Chapter 8 Cardiovascular Pharmacology
ANTI-ISCHEMIC DRUG THERAPY
Anti-ischemic drug therapy during anesthesia is indicated whenever evidence of myocardial ischemia exists. The treatment of ischemia during anesthesia is complicated by the ongoing stress of surgery, blood loss, concurrent organ ischemia, and the patient’s inability to interact with the anesthesiologist. Nonetheless, the fundamental principles of treatment remain the same as in the unanesthetized state. All events of myocardial ischemia involve an alteration in the oxygen supply/demand balance (Table 8-1). The 2007 American College of Cardiology/American Heart Association (ACC/AHA) Guidelines on the Management and Treatment of Patients with Unstable Angina and Non–ST-Segment Elevation Myocardial Infarction provide an excellent framework for the treatment of patients with ongoing myocardial ischemia.1
Table 8-1 Myocardial Ischemia: Factors Governing O2 Supply and Demand
| O2 Supply | O2 Demand |
|---|---|
| Heart rate* | Heart rate* |
| O2 content | Contractility |
| Hemoglobin, percent oxygen saturation, PaO2 | Wall tension |
| Coronary blood flow | Afterload |
| CPP = DBP − LVEDP* | Preload (LVEDP)* |
| Coronary vascular resistance |
CPP = coronary perfusion pressure; DBP = diastolic blood pressure; LVEDP = left ventricular end-diastolic pressure.
* Affects both supply and demand.
Modified from Royster RL: Intraoperative administration of inotropes in cardiac surgery patients. J Cardiothorac Anesth 6(Suppl 5):17, 1990.
Nitroglycerin
Nitroglycerin (NTG) is clinically indicated as initial therapy in nearly all types of myocardial ischemia. Chronic exertional angina, de novo angina, unstable angina, Prinzmetal’s angina (vasospasm), and silent ischemia respond to NTG administration. During intravenous therapy with NTG, if blood pressure (BP) drops and ischemia is not relieved, the addition of phenylephrine will allow coronary perfusion pressure (CPP) to be maintained while allowing higher doses of NTG to be used for ischemia relief. If reflex increases in heart rate (HR) and contractility occur, combination therapy with β-adrenergic blockers may be indicated to blunt this undesired increase in HR. Combination therapy with nitrates and calcium channel blockers may be an effective anti-ischemic regimen in selected patients; however, excessive hypotension and reflex tachycardia may be a problem, especially when a dihydropyridine calcium antagonist is used.
Mechanism of Action
NTG enhances myocardial oxygen delivery and reduces myocardial oxygen demand. NTG is a smooth muscle relaxant that causes vasculature dilation.2 Nitrate-mediated vasodilation occurs with or without intact vascular endothelium. Nitrites, organic nitrites, nitroso compounds, and other nitrogen oxide–containing substances (e.g., nitroprusside) enter the smooth muscle cell and are converted to reactive nitric oxide (NO) or S-nitrosothiols, which stimulate guanylate cyclase metabolism to produce cyclic guanosine monophosphate (cGMP) (Fig. 8-1). A cGMP-dependent protein kinase is stimulated with resultant protein phosphorylation in the smooth muscle. This leads to a dephosphorylation of the myosin light chain and smooth muscle relaxation. Vasodilation is also associated with a reduction of intracellular calcium. Sulfhydryl (SH) groups are required for formation of NO and the stimulation of guanylate cyclase. When excessive amounts of SH groups are metabolized by prolonged exposure to NTG, vascular tolerance occurs. The addition of N-acetylcysteine, an SH donor, reverses NTG tolerance. The mechanism by which NTG compounds are uniquely better venodilators, especially at lower serum concentrations, is unknown but may be related to increased uptake of NTG by veins compared with arteries.3
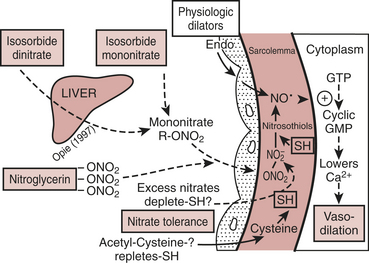
Physiologic Effects
Two important physiologic effects of NTG are systemic and regional venous dilation. Venodilation can markedly reduce venous pressure, venous return to the heart, and cardiac filling pressures. Prominent venodilation occurs at lower doses and does not increase further as the NTG dose increases. Venodilation results primarily in pooling of blood in the splanchnic capacitance system. Mesenteric blood volume increases as ventricular size, ventricular pressures, and intrapericardial pressure decrease.
 ) and improve myocardial ischemia. NTG may preferentially reduce cardiac preload while maintaining systemic perfusion pressure, an important hemodynamic effect in myocardial ischemia. However, in hypovolemic states, higher doses of NTG may markedly reduce systemic BP to dangerous levels. A reflex increase in HR may occur at arterial vasodilating doses.
) and improve myocardial ischemia. NTG may preferentially reduce cardiac preload while maintaining systemic perfusion pressure, an important hemodynamic effect in myocardial ischemia. However, in hypovolemic states, higher doses of NTG may markedly reduce systemic BP to dangerous levels. A reflex increase in HR may occur at arterial vasodilating doses.NTG has several important effects on the coronary circulation (Box 8-1). NTG is a potent epicardial coronary artery vasodilator in both normal and diseased vessels. Stenotic lesions dilate with NTG, reducing the resistance to coronary blood flow (CBF) and improving myocardial ischemia. Smaller coronary arteries may dilate relatively more than larger coronary vessels; however, the degree of dilation may depend on the baseline tone of the vessel. NTG effectively reverses or prevents coronary artery vasospasm.
BOX 8-1 Effects of Nitroglycerin and Organic Nitrates on the Coronary Circulation
Modified frgom Abrams J: Hemodynamic effects of nitroglycerin and long-acting nitrates. Am Heart J 110(part 2):216, 1985.
 decreases
decreasesTotal CBF may initially increase but eventually decreases with NTG despite coronary vasodilation. Autoregulatory mechanisms probably result in decreases in total flow as a result of reductions in wall tension and myocardial oxygen consumption. However, regional myocardial blood flow may improve by vasodilation of intercoronary collateral vessels or reduction of subendocardial compressive forces. Coronary arteriographic studies in humans demonstrate that coronary collateral vessels increase in size after NTG administration. This effect may be especially important when epicardial vessels have subtotal or total occlusive disease. Improvement in collateral flow may also be protective in situations in which coronary artery steal may occur with other potent coronary vasodilator agents. The improvement in blood flow to the subendocardium, the most vulnerable area to the development of ischemia, is secondary to both improvement in collateral flow and reductions in left ventricular end-diastolic pressure (LVEDP), which reduce subendocardial resistance to blood flow. With the maintenance of an adequate CPP (e.g., with administration of phenylephrine), NTG can maximize subendocardial blood flow. The ratio of endocardial to epicardial blood in transmural segments is enhanced with NTG. Inhibition of platelet aggregation also occurs with NTG; however, the clinical significance of this action is unknown.
Summary
Nitroglycerin remains a first-line agent for the treatment of myocardial ischemia. Special care must be taken in patients with signs of hypovolemia or hypotension, because the vasodilating effects of the drug may worsen the clinical condition. Recommendations from the ACC/AHA on intraoperative use of NTG are given in Box 8-2.
BOX 8-2 Recommendations for Intraoperative Nitroglycerin
* Conditions for which there is evidence for and/or general agreement that a procedure be performed or a treatment is of benefit.
† Conditions for which there is a divergence of evidence and/or opinion about the treatment.
‡ Conditions for which there is evidence and/or general agreement that a procedure is not necessary.
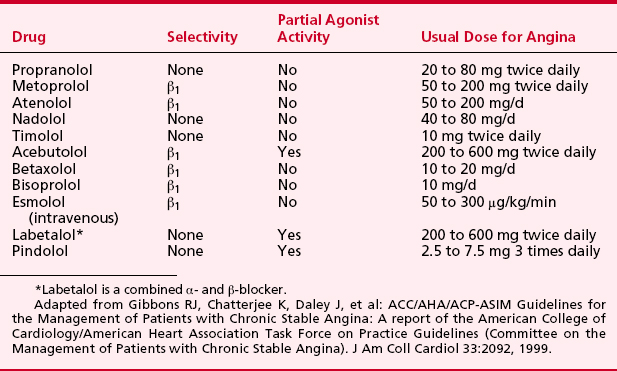
β-Adrenergic Blockers
β-Adrenergic blockers have multiple favorable effects in treating the ischemic heart during anesthesia (Box 8-3). They reduce oxygen consumption by decreasing HR, BP, and myocardial contractility. HR reduction increases diastolic CBF. Increased collateral blood flow and redistribution of blood to ischemic areas may occur with β-blockers. More free fatty acids may be available for substrate consumption by the myocardium. Microcirculatory oxygen delivery improves, and oxygen dissociates more easily from hemoglobin after β-adrenergic blockade. Platelet aggregation is inhibited. β-Blockers should be started early in ischemic patients in the absence of contraindications. Many patients at high risk of perioperative cardiac morbidity should be started on β-blocker therapy before surgery and continued on this therapy for up to 30 days after surgery.
Perioperative administration of β-adrenergic blockers reduces both mortality and morbidity when given to patients at high risk for coronary artery disease who must undergo noncardiac surgery.4 These data suggest that intermediate- and high-risk patients presenting for noncardiac surgery should receive perioperative β-adrenergic blockade to reduce postoperative cardiac mortality and morbidity. Recommendations on the perioperative use of β-adrenergic blockade for noncardiac surgery are given in Box 8-4.
BOX 8-4 Recommendations for Perioperative Medical Therapy
Adapted from Eagle KA, Berger PB, Calkins H, et al: ACC/AHA guideline update for perioperative cardiovascular evaluation for noncardiac surgery-executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1996 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). J Am Coll Cardiol 39:542, 2002.
Physiologic Effects
ANTI-ISCHEMIC EFFECTS
β-Blockade on the ischemic heart may result in a favorable shift in the oxygen demand/supply ratio.5 The reductions in the force of contraction and HR reduce myocardial oxygen consumption and result in autoregulatory decreases in myocardial blood flow. Several studies have shown that blood flow to ischemic regions is maintained with propranolol.
electrophysiologic effects
Generalized slowing of cardiac depolarization results from reducing the rate of diastolic depolarization (phase 4). Action potential duration and the QT interval may shorten with β-adrenergic blockers. The ventricular fibrillation threshold is increased with β-blockers. These antiarrhythmic actions of β-blockers are enhanced in settings of catecholamine excess, such as in pheochromocytoma, acute myocardial infarction, the perioperative period, and hyperthyroidism.
Pharmacology of Intravenous β-Adrenergic Blockers
metoprolol
Metoprolol was the first clinically used cardioselective β-blocker (Table 8-2). Its affinity for β1-receptors is 30 times higher than its affinity for β2-receptors, as demonstrated by radioligand binding. Metoprolol is lipid soluble, with 50% of the drug metabolized during first-pass hepatic metabolism and with only 3% excreted renally. Protein binding is less than 10%. Metoprolol’s serum half-life is 3 to 4 hours.
Summary
β-Adrenergic blockers are first-line agents in the treatment of myocardial ischemia. These agents effectively reduce myocardial work and oxygen demand. There is growing evidence that β-adrenergic-blocking agents may play a significant role in reducing perioperative cardiac morbidity and mortality in noncardiac surgery.6
Calcium Channel Blockers
Calcium channel blockers reduce myocardial oxygen demands by depression of contractility, HR, and/or decreased arterial BP.7 Myocardial oxygen supply may be improved by dilation of coronary and collateral vessels. Calcium channel blockers are used primarily for symptom control in patients with stable angina pectoris. In an acute ischemic situation, calcium channel blockers (verapamil and diltiazem) may be used for rate control in situations when β-blockers cannot be used. The most important effects of calcium channel blockers, however, may be the treatment of variant angina. These drugs can attenuate ergonovine-induced coronary vasoconstriction in patients with variant angina, suggesting protection via coronary dilation. Most episodes of silent myocardial ischemia, which may account for 70% of all transient ischemic episodes, are not related to increases in myocardial oxygen demands (HR and BP) but, rather, intermittent obstruction of coronary flow likely caused by coronary vasoconstriction or spasm. All calcium channel blockers are effective at reversing coronary spasm, reducing ischemic episodes, and reducing NTG consumption in patients with variant or Prinzmetal’s angina. Combinations of NTG and calcium channel blockers, which also effectively relieve and possibly prevent coronary spasm, are at present rational therapy for variant angina. β-Blockers may aggravate anginal episodes in some patients with vasospastic angina and should be used with caution. Preservation of CBF with calcium channel blockers is a significant difference from the predominant β-blocker anti-ischemic effects of reducing myocardial oxygen consumption.
Calcium channel blockers have proven effective in controlled trials of stable angina. However, rapid-acting dihydropyridines such as nifedipine may cause a reflex tachycardia, especially during initial therapy, and exacerbate anginal symptoms. Such proischemic effects probably explain why the short-acting dihydropyridine nifedipine in high doses produced adverse effects in patients with unstable angina. The introduction of long-acting dihydropyridines such as extended-release nifedipine, amlodipine, felodipine, isradipine, nicardipine, and nisoldipine has led to fewer adverse events. These agents should be used in combination with β-blockers. Some patients may have symptomatic relief improved more with calcium channel blockers than with β-blocker therapy.
Physiologic Effects
hemodynamic effects
Systemic hemodynamic effects of calcium channel blockers represent a complex interaction among myocardial depression, vasodilation, and reflex activation of the autonomic nervous system (Table 8-3).
Table 8-3 Calcium Channel Blocker Vasodilator Potency and Inotropic, Chronotropic, and Dromotropic Effects on the Heart
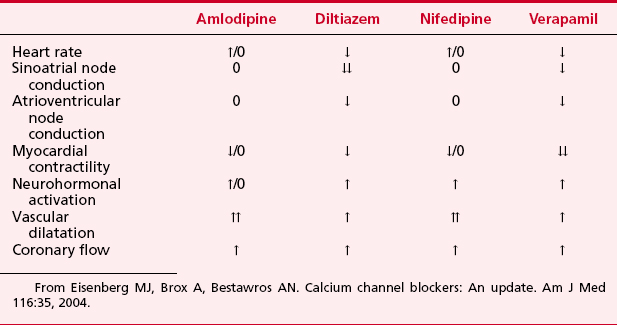
Verapamil is a less potent arterial dilator than the dihydropyridines and results in less reflex sympathetic activation. In vivo, verapamil generally results in moderate vasodilation without significant change in HR, CO, or SV. Verapamil can significantly depress myocardial function in patients with preexisting ventricular dysfunction.
Summary
Calcium antagonists provide excellent symptom control in patients with unstable angina. In the absence of β-adrenergic blockade, the short-acting dihydropyridine nifedipine may increase the risk of myocardial infarction or recurrent angina. When β-adrenergic blockers cannot be used, and HR slowing is indicated, verapamil and diltiazem may offer an alternative.8
DRUG THERAPY FOR SYSTEMIC HYPERTENSION
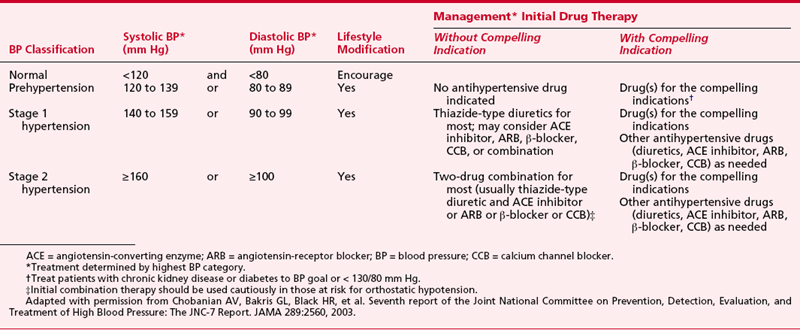
The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC-7 Report) defined systolic BPs (Table 8-4) exceeding 140 mm Hg and diastolic BPs exceeding 90 mm Hg as stage 1 hypertension. BPs less than 120/80 mm Hg were defined as normal and those in between as consistent with “prehypertension.”9
Medical Treatment for Hypertension
More than 80 distinct medications are marketed for treatment of hypertension (Table 8-5). Often, combined therapy with two or more classes of antihypertensive medications may be needed to achieve treatment goals (Table 8-6). Although the specific drug selected for initial therapy now has been deemed less important than in the past, recognition that specific antihypertensive drug classes alleviate end-organ damage, beyond that simply associated with reductions in systemic BP, has led to targeted selection of antihypertensive drug combinations on the basis of coexisting risk factors such as recent myocardial infarction, chronic renal insufficiency, or diabetes.10
| Drug (Trade Name) | Usual Dose Range (mg/d) | Usual Daily Frequency |
|---|---|---|
| Thiazide Diuretics | ||
| Chlorothiazide (Diuril) | 125 to 500 | 1 to 2 |
| Chlorthalidone (generic) | 12.5 to 25 | 1 |
| Hydrochlorothiazide (Microzide, HydroDIURIL†) | 12.5 to 50 | 1 |
| Polythiazide (Renese) | 2 to 4 | 1 |
| Indapamide (Lozol†) | 1.25 to 2.5 | 1 |
| Metolazone (Mykrox) | 0.5 to 1.0 | 1 |
| Metolazone (Zaroxolyn) | 2.5 to 5 | 1 |
| Loop Diuretics | ||
| Bumetanide (Bumex†) | 0.5 to 2 | 2 |
| Furosemide (Lasix†) | 20 to 80 | 2 |
| Torsemide (Demadex†) | 2.5 to 10 | 1 |
| Potassium-Sparing Diuretics | ||
| Amiloride (Midamor†) | 5 to 10 | 1 to 2 |
| Triamterene (Dyrenium) | 50 to 100 | 1 to 2 |
| Aldosterone Receptor Blockers | ||
| Eplerenone (Inspra) | 50 to 100 | 1 |
| Spironolactone (Aldactone†) | 25 to 50 | 1 |
| β-Blockers | ||
| Atenolol (Tenormin†) | 25 to 100 | 1 |
| Betaxolol (Kerlone†) | 5 to 20 | 1 |
| Bisoprolol (Zebeta†) | 2.5 to 10 | 1 |
| Metoprolol (Lopressor†) | 50 to 100 | 1 to 2 |
| Metoprolol extended release (Toprol XL) | 50 to 100 | 1 |
| Nadolol (Corgard†) | 40 to 120 | 1 |
| Propranolol (Inderal†) | 40 to 160 | 2 |
| Propranolol long-acting (Inderal LA†) | 60 to 180 | 1 |
| Timolol (Blocadren†) | 20 to 40 | 2 |
| β-Blockers with Intrinsic Sympathomimetic Activity | ||
| Acebutolol (Sectral†) | 200 to 800 | 2 |
| Penbutolol (Levatol) | 10 to 40 | 1 |
| Pindolol (generic) | 10 to 40 | 2 |
| Combined α-Blockers and β-Blockers | ||
| Carvedilol (Coreg) | 12.5 to 50 | 2 |
| Labetalol (Normodyne, Trandate†) | 200 to 800 | 2 |
| Angiotensin-Converting Enzyme Inhibitors | ||
| Benazepril (Lotensin†) | 10 to 40 | 1 |
| Captopril (Capoten†) | 25 to 100 | 2 |
| Enalapril (Vasotec†) | 5 to 40 | 1 to 2 |
| Fosinopril (Monopril) | 10 to 40 | 1 |
| Lisinopril (Prinivil, Zestril†) | 10 to 40 | 1 |
| Moexipril (Univasc) | 7.5 to 30 | 1 |
| Perindopril (Aceon) | 4 to 8 | 1 |
| Quinapril (Accupril) | 10 to 40 | 1 |
| Ramipril (Altace) | 2.5 to 20 | 1 |
| Trandolapril (Mavik) | 1 to 4 | 1 |
| Angiotensin II Antagonists | ||
| Candesartan (Atacand) | 8 to 32 | 1 |
| Eprosartan (Teveten) | 400 to 800 | 1 to 2 |
| Irbesartan (Avapro) | 150 to 300 | 1 |
| Losartan (Cozaar) | 25 to 100 | 1 to 2 |
| Olmesartan (Benicar) | 20 to 40 | 1 |
| Telmisartan (Micardis) | 20 to 80 | 1 |
| Valsartan (Diovan) | 80 to 320 | 1 to 2 |
| CCBs: Nondihydropyridines | ||
| Diltiazem extended release (Cardizem CD, Dilacor XR, Tiazac†) | 180 to 420 | 1 |
| Diltiazem extended release (Cardizem LA) | 120 to 540 | 1 |
| Verapamil immediate release (Calan, Isoptin†) | 80 to 320 | 2 |
| Verapamil long-acting (Calan SR, Isoptin SR†) | 120 to 480 | 1 to 2 |
| Verapamil controlled onset, extended release (Covera HS, Verelan PM) | 120 to 360 | 1 |
| CCB: Dihydropyridines | ||
| Amlodipine (Norvasc) | 2.5 to 10 | 1 |
| Felodipine (Plendil) | 2.5 to 20 | 1 |
| Isradipine (DynaCirc CR) | 2.5 to 10 | 2 |
| Nicardipine sustained release (Cardene SR) | 60 to 120 | 2 |
| Nifedipine long-acting (Adalat CC, Procardia XL) | 30 to 60 | 1 |
| Nisoldipine (Sular) | 10 to 40 | 1 |
| α1-Blockers | ||
| Doxazosin (Cardura) | 1 to 16 | 1 |
| Prazosin (Minipress†) | 2 to 20 | 2 to 3 |
| Terazosin (Hytrin) | 1 to 20 | 1 to 2 |
| Central α2-Agonists and Other Centrally Acting Drugs | ||
| Clonidine (Catapres†) | 0.1 to 0.8 | 2 |
| Clonidine patch (Catapres-TTS) | 0.1 to 0.3 | 1 weekly |
| Methyldopa (Aldomet†) | 250 to 1000 | 2 |
| Reserpine (generic) | 0.05 to 0.25 | 1 |
| Guanfacine (Tenex†) | 0.5 to 2 | 1 |
| Direct Vasodilators | ||
| Hydralazine (Apresoline†) | 25 to 100 | 2 |
| Minoxidil (Loniten†) | 2.5 to 80 | 1 to 2 |
CCB = calcium channel blocker.
*In some patients treated once daily, the antihypertensive effect may diminish toward the end of the dosing interval (trough effect). BP should be measured just before dosing to determine if satisfactory BP control is obtained. Accordingly, an increase in dosage or frequency may need to be considered. These dosages may vary from those listed in the Physicians’ Drug Reference, 51st ed.
† Available now or soon to become available in generic preparations.
Adapted with permission from Chobanian AV, Bakris GL, Black HR, et al: Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC-7 Report. JAMA 289:2560, 2003.
Table 8-6 Combination Drugs for Hypertension
| Combination Type | Fixed-Dose Combination (mg)* | Trade Name |
|---|---|---|
| ACEIs and CCB | Amlodipine-benazepril hydrochloride (2.5/10, 5/10, 5/20, 10/20) | Lotrel |
| Enalapril-felodipine (5/5) | Lexxel | |
| Trandolapril-verapamil (2/180, 1/240, 2/240, 4/240) | Tarka | |
| ACEIs and diuretics | Benazepril-hydrochlorothiazide (5/6.25, 10/12.5, 20/12.5, 20/25) | Lotensin HCT |
| Captopril-hydrochlorothiazide (25/15, 25/25, 50/15, 50/25) | Capozide | |
| Enalapril-hydrochlorothiazide (5/12.5, 10/25) | Vaseretic | |
| Fosinopril-hydrochlorothiazide (10/12.5, 20/12.5) | Monopril/HCT | |
| Lisinopril-hydrochlorothiazide (10/12.5, 20/12.5, 20/25) | Prinzide, Zestoretic | |
| Moexipril-hydrochlorothiazide (7.5/12.5, 15/25) | Uniretic | |
| Quinapril-hydrochlorothiazide (10/12.5, 20/12.5, 20/25) | Accuretic | |
| ARBs and diuretics | Candesartan-hydrochlorothiazide (16/12.5, 32/12.5) | Atacand HCT |
| Eprosartan-hydrochlorothiazide (600/12.5, 600/25) | Teveten-HCT | |
| Irbesartan-hydrochlorothiazide (150/12.5, 300/12.5) | Avalide | |
| Losartan-hydrochlorothiazide (50/12.5, 100/25) | Hyzaar | |
| Olmesartan medoxomil-hydrochlorothiazide (20/12.5, 40/12.5, 40/25) | Benicar HCT | |
| Telmisartan-hydrochlorothiazide (40/12.5, 80/12.5) | Micardis-HCT | |
| Valsartan-hydrochlorothiazide (80/12.5, 160/12.5, 160/25) | Diovan-HCT | |
| BBs and diuretics | Atenolol-chlorthalidone (50/25, 100/25) | Tenoretic |
| Bisoprolol-hydrochlorothiazide (2.5/6.25, 5/6.25, 10/6.25) | Ziac | |
| Metoprolol-hydrochlorothiazide (50/25, 100/25) | Lopressor HCT | |
| Nadolol-bendroflumethiazide (40/5, 80/5) | Corzide | |
| Propranolol LA-hydrochlorothiazide (40/25, 80/25) | Inderide LA | |
| Timolol-hydrochlorothiazide (10/25) | Timolide | |
| Centrally acting drug and diuretic | Methyldopa-hydrochlorothiazide (250/15, 250/25, 500/30, 500/50) | Aldoril |
| Reserpine-chlorthalidone (0.125/25, 0.25/50) | Demi-Regroton, Regroton | |
| Reserpine-chlorothiazide (0.125/250, 0.25/500) | Diupres | |
| Reserpine-hydrochlorothiazide (0.125/25, 0.125/50) | Hydropres | |
| Diuretic and diuretic | Amiloride-hydrochlorothiazide (5/50) | Moduretic |
| Spironolactone-hydrochlorothiazide (25/25, 50/50) | Aldactazide | |
| Triamterene-hydrochlorothiazide (37.5/25, 75/50) | Dyazide, Maxzide |
BB = β-blocker; ACEI = angiotensin-converting enzyme inhibitor; ARB = angiotensin-receptor blocker; CCB = calcium channel blocker.
* Some drug combinations are available in multiple fixed doses. Each drug dose is reported in milligrams.
Adapted with permission from Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC-7 Report. JAMA 289:2560, 2003.
Management of Severe Hypertension
For purposes of characterizing treatment urgency, severe hypertension is characterized as either a hypertensive emergency with target organ injury (e.g., myocardial ischemia, stroke, pulmonary edema) or a hypertensive urgency with severe elevations in BP not yet associated with target organ damage. Chronic elevations in BP, even when of a severe nature, do not necessarily require urgent intervention and often may be managed with oral antihypertensive therapy on an outpatient basis. In contrast, a hypertensive emergency necessitates immediate therapeutic intervention, most often in an intensive care setting, with intravenous antihypertensive therapy and invasive arterial BP monitoring. In the most extreme cases of malignant hypertension, severe elevations in BP may be associated with retinal hemorrhages, papilledema, and evidence of encephalopathy, which may include headache, vomiting, seizure, and/or coma. Progressive renal failure and cardiac decompensation are additional clinical features characteristic of the most severe hypertensive emergencies.
The favored parenteral drug for rapid treatment of hypertensive emergencies remains sodium nitroprusside (Table 8-7). An NO donor, sodium nitroprusside induces arterial and venous dilation, providing rapid and predictable reductions in systemic BP. Prolonged administration of large doses may be associated with cyanide or thiocyanate toxicity; however, this is rarely a concern in the setting of acute hypertensive emergencies. Although less potent and predictable than sodium nitroprusside, NTG, another NO donor, may be preferable in the setting of myocardial ischemia or after coronary artery bypass grafting (CABG). NTG preferentially dilates venous capacitance beds as opposed to arterioles; however, rapid onset of tolerance limits the efficacy of sustained infusions to maintain BP control. Nicardipine, a parenteral dihydropyridine calcium channel blocker, and fenoldopam, a selective dopamine-1 (D1)-receptor antagonist, have been utilized increasingly in select patient populations after CABG and in the setting of renal insufficiency, respectively.11


Table 8-7 Parenteral Drugs for Treatment of Hypertensive Emergencies
Rights were not granted to include this table in electronic media. Please refer to the printed book.
Reproduced with permission from Chobanian AV, Bakris GL, Black HR, et al. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42:1206, 2003.
PHARMACOTHERAPY FOR ACUTE AND CHRONIC HEART FAILURE
Chronic heart failure is one major cardiovascular disorder that continues to increase in incidence and prevalence, both in the United States and worldwide. It affects nearly 5 million persons in the United States, and roughly 550,000 new cases are diagnosed each year.12 Currently, 1% of those 50 to 59 years of age and 10% of individuals older than 80 have heart failure. Because heart failure is primarily a disease of the elderly, its prevalence is projected to increase twofold to threefold over the next decade, as the median age of the U.S. population continues to increase. The increasingly prolonged survival of patients with various cardiovascular disorders that culminate in ventricular dysfunction (e.g., patients with coronary artery disease are living longer rather than dying acutely with myocardial infarction) further compounds the heart failure epidemic. Despite improvements in the understanding of the neurohormonal mechanisms underlying its pathophysiology and remarkable advances made in pharmacologic therapy, heart failure continues to cost the United States an estimated $38 billion annually in medical expenditures, and it contributes to approximately 250,000 deaths per year. Given the public health impact of the disease and the rapid pace of therapeutic advances, it is essential that the perioperative physician remain aware of contemporary clinical practice for the benefit of those patients with chronic heart failure presenting to the operating room or intensive care unit.
Heart Failure Classification
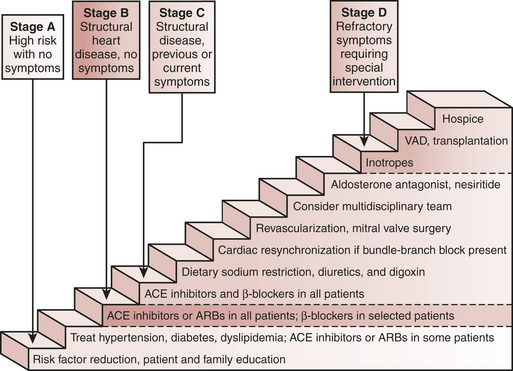
The ACC/AHA updated guidelines for evaluating and managing heart failure include a new, four-stage classification system emphasizing both the evolution and progression of the disease (Box 8-5). It calls attention to patients with preclinical stages of heart failure to focus on halting disease progression. The staging system is meant to complement, not replace, the widely used New York Heart Association (NYHA) classification, a semiquantitative index of functional classification that categorizes patients with heart failure by the severity of their symptoms. The NYHA classification remains useful clinically because it reflects symptoms, which in turn correlate with quality of life and survival. The new classification system for heart failure, recognizing its progressive course and identifying those who are at risk, reinforces the importance of determining the optimal strategy for neurohormonal antagonism in an attempt to improve the natural history of the syndrome.
BOX 8-5 ACC/AHA Four-Stage Classification and Management Recommendations
Adapted from permission from Clinical update: New guidelines for evaluating and managing heart failure. Women’s Health in Primary Care 5(2):105, 2002.
Stage C Structural heart disease with prior or current symptoms of heart failure.
ACE = angiotensin-converting enzyme; LV = left ventricular.
β-Blockers are relatively contraindicated in patients with bronchospastic pulmonary disease.
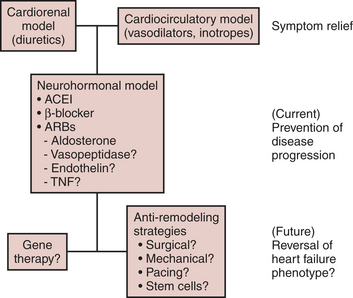
Heart failure remains the final common pathway for coronary artery disease, hypertension, valvular heart disease, and cardiomyopathy, in which the natural history results in symptomatic or asymptomatic left ventricular dysfunction. The neurohormonal responses to impaired cardiac performance (salt and water retention, vasoconstriction, sympathetic stimulation) are initially adaptive but, if sustained, become maladaptive, resulting in pulmonary congestion and excessive afterload. This, in turn, leads to a vicious cycle of increases in cardiac energy expenditure and worsening of pump function and tissue perfusion (Table 8-8). Although the cardiorenal and cardiocirculatory branches of this neurohormonal hypothesis of heart failure were the original foundation for the use of diuretics, vasodilators, and inotropes, respectively, seminal information in the early 1990s emerged from large, randomized clinical trials that showed angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers, but not most other vasodilators, prolonged survival in patients with heart failure. In a similar fashion, the use of β-blockers, despite their negative inotropic effects, improved morbidity and mortality in randomized controlled trials.
Table 8-8 Neurohormonal Effects of Impaired Cardiac Performance on the Circulation
| Response | Short-Term Effects | Long-Term Effects |
|---|---|---|
| Salt and water retention | Augments preload | Pulmonary congestion, edema |
| Vasoconstriction | Maintains blood pressure for perfusion of vital organs | Exacerbates pump dysfunction (excessive afterload), increases cardiac energy expenditure |
| Sympathetic stimulation | Increases heart rate and ejection | Increases energy expenditure |
Modified from Katz AM: Heart failure. In Fozzard HA, Haber E, Jennings RB: The Heart and Cardiovascular System: Scientific Foundations, 2nd ed. New York, Raven, 1992, pp 333-353.
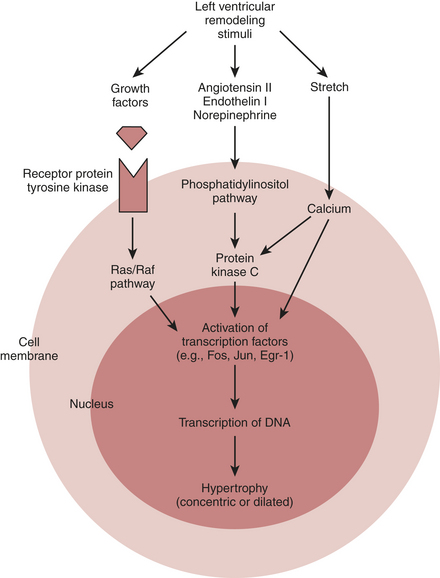
The finding that low-dose aldosterone antagonists added to conventional therapy for heart failure reduced mortality in patients with severe heart failure suggests that there is more to the neurohormonal hypothesis of drug efficacy than cardiorenal and hemodynamic effects alone. Taken together with evidence from basic investigations showing that Ang II is a growth factor and a vasoconstrictor, the clinical data promoted a shift in focus from cardiorenal and cardiocirculatory processes toward cardiac remodeling as the central component in the progression of this neurohormone-mediated cardiac syndrome.13 The renin-angiotensin-aldosterone system (RAAS), excess sympathetic activity, endothelin, and various cytokines all have been implicated as stimuli of proliferative signaling that contribute to maladaptive cardiac growth. Accordingly, ventricular remodeling, or the structural alterations of the heart in the form of dilatation and hypertrophy (Box 8-6), in addition to the counterregulatory hemodynamic responses, lead to progressive ventricular dysfunction and represent the target of current therapeutic interventions (Fig. 8-2).
BOX 8-6 Mechanical Disadvantage Created by Left Ventricular Remodeling
Adapted from Mann DL: Mechanisms and models in heart failure: an combinatorial approach. Circulation 100:999–1008, 1999.
Pathophysiologic Role of the Renin-Angiotensin System in Heart Failure
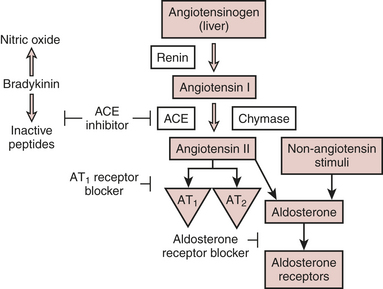
The renin-angiotensin system (RAS) is one of several neuroendocrine systems that are activated in patients with heart failure. The RAS is also an important mediator in the progression of heart failure. In the short term, the juxtaglomerular cells of the kidney release the proteolytic enzyme renin in response to a decrease in BP or renal perfusion (e.g., hemorrhage) generating Ang I from circulating angiotensinogen. ACE cleavage of Ang II from Ang I in the lung produces circulating Ang II. Acutely, Ang II acts as a potent arteriolar and venous vasoconstrictor to return BP and filling pressure to baseline, respectively. Ang II also stimulates the release of aldosterone from the adrenal cortex and antidiuretic hormone from the posterior pituitary. Both contribute to increases in blood volume through their effects on the kidney to promote salt and water reabsorption, respectively. In the long term, elevations in Ang II lead to sodium and fluid retention and increases in systemic vascular resistance, which contribute to symptoms of heart failure, pulmonary congestion, and hemodynamic decompensation (Fig. 8-3).
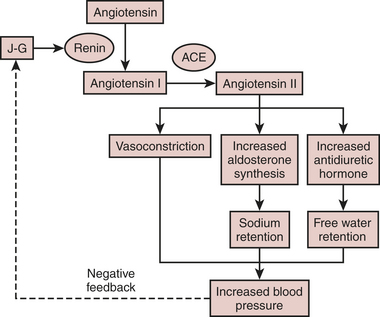
Figure 8-3 Basic pathway of the renin-angiotensin-aldosterone system (RAAS).
(From Jaski BE: Basis of Heart Failure: A Problem Solving Approach. Boston, Kluwer Academic Publishers, 2000, with kind permission of Springer Science and Business Media.)
The effects of Ang II on its receptors AT1 and AT2 are well appreciated. The AT1 receptor is involved in several effects that lead to adverse cardiovascular outcomes. Activation of AT1 receptors promotes aldosterone and vasopressin secretion with concomitant increases in salt and water reabsorption through the kidneys, vasoconstriction, catecholamine release, and cell growth and proliferation of cardiovascular tissue. Stimulation of AT2 receptors, on the other hand, results in natriuresis, vasodilation, release of bradykinin and NO, and cell growth inhibition or apoptosis. The Ang II that is formed locally in the heart acts primarily through AT1 receptors located on myocytes and fibroblasts where it participates in the regulation of cardiac remodeling. Through complex cascades of intracellular signal transduction that activate protein transcription factors within the nucleus initiating the creation of RNA transcripts, the long-term effects of intracardiac Ang II on the AT1 receptor result in cardiomyocyte hypertrophy, fibroblast proliferation, and extracellular matrix deposition (Fig. 8-4). These processes contribute to progressive left ventricular remodeling and left ventricular dysfunction characteristic of heart failure.
Angiotensin-Converting Enzyme Inhibitors
clinical evidence
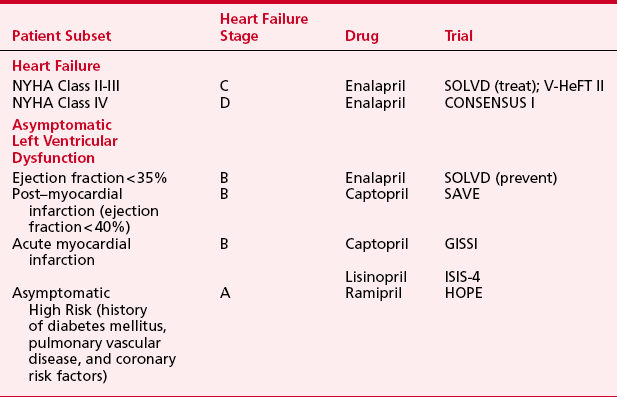
Evidence supporting the beneficial use of ACE inhibitors in patients with heart failure comes from various randomized, placebo-controlled clinical trials (Table 8-9). Initially this class of drugs was evaluated for treatment of symptomatic heart failure (SOLVD, V-HeFT, CONSENSUS). Patients with NYHA class II to IV heart failure treated with ACE inhibitors had reductions in mortality ranging from 16% to 31%. Subsequently, ACE inhibitors were also found to improve outcome for asymptomatic patients with left ventricular systolic dysfunction in the following categories: patients with ejection fractions less than 35% due to cardiomyopathy, patients within 2 weeks after myocardial infarction with ejection fractions less than 40%, and patients presenting within the first 24 hours of myocardial infarction regardless of ejection fraction. Results from the Heart Outcomes Prevention Evaluation (HOPE) study have further expanded the indications for this class of agents to include asymptomatic, high-risk patients to prevent new-onset heart failure.14 In patients with diabetes or peripheral vascular disease and an additional atherosclerotic risk factor, but without clinical heart failure or systolic dysfunction, ramipril (10 mg/day) reduced the heart failure risk by 23%. Together, these data endorse the use of ACE inhibitors as first-line therapy for a broad spectrum of patients, including those with left ventricular systolic dysfunction, with or without symptoms, and in high-risk patients with vascular disease and/or diabetes, in addition to those with the traditional coronary risk factors. Since the beginning of these trials, the rationale for the use of ACE inhibitors has expanded from a reduction in the progression of clinical heart failure through ACE inhibitor–mediated vasodilatory action to acknowledgment that ACE inhibitors also directly affect the cellular mechanisms responsible for progressive myocardial pathology.
mechanisms of action
ACE inhibitors act by inhibiting one of several proteases responsible for cleaving the decapeptide, Ang I, to form the octapeptide Ang II. Because ACE is also the enzyme that degrades bradykinin, ACE inhibitors lead to increased circulating and tissue levels of bradykinin (Fig. 8-5). ACE inhibitors have several useful effects in chronic heart failure. They are potent vasodilators through decreasing Ang II and norepinephrine and increasing bradykinin, NO, and prostacyclin. By reducing the secretion of aldosterone and antidiuretic hormone (ADH), ACE inhibitors also reduce salt and water reabsorption from the kidney. ACE inhibitors reduce release of norepinephrine from sympathetic nerves by acting on AT1 receptors at the nerve terminal. Within tissue, ACE inhibitors inhibit Ang II production and thus attenuate Ang II-mediated cardiomyocyte hypertrophy and fibroblast hyperplasia. Clinical evidence supporting an ACE inhibitor-mediated role in cardiac remodeling comes from comparative studies of enalapril versus placebo (SOLVD trial) and enalapril versus hydralazine isosorbide dinitrate (VHeft II trial).
Angiotensin II Receptor Blockers for Heart Failure
pathophysiology/mechanism of action
Although ACE inhibitors reduce mortality, many patients will not tolerate their side effects. ACE inhibitors incompletely antagonize Ang II. These factors have prompted the development of specific Ang II receptor blockers in the pharmacologic treatment of heart failure. Non–ACE-generated Ang II within the myocardium contributes to left ventricular remodeling and progression of heart failure through AT1 receptor effects. Selective AT1 blockers prevent Ang II from acting on the cell, preventing vasoconstriction, sodium retention, and release of norepinephrine and delaying or preventing left ventricular hypertrophy and fibrosis. AT2 receptors remain unaffected, and their actions, including NO release, remain intact.15
Aldosterone Receptor Antagonists
clinical evidence
Two large-scale trials have demonstrated improved outcomes with aldosterone-receptor antagonism in chronic heart failure. The Randomized Aldactone Evaluation Study (RALES), conducted in more than 1600 symptomatic heart failure (e.g., stage C, NYHA III-IV) patients, showed the efficacy of spironolactone (26 mg/day) (in combination with standard therapy: ACE inhibitor, loop diuretic with or without digoxin and a β-blocker). Eplerenone is a new aldosterone antagonist that lacks some of spironolactone’s common side effects. The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHSUS), conducted in more than 6600 patients with symptomatic heart failure within 3 to 14 days after myocardial infarction, showed that eplerenone (25 to 50 mg/day) in combination with ACE inhibitor, loop diuretic, and β-blocker reduced all-cause mortality (P = .008), death from cardiovascular causes (P = .0002), and hospitalization for cardiovascular events.16,17
β-Adrenergic Receptor Antagonists
How β-Adrenergic Receptor Blockers Influence the Pathophysiology of Heart Failure
clinical practice
The three agents with clinical trial evidence for improved morbidity and mortality in patients with heart failure are carvedilol, metoprolol CR/XL, and bisoprolol.18 Starting doses of β-blockers should be small to minimize worsening of heart failure symptoms, hypotension, and bradycardia. The dose should be doubled every 1 to 2 weeks, as tolerated, until target doses shown to be effective in large trials are achieved. Although it is recommended that β-blocker therapy be continued indefinitely in patients with heart failure, if it is to be electively stopped, a slow downtitration is preferred. Acute withdrawal of β-blocker therapy in the face of high adrenergic tone may result in sudden cardiac death. The adverse effects of β-blocker therapy include fatigue, dizziness, hypotension, and bradycardia. Because the absolute risk of adverse events is small compared with the overall risk reduction of cardiovascular death, few patients have been withdrawn from β-blocker therapy.
Adjunctive Drugs
Digoxin
The efficacy of digoxin for symptomatic heart failure was shown in randomized, controlled trials. The Digitalis Investigators Group (DIG) trial, enrolling more than 6500 patients with an average follow-up of 37 months, showed that digoxin reduced the incidence of heart failure exacerbations. Although the study showed no difference in survival in patients with an ejection fraction less than 45% receiving either digoxin or placebo, the combined endpoint of death or hospitalization for heart failure was significantly reduced in patients who received digoxin (27% vs. 35%; relative risk, 0.72; 95% confidence interval, 0.66 to 0.79). Efficacy of digoxin in patients with mildly symptomatic heart failure was shown in pooled results from the Prospective Randomized Study of Ventricular Function (PROVED) and the Randomized Assessment of Digoxin and Inhibitors of Angiotensin-Converting Enzyme (RADIANCE) trials. Patients randomized to digoxin withdrawal had an increased likelihood of treatment failure compared with those who continued to receive digoxin, suggesting that patients with left ventricular systolic dysfunction benefit from digoxin (or, at least, do not benefit from digoxin withdrawal), even when they have only mild symptoms. Accordingly, digoxin is recommended for symptomatic heart failure unless contraindicated. Together with ACE inhibitors, β-blockers, and diuretics, digoxin should be added to the therapeutic armamentarium. Ideally, serum digoxin concentration should remain between 0.7 and 1.1 ng/mL. In the elderly patient with renal insufficiency, severe conduction abnormalities, or acute coronary syndromes, even a low dose of 0.125 mg/day should be used with extra caution.19
Future Therapy
Among the promising nonpharmacologic therapies for the management of heart failure are the implantable defibrillators and biventricular pacemakers. In the COMPANION trial (The Comparison of Medical Therapy, Pacing, and Defibrillation in Heart Failure), cardiac resynchronization therapy with a pacemaker combined with an implantable defibrillator significantly decreased the likelihood of death from or hospitalization for heart failure when compared with conventional pharmacologic therapy.20
Management of Acute Exacerbations of Chronic Heart Failure
Patients with chronic heart failure, despite good medical management, may experience episodes of pulmonary edema or other signs of acute volume overload. These patients may require hospitalization for intensive management if diuretics fail to relieve their symptoms. Other patients may experience exacerbations of heart failure associated with acute myocardial ischemia or infarction, worsening valvular dysfunction, infections (including myocarditis), or failure to maintain an established drug regimen. Fonarow and associates described a risk stratification system for in-hospital mortality in acutely decompensated heart failure using data from a national registry. Low-, intermediate-, and high-risk patients with mortality ranging from 2.1% to 21.9% were identified using blood urea nitrogen, creatinine, and systolic BP on admission. These patients will require all the standard medications, as outlined in previous sections, and may also require infusions of vasodilators or positive inotropic drugs.21
Nesiritide
As the clinical severity of heart failure increases, the concentrations of BNP in blood also increase. As a result, measurements of BNP in blood have been used to evaluate new onset of dyspnea (to distinguish between lung disease and heart failure). BNP concentrations in blood increase with decreasing left ventricular ejection fraction; therefore, measurements of this mediator have been used to estimate prognosis. BNP concentrations decline in response to therapy with ACE inhibitors, Ang II antagonists, and aldosterone antagonists.
In addition, recombinant BNP has been released as a drug (nesiritide) indicated for patients with acute heart failure and dyspnea with minimal activity. Nesiritide produces arterial and venous dilatation through increasing cGMP. Nesiritide does not increase HR and has no effect on cardiac inotropy. It has a rapid onset of action and a short elimination half-life (15 minutes). In clinical studies, loading doses have ranged from 0.25 to 2 μg/kg and maintenance doses have ranged from 0.005 to 0.03 μg/kg/min. Studies have shown that nesiritide reduces symptoms of acute decompensated heart failure similarly to NTG, without development of acute tolerance. Patients receiving nesiritide experienced fewer adverse events than those receiving NTG. However, the mortality rate at 6 months was higher in the patients receiving nesiritide than in the NTG group.22 Compared with dobutamine, nesiritide was associated with fewer instances of ventricular tachycardia or cardiac arrest.
Inotropes
Nevertheless, placebo-controlled studies suggest that there may be no role whatsoever for discretionary administration of positive inotropes to patients with chronic heart failure. In one study, 951 hospitalized patients with decompensated chronic heart failure who did not require intravenous inotropic support were assigned to receive a 48-hour infusion of either milrinone or saline. Meanwhile, all patients received ACE inhibitors and diuretics as deemed necessary. Total hospital days did not differ between groups; however, those receiving milrinone were significantly more likely to require intervention for hypotension or to have new atrial arrhythmias. A subanalysis of these results found that patients suffering from ischemic cardiomyopathy were particularly subject to adverse events from milrinone (a 42% incidence of death or rehospitalization versus 36% for placebo). At the present, positive inotropic drug support can be recommended only when there is no alternative. Thus, dobutamine and milrinone continue to be used to treat low CO in decompensated heart failure, but only in selected patients.23
Low-Output Syndrome
Acute heart failure is a frequent concern of the cardiac anesthesiologist, particularly at the time of separation from cardiopulmonary bypass (CPB). The new onset of ventricular dysfunction and a low CO state after aortic clamping and reperfusion is a condition with more pathophysiologic similarity to cardiogenic shock than to chronic heart failure and is typically treated with positive inotropic drugs, vasopressors (or vasodilators), if needed, and/or mechanical assistance. The latter more commonly takes the form of intra-aortic balloon counterpulsation and less commonly includes one of the several available ventricular assist devices.
Specific Drugs for Treating the Low-Output Syndrome
Whereas all positive inotropic drugs increase the strength of contraction in noninfarcted myocardium, mechanisms of action differ. These drugs can be divided into those that increase cyclic adenosine monophosphate (cAMP) (directly or indirectly) for their mechanisms of action and those that do not. The agents that do not depend on cAMP form a diverse group, including cardiac glycosides, calcium salts, calcium sensitizers, and thyroid hormone. In contrast to chronic heart failure, cardiac glycosides are not used for this indication, owing to their limited efficacy and narrow margin of safety. Calcium salts continue to be administered for ionized hypocalcemia and hyperkalemia, which are common occurrences during and after cardiac surgery. Increased Ca2+ in buffer solutions bathing cardiac muscle in vitro unquestionably increase inotropy. Calcium sensitizers, specifically levosimendan, function by binding to troponin C in a calcium-dependent fashion. Thus, levosimendan does not impair diastolic function because its affinity for troponin C declines with Ca2+ during diastole. Although several reports have described the successful use of levosimendan in patients recovering from CABG, clinical experience with this agent remains limited and there is no consensus as to how and when this agent should be used, relative to other, better established agents.24
Intravenous thyroid hormone (T3, or liothyronine) has been studied extensively as a positive inotrope in cardiac surgery. There are multiple studies supporting the existence of euthyroid “sick” syndrome with persistent reduced concentrations of T3 in blood after cardiac surgery in both children and adults. There are also data suggesting that after ischemia and reperfusion, T3 increases inotropy faster than and as potently as isoproterenol. Nevertheless, randomized controlled clinical trials have failed to show efficacy of T3 after CABG.
Pharmacologic Treatment of Diastolic Heart Failure
Abnormal diastolic ventricular function is a common cause of clinical heart failure. As many as one in three patients presenting with clinical signs of chronic heart failure has a normal or near-normal ejection fraction (≥40%). The risk of diastolic heart failure increases with age, approaching 50% in patients older than 70 years old. Diastolic heart failure is also more common in females and in patients with hypertension or diabetes mellitus. Although the prognosis of patients with diastolic heart failure is better than for systolic heart failure (5% to 8% vs. 10% to 15% annual mortality, respectively), the complication rate is the same. The 1-year readmission rate for patients with isolated diastolic heart failure approaches 50%.
In contrast to the large randomized trials that have led to the treatment guidelines for systolic heart failure, there are few randomized, double-blind, placebo-controlled, multicenter trials performed in patients with diastolic heart failure. Consequently, the guidelines are based on clinical experience, small clinical studies, and an understanding of the pathophysiologic mechanisms. The general approach to treating diastolic heart failure has three main components. First, treatment should reduce symptoms, primarily by lowering pulmonary venous pressure during rest and exercise by reducing left ventricular volume, maintaining AV synchrony, and increasing the duration of diastole by reducing HR. Second, treatment should target the underlying causes of diastolic heart failure. Specifically, ventricular remodeling should be reversed by controlling hypertension, replacing stenotic aortic valves, and treating ischemia. Third, treatment should target the underlying mechanisms that are altered by the disease processes, mainly neurohormonal activation. Drug treatment of diastolic heart failure with respect to these three goals is shown in Table 8-10.
| Goal | Management Strategy | Drugs/Recommended Doses |
|---|---|---|
| Reduce the congestive state |
ACE = angiotensin-converting enzyme.
Adapted from Aurigemma GP, Gaasch WH: Clinical practice. Diastolic heart failure. N Engl J Med 351:1097, 2004.
Except in the presence of acute diastolic heart failure, positive inotropic and chronotropic agents should be avoided because they may worsen diastolic function by increasing contractile force and HR, or by increasing calcium concentrations in diastole. However, in the short-term management of acute diastolic dysfunction or heart failure (e.g., post CPB), β-adrenergic agonists (e.g., epinephrine) and phosphodiesterase inhibitors (e.g., milrinone) enhance calcium sequestration by the sarcoplasmic reticulum and thereby promote a more rapid and complete myocardial relaxation between beats.25,26
Current Clinical Practice
The pharmacotherapy of heart failure begins with primary prevention of left ventricular dysfunction. Because hypertension and coronary artery disease are leading causes of left ventricular dysfunction, adequate treatment of both hypertension and hypercholesterolemia has been endorsed after encouraging results in prevention trials. Limitation of neurohormonal activation with ACE inhibitors, and possibly β-blockers, should be initiated in diabetic, hypertensive, and hypercholesterolemic patients (AHA/ACC, stage A heart failure) who are at increased risk for cardiovascular events, despite normal contractile function, in order to reduce the onset of new heart failure (HOPE trial). In patients with asymptomatic left ventricular dysfunction (ejection fraction = 40%) (stage B), treatment with ACE inhibitors and β-blockers can blunt the disease progression. In the symptomatic patient with heart failure (stage C), diuretics are titrated to relieve symptoms of pulmonary congestion and peripheral edema and achieve a euvolemic state whereas ACE inhibitors and β-blockers are recommended to blunt disease progression. Although digoxin has no effect on patient survival, it may be considered in stage C if the patient remains symptomatic despite adequate doses of ACE inhibitors and diuretics. In general, the primary treatment objectives for stages A to C heart failure are to (1) improve quality of life, (2) reduce morbidity, and (3) reduce mortality. At this time, the most important factor affecting long-term outcome is blunting of neurohormonal stimulation, because this mediates disease progression. Pharmacologic therapy in stage D, or patients with severe, decompensated heart failure, is based on hemodynamic status to alleviate symptoms with diuretics, vasodilators, and, in palliative circumstances, intravenous inotropic infusions. ACE inhibitors and β-blockers are also incorporated in the treatment regimen to retard disease progression through reductions in ventricular enlargement, vascular hypertrophy, and ventricular arrhythmias27 (Fig. 8-6).
PHARMACOTHERAPY FOR CARDIAC ARRHYTHMIAS
Perhaps the most widely used electrophysiologic and pharmacologic classification of antiarrhythmic drugs is that proposed by Vaughan Williams (Table 8-11). There is, however, substantial overlap in pharmacologic and electrophysiologic effects of specific agents among the classes, and the linkage between observed electrophysiologic effects and the clinical antiarrhythmic effect is often tenuous.28
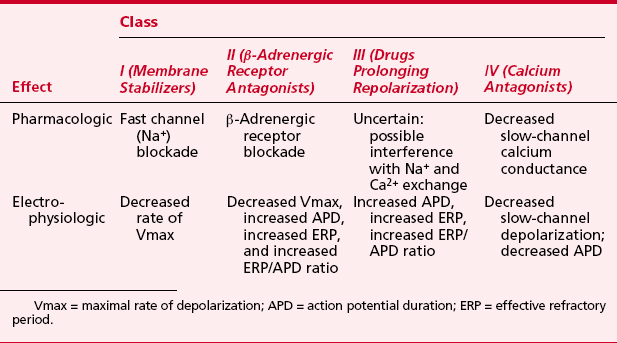
Class I Antiarrhythmic Drugs: Sodium Channel Blockers
Class IA
procainamide
Administered intravenously, procainamide is an effective emergency treatment for ventricular arrhythmias, especially after lidocaine failure, but, recently, amiodarone has become a more popular drug for intravenous suppression of ventricular arrhythmias.29 Dosage is 100 mg, or approximately 1.5 mg/kg given at 5-minute intervals until the therapeutic effect is obtained or a total dose of 1 g or 15 mg/kg is given (Tables 8-12 and 8-13). Arterial pressure and the ECG should be monitored continuously during loading and administration stopped if significant hypotension occurs or if the QRS complex is prolonged by 50% or more. Maintenance infusion rates are 2 to 6 mg/min to maintain therapeutic plasma concentrations of 4 to 8 μg/mL.
Table 8-12 Intravenous Supraventricular Antiarrhythmic Therapy
| Class I | Procainamide (IA)—converts acute atrial fibrillation, suppresses PACs and precipitation of atrial fibrillation/flutter, converts accessory pathway SVT. 100 mg IV loading dose every 5 min until arrhythmia subsides or total dose of 15 mg/kg (rarely needed) with continuous infusion of 2 to 6 mg/min. |
| Class II | Esmolol—converts or maintains slow ventricular response in acute atrial fibrillation. 0.5 to 1 mg/kg loading dose with each 50 μg/kg/min increase in infusion, with infusions of 50 to 300 μg/kg/min. Hypotension and bradycardia are limiting factors. |
| Class III | Amiodarone—converts acute atrial fibrillation to sinus rhythm; 5 mg/kg IV over 15 min. |
| Ibutilide (Convert)—converts acute atrial fibrillation and flutter. | |
| Adults (>60 kg): 1 mg given over 10 min IV, may be repeated once. | |
| Adults (<60 kg) and children: 0.01 mg/kg given over 10 min IV, may be repeated once. | |
| Class IV | Verapamil—slow ventricular response to acute atrial fibrillation, converts AV node reentry SVT. 75 to 150 μg/kg IV bolus. |
| Diltiazem—slow ventricular response in acute atrial fibrillation, converts AV node reentry SVT. 0.25 μg/kg bolus, then 100 to 300 μg/kg/hr infusion. | |
| Others | Adenosine—converts AV node reentry SVT and accessory pathway SVT. Aids in diagnosis of atrial fibrillation and flutter. |
| Adults: 3 to 6 mg IV bolus, repeat with 6- to 12-mg bolus. | |
| Children: 100 μg/kg bolus, repeat with 200-μg/kg bolus. | |
| Increased dosage required with methylxanthines, decreased use required with dipyridamole. | |
| Digoxin—maintenance IV therapy for atrial fibrillation and flutter, slows ventricular response. | |
| Adults: 0.25-mg IV bolus followed by 0.125 mg every 1 to 2 hr until rate controlled, not to exceed 10 μg/kg in 24 hr. | |
| Children (<10 years of age): 10- to 30-μg/kg load given in divided doses over 24 hours. | |
| Maintenance: 25% of loading dose. |
Table 8-13 Intravenous Ventricular Antiarrhythmic Therapy
| Class I |
Adapted from Management of Cardiac Rhythm Disturbances. 46th Annual Refresher Course Lectures and Clinical Update Program. Park Ridge, IL: American Society of Anesthesiologists; 1995:No. 255.
Class IB
lidocaine
The direct electrophysiologic effects of lidocaine produce virtually all of its antiarrhythmic action. Lidocaine depresses the slope of phase 4 diastolic depolarization in Purkinje fibers and increases the ventricular fibrillation threshold. Lidocaine may be ineffective in hypokalemic patients.
Therapeutic plasma levels of lidocaine range from 1.5 to 5 μg/mL; signs of toxicity are frequent with concentrations above 9 μg/mL. An initial bolus dose of 1 to 1.5 mg/kg should be followed immediately by a continuous infusion of 20 to 50 μg/kg/min to prevent the “therapeutic hiatus” produced by the rapid redistribution half-life of lidocaine.30
Class II: β-Adrenergic Receptor Antagonists
Propranolol
The electrophysiologic effects of β-receptor antagonism are decreased automaticity, increased APD, primarily in ventricular muscle, and a substantially increased ERP in the AV node. β-Blockade decreases the rate of spontaneous (phase 4) depolarization in the SA node; the magnitude of this effect depends on the background sympathetic tone. Although resting HR is decreased by β-blockade, the inhibition of the increase of HR in response to exercise or emotional stress is much more marked. Automaticity in the AV node and more distal portions of the conduction system is also depressed. β-Blockade affects the ventricular fibrillation threshold variably, but it consistently reverses the fibrillation threshold-lowering effect of catecholamines.
Esmolol
Esmolol is rapidly metabolized in blood by hydrolysis of its methyl ester linkage. Its half-life in whole blood is 12.5 and 27.1 minutes in dogs and humans, respectively. The acid metabolite possesses a slight degree (1500 times less than esmolol) of β-antagonism. Esmolol is not affected by plasma cholinesterase; the esterase responsible is located in erythrocytes and is not inhibited by cholinesterase inhibitors, but it is deactivated by sodium fluoride. Of importance to clinical anesthesia, no metabolic interactions between esmolol and other ester molecules are known. Specifically, esmolol doses up to 500 μg/kg/min have not modified neuromuscular effects of succinylcholine.
Dosing begins at 25 μg/kg/min and is titrated to effect up to 250 μg/kg/min. Doses higher than this may cause significant hypotension due to reduced CO in patients. Esmolol is especially effective in treating acute onset atrial fibrillation or flutter perioperatively and results in both acute control of the ventricular response and conversion of the arrhythmia back to sinus rhythm.31
Class III: Agents That Block Potassium Channels and Prolong Repolarization
Amiodarone
The drug has a wide spectrum of effectiveness, including supraventricular, ventricular, and preexcitation arrhythmias. It may also be effective against ventricular tachycardia and ventricular fibrillation refractory to other treatment. Amiodarone has been approved by the AHA as the first-line antiarrhythmic agent in cardiopulmonary resuscitation. Amiodarone may be effective prophylactically in preventing atrial fibrillation postoperatively. It also can decrease the number of shocks in patients who have implantable cardioverter defibrillators compared with other antiarrhythmic drugs.32
Despite relatively widespread use of amiodarone, anesthetic complications have infrequently been reported. In two case reports, bradycardia and hypotension were prominent. One of the reports described profound resistance to the vasoconstrictive effects of α-adrenergic agonists. The slow decay of amiodarone in plasma and tissue makes such adverse reactions possible long after discontinuing its administration. Because T3 is reported to reverse electrophysiologic effects of amiodarone, T3 could possibly be used to reverse hemodynamic abnormalities, such as those described in these two case reports, although this theory has not been tested. Epinephrine has been shown to be more effective than dobutamine or isoproterenol in reversing amiodarone-induced cardiac depression.
Other Antiarrhythmic Agents
Digoxin
The main preparation of cardiac glycosides available is digoxin. Digoxin reaches peak effects in 1.5 to 2 hours but has a significant effect within 5 to 30 minutes. For undigitalized patients, the initial dose is 0.5 to 0.75 mg of digoxin, with subsequent doses of 0.125 to 0.25 mg. The usual total digitalizing dose ranges from 0.75 to 1.0 mg by the intravenous route. Digoxin is approximately 25% protein bound, and the therapeutic range of plasma concentrations is 0.5 to 2.0 ng/mL.
Adenosine
For clinical use, adenosine must be administered by a rapid intravenous bolus in a dose of 100 to 200 μg/kg, although continuous intravenous infusions of 150 to 300 μg/kg/min have been used to produce controlled hypotension. For practical purposes, in adults an intravenous dose of 3 to 6 mg is given by bolus followed by a second dose of 6 to 12 mg after 1 minute if the first dose was not effective. This therapy rapidly interrupts narrow-complex tachycardia caused by AV nodal reentry. Comparison with verapamil has shown adenosine to be equally effective as an antiarrhythmic agent but with the advantages of fewer adverse hemodynamic effects, a faster onset of action, and a more rapid elimination so that undesired effects are short-lived.33
Potassium
Treatment of hyperkalemia is based on its magnitude and on the clinical presentation. For life-threatening, hyperkalemia-induced arrhythmias, the principle is rapid reduction of extracellular potassium concentration, a treatment that does not acutely decrease total body potassium content. Calcium chloride, 10 to 20 mg/kg, given by intravenous infusion, will directly antagonize the effects of potassium on the cardiac cell membranes. Sodium bicarbonate, 1 to 2 mEq/kg, or a dose calculated from acid-base measurements to produce moderate alkalinity (pH approximately 7.45 to 7.50), will shift potassium intracellularly. A change in pH of 0.1 unit produces a 0.5 to 1.5 mEq/L change of potassium concentration in the opposite direction. An intravenous infusion of glucose and insulin has a similar effect; glucose at a dose of 0.5 to 2.0 g/kg with insulin in the ratio of 1 unit to 4 g of glucose is appropriate. Sequential measurement of serum potassium is important with this treatment because marked hypokalemia can result.
Acute hypokalemia frequently occurs after CPB as a result of hemodilution, urinary losses, and intracellular shifts, the latter perhaps relating to abnormalities of the glucose-insulin system seen with nonpulsatile hypothermic CPB. With frequent assessment of serum potassium concentrations and continuous ECG monitoring, potassium infusion at rates of up to 10 to 15 mEq/hr may be administered to treat serious hypokalemia.34
Magnesium
Arrhythmias induced by magnesium deficiency may be refractory to treatment with antiarrhythmic drugs and either electrical cardioversion or defibrillation. For this reason, adjunctive treatment of refractory arrhythmias with magnesium has been advocated even when magnesium deficiency has not been documented. Magnesium deficiency is common in cardiac surgery patients owing to the diuretic agents these patients are often receiving and because magnesium levels decrease with CPB because of hemodilution of the pump. Magnesium lacks a counterregulatory hormone to increase magnesium levels during CPB in contrast to the hypocalcemia that is corrected by parathyroid hormone. The results of magnesium administration trials involving CABG have been conflicting. Some studies have shown a benefit and others have not in regard to reducing the incidence of postoperative arrhythmias.35
SUMMARY
Anti-Ischemic Drug Therapy
Drug Therapy for Systemic Hypertension
Pharmacotherapy for Acute and Chronic Heart Failure
Pharmacotherapy for Cardiac Arrhythmias
1. ACC/AHA 2007 Guideline Update for the Management of Patients with Unstable Angina and NonST-Segment Elevation Myocardial Infarction. A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients with Unstable Angina). J Am Coll Cardiol 50:1-157, 2007
2. Abrams J. Mechanisms of action of the organic nitrates in the treatment of myocardial ischemia. Am J Cardiol. 1992;70:30B.
3. Anderson T.J., Meredith I.T., Ganz P., et al. Nitric oxide and nitrovasodilators: Similarities. differences, and potential interactions. J Am Coll Cardiol. 1994;24:555.
4. London M.J., Zaugg M., Schaub M.C., Spahn D.R. Perioperative beta-adrenergic receptor blockade. Anesthesiology. 2004;100:170.
5. ACC/AHA. 2007 Guidelines on perioperative cardiovascular evaluation & care for noncardiac surgery. J Am Coll Cardiol. 2007;50:1707-1732.
6. Giles J., Sear J., Foex P. Effect of chronic beta-blockage on perioperative outcome in patients undergoing noncardiac surgery. Anaesthesia. 2004;59:574.
7. Eisenberg M.J., Brox A., Bestawros A.N. Calcium channel blockers: An update. Am J Med. 2004;116:35.
8. Wijeysundara D.N., Deactie W.S. Calcium Channel Blockers for reducing cardiac morbidity after noncardiac surgery: A meta-analysis. Anesth Analg. 2003;97:634-641.
9. Chobanian A.V., Bakris G.L., Black H.R., et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560.
10. Psaty B.M., Lumley T., Furberg C.D., et al. Health outcomes associated with various antihypertensive therapies used as first-line agents: A network meta-analysis. JAMA. 2003;289:2534.
11. Williams B. Recent hypertension trials. J Am Coll Cardiol. 2005;45:813.
12. American Heart Association: Heart Disease and Stroke Statistics, 2004. Dallas, TX, American Heart Association. Available at: www.americanheart.org
13. Cohn J.N., Ferrari R., Sharpe N. Cardiac remodeling-concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569.
14. Arnold J.M., Yusuf S., Young J., et al. Prevention of Heart Failure in Patients in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation. 2003;107:1284.
15. Mann D.L., Deswal A., Bozkurt B., Torre-Amione G. New therapeutics for chronic heart failure. Annu Rev Med. 2002;53:59.
16. Pitt B., Remme W., Zannad F., et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309.
17. Weber K.T. Efficacy of aldosterone receptor antagonism in heart failure. Potential mechanisms. Current Heart Failure Reports. 2004;I:51.
18. Dulin B., Abraham W.T. Pharmacology of carvedilol. Am J Cardiol. 2004;93:3B-6B.
19. Rahimtoola S.H. Digitalis therapy for patients in clinical heart failure. Circulation. 2004;109:2942.
20. Bristow M.R., Saxon L.A., Boehmer J., et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140.
21. Fonarow G.C., Dams K., Abraham W., et al. Risk stratification for in-hospital mortality in acute decompensated heart failure. JAMA. 2005;293:572.
22. Elkayam U., Akhter M.W., Singh H., et al. Comparison of effects on left ventricular filling pressure of intravenous nesiritide and high-dose nitroglycerin in patients with decompensated heart failure. Am J Cardiol. 2004;93:237.
23. DiDomenico R.J., Park H.Y., Southworth M.R., et al. Guidelines for acute decompensated heart failure treatment. Ann Pharmacother. 2004;38:649.
24. Lekmann A., Boldt J. New pharmacologic approaches for the perioperative treatment of ischemic cardiogenic shock. J Cardiothorac Vasc Anesth. 2005;19:97.
25. Wang J., Kurrel meyer K.M., Torre-Amione G., et al. Nagueh SF: Systolic and diastolic dyssynchony in patients with diastolic heart failure & the effect of medical treatment. J Am Coll Cardiol. 2007;49::88-96.
26. Lobato E., Willert J., Looke T., et al. Effects of milrinone versus epinephrine on left ventricular relaxation after cardiopulmonary bypass following myocardial revascularization. J Cardiothorac Vasc Anesth. 19, 2005.
27. Liu P., Konstam M., Force T. Highlights of the 2004 Scientific Sessions of the Heart Failure Society of America. J Am Coll Cardiol. 2005;45:617.
28. Singh S., Patrick J. Antiarrhythmic drugs. Curr Treat Options Cardiovasc Med. 2004;6:357.
29. Dimarco J., Gersh B., Opie L. Antiarrhythmic drugs and strategies. In: Opie L., Horsh G., editors. Drugs for the Heart. 6th ed. Philadelphia: Elsevier; 2005:218-274.
30. Dorian P., Cass D., Schwartz G., et al. Amiodarone compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346:884.
31. Kaplan J.A. Role of ultrashort-acting beta-blockers in the perioperative period. J Cardiothorac Anesth. 1988;2:683.
32. Dorian P., Mangat I. Role of amiodarone in the era of the implantable cardioverter defibrillator. J Cardiovasc Electrophysiol. 2003;14(9 Suppl):S78-S81.
33. Hood M.A., Smith W.M. Adenosine versus verapamil in the treatment of supraventricular tachycardia: A randomized double-crossover trial. Am Heart J. 1992;123:1543.
34. Mohnle P., Schwann N., Vaughn W., et al. Perturbations in laboratory values after coronary artery bypass surgery with cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2005;19:19.
35. Geertman H., van der Starre P., Sie H., et al. Magnesium in addition to sotalol does not influence the incidence of postoperative atrial tachyarrhythmias after coronary artery bypass surgery. J Cardiothorac Vasc Anesth. 2004;18:309.