Chapter 3 Cardiovascular Pharmacology
In this chapter the pharmacology of cardiovascular drugs that are used in the intensive care unit (ICU) is reviewed. Specific indications for particular drugs are discussed in other relevant chapters. Guidelines for the reintroduction of medications following routine cardiac surgery are provided in Chapter 17.
INOTROPES AND VASOPRESSORS
Infusions of vasoactive drugs are prescribed in different ways in different institutions. Three common methods are micrograms per kilogram per minute (μg/kg/min), micrograms per minute (μg/min), and milligrams per hour (mg/hr). In this book μg/kg/min is used. A conversion among the methods is provided in Appendix 1.
Sympathomimetics
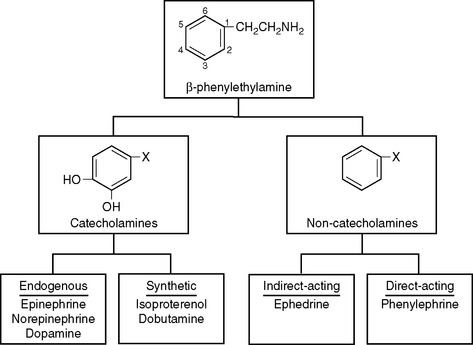
All sympathomimetics are derived from β-phenylethylamine. The presence of hydroxyl groups on the 3- and 4-carbons in the benzene ring designates a compound as a catecholamine, which may be endogenous or synthetic (Fig. 3-1). The noncatecholamine sympathomimetics include a diverse range of drugs, such as the asthma medication albuterol and the central nervous system stimulant amphetamine. Two commonly used vasoactive noncatecholamine sympathomimetics are ephedrine and phenylephrine.
Mechanism of Action
Sympathomimetics bind to and stimulate adrenergic receptors that are located on cell membranes. In 1948, Alquist described two adrenergic receptor subtypes, alpha (α) and beta (β), based on their relative responsiveness to norepinephrine, epinephrine, and isoproterenol.1 In the 1970s this classification was refined to include α1, α2, β1, and β2 receptor subtypes. Subsequently, further divisions of each receptor subtype have been discovered, but clinically useful drugs to exploit these expanded classifications have not been developed.
Adrenergic receptors are part of a family of receptors known as G protein coupled receptors. Receptor stimulation by an agonist (see Chapter 4) facilitates the binding of the nucleotide guanosine triphosphate to a G protein, which activates it. The activated G protein then stimulates or inhibits one of a number of second messenger systems. Two second messenger systems mediate the actions of adrenergic receptors:
β Receptors.
β2 Receptors are found on the heart and throughout the vasculature, particularly the arterioles of skeletal muscle, the coronary circulation, and the liver. Their stimulation leads to vasodilation and enhanced diastolic relaxation (lusitropy). Outside of the cardiovascular system, β2 receptor activation causes bronchodilation, uterine and bladder relaxation, and decreased gastrointestinal motility.
Individual Sympathomimetics
Norepinephrine.
Norepinephrine causes potent stimulation at α and β1 receptors, but unlike epinephrine, it has minimal effect at β2 receptors. Blood pressure is reliably increased but the effect on cardiac output is variable. Although β1 receptor stimulation has a direct inotropic effect, in the setting of hypovolemia or impaired ventricular function, increased left ventricular afterload due to α1 receptor stimulation can cause cardiac output to fall. Similarly, the effect on heart rate is variable: direct β1 stimulation has a chronotropic effect but increased blood pressure can cause baroreceptor-mediated bradycardia.
Norepinephrine is useful following cardiac surgery to counter the vasodilatory effects of cardiopulmonary bypass and sedation. However, doses above 0.05 to 0.1 μg/kg/min should be avoided in patients with impaired ventricular function unless cardiac output is being measured. Norepinephrine is commonly combined with an inodilator such as dobutamine or milrinone. Norepinephrine is typically commenced at a dose of 0.01 to 0.05 μg/kg/min and titrated to blood pressure. There is no maximum dose, but infusions greater than 0.1 to 0.2 μg/kg/min are rarely needed in cardiac surgery patients except in the presence of vasoplegic syndrome (Chapter 2) or septic shock, in which case doses as high as 0.5 to 1 μg/kg/min may be required. Troublesome metabolic effects, particularly lactic acidosis, are much less common with norepinephrine than with epinephrine.2
Dopamine.
Dopamine is a precursor to norepinephrine and is itself an important neurotransmitter in the peripheral and central nervous systems. Dopamine stimulates α and β receptors and type 1 and 2 dopamine (DA) receptors. DA-1 receptors are found in the renal, mesenteric, and cerebral circulations,3 and their stimulation results in vasodilation. DA-1 receptors are also found in the renal tubule, where they mediate natriuresis. DA-2 receptors are analogous to α2 receptors in that they are found presynaptically and inhibit the release of norepinephrine. Dopamine also has an indirect mechanism of action.
At low doses (<3 μg/kg/min), dopaminergic effects predominate. At higher doses, initially β receptor effects predominate; then α receptor effects predominate. The widely accepted dose range is 3 to 10 μg/kg/min for β effects and more than 10 μg/kg/min for α effects. However, these dose ranges must be viewed with skepticism. There is huge individual variability in the pharmacokinetics of dopamine such that dramatically different plasma concentrations may occur in different patients who are receiving the same dose.4 Furthermore, the clinical effects of a given plasma concentration are dependent on the functional activity of the adrenergic receptors. β Receptors are desensitized in a variety of clinical settings, including after cardiac surgery and with heart failure.5–7 Because of its indirect action, dopamine has reduced effectiveness in patients with heart failure or shock. Despite these caveats, it is generally true that as the dose of dopamine increases, there is a progressive increase in blood pressure and heart rate.
Dopamine at a dose of 1 to 3 μg/kg/min has been termed “renal-dose dopamine” and has traditionally been used to provide selective renal vasodilation in patients at risk for renal dysfunction. However, it is now clear that although low-dose dopamine may increase blood flow to the renal cortex, blood flow to the renal medulla may actually decrease.8 Given the relatively hypoxic environment of the renal medulla under normal circumstances (Chapter 1), this effect is potentially harmful. Furthermore, the increase in urine output that occurs with low-dose dopamine is due primarily to a direct tubular natriuretic effect rather than to renal vasodilation. In a well-conducted, large, randomized trial, low-dose dopamine did not reduce the incidence of acute renal failure in patients with early renal dysfunction.9 Dopamine has a number of other potentially detrimental effects, including inhibition of hypoxic ventilatory drive, impairment of ventilation-perfusion matching in the lung, and suppression of the secretion of some anterior pituitary hormones, such as prolactin, growth hormone, and thyrotropin.8
Dobutamine.
Dobutamine is useful for treating low cardiac output following cardiac surgery,10 and it may be used in combination with norepinephrine for treating septic shock.11 The dose range is 1 to 10 μg/kg/min. At higher doses tachyarrhythmias become common. Metabolic side effects are minimal.
Phenylephrine.
Phenylephrine is a noncatecholamine, direct-acting α receptor agonist that does not possess any significant β receptor activity. Bolus doses of 50 to 100 μg are commonly used during induction of anesthesia in patients with aortic stenosis to counteract the vasodilation produced by anesthetic drugs. In this situation phenylephrine has the advantage of increasing coronary perfusion pressure without increasing heart rate. Cardiac output may fall due to increased afterload and baroreceptor-mediated reflex bradycardia. Phenylephrine has a slightly longer duration of action than norepinephrine. The drug may be administered as a continuous infusion at a dose of 0.1 to 1 μg/kg/min.
Phosphodiesterase Type III Inhibitors
The phosphodiesterases (PDEs) are a family of enzymes that catalyze the breakdown of cyclic nucleotides, including cAMP and cGMP. There are multiple subtypes of PDE that have varying tissue distributions and actions.12 Caffeine and theophylline are nonspecific PDE inhibitors that are used as bronchodilators. Papaverine is a vasodilator and nonspecific PDE inhibitor that is used by cardiac surgeons during coronary artery bypass graft (CABG) surgery to prevent spasm of the internal mammary artery.
Drugs that selectively inhibit PDE subtype III function as inodilators. The commercially available PDE-III inhibitors all have a similar pharmacologic profile: they increase contractility and cause pulmonary and systemic (arteriolar and venous) vasodilation. As such, PDE-III inhibitors are useful for treating low cardiac output, particularly in the presence of pulmonary edema or pulmonary hypertension. They are potent vasodilators of coronary grafts13 and cause less tachycardia and atrial fibrillation than dobutamine.10 The inotropic effect is independent of the β1 receptor, which is advantageous in patients with β1 receptor desensitization (see earlier discussion). Also, by combining a PDE-III inhibitor with a β1 receptor agonist, a dual mechanism of action is exploited. Arteriolar and venous dilation can cause modest hypotension. This can be treated with either fluid or low-dose norepinephrine, depending on the status of the patient’s intravascular volume. In patients with cardiogenic shock, the combination of a PDE-III and norepinephrine provides support for both cardiac output and blood pressure, but without the troublesome tachycardia and metabolic disturbance that can occur with epinephrine.
PDE-III inhibitors are available as intravenous formulations for short-term use. Unlike the sympathomimetics, they have durations of action measured in hours, so their effects cannot be readily judged. Oral formulations of PDE-III inhibitors for the treatment of chronic heart failure have been studied but have resulted in higher mortality rates.14
Miscellaneous Vasoactive Drugs
Calcium.
An intravenous bolus dose of calcium chloride of 5 mg/kg (or 0.035 mmol/kg) increases blood pressure but has little effect on myocardial contractility.15 The duration of effect following a bolus dose is 5 to 10 minutes. The pressor effect is much more pronounced in the presence of hypocalcemia. Calcium chloride does not improve outcome after cardiac arrest16 and is no longer included in routine resuscitation protocols. However, calcium is useful in the management of hyperkalemia because it reduces potassium-induced arrhythmias, and in the treatment of hypocalcemia.
Vasopressin.
Vasopressin (V; antidiuretic hormone) is a peptide hormone released from the posterior pituitary in response to an increase in serum osmolarity or hypovolemia (Chapters 1 and Chapter 32). Stimulation of V1 receptors within vascular smooth muscle results in vasoconstriction (via the IP3/DAG second-messenger system), whereas stimulation of V2 receptors in the kidney results in water retention (via the cAMP second-messenger system). An additional action of vasopressin is to increase the release of the von Willebrand factor from the vascular endothelium, which increases platelet aggregation. The elimination half-time of vasopressin is 10 to 30 minutes.
Exogenously administered vasopressin is used to treat catecholamine-resistant vasodilatory shock. In early shock, endogenous stores of vasopressin are released from the posterior pituitary, but as shock progresses, these stores become depleted. In patients with advanced vasodilatory shock, vasopressin infusion at 4 units/hr combined with norepinephrine has been shown to be superior to norepinephrine alone in terms of hemodynamics and markers of splanchnic perfusion.17 However, higher doses of vasopressin, sufficient to replace rather than augment norepinephrine, can cause a marked reduction in cardiac output and may worsen splanchnic perfusion.18 In animal models, vasopressin is associated with relatively less vasoconstriction within the coronary, cerebral, and pulmonary circulations than is associated with catecholamines.19,20 Despite an antidiuretic effect, in patients with vasodilatory shock urine output may actually improve with vasopressin,21 presumably due to an improvement in renal blood flow. As with all vasoconstrictors, precipitous reductions in cardiac output can occur with vasopressin, particularly in the settings of hypovolemia and impaired ventricular function. Current recommendations are that vasopressin, in a dose of 0.01 to 0.04 units/min, should be considered in patients with vasodilatory shock who have adequate volume resuscitation and are refractory to high doses of catecholamines.22
Levosimendan.
Levosimendan is an inotropic drug that acts by stabilizing the interaction between calcium and tropo-nin, so-called calcium sensitization. In addition, levosimendan functions as a vasodilator, partly through PDE-III inhibition and partly by opening adenosine triphosphate gated potassium channels. Because the effect of levosimendan is not mediated by β1 receptors or increased cAMP, it does not increase myocardial oxygen consumption and is not arrhythmogenic. Levosimendan may be a particularly useful inotropic agent in patients who are receiving β blockers. In the treatment of heart failure, levosimendan is more effective than dobutamine in improving cardiac output and, in one study, was associated with better mortality compared to dobutamine.23
Levosimendan has an elimination half-time of 1 hour, but it has pharmacologically active metabolites with elimination half-times of up to 80 hours. The drug is usually administered as a continuous infusion of 0.1 to 0.2 μg/kg/min for 24 hours, which may be preceded by a loading dose of 12 to 24 μg/kg. Following a 24-hour infusion, a positive inotropic effect is sustained for several days. Side effects are typically modest and include mild tachycardia and hypotension. So far, levosimendan has been used primarily in patients with decompensated heart failure, but there is some limited experience in its use in patients after cardiac surgery.24
β Blockers
β Blockers tend to be either lipid-soluble (e.g., metoprolol) or water-soluble (e.g., atenolol). Lipid-soluble agents typically undergo extensive hepatic metabolism and have a low oral bioavailability. Some lipid-soluble β blockers are metabolized by the cytochrome P-450 (CYP) 2D6 enzyme system, so their metabolism is susceptible to inhibition by other drugs (see Table 4-3). In contrast, water-soluble agents have high oral bioavailability and tend to be eliminated unchanged by the kidney. In patients with hepatic impairment, a water-soluble β blocker is appropriate, whereas in patients with renal impairment, a lipid-soluble β blocker may be more appropriate.
β Blockers have antihypertensive, antiarrhythmic, and antiischemic actions, and they inhibit ventricular remodeling. Treatment with β blockers is associated with reduced mortality rates in patients with coronary artery disease25–28 and chronic heart failure (see Chapter 19) and in high-risk patients undergoing noncardiac surgery.29 In patients undergoing CABG surgery, preoperative treatment with β blockers is associated with reduced perioperative mortality rates.30 Acute cessation of chronic β blocker treatment can precipitate myocardial ischemia.31
In the cardiothoracic ICU, β blockers are used in the following circumstances:
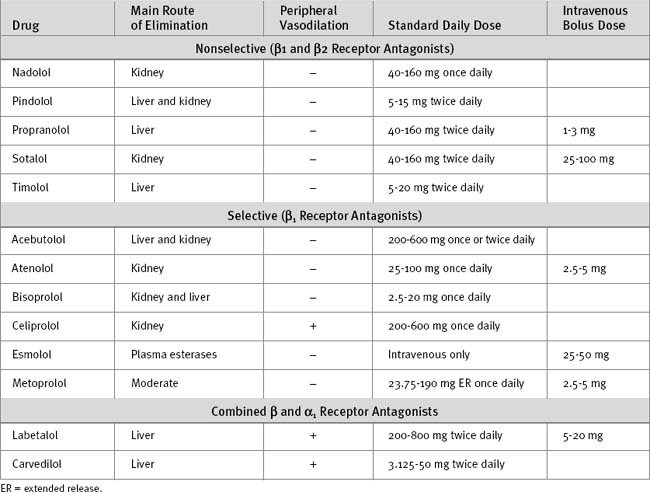
In patients with ventricular dysfunction, introduction of a β blocker may initially worsen the symptoms of heart failure. Ventricular remodeling and improved ejection fraction develop slowly over several months. Thus, β blockers should be avoided in patients who have only recently discontinued inotropic support or who are fluidoverloaded. Introduction of β blockers for the treatment of heart failure is generally not appropriate in the ICU. Characteristics of commonly used β blockers are listed in Table 3-2.
Labetalol.
Labetalol is an α1 blocker and a nonselective β blocker that is formulated for intravenous and oral use. Labetalol is a useful drug for the treatment of postoperative hypertension. Intravenously, labetalol may be administered as repeated bolus doses of 5 to 10 mg. Labetalol undergoes significant hepatic metabolism and has low oral bioavailability; thus, the oral dose is much higher than the intravenous dose.
Carvedilol.
As with labetalol, carvedilol has both α1 receptor and nonselective β blocking properties. In addition, carvedilol has unique antiproliferative and antioxidant properties.32 Carvedilol is widely used in treating chronic heart failure and patients with impaired left ventricular function following myocardial infarction. The starting dose for patients with heart failure is 3.125 to 6.25 mg twice daily; the dose is increased every 2 weeks. The target maintenance dose is at least 25 mg twice daily.32
Nitrates
Nitroglycerin.
Nitroglycerin is a very short-acting organic nitrate that is available in several formulations. As a sublingual spray or tablet, nitroglycerin is used in treating acute angina. As a transdermal patch or topical ointment, nitroglycerin is used in the prevention of angina and the treatment of congestive cardiac failure. In the ICU, nitroglycerin is typically administered as a continuous intravenous infusion for the:
The dose range is 0.25 to 5 μg/kg/min, and the clinical effect dissipates within a few minutes after the infusion is stopped. At lower doses (<2 μg/kg/min) the main effect is dilation of veins, coronary arteries, and pulmonary arterioles. Thus, low-dose nitroglycerin is useful in the treatment of myocardial ischemia, pulmonary hypertension, and congestive cardiac failure. By selectively dilating large coronary arteries, nitroglycerin, unlike sodium nitroprusside, does not cause coronary steal. At higher doses, systemic vasodilation becomes more prominent,33 which can result in hypotension and cause paradoxical worsening of myocardial ischemia. However, in patients with severe hypertension, nitroglycerin is often ineffective in controlling blood pressure.
Calcium Channel Blockers
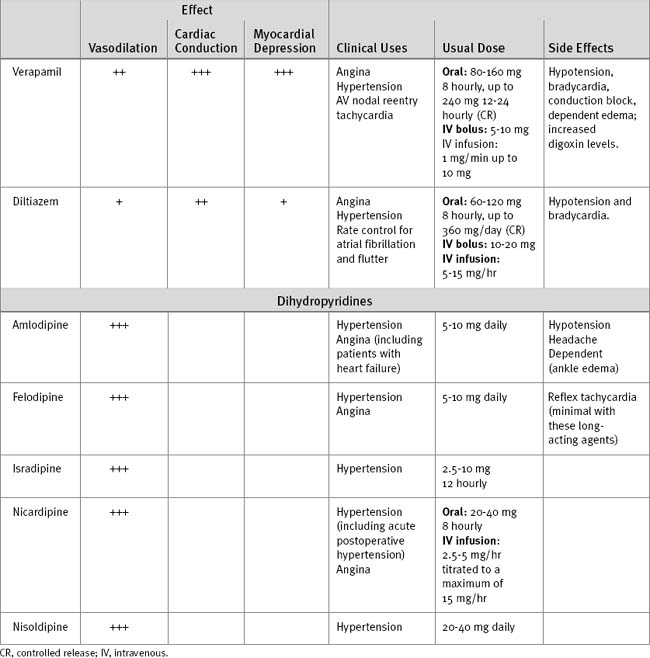
Calcium channel blockers reduce the intracellular calcium concentration within vascular smooth muscle and myocytes, causing, to a variable degree, inhibition of cardiac conduction, reduced myocardial contractility, and arteriolar vasodilation. All agents are available orally. In addition, diltiazem, verapamil, nifedipine, and nicardipine are available as intravenous formulations. Some calcium antagonists undergo hepatic metabolism by the CYP3A enzyme system, and therefore their metabolism may be enhanced or inhibited by other drugs (see Table 4-3). Calcium channel blockers are widely used in treating angina, hypertension, and cardiac arrhythmias. In the ICU, calcium channel blockers are indicated for treatment of postoperative hypertension, for preventing spasm within coronary artery bypass grafts, and for treating cardiac arrhythmias.
The main side effects of chronically administered dihydropyridine calcium channel blockers are headache and dependent (ankle) edema; reflex tachycardia can occur but is usually modest with the long-acting agents. Short-acting formulations such as the original capsule form of nifedipine cause rapid reflex adrenergic activation and are associated with increased mortality rates in patients with myocardial infarction.34 For this reason, short-acting formulations are no longer used.
Two other types of calcium channel blockers are in clinical use: verapamil, a phenylalkylamine; and diltiazem, a benzothiapine. Besides causing vasodilation, these drugs have effects on myocardial contractility and cardiac conduction. Diltiazem is discussed later under the heading Antiarrhythmic Drugs. The characteristics of selected calcium channel blockers are provided in Table 3-3.
Miscellaneous Vasodilators
Hydralazine.
Hydralazine is an arteriolar vasodilator that is available for the acute control of hypertension. The intravenous dose is 2.5 to 10 mg, with an onset of action within 15 minutes and a duration of action of 2 to 4 hours. Reflex tachycardia is common but may be prevented by coadministration of a β blocker. Hydralazine is available for oral use but is rarely given by this route because of problems involving reflex tachycardia, sodium and water retention, metabolic variability, and immunologic reactions.
Fenoldopam.
Fenoldopam is a specific DA-1 receptor agonist that causes natriuresis and vasodilation within mesenteric, renal, coronary, and cerebral circulations. For the treatment of hypertension, doses of 0.1 to 1.6 μg/kg/min have been studied.35 The initial dose should be low (0.1 to 0.2 μg/kg/min) and slowly increased, as this results in less reflex tachycardia than starting at higher doses. The clinical effect dissipates within a few minutes of stopping an infusion. Fenoldopam has an efficacy similar to that of nitroprusside in the treatment of hypertension following cardiac surgery,36,37 but it is significantly more expensive. Fenoldopam has been investigated as a renal protective agent in cardiac surgery patients. The results of these trials have been conflicting38,39 and, on balance, current evidence does not support the use of fenoldopam for this purpose.
Nesiritide.
Nesiritide is a synthetic analog of brain (B-type) natriuretic peptide (see Chapter 1). It acts via the cGMP second-messenger system, causing vasodilation of veins, coronary arteries and, to a lesser extent, systemic arterioles.40 It also has a mild natriuretic effect. In the treatment of decompensated heart failure, nesiritide is more effective than nitroglycerin in reducing pulmonary capillary wedge pressure and improving dyspnea.41 Side effects are few and include mild hypotension. Nesiritide is administered as a loading dose of 2 μg/kg followed by an infusion of 0.01 μg/kg/min. Currently, there is only limited experience with nesiritide in cardiac surgery patients.42
Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers
Angiotensin-converting Enzyme Inhibitors.
ACE inhibitors are indicated for a range of cardiovascular disorders, including hypertension, coronary artery disease,43,44 and asymptomatic45 and symptomatic left ventricular dysfunction.46,47 ACE inhibitors also improve survival rates in patients with risk factors for, but without documented evidence of, coronary artery disease.44
ACE inhibitors are useful in treating postoperative hypertension, particularly in patients receiving ACE inhibitors preoperatively. However, this class of drug must be used very carefully in the ICU, especially in patients with preexisting renal dysfunction or perioperative hemodynamic instability. The initial dose of ACE inhibitor should be low (e.g., enalapril 1 to 2 mg/day) and only slowly increased. In patients with impaired ventricular function or renal impairment, reintroduction of ACE inhibitor therapy should be delayed for 1 to 2 days after surgery.48 The doses of various ACE inhibitors are listed in Table 3-4.
Table 3-4 Dosages of Selected ACE Inhibitors and Angiotensin Receptor Blockers
| Drug | Initial Dose (mg) | Maintenance Dose |
|---|---|---|
| ACE Inhibitors | ||
| Captopril | 6.25 mg three times daily | 25-50 mg three times daily |
| Enalapril | 2.5 mg daily | 10-20 mg daily or twice daily |
| Lisinopril | 2.5 or 5 mg daily | 10-40 mg daily |
| Quinapril | 2.5 or 5 mg daily | 10-40 mg daily |
| Perindopril | 2 mg daily | 4-8 mg daily |
| Ramipril | 1.25 or 2.5 mg daily | 2.5-10 mg daily |
| Cilazapril | 0.5 mg daily | 1-5 mg daily |
| Fosinopril | 10 mg daily | 20-40 mg daily |
| Angiotensin Receptor-Blocking Drugs | ||
| Losartan | 25 or 50 mg once daily | 50-100 mg daily |
| Candesartan | 8 or 16 mg once daily | 8-32 mg daily |
| Telmisartan | 20 or 40 mg daily | 80-120 mg daily |
Angiotensin Receptor Blockers.
The angiotensin receptor blockers (ARBs; see Table 3-4) have an action, therapeutic application, and side-effect profile similar to those of the ACE inhibitors, except that ARBs are not associated with angioedema and cough and are used primarily for treating hypertension. ARBs may be combined with an ACE inhibitor in patients with resistant hypertension or chronic heart failure, and they may have a specific role in the treatment of diastolic heart failure (Chapter 19).
Antiarrhythmic Drugs
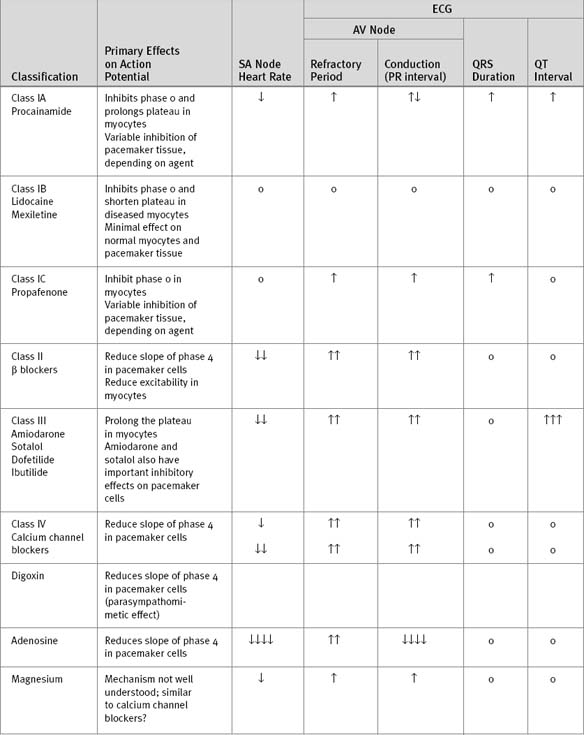
Antiarrhythmic drugs may be classified on the basis of their electrophysiologic properties using the system proposed by Vaughan Williams and Brahma Singh (Table 3-5). Digoxin, adenosine, and magnesium do not fit into this classification system. The electrophysiologic basis of cardiac action potentials is described in Chapter 1, and the treatment of specific arrhythmias, in Chapter 21.
Class I.
Class I agents (known as membrane-stabilizing drugs) block fast sodium channels responsible for phase 0 of the action potential (see Fig. 1.1). Class I agents are further subdivided on the basis of their effect on the duration of the action potential: class IA agents prolong the action potential (class III activity); class IB agents shorten the action potential; class IC agents have no effect on the duration of action potential. Class I drugs act by reducing automaticity, inhibiting retrograde conduction within reentry circuits, and prolonging the refractory period (class IA only). However, the blockade of fast sodium channels reduces the speed of conduction of the action potential, which can precipitate reentry arrhythmias. Class IA agents can also cause torsades de pointes ventricular tachycardia (see subsequent material). Thus, proarrhythmia is a significant problem with class IC agents. The class I drugs encainide and flecainide have been shown to increase mortality rates in patients after myocardial infarction.49
Class III.
Class III drugs block potassium channels responsible for phase 3 (repolarization) of the action potential. Thus, class III drugs prolong the action potential and increase the QT interval. Prolongation of the action potential increases myocyte refractoriness, which can interrupt reentry circuits. However, it also predisposes to early after-depolarizations (Chapter 21), which can precipitate torsades de pointes ventricular tachycardia. All class III (and class IA) drugs have the potential to cause torsades de pointes and should be avoided in patients with QT prolongation.
Amiodarone (Class III).
is a class III antiarrhythmic drug that also has class I, II, and IV activity. Acutely, amiodarone functions mainly as a b blocker. It is available in both oral and intravenous preparations. Amiodarone has proven efficacy for prophylaxis and pharmacologic cardioversion of a wide range of arrhythmias, including atrial fibrillation and life-threatening ventricular arrhythmias (see Chapter 21). Compared to other class III agents, amiodarone has a low incidence of proarrhythmia, with a rate of torsades de pointes ventricular tachycardia of less than 1%. Furthermore, amiodarone causes less hypotension than many other intravenously administered agents and can therefore be used with relative safety in patients who are inotrope-dependent or who have impaired ventricular function. Rapid intravenous administration can cause transient (but marked) hypotension; this effect is due primarily to the Tween 80 solvent. When given as a slow intravenous bolus (e.g., 300 mg over 15 to 30 min) or as a continuous infusion, amiodarone is remarkably well tolerated.
Amiodarone is highly lipid soluble with a very large steady-state volume of distribution. It is rapidly concentrated within myocardial tissue but only slowly distributed to fat. The use of the term “loading dose” with respect to amiodarone is confusing. To fully load the steady-state volume of distribution requires more than 10 g of amiodarone; an amount of drug that must be given over days to weeks to avoid plasma levels that are toxic. In the acute setting, the term “loading dose” refers to the initial volume of distribution (see Chapter 4, Pharmacokinetic Considerations, and Fig. 4-1). Once the initial volume of distribution has been loaded, a reduced dose of drug is administered, either orally or by continuous infusion, to maintain plasma levels as the drug is redistributed to peripheral compartments. Once a steady state has been achieved, a much lower maintenance dose is required. Dose regimes for intravenous and oral amiodarone are provided in Table 3-6.
| Indication | Dosing Regime |
|---|---|
| Acute suppression of life-threatening arrhythmias in the ICU | Option 1 |
| 5 mg/kg IV over 30 min, followed by infusion of 1 mg/min | |
| Option 2 | |
| 150-300 mg over 10-30 min, followed by infusion of 2 mg/min for 4 hr, followed by infusion of 1 mg/min | |
| Intravenous prophylaxis of atrial and ventricular arrhythmias in critically unwell patients | 2 mg/min for 4 hours followed by 1 mg/min |
| Oral postoperative prophylaxis of atrial fibrillation | 400 mg three times daily for 2 days, followed by 200 mg three times daily for 1 week, then stop |
| Oral loading dose with amiodarone | 400 mg twice daily for 7 days, followed by |
| 400 mg daily for 6 weeks | |
| Maintenance oral dose | 200 mg daily |
Acutely, amiodarone is associated with relatively few side effects. One concern is a possible relationship between amiodarone and acute respiratory distress syndrome (ARDS). This association has been observed in critically unwell patients50 and following cardiac51 and thoracic52 surgery. However, these studies are retrospective and observational, and the association has not been confirmed by all investigators.53 Given the clear benefits of amiodarone in cardiac surgery patients and the lack of a definitive association between it and ARDS, it is reasonable to continue to use the drug for the treatment and prevention of perioperative arrhythmias.
In contrast to acute treatment, chronic treatment with amiodarone is associated with a number of side effects (Table 3-7). Pulmonary fibrosis is a rare but potentially fatal complication that usually, but not always, resolves after discontinuation of the drug. The high iodine content of amiodarone causes thyroid dysfunction (hypo- and hyperthyroidism) in up to 10% of patients in long-term treatment. Mild hypothyroidism may be managed with thyroxine without the need to discontinue treatment.
Table 3-7 Side Effects of Chronic Amiodarone Therapy
| Atropine-resistant bradycardia or heart block |
| Pulmonary fibrosis |
| Hypo- and hyperthyroidism |
| Elevated hepatic transaminases |
| Corneal deposits causing peripheral visual halos |
| Photodermatitis and photosensitivity |
| Gray-blue skin discoloration |
| Tremor and ataxia |
| Parasthesia |
| Proximal myopathy |
Amiodarone interacts with many hepatically metabolized drugs and also displaces some protein-bound drugs, notably warfarin and digoxin. Amiodarone inhibits a number of subtypes of the CYP enzyme system, including 2C9 (responsible for warfarin metabolism), 2D6, and 3A. Substrate drugs for CYP2D6 and CYP3A are listed in Table 4-3. The maintenance dose of warfarin may need to be greatly reduced in patients receiving amiodarone. Amiodarone should be avoided in patients with documented hypersensitivity to iodine and in patients with prolonged QT intervals or histories of torsades de pointes ventricular tachycardia.
Sotalol (Class III).
This is a nonselective β blocker with important class III activity.54 Higher doses are required for the class III effect than for b blockade. Sotalol is effective against a wide range of arrhythmias, including atrial fibrillation and recurrent ventricular tachycardia (Chapter 21). However, in patients with impaired ventricular function, sotalol can cause considerable hypotension, particularly when given intravenously. Proarrhythmia is a greater concern with sotalol than with amiodarone, with an incidence of torsades de pointes of about 2%. Sotalol should be avoided in a patient who has a corrected QT (QTc) interval greater than 0.45 seconds and should be discontinued if the QTc exceeds 0.55 seconds during treatment.
Ibutilide (Class III).
This is a class III antiarrhythmic drug available for intravenous use.55 It is structurally similar to sotalol but, unlike sotalol, does not usually cause hypotension in patients with impaired ventricular function. Ibutilide may be used for the pharmacologic cardioversion of atrial fibrillation, atrial flutter, and ventricular tachycardia. The use of ibutilide is limited by its relatively high incidence of torsades de pointes ventricular tachycardia. The dose (for patients >60 kg) is 1 mg over 10 minutes; if unsuccessful, the dose may be repeated 10 minutes later.55,56
Ibutilide should be avoided in a patient with a QTc greater than 0.45 seconds and in a patient with hypokalemia or hypomagnesemia.55 The drug should be administered under continuous ECG monitoring, and the infusion should be stopped when the patient reverts to sinus rhythm or if the QTc exceeds 0.55 seconds. ECG monitoring should continue for 4 hours following treatment.
Dofetilide (Class III).
This is an orally administered class III antiarrhythmic agent that is indicated for the pharmacologic cardioversion of atrial fibrillation and flutter. Dofetilide can be safely used in a patient with impaired ventricular function57 but is associated with a high incidence of torsades de pointes ventricular tachycardia. The drug should be avoided in a patient with a QTc greater than 0.45 seconds. The dose is 250 to 500 μg twice daily, but this must be decreased in a patient with renal failure. Dofetilide is contraindicated in a patient with a creatinine clearance less than 20 ml/min.56
Diltiazem (Class IV).
This is indicated for rate control of atrial fibrillation and for the pharmacologic cardioversion of AV nodal reentry tachycardia. Intravenously, diltiazem may be administered as repeated bolus doses of 10 to 20 mg or as a continuous infusion of 5 to 15 mg/hr. Orally, diltiazem may be commenced at 30 to 60 mg every 6 hours, increasing up to a maximum of 360 mg per day. Alternatively, a slow-release formulation may be used at a dose of 180 to 360 mg daily. Diltiazem has a very low rate of proarrhythmia but can cause marked bradycardia and heart block, particularly when administered with a β blocker or digoxin. Diltiazem has less effect on cardiac contractility than verapamil but must be used cautiously in patients with impaired ventricular function. Diltiazem is both a substrate and an inhibitor of the CYP3A enzyme system (see Table 4-3) and is therefore involved in important drug interactions.
Lidocaine (Class IB).
This is an intravenously administered local anesthetic drug that has been used for many years for the treatment of ventricular tachycardia. In patients with myocardial infarction, lidocaine suppresses ventricular extrasystoles but does not reduce the likelihood of developing ventricular fibrillation; therefore, lidocaine is not indicated for suppression of ectopy. For the treatment of intractable ventricular tachycardia, lidocaine is usually less effective than amiodarone (see Chapter 21).
Because of high first-pass metabolism, lidocaine must be administered intravenously. (Mexiletine is an orally administered drug with pharmacologic properties virtually identical to those of lidocaine.) The elimination half-time of lidocaine is about 4 hours, but the duration of effect following an intravenous bolus dose is only a few minutes because of redistribution. Lidocaine is usually given as a bolus dose of 1 mg/kg over 1 to 2 minutes, which may be supplemented with one or two further doses of 0.5 mg/kg separated by 15 minutes. Following this, lidocaine may be administered as an intravenous infusion at 1 to 4 mg/min, ensuring that the total dose does not exceed 300 mg in the first hour. Serum levels should be obtained after 12 to 24 hours, aiming for a concentration of 6 to 21 μmol/l, (1.5 to 5 μg/ml). High serum levels produce neurologic symptoms, which include paresthesia, dysphoria, and agitation. The infusion rate should be reduced in patients with low cardiac output (because of a reduced rate of redistribution) and in patients who develop neurologic symptoms. Although lidocaine has no sedative properties itself, it augments the sedative effect of other hypnotic drugs.
Magnesium sulfate.
This is a safe and efficacious antiarrhythmic agent that has a broad application in the cardiothoracic ICU (Chapter 21). It is useful in preventing and treating postoperative atrial and ventricular arrhythmias and is specifically indicated for torsades de pointes ventricular tachycardia (including that which is drug induced) and multifocal atrial tachycardia.
Diuretics
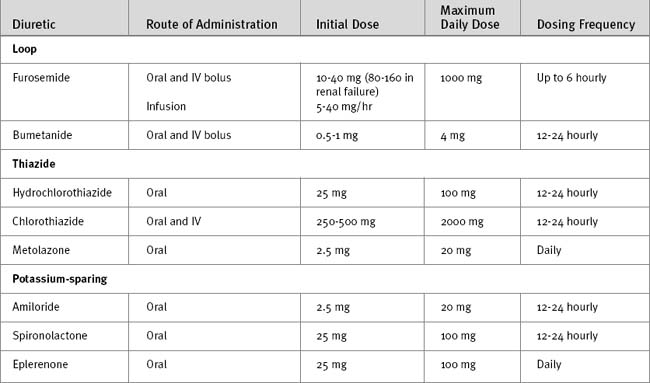
Diuretic drugs cause sodium loss (natriuresis) and water loss (diuresis) by the kidney and, as such, are important in the management of hypertension and edematous states. Diuretic drugs are classified by their mechanism of action within the kidney as loop diuretics, thiazide diuretics, or potassium-sparing diuretics. Doses of commonly encountered diuretics are listed in Table 3-8.
Loop Diuretics.
The main adverse effect of loop diuretics is an excessive clinical effect: hypovolemia, hypokalemia, hypochloremic metabolic alkalosis, and hypomagnesemia. Other side effects include deafness, hyperuricemia (and gout), and allergic skin rashes. The effect of loop diuretics on serum sodium concentration is difficult to predict. The urinary sodium concentration in a patient on a furosemide infusion is typically less than 100 mmol/l; thus, acutely, hypernatremia can occur. However, if urinary losses are replaced with a low-sodium solution (e.g., intravenous 5% dextrose or oral water), hyponatremia will develop. Chronic furosemide use typically results in hyponatremia. Furosemide and, to a lesser extent, bumetanide can cause deafness. This risk is greatest in patients with renal impairment who are receiving high doses of furosemide, by either infusion or rapidly administered intravenous bolus doses. The role of loop diuretics in the treatment of systemic edema and renal failure is debatable and is discussed in Chapters 32 and Chapter 33, respectively.
Thiazide Diuretics.
Thiazides diuretics (e.g., chlorothiazide, hydrochlorothiazide, metolazone) inhibit sodium and chloride reabsorption in the distal nephron. Thiazide diuretics promote potassium and magnesium excretion but, unlike loop diuretics, inhibit calcium excretion. Thiazide diuretics are less potent than loop diuretics and are ineffective when the glomerular filtration rate falls below about 30 ml/min.58 Thiazide diuretics are used in the treatment of hypertension and mild heart failure. All thiazide diuretics are administered orally except chlorothiazide, which is also available for intravenous use. Thiazide diuretics are formulated with other agents such as ACE inhibitors for the treatment of hypertension. In the cardiothoracic ICU, thiazides, particularly metolazone, are occasionally useful as cotreatment in patients who are refractory to loop diuretics. Metolazone has a long duration of action (12 to 24 hours) and is slightly more efficacious than other thiazide diuretics because it has an additional diuretic effect in the proximal nephron. Adverse effects of thiazides include excessive clinical effects (hypovolemia, hyponatremia, hypokalemia, hypochloremic metabolic alkalosis), hyperuricemia, hyperlipidemia, hyperglycemia, photosensitivity, and allergic skin rashes.
Potassium-sparing Diuretics.
Aldosterone antagonists (spironolactone, eplerenone) inhibit the action of aldosterone in the collecting duct; as such, these agents cause modest diuresis and natriuresis but inhibit potassium and hydrogen ion secretion. Aldosterone antagonists are most useful in conditions associated with increased aldosterone secretion, notably congestive cardiac failure and hepatic cirrhosis. Spironolactone and eplerenone have been shown to reduce mortality rates in patients with severe heart failure.59,60 Both agents are administered orally and are suitable for once-daily dosing.
The side effects of aldosterone antagonists include hyperkalemia, hyperchloremic metabolic acidosis, gynecomastia, acute renal failure, and kidney stones. Hyperkalemia, with the potential for cardiac arrest, is the most feared complication of aldosterone antagonists. Toxicity is greatest in patients with renal impairment and those receiving ACE inhibitors or nonsteroidal antiinflammatory drugs. Following the initiation of treatment with an aldosterone antagonist, potassium supplements should be stopped and serum potassium and creatinine carefully monitored.
Antiplatelet Drugs
A number of antiplatelet drugs are encountered in the cardiothoracic ICU, all of which, to varying degrees, can increase bleeding. All antiplatelet drugs cause prolongation of the bleeding time but do not cause any abnormalities in routine tests of coagulation. Antiplatelet drugs vary with respect to their duration of effect (5 to 7 days for aspirin and clopidogrel; 4 to 12 hours for glycoprotein IIb/IIIa receptor antagonists), and their impact on postsurgical bleeding (minor for aspirin; intermediate for clopidogrel; high for glycoprotein IIb/IIIa receptor blockers). Recommendations for antiplatelet drugs in acute coronary syndromes are summarized in Table 18.12. Other drugs relating to bleeding and coagulation are discussed in Chapter 30.
Aspirin.
Low-dose aspirin (83 mg/day) is indicated in a wide range of thromboembolic conditions, including acute coronary syndromes, stable or unstable angina, ischemic neurologic events, and the implantation of bioprosthetic valves and following coronary artery bypass graft surgery. However, aspirin is a relatively weak antiplatelet agent, inhibiting only thromboxane-induced aggregation. Platelet aggregation can still be induced by other stimuli, notably thrombin. Low-dose aspirin is relatively safe and does not appear to increase postoperative bleeding, even in patients undergoing repeat sternotomy.61 Thus, in most cardiac surgical units, aspirin is continued throughout the perioperative period.
Thienopyridines: Ticlopidine and Clopidogrel.
Ticlopidine and clopidogrel are structurally related thienopyridine compounds that irreversibly inhibit adenosine-diphosphate-induced platelet aggregation. Clopidogrel has a more rapid action and a better safety profile than ticlopidine. Maximal inhibition of platelet aggregation occurs 3 to 5 days after taking a standard dose of clopridogrel (75 mg) but within 6 hours of taking a 300 mg loading dose.62 Platelet function takes 5 to 7 days to return to normal after the drug is withheld.
Clopidogrel improves survival in patients with unstable angina and non-ST segment myocardial infarction63 and reduces myocardial infarction and other adverse outcomes in patients undergoing percutaneous coronary intervention (PCI).64 The optimal duration of treatment has not been determined, but the major benefit occurs in the first 30 days.63 Clopidogrel is also used by some surgeons following off-pump CABG surgery to improve graft patency. Patients receiving both clopidogrel and aspirin prior to CABG surgery are more likely than those treated with aspirin alone to have major bleeding that requires transfusion or reoperation.62 Thus, if possible, clopidogrel should be stopped at least 5 days prior to surgery.65
Glycoprotein IIB/IIIA Receptor Blockers.
Glycoprotein IIb/IIIa receptor blockers (tirofiban, lamifiban, epifibatide, abciximab) inhibit platelet aggregation by preventing the binding of fibrinogen and the von Willebrand factor (and other adhesive molecules) to glycoprotein IIb/IIIa receptors on the surface of activated platelets. Glycoprotein IIb/IIIa receptor blockers are used in the treatment of acute coronary syndromes, particularly if PCI is undertaken (Chapter 18). All currently available agents are administered as an intravenous bolus followed by an infusion for 1 to 3 days. Inhibition of platelet aggregation occurs within 30 minutes of a bolus dose and persists for up to 12 hours following discontinuation of the infusion.
Lipid-lowering Drugs
Statins.
Statins are indicated primarily in the treatment of dyslipidemia, particularly in patients with coronary artery disease and after CABG surgery.66 There is also evidence that statins have antiinflammatory effects that provide an additional benefit over and above their lipid-lowering effect.67,68 Statins inhibit the development of inflammatory coronary artery disease following heart transplantation69 and appear to have a protective effect in patients who develop septic shock.70–72
1 Alquist RP. A study of adrenotropic receptors. Am J Physiol. 1948;53:506.
2 Totaro RJ, Raper RF. Epinephrine-induced lactic acidosis following cardiopulmonary bypass. Crit Care Med. 1997;25:1693-1699.
3 Goldberg LI, Rajfer SI. Dopamine receptors: applications in clinical cardiology. Circulation. 1985;72:245-248.
4 MacGregor DA, Smith TE, Prielipp RC, et al. Pharmacokinetics of dopamine in healthy male subjects. Anesthesiology. 2000;92:338-346.
5 Schwinn DA, Leone BJ, Spahn DR, et al. Desensitization of myocardial beta-adrenergic receptors during cardiopulmonary bypass: evidence for early uncoupling and late downregulation. Circulation. 1991;84:2559-2567.
6 Booth JV, Landolfo KP, Chesnut LC, et al. Acute depression of myocardial beta-adrenergic receptor signaling during cardiopulmonary bypass: impairment of the adenylyl cyclase moiety. Duke Heart Center Perioperative Desensitization Group. Anesthesiology. 1998;89:602-611.
7 Vatner DE, Asai K, Iwase M, et al. Beta-adrenergic receptor-G protein-adenylyl cyclase signal transduction in the failing heart. Am J Cardiol. 1999;83:80H-85H.
8 Debaveye YA, van den Berghe GH. Is there still a place for dopamine in the modern intensive care unit ? Anesth Analg. 2004;98:461-468.
9 Bellomo R, Chapman M, Finfer S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000;356:2139-2143.
10 Feneck RO, Sherry KM, Withington PS, et al. Comparison of the hemodynamic effects of milrinone with dobutamine in patients after cardiac surgery. J Cardiothorac Vasc Anesth. 2001;15:306-315.
11 Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med. 1997;23:282-287.
12 Francis SH, Turko IV, Corbin JD. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog Nucleic Acid Res Mol Biol. 2001;65:1-52.
13 Lobato EB, Janelle GM, Urdaneta F, et al. Comparison of milrinone versus nitroglycerin, alone and in combination, on grafted internal mammary artery flow after cardiopulmonary bypass: effects of alpha-adrenergic stimulation. J Cardiothorac Vasc Anesth. 2001;15:723-727.
14 Packer M, Kukin ML, Sollano JA, et al. Effect of oral milrinone on mortality in severe chronic heart failure. N Engl J Med. 1991;325:1468-1475.
15 Royster RL, Butterworth JF, Prielipp RC, et al. A randomized, blinded, placebo-controlled evaluation of calcium chloride and epinephrine for inotropic support after emergence from cardiopulmonary bypass. Anesth Analg. 1992;74:3-13.
16 Thompson BM, Steuven HS, Tonsfeldt DJ, et al. Calcium: limited indications, some danger. Circulation. 1986;74:IV90-IV93.
17 Dunser MW, Mayr AJ, Ulmer H, et al. Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003;107:2313-2319.
18 Klinzing S, Simon M, Reinhart K, et al. High-dose vasopressin is not superior to norepinephrine in septic shock. Crit Care Med. 2003;31:2646-2650.
19 Vanhoutte PM, Katusic ZS, Shepherd JT. Vasopressin induces endothelium-dependent relaxations of cerebral and coronary, but not of systemic, arteries. J Hypertens. 1984;2:421-422.
20 Evora PR, Pearson PJ, Schaff HV. Arginine vasopressin induces endothelium-dependent vasodilatation of the pulmonary artery: V1-receptor-mediated production of nitric oxide. Chest. 1993;103:1241-1245.
21 Patel BM, Chittock DR, Russell JA, et al. Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology. 2002;96:576-582.
22 Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Intens Care Med. 2004;30:536-555.
23 Follath F, Cleland JG, Just H, et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomised double-blind trial. Lancet. 2002;360:196-202.
24 Labriola C, Siro-Brigiani M, Carrata F, et al. Hemodynamic effects of levosimendan in patients with low-output heart failure after cardiac surgery. Int J Clin Pharmacol Therapeut. 2004;42:204-211.
25 Dargie HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet. 2001;357:1385-1390.
26 Anonymous. Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. N Engl J Med. 1981;304:801-807.
27 Olsson G, Wikstrand J, Warnold I, et al. Metoprolol-induced reduction in postinfarction mortality: pooled results from five double-blind randomized trials. Eur Heart J. 1992;13:28-32.
28 Bunch TJ, Muhlestein JB, Bair TL, et al. Effect of beta-blocker therapy on mortality rates and future myocardial infarction rates in patients with coronary artery disease but no history of myocardial infarction or congestive heart failure. Am J Cardiol. 2005;95:827-831.
29 Mangano DT, Layug EL, Wallace A, et al. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med. 1996;335:1713-1720.
30 Ferguson TBJr, Coombs LP, Peterson ED, Society of Thoracic Surgeons National Adult Cardiac Surgery D. Preoperative beta-blocker use and mortality and morbidity following CABG surgery in North America. JAMA. 2002;287:2221-2227.
31 Psaty BM, Koepsell TD, Wagner EH, et al. The relative risk of incident coronary heart disease associated with recently stopping the use of beta-blockers. JAMA. 1999;263:1653-1657.
32 Keating GM, Jarvis B. Carvedilol: a review of its use in chronic heart failure. Drugs. 2003;63:1697-1741.
33 Gerson JI, Allen FB, Seltzer JL. Arterial and venous dilation by nitroprusside and nitroglycerin —is there a difference ? Anesth Analges. 1982;61:256-260.
34 Psaty BM, Heckbert SR, Koepsell TD, et al. The risk of myocardial infarction associated with antihypertensive drug therapies. JAMA. 1995;274:620-625.
35 Murphy MB, Murray C, Shorten GD. Fenoldopam: a selective peripheral dopamine-receptor agonist for the treatment of severe hypertension. N Engl J Med. 2001;345:1548-1557.
36 Hill AJ, Feneck RO, Walesby RK. A comparison of fenoldopam and nitroprusside in the control of hypertension following coronary artery surgery. J Cardiothorac Vasc Anesth. 1993;7:279-284.
37 Tanaka KA, Szlam F, Katori N, et al. In vitro effects of antihypertensive drugs on thromboxane agonist (U46619)-induced vasoconstriction in human internal mammary artery. Br J Anaesth. 2004;93:257-262.
38 Caimmi P-P, Pagani L, Micalizzi E, et al. Fenoldopam for renal protection in patients undergoing cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2003;17:491-494.
39 Bove T, Landoni G, Calabro MG, et al. Renoprotective action of fenoldopam in high-risk patients undergoing cardiac surgery: a prospective, double-blind, randomized clinical trial. Circulation. 2005;111:3230-3235.
40 Keating GM, Goa KL. Nesiritide: a review of its use in acute decompensated heart failure. Drugs. 2003;63:47-70.
41 Publication Committee for the VMAC Investigators. Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531-1540.
42 Moazami N, Damiano RJ, Bailey MS, et al. Nesiritide (BNP) in the management of postoperative cardiac patients. Ann Thorac Surg. 2003;75:1974-1976.
43 Kober L, Torp-Pedersen C, Carlsen JE, et al. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. N Engl J Med. 1995;333:1670-1676.
44 Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145-153.
45 SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. The SOLVD Investigators. N Eng J Med. 1992;327:685-691.
46 Flather MD, Yusuf S, Kober L, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. ACE-Inhibitor Myocardial Infarction Collaborative Group. Lancet. 2000;355:1575-1581.
47 SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med. 1991;325:293-302.
48 Manche A, Galea J, Busuttil W. Tolerance to ACE inhibitors after cardiac surgery. Eur J Cardiothorac Surg. 1999;15:55-60.
49 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-412.
50 Donaldson L, Grant IS, Naysmith MR, et al. Acute amiodarone-induced lung toxicity. Intensive Care Med. 1998;24:626-630.
51 Greenspon AJ, Kidwell GA, Hurley W, et al. Amiodarone-related postoperative adult respiratory distress syndrome. Circulation. 1991;84:III407-III415.
52 van Mieghem W, Coolen L, Malysse I, et al. Amiodarone and the development of ARDS after lung surgery. Chest. 1994;105:1642-1645.
53 Rady MY, Ryan T, Starr NJ. Preoperative therapy with amiodarone and the incidence of acute organ dysfunction after cardiac surgery. Anesth Analg. 1997;85:489-497.
54 Anderson JL, Prystowsky EN. Sotalol: an important new antiarrhythmic. Am Heart J. 1999;137:388-409.
55 Murray KT. Ibutilide. Circulation. 1998;97:493-497.
56 Fuster V, Ryden LE, Asinger RW, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: executive summary. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients With Atrial Fibrillation): developed in Collaboration with the North American Society of Pacing and Electrophysiology. J Am Coll Cardiol. 2001;38:1231-1266.
57 Torp-Pedersen C, Moller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med. 1999;341:857-865.
58 Remme WJ, Swedberg K. Guidelines for the diagnosis and treatment of chronic heart failure. Eur Heart J. 2001;22:1527-1560.
59 Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709-717.
60 Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309-1321.
61 Tuman KJ, McCarthy RJ, O’Connor CJ, et al. Aspirin does not increase allogeneic blood transfusion in reoperative coronary artery surgery. Anesth Analg. 1996;83:1178-1184.
62 Lange RA, Hillis LD. Antiplatelet therapy for ischemic heart disease. N Engl J Med. 2004;350:277-280.
63 Yusuf S, Zhao F, Mehta SR, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494-502.
64 Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527-533.
65 Patrono C, Bachmann F, Baigent C, et al. Expert consensus document on the use of antiplatelet agents. The task force on the use of antiplatelet agents in patients with atherosclerotic cardiovascular disease of the European society of cardiology. Eur Heart J. 2004;25:166-181.
66 Post Coronary Artery Bypass Graft Trial Investigators. The effect of aggressive lowering of low-density lipoprotein cholesterol levels and low-dose anticoagulation on obstructive changes in saphenous-vein coronary-artery bypass grafts. The Post Coronary Artery Bypass Graft Trial Investigators. N Engl J Med. 1997;336:153-162.
67 Plenge JK, Hernandez TL, Weil KM, et al. Simvastatin lowers C-reactive protein within 14 days: an effect independent of low-density lipoprotein cholesterol reduction. Circulation. 2002;106:1447-1452.
68 Albert MA, Danielson E, Rifai N, et al. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64-70.
69 Kobashigawa JA, Katznelson S, Laks H, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med. 1995;333:621-627.
70 Almog Y. Statins, inflammation, and sepsis: hypothesis. Chest. 2003;124:740-743.
71 Almog Y, Shefer A, Novack V, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004;110:880-885.
72 Steiner S, Speidl WS, Pleiner J, et al. Simvastatin blunts endotoxin-induced tissue factor in vivo. Circulation. 2005;111:1841-1846.