[level-membership-for-emergency-medicine-category]
148 Cardiovascular Drugs
 Key Points
Key Points• Cardiovascular drugs are responsible for many fatalities.
• β-Receptor antagonists, calcium channel antagonists, and digoxin primarily cause toxicity by disruption of intracellular calcium homeostasis and lead to hypotension and dysrhythmias.
• With sustained-release forms of calcium channel antagonists and beta-receptor antagonists, toxicity has a delayed peak and longer duration, which can lead to cardiovascular collapse and arrest if treatment is delayed or there is insufficient cardiovascular moritoring.
• Diagnostic testing should include continuous cardiac monitoring; electrocardiogram; measurement of appropriate serum drug concentrations, electrolytes, and glucose; and investigation of coingestants.
• The call to the pharmacist to obtain the hyperinsulinemia-euglycemia infusion in calcium channel antagonist overdose should be made when the norepinephrine infusion is begun.
Epidemiology
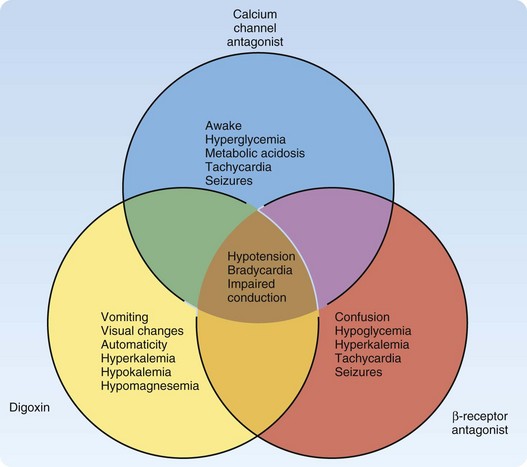
The 2009 Annual Report of the American Association of Poison Control Centers National Poison Center Database System reported cardiovascular drugs as the second most common cause of fatalities overall (10%), and they were the second fastest in rate of exposure increase.1 Cardiovascular drugs as a category were ranked as the fifth leading cause of death (44 total deaths: 5 from β-receptor antagonists, 16 from calcium channel antagonists, and 23 from cardiac glycosides). These specific cardiovascular drugs share the clinical effects of hypotension, bradycardia, and conduction disturbances. However, unique differences can help distinguish them in an unknown overdose (Fig. 148.1). Other pharmaceuticals included in the category of cardiovascular agents are angiotensin-converting enzyme inhibitors, antiarrhythmics, clonidine, and other antihypertensives; they are not discussed in this chapter.
Calcium Channel Antagonists
Pathophysiology
Calcium channel antagonists block the intracellular flow of calcium ions through L-type voltage-gated calcium channels in myocardial, smooth muscle, and pancreatic beta-islet cells. These mechanisms of action result in cardiovascular toxicity both directly and indirectly. Depending on the selectivity of the calcium channel antagonist, the direct cardiovascular toxicity is a combination of the effects on the cardiac conduction system, myocardial contractility, and vascular smooth muscle vasodilation. The dihydropyridine class (e.g., amlodipine, nifedipine) preferentially acts on the peripheral vasculature, thereby potentially leading to hypotension and reflex tachycardia. Verapamil operates on the sinoatrial and atrioventricular (AV) nodes and on the myocardium. Diltiazem acts to a lesser extent than verapamil on the cardiac tissue and nodes, and it also dilates peripheral vasculature (Table 148.1). The degree of contribution from each mechanism of cardiovascular toxicity can be difficult to predict. Despite the differences in therapeutic mechanisms, the distinctions among families of calcium channel antagonists are often blurred during an overdose, and the patient generally suffers from negative chronotropic, inotropic, and dromotropic effects.2
| CLASS | ACTION(S) | EXAMPLE(S) |
|---|---|---|
| Phenylalkylamines | Act on sinoatrial and atrioventricular nodes and the myocardium | Verapamil (Calan) |
| Benzothiazepines | Dilate peripheral vasculature and act to a lesser degree than verapamil on cardiac tissues and nodes | Diltiazem (Cardizem, Tiazac) |
| Dihydropyridines | Act on peripheral vasculature, leading to hypotension and reflex tachycardia |
Presenting Signs and Symptoms
Verapamil and diltiazem overdoses cause bradycardia and numerous conduction abnormalities in and below the AV node. The decreased blood pressure results from vasodilation and decreased cardiac contractility from the negative inotropic and chronotropic effects.2
Differential Diagnosis and Medical Decision Making
The differential diagnosis (see also Fig. 148.1) for overdose of calcium channel antagonists includes other cardiovascular drugs such as beta-blockers, clonidine, digitalis, and other antidysrhythmics. The emergency physician should also consider myocardial infarction and other causes of cardiogenic shock. The potency of the effect of calcium channel antagonists on the cardiovascular system is astounding. Significant cardiovascular toxicity can occur after supratherapeutic ingestion of calcium channel antagonists. Ingestion of double the therapeutic dose should instigate medical evaluation and treatment. Immediate-release calcium channel antagonists should have some clinical effect within 6 hours. Sustained-release calcium channel antagonists should result in clinical manifestations within 1 to 14 hours.3
Diagnostic testing is contingent on the necessity of treatment for hemodynamic instability. Once the patient’s airway, breathing, and cardiovascular status have been assessed and stabilized, testing should start with a 12-lead electrocardiogram (ECG) and chest radiography. Rapid determination of hyperglycemia and metabolic acidosis with capillary glucose and arterial blood gas analysis may demonstrate a severe calcium channel antagonist overdose. An elevated serum lactate concentration may be another marker of severe calcium channel antagonist overdose.4 Testing for serum concentrations of calcium channel antagonists is not clinically useful or available to guide treatment. Otherwise, standard laboratory testing for a general overdose is a good comprehensive approach.
Treatment
Whole-bowel irrigation has been suggested for overdose of calcium channel antagonists because many of these drugs are sustained-release preparations. Whole-bowel irrigation is not indicated for a patient with hemodynamic instability because a significant amount of the drug has already been absorbed,5 and, therefore, the opportunity for prevention has passed. In addition, challenging a hypoperfused gastrointestinal system can have disastrous consequences, such as functional and physical obstruction by a calcium channel antagonist bezoar,6–9 as well as perforation. Generally speaking, no evidence indicates that any gastric decontamination procedure improves outcome in the patient with an overdose, and the risks must be assessed against the benefits.
An antidotal treatment regimen is provided in Figure 148.2. This regimen emphasizes elemental calcium, either as calcium gluconate (30 mL of a 10% solution, or 3 g of calcium gluconate; 14 mEq elemental calcium) or calcium chloride (10 mL of a 10% solution, or 1 g; 13.5 mEq of elemental calcium). Calcium chloride should be administered through central venous access because it is an acidifying salt, which could cause necrosis of the peripheral vasculature. If the intravenous calcium boluses appear to have improved hemodynamic status, close monitoring for recrudescence of toxicity must be maintained, and further boluses must be given as necessary. An intravenous infusion of calcium is warranted only when it effectively treats the hypotension, and further boluses are required to support the blood pressure (Table 148.2). The serum calcium concentration should be monitored, but antidotal treatment rarely gives rise to clinically significant hypercalcemia.
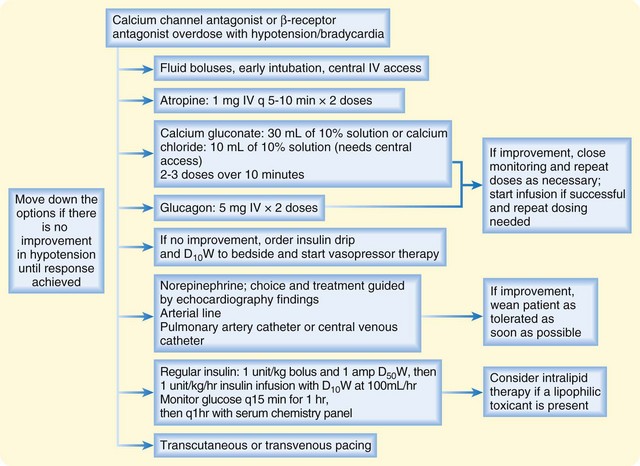
Fig. 148.2 Treatment of overdose with calcium channel antagonist or β-receptor antagonist.
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly).
Table 148.2 Antidotes, Treatments, Facts, and Formulas for Cardiovascular Drugs
| ANTIDOTE OR TREATMENT | DOSING | ADVERSE EFFECTS and signs of improving perfusion |
|---|---|---|
| Atropine |
Consultation with a clinical toxicologist is recommended
Give a bolus of 1.0 units/kg IV with 1 ampule of D50W
Immediately start an infusion of 1.0 units/kg/hr with a glucose infusion of D10W at 100 mL/hr
Titrate 0.5 units/kg every 30 min until desired effect; 1 ampule of D50W can be given during every increase in infusion; titrate to desired effect when blood pressure has reached desired value and signs of improving perfusion
When the blood pressure has reached the desired value with the insulin infusion, taper and wean pressor agent therapy
Monitor serum glucose concentration every 15 min for the first 60 min; when stable, monitor every 60 min thereafter
Monitor serum potassium and other electrolyte concentrations every 60 min
For acute overdose (unknown amount in adult or child): 5-10 vials IV as a bolus; repeat as needed every 30 min
For chronic overdose in adult or child: 1-2 vials IV as a bolus; repeat as needed every 30 min
If amount ingested is known: 1 vial binds 0.5 mg of digoxin
Dosage calculated from serum digoxin concentration:
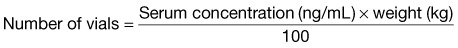
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly); MAP, mean arterial pressure.
Hyperinsulinemia euglycemia (HIE) therapy and catecholamines with inotropic and vasopressor activity are the next line of treatment for refractory hypotension in calcium channel antagonist overdose, but inotropics and vasopressors will be discussed first. A multicenter study compared dopamine and norepinephrine agents in all patients categorized as being in shock, regardless of cause. No difference was seen in the outcome (death at 28 days) for all patients in the study, but a predetermined subgroup analysis found greater mortality in patients with cardiogenic shock who were treated with dopamine.10 Because calcium channel antagonists can cause cardiogenic shock, norepinephrine is probably a good first choice. If clinically significant hypotension persists, adding more agents may be necessary. Cardiovascular data from diagnostic modalities such as transthoracic echocardiogram, pulmonary artery catheter, arterial catheter, and central venous catheter should dictate which cardiovascular agent is the most appropriate choice. Vasopressin has been used in human cases when peripheral vasoconstriction is indicated.11 Worsening of the cardiac index was demonstrated when vasopressin was used in an animal model to treat hypotension induced by calcium channel antagonist.12
In 1999, Yuan et al.13 described the first published use of high-dose insulin-euglycemia (HIE) therapy, in four patients with verapamil overdose and in one patient with amlodipine and atenolol overdose. HIE promotes inotropy by improving myocardial energy production. In addition, insulin has antiinflammatory attributes that protect against apoptosis and ischemic reperfusion injury.2
Subsequently, numerous case reports, reviews, and HIE regimens were published.2,14–19 HIE therapy has successfully reversed cardiogenic shock from a polydrug overdose,20 and historically it was used in multiple nontoxicologic conditions, such as acute myocardial infarction, post–cardiac surgery status, and septic shock.21 The superiority of HIE therapy for cardiogenic shock resulting from calcium channel antagonist toxicity was also demonstrated dramatically in animal models.22–25
Failure of HIE therapy often occurs when it is started as rescue therapy and when the dose is inadequate.26 A small prospective observational study of patients treated with HIE therapy supports it safety.27 HIE therapy should be started when boluses of calcium, glucagon, atropine, and intravenous fluids have failed and the physician is considering a pressor agent to improve refractory hypotension. The call to the pharmacist to obtain the HIE infusion should be made when the norepinephrine infusion is begun.
A relatively new and novel antidote is increasingly being used to treat highly lipophilic toxicants, such as verapamil. Intralipid 20% was initially used for local anesthetic toxicity. In patients and animal models, dysrhythmias, hypotension, and even cardiac arrest from bupivacaine toxicity were reversed rapidly with intralipid bolus. Three theories on how this works have been proposed. One theory is the lipid forms a “sink” in the vascular compartment that pulls the toxicant from the tissues, where toxicity is occurring, and it becomes trapped and eliminated. The second is that bupivacaine inhibits transport of fatty acids into mitochondria required for energy production, and the exogenous lipids overcome this inhibition. The third is that fatty acids increase calcium in cardiac myocytes and therefore increase inotropy. Generally, boluses are used, but an infusion is sometimes necessary. Unfortunately, the safety of the boluses is unknown, but the complications of intralipid use are generally from prolonged total parenteral infusions.28
Young et al.29 reported a case of a 32-year-old man who ingested 13.44 g of verapamil and bupropion, zolpidem, quetiapine, clonazepam, and benazepril. This patient had refractory hypotension after treatment with intravenous fluids, glucagon, calcium, and norepinephrine. He was then administered 100 mL of 20% intralipid over 20 minutes and then 0.5 mL/kg/hour for almost 24 hours. His blood pressure improved enough in 1 hour to begin weaning the norepinephrine, and the glucagon infusion was discontinued 2 hours after intralipid administration.29 Consultation with a medical toxicologist can be very helpful when considering HIE or intralipid therapy.
Disposition
Asymptomatic patients who have ingested an immediate-release calcium channel antagonist can be monitored for 6 hours in the emergency department. After ingestion of a sustained-release calcium channel antagonist, the asymptomatic patient should undergo cardiovascular monitoring for 18 to 24 hours.3 All symptomatic patients with cardiovascular instability after cardiovascular drug overdose should be admitted to the intensive care unit for cardiovascular monitoring, diagnostic studies, and treatment until the effects have resolved.
Special Considerations: Pediatric Overdose
The 2009 National Poison Center Database System data noted one pediatric fatality of a suicidal 16-year-old girl, who ingested verapamil and an unknown drug.1 The clinical consequences of accidental pediatric calcium channel antagonist ingestions depend on the dose.
One must consider that the major flaw of studies attempting to demonstrate a dose response of accidental pediatric drug ingestions is that many of the reports are nonexposures. The caretaker of the child may report the exposure to a poison center if a pill is missing and the child is implicated by his or her presence in the vicinity. If the child did not take the drug, the case may be referred to as having a good outcome, and the “dose” considered safe.30
A guideline for pediatric ingestions of calcium channel antagonists states that immediate referral to a health care center is necessary if the dose exceeded the usual therapeutic dose or was considered equal to or greater than the lowest toxic dose (whichever is lower).3 At these doses, significant bradycardia or hypotension may occur. Accidental single ingestions of calcium channel antagonists in children are considered lethal enough to be fatal.31
Cardiovascular monitoring for 6 to 8 hours for immediate-release medications and for at least 24 hours for sustained-release medications should reveal delayed toxicity. All symptomatic children should be admitted for cardiovascular monitoring and treated with standard therapy.3
Tips and Tricks
Calcium Channel Antagonist Toxicity
• The patient who presents with undifferentiated hypotension and bradycardia may paradoxically be relatively alert as a result of calcium channel antagonist toxicity, which may cause cerebrovascular vasodilation that is cerebroprotective.
• Insulin is an ideal inotropic agent because it can increase the contractility of the heart without raising oxygen demand.
• Monitor electrolyte concentrations and excess intravenous fluid closely.
• An echocardiogram, central venous pressure monitor, and pulmonary artery catheter can be helpful for diagnosing and treating the multiple components of the shock that occurs during calcium channel antagonist toxicity.
• Continue to monitor serum glucose concentrations after the insulin infusion is discontinued until consistent euglycemia is achieved.
 Priority Actions
Priority Actions
Treatment of Calcium Channel Antagonist Toxicity
• Consult with a medical toxicologist.
• Intubate hemodynamically unstable patients electively, before emergency intubation is required because of cardiopulmonary arrest.
• Obtain central venous access for fluid resuscitation and administration of pharmaceutical infusions.
• Gastric decontamination is critical in the patient presenting with hemodynamic stability, but it should not be used if the patient is unstable.
• Use hyperinsulinemia/euglycemia (HIE) therapy early (in conjunction with medical toxicology consultation) after intravenous boluses of normal saline, atropine, calcium, and glucagon have failed.
• Titrate the HIE therapy in the same fashion as other standard inotropic medications to obtain a mean arterial pressure of 65 to 75 mm Hg and sings of improving perfusion. Tight glucose control is not the target of treatment.
• Maintain the insulin infusion and taper and stop vasopressor therapy when the mean arterial pressure has achieved 65 to 75 mm Hg and signs of organ perfusion are present.
• If hypoglycemic episodes occur in the setting of hemodynamic stability, decrease the rate of the insulin infusion. If hemodynamic instability occurs, increase the rate of the dextrose infusion.
β-Receptor Antagonists
Pathophysiology
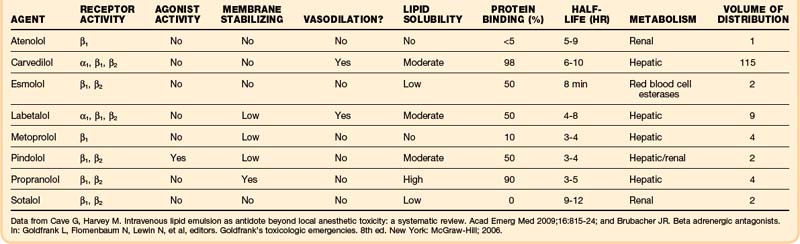
β-Receptor antagonism is not the only pharmacologic mechanism of toxicity seen after β-receptor antagonist overdose. α-Receptor antagonism, sodium or potassium channel antagonism, central nervous system penetration leading to seizures with altered mental status, and sympathomimetic stimulation may also occur (Table 148.3).
Presenting Signs and Symptoms
Immediate-release products should cause signs and symptoms within 6 hours. Unfortunately, most β-receptor antagonists are of modified-release formulation. They have some pharmacologic effect during the first 6 hours after ingestion, but the peak serum concentration is delayed, and the pharmacologic effect may last longer than with immediate-release formulations.32
Sotalol is infamous for antagonizing the delayed rectifier potassium channels in the myocardium. The results are a prolonged QT interval and a higher risk of torsades de pointes (polymorphic tachycardia), monomorphic ventricular tachycardia, ventricular fibrillation, and asystole. These clinical effects can also be delayed and prolonged. In one case report, the onset of ventricular dysrhythmias occurred 4 to 9 hours after ingestion and did not normalize until 100 hours from the time of ingestion.33
Tips and Tricks
β-Receptor Antagonist Toxicity
• Obtain a capillary blood glucose measurement to detect euglycemia or hypoglycemia.
• Identify the unique toxicity of each β-receptor antagonist.
• Do not start infusions of glucagon or calcium until multiple boluses have successfully reversed the toxicity.
• Avoid isoproterenol as an antidotal agent.
• Hyperinsulinemia/euglycemia therapy works to treat hypotension from β-receptor antagonist toxicity.
• Atenolol and sotalol have characteristics that allow the use of hemodialysis to enhance elimination.
• Monitor electrolyte concentrations and excess intravenous fluid closely.
• An echocardiogram, central venous pressure monitor, and pulmonary artery catheter can be helpful for diagnosing and treating the multiple components of the shock that occurs during β-receptor antagonist toxicity.
• If the beta-blocker is lipophilic (e.g., propranolol), intralipid therapy may be effective at reversing toxicity.
Differential Diagnosis and Medical Decision Making
The differential diagnosis (see also Fig. 148.1) of beta-blocker toxicity includes the following: congestive heart failure and pulmonary edema, cardiogenic or hemorrhagic shock, epidural hematoma, epidural and subdural infections, meningitis, other causes of electrolyte abnormalities, and overdoses of cardiac glycosides, calcium channel blockers, carbamazepine, carbon monoxide, cocaine, and antidepressants.
Treatment
The end result of overdose of either β-receptor or calcium channel antagonists is shock, despite their different mechanisms of action. Because these agents have a final common pathway, antidotal therapy is analogous. Treatment follows the same algorithm as shown in Figure 148.2. Atropine can be used initially, followed by intravenous calcium, glucagon, HIE, and catecholamines with chronotropic and inotropic effects for refractory bradycardia and AV block. Unfortunately, severe bradycardia and AV block are often refractory to pharmaceutical efforts, and transcutaneous or transvenous pacing may be required.
Glucagon increases cardiac contractility by bypassing the antagonized β-receptors through activation of cAMP by agonism at the glucagon receptors. This activation increases contractility by activating the phosphorylation cascade, which leads to contraction of actin and myosin. Glucagon also stimulates release of endogenous insulin, a beneficial side effect. Unfortunately, glucagon is often ineffective at reversing β-receptor antagonist toxicity.34
Vasopressin was used in an experimental animal model poisoned by propranolol.35 The investigators discovered equally dismal survival rates for treatment with glucagon and vasopressin.
HIE therapy is a therapeutic approach for β-receptor antagonist toxicity. Animal models demonstrated the superiority of HIE therapy over glucagon, epinephrine, and saline solution for the reversal of the toxic effects of propranolol.36 Yuan et al.13 reported effective reversal of the toxic effects of a β-receptor and calcium channel antagonist coingestion with this treatment. Despite the absence of the diabetic ketoacidosis metabolic state produced by calcium channel antagonist, HIE therapy is believed to be just as effective in β-receptor antagonist intoxication. Mechanistically, this antidote improves inotropy by promoting aerobic utilization of glucose by the myocardial myocytes, inhibition of fatty acid metabolism, decreased lactate production, and improvement of myocardial oxygen utilization without increasing oxygen demand. The mild hypokalemia that occurs also may beneficial.17
A case of cardiac arrest induced by nebivolol, diazepam, and baclofen was reversed with a bolus of intralipid 20% and HIE therapy.37 Intralipid therapy should be considered in lipophilic β-receptor antagonist overdose.
Disposition
 Priority Actions
Priority Actions
Treatment of β-Receptor Antagonist Toxicity
• Supportive care of airway, breathing, and circulation is the first critical step.
• Gastric decontamination is critical in the patient presenting with hemodynamic stability.
• Administer doses of calcium and glucagon adequate to elicit a clinical response.
• Consider HIE therapy for cardiogenic shock.
• Consider norepinephrine as the first choice for vasopressor therapy.
• Electrical pacing is an option for symptomatic β-receptor antagonist bradycardia.
Digoxin
Pathophysiology
Digoxin is a cardiac glycoside that was historically used for the treatment of congestive heart failure and for rate control in atrial fibrillation. Despite a reduction in popularity, digoxin is still clinically effective for many patients. Natural cardiac glycosides provide another possible exposure source (Box 148.1).
Several clinically important points must be remembered about the pharmacology of digoxin (Table 148.4). The drug is well absorbed, and the onset of action occurs within minutes to hours of administration. The serum digoxin concentration initially is supratherapeutic, until equilibrium between the serum and tissues has occurred. The optimum time for measurement of the serum digoxin concentration is at least 6 hours after ingestion. The volume of distribution is large and the enterohepatic circulation is small, thus rendering methods for enhancing elimination clinically ineffective. These properties effectively make the elimination half-life approximately 36 to 48 hours.38,39 Most digoxin is eliminated as the parent compound in the urine. Consequently, a decrease in renal function often leads to acute-on-chronic digoxin toxicity in the geriatric patient. The clinician must keep in mind that these pharmacokinetic data are from controlled clinical situations. True toxicokinetic data are difficult to forecast in the patient with digoxin overdose because of inaccuracies about the timing and amount of the dose, comorbidities, drug interactions, and unpredictable variables in the human metabolism of digoxin.
| Onset of action |
Data from Cave G, Harvey M. Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: a systematic review. Acad Emerg Med 2009;16:815-24; And Wax PM, Erdman AR, Chyka PA, et al. Beta-blocker ingestion: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila) 2005;43:131-46.
Presenting Signs and Symptoms
Patients may be asymptomatic from minutes to hours after ingestion of digoxin. Gastrointestinal symptoms are common, and patients have nausea and vomiting. Patients may complain of changes in vision, especially chromatopsia and xanthopsia. Some mild confusion, weakness, and dizziness also can occur from the direct effect of the digoxin. The cardiac effects are usually represented by symptoms such as palpitations, chest pain, dizziness, and dyspnea. The cardiac signs in acute toxicity are a mixture of bradydysrhythmias, tachydysrhythmias with conductive blockade, and hypotension. Cardiac conduction can be impaired anywhere along the pathway from the sinus node to the AV node and the His-Purkinje fibers. Commonly reported dysrhythmias are listed in Box 148.2. Bidirectional ventricular tachycardia is considered pathognomonic for digoxin toxicity.
Box 148.2 Dysrhythmias Associated with Digoxin Toxicity
Tips and Tricks
Digoxin Toxicity
• If the patient has life-threatening digoxin toxicity, treatment with the digoxin-specific Fab fragments is the first priority.
• Acute-on-chronic and acute digoxin toxicities have the same manifestations, but acute and chronic digoxin toxicities have distinct symptoms, dysrhythmias, and laboratory findings.
• The digoxin serum concentration may be only slightly elevated or normal in chronic digoxin toxicity.
• Digoxin toxicity can manifest as almost any dysrhythmia.
• The equilibrium concentration of serum digoxin occurs 6 hours after ingestion, and is inaccurate after administration of digoxin-specific Fab fragments.
• Recurrence of toxicity is typically related to underdosing of digoxin-specific Fab fragments.
• The indications and treatment of digoxin toxicity and concomitant renal impairment are the same.
• No method of enhancing elimination, including hemodialysis, is effective.
Differential Diagnosis and Medical Decision Making
Acute digoxin toxicity: A patient who is naive to the medication is exposed to a single acute ingestion.
Acute-on-chronic digoxin toxicity: The serum digoxin concentration increases because of renal failure or an inadvertent or intentional increase in dose. The clinical presentations for this and acute toxicity are similar, so management is similar.
Chronic digoxin toxicity: A patient has clinical signs of digoxin toxicity with a mildly elevated or therapeutic serum digoxin concentration.
The differential diagnosis includes other cardiovascular drugs (see Fig. 148.1), acute renal failure, hypercalcemia, hyperkalemia and hypokalemia, hypernatremia and hyponatremia, and hypomagnesemia.
Laboratory testing should be performed expeditiously in any digoxin overdose, with the focus on serum potassium, magnesium, and digoxin concentrations. Rapid assessment of serum potassium concentration helps determine the severity of the toxicity. In acute digoxin poisoning, the serum potassium value is elevated. If this value is 5 mEq/L or greater, digoxin-specific Fab fragment therapy should be considered.40 In chronic digoxin toxicity, the serum potassium concentration is often low, usually because of concomitant ingestion of a diuretic. The hypokalemia, in effect, worsens the inhibition of the Na+,K+ ATP-ase pump.
A serum digoxin concentration measured in blood collected after administration of digoxin-specific Fab fragments is clinically not useful and is uninterpretable. The assay often measures the antidote, the drug, and the combination of the two, and it interprets one or all of them as the serum digoxin concentration; the result may be higher than, lower than, or within the therapeutic range for serum digoxin.41,42
Screening for accompanying hypomagnesemia is important because this condition may lead to refractory hypokalemia, blockade of inward calcium channels and intracellular binding sites, blockade of extracellular movement of potassium, decrease in myocardial irritability, and a prolonged QT interval. Hypomagnesemia increases myocardial uptake of digoxin and worsens dysfunction of the Na+,K+-ATP-ase pump.43
Treatment
The successful use of digoxin-specific Fab fragments to treat digoxin intoxication was first described in 1976.44 Since that time, multiple studies have demonstrated its safety and efficacy.45,46 The best antidote to administer for digoxin toxicity is digoxin-specific Fab fragments, whether for hypotension, dysrhythmias, serum digoxin concentration, or hyperkalemia.
Two commercial formulations of digoxin-specific Fab fragments are available, Digibind and DigiFab. Literature for both products warns against anaphylaxis and administration of the agents to people with papain, chymopapain, or papaya allergies. Other adverse events associated with administration of digoxin-specific Fab fragments occur from removal of the therapeutic benefit of the digoxin—for example, recurrence of congestive heart failure46 or atrial fibrillation with a rapid ventricular response.

The number obtained is the number of vials needed to treat the patient.
When the amount ingested is known, the digoxin-specific Fab fragment dose can be calculated by knowing that 1 vial will bind 0.5 mg, or multiply the amount ingested by 2 (see Table 148.4 for determining dose). In the noncritical patient, digoxin-specific Fab fragments should be reconstituted with 4 mL of saline, used immediately or within 4 hours if refrigerated, and infused over 30 minutes. Indications for digoxin-specific Fab fragments are listed in Box 148.3. A clinical response should be seen within 60 minutes.46
Many patients presenting with acute-on-chronic digoxin toxicity have renal failure. Digoxin-specific Fab fragments should not be withheld in these patients because of concern about the inability of the kidney to remove the digoxin-Fab complex. This complex cannot be removed by dialysis either. Mild recrudescence of toxicity has been reported when the Fab fragments become unbound and are eliminated faster than digoxin.47 However, multiple publications cite inadequate dosing as a much greater risk factor for recrudescence, and the transient rise in serum digoxin concentration has been within the therapeutic range and not clinically significant.47,48 Simply administering another dose of digoxin-specific Fab fragments or giving conscientious supportive care may be the only additional therapy required. These patients typically have transient renal impairment, and once it has resolved, the digoxin, Fab fragments, and Fab-digoxin complex are removed. Finally, the use of digoxin-specific Fab fragments can be cost effective.49
Transcutaneous pacing and transvenous pacing have been used to treat symptomatic bradycardia, but transvenous pacing must be used with caution. One study reported a higher mortality rate in patients receiving transvenous pacing because of dysrhythmias.50 In addition, iatrogenic complications (36%) were seen in patients receiving transvenous pacing.
This danger of transvenous pacing in the setting of digoxin toxicity has come into question, however. A retrospective review of 70 patients was divided into two groups. One group received transvenous pacing, and the other did not. No deaths occurred in the paced group, and 2 patients in the nonpaced group died of ventricular dysrhythmias, but no statistically significant difference was seen. Transvenous pacing was not an independent predictor of prognosis. The investigators reported no complications of venous thromboembolism, cardiac rupture, or infection. The investigators speculated that the previous study had poor outcomes from overdrive transvenous pacing, which can stimulate ventricular dysrhythmias. In their study, the heart rate was limited to 60 beats/minute.51
The use of intravenous calcium in the treatment of hyperkalemia in the digoxin-poisoned patient was considered a contraindication during much of the twentieth century. The concern about calcium administration in digoxin toxicity is an additive toxic effect. During digoxin toxicity, patients already have a dysfunction in intracellular calcium regulation along with an elevated calcium concentration. If more calcium is added to this hypercontractile state, a condition referred to as “stone heart” could be produced. This concern was supported by an early human case series and two animal studies, all published before 1940.52–54
More recently, a case report and animal study found no synergistic effect of calcium and digoxin toxicity resulting dysrhythmia or death during treatment of hyperkalemia.55,56 A retrospective review of 159 patients with digoxin toxicity over 17.5 years compared dysrhythmia in 1 hour from calcium or mortality in the patients treated with calcium versus no calcium. The investigators discovered no dysrhythmias and similar mortality in both groups and increased mortality in patients with higher serum potassium.57
An excellent review of the literature on this topic specified that the rate and amount of calcium administered is probably more contributory to dysrhythmias or the stone heart. The reviewers reasonably concluded that if the patient has signs of hyperkalemia toxicity, such as loss of P waves, peaked T waves, or a widened QRS complex, treatment with calcium should be undertaken. If the patient has manifestations of digoxin toxicity such as ectopic beats or ventricular tachycardia, then digoxin-specific Fab fragments should be the first-line treatment.58 Ultimately, the anxiety created by calcium combined with digoxin toxicity is probably much greater than the true risk. This risk can be minimized even more by using other methods to decrease the serum potassium. If the patient is hemodynamically stable, administration of digoxin-specific Fab fragments should be the first choice to treat the hyperkalemia and all the other components of digoxin toxicity.
 Priority Actions
Priority Actions
Treatment of Digoxin Toxicity
• Supportive care of airway, breathing, and circulation is the first critical step.
• Gastric decontamination is critical in the patient presenting with hemodynamic stability, but refrain from challenging the gastrointestinal system of a patient with hemodynamic instability.
• Assess serum potassium and magnesium levels immediately.
• Digoxin-specific Fab fragments are the definitive treatment for all clinical manifestations of digoxin toxicity.
• Dosage of digoxin-specific Fab fragments can be (1) empirical (5-10 vials) in the hemodynamically unstable patient, (2) calculated according to the amount of digoxin bound by 1 vial, or (3) calculated from the serum digoxin concentration.
• Decrease the current for cardioversion in patients with digoxin toxicity.
• Use a small number of vials (1-2) to treat patients with chronic digoxin toxicity.
1 Bronstein AC, Spyker DS, Cantelina LR, et al. 2009 Annual report of the American Association of Poison Control Centers’ National Poison Center Database (NPDS): 27th annual report. Clin Toxicol (Phila). 2010;48:979–1178.
2 Shepherd G, Klein-Schwartz W. High-dose insulin therapy for calcium-channel blocker overdose. Ann Pharmacother. 2005;39:923–930.
3 Olson KR, Erdman AR, Woolf AD, et al. Calcium channel blocker ingestion: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2005;43:797–822.
4 Levine M, Boyer EW, Pozner CN, et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med. 2007;35:2071–2075.
5 Position paper: whole bowel irrigation. J Toxicol Clin Toxicol. 2004;42:843–854.
6 Wax PM. Intestinal infarction due to nifedipine overdose. J Toxicol Clin Toxicol. 1995;33:725–728.
7 Sporer KA, Manning JJ. Massive ingestion of sustained-release verapamil with a concretion and bowel infarction. Ann Emerg Med. 1993;22:603–605.
8 Fauville JP, Hantson P, Honore P, et al. Severe diltiazem poisoning with intestinal pseudo-obstruction: case report and toxicological data. J Toxicol Clin Toxicol. 1995;33:273–277.
9 Cumpston KL, Aks SE, Sigg T, Pallasch E. Whole-bowel irrigation and the hemodynamically unstable calcium channel blocker overdose: primum non nocere. J Emerg Med. 2010;38:171–174.
10 DeBacker D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:789–799.
11 Leone M, Charvet A, Boyle WA. Terlipressin: a new therapeutic for calcium channel blocker overdose. J Crit Care. 2005;20:114–115.
12 Sztajnkrycer MD, Bond GR, Johnson SB, Weaver AL. Use of vasopressin in a canine model of severe verapamil poisoning: a preliminary descriptive study. Acad Emerg Med. 2004;11:1253–1261.
13 Yuan TH, Kerns WP, 2nd., Tomaszewski CA, et al. Insulin-glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37:463–474.
14 Lheureux PE, Zahir S, Gris M, et al. Bench-to-bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium-channel blockers. Crit Care. 2006;10:212.
15 Levine MD, Boyer E. Hyperinsulinemia-euglycemia therapy: a useful tool in treating calcium channel blocker poisoning. Crit Care. 2006;10:149.
16 Ortiz-Munoz L, Rodriguez-Ospina LF, Figueroa-Gonzalez M. Hyperinsulinemic-euglycemic therapy for intoxication with calcium channel blockers. Bol Asoc Med P R. 2005;97:182–189.
17 Megarbane B, Karyo S, Baud FJ. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium channel antagonist and beta-blocker poisoning. Toxicol Rev. 2004;23:215–222.
18 Boyer EW, Duic PA, Evans A. Hyperinsulinemia/euglycemia therapy for calcium channel blocker poisoning. Pediatr Emerg Care Feb. 2002;18:36–37.
19 Smith S. Drugs and pharmaceuticals: management of intoxication and antidotes. In: Luch A, ed. Molecular, clinical and environmental toxicology. Vol 2: Clinical toxicology. Basel: Birkauser; 2010:433–434.
20 Holger JS, Engebretsen KM, Marini JJ. High dose insulin in toxic cardiogenic shock. Clin Toxicol (Phila). 2009;47:303–307.
21 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367.
22 Kline JA, Leonova E, Raymond RM. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med. 1995;23:1251–1263.
23 Kline JA, Leonova E, Williams TC, et al. Myocardial metabolism during graded intraportal verapamil infusion in awake dogs. J Cardiovasc Pharmacol. 1996;27:719–726.
24 Kline JA, Raymond RM, Schroeder JD, Watts JA. The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol. 1997;145:357–362.
25 Kline JA, Tomaszewski CA, Schroeder JD, Raymond RM. Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharmacol Exp Ther. 1993;267:744–750.
26 Cumpston KL, Mycyk M, Pallasch E, et al. Failure of hyperinsulinemia/euglycemia therapy in a severe diltiazem overdose. Clin Toxicol (Phila). 2002;40:618.
27 Greene SK, Gawarammana I, Wood DM, et al. Relative safety of hyperinsulinaemia/euglycaemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med. 2007;33:2019–2024.
28 Cave G, Harvey M. Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: a systematic review. Acad Emerg Med. 2009;16:815–824.
29 Young AC, Velez LI, Kleinschmidt KC. Intravenous fat emulsion therapy for intentional sustained-release verapamil overdose. Resuscitation. 2009;80:591–593.
30 Osterhoudt KC, Henretig FM. How much confidence that calcium channel blockers are safe? Vet Hum Toxicol. 1998;40:239.
31 Belson MG, Gorman SE, Sullivan K, Geller RJ. Calcium channel blocker ingestions in children. Am J Emerg Med. 2000;18:581–586.
32 Wax PM, Erdman AR, Chyka PA, et al. Beta-blocker ingestion: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 2005;43:131–146.
33 Neuvonen PJ, Elonen E, Vuorenmaa T, Laakso M. Prolonged Q-T interval and severe tachyarrhythmias, common features of sotalol intoxication. Eur J Clin Pharmacol. 1981;20:85–89.
34 Boyd R, Ghosh A. Towards evidence based emergency medicine: best BETs from the Manchester Royal Infirmary. Glucagon for the treatment of symptomatic beta blocker overdose. Emerg Med J. 2003;20:266–267.
35 Holger JS, Engebretsen KM, Obetz CL, et al. A comparison of vasopressin and glucagon in beta-blocker induced toxicity. Clin Toxicol (Phila). 2006;44:45–51.
36 Kerns W, 2nd., Schroeder D, Williams C, et al. Insulin improves survival in a canine model of acute beta-blocker toxicity. Ann Emerg Med. 1997;29:748–757.
37 Stellpflug SJ, Harris CR, Engebretsen KM, et al. Intentional overdose with cardiac arrest treated with intravenous fat emulsion and high-dose insulin. Clin Toxicol (Phila). 2010;48:227–229.
38 Leikin JB, Paloucek F. Digoxin. Leikin and Paloucek’s poisoning and toxicology handbook. 3rd ed. Hudson, Ohio: Lexi-Comp; 2002.
39 Hack JB, Levin N. Cardioactive steroids. Goldfrank L, Flomenbaum N, Lewin N, et al. Goldfrank’s toxicologic emergencies, 8th ed, New York: McGraw-Hill, 2006.
40 Bismuth C, Gaultier M, Conso F, Efthymiou ML. Hyperkalemia in acute digitalis poisoning: prognostic significance and therapeutic implications. Clin Toxicol. 1973;6:153–162.
41 Ujhelyi MR, Robert S. Pharmacokinetic aspects of digoxin-specific Fab therapy in the management of digitalis toxicity. Clin Pharmacokinet. 1995;28:483–493.
42 Wenger TL, Butler VP, Jr., Haber E, Smith TW. Treatment of 63 severely digitalis-toxic patients with digoxin-specific antibody fragments. J Am Coll Cardiol. 1985;5(suppl A):118A–123A.
43 French JH, Thomas RG, Siskind AP, et al. Magnesium therapy in massive digoxin intoxication. Ann Emerg Med. 1984;13:562–566.
44 Smith TW, Haber E, Yeatman L, Butler VP, Jr. Reversal of advanced digoxin intoxication with Fab fragments of digoxin-specific antibodies. N Engl J Med. 1976;294:797–800.
45 Antman EM, Wenger TL, Butler VP, Jr., et al. Treatment of 150 cases of life-threatening digitalis intoxication with digoxin-specific Fab antibody fragments: final report of a multicenter study. Circulation. 1990;81:1744–1752.
46 Hickey AR, Wenger TL, Carpenter VP, et al. Digoxin immune Fab therapy in the management of digitalis intoxication: safety and efficacy results of an observational surveillance study. J Am Coll Cardiol. 1991;17:590–598.
47 Mehta RN, Mehta NJ, Gulati A. Late rebound digoxin toxicity after digoxin-specific antibody Fab fragments therapy in anuric patient. J Emerg Med. 2002;22:203–206.
48 Mycyk MB, Bryant SM, Cumpston KL. Late rebound digoxin toxicity after digoxin-specific antibody Fab fragments therapy in anuric patient. J Emerg Med. 2003;24:91.
49 Mauskopf JA, Wenger TL. Cost-effectiveness analysis of the use of digoxin immune Fab (ovine) for treatment of digoxin toxicity. Am J Cardiol. 1991;68:1709–1714.
50 Taboulet P, Baud FJ, Bismuth C, Vicaut E. Acute digitalis intoxication: is pacing still appropriate? Clin Toxicol. 1993;31:261–273.
51 Chen JY, Liu PY, Chen JH, Lin LJ. Safety of transvenous temporary cardiac pacing in patients with accidental digoxin overdose and symptomatic bradycardia. Cardiology. 2004;102:152–155.
52 Bower JO, Mengle H. The additive effect of calcium and digitalis. JAMA. 1936;106:1151–1153.
53 Gold H, Edwards D. The effects of ouabain on heart in the presence of hypercalcemia. Am Heart J. 1927;3:45–50.
54 Smith PK, Winkler A, Hoff HE. L calcium and digitalis synergism: the toxicity of calcium salts injected intravenously into digitalized animals. Arch Intern Med. 1939;64:322–328.
55 Van Deusen SK, Birkhahn RH, Gaeta TJ. Treatment of hyperkalemia in a patient with unrecognized digitalis toxicity. J Toxicol Clin Toxicol. 2003;41:373–376.
56 Hack JB, Woody JH, Lewis DE, et al. The effect of calcium chloride in treating hyperkalemia due to acute digoxin toxicity in a porcine model. J Toxicol Clin Toxicol. 2004;42:337–342.
57 Levine M, Nikkanen H, Pallin DJ. Intravenous calcium administration in digoxin toxic patients. Ann Emerg Med. 2007;50:S27.
58 Erickson CP, Olson KR. Case files of the Medical Toxicology Fellowship of the California Poison Control System-San Francisco: calcium plus digoxin-more taboo than toxic? J Med Toxicol. 2008;4:33–39.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
148 Cardiovascular Drugs
• Cardiovascular drugs are responsible for many fatalities.
• β-Receptor antagonists, calcium channel antagonists, and digoxin primarily cause toxicity by disruption of intracellular calcium homeostasis and lead to hypotension and dysrhythmias.
• With sustained-release forms of calcium channel antagonists and beta-receptor antagonists, toxicity has a delayed peak and longer duration, which can lead to cardiovascular collapse and arrest if treatment is delayed or there is insufficient cardiovascular moritoring.
• Diagnostic testing should include continuous cardiac monitoring; electrocardiogram; measurement of appropriate serum drug concentrations, electrolytes, and glucose; and investigation of coingestants.
• The call to the pharmacist to obtain the hyperinsulinemia-euglycemia infusion in calcium channel antagonist overdose should be made when the norepinephrine infusion is begun.
Epidemiology
The 2009 Annual Report of the American Association of Poison Control Centers National Poison Center Database System reported cardiovascular drugs as the second most common cause of fatalities overall (10%), and they were the second fastest in rate of exposure increase.1 Cardiovascular drugs as a category were ranked as the fifth leading cause of death (44 total deaths: 5 from β-receptor antagonists, 16 from calcium channel antagonists, and 23 from cardiac glycosides). These specific cardiovascular drugs share the clinical effects of hypotension, bradycardia, and conduction disturbances. However, unique differences can help distinguish them in an unknown overdose (Fig. 148.1). Other pharmaceuticals included in the category of cardiovascular agents are angiotensin-converting enzyme inhibitors, antiarrhythmics, clonidine, and other antihypertensives; they are not discussed in this chapter.
Calcium Channel Antagonists
Pathophysiology
Calcium channel antagonists block the intracellular flow of calcium ions through L-type voltage-gated calcium channels in myocardial, smooth muscle, and pancreatic beta-islet cells. These mechanisms of action result in cardiovascular toxicity both directly and indirectly. Depending on the selectivity of the calcium channel antagonist, the direct cardiovascular toxicity is a combination of the effects on the cardiac conduction system, myocardial contractility, and vascular smooth muscle vasodilation. The dihydropyridine class (e.g., amlodipine, nifedipine) preferentially acts on the peripheral vasculature, thereby potentially leading to hypotension and reflex tachycardia. Verapamil operates on the sinoatrial and atrioventricular (AV) nodes and on the myocardium. Diltiazem acts to a lesser extent than verapamil on the cardiac tissue and nodes, and it also dilates peripheral vasculature (Table 148.1). The degree of contribution from each mechanism of cardiovascular toxicity can be difficult to predict. Despite the differences in therapeutic mechanisms, the distinctions among families of calcium channel antagonists are often blurred during an overdose, and the patient generally suffers from negative chronotropic, inotropic, and dromotropic effects.2
| CLASS | ACTION(S) | EXAMPLE(S) |
|---|---|---|
| Phenylalkylamines | Act on sinoatrial and atrioventricular nodes and the myocardium | Verapamil (Calan) |
| Benzothiazepines | Dilate peripheral vasculature and act to a lesser degree than verapamil on cardiac tissues and nodes | Diltiazem (Cardizem, Tiazac) |
| Dihydropyridines | Act on peripheral vasculature, leading to hypotension and reflex tachycardia |
Presenting Signs and Symptoms
Verapamil and diltiazem overdoses cause bradycardia and numerous conduction abnormalities in and below the AV node. The decreased blood pressure results from vasodilation and decreased cardiac contractility from the negative inotropic and chronotropic effects.2
Differential Diagnosis and Medical Decision Making
The differential diagnosis (see also Fig. 148.1) for overdose of calcium channel antagonists includes other cardiovascular drugs such as beta-blockers, clonidine, digitalis, and other antidysrhythmics. The emergency physician should also consider myocardial infarction and other causes of cardiogenic shock. The potency of the effect of calcium channel antagonists on the cardiovascular system is astounding. Significant cardiovascular toxicity can occur after supratherapeutic ingestion of calcium channel antagonists. Ingestion of double the therapeutic dose should instigate medical evaluation and treatment. Immediate-release calcium channel antagonists should have some clinical effect within 6 hours. Sustained-release calcium channel antagonists should result in clinical manifestations within 1 to 14 hours.3
Diagnostic testing is contingent on the necessity of treatment for hemodynamic instability. Once the patient’s airway, breathing, and cardiovascular status have been assessed and stabilized, testing should start with a 12-lead electrocardiogram (ECG) and chest radiography. Rapid determination of hyperglycemia and metabolic acidosis with capillary glucose and arterial blood gas analysis may demonstrate a severe calcium channel antagonist overdose. An elevated serum lactate concentration may be another marker of severe calcium channel antagonist overdose.4 Testing for serum concentrations of calcium channel antagonists is not clinically useful or available to guide treatment. Otherwise, standard laboratory testing for a general overdose is a good comprehensive approach.
Treatment
Whole-bowel irrigation has been suggested for overdose of calcium channel antagonists because many of these drugs are sustained-release preparations. Whole-bowel irrigation is not indicated for a patient with hemodynamic instability because a significant amount of the drug has already been absorbed,5 and, therefore, the opportunity for prevention has passed. In addition, challenging a hypoperfused gastrointestinal system can have disastrous consequences, such as functional and physical obstruction by a calcium channel antagonist bezoar,6–9 as well as perforation. Generally speaking, no evidence indicates that any gastric decontamination procedure improves outcome in the patient with an overdose, and the risks must be assessed against the benefits.
An antidotal treatment regimen is provided in Figure 148.2. This regimen emphasizes elemental calcium, either as calcium gluconate (30 mL of a 10% solution, or 3 g of calcium gluconate; 14 mEq elemental calcium) or calcium chloride (10 mL of a 10% solution, or 1 g; 13.5 mEq of elemental calcium). Calcium chloride should be administered through central venous access because it is an acidifying salt, which could cause necrosis of the peripheral vasculature. If the intravenous calcium boluses appear to have improved hemodynamic status, close monitoring for recrudescence of toxicity must be maintained, and further boluses must be given as necessary. An intravenous infusion of calcium is warranted only when it effectively treats the hypotension, and further boluses are required to support the blood pressure (Table 148.2). The serum calcium concentration should be monitored, but antidotal treatment rarely gives rise to clinically significant hypercalcemia.
Fig. 148.2 Treatment of overdose with calcium channel antagonist or β-receptor antagonist.
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly).
Table 148.2 Antidotes, Treatments, Facts, and Formulas for Cardiovascular Drugs
| ANTIDOTE OR TREATMENT | DOSING | ADVERSE EFFECTS and signs of improving perfusion |
|---|---|---|
| Atropine |
Consultation with a clinical toxicologist is recommended
Give a bolus of 1.0 units/kg IV with 1 ampule of D50W
Immediately start an infusion of 1.0 units/kg/hr with a glucose infusion of D10W at 100 mL/hr
Titrate 0.5 units/kg every 30 min until desired effect; 1 ampule of D50W can be given during every increase in infusion; titrate to desired effect when blood pressure has reached desired value and signs of improving perfusion
When the blood pressure has reached the desired value with the insulin infusion, taper and wean pressor agent therapy
Monitor serum glucose concentration every 15 min for the first 60 min; when stable, monitor every 60 min thereafter
Monitor serum potassium and other electrolyte concentrations every 60 min
For acute overdose (unknown amount in adult or child): 5-10 vials IV as a bolus; repeat as needed every 30 min
For chronic overdose in adult or child: 1-2 vials IV as a bolus; repeat as needed every 30 min
If amount ingested is known: 1 vial binds 0.5 mg of digoxin
Dosage calculated from serum digoxin concentration:
D10W, 10% dextrose in water; D50W, 50% dextrose in water; IV, intravenous(ly); MAP, mean arterial pressure.
Hyperinsulinemia euglycemia (HIE) therapy and catecholamines with inotropic and vasopressor activity are the next line of treatment for refractory hypotension in calcium channel antagonist overdose, but inotropics and vasopressors will be discussed first. A multicenter study compared dopamine and norepinephrine agents in all patients categorized as being in shock, regardless of cause. No difference was seen in the outcome (death at 28 days) for all patients in the study, but a predetermined subgroup analysis found greater mortality in patients with cardiogenic shock who were treated with dopamine.10 Because calcium channel antagonists can cause cardiogenic shock, norepinephrine is probably a good first choice. If clinically significant hypotension persists, adding more agents may be necessary. Cardiovascular data from diagnostic modalities such as transthoracic echocardiogram, pulmonary artery catheter, arterial catheter, and central venous catheter should dictate which cardiovascular agent is the most appropriate choice. Vasopressin has been used in human cases when peripheral vasoconstriction is indicated.11 Worsening of the cardiac index was demonstrated when vasopressin was used in an animal model to treat hypotension induced by calcium channel antagonist.12
In 1999, Yuan et al.13 described the first published use of high-dose insulin-euglycemia (HIE) therapy, in four patients with verapamil overdose and in one patient with amlodipine and atenolol overdose. HIE promotes inotropy by improving myocardial energy production. In addition, insulin has antiinflammatory attributes that protect against apoptosis and ischemic reperfusion injury.2
Subsequently, numerous case reports, reviews, and HIE regimens were published.2,14–19 HIE therapy has successfully reversed cardiogenic shock from a polydrug overdose,20 and historically it was used in multiple nontoxicologic conditions, such as acute myocardial infarction, post–cardiac surgery status, and septic shock.21 The superiority of HIE therapy for cardiogenic shock resulting from calcium channel antagonist toxicity was also demonstrated dramatically in animal models.22–25
Failure of HIE therapy often occurs when it is started as rescue therapy and when the dose is inadequate.26 A small prospective observational study of patients treated with HIE therapy supports it safety.27 HIE therapy should be started when boluses of calcium, glucagon, atropine, and intravenous fluids have failed and the physician is considering a pressor agent to improve refractory hypotension. The call to the pharmacist to obtain the HIE infusion should be made when the norepinephrine infusion is begun.
[/not-level-membership-for-emergency-medicine-category]
