[level-membership-for-anesthesiology-category]
10 Cardiovascular Pharmacology
Drug Therapy for Systemic Hypertension
Pharmacotherapy for Acute and Chronic Heart Failure
Pharmacotherapy for Cardiac Arrhythmias
Anti-ischemic drug therapy
Anti-ischemic drug therapy during anesthesia is indicated whenever evidence of myocardial ischemia exists. The treatment of ischemia during anesthesia is complicated by the ongoing stress of surgery, blood loss, concurrent organ ischemia, and the patient’s inability to interact with the anesthesiologist. Nonetheless, the fundamental principles of treatment remain the same as in the unanesthetized state. All events of myocardial ischemia involve an alteration in the oxygen supply/demand balance (Table 10-1). The American College of Cardiology/American Heart Association (ACC/AHA) Guidelines on the Management and Treatment of Patients with Unstable Angina and Non-ST-Segment Elevation Myocardial Infarction provide an excellent framework for the treatment of patients with ongoing myocardial ischemia.1 These guidelines detail the initial evaluation, management, hospital care, and coronary revascularization strategies in the nonanesthetized patient with an acute coronary syndrome. In the anesthetized patient with evidence of myocardial ischemia, initiation of anti-ischemic drug therapy is indicated. This section reviews the common agents used for this purpose (see Chapter 18).
TABLE 10-1 Myocardial Ischemia: Factors Governing O2 Supply and Demand
| O2 Supply | O2 Demand |
|---|---|
| Heart rate* | Heart rate* |
| O2 content | Contractility |
| Hgb, SAT%, Pao2 | Wall tension |
| Coronary blood flow | Afterload |
| CPP = DP − LVEDP* | Preload (LVEDP)* |
| CVR |
CPP, coronary perfusion pressure; CVR, coronary vascular resistance; DP, diastolic blood pressure; Hgb, hemoglobin; LVEDP, left ventricular end-diastolic pressure; SAT%, percent oxygen saturation.
* Affects both supply and demand.
Modified from Royster RL: Intraoperative administration of inotropes in cardiac surgery patients. J Cardiothorac Anesth 6(suppl 5):17, 1990.

Nitroglycerin
Nitroglycerin (NTG) is clinically indicated as initial therapy in nearly all types of myocardial ischemia.2 Chronic exertional angina, de novo angina, unstable angina, Prinzmetal’s angina (vasospasm), and silent ischemia respond to NTG administration.2–6 NTG therapy decreases the incidence of anginal attacks and improves exercise tolerance before angina symptoms.7 During therapy with intravenous (IV) NTG, if blood pressure (BP) declines and ischemia is not relieved, the addition of phenylephrine will allow coronary perfusion pressure (CPP) to be maintained while allowing greater doses of NTG to be used for ischemia relief.8 If reflex increases in heart rate (HR) and contractility occur, combination therapy with β-adrenergic blockers may be indicated to blunt this undesired increase in HR. Combination therapy with nitrates and calcium channel blockers may be an effective anti-ischemic regimen in selected patients; however, excessive hypotension and reflex tachycardia may be a problem, especially when a dihydropyridine calcium antagonist is used.9
Mechanism of Action
NTG enhances myocardial oxygen delivery and reduces myocardial oxygen demand. NTG is a smooth muscle relaxant that causes vasculature dilation. Nitrate-mediated vasodilation occurs with or without intact vascular endothelium.10 Nitrites, organic nitrites, nitroso compounds, and other nitrogen oxide–containing substances (e.g., nitroprusside) enter the smooth muscle cell and are converted to reactive nitric oxide (NO) or S-nitrosothiols, which stimulate guanylate cyclase metabolism to produce cyclic guanosine monophosphate (cGMP)11–13 (Figure 10-1). A cGMP-dependent protein kinase is stimulated with resultant protein phosphorylation in the smooth muscle. This leads to a dephosphorylation of the myosin light chain and smooth muscle relaxation.14,15 Vasodilation is also associated with a reduction of intracellular calcium.16 Sulfhydryl (SH) groups are required for formation of NO and the stimulation of guanylate cyclase. When excessive numbers of SH groups are metabolized by prolonged exposure to NTG, vascular tolerance occurs.17 The addition of N-acetylcysteine, an SH donor, reverses NTG tolerance.18 The mechanism by which NTG compounds are uniquely better venodilators, especially at lower serum concentrations, is unknown but may be related to increased uptake of NTG by veins compared with arteries.19
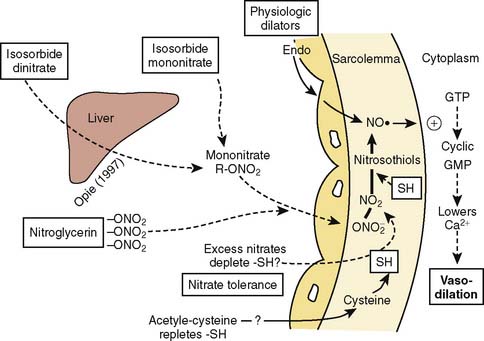
Physiologic Effects
Two important physiologic effects of NTG are systemic and regional venous dilation (Figure 10-2). Venodilation can markedly reduce venous pressure, venous return to the heart, and cardiac filling pressures. Prominent venodilation occurs at lower doses and does not increase further as the NTG dose increases.20 Venodilation results primarily in pooling of blood in the splanchnic capacitance system.21 Mesenteric blood volume increases as ventricular size, ventricular pressures, and intrapericardial pressure decrease.21
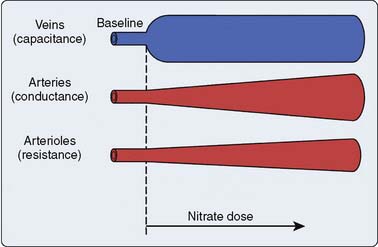
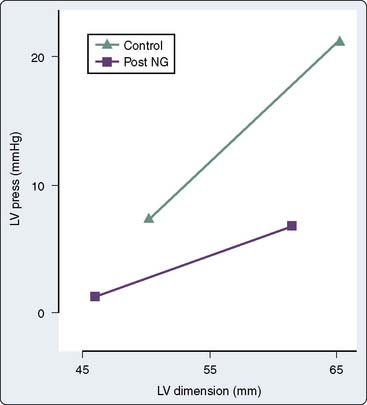
NTG increases the distensibility and conductance of large arteries without changing systemic vascular resistance (SVR) at low doses.22 Improved compliance of the large arteries does not necessarily imply afterload reduction. At greater doses, NTG dilates smaller arterioles and resistance vessels, reducing afterload and BP23 (see Figure 10-2). Reductions in cardiac dimension and pressure reduce myocardial oxygen consumption (MVO2) and improve myocardial ischemia24 (Figure 10-3). NTG may preferentially reduce cardiac preload, while maintaining systemic perfusion pressure, an important hemodynamic effect in myocardial ischemia. However, in hypovolemic states, greater doses of NTG may markedly reduce systemic BP to dangerous levels. A reflex increase in HR may occur at arterial vasodilating doses.
NTG causes vasodilation of pulmonary arteries and veins and predictably decreases right atrial (RAP), pulmonary artery (PAP), and pulmonary capillary wedge pressures (PCWP).23 Pulmonary artery hypertension may be reduced in various disease states and in congenital heart disease with NTG.25,26 Renal arteries, cerebral arteries, and cutaneous vessels also dilate with NTG.27 Blood flow to the kidney and brain may decrease if adequate renal and cerebral perfusion pressures are not maintained.
NTG has several important effects on the coronary circulation (Box 10-1). NTG is a potent epicardial coronary artery vasodilator in both normal and diseased vessels. Stenotic lesions dilate with NTG, reducing the resistance to coronary blood flow (CBF) and improving myocardial ischemia.28,29 Smaller coronary arteries may dilate relatively more than larger coronary vessels; however, the degree of dilation may depend on the baseline tone of the vessel.30 NTG effectively reverses or prevents coronary artery vasospasm.31
BOX 10-1 Effects of Nitroglycerin and Organic Nitrates on the Coronary Circulation
(Modified from Abrams J: Hemodynamic effects of nitroglycerin and long-acting nitrates. Am HeartJ 110[pt 2]:216, 1985.)
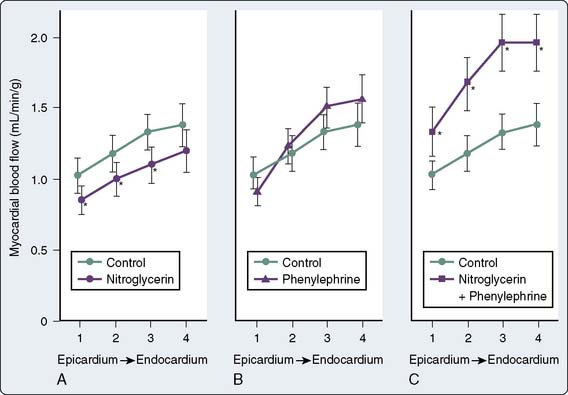
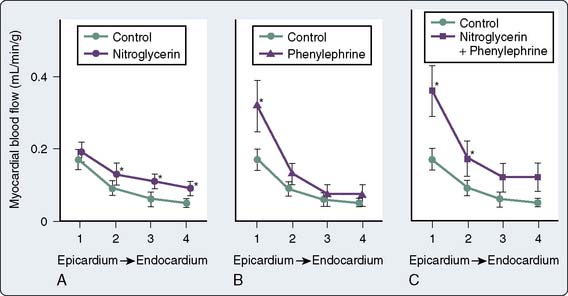
Total CBF may initially increase but eventually decreases with NTG despite coronary vasodilation32 (Figure 10-4). Autoregulatory mechanisms probably result in decreases in total flow as a result of reductions in wall tension and myocardial oxygen consumption.23 However, regional myocardial blood flow may improve by vasodilation of intercoronary collateral vessels or reduction of subendocardial compressive forces33 (Figure 10-5). Coronary arteriographic studies in humans demonstrate that coronary collateral vessels increase in size after NTG administration.34 This effect may be especially important when epicardial vessels have subtotal or total occlusive disease.35 Improvement in collateral flow also may be protective in situations in which coronary artery steal may occur with other potent coronary vasodilator agents. The improvement in blood flow to the subendocardium, the most vulnerable area to the development of ischemia, is secondary to both improvement in collateral flow and reductions in left ventricular end-diastolic pressure (LVEDP), which reduce subendocardial resistance to blood flow.36 With the maintenance of an adequate CPP (e.g., with administration of phenylephrine), NTG can maximize subendocardial blood flow8 (see Figures 10-4 and 10-5). The ratio of endocardial to epicardial blood in transmural segments is enhanced with NTG.36 Inhibition of platelet aggregation also occurs with NTG; however, the clinical significance of this action is unknown.37
Pharmacology
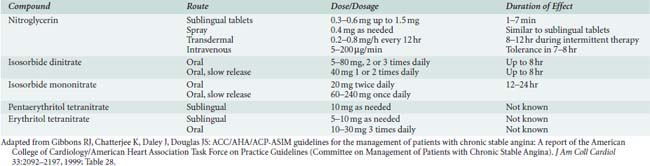
Organic nitrates are biotransformed by reduction hydrolysis catalyzed by the hepatic enzyme glutathione-organic nitrate reductase.15 The rate of hepatic denitrification is characteristic of each nitrate and is further dependent on hepatic blood flow or presence of hepatic disease.15 Common organic nitrates for clinical use are shown in Table 10-2.
Sublingual Nitroglycerin
Sublingual NTG (0.15- to 0.6-mg tablets) achieves blood levels adequate to cause hemodynamic changes within several minutes; physiologic effects last 30 to 45 minutes.38 Sublingual bioavailability is approximately 80% and bypasses the high first-pass biodegradation in the liver (90%) by nitrate reductase to glycerol dinitrate and nitrite, which are excreted renally. Plasma half-life of sublingual NTG is 4 to 7 minutes. NTG spray has pharmacokinetics and pharmacodynamics equivalent to those of a 0.4-mg sublingual tablet; however, it has a longer shelf half-life compared with the tablets, which decompose in air and warm temperatures.39 A tablet that adheres to the buccal area between the upper lip and teeth has rapid onset and has the advantage of longer half-life than sublingual tablets.40 Although NTG is readily absorbed through the gastric mucosa, the high rate of liver metabolism makes oral administration highly unpredictable.
Nitroglycerin Ointment and Patches
NTG ointment (2%) is readily absorbed through the skin, with this method of administration providing longer-lasting effects.41 Adequate NTG blood levels are reached within 20 to 30 minutes, and duration of action is 4 to 6 hours.41 Ointment is administered in inches (15 mg/inch), but the surface area of application and not the amount administered determines the blood level achieved. NTG ointment is messy, requires application four times a day, and is most appropriate for nursing administration in special care units.42
NTG patches contain either liquid NTG or NTG bonded to a polymer gel and slowly released to the skin through a semipermeable membrane.43 The pharmacokinetics approach that of a consistent IV infusion.43 Blood levels are reached within 20 to 30 minutes, and a steady state is reached within 2 hours. Blood levels may be maintained up to 24 hours and are largely determined by patch size. Patches or disks contain an NTG concentration per square centimeter, and dosages of 0.2 to 0.8 mg/hr usually are required for relief of myocardial ischemia. Although convenient for patients, tolerance may be a problem with these sustained-release preparations.41 Intermittent therapy is recommended to avoid tolerance.44
Intravenous Nitroglycerin
NTG has been available since the early 1980s as an injectable drug with stable shelf half-life in a 400-μg/mL solution of D5W (5% dextrose in water). Blood levels are achieved instantaneously, and arterial dilating doses with resulting hypotension may quickly occur. If the volume status of the patient is unknown, initial dosages of 5 to 10 μg/min are recommended. The dosage necessary for relieving myocardial ischemia may vary from patient to patient, but relief is usually achieved with 75 to 150 μg/min. In a clinical study of 20 patients with rest angina, a mean dosage of 72 μg/min reduced or abolished ischemic episodes in 85% of patients.45 However, doses as high as 300 to 400 μg/min may be necessary for ischemic relief in some patients. Arterial dilation becomes clinically apparent at doses around 50 μg/min. Drug offset after discontinuation of an infusion is rapid (2 to 5 minutes). The dosage of NTG available is less when administered in plastic bags and polyvinylchloride tubing because of NTG absorption by the bag and tubing, although this is not a significant clinical problem because the drug is titrated to effect.46
Adverse Effects
The metabolism of NTG by liver nitrate reductase produces a nitrite that oxidizes the ferrous iron of hemoglobin to the ferric form of methemoglobin. The ferric iron does not bind or release oxygen.47 Methemoglobin is formed normally and is reduced by enzyme systems within the red blood cell.48 Normally, methemoglobin levels do not exceed 1%, but may increase when direct oxidants are present in the serum (nitrates, sulfonamides, aniline dye derivates). Methemoglobinemia with levels up to 20% is not a clinical problem. Documented increases in methemoglobin blood levels occur with IV NTG, averaging 1.5% in one study of 50 patients receiving NTG for longer than 48 hours.49 NTG dosages of 5 mg/kg/day orally should be avoided to prevent significant methemoglobinemia.50 However, rare instances of smaller doses causing clinically significant problems have been reported.51 Nitrates are effective in producing methemoglobin to bind cyanide in sodium nitroprusside toxicity.
Several mechanisms of nitrate tolerance have been proposed, including a depletion of SH groups, neurohumoral activation, volume expansion, and/or downregulation of nitrate receptors.52–57 Tolerance may occur with all forms of nitrate administration that maintain continuous blood levels of the drug.17,58–61 Discontinuation of the drug after prolonged exposure may result in a rebound phenomenon, possibly resulting in coronary vasospasm and myocardial ischemia or infarction.62 Tolerance to NTG apparently does not occur in all patients.63 If tolerance develops after prolonged exposure, physiologic responsiveness may be achieved with greater dosages of NTG, an important observation during NTG administration in cardiac surgery.64 Intermittent dosing with a nitrate-free interval each day or night can maintain NTG responsiveness.44,65
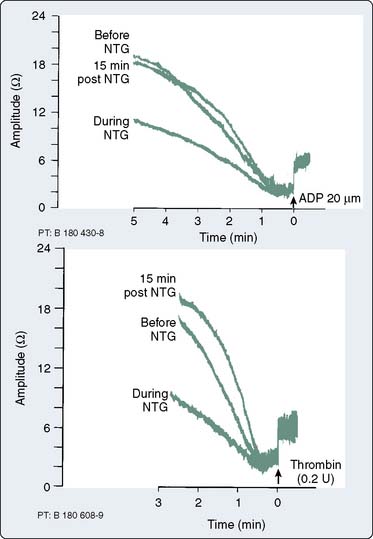
NTG interferes with platelet aggregation.66 The ability of the platelet to adhere to damaged intima is reduced.67 Primary and secondary wave aggregation of platelets is also attenuated.68 Previously formed platelet plugs are disaggregated.69 A clinical study of 10 patients with coronary artery disease (CAD) demonstrated that a mean dosage of NTG (1.19 μg/kg/min) inhibited platelet aggregation by 50%, with a return to baseline platelet aggregation 15 minutes after the infusion was discontinued70 (Figure 10-6). NO production increases cGMP, which modulates intracellular platelet calcium and reduces platelet secretion of proaggregatory factors.71 The clinical significance of these actions remains unclear. As with other potent vasodilators, NTG may increase intrapulmonary shunting of blood and reduce arterial oxygen tension.
NTG may induce resistance to the anticoagulant effects of heparin.72 During simultaneous infusions of NTG and heparin, an increase in the NTG infusion caused the activated partial thromboplastin time to decrease.73 Becker et al74 reported NTG-induced heparin resistance at NTG infusion rates greater than 350 μg/min. The authors suggested a qualitative problem with antithrombin III (AT III) because AT III levels did not decrease. Others have suggested that NTG interferes with AT III binding to heparin by N-desulfation of the heparin molecule at the AT III binding sites.75 N-desulfation of heparin reduces its anticoagulant activity.76
NTG is contraindicated in patients who have used sildenafil, vardenafil, or tadalafil, or in patients who are hypotensive. These drugs for erectile dysfunction inhibit the phosphodiesterase (PDE5) that degrades cGMP, and the cGMP mediates vascular smooth muscle relaxation by NO. NTG-mediated vasodilation is markedly enhanced and prolonged, resulting in cases of profound hypotension, myocardial infarction (MI), and death.77 Small doses of NTG have been used, but the amount of time that must elapse after a patient’s last dose of one of these medications before regular doses of nitrates may be safely administered is unclear.78–80
Summary
NTG remains a first-line agent for the treatment of myocardial ischemia. Special care must be taken in patients with signs of hypovolemia or hypotension because the vasodilating effects of the drug may worsen the clinical condition. Recent ACC/AHA Guidelines address the prophylactic intraoperative use of NTG and suggest that its usefulness in preventing myocardial ischemia and cardiac morbidity in high-risk patients undergoing noncardiac surgery is unclear.81

β-Adrenergic Blockers
β-Adrenergic blockers have multiple favorable effects in treating the ischemic heart during anesthesia (Box 10-2). β-Adrenergic blockers reduce oxygen consumption by decreasing HR, BP, and myocardial contractility. HR reduction increases diastolic CBF. Increased collateral blood flow and redistribution of blood to ischemic areas may occur with β-blockers. More free fatty acids may be available for substrate consumption by the myocardium. Microcirculatory oxygen delivery improves, and oxygen dissociates more easily from hemoglobin after β-adrenergic blockade. Platelet aggregation is inhibited. β-Blockers should be started early in patients with ischemia in the absence of contraindications.1 Many patients at high risk for perioperative cardiac morbidity should be started on β-blockers before surgery and continued for up to 30 days after surgery.82–84 The choice of which β-blocker for any individual patient is based on clinician familiarity and desired pharmacologic profile. There is no evidence that one specific agent is superior to another; however, β-blockers without intrinsic sympathomimetic activity (ISA) are preferable when treating acute myocardial ischemia.
β-Blockers administered during MI reduce myocardial infarct size.85 In addition, a reduction in morbidity has been shown to occur with acute IV metoprolol during MI.85 Similar findings with reductions in mortality extending up to 3 years after MI have been shown in numerous trials with β-adrenergic blockers86,87 (Figure 10-7). The mechanisms for mortality reduction are unclear. In the absence of contraindications, β-blockers should be a routine part of care in patients with all forms of CAD, including unstable angina and recent MI.
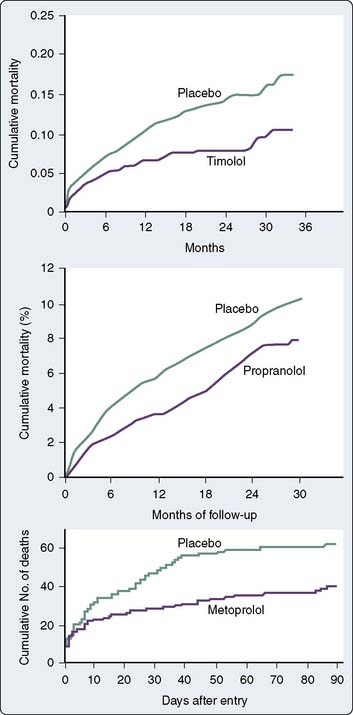
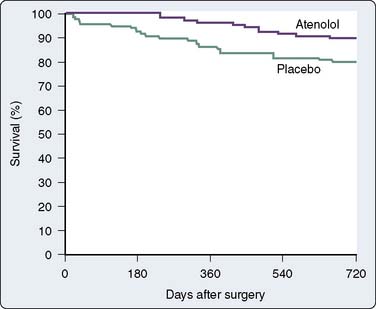
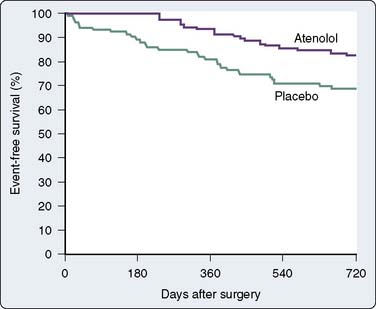
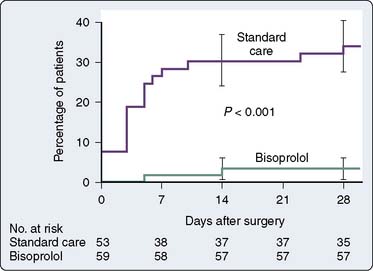
Data confirm the important role of β-blockade in treating patients after acute MI and in reducing mortality in high-risk populations. Immediate β-blockade after thrombolytic therapy in patients with acute MI significantly decreased recurrent early myocardial ischemia and reinfarction.88 Early β-blockade is indicated in the treatment of MI89,90 (Box 10-3). In fact, β-blocker therapy after MI may be greatly underused in patients older than 65 years.91 Atenolol has been found to reduce ischemia and adverse outcome in patients with mildly symptomatic ischemia.92 Multiple studies have shown that perioperative administration of β-adrenergic blockers reduces both mortality and morbidity when given to patients at high risk for CAD who must undergo noncardiac surgery82–84,93,94 (Figures 10-8 to 10-10). These data suggest that intermediate- and high-risk patients presenting for noncardiac surgery should receive perioperative β-adrenergic blockade to reduce postoperative cardiac mortality and morbidity. However, in the Perioperative Ischemic Evaluation Study (POISE) trial, the use of higher dose metoprolol started in patients on the day of noncardiac surgery was associated with increased risk for severe stroke and greater total mortality.95 These findings have led to increased scrutiny of perioperative β-blockade usage. Recent ACC/AHA recommendations on the perioperative use of β-adrenergic blockade for noncardiac surgery are given in Box 10-4.81
BOX 10-3 ACC/AHA Guidelines for Early Use of β-Adrenoceptor Blocking Agents after Stemi
Reproduced from Antman EM, Hand M, Armstrong PW, et al: 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Group to Review New Evidence and Update the ACC/AHA 2004 Guidelines for the Management of Patients with ST-Elevation Myocardial Infarction). Circulation 117:296, 2008, by permission.90
Early Therapy
Class I*
Class IIa:
Class III
* Risk factors for cardiogenic shock (the greater the number of risk factors present, the higher the risk of developing cardiogenic shock) are age older than 70 years, systolic blood pressure less than 120 mm Hg, sinus tachycardia greater than 110 bpm or heart rate less than 60 bpm, and increased time since onset of symptoms of STEMI.
BOX 10-4 Recommendations for Perioperative β-Blocker Therapy
Adapted from Fleisher LA, Beckman JA, Brown KA, et al: 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 120:e169, 2009, by permission.
Class IIa
Class IIb
Class III
* Clinical risk factors include history of ischemic heart disease, history of compensated or prior heart failure, history of cerebrovascular disease, diabetes mellitus, and renal insufficiency (defined in the Revised Cardiac Risk Index as a preoperative serum creatinine concentration greater than 2 mg/dL).
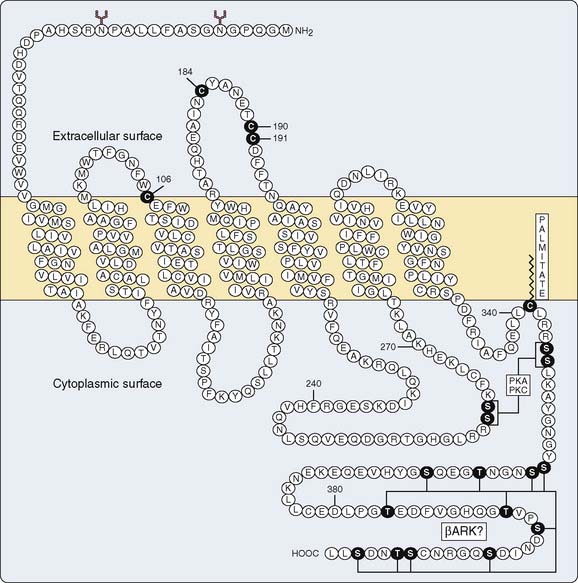
β Receptor
The β receptor was conceptualized by Ahlquist,96 who divided various physiologic effects of catecholamine stimulation into α and β responses. The β receptor has been identified biochemically as a polypeptide chain of approximately 50,000 to 60,000 kDa.97 The receptor’s structure is common to most receptor proteins that have been identified: seven transmembrane crossings with two extramembranous terminal ends98 (Figure 10-11). All receptors that transduce a signal through G proteins share this basic structure.99 There are three extracellular and intracellular loops connecting the intramembranous portion of the receptor.98 Agonist-antagonist binding occurs at the intramembranous portion, whereas the intracellular loops modulate interaction with the G-protein complex.100,101 The terminal intracellular end contains amino acid residues that undergo phosphorylation, which relates to desensitization and downregulation of the receptor.102
Receptor stimulation activates a G protein, which stimulates adenylyl cyclase. The G-protein complex is composed of both stimulatory (Gs) and inhibitory (Gi) intermediary proteins.99 Adenylyl cyclase converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP), which phosphorylates a protein kinase and produces the appropriate cellular response. A typical cascade of this sequence leading to increases in myocardial contractility from β-receptor stimulation is illustrated in Figure 10-12.
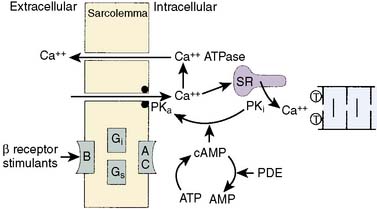
β-Receptor numbers in any tissue may decrease with chronic stimulation (downregulation) or increase with chronic blockade (upregulation). The process of desensitization of the adrenergic response in chronic stimulation (i.e., congestive heart failure [CHF]) may involve downregulation of the receptors but may involve either the G-protein complex or adenylyl cyclase. Desensitization may occur quickly, whereas downregulation with actual internalization of the receptor within the cell may take days to weeks.103 Myocardial ischemia increases β-receptor density, although it remains controversial whether this upregulation results in greater adrenergic response.104 Several studies have demonstrated that high-affinity β receptors in nonischemic tissue were shifted to a low-affinity state during ischemia.105,106 Also, the levels of Gs and its activity are reduced during myocardial ischemia.107 However, stimulation of these receptors with isoproterenol during ischemia does result in increases in cAMP production.104,108
There are two types of β receptors with a multitude of responses109 (Table 10-3). Both β1– and β2-receptor stimulation primarily involve cardiac function (Figure 10-13). Responses of isolated human atrial tissue demonstrated greater inotropic response to β1– than to β2-receptor stimulation.110 Endogenous norepinephrine produces inotropic responses in human atrial appendages and ventricular papillary muscle by β1-receptor stimulation, whereas epinephrine produces its maximal inotropic effects on the atria by β2-receptor stimulation and up to 50% of its maximal inotropic response in the ventricle by β2-receptor stimulation.111,112 Sinus node, atrioventricular (AV) node, the left and right bundle branches, and the Purkinje system contain higher densities of β2 receptors.113 Clearly, both receptor subtypes have cardiac inotropic, chronotropic, and dromotropic properties.
TABLE 10-3 Physiologic Effects of β1– and β2-Receptor Stimulation
| Physiologic Effect | β1 Response | β2 Response |
|---|---|---|
| Cardiac | ||
| Increased heart rate | ++ | ++ |
| Increased contractility | ||
| Atrium | + | ++ |
| Ventricle | ++ | ++ |
| Increased automaticity and conduction velocity | ||
| Nodal tissue | ++ | ++ |
| His-Purkinje | ++ | ++ |
| Arterial relaxation | ||
| Coronary | ++ | |
| Skeletal muscle | ++ | |
| Pulmonary | + | |
| Abdominal | + | |
| Renal | + | + |
| Venous relaxation | ++ | |
| Smooth muscle relaxation | ||
| Tracheal and bronchial | + | |
| Gastrointestinal | + | |
| Bladder | + | |
| Uterus | + | |
| Splenic capsule | + | |
| Ciliary muscle | + | |
| Metabolic | ||
| Renin release | ++ | |
| Lipolysis | ++ | + |
| Insulin secretion | + | |
| Glycogenolysis, gluconeogenesis | ++ | |
| Cellular K+ uptake | + | |
| Antidiuretic hormone secretion (pituitary) | + |
Modified from Lefkowitz RJ, Hoffman BB, Taylor P: Neurohumoral transmission: The autonomic and somatic motor nervous systems. In Gilman AG, Rall TW, Niew AS, Taylor P (eds): Goodman and Gilman’s the pharmacological basis of therapeutics. New York: Pergamon Press, 1990, pp 84–121, by permission of McGraw-Hill Companies.
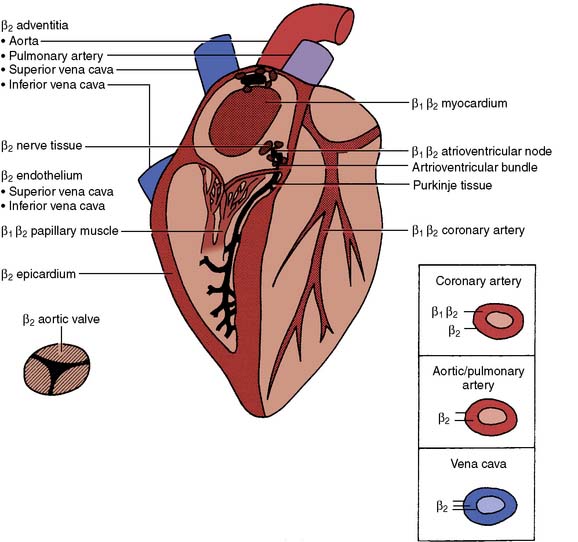
β2-Adrenoceptors comprise 93% of the total population of β receptors in arterioles and 100% of receptors in epicardium, vena cava, aorta, and pulmonary artery.112 β2 receptors are found on the intimal surface of human internal mammary artery, but not on the saphenous vein,114 the two vessels most commonly used for coronary artery bypass graft surgery (CABG). β2-Stimulation results in vascular smooth muscle relaxation and vasodilation.
β3-Adrenoreceptors are found on visceral adipocytes, the gallbladder, and colon. Stimulation of β3 receptors is thought to mediate lipolytic and thermic responses in brown and white adipose tissue.115
Physiologic Effects
Anti-ischemic Effects
β-Blockade on the ischemic heart may result in a favorable shift in the O2 demand/supply ratio (see Table 10-1). The reductions in the force of contraction and HR reduce myocardial oxygen consumption and result in autoregulatory decreases in myocardial blood flow. Several studies have shown that blood flow to ischemic regions with propranolol is maintained; however, this is probably secondary to maintenance of α-vasoconstrictor tone of epicardial vessels and of a pressure gradient to the vasodilated endocardial areas of ischemia.116,117 Reductions in blood flow in some patients with vasospastic angina may worsen with the administration of propranolol.118 Intracoronary infusion of propranolol does not worsen stenotic lesions and actually increases the luminal size of the stenosis at rest and during exercise.119
Antihypertensive Effects
The exact mechanisms involved in BP reduction are not clear. Both β1– and β2-receptor blockers inhibit myocardial contractility and reduce HR; both effects should reduce BP. No acute decrease in BP occurs during acute administration of propranolol.120 However, chronic BP reduction has been attributed to a chronic reduction in cardiac output (CO).121 Reductions in high levels of plasma renin have been suggested as effective therapy in controlling essential hypertension.122 However, the relation between renin levels and hypertension is not established, and the decrease in BP in patients has no relation to the change in renin levels.123,124 Stimulation of prejunctional β receptors results in norepinephrine release from postganglionic sympathetic fibers and increases in vascular tone to most major organ systems.125 Prejunctional β-blockade reduces norepinephrine release, sympathetic nerve traffic, and vascular tone.125
Electrophysiologic Effects
Several β-blockers have potent local anesthetic activity at greater serum levels because of sodium-channel–blocking activity and result in depression of phase 0 of the cardiac action potential.126 However, this membrane-stabilizing or quinidine-like effect is of questionable clinical relevance because it is observed at concentrations far exceeding therapeutic levels.127 Generalized slowing of cardiac depolarization results from reducing the rate of diastolic depolarization (phase 4). Action potential duration (APD) and the QT interval may shorten with β-adrenergic blockers.126 The ventricular fibrillation (VF) threshold is increased with β-blockers.128 These antiarrhythmic actions of β-blockers are enhanced in settings of catecholamine excess as in pheochromocytoma, acute MI, the perioperative period, and hyperthyroidism.
Metabolic Effects
Although β2-blockers are reported to reduce insulin release, the clinical significance of this reduction is questionable.129 Catecholamines, however, promote glycogenolysis and mobilization of glucose in response to hypoglycemia. In the diabetic patient, nonselective β-blockade may impede this process, thereby worsening recovery from a hypoglycemic episode. Also, the usual hypoglycemic symptoms of tachycardia and anxiety may be suppressed when taking β-blockers, thus delaying detection. In fact, bradycardia and hypertension have been documented as side effects of hypoglycemia in the diabetic patient receiving propranolol because of unopposed β-receptor stimulation with catecholamine release.130
Stimulation of β2-receptors increases the movement of potassium into skeletal muscle cells, reduces aldosterone secretion, and increases renal potassium loss, effects resulting in reduction of serum potassium. β2-Receptor blockers aid in the maintenance of serum potassium levels by blocking the adrenergic-stimulated movement of potassium intracellularly.131 β2-Receptor blockers may cause mild increases in serum potassium concentration, which may be significant in patients with renal insufficiency.132
Inhibition of catecholamine-stimulated lipolysis may occur with β-blockers, which reduces the availability of free fatty acids to activate contracting muscle, such as the heart.133 β-Adrenergic blockers produce increases in serum triglycerides, decreases in high-density lipoprotein cholesterol, and little change in low-density lipoprotein cholesterol. A proposed mechanism is an increase in the relative ratio of α- to β-activity receptors.134 Increases in β-receptor activity result in increases in lipoprotein lipase and triglyceride levels.135 These effects on blood lipids are concerns for patients receiving chronic therapy. However, animal studies have shown that β-blockers actually have a retarding effect on the development of atherosclerosis.136 β-Blockers with ISA produce the smallest changes in the lipid profile.137
Intrinsic Sympathomimetic Activity
Several β-blockers (acebutolol, carteolol, penbutolol, pindolol) have agonist and antagonist properties, and are characterized as having ISA.138 These agents are actually agonists that elicit a submaximal response and block the effects of endogenous catecholamines in a competitive fashion.138 CO and HR are reduced less with ISA drugs.139 Peripheral blood flow also is reduced less, making ISA agents attractive in patients with peripheral vascular disease.140 ISA drugs also cause less bronchoconstriction and are advantageous in chronic obstructive pulmonary disease. Theoretically, differences in changes in β-receptor density should differ with ISA. The effects of ISA drugs reduce β-receptor density (similar to pure agonists), whereas non-ISA agents increase β-receptor density.141 Although controversial, ISA drugs appear to have a role in mortality reduction after MI, similar to non-ISA β-blockers.142
General Pharmacology
Lipid-soluble β-blockers (propranolol, labetalol, metoprolol) are well absorbed after oral administration and attain high concentrations in the brain.143 Lipid-soluble agents have a high incidence of central nervous system (CNS) side effects, such as depression, sleep disturbances, and impotence. First-pass hepatic metabolism after oral ingestion can be very high but varies from patient to patient and affects daily dosing schedules.144 Cirrhosis, CHF, and cigarette smoking may reduce hepatic metabolism.145 Lipophilic agents are highly protein bound. The hepatic metabolism of lipophilic agents is independent of protein binding, which is different from most drugs in which hepatic metabolism occurs only with the unbound drug.146
Pindolol and timolol have intermediate lipid solubility properties and are metabolized partially by the liver (50%) and excreted through the kidneys (50%). Acebutolol, which is water soluble, has an active metabolite, diacetolol, which is water soluble and excreted renally.147 The plasma elimination of acebutolol is more rapid than that of diacetolol. For further information on the available oral and IV β-adrenergic blockers for treatment of myocardial ischemia, see Table 10-4. Additional β-blockers with other indications, such as carvedilol (for CHF) and sotalol (for arrhythmias), are covered later in this chapter.
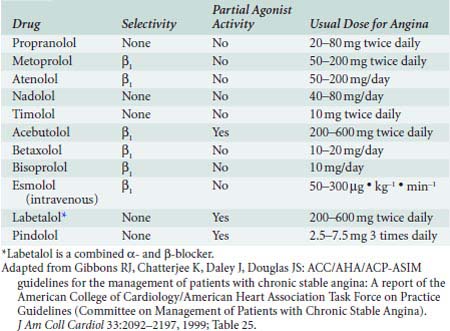
Pharmacology of Intravenous β-Adrenergic Blockers
Propranolol
Propranolol has an equal affinity for β1 and β2 receptors, lacks ISA, and has no β-adrenergic receptor activity. It is the most lipid-soluble β-blocker and generally has the most CNS adverse effects. First-pass liver metabolism (90%) is very high, requiring much greater oral doses than IV doses for pharmacodynamic effect.148 Although propranolol has an active metabolite (4-hydroxypropranolol), the metabolite’s half-life is much shorter and does not add to the clinical effect.149 Serum half-life of the drug after IV dosing is 3 to 4 hours.150
Because of the high hepatic extraction of propranolol, factors that affect hepatic blood flow markedly affect propranolol plasma levels. Because propranolol reduces hepatic blood flow, it can reduce its own metabolism, as well as the metabolism of other drugs.151 This must be taken into consideration during anesthetic procedures in patients with liver disease, reduced CO states, and right ventricular (RV) heart failure (HF).
Propranolol serum levels of 100 ng/mL produce a maximum β-blocking effect in relation to reducing exercise-induced tachycardia.144 Propranolol still produces a 50% reduction of exercise-induced tachycardia at serum levels of 12 ng/mL.152 Reductions in HR with propranolol occur at lower serum levels than depression of myocardial contractility.153 Accordingly, as drug levels decrease after discontinuation of therapy, reductions in the chronotropic response last much longer than reductions in inotropy.153 This is an important concept in treating tachycardias in patients with significant ventricular dysfunction and CHF.
The usual IV dose of propranolol initially is 0.5 to 1.0 mg titrated to effect. A titrated dose resulting in maximum pharmacologic serum levels is 0.1 mg/kg. The use of continuous infusions of propranolol has been reported after noncardiac surgery in patients with cardiac disease.154 A continuous infusion of 1 to 3 mg/hr can prevent tachycardia and hypertension but must be used with caution because of the potential of cumulative effects.
Metoprolol
Metoprolol was the first clinically used cardioselective β-blocker. Its affinity for β1 receptors is 30 times greater than its affinity for β2 receptors, as demonstrated by radioligand binding.155 Metoprolol is lipid soluble, with 50% of the drug metabolized during first-pass hepatic metabolism and with only 3% excreted renally.156 Protein binding is less than 10%. Metoprolol’s serum half-life is 3 to 4 hours. Because of its lipophilic properties, metoprolol has been shown in animal studies to diffuse into ischemic tissue better than atenolol, a hydrophilic β-receptor blocker.155
Esmolol
Esmolol’s chemical structure is similar to that of metoprolol and propranolol, except it has a methylester group in the para position of the phenyl ring, making it susceptible to rapid hydrolysis by red blood cell esterases (9-minute half-life).157 Esmolol is not metabolized by plasma cholinesterase. Hydrolysis results in an acid metabolite and methanol with clinically insignificant levels.158 Ninety percent of the drug is eliminated in the form of the acid metabolite, normally within 24 hours.158 A loading dose of 500 μg/kg given intravenously followed by a 50-to 300-μg/kg/min infusion will reach steady-state concentrations within 5 minutes. Without the loading dose, steady-state concentrations are reached in 30 minutes.158
Esmolol is cardioselective, blocking primarily β1 receptors. It lacks ISA and membrane-stabilizing effects and is mildly lipid soluble. Esmolol produced significant reductions in BP, HR, and cardiac index after a loading dose of 500 μg/kg and an infusion of 300 μg/kg/min in patients with CAD, and the effects were completely reversed 30 minutes after discontinuation of the infusion.159 Initial therapy during anesthesia may require significant reductions in both the loading and infusion doses.
Hypotension is a common side effect of IV esmolol. The incidence of hypotension was greater with esmolol (36%) than with propranolol (6%) at equal therapeutic end points.160 The cardioselective drugs may cause more hypotension because of β1-induced myocardial depression and the failure to block β2 peripheral vasodilation. Esmolol appears safe in patients with bronchospastic disease. In another comparative study with propranolol, esmolol and placebo did not change airway resistance, whereas 50% of patients treated with propranolol experienced development of clinically significant bronchospasm.161 Phlebitis may occur at the site of IV administration after prolonged infusion.162
Esmolol inhibits human plasma cholinesterase during in vitro studies, in which clinically insignificant prolongation of duration of succinylcholine action by esmolol was reported.163 Digoxin levels may increase slightly with concomitant esmolol administration.164 In addition, both esmolol and landiolol, a new short-acting β1-receptor blocker, also suppress the bispectral index during general anesthesia.165
Labetalol
Labetalol is an equal mixture of four stereoisomers with varying α- and β-blocking properties. Labetalol provides selective α1-receptor blockade and nonselective β1– and β2-blockade. The potency of β-adrenergic blockade is 5- to 10-fold greater than α1-adrenergic blockade.15,165 Labetalol has partial β2-agonist effects that promote vasodilation.166 Labetalol is moderately lipid soluble and is completely absorbed after oral administration.167 First-pass hepatic metabolism is significant with production of inactive metabolites.167 Renal excretion of the unchanged drug is minimal. Elimination half-life is approximately 6 hours.167
In contrast with other β-blockers, clinically, labetalol should be considered a peripheral vasodilator that does not cause a reflex tachycardia. BP and systolic vascular resistance decrease after an IV dose.168 Stroke volume (SV) and CO remain unchanged, with HR decreasing slightly.169 The reduction in BP is dose related, and acutely hypertensive patients usually respond within 3 to 5 minutes after a bolus dose of 100 to 250 μg/kg.170 However, the more critically ill or anesthetized patients should have their BP titrated beginning with 5- to 10-mg IV increments. Reduction in BP may last as long as 6 hours after IV dosing.
Significant Adverse Effects
CHF can be precipitated by β-adrenergic blockers, especially when other cardiac drugs with myocardial depressant properties are used, such as calcium blockers and disopyramide. β-Blockers blunt the usual reflex in sympathetic activity with these other depressant agents.171 Similarly, these effects are additive on the conduction system, and heart block can occur. Propranolol reduces the clearance of many drugs that depend on hepatic metabolism by reducing hepatic blood flow (e.g., lidocaine).172
Sudden withdrawal of β-adrenergic blockers can precipitate a state of enhanced adrenergic activity, resulting in tachycardia, hypertension, arrhythmias, myocardial ischemia, and infarction.173 Most studies indicated that this period of hypersensitivity occurs from 2 to 6 days after withdrawal of β -blockade. This corresponds to an increase in human lymphocyte β receptors during this period. Continuing β-receptor blockers before cardiac surgery results in a more stable anesthetic induction, intubation, and sternotomy sequence than performing anesthesia and surgery during a period of withdrawal hypersensitivity.83,174 Furthermore, reinstitution of small doses of β-adrenergic blockade after cardiac surgery smooths the postoperative course and reduces the incidence of tachyarrhythmias.175
Summary
β-Adrenergic blockers are first-line agents in the treatment of myocardial ischemia. These agents effectively reduce myocardial work and oxygen demand. There is growing evidence that β-adrenergic–blocking agents may play a significant role in reducing perioperative cardiac morbidity and mortality in noncardiac surgery.176

Calcium Channel Blockers
Calcium channel blockers reduce myocardial oxygen demands by depression of contractility, HR, and/or decreased arterial BP.177 Myocardial oxygen supply may be improved by dilation of coronary and collateral vessels. Calcium channel blockers are used primarily for symptom control in patients with stable angina pectoris. In an acute ischemic situation, calcium channel blockers (verapamil and diltiazem) may be used for rate control in situations when β-blockers cannot be used. The most important effects of calcium channel blockers, however, may be the treatment of variant angina. These drugs can attenuate ergonovine-induced coronary vasoconstriction in patients with variant angina, suggesting protection via coronary dilation.178 Most episodes of silent myocardial ischemia, which may account for 70% of all transient ischemic episodes, are not related to increases in myocardial oxygen demands (HR and BP) but, rather, intermittent obstruction of coronary flow likely caused by coronary vasoconstriction or spasm.179 All calcium channel blockers are effective at reversing coronary spasm, reducing ischemic episodes, and reducing NTG consumption in patients with variant or Prinzmetal’s angina.180 Combinations of NTG and calcium channel blockers, which also effectively relieve and possibly prevent coronary spasm, are currently rational therapy for variant angina. β-Blockers may aggravate anginal episodes in some patients with vasospastic angina and should be used with caution.181 Preservation of CBF with calcium channel blockers is a significant difference from the predominant β-blocker anti-ischemic effects of reducing myocardial oxygen consumption.
Calcium channel blockers have proved effective in controlled trials of stable angina.182–185 However, rapid-acting dihydropyridines such as nifedipine may cause a reflex tachycardia, especially during initial therapy, and exacerbate anginal symptoms. Such proischemic effects probably explain why the short-acting dihydropyridine nifedipine in high doses produced adverse effects in patients with unstable angina. The introduction of long-acting dihydropyridines such as extended-release nifedipine, amlodipine, felodipine, isradipine, nicardipine, and nisoldipine has led to fewer adverse events. These agents should be used in combination with β-blockers. Some patients may have symptomatic relief improved more with calcium channel blockers than with β-blocker therapy, although currently it is difficult to predict which patients respond better by other than empiric observation.
The causes of unstable angina may involve coronary vasospasm, accelerated atherosclerotic process, or enhanced platelet aggregation with fibrin clot formation. Calcium channel blockers have favorable effects in all three of the processes and are effective in the relief of symptoms of unstable angina.186 There are no significant clinical differences in the response of patients with unstable angina to β-adrenergic blockers and calcium channel blockers.187
Calcium Channel
Calcium channels are functional pores in membranes through which calcium flows down an electrochemical gradient when the channels are open. Calcium channels exist in cardiac muscle, smooth muscle, and probably many other cellular membranes. These channels also are present in cellular organelle membranes such as the sarcoplasmic reticulum (SR) and mitochondria. Calcium functions as a primary generator of the cardiac action potential and an intracellular second messenger to regulate various intracellular events.188
There are three types of voltage-dependent channels: the T (transient), L (long-lasting), and N (neuronal) channels.189 The T and L channels are located in cardiac and smooth muscle tissue, whereas the N channels are located only in neural tissue. The T channel is activated at low voltages (−50 mV) in cardiac tissue, plays a major role in cardiac depolarization (phase 0), and is not blocked by calcium antagonists.190,191 The L channels are the classic “slow” channels, are activated at greater voltages (−30 mV), and are responsible for phase 2 of the cardiac action potential. These channels are blocked by calcium antagonists.190,191
As mentioned previously, receptor-operated channels regulate calcium entry through voltage-regulated channels or through channels regulated by the receptor system per se. The β-adrenergic receptor operates an L-type voltage-dependent channel, which is activated by phosphorylation of a protein kinase by cAMP generated by the β-adrenergic receptor.191 β1-Receptor stimulation activates a G protein, which causes phospholipase C to hydrolyze phosphatidylinositol diphosphate to diacylglycerol (DAG) and inositol triphosphate (IP3).192 DAG activates protein kinase C that likely phosphorylates an L-type channel allowing calcium entry, whereas IP3 is a second messenger that interacts with the SR and directly promotes calcium release from the SR through an intracellular calcium channel.193 A receptor- operated channel also may increase calcium entry stimulated directly by a G protein. Both α2-receptor and β-receptor stimulation may activate G-protein–regulated channels, which are not voltage dependent194,195 (see Chapters 7 and 9).
Calcium channel blockers interact with the L-type calcium channel and are composed of drugs from four different classes: (1) the 1,4-dihydropyridine (DHP) derivatives (nifedipine, nimodipine, nicardipine, isradipine, amlodipine, and felodipine); (2) the phenylalkyl-amines, (verapamil); (3) the benzothiazepines (diltiazem); and (4) a diarylaminopropylamine ether (bepridil).15 The L-type calcium channel has specific receptors, which bind to each of the different chemical classes of calcium channel blockers.196 The binding to calcium blocker receptors by dihydropyridine derivatives (nifedipine) is voltage dependent.197 Calcium channels transform from a closed resting form that can potentially open, to an activated open form, to an inactive conformation that cannot open, and finally back to the closed resting form. Nifedipine binds preferentially to the inactive receptor that has just recently undergone activation and cannot open. Nifedipine essentially acts as a plug to block the channel. Verapamil binds to the L-type channel preferentially when it is active or open.198 The greater the period of activation of the channel, the more effective is the blockade (use dependent). Any repetitive activity, such as cardiac pacemaker activity, is sensitive to use-dependent agents.
Physiologic Effects
Hemodynamic Effects
Systemic hemodynamic effects of calcium channel blockers in vivo represent a complex interaction among myocardial depression, vasodilation, and reflex activation of the autonomic nervous system (Table 10-5).
TABLE 10-5 Calcium Channel Blocker Vasodilator Potency and Inotropic, Chronotropic, and Dromotropic Effects on the Heart
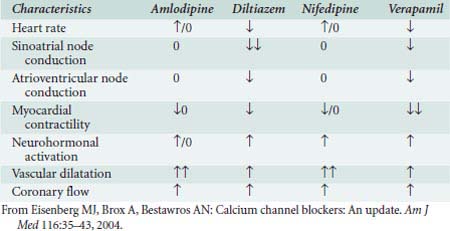
Nifedipine, like all dihydropyridines, is a potent arterial dilator with few venodilating effects.199 Reflex activation of the sympathetic nervous system (SNS) may increase HR. The intrinsic negative inotropic effect of nifedipine is offset by potent arterial dilation, which results in decline of BP and increase in CO in patients.200 Dihydropyridines are excellent antihypertensive agents because of their arterial vasodilatory effects. Antianginal effects result from reduced myocardial oxygen requirements secondary to the afterload-reducing effect and to coronary vascular dilation, resulting in improved myocardial oxygen delivery.
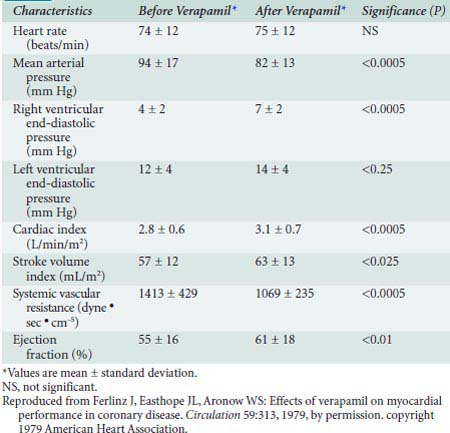
Verapamil is a less potent arterial dilator than the dihydropyridines and results in less reflex sympathetic activation. In vivo, verapamil generally results in moderate vasodilation without significant change in HR, CO, or SV201 (Table 10-6). IV administration of verapamil in the catheterization laboratory causes increases in RV pressure and LVEDP, decreases in SVR, improvement in ejection fraction (EF), and little change in PAPs.202 Verapamil can significantly depress myocardial function in patients with preexisting ventricular dysfunction.203
TABLE 10-6 Hemodynamic Effects of Intravenous Administration of Verapamil in 20 Patients with Coronary Artery Disease
Diltiazem is a less potent vasodilator and has fewer negative inotropic effects compared with verapamil. Studies in patients show reductions in SVR and BPs, with increases in CO, PAWP, and EF204 (Figure 10-14). Diltiazem attenuates baroreflex increases in HR secondary to NTG and decreases in HR secondary to phenylephrine.205 Regional blood flow to the brain and kidney increases, whereas skeletal muscle flow does not change.206 In contrast with verapamil, diltiazem is not as likely to aggravate CHF, although it should be used carefully in these patients.207
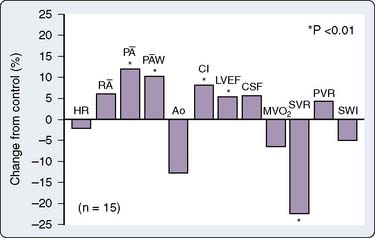
 , mean pulmonary artery pressure; P
, mean pulmonary artery pressure; P W, mean pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; R
W, mean pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; R , mean right atrial pressure; SVR, systemic vascular resistance; SWI, stroke work index.
, mean right atrial pressure; SVR, systemic vascular resistance; SWI, stroke work index.Coronary Blood Flow
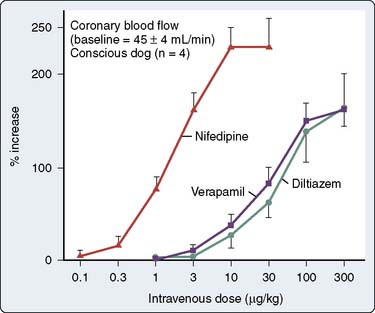
Coronary artery dilation occurs with the calcium channel blockers with increases in total CBF (Figure 10-15). Nifedipine is the most potent coronary vasodilator, especially in epicardial vessels, which are prone to coronary vasospasm. Diltiazem is effective in blocking coronary artery vasoconstriction caused by a variety of agents, including α-agonists, serotonin, prostaglandin, and acetylcholine.178,208,209
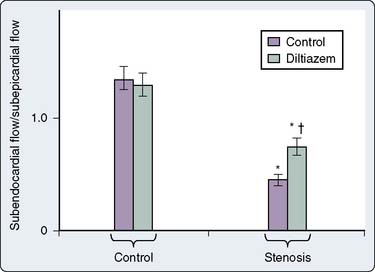
Calcium channel blockers also may dilate the coronary artery at the stenotic site, thus reducing the pressure gradient across the coronary lesion.178 Diltiazem preferentially dilates coronary arteries compared with other peripheral vessels.210 Animal studies demonstrate that nifedipine, verapamil, and diltiazem increase coronary collateral flow distal to coronary ligation in animals and improve subendocardial flow relative to subepicardial flow211–214 (Figure 10-16).
Electrophysiologic Effects
Calcium channel blockers exert their primary electrophysiologic effects on tissue of the conducting system that is dependent on calcium for generation of the action potential, primarily at the sinoatrial (SA) and AV nodes. They do not alter the effective refractory period (ERP) of atrial, ventricular, or His-Purkinje tissue. Diltiazem and verapamil exert these electrophysiologic effects in vivo and in vitro, whereas the electrophysiologic depression of dihydropyridines (nifedipine) is completely attenuated by reflex sympathetic activation. Nifedipine actually can enhance SA and AV node conduction, whereas verapamil and diltiazem slow conduction velocity and prolong refractoriness of nodal tissue.215–217
Atherosclerosis
Calcium is involved in the generation of atherosclerotic plaque and in damaged atherosclerotic tissue (calcification).218 Verapamil and nifedipine have been found to have antiatherogenic effects.218 Nifedipine has been shown to retard angiographic progression of CAD in humans.219 Diltiazem also may reduce atherosclerotic progression after heart transplantation.220 Diltiazem suppresses aortic atherosclerosis, but not that of coronary arteries, inhibits spontaneous calcinosis in hypertensive rats, and prevents vitamin D–induced calcinosis in arterial elastic tissue.221–223 Diltiazem also suppresses necrosis of aortic smooth muscle cells by hyperlipidemia serum224 and intimal thickening in rabbit carotid arteries.225
Platelet Aggregation
Calcium antagonists, nitrates, and β-adrenergic blockers all inhibit platelet aggregation. This could be a most important effect of all anti-ischemic drugs, especially in the treatment of chronic disease. Calcium is a mediator involved in the release of platelet aggregatory factors, such as ADP, and verapamil inhibits calcium-induced release of these factors.226 Diltiazem inhibition of platelet aggregation correlates with changes in intracellular calcium levels.227 In vivo, diltiazem inhibits platelet aggregation after 24 hours in healthy volunteers.228 Similar antiaggregatory effects of diltiazem were seen in patients with unstable angina, but no inhibitory effect of platelet aggregation was found with verapamil. Diltiazem metabolites are even more effective in inhibiting platelet aggregation than diltiazem.229
Metabolic Effects
Nifedipine may be associated with decreases in serum glucose levels in patients with diabetes, but glucose levels in healthy volunteers generally increase slightly with nifedipine and in hypertensive patients with diltiazem.230,231 Diltiazem reportedly has no effect on insulin, glucagon, growth hormone, or cortisol levels.232,233 However, nifedipine apparently delays insulin release in patients with diabetes.233
Pharmacology
Table 10-7 illustrates pharmacokinetic parameters for the U.S. Food and Drug Administration–approved anti-ischemic calcium channel blockers.
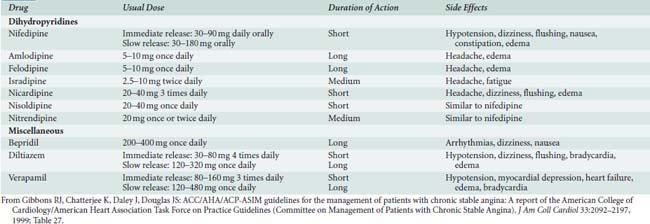
Nifedipine
Nifedipine was the first dihydropyridine derivative to be used clinically. Other dihydropyridines available for clinical use include nicardipine, isradipine, amlodipine, felodipine, and nimodipine.234 In contrast with the other calcium channel blockers, nimodipine is highly lipid soluble and penetrates the blood–brain barrier. It is indicated for vascular spasm after intracerebral bleeding.
Nicardipine
Nicardipine is a dihydropyridine agent with a longer half-life than nifedipine and with vascular selectivity for coronary and cerebrovascular beds. Nicardipine may be the most potent overall relaxant of vascular smooth muscle among the dihydropyridines.235 Peak plasma levels are reached 1 hour after oral administration, with bioavailability of 35%.236 Plasma half-life is approximately 8 to 9 hours. Although the drug undergoes extensive hepatic metabolism with less than 1% of the drug excreted renally, greater renal elimination occurs in some patients.237 Plasma levels may increase in patients with renal failure; reduction of the dose is recommended in these patients.236
Nicardipine is a potent cerebrovascular vasodilator and may prevent ischemia-related neuronal necrosis in animal studies.238,239 Marked improvements in CBF occur with nicardipine.240 Although coronary vasodilation is one explanation, positive inotropic effects with either PDE activity or calcium channel agonist (as well as antagonist) activity resulting in autoregulatory increases in blood flow may be an additional mechanism.241
Verapamil
The structure of verapamil is similar to that of papaverine. Verapamil exhibits significant first-pass hepatic metabolism, with a bioavailability of only 10% to 20%.242 One hepatic metabolite, norverapamil, is active and has a potency approximately 20% of that of verapamil.242 Peak plasma levels are reached within 30 minutes. Bioavailability markedly increases in hepatic insufficiency, mandating reduced doses. 242 IV verapamil achieves hemodynamic and dromotropic effects within minutes, peaking at 15 minutes and lasting up to 6 hours. Accumulation of the drug occurs with prolonged half-life during long-term oral administration.243
Diltiazem
After oral dosing, the bioavailability of diltiazem is greater than verapamil’s, varying between 25% and 50%.244 Peak plasma concentration is achieved between 30 and 60 minutes, and elimination half-life is 2 to 6 hours.244 Protein binding is approximately 80%. As with verapamil, hepatic clearance is flow dependent and major hepatic metabolism occurs, with metabolites having 40% of the clinical activity of diltiazem.245,246 Hepatic disease may require decreased dosing, whereas renal failure does not affect dosing.247
Significant Adverse Effects
Most significant adverse hemodynamic effects can be predicted from the calcium channel blockers’ primary effects of vasodilation, and negative inotropy, chronotropy, and dromotropy. Hypotension, HF, bradycardia and asystole, and AV nodal block have occurred with calcium channel blockers.248 These side effects are more likely to occur with combination therapy with β-blockers or digoxin, in the presence of hypokalemia.248
Verapamil increases digoxin levels, whereas diltiazem has variable effects and nifedipine no effect on digoxin levels.249–251 Cimetidine and ranitidine increase calcium blockers’ serum levels either by liver enzyme induction or reductions of hepatic blood flow.252 The physiologic effects of calcium blockers may be additive to those of anesthetic agents in animal studies, but clinically significant effects are variable.253,254 The cautious use of IV verapamil with a β-adrenergic receptor antagonist is necessary because of the increased risk for AV block or severe myocardial depression.
Paradoxic aggravation of myocardial ischemia may be seen with the short-acting dihydropyridines (nifedipine).255 This may be secondary to decreased CPP with associated hypotension, selective vasodilation in the nonischemic region (coronary steal), or increased oxygen demand as a result of reflex sympathetic stimulation and tachycardia.
Case reports of a withdrawal syndrome similar to β-blocker withdrawal have been presented. Five of 143 patients had significant ST-segment changes after diltiazem or verapamil withdrawal.256 MI and coronary spasm have been reported after diltiazem withdrawal.257,258 One study comparing propranolol with verapamil withdrawal in patients with stable angina found that 2 of 20 patients had severe exacerbation of their angina with propranolol withdrawal, and no patients had hemodynamic or symptomatic evidence of a withdrawal phenomenon with verapamil.259
Drug Therapy for Systemic Hypertension
Systemic hypertension, long recognized as a leading cause of cardiovascular morbidity and mortality, accounts for enormous health-related expenditures. Nearly a fourth of the U.S. population has hypertensive vascular disease; however, 30% of these individuals are unaware of their condition, and another 30% to 50% are inadequately treated.260,261 On a worldwide basis, nearly 1 billion individuals are hypertensive.262 Based on data from the Framingham Heart Study, normotensive patients at age 55 can expect a 90% lifetime risk for subsequent development of hypertension.263 Furthermore, hypertension management comprises the most common reason underlying adult visits to primary care physicians, and antihypertensive drugs are the most prescribed medication class.264
Despite the asymptomatic nature of hypertensive disease, with symptom onset delayed 20 to 30 years after development of systemic hypertension, substantial incontrovertible evidence demonstrates a direct association between systemic hypertension and increased morbidity and mortality. The World Health Organization estimates that hypertension underlies one in eight deaths worldwide, making elevated BP the third leading cause of mortality.262 In fact, hypertension accounts for the single most treatable risk factor for MI, stroke, peripheral vascular disease, CHF, renal failure, and aortic dissection.260 In prospective randomized trials, over the course of adult lifetimes, successful treatment of hypertension has been associated with 35% to 40% reductions in the incidence of stroke, 50% reductions in CHF, and 25% reductions in MIs.260,262 Improved treatment of hypertension has been credited with the major reductions in stroke and cardiovascular mortality occurring since the 1970s in the United States.
Pathophysiologic mechanisms underlying predisposition to hypertension remain, for the most part, unclear. Undoubtedly, both genetic and environmental factors play contributory roles.265 Concordance for hypertension is greater between monozygotic or dizygotic twins, and even siblings within a single family, than that observed between unrelated individuals, supporting a genetic component. However, by some estimates, genetic predeterminants account for only 30% to 40% of hypertensive disease.265 As for environmental factors, a direct association between body mass index and hypertension has been reported, and dietary sodium intake is associated with long-term risk for development of hypertension.266,267
Definitions for hypertension are somewhat arbitrary, although derived from clinical trials suggesting systemic pressures at which the benefits of treatment outweigh the risk for adverse effects related to antihypertensive therapy. BP varies with normal distribution across the population at large, and aging is associated with progressive increases in systolic pressure. After 50 years of age, reductions in diastolic pressure are commonly observed, resulting in widening of pulse pressure with age. Published evidence suggests that systolic and pulse pressures are better predictors for morbidity and mortality than diastolic pressure.264 Most recently, the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7 Report) defined systolic BPs (Table 10-8) exceeding 140 mm Hg and diastolic BPs exceeding 90 mm Hg as stage 1 hypertension. BPs less than 120/80 mm Hg were defined as normal, and those in between as consistent with “prehypertension.”260
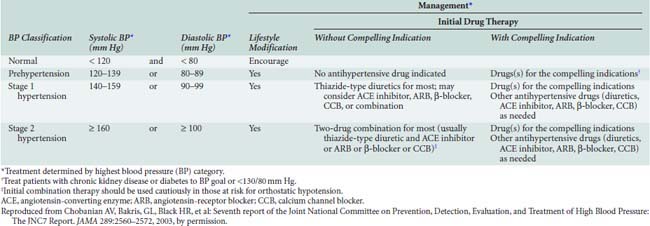
Although antihypertensive drug therapy is widely regarded as essential for BPs greater than 140/90 mm Hg, recent evidence suggests benefits to more aggressive BP reduction for certain patient subsets. The association between systemic BP and cardiovascular risk has been described as a “J-curve,” with progressive cardiovascular risk reductions accompanying BP reductions until a critical threshold—after which the potential for myocardial ischemia and/or other organ injury increases.268
Risk for cardiovascular disease appears to increase at BPs greater than 115/75 mm Hg, with a doubling in risk associated with each 20/10-mm Hg increment in systemic pressure.260 Thus, the most recent JNC-7 report recommends drug therapy for “prehypertensive” disease in patients with “compelling indications” such as chronic renal disease or diabetes. Antihypertensive therapy generally is targeted to achieve systemic BPs less than 140/90 mm Hg; however, for high-risk patients such as those with diabetes, renal, or cardiovascular disease, lower BP targets are suggested, typically more than 130/80 mm Hg.260

Medical Treatment for Hypertension
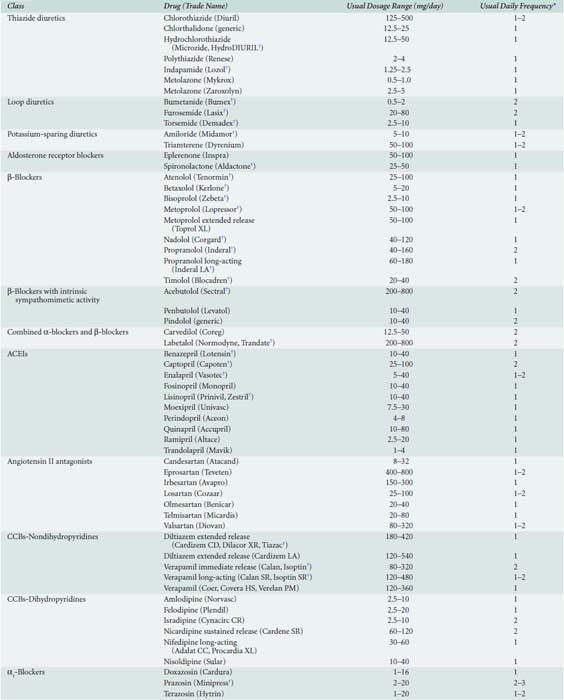
Currently, nearly 80 distinct medications are marketed for treatment of hypertension264 (Table 10-9). Often, combined therapy with two or more classes of antihypertensive medications may be needed to achieve treatment goals269 (Table 10-10). Although the specific drug selected for initial therapy now has been deemed less important than in the past, recognition that specific antihypertensive drug classes alleviate end-organ damage, beyond that simply associated with reductions in systemic BP, has led to targeted selection of antihypertensive drug combinations on the basis of coexisting risk factors such as recent MI, chronic renal insufficiency, or diabetes.
| Combination Type | Fixed-Dose Combination, mg* | Trade Name |
|---|---|---|
| ACEIs and CCBs | Amlodipine-benazepril hydrochloride (2.5/10, 5/10, 5/20, 10/20) | Lotrel |
| Enalapril-felodipine (5/5) | Lexxel | |
| Trandolapril-verapamil (2/180, 1/240, 2/240, 4/240) | Tarka | |
| ACEIs and diuretics | Benazepril-hydrochlorothiazide (5/6.25, 10/12.5, 20/12.5, 20/25) | Lotensin HCT |
| Captopril-hydrochlorothiazide (25/15, 25/25, 50/15, 50/25) | Capozide | |
| Enalapril-hydrochlorothiazide (5/12.5, 10/25) | Vaseretic | |
| Fosinopril-hydrochlorothiazide (10/12.5, 20/12.5) | Monopril/HCT | |
| Lisinopril-hydrochlorothiazide (10/12.5, 20/12.5, 20/25) | Prinzide, Zestoretic | |
| Moexipril-hydrochlorothiazide (7.5/12.5, 15/25) | Uniretic | |
| Quinapril-hydrochlorothiazide (10/12.5, 20/12.5, 20/25) | Accuretic | |
| ARBs and diuretics | Candesartan-hydrochlorothiazide (16/12.5, 32/12.5) | Atacand HCT |
| Eprosartan-hydrochlorothiazide (600/12.5, 600/25) | Teveten-HCT | |
| Irbesartan-hydrochlorothiazide (150/12.5, 300/12.5) | Avalide | |
| Losartan-hydrochlorothiazide (50/12.5, 100/25) | Hyzaar | |
| Olmesartan medoxomil-hydrochlorothiazide (20/12.5, 40/12.5, 40/25) | Benicar HCT | |
| Telmisartan-hydrochlorothiazide (40/12.5, 80/12.5) | Micardis-HCT | |
| Valsartan-hydrochlorothiazide (80/12.5, 160/12.5, 160/25) | Diovan-HCT | |
| BBs and diuretics | Atenolol-chlorthalidone (50/25, 100/25) | Tenoretic |
| Bisoprolol-hydrochlorothiazide (2.5/6.25, 5/6.25, 10/6.25) | Ziac | |
| Metoprolol-hydrochlorothiazide (50/25, 100/25) | Lopressor HCT | |
| Nadolol-bendroflumethiazide (40/5, 80/5) | Corzide | |
| Propranolol LA-hydrochlorothiazide (40/25, 80/25) | Inderide LA | |
| Timolol-hydrochlorothiazide (10/25) | Timolide | |
| Centrally acting drug and diuretic | Methyldopa-hydrochlorothiazide (250/15, 250/25, 500/30, 500/50) | Aldoril |
| Reserpine-chlorthalidone (0.125/25, 0.25/50) | Demi-Regroton, Regroton | |
| Reserpine-chlorothiazide (0.125/250, 0.25/500) | Diupres | |
| Reserpine-hydrochlorothiazide (0.125/25, 0.125/50) | Hydropres | |
| Diuretic and diuretic | Amiloride-hydrochlorothiazide (5/50) | Moduretic |
| Spironolactone-hydrochlorothiazide (25/25, 50/50) | Aldactazide | |
| Triamterene-hydrochlorothiazide (37.5/25, 75/50) | Dyazide, Maxzide |
ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin-receptor blocker; BB, β-blocker; CCB, calcium channel blocker.
* Some drug combinations are available in multiple fixed doses. Each drug dose is reported in milligrams.
Reproduced from Chobanian AV, Bakris GL, Black HR, et al: Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42:1206–1252, 2003, by permission.
Diuretics
Thiazide diuretic therapy comprises the cornerstone of most antihypertensive regimens.270,271 Three classes of diuretics have proved efficacious in reducing systemic BP—the thiazide and related sulfonamide compounds, loop diuretics, and potassium-sparing agents. All classes of diuretics initially reduce BP by increasing urinary excretion of sodium, with resultant reductions in plasma volume and CO; however, over a period of 6 to 8 weeks, diuretic therapy leads to reductions in SVR—hypothesized to relate to activation of vascular endothelial potassium channels.
Thiazide diuretics remain the first-choice medical therapy for the majority of patients with hypertension.272 Although the natriuretic effect achieved by blockade of sodium and chloride transport in the distal convoluted tubule is relatively weak, thiazide diuretics generally produce 10-mm Hg reductions in BP and have proved efficacious in numerous randomized trials to reduce morbidity and mortality related to hypertensive vascular disease.264,269
β-Blockers
β-Adrenergic receptor blockers, which reduce sympathetic stimulation of the heart and vasculature, comprise another common antihypertensive therapy, particularly in settings of CAD or CHF.273–277 More specifically, β-blockers inhibit myocardial and peripheral β1-adrenergic receptors to reduce CO. As well, β-blockers reduce renin release from renal juxtaglomerular cells and norepinephrine release by inhibition of prejunctional β2-adrenergic receptors in the peripheral vasculature. β-Blockers traditionally are classified on the basis of cardioselectivity, lipid solubility, and intrinsic sympathetic activity, with first-generation agents such as propranolol nonselectively blocking both β1– and β2-adrenergic receptors. Second-generation agents (e.g., metoprolol or atenolol) exhibit a relatively cardioselective preference for β1-adrenergic receptor blockade at low doses. Pindolol, a novel β-blocker with ISA, stimulates vasodilation by activation of β2-adrenergic receptors. Combination agents, such as labetalol, provide mixed β- and α1-adrenergic blocking properties resulting in inhibition of sympathetic activity, as well as direct vasodilatory effects.
Angiotensin-Converting Enzyme Inhibitors
Angiotensin-converting enzyme (ACE) inhibitors reduce peripheral vascular resistance by inhibiting conversion of angiotensin I (Ang I) to the highly vasoconstrictive angiotensin II (Ang II).278 Recent evidence suggests that much of the long-term antihypertensive effect of ACE inhibitors derives from protective effects on bradykinin de-gradation. Bradykinin, a potent vasodilator under normal conditions, undergoes degradation by the action of ACE. Therefore, ACE inhibition results in increased plasma and tissue concentrations of bradykinin.
Angiotensin II Antagonists
Ang II antagonists, or angiotensin-receptor blockers, are perhaps the best tolerated of all current antihypertensive therapies and rapidly are becoming a favored drug therapy for management of hypertensive vascular disease.278 Ang II antagonists bind and competitively inhibit Ang II AT1 receptors, thereby directly inhibiting the vasoconstrictive effects of Ang II.279
Calcium Channel Blockers
Calcium channel blockers, commonly classified as either dihydropyridines or nondihydropyridines, share a common mechanism of action in that they bind various sites on the α1 subunit of the L-type voltage-dependent calcium channel to partially inhibit calcium entry into cells. Although all calcium channel blockers induce arterial vasodilation, the dihydropyridines more frequently induce a reflex tachycardia, whereas the nondihydropyridines (e.g., diltiazem and verapamil) may impair cardiac conduction or contractility, or both.280
Common side effects reported with calcium channel blockers, and relating to arterial vasodilation, include ankle edema, flushing, and headaches. Nondihydropyridines are most often contraindicated in patients with preexisting HF or cardiac conduction defects because of their potential for precipitating HF, heart block, or both. Controversy surrounding the safety of calcium channel blockers as antihypertensive therapy has been substantially negated by recent prospective randomized trials demonstrating the safety and efficacy of long-acting calcium channel blockers for treatment of hypertensive cardiovascular disease.281–283 Administration of short-acting dihydropyridines (e.g., nifedipine) for rapid control of hypertension has been associated with an increased incidence of acute coronary events and is contraindicated in the acute treatment of severe hypertension.
α1-Blockers
Adverse effects associated with α1-blockers include orthostatic hypotension, fluid retention, and reflex tachycardia.284 Although rarely administered as monotherapy, α1-blockers continue to prove useful in combination with other antihypertensives because of their lack of metabolic side effects and propensity to dilate urethral smooth muscle, alleviating symptoms of prostatism.264

Central α2-Agonists and Other Centrally Acting Drugs
Centrally acting antihypertensive agents, such as clonidine and methyldopa, stimulate α2-adrenergic and imidazoline receptors within the CNS to reduce sympathetic outflow and SVR. In the case of clonidine, activation of presynaptic α-adrenergic receptors inhibits norepinephrine release and subsequent catecholamine generation.285 Reserpine differs from the α2-agonists in that it inhibits reuptake of norepinephrine by storage vesicles in the postganglionic adrenergic neurons, depleting norepinephrine. As opposed to the α2-agonists, reserpine’s peripheral effects predominate over central activity.

Novel Approaches to Antihypertensive Therapy
Despite the array of antihypertensive medications currently available, heterogeneity of treatment effects and adverse effects of current drugs on quality-of-life measures suggest the need for alternative approaches to antihypertensive therapy.286 Promising approaches include both aldosterone-receptor blockers and renin inhibitors. With the exception of potential for hyperkalemia, aldosterone-receptor inhibitors have proved well tolerated, and renin inhibitors administered in concert with Ang II inhibitors alleviated the compensatory increase in plasma renin activity. Third-generation β-blockers incorporating vasodilatory activity offer particular promise for treating hypertension in the setting of concomitant HF. Further modifications to endothelin-receptor antagonists and dual vasopeptidase inhibitors offer novel antihypertensive approaches for the future. Ultimately, gene therapy may prove the definitive solution to essential hypertension. It appears likely that environmental factors interacting with multiple genetic polymorphisms contribute to overall risk for hypertension. Gene therapy offers a viable approach to long-term management of hypertension but will prove dependent on further identification of target genes, improvements in gene transfer efficiency, and development of safer transfer vectors. Near term, advances in personalized medicine offer potential for DNA testing of genetic polymorphisms to identify antihypertensive drugs most likely to benefit specific patients.287

Management of Severe Hypertension
A common therapeutic approach to the hypertensive emergency includes a limited reduction in BP, of perhaps 10%, over the initial 1 to 2 hours of therapy, followed by further reductions to a target diastolic pressure (e.g., 110 mm Hg) during the initial 12 hours of therapy. Further reductions in BP to acceptable target levels should proceed over a period of days to minimize potential for inducing ischemic injury in the setting of altered autoregulatory flow to target organ vascular beds.264,288
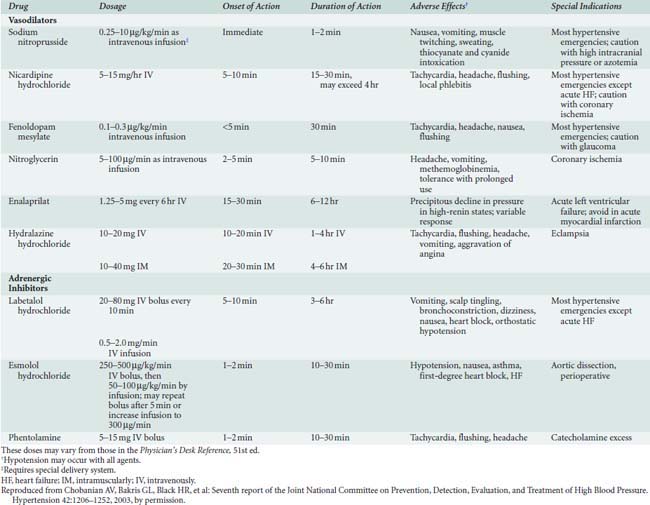
The favored parenteral drug for rapid treatment of hypertensive emergencies remains sodium nitroprusside264 (Table 10-11). An NO donor, sodium nitroprusside induces arterial and venous dilation, providing rapid and predictable reductions in systemic BP. Prolonged administration of large doses may be associated with cyanide or thiocyanate toxicity; however, rarely is this a concern in the setting of acute hypertensive emergencies. Although less potent and predictable than sodium nitroprusside, NTG, another NO donor, may be preferable in the setting of myocardial ischemia or after CABG. NTG preferentially dilates venous capacitance beds as opposed to arterioles; however, rapid onset of tolerance limits the efficacy of sustained infusions to maintain BP control. Nicardipine, a parenteral dihydropyridine calcium channel blocker, and fenoldopam, a selective dopamine1-receptor antagonist, have been utilized increasingly in select patient populations after CABG and in the setting of renal insufficiency, respectively.

Compelling Indications for Specific Antihypertensive Drug Selection
Despite compelling evidence supporting the preferential administration of thiazide diuretics as agents of choice for treatment of hypertension, most patients will require addition of a second nondiuretic antihypertensive drug to achieve desired target BPs.264 Selection of a complementary antihypertensive agent necessitates a thorough understanding of the mechanisms of action to optimize the additive effect of combined therapy. For example, β-blockers, ACE inhibitors, and Ang II antagonists inhibit renin release. Therefore, combinations of these agents are unlikely to achieve a maximal additive antihypertensive response. Similarly, both the diuretics and dihydropyridine calcium channel blockers produce peripheral vasodilation such that combination with other antihypertensive drug classes may prove more efficacious. Increasingly, data derived from ongoing outcomes-based trials of antihypertensive therapy in patients with coexisting diseases suggest that specific antihypertensive drugs reduce morbidity beyond that expected for BP reduction alone264 (Table 10-12).
TABLE 10-12 Clinical Trial and Guideline Basis for Compelling Indications for Individual Drug Classes
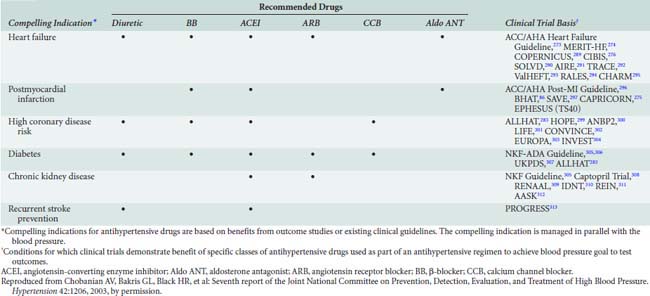
For example, in patients with ischemic heart disease, and most especially a recent MI, β-blockers have been demonstrated to reduce mortality and morbidity related to subsequent cardiac events.86,275,277,305 Although precise mechanisms remain unclear, reductions in overall sympathetic stimulation and antiarrhythmic effects presumably play contributory roles. In the setting of renal insufficiency, ACE inhibitors and Ang II antagonists delay progression of both diabetic and nondiabetic-associated renal disease.305,308–310 Calcium channel blockers have proved efficacious in the setting of peripheral vascular diseases, such as Raynaud syndrome, and α1-blockers favorably influence symptoms of prostatism. In the setting of hypertension and pregnancy, methyldopa and hydralazine remain preferred antihypertensives for management of chronic hypertension and preeclampsia, respectively.260

Perioperative Implications of Hypertension
There is little evidence to suggest that mild-to-moderate degrees of hypertension adversely affect morbidity and mortality in the perioperative setting. In fact, ACC/AHA guidelines state that mild-to-moderate hypertension does not represent an independent risk factor for perioperative cardiovascular complications.314 However, given the strong association between hypertension and cardiovascular diseases, a preoperative diagnosis of hypertension necessitates preoperative assessment for evidence of target-organ damage (e.g., CAD, renal dysfunction, stroke, peripheral vascular disease).315 In contrast with isolated hypertension, secondary cardiovascular diseases, in many cases, do pose increased risk for perioperative morbidity and mortality. In patients with a preoperative diagnosis of hypertension, an electrocardiogram (ECG) is performed (to assess for evidence of ischemia, prior MI, or left ventricular [LV] hypertrophy), and a urinalysis and plasma creatinine determination may be obtained to assess renal function. Specific antihypertensive therapies may necessitate additional evaluations such as assessment of plasma potassium and sodium in patients taking diuretics.
In cases of mild-to-moderate hypertension, few controlled trials assessing the association between preoperative hypertension and perioperative morbidity and mortality are available. Most investigations have been observational in nature with no correction for potentially confounding risk factors. In many cases, the number of study participants has been inadequate to ensure statistical power to assess for relevant associations between outcomes and a preoperative diagnosis of hypertension. Howell et al288 published a meta-analysis summarizing 30 studies including more than 12,995 patients for whom an association between hypertension and perioperative complications could be assessed. These authors calculated an odds ratio of 1.31, suggesting a slightly increased risk for perioperative cardiovascular complications in patients with preexisting hypertension; however, given limitations of the dataset, the authors further concluded that such a small odds ratio in the setting of a “low perioperative event rate” likely represents a clinically insignificant association between preexisting hypertension and cardiac risk. Other investigators have reported similar small associations between isolated systolic hypertension before surgery and subsequent perioperative morbidity.
As opposed to the apparently low perioperative risk posed by mild-to-moderate hypertension, cases of severe hypertension (diastolic BP > 110 mm Hg) frequently lead to questions whether elective surgery should be postponed to allow for titration of antihypertensive therapy to acceptable systemic BPs. Again, although there are little data to support conclusive recommendations regarding acceptable preoperative BPs or the length of antihypertensive therapy necessary to achieve a new “steady state,” patients with poorly controlled BP experience more labile BP responses in the perioperative setting with greater potential for hypertensive or hypotensive episodes, or both.315–317 Guidelines published by the ACC/AHA suggest that systemic BPs exceeding 180 mm Hg systolic and/or 110 mm Hg diastolic should be controlled before surgery.314 In a review, Howell and colleagues288 concluded that surgery need not be canceled in the setting of severe hypertension; however, a careful preoperative assessment for target organ damage (e.g., cardiovascular, renal, cerebrovascular disease) should be performed before surgery, and intraoperative arterial pressures should be maintained within 20% of preoperative BPs. At a minimum, in patients with severe hypertension, invasive monitoring of arterial pressure with strict control of perioperative BPs, continuing into the postoperative setting, appears justified; given the predilection for CAD in these patients, perioperative therapy with β-blockers may be indicated.93,94
Pharmacotherapy for acute and chronic heart failure
Chronic HF is one major cardiovascular disorder that continues to increase in incidence and prevalence, both in the United States and worldwide. It affects nearly 550 million persons in the United States, and roughly 600,000 new cases are diagnosed each year.318,319 Currently, 1% to 2% of those 40 to 59 years of age and 12% to 14% of individuals older than 80 have HF.318 Because HF is primarily a disease of the elderly, its prevalence is projected to increase twofold to threefold over the next decade, as the median age of the U.S. population continues to increase.320 The increasingly prolonged survival of patients with various cardiovascular disorders that culminate in ventricular dysfunction (e.g., patients with CAD are living longer rather than dying acutely with MI), and the greater diagnostic awareness further compound the HF epidemic. Despite improvements in the understanding of the neurohormonal mechanisms underlying its pathophysiology and remarkable advances made in pharmacologic therapy, HF continues to cost the United States an estimated $38 billion annually in medical expenditures,321 and it contributes to approximately 292,000 deaths per year.318 Given the public health impact of the disease and the rapid pace of therapeutic advances, it is essential that the perioperative physician remain aware of contemporary clinical practice for the benefit of those patients with chronic HF presenting to the operating room or intensive care unit.
Accordingly, the pharmacologic management of HF as it relates to guidelines published by the ACC/AHA is reviewed.322 The focus is primarily on chronic HF associated with LV systolic dysfunction, although chronic HF with preserved systolic function (or diastolic heart failure [DHF]) and acute HF also are discussed. For each drug commonly used in clinical practice, the general format of presentation includes whether it alters the progression of myocardial damage (for agents administered chronically), its mechanism(s) of action, current clinical data on trials of its use for HF, and its current place in the treatment of HF. New, non-neurohormonal, pharmacologic options in HF management also are reviewed.

Heart Failure Classification
The ACC/AHA updated guidelines for evaluating and managing HF include a new, four-stage classification system emphasizing both the evolution and progression of the disease (Figure 10-17). It calls attention to patients with preclinical stages of HF to focus on halting disease progression. The staging system is meant to complement, not replace, the widely used New York Heart Association (NYHA) classification, a semiquantitative index of functional classification that categorizes patients with HF by the severity of their symptoms (Box 10-5). NYHA classification remains useful clinically because it reflects symptoms, which, in turn, correlate with quality of life and survival.323 The new classification system for HF, recognizing its progressive course and identifying those who are at risk (e.g., the first two stages, A and B, clearly are not HF), reinforces the importance of determining the optimal strategy for neurohormonal antagonism in an attempt to improve the natural history of the syndrome. In this new staging approach, patients would only be expected to either not advance at all or to advance from one stage to the next, unless progression of the disease was halted by treatment. This is in contrast with the NYHA functional classification in which the severity of symptoms characteristically fluctuates depending on changes in diet and medications, even in the absence of measurable changes in LV structure and function.318,322 The new HF staging approach may be comparable with that used in such disorders as cancer.
BOX 10-5 New York Heart Association Functional Class
Although the pathophysiology of HF has been addressed elsewhere, it is important to mention key concepts as the basis of current pharmacologic therapy. To start, HF remains the final common pathway for CAD, hypertension, valvular heart disease, and cardiomyopathy, in which the natural history results in symptomatic or asymptomatic LV dysfunction (specifically, LV systolic dysfunction for the purposes of this discussion). The neurohormonal responses to impaired cardiac performance (salt and water retention, vasoconstriction, sympathetic stimulation) are initially adaptive but, if sustained, become maladaptive, resulting in pulmonary congestion and excessive afterload. This, in turn, leads to a vicious cycle of increases in cardiac energy expenditure and worsening of pump function and tissue perfusion (Table 10-13). Although the cardiorenal and cardiocirculatory branches of this neurohormonal hypothesis of HF were the original foundation for the use of diuretics, vasodilators, and inotropes, respectively, seminal information in the early 1990s emerged from large, randomized clinical trials that showed ACE inhibitors324,325 and angiotensin receptor blockers,293,326 but not most other vasodilators,327 prolonged survival in patients with HF. In a similar fashion, the use of β-blockers, despite their negative inotropic effects, improved morbidity and mortality in randomized, controlled trials.289,328,329
TABLE 10-13 Neurohormonal Effects of Impaired Cardiac Performance and Their Effects on the Circulation
| Response | Short-Term Effects | Long-Term Effects |
|---|---|---|
| Salt and water retention | Augments preload | Pulmonary congestion, anasarca |
| Vasoconstriction | Maintains blood pressure for perfusion of vital organs (brain, heart) | Exacerbates pump dysfunction (excessive afterload), increases cardiac energy expenditure |
| Sympathetic stimulation | Increases heart rate and ejection | Increases energy expenditure |
The finding that low-dose aldosterone antagonists added to conventional therapy for HF reduce mortality in patients with severe HF suggests that there is more to the neurohormonal hypothesis of drug efficacy than cardiorenal and hemodynamic effects alone.294,298 Taken together with evidence from basic investigations showing that Ang II is a growth factor and a vasoconstrictor,330 the clinical data promoted a shift in focus from cardiorenal and cardiocirculatory processes toward cardiac remodeling as the central component in the progression of this neurohormone-mediated cardiac syndrome.331 The renin-angiotensin-aldosterone system, excess sympathetic activity, endothelin, and various cytokines all have been implicated as stimuli of proliferative signaling that contribute to maladaptive cardiac growth. Accordingly, ventricular remodeling, or the structural alterations of the heart in the form of dilatation and hypertrophy (Boxes 10-6 and 10-7), in addition to the counter-regulatory hemodynamic responses, lead to progressive ventricular dysfunction and represent the target of current therapeutic interventions (Figure 10-18).
BOX 10-6 Pathobiology of Left Ventricular Remodeling
From Mann DL: Mechanisms and models in heart failure: An approach. Circulation 100:999–1088, 1999.
BOX 10-7 Mechanical Disadvantage Created by Left Ventricular Remodeling
From Mann DL: Mechanisms and models in heart failure: An approach. Circulation 100:999–1088, 1999.

Pathophysiologic Role of the Renin-Angiotensin System in Heart Failure
The renin-angiotensin system (RAS) is one of several neuroendocrine systems that are activated in patients with HF. The RAS is also an important mediator in the progression of HF. In the short term, the juxtaglomerular cells of the kidney release the proteolytic enzyme, renin, in response to a decrease in BP or renal perfusion (e.g., hemorrhage), generating Ang I from circulating angiotensinogen. ACE cleavage of Ang II from Ang I in the lung produces circulating Ang II. Acutely, Ang II acts as a potent arteriolar and venous vasoconstrictor to return BP and filling pressure to baseline, respectively. Ang II also stimulates the release of aldosterone from the adrenal cortex and antidiuretic hormone (ADH) from the posterior pituitary. Both contribute to increases in blood volume through their effects on the kidney to promote salt and water reabsorption, respectively. In the long term, increases in Ang II lead to sodium and fluid retention, and increases in SVR, which contribute to symptoms of HF, pulmonary congestion, and hemodynamic decompensation (Figure 10-19).
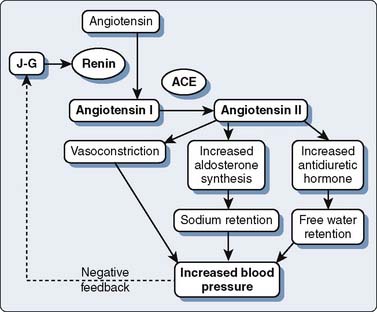
Figure 10-19 Basic pathway of the renin-angiotensin-aldosterone system. ACE, angiotensin-converting enzyme.
(From Jaski BE: Basis of heart failure: A problem solving approach. Boston: Kluwer Academic Publishers, 2000, by permission of Springer Science and Business Media.)
In addition to these cardiorenal and cardiocirculatory effects, most of the hormones and receptors of the RAS are expressed in the myocardium, where they contribute to maladaptive growth or remodeling, key factors in the progression of HF. Increased expression of messenger RNA for angiotensinogen, ACE, and Ang II has been identified in the failing human heart.332 Correspondingly, increased coronary sinus Ang II concentrations were measured in patients with dilated and ischemic cardiomyopathy, signifying a paracrine or autocrine action of the RAS. Moreover, progressive increases in coronary sinus Ang II production correlated with increases in NYHA functional classification of HF.332 Taken together, these data provide evidence that intracardiac RAS is involved in the evolution of the disease process.
The effects of Ang II on its receptors, AT1 and AT2, are well appreciated. The AT1 receptor is involved in several effects that lead to adverse cardiovascular outcomes. Activation of AT1 receptors promotes aldosterone and vasopressin secretion with concomitant increases in salt and water reabsorption through the kidneys, vasoconstriction, catecholamine release, and cell growth and proliferation of cardiovascular tissue (Table 10-14). Stimulation of AT2 receptors, in contrast, results in natriuresis, vasodilation, release of bradykinin and NO, and cell growth inhibition or apoptosis. The Ang II that is formed locally in the heart acts primarily through AT1 receptors located on myocytes and fibroblasts, where it participates in the regulation of cardiac remodeling. Through complex cascades of intracellular signal transduction that activate protein transcription factors within the nucleus, initiating the creation of RNA transcripts, the long-term effects of intracardiac Ang II on the AT1 receptor result in cardiomyocyte hypertrophy, fibroblast proliferation, and extracellular matrix deposition333 (Figure 10-20). These processes contribute to progressive LV remodeling and LV dysfunction characteristic of HF.
TABLE 10-14 Cellular and Physiologic Effects of AT1 and AT2 Stimulation That Lead to Tissue Remodeling
| AT1 Receptor | AT2 Receptor |
|---|---|
| Vasoconstriction Cell growth and proliferation Positive inotropy Aldosterone secretion Catecholamine release |
Vasodilation Cell growth inhibition/apoptosis Negative chronotropy Natriuresis Bradykinin release |
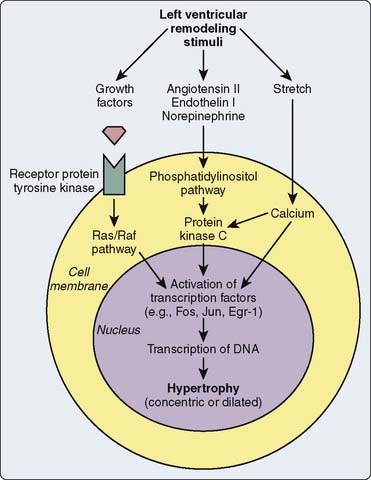
Angiotensin-Converting Enzyme Inhibitors
Clinical Evidence
Evidence supporting the beneficial use of ACE inhibitors in HF patients comes from various randomized, placebo-controlled clinical trials (Table 10-15). Initially, this class of drugs was evaluated for treatment of symptomatic HF (Studies Of Left Ventricular Dysfunction [SOLVD], Vasodilator-Heart Failure Trial [V-HeFT], Cooperative North Scandinavian Enalapril Survival Study [CONSENSUS]). Patients with NYHA Class II to IV HF treated with ACE inhibitors had reductions in mortality rate ranging from 16% to 31%. Subsequently, ACE inhibitors also were found to improve outcome for asymptomatic patients with LV systolic dysfunction in the following categories: patients with EFs less than 35% because of cardiomyopathy,333 patients within 2 weeks after MI with EFs less than 40%,334 and patients presenting within the first 24 hours of MI regardless of EF.335 Recent results from the Heart Outcomes Prevention Evaluation (HOPE) study have further expanded the indications for this class of agents to include asymptomatic, high-risk patients to prevent new-onset HF.336 In patients with diabetes or peripheral vascular disease and an additional atherosclerotic risk factor, but without clinical HF or systolic dysfunction, ramipril (10 mg/day) reduced the HF risk by 23%. More recently, the Prevention of Events with Angiotensin Converting Enzyme (PEACE) trial evaluated the use of angiotensin converting enzyme-inhibitors (ACE-I) in addition to modern conventional therapy in patients with stable CAD with preserved LV function. The results of this trial did not demonstrate any decrease in mortality from cardiovascular causes or nonfatal MI compared with placebo.337 As a result, the ACC/AHA committee has changed the level of recommendation for the use of ACE-I from Class I to Class IIa for patients with Stage A HF.322 Together, these data endorse the use of ACE inhibitors as first-line therapy for a broad spectrum of patients including those with LV systolic dysfunction, with or without symptoms, and in high-risk patients with vascular disease, diabetes, or both, in addition to those with the traditional coronary risk factors. Indeed, the addition of ACE-I in patients with stable CAD, preserved LV systolic function, and at low risk for cardiovascular events also can be useful (Class IIa recommendation). Since the commencement of these trials, the rationale for the use of ACE inhibitors has expanded from a reduction in the progression of clinical HF through ACE inhibitor–mediated vasodilatory action, to acknowledgment that ACE inhibitors also directly affect the cellular mechanisms responsible for progressive myocardial pathology.
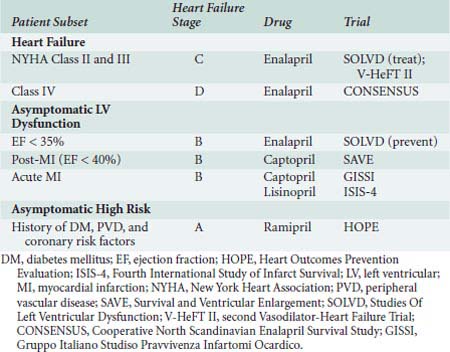
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
10 Cardiovascular Pharmacology
Drug Therapy for Systemic Hypertension
Pharmacotherapy for Acute and Chronic Heart Failure
Pharmacotherapy for Cardiac Arrhythmias
Anti-ischemic drug therapy
Anti-ischemic drug therapy during anesthesia is indicated whenever evidence of myocardial ischemia exists. The treatment of ischemia during anesthesia is complicated by the ongoing stress of surgery, blood loss, concurrent organ ischemia, and the patient’s inability to interact with the anesthesiologist. Nonetheless, the fundamental principles of treatment remain the same as in the unanesthetized state. All events of myocardial ischemia involve an alteration in the oxygen supply/demand balance (Table 10-1). The American College of Cardiology/American Heart Association (ACC/AHA) Guidelines on the Management and Treatment of Patients with Unstable Angina and Non-ST-Segment Elevation Myocardial Infarction provide an excellent framework for the treatment of patients with ongoing myocardial ischemia.1 These guidelines detail the initial evaluation, management, hospital care, and coronary revascularization strategies in the nonanesthetized patient with an acute coronary syndrome. In the anesthetized patient with evidence of myocardial ischemia, initiation of anti-ischemic drug therapy is indicated. This section reviews the common agents used for this purpose (see Chapter 18).
TABLE 10-1 Myocardial Ischemia: Factors Governing O2 Supply and Demand
| O2 Supply | O2 Demand |
|---|---|
| Heart rate* | Heart rate* |
| O2 content | Contractility |
| Hgb, SAT%, Pao2 | Wall tension |
| Coronary blood flow | Afterload |
| CPP = DP − LVEDP* | Preload (LVEDP)* |
| CVR |
CPP, coronary perfusion pressure; CVR, coronary vascular resistance; DP, diastolic blood pressure; Hgb, hemoglobin; LVEDP, left ventricular end-diastolic pressure; SAT%, percent oxygen saturation.
* Affects both supply and demand.
Modified from Royster RL: Intraoperative administration of inotropes in cardiac surgery patients. J Cardiothorac Anesth 6(suppl 5):17, 1990.
Nitroglycerin
Nitroglycerin (NTG) is clinically indicated as initial therapy in nearly all types of myocardial ischemia.2 Chronic exertional angina, de novo angina, unstable angina, Prinzmetal’s angina (vasospasm), and silent ischemia respond to NTG administration.2–6 NTG therapy decreases the incidence of anginal attacks and improves exercise tolerance before angina symptoms.7 During therapy with intravenous (IV) NTG, if blood pressure (BP) declines and ischemia is not relieved, the addition of phenylephrine will allow coronary perfusion pressure (CPP) to be maintained while allowing greater doses of NTG to be used for ischemia relief.8 If reflex increases in heart rate (HR) and contractility occur, combination therapy with β-adrenergic blockers may be indicated to blunt this undesired increase in HR. Combination therapy with nitrates and calcium channel blockers may be an effective anti-ischemic regimen in selected patients; however, excessive hypotension and reflex tachycardia may be a problem, especially when a dihydropyridine calcium antagonist is used.9
Mechanism of Action
NTG enhances myocardial oxygen delivery and reduces myocardial oxygen demand. NTG is a smooth muscle relaxant that causes vasculature dilation. Nitrate-mediated vasodilation occurs with or without intact vascular endothelium.10 Nitrites, organic nitrites, nitroso compounds, and other nitrogen oxide–containing substances (e.g., nitroprusside) enter the smooth muscle cell and are converted to reactive nitric oxide (NO) or S-nitrosothiols, which stimulate guanylate cyclase metabolism to produce cyclic guanosine monophosphate (cGMP)11–13 (Figure 10-1). A cGMP-dependent protein kinase is stimulated with resultant protein phosphorylation in the smooth muscle. This leads to a dephosphorylation of the myosin light chain and smooth muscle relaxation.14,15 Vasodilation is also associated with a reduction of intracellular calcium.16 Sulfhydryl (SH) groups are required for formation of NO and the stimulation of guanylate cyclase. When excessive numbers of SH groups are metabolized by prolonged exposure to NTG, vascular tolerance occurs.17 The addition of N-acetylcysteine, an SH donor, reverses NTG tolerance.18 The mechanism by which NTG compounds are uniquely better venodilators, especially at lower serum concentrations, is unknown but may be related to increased uptake of NTG by veins compared with arteries.19
Physiologic Effects
Two important physiologic effects of NTG are systemic and regional venous dilation (Figure 10-2). Venodilation can markedly reduce venous pressure, venous return to the heart, and cardiac filling pressures. Prominent venodilation occurs at lower doses and does not increase further as the NTG dose increases.20 Venodilation results primarily in pooling of blood in the splanchnic capacitance system.21 Mesenteric blood volume increases as ventricular size, ventricular pressures, and intrapericardial pressure decrease.21
NTG increases the distensibility and conductance of large arteries without changing systemic vascular resistance (SVR) at low doses.22 Improved compliance of the large arteries does not necessarily imply afterload reduction. At greater doses, NTG dilates smaller arterioles and resistance vessels, reducing afterload and BP23 (see Figure 10-2). Reductions in cardiac dimension and pressure reduce myocardial oxygen consumption (MVO2) and improve myocardial ischemia24 (Figure 10-3). NTG may preferentially reduce cardiac preload, while maintaining systemic perfusion pressure, an important hemodynamic effect in myocardial ischemia. However, in hypovolemic states, greater doses of NTG may markedly reduce systemic BP to dangerous levels. A reflex increase in HR may occur at arterial vasodilating doses.
NTG causes vasodilation of pulmonary arteries and veins and predictably decreases right atrial (RAP), pulmonary artery (PAP), and pulmonary capillary wedge pressures (PCWP).23 Pulmonary artery hypertension may be reduced in various disease states and in congenital heart disease with NTG.25,26 Renal arteries, cerebral arteries, and cutaneous vessels also dilate with NTG.27 Blood flow to the kidney and brain may decrease if adequate renal and cerebral perfusion pressures are not maintained.
NTG has several important effects on the coronary circulation (Box 10-1). NTG is a potent epicardial coronary artery vasodilator in both normal and diseased vessels. Stenotic lesions dilate with NTG, reducing the resistance to coronary blood flow (CBF) and improving myocardial ischemia.28,29 Smaller coronary arteries may dilate relatively more than larger coronary vessels; however, the degree of dilation may depend on the baseline tone of the vessel.30 NTG effectively reverses or prevents coronary artery vasospasm.31
BOX 10-1 Effects of Nitroglycerin and Organic Nitrates on the Coronary Circulation
(Modified from Abrams J: Hemodynamic effects of nitroglycerin and long-acting nitrates. Am HeartJ 110[pt 2]:216, 1985.)
Total CBF may initially increase but eventually decreases with NTG despite coronary vasodilation32 (Figure 10-4). Autoregulatory mechanisms probably result in decreases in total flow as a result of reductions in wall tension and myocardial oxygen consumption.23 However, regional myocardial blood flow may improve by vasodilation of intercoronary collateral vessels or reduction of subendocardial compressive forces33 (Figure 10-5). Coronary arteriographic studies in humans demonstrate that coronary collateral vessels increase in size after NTG administration.34 This effect may be especially important when epicardial vessels have subtotal or total occlusive disease.35 Improvement in collateral flow also may be protective in situations in which coronary artery steal may occur with other potent coronary vasodilator agents. The improvement in blood flow to the subendocardium, the most vulnerable area to the development of ischemia, is secondary to both improvement in collateral flow and reductions in left ventricular end-diastolic pressure (LVEDP), which reduce subendocardial resistance to blood flow.36 With the maintenance of an adequate CPP (e.g., with administration of phenylephrine), NTG can maximize subendocardial blood flow8 (see Figures 10-4 and 10-5). The ratio of endocardial to epicardial blood in transmural segments is enhanced with NTG.36 Inhibition of platelet aggregation also occurs with NTG; however, the clinical significance of this action is unknown.37
Pharmacology
Organic nitrates are biotransformed by reduction hydrolysis catalyzed by the hepatic enzyme glutathione-organic nitrate reductase.15 The rate of hepatic denitrification is characteristic of each nitrate and is further dependent on hepatic blood flow or presence of hepatic disease.15 Common organic nitrates for clinical use are shown in Table 10-2.
Sublingual Nitroglycerin
Sublingual NTG (0.15- to 0.6-mg tablets) achieves blood levels adequate to cause hemodynamic changes within several minutes; physiologic effects last 30 to 45 minutes.38 Sublingual bioavailability is approximately 80% and bypasses the high first-pass biodegradation in the liver (90%) by nitrate reductase to glycerol dinitrate and nitrite, which are excreted renally. Plasma half-life of sublingual NTG is 4 to 7 minutes. NTG spray has pharmacokinetics and pharmacodynamics equivalent to those of a 0.4-mg sublingual tablet; however, it has a longer shelf half-life compared with the tablets, which decompose in air and warm temperatures.39 A tablet that adheres to the buccal area between the upper lip and teeth has rapid onset and has the advantage of longer half-life than sublingual tablets.40 Although NTG is readily absorbed through the gastric mucosa, the high rate of liver metabolism makes oral administration highly unpredictable.
Nitroglycerin Ointment and Patches
NTG ointment (2%) is readily absorbed through the skin, with this method of administration providing longer-lasting effects.41 Adequate NTG blood levels are reached within 20 to 30 minutes, and duration of action is 4 to 6 hours.41 Ointment is administered in inches (15 mg/inch), but the surface area of application and not the amount administered determines the blood level achieved. NTG ointment is messy, requires application four times a day, and is most appropriate for nursing administration in special care units.42
NTG patches contain either liquid NTG or NTG bonded to a polymer gel and slowly released to the skin through a semipermeable membrane.43 The pharmacokinetics approach that of a consistent IV infusion.43 Blood levels are reached within 20 to 30 minutes, and a steady state is reached within 2 hours. Blood levels may be maintained up to 24 hours and are largely determined by patch size. Patches or disks contain an NTG concentration per square centimeter, and dosages of 0.2 to 0.8 mg/hr usually are required for relief of myocardial ischemia. Although convenient for patients, tolerance may be a problem with these sustained-release preparations.41 Intermittent therapy is recommended to avoid tolerance.44
Intravenous Nitroglycerin
NTG has been available since the early 1980s as an injectable drug with stable shelf half-life in a 400-μg/mL solution of D5W (5% dextrose in water). Blood levels are achieved instantaneously, and arterial dilating doses with resulting hypotension may quickly occur. If the volume status of the patient is unknown, initial dosages of 5 to 10 μg/min are recommended. The dosage necessary for relieving myocardial ischemia may vary from patient to patient, but relief is usually achieved with 75 to 150 μg/min. In a clinical study of 20 patients with rest angina, a mean dosage of 72 μg/min reduced or abolished ischemic episodes in 85% of patients.45 However, doses as high as 300 to 400 μg/min may be necessary for ischemic relief in some patients. Arterial dilation becomes clinically apparent at doses around 50 μg/min. Drug offset after discontinuation of an infusion is rapid (2 to 5 minutes). The dosage of NTG available is less when administered in plastic bags and polyvinylchloride tubing because of NTG absorption by the bag and tubing, although this is not a significant clinical problem because the drug is titrated to effect.46
Adverse Effects
The metabolism of NTG by liver nitrate reductase produces a nitrite that oxidizes the ferrous iron of hemoglobin to the ferric form of methemoglobin. The ferric iron does not bind or release oxygen.47 Methemoglobin is formed normally and is reduced by enzyme systems within the red blood cell.48 Normally, methemoglobin levels do not exceed 1%, but may increase when direct oxidants are present in the serum (nitrates, sulfonamides, aniline dye derivates). Methemoglobinemia with levels up to 20% is not a clinical problem. Documented increases in methemoglobin blood levels occur with IV NTG, averaging 1.5% in one study of 50 patients receiving NTG for longer than 48 hours.49 NTG dosages of 5 mg/kg/day orally should be avoided to prevent significant methemoglobinemia.50 However, rare instances of smaller doses causing clinically significant problems have been reported.51 Nitrates are effective in producing methemoglobin to bind cyanide in sodium nitroprusside toxicity.
Several mechanisms of nitrate tolerance have been proposed, including a depletion of SH groups, neurohumoral activation, volume expansion, and/or downregulation of nitrate receptors.52–57 Tolerance may occur with all forms of nitrate administration that maintain continuous blood levels of the drug.17,58–61 Discontinuation of the drug after prolonged exposure may result in a rebound phenomenon, possibly resulting in coronary vasospasm and myocardial ischemia or infarction.62 Tolerance to NTG apparently does not occur in all patients.63 If tolerance develops after prolonged exposure, physiologic responsiveness may be achieved with greater dosages of NTG, an important observation during NTG administration in cardiac surgery.64 Intermittent dosing with a nitrate-free interval each day or night can maintain NTG responsiveness.44,65
NTG interferes with platelet aggregation.66 The ability of the platelet to adhere to damaged intima is reduced.67 Primary and secondary wave aggregation of platelets is also attenuated.68 Previously formed platelet plugs are disaggregated.69 A clinical study of 10 patients with coronary artery disease (CAD) demonstrated that a mean dosage of NTG (1.19 μg/kg/min) inhibited platelet aggregation by 50%, with a return to baseline platelet aggregation 15 minutes after the infusion was discontinued70 (Figure 10-6). NO production increases cGMP, which modulates intracellular platelet calcium and reduces platelet secretion of proaggregatory factors.71 The clinical significance of these actions remains unclear. As with other potent vasodilators, NTG may increase intrapulmonary shunting of blood and reduce arterial oxygen tension.
NTG may induce resistance to the anticoagulant effects of heparin.72 During simultaneous infusions of NTG and heparin, an increase in the NTG infusion caused the activated partial thromboplastin time to decrease.73 Becker et al74 reported NTG-induced heparin resistance at NTG infusion rates greater than 350 μg/min. The authors suggested a qualitative problem with antithrombin III (AT III) because AT III levels did not decrease. Others have suggested that NTG interferes with AT III binding to heparin by N-desulfation of the heparin molecule at the AT III binding sites.75 N-desulfation of heparin reduces its anticoagulant activity.76
NTG is contraindicated in patients who have used sildenafil, vardenafil, or tadalafil, or in patients who are hypotensive. These drugs for erectile dysfunction inhibit the phosphodiesterase (PDE5) that degrades cGMP, and the cGMP mediates vascular smooth muscle relaxation by NO. NTG-mediated vasodilation is markedly enhanced and prolonged, resulting in cases of profound hypotension, myocardial infarction (MI), and death.77 Small doses of NTG have been used, but the amount of time that must elapse after a patient’s last dose of one of these medications before regular doses of nitrates may be safely administered is unclear.78–80
Summary
NTG remains a first-line agent for the treatment of myocardial ischemia. Special care must be taken in patients with signs of hypovolemia or hypotension because the vasodilating effects of the drug may worsen the clinical condition. Recent ACC/AHA Guidelines address the prophylactic intraoperative use of NTG and suggest that its usefulness in preventing myocardial ischemia and cardiac morbidity in high-risk patients undergoing noncardiac surgery is unclear.81
β-Adrenergic Blockers
β-Adrenergic blockers have multiple favorable effects in treating the ischemic heart during anesthesia (Box 10-2). β-Adrenergic blockers reduce oxygen consumption by decreasing HR, BP, and myocardial contractility. HR reduction increases diastolic CBF. Increased collateral blood flow and redistribution of blood to ischemic areas may occur with β-blockers. More free fatty acids may be available for substrate consumption by the myocardium. Microcirculatory oxygen delivery improves, and oxygen dissociates more easily from hemoglobin after β-adrenergic blockade. Platelet aggregation is inhibited. β-Blockers should be started early in patients with ischemia in the absence of contraindications.1 Many patients at high risk for perioperative cardiac morbidity should be started on β-blockers before surgery and continued for up to 30 days after surgery.82–84 The choice of which β-blocker for any individual patient is based on clinician familiarity and desired pharmacologic profile. There is no evidence that one specific agent is superior to another; however, β-blockers without intrinsic sympathomimetic activity (ISA) are preferable when treating acute myocardial ischemia.
β-Blockers administered during MI reduce myocardial infarct size.85 In addition, a reduction in morbidity has been shown to occur with acute IV metoprolol during MI.85 Similar findings with reductions in mortality extending up to 3 years after MI have been shown in numerous trials with β-adrenergic blockers86,87 (Figure 10-7). The mechanisms for mortality reduction are unclear. In the absence of contraindications, β-blockers should be a routine part of care in patients with all forms of CAD, including unstable angina and recent MI.
Data confirm the important role of β-blockade in treating patients after acute MI and in reducing mortality in high-risk populations. Immediate β-blockade after thrombolytic therapy in patients with acute MI significantly decreased recurrent early myocardial ischemia and reinfarction.88 Early β-blockade is indicated in the treatment of MI89,90 (Box 10-3). In fact, β-blocker therapy after MI may be greatly underused in patients older than 65 years.91 Atenolol has been found to reduce ischemia and adverse outcome in patients with mildly symptomatic ischemia.92 Multiple studies have shown that perioperative administration of β-adrenergic blockers reduces both mortality and morbidity when given to patients at high risk for CAD who must undergo noncardiac surgery82–84,93,94 (Figures 10-8 to 10-10). These data suggest that intermediate- and high-risk patients presenting for noncardiac surgery should receive perioperative β-adrenergic blockade to reduce postoperative cardiac mortality and morbidity. However, in the Perioperative Ischemic Evaluation Study (POISE) trial, the use of higher dose metoprolol started in patients on the day of noncardiac surgery was associated with increased risk for severe stroke and greater total mortality.95 These findings have led to increased scrutiny of perioperative β-blockade usage. Recent ACC/AHA recommendations on the perioperative use of β-adrenergic blockade for noncardiac surgery are given in Box 10-4.81
BOX 10-3 ACC/AHA Guidelines for Early Use of β-Adrenoceptor Blocking Agents after Stemi
Reproduced from Antman EM, Hand M, Armstrong PW, et al: 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Group to Review New Evidence and Update the ACC/AHA 2004 Guidelines for the Management of Patients with ST-Elevation Myocardial Infarction). Circulation 117:296, 2008, by permission.90
Early Therapy
Class I*
Class IIa:
Class III
* Risk factors for cardiogenic shock (the greater the number of risk factors present, the higher the risk of developing cardiogenic shock) are age older than 70 years, systolic blood pressure less than 120 mm Hg, sinus tachycardia greater than 110 bpm or heart rate less than 60 bpm, and increased time since onset of symptoms of STEMI.
BOX 10-4 Recommendations for Perioperative β-Blocker Therapy
Adapted from Fleisher LA, Beckman JA, Brown KA, et al: 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 120:e169, 2009, by permission.
Class IIa
Class IIb
Class III
* Clinical risk factors include history of ischemic heart disease, history of compensated or prior heart failure, history of cerebrovascular disease, diabetes mellitus, and renal insufficiency (defined in the Revised Cardiac Risk Index as a preoperative serum creatinine concentration greater than 2 mg/dL).
β Receptor
The β receptor was conceptualized by Ahlquist,96 who divided various physiologic effects of catecholamine stimulation into α and β responses. The β receptor has been identified biochemically as a polypeptide chain of approximately 50,000 to 60,000 kDa.97 The receptor’s structure is common to most receptor proteins that have been identified: seven transmembrane crossings with two extramembranous terminal ends98 (Figure 10-11). All receptors that transduce a signal through G proteins share this basic structure.99 There are three extracellular and intracellular loops connecting the intramembranous portion of the receptor.98 Agonist-antagonist binding occurs at the intramembranous portion, whereas the intracellular loops modulate interaction with the G-protein complex.100,101 The terminal intracellular end contains amino acid residues that undergo phosphorylation, which relates to desensitization and downregulation of the receptor.102
Receptor stimulation activates a G protein, which stimulates adenylyl cyclase. The G-protein complex is composed of both stimulatory (Gs) and inhibitory (Gi) intermediary proteins.99 Adenylyl cyclase converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP), which phosphorylates a protein kinase and produces the appropriate cellular response. A typical cascade of this sequence leading to increases in myocardial contractility from β-receptor stimulation is illustrated in Figure 10-12.
β-Receptor numbers in any tissue may decrease with chronic stimulation (downregulation) or increase with chronic blockade (upregulation). The process of desensitization of the adrenergic response in chronic stimulation (i.e., congestive heart failure [CHF]) may involve downregulation of the receptors but may involve either the G-protein complex or adenylyl cyclase. Desensitization may occur quickly, whereas downregulation with actual internalization of the receptor within the cell may take days to weeks.103 Myocardial ischemia increases β-receptor density, although it remains controversial whether this upregulation results in greater adrenergic response.104 Several studies have demonstrated that high-affinity β receptors in nonischemic tissue were shifted to a low-affinity state during ischemia.105,106 Also, the levels of Gs and its activity are reduced during myocardial ischemia.107 However, stimulation of these receptors with isoproterenol during ischemia does result in increases in cAMP production.104,108
There are two types of β receptors with a multitude of responses109 (Table 10-3). Both β1– and β2-receptor stimulation primarily involve cardiac function (Figure 10-13). Responses of isolated human atrial tissue demonstrated greater inotropic response to β1– than to β2-receptor stimulation.110 Endogenous norepinephrine produces inotropic responses in human atrial appendages and ventricular papillary muscle by β1-receptor stimulation, whereas epinephrine produces its maximal inotropic effects on the atria by β2-receptor stimulation and up to 50% of its maximal inotropic response in the ventricle by β2-receptor stimulation.111,112 Sinus node, atrioventricular (AV) node, the left and right bundle branches, and the Purkinje system contain higher densities of β2 receptors.113 Clearly, both receptor subtypes have cardiac inotropic, chronotropic, and dromotropic properties.
TABLE 10-3 Physiologic Effects of β1– and β2-Receptor Stimulation
| Physiologic Effect | β1 Response | β2 Response |
|---|---|---|
| Cardiac | ||
| Increased heart rate | ++ | ++ |
| Increased contractility | ||
| Atrium | + | ++ |
| Ventricle | ++ | ++ |
| Increased automaticity and conduction velocity | ||
| Nodal tissue | ++ | ++ |
| His-Purkinje | ++ | ++ |
| Arterial relaxation |
