaucoma: recent and future directions. Curr Opin Ophthalmol. 2011;22:78–86.
215 Dong CJ, Guo Y, Agey P, et al. Alpha2 adrenergic modulation of NMDA receptor function as a major mechanism of RGC protection in experimental glaucoma and retinal excitotoxicity. Invest Ophthalmol Vis Sci. 2008;49:4515–4522.
216 Russo R, Cavaliere F, Rombola L, et al. Rational basis for the development of coenzyme Q10 as a neurotherapeutic agent for retinal protection. Prog Brain Res. 2008;173:575–582.
217 Pease ME, Zack DJ, Berlinicke C, et al. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50:2194–2200.
218 Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin Biol Ther. 2006;6:717–726.
219 Kashimoto R, Kurimoto T, Miyoshi T, et al. Cilostazol promotes survival of axotomized retinal ganglion cells in adult rats. Neurosci Lett. 2008;436:116–119.
220 Koriyama Y, Tanii H, Ohno M, et al. A novel neuroprotective role of a small peptide from flesh fly, 5-S-GAD in the rat retina in vivo. Brain Res. 2008;1240:196–203.
221 Bazan NG. Neurotrophins induce neuroprotective signaling in the retinal pigment epithelial cell by activating the synthesis of the anti-inflammatory and anti-apoptotic neuroprotectin D1. Adv Exp Med Biol. 2008;613:39–44.
222 Mukherjee PK, Marcheselli VL, Barreiro S, et al. Neurotrophins enhance retinal pigment epithelial cell survival through neuroprotectin D1 signaling. Proc Natl Acad Sci U S A. 2007;104:13152–13157.
223 Wang ZY, Shen LJ, Tu L, et al. Erythropoietin protects retinal pigment epithelial cells from oxidative damage. Free Radic Biol Med. 2009;46:1032–1041.
224 Wrona M, Rozanowska M, Sarna T. Zeaxanthin in combination with ascorbic acid or alpha-tocopherol protects ARPE-19 cells against photosensitized peroxidation of lipids. Free Radic Biol Med. 2004;36:1094–1101.
225 Bryckaert M, Guillonneau X, Hecquet C, et al. Both FGF1 and bcl-x synthesis are necessary for the reduction of apoptosis in retinal pigmented epithelial cells by FGF2: role of the extracellular signal-regulated kinase 2. Oncogene. 1999;18:7584–7593.
226 Elner SG, Yoshida A, Bian ZM, et al. Human RPE cell apoptosis induced by activated monocytes is mediated by caspase-3 activation. Trans Am Ophthalmol Soc. 2003;101:77–89. discussion 89–91
227 Ferrington DA, Tran TN, Lew KL, et al. Different death stimuli evoke apoptosis via multiple pathways in retinal pigment epithelial cells. Exp Eye Res. 2006;83:638–650.
228 Sreekumar PG, Kannan R, Kitamura M, et al. alphaB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS ONE. 2010;5:e12578.
229 Alge CS, Priglinger SG, Neubauer AS, et al. Retinal pigment epithelium is protected against apoptosis by alphaB-crystallin. Invest Ophthalmol Vis Sci. 2002;43:3575–3582.
230 AREDS. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
231 Imai H, Singh RS, Fort PE, et al. Neuroprotection for diabetic retinopathy. Dev Ophthalmol. 2009;44:56–68.
232 Bao XH, Naomoto Y, Hao HF, et al. Autophagy: Can it become a potential therapeutic target? Int J Mol Med. 2010;25:493–503.
233 Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–1518.
234 Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539.
235 Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939.
236 Petiot A, Ogier-Denis E, Blommaart EF, et al. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998.
237 Sarkar S, Rubinsztein DC. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy. 2006;2:132–134.
238 Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652.
239 Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736.
240 Winslow AR, Rubinsztein DC. Autophagy in neurodegeneration and development. Biochim Biophys Acta. 2008;1782:723–729.
241 Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901.
242 Kim JS, Nitta T, Mohuczy D, et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47:1725–1736.
243 Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666.
244 Carew JS, Nawrocki ST, Giles FJ, et al. Targeting autophagy: a novel anticancer strategy with therapeutic implications for imatinib resistance. Biologics. 2008;2:201–204.
245 Dalby KN, Tekedereli I, Lopez-Berestein G, et al. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329.
246 Spalton DJ. Retinopathy and antimalarial drugs – the British experience. Lupus. 1996;5(Suppl 1):S70–S72.
247 Kaminskyy V, Zhivotovsky B. Proteases in autophagy. Biochim Biophys Acta. 2011;1824:44–50.
248 Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580.
249 Stepkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. 2011;50:1186–1195.
250 Goligorsky MS. SIRTing out the link between autophagy and ageing. Nephrol Dial Transplant. 2010;25:2434–2436.
251 Hariharan N, Maejima Y, Nakae J, et al. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107:1470–1482.
252 Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38.
253 Massey AC, Kaushik S, Sovak G, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103:5805–5810.
254 Kaushik S, Massey AC, Mizushima N, et al. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179–2192.
255 Iwata A, Christianson JC, Bucci M, et al. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A. 2005;102:13135–13140.
256 Ding Q, Dimayuga E, Martin S, et al. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86:489–497.
257 Cuervo AM, Palmer A, Rivett AJ, et al. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800.
258 Massey AC, Follenzi A, Kiffin R, et al. Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy. 2008;4:442–456.
259 Araki M. Regeneration of the amphibian retina: role of tissue interaction and related signaling molecules on RPE transdifferentiation. Dev Growth Differ. 2007;49:109–120.
260 Karl MO, Reh TA. Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol Med. 2010;16:193–202.
261 Bernardos RL, Barthel LK, Meyers JR, et al. Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J Neurosci. 2007;27:7028–7040.
262 Raymond PA, Hitchcock PF. How the neural retina regenerates. Results Probl Cell Differ. 2000;31:197–218.
263 Fischer AJ, Reh TA. Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4:247–252.
264 Ooto S, Akagi T, Kageyama R, et al. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A. 2004;101:13654–13659.
265 Sengupta N, Caballero S, Sullivan SM, et al. Regulation of adult hematopoietic stem cells fate for enhanced tissue-specific repair. Mol Ther. 2009;17:1594–1604.
266 Carr AJ, Vugler AA, Hikita ST, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS ONE. 2009;4:e8152.
267 Du H, Lim SL, Grob S, et al. Induced pluripotent stem cell therapies for geographic atrophy of age-related macular degeneration. Semin Ophthalmol. 2011;26:216–224.
268 Vugler A, Lawrence J, Walsh J, et al. Embryonic stem cells and retinal repair. Mech Dev. 2007;124:807–829.
269 Lu B, Malcuit C, Wang S, et al. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells. 2009;27:2126–2135.
270 Fong CY, Gauthaman K, Bongso A. Teratomas from pluripotent stem cells: A clinical hurdle. J Cell Biochem. 2010;111:769–781.
271 Gore A, Li Z, Fung HL, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67.
272 Binder S, Stanzel BV, Krebs I, et al. Transplantation of the RPE in AMD. Prog Retin Eye Res. 2007;26:516–554.
273 da Cruz L, Chen FK, Ahmado A, et al. RPE transplantation and its role in retinal disease. Prog Retin Eye Res. 2007;26:598–635.
274 MacLaren RE, Pearson RA, MacNeil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–207.
275 Bi YY, Feng DF, Pan DC. Stem/progenitor cells: a potential source of retina-specific cells for retinal repair. Neurosci Res. 2009;65:215–221.
276 Ikeda H, Osakada F, Watanabe K, et al. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc Natl Acad Sci U S A. 2005;102:11331–11336.
277 Osakada F, Ikeda H, Sasai Y, et al. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat Protoc. 2009;4:811–824.
278 Osakada F, Ooto S, Akagi T, et al. Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27:4210–4219.
279 Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4:73–79.
280 Bull ND, Martin KR. Using stem cells to mend the retina in ocular disease. Regen Med. 2009;4:855–864.
281 Enzmann V, Yolcu E, Kaplan HJ, et al. Stem cells as tools in regenerative therapy for retinal degeneration. Arch Ophthalmol. 2009;127:563–571.
282 Machalinska A, Baumert B, Kuprjanowicz L, et al. Potential application of adult stem cells in retinal repair – challenge for regenerative medicine. Curr Eye Res. 2009;34:748–760.
283 Lamba DA, Karl MO, Reh TA. Strategies for retinal repair: cell replacement and regeneration. Prog Brain Res. 2009;175:23–31.