[level-membership-for-basic-science-category]
CHAPTER 6: Developmental Genetics
CHAPTER 7: Patterns of Inheritance
CHAPTER 8: Mathematical and Population Genetics
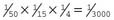 .
.CHAPTER 9: Polygenic and Multifactorial Inheritance
CHAPTER 10: Hemoglobin and the Hemoglobinopathies
CHAPTER 11: Biochemical Genetics
CHAPTER 13: Immunogenetics
CHAPTER 14: Cancer Genetics
CHAPTER 15: Genetic Factors in Common Diseases
CHAPTER 16: Congenital Abnormalities and Dysmorphic Syndromes
CHAPTER 17: Genetic Counseling
CHAPTER 18: Chromosome Disorders
CHAPTER 19: Single-Gene Disorders
CHAPTER 20: Screening for Genetic Disease
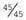 + 5 = 90%. The specificity is the proportion of true negatives detected by the test, i.e., 99,190 (the unaffected cases who test negative)/99,190 + 760 (the unaffected cases who test positive) = 99.2%.
+ 5 = 90%. The specificity is the proportion of true negatives detected by the test, i.e., 99,190 (the unaffected cases who test negative)/99,190 + 760 (the unaffected cases who test positive) = 99.2%.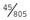 = 5.6%.
= 5.6%.CHAPTER 21: Prenatal Testing and Reproductive Genetics
CHAPTER 22: Risk Calculation
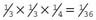 .
.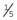 , or 20%, chance of being a carrier.
, or 20%, chance of being a carrier.| Probability | Is a Carrier | Is Not a Carrier |
|---|---|---|
| Prior | 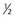 |
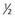 |
| Conditional (2 normal sons) | 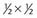 |
1 |
| Joint | 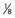 |
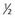 |
| Posterior | 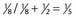 |
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 6: Developmental Genetics
CHAPTER 7: Patterns of Inheritance
CHAPTER 8: Mathematical and Population Genetics
CHAPTER 9: Polygenic and Multifactorial Inheritance
CHAPTER 10: Hemoglobin and the Hemoglobinopathies
CHAPTER 11: Biochemical Genetics
