[level-membership-for-emergency-medicine-category]Chapter 152
Cardiovascular Drugs
Cardiovascular drugs are a common cause of poisoning in the United States; in 2010, 97,336 exposures to cardiovascular drugs were reported to U.S. Poison Control Centers.1 Cardiovascular drugs are the second most common cause, accounting for more than 10%, of all poisoning deaths in the United States. Of the scores of cardiovascular drugs, three classes—cardioactive steroids (primarily digoxin), beta-adrenergic blockers, and calcium channel blockers—account for the majority of fatalities.
Cardioactive Steroids (Digoxin)
Digoxin is derived from the Balkan foxglove plant, Digitalis lanata (Fig. 152-1), and the trade name for digoxin (Lanoxin) is derived from the Latin name of this plant.2 Digitoxin, which is no longer in clinical use, comes from Digitalis purpurea.2 Although William Withering was the first to describe the medicinal use of the foxglove plant,3 the ancient Egyptians reference medicinal use of foxglove. Despite centuries of experience with digitalis, chronic and acute poisonings still occur. Controversy about the therapeutic benefit of cardioactive steroids is not new. Benjamin Rush wrote in 1797, “I suspect the cases in which [digitalis preparations] were useful to have been either so few or doubtful and that the cases they had done harm were so much more numerous and unequivocal as justly to banish them from the Materia Medica.”4 Medication errors and toxic effects account for the most common causes (44%) of preventable iatrogenic cardiac arrests.

Principles of Disease
Digoxin is used therapeutically (1) to increase the force of myocardial contraction to increase cardiac output in patients with heart failure and (2) to decrease atrioventricular (AV) conduction to slow the ventricular rate in atrial fibrillation. The basis for its first effect is inhibition of membrane sodium-potassium–adenosine triphosphatase (Na+,K+-ATPase), which increases intracellular sodium and extracellular potassium concentrations. This increase in intracellular sodium concentration results in dysfunction of the sodium-calcium ion exchanger, which normally extrudes intracellular calcium after systole. This subsequent increase in intracellular calcium concentration results in a larger amount of calcium pumped into the sarcoplasmic reticulum, so that on calcium-induced calcium release during subsequent action potentials, a larger amount of calcium is released into the cell, causing a more powerful contraction and thus increased cardiac output. Any molecule with this effect is classified as a cardioactive steroid. Cardiac glycosides (such as digoxin) are merely cardioactive steroids with additional sugar moieties attached to their steroid nucleus. At therapeutic doses, the effects of digoxin on serum electrolyte levels are minimal. With toxic levels, digoxin paralyzes the Na+,K+-ATPase pump, potassium cannot be transported into cells, and serum potassium concentration can rise as high as 13.5 mmol/L.5
Digoxin also exerts three primary effects on Purkinje fibers: (1) decreased resting potential, resulting in slowed phase 0 depolarization and conduction velocity; (2) decreased action potential duration, which increases sensitivity of muscle fibers to electrical stimuli; and (3) enhanced automaticity resulting from increased rate of phase 4 repolarization and delayed afterdepolarizations. These mechanisms account for an increase in premature ventricular contractions, the most common manifestation of digoxin toxicity. At toxic extremes, these effects result in a dangerous sensitivity to mechanical and electrical stimulation. Interventions with pacemaker wires, catheters, and cardioversion can result in asystole, ventricular tachycardia, and ventricular fibrillation.6
Unlike most cardiovascular drugs, digoxin can produce virtually any dysrhythmia or conduction block, and bradycardias are as common as tachycardias (Box 152-1). Unfortunately, none is unique to digoxin, and because they can all occur in the setting of ischemic and other heart disease, digoxin toxicity remains a clinical rather than an electrocardiographic diagnosis.
The volume of distribution (Vd) of digoxin is 5 L/kg for adults but varies from 3.5 L/kg in premature infants to 16.3 L/kg in older infants.7 This indicates that only a small fraction of digoxin remains in the intravascular space, and the drug is highly concentrated in cardiac tissue. The myocardial-to-serum ratio at equilibrium ranges from 15 : 1 to 30 : 1. The Vd for digitoxin is only 0.5 L/kg, giving it a different pharmacokinetic profile.
Multiple drugs and disease states can negatively alter absorption, Vd, protein binding, and elimination and render the heart more susceptible to digoxin toxicity. The factors listed in Box 152-2 are especially important risk factors in chronic intoxication.
Clinical Features
The symptoms and signs of chronic digoxin toxicity are nonspecific. The most common symptoms, in more than 80% of cases, are nausea, anorexia, fatigue, and visual disturbance, but a variety of gastrointestinal, neurologic, and ophthalmic disturbances also occur (Box 152-3). Digoxin intoxication should be considered in any patient receiving maintenance therapy who has consistent symptoms, especially with new conduction disturbances or dysrhythmias.
There are significant differences between acute and chronic toxicity (Table 152-1). Chronic poisoning has an insidious onset and is accompanied by a higher mortality rate that is probably due in part to underlying heart disease and chronic accumulation of the toxin. In cases of chronic intoxication, the LL50 (the level with a 50% mortality) is only 6 ng/mL.8 The LL50 for acute intoxication is not known, but it is certainly much higher, especially in children. The association of hyperkalemia with acute toxicity is obvious given the mechanism of digoxin; either hypokalemia or hyperkalemia may occur with chronic toxicity.
Table 152-1
Chronic versus Acute Digitalis Intoxication
| CHRONIC | ACUTE |
| Higher mortality (LL50 6 ng/mL) | Lower mortality |
| Ventricular dysrhythmias more common | Bradycardia and atrioventricular block more common |
| Usually elderly patients | Usually younger patients |
| Often need Fab fragment therapy | Often do well without Fab (Caution: many exceptions) |
| Underlying heart disease increases morbidity and mortality | Absence of heart disease decreases morbidity and mortality |
Diagnostic Strategies
Diagnosis and management rely heavily on serum digoxin levels, but it is the steady state, rather than peak concentration, that correlates with tissue toxicity and is used to calculate antidote dosages. Peak concentrations after an oral dose of digoxin occur in 1.5 to 2 hours, with a range of 0.5 to 6 hours.9 Steady-state serum concentrations are not achieved until after distribution, or 6 to 8 hours after a dose or overdose, and may be only one fourth to one fifth of the peak concentration. The ideal serum digoxin concentration for patients with heart failure is considered to be 0.7 to 1.1 ng/mL,10 although laboratory “normals” are often reported up to 2.0 ng/mL. Serum steady-state digoxin levels of 1.1 to 3.0 ng/mL are equivocal; that is, levels as low as 1.1 ng/mL have been associated with increased mortality,11 and patients with levels up to 3.0 ng/mL can be asymptomatic.12 The incidence of digoxin-incited dysrhythmia reaches 10% at a level of 1.7 ng/mL and rises to 50% at a level of 2.5 ng/mL. Determination of a level too soon after the last maintenance dose falsely suggests toxicity, especially in cases of chronic intoxication, in which significant morbidity and mortality can occur at levels of 2 to 6 ng/mL.10 After an acute massive overdose in a patient who is rapidly becoming symptomatic, however, it may be impractical to wait 6 to 8 hours for the first reading.13 It is unlikely that early levels exceeding 10 to 20 ng/mL will fade to clinical insignificance at 6 to 8 hours after ingestion.
Management
The mortality rate before Fab fragment therapy was 23% despite all of the interventions described.14 Fab fragment treatment is well established in both chronic and acute poisonings, with a 90% response rate.15 Nonresponders usually receive too little antibody or receive it too late. Other nonresponders are compromised by underlying heart or multisystem disease.
Digitalis antibodies are derived from sheep, but allergic reactions occur in less than 1% of cases.15 Reactions have included erythema, urticaria, and facial edema, all of which are responsive to the usual treatment. Other expected reactions when Fab fragments neutralize digitalis include hypokalemia, exacerbation of congestive heart failure, and increase in ventricular rate with atrial fibrillation. Two Fab fragment preparations were previously available; however, Digibind has been discontinued, leaving DigiFab as the only available product in the United States. If vials of Digibind are still available at a given institution, they require a 0.22-µm membrane filter for proper use; such a filter is not required for DigiFab.
Fab fragment treatment is best reserved for cases of serious cardiovascular toxicity rather than for routine or prophylactic administration with higher than expected serum levels. In acute poisoning, antibody treatment should be used for a serum potassium level above 5.0 mEq/L or unstable dysrhythmias, such as symptomatic sinus bradycardia, ventricular dysrhythmias, or second- or third-degree heart block unresponsive to atropine. Although toxicity increases with greater body load, there is no clear correlation with amount ingested, especially in children, and many patients with large ingestions or high serum levels become only mildly symptomatic.8 Fab fragment therapy should be used before transvenous pacing, which carries significant risk.
The median time to initial response is 19 minutes after completion of the Fab infusion, but complete resolution of digitalis toxic rhythms may require hours.16,17 Late administration of Fab fragments has resuscitated 54% of patients who have suffered cardiac arrest.18 This antidote should be considered whenever hemodynamic compromise attends a digitalis toxic dysrhythmia or heart block (Box 152-4).19
Current formulas for calculation of DigiFab doses are found in the package insert. There are at least three approaches. The first is empirical. A patient has a history of digitalis ingestion, consistent symptoms, and life-threatening dysrhythmias. There is no time to assess serum digoxin levels, and 10 vials should be administered over 30 minutes for the average acute ingestion, 4 to 6 vials for the average chronic ingestion. In cardiac arrest, 20 vials can be administered undiluted by intravenous bolus. The second approach uses a simple calculation when the ingested dose is known with reasonable certainty. One vial of Digibind or DigiFab contains 38 mg or 40 mg, respectively, of Fab fragments, which bind 0.5 mg of digoxin or digitoxin (Box 152-5). A third approach is to base the dosage on the steady-state serum digoxin or digitoxin level after 6 to 8 hours (Boxes 152-6 and 152-7). Because most assays measure both bound and unbound drug, digoxin levels will be elevated for up to 1 week, with values often greater than 100 ng/mL once Fab fragments have been administered. Newer methods can measure free digoxin, but it is more meaningful to follow the patient clinically.
BOX 152-5 Sample Calculation of Digibind or DigiFab Based on Ingested Dose of Digoxin or Digitoxin*
Case: A toxic-appearing 40-year-old woman has ingested fifty 0.25-mg digoxin tablets.
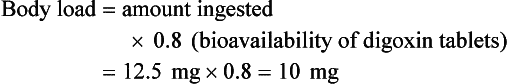
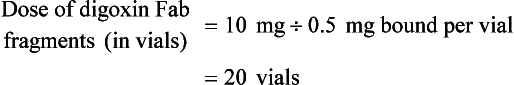
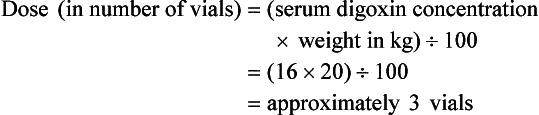
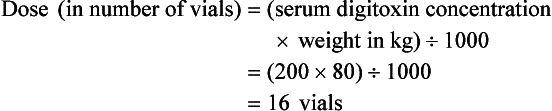
Electrolyte Correction
In acute poisoning, serum potassium concentration may begin to rise rapidly within 1 to 2 hours of ingestion. Potassium should be withheld, even if mild hypokalemia is measured initially. The initial serum potassium concentration may in fact be a better predictor of mortality than the initial digoxin concentration. In a study of 91 patients with acute digoxin poisoning, nearly 50% of the patients with serum potassium concentrations between 5.0 and 5.5 mmol/L died. No patients with a potassium level of less than 5.0 mEq/L died, and all 10 patients with serum potassium concentrations exceeding 5.5 mmol/L perished.20 Because of these findings, a serum potassium concentration greater than 5 mmol/L alone warrants treatment with Fab fragments. Even with Fab fragments, severe hyperkalemia should be treated with intravenous administration of glucose, insulin, and sodium bicarbonate as needed.
The decision to administer calcium to patients with hyperkalemia and digoxin poisoning represents a clinical dilemma. Classic teaching is that in the setting of the increased intracellular calcium concentration from digoxin poisoning, administration of exogenous calcium will result in “stone heart” from excessive intracellular calcium.21 This concept has been in the literature since 1927,22 based on animal studies.23 Documented cases of cardiac arrest after calcium administration are exceedingly rare in the literature, and the temporal relationship is dubious.24 More recent data indicate that the intravenous administration of calcium can be safe for hyperkalemia in the setting of digoxin toxicity.25 Unequivocally, however, the best treatment of hyperkalemia due to acute digoxin toxicity is Fab fragments. The treatment of hyperkalemia in the setting of chronic digoxin toxicity and renal failure is less clear; however, the evidence that calcium salts will be harmful is dubious at best. Calcium salts should be administered during several minutes through a secure peripheral intravenous site or through a central venous catheter. Indications are identical to those for hyperkalemia, including but not exclusive to a widened QRS duration on the electrocardiogram. A reasonable starting dose would be 3 g of calcium gluconate or 1 g of calcium chloride. Calcium salts should be followed immediately with other pharmacologic measures to lower serum potassium concentration.
Many patients receiving diuretic therapy are also magnesium depleted, even when the measured serum magnesium level is normal. If significant magnesium depletion is suspected (e.g., if electrocardiographic changes such as QTc prolongation are present), 1 to 2 g of magnesium sulfate can be given during 10 to 20 minutes (child: 25 mg/kg), followed by a constant infusion of 1 to 2 g/hr. Patients should be closely monitored for respiratory depression, which is usually preceded by progressive loss of deep tendon reflexes. Hypermagnesemia can exacerbate digitalis toxicity, but magnesium has been reported to reverse digoxin-induced tachydysrhythmias.26 It is prudent to infuse magnesium slowly and to stop the infusion if heart block or bradycardia develops. Avoid magnesium in patients with renal failure. The role of magnesium in bradydysrhythmias and conduction blocks is less clear but probably dangerous because hypermagnesemia can impair impulse formation and AV conduction.
Pacing
Transvenous pacing has been a mainstay of treatment for several decades, but the catheter may induce ventricular tachydysrhythmias in a myocardium made irritable by digoxin. Iatrogenic accidents of cardiac pacing are frequent (14 of 39, 36%) and often fatal (5 of 39, 13%).27 We recommend avoidance of transvenous pacing unless external pacing fails. Pacing usually is required only temporarily while waiting for Fab fragments to take effect. Cardioversion for tachydysrhythmia also may be hazardous in the setting of digoxin intoxication. If it is deemed necessary, such as when the tachydysrhythmia is thought to be causing severe hypotension, we recommend use of lower than usual energy settings, such as 25 to 50 J, although there is no proof that this is less hazardous.
Phenytoin and Lidocaine
Phenytoin and lidocaine are believed to be the safest of the antidysrhythmic drugs for control of tachydysrhythmias in the setting of digoxin toxicity. Indications include unstable tachydysrhythmias when Fab fragments are unavailable and unstable tachydysrhythmias that occur while waiting for Fab fragments to take effect. Phenytoin may enhance AV conduction. Phenytoin has been infused at 25 to 50 mg/min to a loading dose of 15 to 20 mg/kg; it may also be administered in 100-mg boluses every 5 minutes until arrhythmias improve or 1000 mg is administered. Lidocaine can be given initially at a dosage of 1 to 1.5 mg/kg during several minutes, followed by an infusion of 1 to 4 mg/min (30-50 µg/kg/min).28 Most other cardiac drugs (isoproterenol, procainamide, amiodarone, beta-blockers, calcium antagonists) may worsen dysrhythmias or depress AV conduction. Digoxin immune Fab fragments are the preferred therapy for dysrhythmias.
Pediatric Considerations
Children at greatest risk are those receiving chronic digoxin therapy for heart disease. Children with healthy hearts can tolerate massive acute oral ingestions without Fab treatment.28 This excludes therapeutic errors, children who are taking digoxin therapeutically, and children with heart disease.
Signs and symptoms in children with digoxin poisoning are somewhat different (Table 152-2). Vomiting, somnolence, and obtundation are more common than in adults.18 CNS depression, in the absence of a history, might lead the clinician to suspect narcotic or sedative-hypnotic overdose or nontoxicologic causes, such as head injury, metabolic disorder, and CNS infection. Conduction disturbances and bradycardias are more common than ventricular dysrhythmias in children, especially with acute ingestion.9,18
Table 152-2
Age Differences in Digitalis Intoxication
| ADULT | PEDIATRIC |
| Toxic at lower levels | Asymptomatic at higher levels |
| Nausea, fatigue, and visual disturbances most common | Obtundation and vomiting more common than in adults |
| Tachydysrhythmias as common as blocks and bradydysrhythmias | Bradydysrhythmias and blocks most common |
| Allergic reactions to Fab fragments uncommon (<1%) | Allergic reactions extremely rare |
| Vd less variable (5-7.5 L/kg) | Vd more variable (3.5-6.0 L/kg in premature infants, 8.0-16.3 L/kg in infants 2-24 months) |
Beta-Adrenergic Blockers
Beta-adrenergic blocking drugs became widely used in Europe in the 1960s for treatment of dysrhythmias. Their antihypertensive effects were later appreciated, and, by the 1970s, they were one of the most widely prescribed classes of drugs in the United States. Current indications include supraventricular dysrhythmias, hypertension (although the drugs have fallen out of favor for this indication), angina, thyrotoxicosis, migraine, and glaucoma. Of the numerous beta-blockers available, propranolol is the most deadly. In 2010, U.S. Poison Control Centers received more than 23,000 calls regarding beta-blocker exposures.1
Principles of Disease
Beta-blockers structurally resemble isoproterenol, a pure beta-agonist. They competitively inhibit endogenous catecholamines such as epinephrine at beta-adrenergic receptors, blocking the catecholamine effects on augmentation of myocardial contraction (inotropy), enhancing cardiac conduction (dromotropy), and accelerating heart rate (chronotropy). These are all beta1 effects. Complex beta2 effects include vascular (smooth muscle relaxation and vasodilation), liver (glycogenolysis, gluconeogenesis), lung (bronchodilation), adipose tissue (release of free fatty acids), and uterus (smooth muscle relaxation) effects. Equally important properties, which vary from one beta-blocker to another, include cardioselectivity (beta1 selectivity), membrane-stabilizing effect (fast cardiac sodium channel blocking properties), lipophilicity, and intrinsic sympathomimetic activity (Table 152-3). Although cardioselectivity is lost in overdose, cardioselective beta-blockers such as atenolol, metoprolol, and esmolol still have a lower mortality rate than that of propranolol.
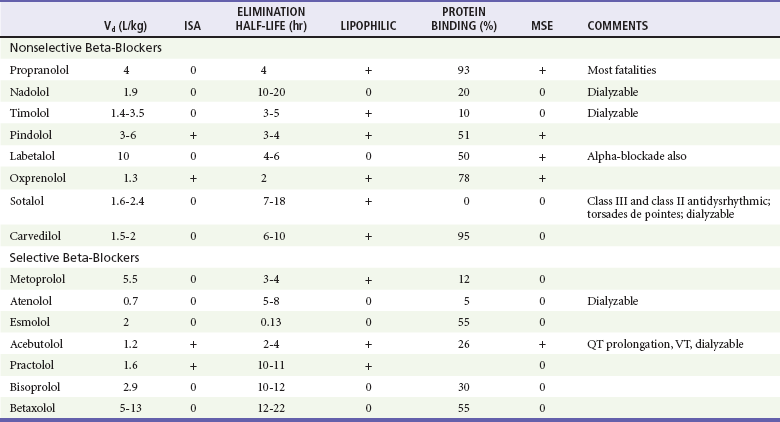
Beta-blockers are rapidly absorbed after oral ingestion, and the peak effect of normal-release preparations occurs in 1 to 4 hours. Hepatic metabolism on first pass results in significantly less bioavailability after oral dosing than with intravenous injection (e.g., 1 : 40 for propranolol). Because volume of distribution for most beta-blockers generally exceeds 1 L/kg, hemodialysis is not efficacious for most beta-blockers. Protein binding varies from 0% for sotalol to 93% for propranolol. Elimination half-lives vary from 8 to 9 minutes for esmolol to as long as 24 hours for nadolol and others (see Table 152-3).
Clinical Features
The most common initial sign remains bradycardia, which should suggest the possibility of cardiac drug overdose. Hypotension and unconsciousness are the second and third most common signs (Box 152-8). Much of propranolol’s unique toxicity derives from its lipophilic nature, which allows it to penetrate the CNS, causing obtundation, respiratory depression, and seizures. Seizures probably result from a combination of hypotension, hypoglycemia, hypoxia, and direct CNS toxicity. Other beta-blockers are not lipophilic and do not have these effects. Surprisingly, bronchospasm is rarely a problem in cases of beta-blocker overdose, even with nonselective beta-blockers. The few cases of symptomatic bronchospasm respond to the usual bronchodilator nebulizations.
Diagnostic Strategies
Diagnosis and management depend entirely on the clinical picture. Blood levels of beta-blockers correlate poorly with severity of intoxication and are not readily available. Most urine toxicology screens do not identify antidysrhythmic drugs and are not helpful.29 Hypoglycemia is common in children, and bedside determination of glucose concentration should be done if CNS depression is noted. Known access of the patient to a beta-blocker and consistent clinical features such as obtundation, seizures, hypotension, bradydysrhythmias, and occasionally tachydysrhythmias should lead the clinician to consider beta-blocker intoxication. Whereas some authors believe that a serum lactate concentration can help predict mortality in overdose patients,30 this has not held true in pure beta-blocker ingestions.31
Management
Glucagon
Because glucagon has both inotropic and chronotropic effects and does not depend on beta receptors for its action, it has long been used for beta-blocker toxicity. It stimulates the production of intracellular cyclic adenosine monophosphate independently of the beta-adrenergic receptor.32 Furthermore, it helps counteract the hypoglycemia induced by beta-blocker overdose. The dosing of glucagon is not well studied, but the drug is frequently used as a 5- to 10-mg IV bolus (0.05-0.1 mg/kg for children). If a response occurs to glucagon, the “response dose” should be started as an infusion at a rate of response dose during 1 hour. Glucagon has a very short (20-minute) half-life, and unfortunately its effect is often transient. Vomiting is a common complication of glucagon, so the airway should be secured or monitored closely to prevent aspiration. With cumulative large doses, glucagon should be diluted in 5% glucose in water for constant infusion. Side effects also include hypokalemia and allergic reactions. The response to glucagon alone is often inadequate, and glucagon is likely to be less effective than high-dose insulin.33 Furthermore, hospitals may not be adequately stocked for treatment beyond a few hours.34 From clinical experience, we find glucagon most useful as a transient therapy to bridge patients to high-dose insulin.
High-Dose Insulin
Despite glucagon’s longer history for treatment of beta-blocker toxicity, high-dose insulin (HDI) is probably a better agent. HDI is not a vasopressor; it is a profound inotrope35 with vasodilating properties.36,37 The mechanism for HDI is not fully known, but it probably involves both optimization of the use of carbohydrates for fuel by cardiac myocytes and modulation of intracellular calcium.38 HDI improves cardiac output significantly in beta-blocker toxicity from an increase in stroke volume more than heart rate. Dosing of HDI is not agreed on; dosing in humans successfully treated with HDI has ranged from 0.5 to 21.8 U/kg/hr.39 In the largest human case series to date of patients receiving HDI for poison-induced cardiogenic shock, 11 of 12 patients survived; the only death occurred when HDI was terminated early because of a protocol violation.40 Mean maximal HDI dosing in this series was 8.7 U/kg/hr. Previous recommendations for dosing of HDI were a bolus of 1 U/kg of regular insulin IV, followed by a drip at 1 U/kg/hr.40 On the basis of animal data,41 some authors start HDI at 10 U/kg/hr in severely toxic patients with the belief that the glucose use from insulin saturates before the inotropic effect. Regardless of HDI dosing, patients should receive a bolus of dextrose before insulin and a dextrose infusion should be started thereafter, such as a bolus of 25 g glucose (1 traditional “amp” of D50) if the serum glucose concentration is below 200 mg/dL (11.1 mmol/L), followed by concentrated dextrose solutions (such as D25, D50, or even D70) through a central line because patients receiving HDI therapy are at risk of fluid overload. Glucose concentration should be monitored as frequently as every 10 minutes until a steady state of glucose use is achieved. Potassium concentration should also be monitored closely and potassium replaced as needed because patients may become hypokalemic.40
Vasopressors and Other Inotropes
If hypotension or bradycardia persists, other cardioactive drugs are indicated. The use of vasopressors is not without risk; HDI is probably safer.37 Vasopressors may be useful to transiently maintain mean arterial pressure (MAP) high enough to ensure adequate cerebral perfusion pressure (CPP). A MAP of 65 mm Hg is often quoted as the minimum MAP required to ensure adequate CPP.42 If the patient is not intubated and has a normal mental status, lower MAP readings are probably adequate. The more difficult situation of an intubated patient with a MAP slightly below 65 mm Hg represents a clinical dilemma. It is our practice to accept an MAP of 60 mm Hg if all other signs point to adequate organ perfusion.
A single drug of choice after HDI and glucagon has not emerged, but some clinicians favor isoproterenol (isoprenaline in Europe), dopamine, or epinephrine.28 Other catecholamines that have been successfully used include norepinephrine, dobutamine, and phenylephrine. Often, norepinephrine or dopamine is added to beta-agonists such as isoproterenol that lack vasopressor activity. In the selection of cardioactive medications to supplement HDI and glucagon, we recommend early assessment of systemic vascular resistance and cardiac output by either indirect measurements with a pulse contour analysis–based hemodynamic monitoring tool such as the FloTrac device (Edwards Lifesciences; Irvine, Calif.) or Swan-Ganz catheter and choosing of appropriate drugs if either cardiogenic (inotropic drugs such as norepinephrine, dobutamine, dopamine) or distributive shock (pressors such as norepinephrine, phenylephrine, vasopressin) exists. Refractory cases of bradycardia may respond to an external or transvenous pacemaker. A pacemaker is particularly useful when the heart rate is the predominant cause of decreased cardiac output.
Intravenous Fat Emulsion (Intralipid)
Intravenous fat emulsion (IFE) is a promising new therapy for poison-induced cardiogenic shock. This therapy was first described for treatment of toxicity from local anesthetics such as bupivacaine.43 IFE is proposed to work by three separate mechanisms: (1) the lipid sink, (2) modulation of intracellular metabolism, and (3) activation of ion channels.44 The lipid sink theory posits that fat-soluble drugs are soaked up and removed from the site of toxicity, effectively increasing the volume of distribution for a fat-soluble drug. This is the predominant theory behind the use of IFE. A second theory involves optimization of cardiac metabolism. The heart under physiologic circumstances prefers free fatty acids; in times of stress it switches to glucose metabolism for energy. A dose of IFE theoretically provides a large supply of free fatty acids to optimize energy use in the heart. The final proposed mechanism involves direct activation of cardiac calcium channels. Animal evidence exists to support the use of IFE for both lipid-soluble beta-blockers (such as propranolol)45 and non–lipid-soluble beta-blockers (such as atenolol).46 Dramatic human case reports of successful resuscitations exist for propranolol,47 nebivolol,39 and metoprolol.48 Although IFE was originally indicated for patients in cardiac arrest only, in 2011 the American College of Medical Toxicology released a position statement stating that “in circumstances where there is serious hemodynamic, or other, instability from a xenobiotic with a high degree of lipid solubility, lipid resuscitation therapy (IFE) is viewed as a reasonable consideration for therapy, even if the patient is not in cardiac arrest.”49 Dosing for IFE is not agreed on. We recommend an initial bolus of 1.5 mL/kg of 20% lipid solution given during 2 to 3 minutes, followed immediately by an infusion of 0.25 mL/kg/min.49 The bolus can be repeated up to two additional times for refractory patients. Response is typically within minutes of the bolus. We use IFE in patients with persistent bradycardia or hypotension not responding to intravenous fluids, calcium, and HDI.
Extracorporeal Elimination and Circulatory Assistance
Hemodialysis or hemoperfusion may be beneficial for atenolol, acebutolol, nadolol, sotalol, and timolol, the beta-blockers with lower Vd, lower protein binding, and greater hydrophilicity.50
Unlike overdoses of acetaminophen and iron, overdoses of cardiovascular drugs do not destroy tissue, and if circulation can be supported, complete recovery can be expected. An intra-aortic balloon pump or cardiopulmonary bypass can be lifesaving in cases of refractory hypotension.51,52 A percutaneous left ventricular assist device, although it has not been studied in beta-blocker toxicity, may also be useful. The relatively short half-lives (hours rather than days) of beta-blocking and calcium-blocking drugs fall within the temporal range of such interventions. To be successful, such heroic measures should be taken before prolonged hypotension leads to multiorgan ischemic injury (Box 152-9). Because most patients recover with just supportive care, these expensive and invasive interventions should be reserved for drugs and circumstances, such as propranolol, verapamil, and mixed cardiotoxic overdoses, that are associated with higher rates of mortality.
Pediatric Considerations
Severe pediatric beta-blocker poisonings are rare. In the cases reported, CNS, cardiac, and metabolic toxicities are similar.53 Symptomatic hypoglycemia, however, is much more common in children and occurs even after therapeutic doses. Therefore, serum glucose concentration should be measured in children. Risk factors include young age, fasting state, and diabetes mellitus. Obtunded children should receive empirical glucose, 1 to 2 mL/kg of 25% glucose IV. In general, 5% glucose infusions have been sufficient to maintain euglycemia, especially with concomitant use of glucagon and catecholamines, which stimulate glucose release. Because glycogen mobilization is a beta2 effect, hypoglycemia may be less common with the cardioselective (beta1) blockers.
Children generally fare well after beta-blocker ingestion, with symptoms in only 8 of 378 (2%) potential beta-blocker exposures in children.50
Calcium Channel Blockers
Verapamil and nifedipine, the earliest calcium channel antagonists, were introduced in Europe in the 1970s and in the United States in the early 1980s. Calcium antagonists have found many clinical applications: angina pectoris, hypertension, supraventricular dysrhythmias, hypertrophic cardiomyopathy, and migraine prophylaxis. Most fatalities occur with verapamil, but severe toxicity and death have been reported for most drugs of this class. In 2010, U.S. Poison Control Centers received more than 10,000 calls about calcium channel blockers.1
Pathophysiology
Calcium channel antagonists block the slow L-type calcium channels in the myocardium and vascular smooth muscle, leading to coronary and peripheral vasodilation. They also reduce cardiac contractility, depress SA nodal activity, and slow AV conduction. In cases of overdose, verapamil has the deadliest profile, combining severe myocardial depression and peripheral vasodilation. Both verapamil and diltiazem act on the heart and blood vessels, whereas dihydropyridines such as nifedipine cause primarily vasodilation and subsequent reflex tachycardia. As with beta-blockers, selectivity is lost after overdose, and toxicity is fourfold; the calcium antagonists have negative effects on heart rate, contractility, conduction, and vascular tone, with the exception of dihydropyridines, which tend to result in tachycardia even in severe toxicity.54
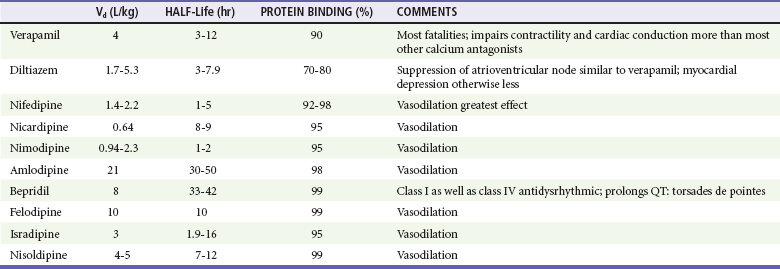
All calcium channel blockers are rapidly absorbed, although first-pass hepatic metabolism significantly reduces bioavailability (Table 152-4). Onset of action and toxicity ranges from less than 30 minutes to 60 minutes. Peak effect of nifedipine can occur as early as 20 minutes after ingestion, but peak effect of sustained-release verapamil can be delayed for many hours. High protein binding and Vd greater than 1 to 2 L/kg make hemodialysis or hemoperfusion ineffective. Fortunately (except with sustained-release preparations), their half-lives are relatively short, limiting toxicity to 24 to 36 hours.
Diagnostic Strategies
Serum levels of calcium antagonists are not readily available, nor do urine toxicology screens reliably detect this class of drugs. Glucose and electrolytes (including calcium and magnesium) should be measured. Hyperglycemia secondary to insulin inhibition occurs occasionally, but the elevation is usually mild (150-300 mg/L), is usually short-lived (<24 hours), and generally requires no treatment. Some authors suggest that hyperglycemia is a predictor of poor outcomes in verapamil and diltiazem toxicity.55 A metabolic (lactic) acidosis occurs with hypotension and hypoperfusion.
Differential Considerations
As with beta-blockade, CNS depressive effects are common and include lethargy, confusion, and coma. Unlike beta-blockers, calcium antagonists seldom induce seizures. Pulmonary effects include noncardiogenic pulmonary edema, and apnea can also occur. As with digitalis and beta-blocker overdose, nausea and vomiting are common (Box 152-10).
Management
Initial management includes rapid establishment of vascular access, supplemental oxygen, cardiac monitoring, and frequent blood pressure measurement. Gastrointestinal decontamination is controversial. For this class of highly lethal overdoses, activated charcoal at 1 g/kg can be considered if the airway is secure and if the patient presents in less than 1 or 2 hours. Multidose activated charcoal can be considered for sustained-release preparations and if a bezoar is suspected. Some authors have argued against the use of whole-bowel irrigation56; however, calcium channel blockers cause significant morbidity and mortality, and risks should be weighed against potential benefits. Gastric lavage may be considered for patients presenting early, but pills are often large, and safe performance of this procedure requires a secure airway. No evidence exists that gastrointestinal decontamination improves mortality in general overdoses or in calcium channel blocker toxicity. With the lack of currently available evidence, gastric decontamination can neither be recommended nor criticized and has its proponents and detractors.
Hypotension and Bradycardia
As with beta-blocker poisoning, a monotherapeutic approach will probably succeed only for trivial overdoses. If patients treated with isotonic fluids, calcium, and atropine have persistent hypotension or bradycardia, we next recommend HDI. HDI is thought to act by improving myocardial carbohydrate metabolism, thereby augmenting myocardial contraction. Serum glucose and potassium levels should be checked frequently to ensure that normal levels are maintained, although transient hyperglycemia is acceptable. Enthusiasm for HDI treatment is growing, and this therapy may prove safer and more efficacious than vasopressors.57 See the earlier discussion for our dosing recommendations.
Most severely poisoned patients require the addition of catecholamines to accelerate the heart rate (chronotropy), to enhance AV conduction (dromotropy), and to restore tone to peripheral vessels (vasotropy). As with beta-blocker toxicity, in the selection of cardioactive medications to supplement HDI and glucagon, we recommend early assessment of systemic vascular resistance and cardiac output by either indirect measurements with a pulse contour analysis–based hemodynamic monitoring tool such as the FloTrac device (Edwards Lifesciences) or Swan-Ganz catheter and choosing of appropriate drugs for cardiogenic (epinephrine, norepinephrine, dopamine, dobutamine) or distributive shock (phenylephrine, vasopressin). Dosing of catecholamines in calcium channel blocker toxicity is not different from that in other clinical situations requiring their use. As long as dysrhythmias are not induced, catecholamine infusions should be aggressively titrated to effect (Box 152-11).
Glucagon has also been used for its inotropic and chronotropic effects in doses similar to those advocated for beta-blocker poisoning. Anecdotal human cases exist of calcium channel blocker poisonings responding to glucagon.58 No controlled human studies exist. It is our practice to omit the use of glucagon in the treatment of calcium channel blocker toxicity.
IFE (e.g., Intralipid) has been described in the use of calcium channel blockers. The proposed mechanisms are discussed earlier. Animal evidence exists that IFE may be effective especially for the treatment of verapamil toxicity.59 Dramatic human case reports exist of successful resuscitation of toxicity from diltiazem48 and verapamil.60 Dosing and indications are identical to those for beta-blocker toxicity.
Pediatric Considerations
Nifedipine and probably other drugs in its class join the short list of medications that can kill a child with ingestion of a single tablet.61 Seizures may be more common in children than in adults and should be treated with diazepam, lorazepam, or calcium.
Overall, death after calcium antagonist ingestion in children is rare. The intravenous route of administration, as with digitalis, is much more dangerous. Even therapeutic doses of intravenous verapamil are considered contraindicated in infants with supraventricular tachycardia because of case reports of cardiovascular collapse and cardiac arrest after injection.62
Nitrates and Nitrites
Nitrates are occasionally found in rural well water contaminated by livestock or fertilizer runoff. Oral nitrates may be converted to nitrites in the gastrointestinal tract, especially in infants, whose hemoglobin is also more susceptible to oxidation. However, most exposures are encountered in young adults, usually male, who inhale various alkyl nitrites (amyl, butyl, isobutyl, or ethyl nitrite) in the hope of enhancing or prolonging sexual pleasure. Because of the sound they make when broken open, these products are best known to abusers as poppers. The popularity of poppers has waned in recent years as sales of sildenafil and related products have soared. Nitrites and nitrates are both potent vasodilators, and excessive use can cause headache, skin flushing, and orthostatic hypotension. Nitrites are also oxidizing agents that convert hemoglobin to methemoglobin, impairing oxygen delivery. Although most exposure is by inhalation, unintentional ingestion may occur because nitrites are also used legitimately as food preservatives. A family of five had methemoglobinemia after consuming a meal seasoned with sodium nitrite mislabeled as table salt.63
Key Concepts
 Consider digoxin intoxication in any patient with gastrointestinal or visual disturbance and a new dysrhythmia or conduction disturbance.
Consider digoxin intoxication in any patient with gastrointestinal or visual disturbance and a new dysrhythmia or conduction disturbance.
 Dose digitalis Fab antibody fragments by body load of digitalis, not by body weight of the patient.
Dose digitalis Fab antibody fragments by body load of digitalis, not by body weight of the patient.
 Use digitalis Fab fragments before pacing or other antidysrhythmic drugs.
Use digitalis Fab fragments before pacing or other antidysrhythmic drugs.









References
1. Bronstein, AC, et al. 2010 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 27th Annual Report. Clin Toxicol (Phila). 2011;49:910–941.
2. Hollman, A. Drugs for atrial fibrillation. Digoxin comes from Digitalis lanata. BMJ. 1996;312:912.
3. Withering, W. An account of the foxglove and some of its medical uses: With practical remarks on dropsy and other diseases. Med Classics. 1937;2:295–443.
4. Rush, B. Medical Enquiries and Observations. Philadelphia: T Dobson; 1797.
5. Reza, MJ, Kovick, RB, Shine, KI, Pearce, ML. Massive intravenous digoxin overdose. N Engl J Med. 1974;291:777–778.
6. French, JH, Thomas, RG, Siskind, AP, Brodsky, M, Iseri, LT. Magnesium therapy in massive digoxin intoxication. Ann Emerg Med. 1984;13:562–566.
7. Wells, TG, Young, RA, Kearns, GL. Age-related differences in digoxin toxicity and its treatment. Drug Saf. 1992;7:135–151.
8. Ordog, GJ, Benaron, S, Bhasin, V, Wasserberger, J, Balasubramanium, S. Serum digoxin levels and mortality in 5,100 patients. Ann Emerg Med. 1987;16:32–39.
9. Lewander, WJ, et al. Acute pediatric digoxin ingestion: A ten-year experience. Am J Dis Child. 1986;140:770–773.
10. Rahimtoola, SH. Digitalis therapy for patients in clinical heart failure. Circulation. 2004;109:2942–2946.
11. Rathore, SS, Curtis, JP, Wang, Y, Bristow, MR, Krumholz, HM. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA. 2003;289:871–878.
12. Smith, TW, Haber, E. Digoxin intoxication: The relationship of clinical presentation to serum digoxin concentration. J Clin Invest. 1970;49:2377–2386.
13. Piergies, AA, Worwag, EM, Atkinson, AJ. Pharmacoepidemiology and drug utilization: A concurrent audit of high digoxin levels. Clin Pharmacol Ther. 1994;55:353–358.
14. Antman, EM, Wenger, TL, Bulter, VP, Jr., Haber, E, Smith, TW. Treatment of 150 cases of life-threatening digitalis intoxication with digoxin-specific Fab antibody fragments: Final report of a multicenter study. Circulation. 1990;81:1744–1752.
15. Hickey, AR, et al. Digoxin immune Fab therapy in the management of digitalis intoxication: Safety and efficacy results of an observational surveillance study. J Am Coll Cardiol. 1991;17:590–598.
16. Weinstein, RS, Cole, S, Knaster, HB, Dahlbert, T. Beta-blocker overdose with propranolol and with atenolol. Ann Emerg Med. 1985;14:161–163.
17. Louis, WJ, McNeil, JJ, Drummer, OH. Pharmacology of combined alpha-beta blockade I. Drugs. 1984;28:16–34.
18. Schaumann, W, Kaufmann, B, Neubert, P, Smolarz, A. Kinetics of the Fab fragments of digoxin antibodies and of bound digoxin in patients with severe digoxin intoxication. Eur J Clin Pharmacol. 1986;30:527–533.
19. Woolf, AD, Wenger, T, Smith, TW, Lovejoy, FH, Jr. The use of digoxin-specific Fab fragments for severe digitalis intoxication in children. N Engl J Med. 1992;326:1739–1744.
20. Bismuth, C, Gaultier, M, Conso, F, Efthymiou, ML. Hyperkalemia in acute digitalis poisoning: Prognostic significance and therapeutic implications. J Toxicol Clin Toxicol. 1973;6:153–162.
21. Khatter, JC, Agbanyo, M, Navaratnam, S, Nero, B, Hoeschen, RJ. Digitalis cardiotoxicity: Cellular calcium overload a possible mechanism. Basic Res Cardiol. 1989;84:553–563.
22. Gold, H, Edwards, DJ. The effects of ouabain on the heart in the presence of hypercalcemia. Am Heart J. 1927;3:45–50.
23. Nola, GT, Pope, S, Harrison, DC. Assessment of the synergistic relationship between serum calcium and digitalis. Am Heart J. 1970;79:499–507.
24. Bower, JO, Mengle, HAK. The additive effect of calcium and digitalis. JAMA. 1936;106:1151–1153.
25. Levine, M, Nikkanen, H, Pallin, DJ. The effects of intravenous calcium in patients with digoxin toxicity. J Emerg Med. 2011;40:41–46.
26. Kinlay, S, Buckley, NA. Magnesium sulfate in the treatment of ventricular arrhythmias due to digoxin toxicity. J Toxicol Clin Toxicol. 1995;33:55–59.
27. Taboulet, P, Baud, FJ, Bismuth, C, Vicaut, E. Acute digitalis intoxication—is pacing still appropriate? J Toxicol Clin Toxicol. 1993;31:261–273.
28. Hazinski MF, Cummins RO, Field JM, eds. Handbook of Emergency Cardiovascular Care for Healthcare Providers. Dallas: American Heart Association, 2002.
29. Maurer, HH. Identification of antiarrhythmic drugs and their metabolites in urine. Arch Toxicol. 1990;64:218–230.
30. Manini, AF, Kumar, A, Olsen, D, Vlahov, D, Hoffman, RS. Utility of serum lactate to predict drug-overdose fatality. Clin Toxicol (Phila). 2010;48:730–736.
31. Mégarbane, B, Deye, N, Malissin, I, Baud, FJ. Usefulness of the serum lactate concentration for predicting mortality in acute beta-blocker poisoning. Clin Toxicol (Phila). 2010;48:974–978.
32. Bailey, B. Glucagon in β-blocker and calcium channel blocker overdoses: A systematic review. J Toxicol Clin Toxicol. 2003;41:595–602.
33. Kerns, W, Schroeder, D, Williams, C, Tomaszewski, C, Raymond, R. Insulin improves survival in a canine model of acute beta blocker toxicity. Ann Emerg Med. 1997;29:748–757.
34. Dart, RC, et al. Expert consensus guidelines for stocking of antidotes in hospitals that provide emergency care. Ann Emerg Med. 2009;54:386–394.
35. Gradinac, S, Coleman, GM, Taegtmeyer, H, Sweeney, MS, Frazier, OH. Improved cardiac function with glucose-insulin-potassium after aortocoronary bypass grafting. Ann Thorac Surg. 1989;48:484–489.
36. Anderson, EA, Hoffman, RP, Balon, TW, Sinkey, CA, Mark, AL. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Invest. 1991;87:2246–2252.
37. Holger, JS, et al. Insulin versus vasopressin and epinephrine to treat beta blocker toxicity. Clin Toxicol (Phila). 2007;45:396–401.
38. Ablorh, N-A, Nitu, F, Engebretsen, KE, Thomas, DD, Holger, JS. Insulin-dependent rescue from cardiogenic shock is not mediated by phospholamban phosphorylation. Clin Toxicol (Phila). 2009;47:296–302.
39. Stellpflug, SJ, Harris, CR, Engebretsen, KM, Cole, JB, Holger, JS. Intentional overdose with cardiac arrest treated with intravenous fat emulsion and high-dose insulin. Clin Toxicol (Phila). 2010;48:227–229.
40. Holger, JS, Stellpflug, SJ, Cole, JB, Harris, CR, Engebretsen, KM. High-dose insulin: A consecutive case series. Clin Toxicol (Phila). 2011;49:653–658.
41. Cole, JB, et al. 10 U/kg/hr of high dose insulin is superior to 1 U/kg/hr in a blinded, randomized, controlled trial in poison-induced cardiogenic shock [abstract 1]. Clin Toxicol (Phila). 2011;49:515.
42. Rivers, E, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377.
43. Rosenblatt, MA, Abel, M, Fischer, GW, Itzkovich, CJ, Eisenkraft, JB. Successful use of a 20% lipid emulsion to resuscitate a patient after presumed bupivacaine-related cardiac arrest. Anesthesiology. 2006;105:217–218.
44. Jamaty, C, et al. Lipid emulsions in the treatment of acute poisoning: A systematic review of human and animal studies. Clin Toxicol (Phila). 2010;48:1–27.
45. Cave, G, Harvey, MG, Castle, CD. The role of fat emulsion therapy in rodent model of propranolol toxicity: A preliminary study. J Med Toxicol. 2006;2:4–7.
46. Cave, G, Harvey, M. Lipid emulsion may augment early blood pressure recovery in a rabbit model of atenolol toxicity. J Med Toxicol. 2009;5:50–51.
47. Dean, P, Ruddy, JP, Marshall, S. Intravenous lipid emulsion in propranolol overdose. Anaesthesia. 2010;65:1148–1150.
48. Stellpflug, SJ, Fritzlar, SJ, Cole, JB, Engebretsen, KM, Holger, JS. Cardiotoxic overdose treated with intravenous fat emulsion and high-dose insulin in the setting of hypertrophic cardiomyopathy. J Med Toxicol. 2011;7:151–153.
49. ACMT Position Statement. Interim guidance for the use of lipid resuscitation therapy. J Med Toxicol. 2011;7:81–82.
50. Saitz, R, Williams, BW, Farber, HW. Atenolol-induced cardiovascular collapse treated with hemodialysis. Crit Care Med. 1991;19:116–118.
51. Lane, AS, Woodward, AC, Goldman, MR. Massive propranolol overdose poorly responsive to pharmacologic therapy: Use of the intraaortic balloon pump. Ann Emerg Med. 1987;16:1381–1383.
52. McVey, FK, Corke, CF. Extracorporeal circulation in the management of massive propranolol overdose. Anaesthesia. 1991;46:744–746.
53. Belson, MG, Sullivan, K, Geller, RJ. Beta-adrenergic antagonist exposures in children. Vet Hum Toxicol. 2001;43:361–365.
54. Schoffstall, JM, Spivey, WH, Gambone, LM, Shaw, RP, Sit, SP. Effects of calcium channel blocker overdose–induced toxicity in the conscious dog. Ann Emerg Med. 1991;20:1104–1108.
55. Levine, M, et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med. 2007;35:2071–2075.
56. Cumpston, KL, Aks, SE, Sigg, T, Pallasch, E. Whole bowel irrigation and the hemodynamically unstable calcium channel blocker overdose: Primum non nocere. J Emerg Med. 2010;38:171–174.
57. Kline, JA, Tomaszewski, CA, Schroeder, JD, Raymond, RM. Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharm Exp Ther. 1993;267:744–750.
58. Doyer, S, Roberts, JR. The use of glucagon in a case of calcium channel blocker overdose. Ann Emerg Med. 1993;22:1229–1233.
59. Bania, TC, Chu, J, Perez, E, Su, M, Hahn, IH. Hemodynamic effects of intravenous fat emulsion plus standard therapy in a model of severe verapamil toxicity. Acad Emerg Med. 2007;14:105–111.
60. Young, AC, Velez, LI, Kleinschmidt, KC. Intravenous fat emulsion therapy for intentional sustained-release verapamil overdose. Resuscitation. 2009;80:591–593.
61. Passal, DB, Crespin, FHJr. Verapamil poisoning in an infant. Pediatrics. 1984;73:543–545.
62. Ralston M, et al, eds. Pediatric Advanced Life Support Provider Manual. Dallas: American Heart Association, 2006.
63. Methemoglobinemia following unintentional ingestion of sodium nitrite—New York. MMWR Morb Mortal Wkly Rep. 2002;51:293.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 152
Cardiovascular Drugs
Cardiovascular drugs are a common cause of poisoning in the United States; in 2010, 97,336 exposures to cardiovascular drugs were reported to U.S. Poison Control Centers.1 Cardiovascular drugs are the second most common cause, accounting for more than 10%, of all poisoning deaths in the United States. Of the scores of cardiovascular drugs, three classes—cardioactive steroids (primarily digoxin), beta-adrenergic blockers, and calcium channel blockers—account for the majority of fatalities.
Cardioactive Steroids (Digoxin)
Digoxin is derived from the Balkan foxglove plant, Digitalis lanata (Fig. 152-1), and the trade name for digoxin (Lanoxin) is derived from the Latin name of this plant.2 Digitoxin, which is no longer in clinical use, comes from Digitalis purpurea.2 Although William Withering was the first to describe the medicinal use of the foxglove plant,3 the ancient Egyptians reference medicinal use of foxglove. Despite centuries of experience with digitalis, chronic and acute poisonings still occur. Controversy about the therapeutic benefit of cardioactive steroids is not new. Benjamin Rush wrote in 1797, “I suspect the cases in which [digitalis preparations] were useful to have been either so few or doubtful and that the cases they had done harm were so much more numerous and unequivocal as justly to banish them from the Materia Medica.”4 Medication errors and toxic effects account for the most common causes (44%) of preventable iatrogenic cardiac arrests.
Principles of Disease
Digoxin is used therapeutically (1) to increase the force of myocardial contraction to increase cardiac output in patients with heart failure and (2) to decrease atrioventricular (AV) conduction to slow the ventricular rate in atrial fibrillation. The basis for its first effect is inhibition of membrane sodium-potassium–adenosine triphosphatase (Na+,K+-ATPase), which increases intracellular sodium and extracellular potassium concentrations. This increase in intracellular sodium concentration results in dysfunction of the sodium-calcium ion exchanger, which normally extrudes intracellular calcium after systole. This subsequent increase in intracellular calcium concentration results in a larger amount of calcium pumped into the sarcoplasmic reticulum, so that on calcium-induced calcium release during subsequent action potentials, a larger amount of calcium is released into the cell, causing a more powerful contraction and thus increased cardiac output. Any molecule with this effect is classified as a cardioactive steroid. Cardiac glycosides (such as digoxin) are merely cardioactive steroids with additional sugar moieties attached to their steroid nucleus. At therapeutic doses, the effects of digoxin on serum electrolyte levels are minimal. With toxic levels, digoxin paralyzes the Na+,K+-ATPase pump, potassium cannot be transported into cells, and serum potassium concentration can rise as high as 13.5 mmol/L.5
Digoxin also exerts three primary effects on Purkinje fibers: (1) decreased resting potential, resulting in slowed phase 0 depolarization and conduction velocity; (2) decreased action potential duration, which increases sensitivity of muscle fibers to electrical stimuli; and (3) enhanced automaticity resulting from increased rate of phase 4 repolarization and delayed afterdepolarizations. These mechanisms account for an increase in premature ventricular contractions, the most common manifestation of digoxin toxicity. At toxic extremes, these effects result in a dangerous sensitivity to mechanical and electrical stimulation. Interventions with pacemaker wires, catheters, and cardioversion can result in asystole, ventricular tachycardia, and ventricular fibrillation.6
Unlike most cardiovascular drugs, digoxin can produce virtually any dysrhythmia or conduction block, and bradycardias are as common as tachycardias (Box 152-1). Unfortunately, none is unique to digoxin, and because they can all occur in the setting of ischemic and other heart disease, digoxin toxicity remains a clinical rather than an electrocardiographic diagnosis.
The volume of distribution (Vd) of digoxin is 5 L/kg for adults but varies from 3.5 L/kg in premature infants to 16.3 L/kg in older infants.7 This indicates that only a small fraction of digoxin remains in the intravascular space, and the drug is highly concentrated in cardiac tissue. The myocardial-to-serum ratio at equilibrium ranges from 15 : 1 to 30 : 1. The Vd for digitoxin is only 0.5 L/kg, giving it a different pharmacokinetic profile.
Multiple drugs and disease states can negatively alter absorption, Vd, protein binding, and elimination and render the heart more susceptible to digoxin toxicity. The factors listed in Box 152-2 are especially important risk factors in chronic intoxication.
Clinical Features
The symptoms and signs of chronic digoxin toxicity are nonspecific. The most common symptoms, in more than 80% of cases, are nausea, anorexia, fatigue, and visual disturbance, but a variety of gastrointestinal, neurologic, and ophthalmic disturbances also occur (Box 152-3). Digoxin intoxication should be considered in any patient receiving maintenance therapy who has consistent symptoms, especially with new conduction disturbances or dysrhythmias.
There are significant differences between acute and chronic toxicity (Table 152-1). Chronic poisoning has an insidious onset and is accompanied by a higher mortality rate that is probably due in part to underlying heart disease and chronic accumulation of the toxin. In cases of chronic intoxication, the LL50 (the level with a 50% mortality) is only 6 ng/mL.8 The LL50 for acute intoxication is not known, but it is certainly much higher, especially in children. The association of hyperkalemia with acute toxicity is obvious given the mechanism of digoxin; either hypokalemia or hyperkalemia may occur with chronic toxicity.
Table 152-1
Chronic versus Acute Digitalis Intoxication
| CHRONIC | ACUTE |
| Higher mortality (LL50 6 ng/mL) | Lower mortality |
| Ventricular dysrhythmias more common | Bradycardia and atrioventricular block more common |
| Usually elderly patients | Usually younger patients |
| Often need Fab fragment therapy | Often do well without Fab (Caution: many exceptions) |
| Underlying heart disease increases morbidity and mortality | Absence of heart disease decreases morbidity and mortality |
Diagnostic Strategies
Diagnosis and management rely heavily on serum digoxin levels, but it is the steady state, rather than peak concentration, that correlates with tissue toxicity and is used to calculate antidote dosages. Peak concentrations after an oral dose of digoxin occur in 1.5 to 2 hours, with a range of 0.5 to 6 hours.9 Steady-state serum concentrations are not achieved until after distribution, or 6 to 8 hours after a dose or overdose, and may be only one fourth to one fifth of the peak concentration. The ideal serum digoxin concentration for patients with heart failure is considered to be 0.7 to 1.1 ng/mL,10 although laboratory “normals” are often reported up to 2.0 ng/mL. Serum steady-state digoxin levels of 1.1 to 3.0 ng/mL are equivocal; that is, levels as low as 1.1 ng/mL have been associated with increased mortality,11 and patients with levels up to 3.0 ng/mL can be asymptomatic.12 The incidence of digoxin-incited dysrhythmia reaches 10% at a level of 1.7 ng/mL and rises to 50% at a level of 2.5 ng/mL. Determination of a level too soon after the last maintenance dose falsely suggests toxicity, especially in cases of chronic intoxication, in which significant morbidity and mortality can occur at levels of 2 to 6 ng/mL.10 After an acute massive overdose in a patient who is rapidly becoming symptomatic, however, it may be impractical to wait 6 to 8 hours for the first reading.13 It is unlikely that early levels exceeding 10 to 20 ng/mL will fade to clinical insignificance at 6 to 8 hours after ingestion.
Management
The mortality rate before Fab fragment therapy was 23% despite all of the interventions described.14 Fab fragment treatment is well established in both chronic and acute poisonings, with a 90% response rate.15 Nonresponders usually receive too little antibody or receive it too late. Other nonresponders are compromised by underlying heart or multisystem disease.
Digitalis antibodies are derived from sheep, but allergic reactions occur in less than 1% of cases.15 Reactions have included erythema, urticaria, and facial edema, all of which are responsive to the usual treatment. Other expected reactions when Fab fragments neutralize digitalis include hypokalemia, exacerbation of congestive heart failure, and increase in ventricular rate with atrial fibrillation. Two Fab fragment preparations were previously available; however, Digibind has been discontinued, leaving DigiFab as the only available product in the United States. If vials of Digibind are still available at a given institution, they require a 0.22-µm membrane filter for proper use; such a filter is not required for DigiFab.
Fab fragment treatment is best reserved for cases of serious cardiovascular toxicity rather than for routine or prophylactic administration with higher than expected serum levels. In acute poisoning, antibody treatment should be used for a serum potassium level above 5.0 mEq/L or unstable dysrhythmias, such as symptomatic sinus bradycardia, ventricular dysrhythmias, or second- or third-degree heart block unresponsive to atropine. Although toxicity increases with greater body load, there is no clear correlation with amount ingested, especially in children, and many patients with large ingestions or high serum levels become only mildly symptomatic.8 Fab fragment therapy should be used before transvenous pacing, which carries significant risk.
The median time to initial response is 19 minutes after completion of the Fab infusion, but complete resolution of digitalis toxic rhythms may require hours.16,17 Late administration of Fab fragments has resuscitated 54% of patients who have suffered cardiac arrest.18 This antidote should be considered whenever hemodynamic compromise attends a digitalis toxic dysrhythmia or heart block (Box 152-4).19
Current formulas for calculation of DigiFab doses are found in the package insert. There are at least three approaches. The first is empirical. A patient has a history of digitalis ingestion, consistent symptoms, and life-threatening dysrhythmias. There is no time to assess serum digoxin levels, and 10 vials should be administered over 30 minutes for the average acute ingestion, 4 to 6 vials for the average chronic ingestion. In cardiac arrest, 20 vials can be administered undiluted by intravenous bolus. The second approach uses a simple calculation when the ingested dose is known with reasonable certainty. One vial of Digibind or DigiFab contains 38 mg or 40 mg, respectively, of Fab fragments, which bind 0.5 mg of digoxin or digitoxin (Box 152-5). A third approach is to base the dosage on the steady-state serum digoxin or digitoxin level after 6 to 8 hours (Boxes 152-6 and 152-7). Because most assays measure both bound and unbound drug, digoxin levels will be elevated for up to 1 week, with values often greater than 100 ng/mL once Fab fragments have been administered. Newer methods can measure free digoxin, but it is more meaningful to follow the patient clinically.
BOX 152-5 Sample Calculation of Digibind or DigiFab Based on Ingested Dose of Digoxin or Digitoxin*
Case: A toxic-appearing 40-year-old woman has ingested fifty 0.25-mg digoxin tablets.
Electrolyte Correction
In acute poisoning, serum potassium concentration may begin to rise rapidly within 1 to 2 hours of ingestion. Potassium should be withheld, even if mild hypokalemia is measured initially. The initial serum potassium concentration may in fact be a better predictor of mortality than the initial digoxin concentration. In a study of 91 patients with acute digoxin poisoning, nearly 50% of the patients with serum potassium concentrations between 5.0 and 5.5 mmol/L died. No patients with a potassium level of less than 5.0 mEq/L died, and all 10 patients with serum potassium concentrations exceeding 5.5 mmol/L perished.20 Because of these findings, a serum potassium concentration greater than 5 mmol/L alone warrants treatment with Fab fragments. Even with Fab fragments, severe hyperkalemia should be treated with intravenous administration of glucose, insulin, and sodium bicarbonate as needed.
The decision to administer calcium to patients with hyperkalemia and digoxin poisoning represents a clinical dilemma. Classic teaching is that in the setting of the increased intracellular calcium concentration from digoxin poisoning, administration of exogenous calcium will result in “stone heart” from excessive intracellular calcium.21 This concept has been in the literature since 1927,22 based on animal studies.23 Documented cases of cardiac arrest after calcium administration are exceedingly rare in the literature, and the temporal relationship is dubious.24 More recent data indicate that the intravenous administration of calcium can be safe for hyperkalemia in the setting of digoxin toxicity.25 Unequivocally, however, the best treatment of hyperkalemia due to acute digoxin toxicity is Fab fragments. The treatment of hyperkalemia in the setting of chronic digoxin toxicity and renal failure is less clear; however, the evidence that calcium salts will be harmful is dubious at best. Calcium salts should be administered during several minutes through a secure peripheral intravenous site or through a central venous catheter. Indications are identical to those for hyperkalemia, including but not exclusive to a widened QRS duration on the electrocardiogram. A reasonable starting dose would be 3 g of calcium gluconate or 1 g of calcium chloride. Calcium salts should be followed immediately with other pharmacologic measures to lower serum potassium concentration.
Many patients receiving diuretic therapy are also magnesium depleted, even when the measured serum magnesium level is normal. If significant magnesium depletion is suspected (e.g., if electrocardiographic changes such as QTc prolongation are present), 1 to 2 g of magnesium sulfate can be given during 10 to 20 minutes (child: 25 mg/kg), followed by a constant infusion of 1 to 2 g/hr. Patients should be closely monitored for respiratory depression, which is usually preceded by progressive loss of deep tendon reflexes. Hypermagnesemia can exacerbate digitalis toxicity, but magnesium has been reported to reverse digoxin-induced tachydysrhythmias.26 It is prudent to infuse magnesium slowly and to stop the infusion if heart block or bradycardia develops. Avoid magnesium in patients with renal failure. The role of magnesium in bradydysrhythmias and conduction blocks is less clear but probably dangerous because hypermagnesemia can impair impulse formation and AV conduction.
Pacing
Transvenous pacing has been a mainstay of treatment for several decades, but the catheter may induce ventricular tachydysrhythmias in a myocardium made irritable by digoxin. Iatrogenic accidents of cardiac pacing are frequent (14 of 39, 36%) and often fatal (5 of 39, 13%).27 We recommend avoidance of transvenous pacing unless external pacing fails. Pacing usually is required only temporarily while waiting for Fab fragments to take effect. Cardioversion for tachydysrhythmia also may be hazardous in the setting of digoxin intoxication. If it is deemed necessary, such as when the tachydysrhythmia is thought to be causing severe hypotension, we recommend use of lower than usual energy settings, such as 25 to 50 J, although there is no proof that this is less hazardous.
Phenytoin and Lidocaine
Phenytoin and lidocaine are believed to be the safest of the antidysrhythmic drugs for control of tachydysrhythmias in the setting of digoxin toxicity. Indications include unstable tachydysrhythmias when Fab fragments are unavailable and unstable tachydysrhythmias that occur while waiting for Fab fragments to take effect. Phenytoin may enhance AV conduction. Phenytoin has been infused at 25 to 50 mg/min to a loading dose of 15 to 20 mg/kg; it may also be administered in 100-mg boluses every 5 minutes until arrhythmias improve or 1000 mg is administered. Lidocaine can be given initially at a dosage of 1 to 1.5 mg/kg during several minutes, followed by an infusion of 1 to 4 mg/min (30-50 µg/kg/min).28
[/not-level-membership-for-emergency-medicine-category]
