[level-membership-for-critical-care-medicine-category]
Objectives
• Describe the etiology and pathophysiology of atherosclerotic coronary artery disease.
• Identify the pathophysiology and clinical manifestations of acute heart failure.
• Discuss the nursing priorities for managing a patient with an acute cardiovascular disorder.

Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at http://evolve.elsevier.com/Urden/priorities/.
Cardiovascular disease remains the leading cause of mortality in the United States. The estimated direct and indirect national cost of cardiovascular disease in 2010 was $503.2 billion.1 Cardiovascular diseases are the leading cause of death for women and men. There are almost 80 million adults in the United States living with cardiovascular disease. Cardiovascular disease accounts for 1 of every 2.9 deaths in the United States.1 In terms of disability, the number of disability-adjusted life-years (DALYs) attributable to cardiovascular disease on a global basis is projected to double by the year 2020. An understanding of the pathology of cardiovascular disease processes and clinical management allows the critical care nurse to accurately anticipate and plan interventions. This chapter focuses on cardiac disorders commonly seen in the critical care environment.
Coronary Artery Disease
Description and Etiology
The biggest contributor to cardiovascular-related morbidity and mortality is coronary artery disease (CAD). Atherosclerosis is a progressive disease that affects arteries throughout the body. In the heart, atherosclerotic changes are clinically known as CAD. This disease process is also known by the term coronary heart disease (CHD) because other heart structures ultimately become involved in the disease process. The atherosclerotic vascular changes that lead to CAD may begin in childhood.2 Research and epidemiological data collected during the past 50 years have demonstrated a strong association between specific risk factors and the development of CAD.1 These risk factors are further delineated as nonmodifiable and modifiable coronary risk factors (Box 12-1).
Risk Factors for Coronary Artery Disease
Age, Gender, and Race
The severe effects of CAD occur as a person ages. In general, CAD symptoms are seen in middle and old age.1 Traditionally, CAD has been regarded as a male disease, but it is increasingly obvious that in modern society it affects both genders.1 The average age for a person having a first heart attack is 64.5 years for men and 70.3 years for women.1 Starting at age 75 years, the prevalence of cardiovascular disease is higher among women than men.2,3 CAD rates for postmenopausal women are two to three times higher than those for premenopausal women of the same age.1 People of color and multiracial populations of both genders have higher CAD mortality rates than do white populations of similar socioeconomic status.1
Family History
A positive family history is one in which a close blood relative has had a myocardial infarction (MI) or stroke before the age of 60. This family history suggests a genetic or lifestyle predisposition to the development of CAD. Individuals with a family history had a 50% greater risk of having an acute MI in the INTERHEART study.4 This was a large, international, standardized case-control study of similar cohorts in 52 countries that was designed to examine the importance of risk factors for CAD on a worldwide basis.4
Hyperlipidemia
Hyperlipidemia is a leading factor responsible for severe atherosclerosis and the development of CAD. Determining total serum cholesterol and triglyceride levels is a helpful start in the evaluation process.3,5 A lipid panel blood test can measure the following values:
• High-density lipoprotein (HDL) cholesterol
• Low-density lipoprotein (LDL) cholesterol
Treatment of hyperlipidemia has advanced beyond the concept of lowering total cholesterol to treatment of specific lipoprotein abnormalities.3,6–8 The target levels for specific serum lipids are listed in Table 12-1.
TABLE 12-1
LIPID GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE
| LIPID | TARGET VALUE* (mg/dl) |
| Total cholesterol | <200 |
| HDL cholesterol | |
| Men | >40 |
| Women | >50 |
| LDL cholesterol | |
| Very high risk | <70 |
| High risk | <100 |
| Low risk | <130 |
| VLDL cholesterol | <30 |
| Triglycerides | <150 |
HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.
*Values outside the target range increase the risk for coronary artery disease.
Total Cholesterol
The total cholesterol is the sum of the HDL, LDL, and VLDL cholesterol in the bloodstream. It is used as a starting point for lipid testing. A total cholesterol level higher than 200 mg/dL is an indication to investigate the lipid profile and other risk factors for CAD. More than 102 million American adults (46.8% of the adult population) have a total blood cholesterol level at or above 200 mg/dL.1
High-Density Lipoprotein Cholesterol
HDL cholesterol is frequently described as the “good cholesterol” because higher serum levels exert a protective effect against acute atherosclerotic events. All the reasons are not completely understood, but one recognized physiological effect is the ability of HDL to promote the efflux of cholesterol from cells. This process may minimize the accumulation of foam cells in the artery wall and decrease the risk of developing atherosclerosis.9 High HDL cholesterol levels confer antiinflammatory and antioxidant benefits on the arterial wall.9 In contrast, a low HDL cholesterol level is an independent risk factor for the development of CAD and other atherosclerotic conditions. HDL cholesterol is generally higher in women, and levels can be raised by physical exercise and by smoking cessation. In patients with low HDL cholesterol levels, when lifestyle changes are ineffective, the HDL level can be raised by drugs such as extended-release nicotinic acid (niacin) and fibrates. In 2006 16.2% of the adult U.S. population had HDL cholesterol levels below 40 mg/dL.1
Low-Density Lipoprotein Cholesterol
LDL cholesterol is usually described as the “bad cholesterol” because high levels are associated with an increased risk of acute coronary syndrome, stroke, and peripheral arterial disease (PAD). High LDL levels initiate the atherosclerotic process by infiltrating the vessel wall and binding to the matrix of cells beneath the endothelium.9 LDL cholesterol also exerts an inflammatory effect on the arterial vessel wall.9 A high LDL cholesterol level is initially managed by nonpharmacological lifestyle changes such as weight loss, smoking cessation, low-fat diet, physical exercise, and attainment of a normal body size as measured by the body mass index (BMI). If lifestyle changes are insufficient to reduce the LDL cholesterol level in the bloodstream, the drug category of choice is a statin. Numerous research studies have conclusively demonstrated that lowering the LDL cholesterol with statins for primary or secondary prevention is highly effective in lowering mortality due to CAD.1,3 Therapy should be tailored to treat the individual cardiovascular risk profile. The target LDL cholesterol is determined according to the individual’s risk profile as described in Table 12-1. Patients at highest risk are advised to maintain LDL cholesterol below 70 mg/dL; those at high risk, a level below 100 mg/dL; and those at low risk, LDL below 130 mg/dL. Controversy exists about whether the tiered LDL goals in the current guidelines are sufficiently low to prevent coronary atherosclerosis developing in persons without CAD. Some cardiologists advocate a reduction of the LDL goal to a 50 to 70 mg/dL range for everyone, not just those with a known cardiovascular disease.10 In 2006 32.6% of the adult U.S. population had LDL cholesterol levels of 130 mg/dL or higher.1
Very-Low-Density Lipoprotein Cholesterol
VLDL cholesterol is not usually measured, although a normal value is about 30 mg/dL.6 The value can be estimated by subtracting the sum of the HDL and LDL from the total cholesterol. When triglyceride levels are elevated, the VLDL cholesterol level also is high.
Triglycerides
Triglycerides are serum lipids that constitute an additional atherogenic risk factor. Triglycerides are carried by VLDL cholesterol in the bloodstream.7 An optimal triglyceride level is below 150 mg/dL, and the more elevated the triglyceride serum level, the higher the risk of developing CAD. A value between 150 and 199 mg/dL is borderline high; a value between 200 and 500 mg/dL is high; and a value above 500 mg/dL signals a high risk of atherogenic complications and a strong risk for the presence or development of type 2 diabetes.
Lipoprotein(a)
LDL cholesterol can be further analyzed by the category of lipid particles that make up the total LDL value. Researchers have investigated the function of several lipid particles to determine their role in the development of premature atherosclerotic CAD. One particle that has been extensively studied is lipoprotein(a), which is abbreviated Lp(a) and described verbally as “LP little a.” Lp(a) is manufactured in the liver and circulates in the bloodstream bound to a large glycoprotein called apolipoprotein(a), abbreviated as apo(a).11 The Lp(a)-apo(a) lipid particle concentration is elevated in the presence of inflammation, and it stimulates atheroma and clot formation in inflamed arteries.11 This effect is thought to occur because the apo(a) is structurally similar to plasminogen, a protein essential for clot formation.
Lp(a) levels are 90% genetically determined. Elevated Lp(a) plasma levels constitute the most frequently encountered genetic lipid disorder in families with premature CAD.11 Testing for Lp(a) is reserved for high-risk patient populations such as those with a strong family history of premature atherosclerotic disease and for patients with premature CAD who do not exhibit the expected cardiac risk factors. Reduction of Lp(a) levels to below 30 mg/dL is the therapeutic goal. This is usually achieved by ingesting high doses (1 to 2 grams/day) of extended-release nicotinic acid (niacin). The Lp(a) level is not reduced by statins, drugs that traditionally lower LDL levels, or by physical exercise, a low-fat diet, weight loss, or tight control of blood glucose levels.11 Lifestyle changes are recommended, and research is ongoing, but a cure is not yet discernible.
High-Fat Diet
A diet rich in saturated fats leads to elevated cholesterol levels in the blood. The first line of treatment to lower elevated serum cholesterol is a low-fat, high-fiber diet and increased physical exercise.7,8 If these measures are not effective, lipid-lowering drugs are indicated.1 This approach sounds simple, but less than one half of the people who qualify for lipid reduction therapy are taking their medications; only one third of treated patients reach their LDL target; and fewer than 20% of patients with CAD are at their LDL goal.1 Although lipid-lowering drugs are very helpful for some, they are not a panacea for everyone.
Obesity
Obesity is a disease of modern times. Global estimates are more than 1 billion overweight adults, and at least 300 million of these people are obese. Obesity is often associated with a sedentary lifestyle. A high risk of coronary heart disease is among the well-established adverse health effects associated with excess weight. Hypertension, hypercholesterolemia, and diabetes are among the clinical conditions that are important mediators of this association.12 In the United States in 2001, 122 million adults were overweight or obese.13 In 2006 144 million adults, or 66% of the U.S. adult population, were overweight or obese.1 Obesity is defined using the body mass index (BMI).
The BMI is calculated as the weight in kilograms divided by the square of the height in meters (kg/m2); it assesses body weight relative to height. BMI is used to evaluate the threat of excess pounds as a risk factor for CAD and permits comparisons of people of different gender, age, height, and body type.1 BMI is calculated as the weight in kilograms divided by the square of the height in meters (kg/m2). The BMI calculation in metric units is shown in Box 12-2. A normal BMI is between 18.5 and 25 kg/m2. A BMI between 25 and 30 kg/m2 indicates the person is overweight. A BMI greater than 30 kg/m2 is the definition of obesity.1
The distribution pattern of fat on the body is a CAD risk factor. The more weight carried in the abdominal area, producing a large waist, the greater the risk of CAD. Excess abdominal adiposity (apple body shape) indicates additional fat around the abdominal organs compared with individuals who have a smaller waist and larger hips (pear body shape). A waist size greater than 40 inches in men and 35 inches in women increases their risk for CAD. Physical exercise assists with weight reduction, lowers the risk for CAD, and decreases the risk of developing type 2 diabetes.
Physical Inactivity
Regular vigorous physical activity using large muscle groups promotes physiological adaptation to aerobic exercise that can prevent the development of CAD and reduce symptoms in patients with established cardiovascular disease.14 Exercise also reduces the incidence of many other diseases, including type 2 diabetes, osteoporosis, obesity, depression, and cancers of the colon and breast.14 Many research trials have demonstrated the positive effects of physical activity on the other major cardiac risk factors.14 Exercise alters the lipid profile by decreasing LDL cholesterol and triglyceride levels and increasing HDL cholesterol levels.14 Exercise reduces insulin resistance at the cellular level, lowering the risk for developing type 2 diabetes, especially if combined with a weight-loss program.14 Epidemiological studies indicate that physical athletics as a young person do not confer protection in later years. A sedentary lifestyle has negative effects, regardless of age, gender, BMI, smoking status, presence or absence of hypertension, or abnormal lipoprotein profile. Lifelong physical activity is necessary to prevent atherosclerotic CAD and stroke.14 In 2005 the prevalence of adults not engaging in any physical activity during leisure or work time was 10.3% in the United States.1 In affluent countries, only one third of the population exercises moderately for the recommended 30 minutes, five times per week.
Hypertension
Normal blood pressure is described as a systolic blood pressure (SBP) below 120 mm Hg and a diastolic blood pressure (DBP) below 80 mm Hg. Hypertension is defined as an SBP greater than 140 mm Hg or DBP higher than 90 mm Hg. Controlled hypertension describes a situation in which administration of antihypertensive medications maintains the patient’s blood pressure within the normal range.
Hypertension is a cardiac risk factor because the high SBP damages the arterial endothelium, leading to vascular inflammation that encourages formation of plaque. Hypertension is a complex, multifactorial disease process. Hypertension is divided into stages for the purposes of treatment, as shown in Table 12-2.
TABLE 12-2
BLOOD PRESSURE GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE
| CATEGORY | SYSTOLIC BP* (mm Hg) | DIASTOLIC BP* (mm Hg) |
| Normal (optimal)* | <120 | <80 |
| Prehypertension | 120-139 | 80-89 |
| Stage 1 hypertension | 140-159 | 90-99 |
| Stage 2 hypertension | ≥160 | ≥100 |
BP, blood pressure; CAD, coronary artery disease.
*Values greater than normal increase the risk for CAD and heart failure.
Prehypertension is an SBP of 120 to 139 mm Hg or DBP above 85 mm Hg.1,13 In the United States, one in five adults is prehypertensive. Hypertension is diagnosed when the blood pressure is above 140/90 mm Hg. Hypertension affects another one in four adults in the United States. After the blood pressure is above 140/80 mm Hg, hypertension is described as stage 1 or stage 2 depending on the severity (see Table 12-2).
Hypertension is often described as the “silent killer,” because 30% of those affected are unaware they have seriously elevated blood pressure.1 A higher percentage of men than women have hypertension until age 45, but from the ages of 45 to 54 years, the percentage of men and women with hypertension is similar.1 It is essential that patients understand that sustained elevation of blood pressure leads inexorably toward atherosclerosis, heart failure, kidney failure, stroke, and heart attack.13 So widespread is hypertension in industrialized societies that even a normotensive person at age 55 has a 90% lifetime risk of developing hypertension. This implies that even normotensive persons should adopt interventions to maintain a normal blood pressure.13 The estimated direct and indirect cost of hypertension in the United States for 2010 is $76.6 billion.1
According to recent guidelines, the goal of treatment for the hypertensive person without other risk factors is to achieve a blood pressure below 140/80 mm Hg. For the hypertensive person who already has diabetes or kidney disease, the target blood pressure is below 130/80 mm Hg. A normal blood pressure is below 120/80 mm Hg.13
Lifestyle interventions that can normalize blood pressure include physical exercise, a low-salt diet, limiting alcohol intake, and achieving normal body weight. Most patients are started on a diuretic, and if this is insufficient, they may be placed on an angiotensin-converting enzyme inhibitor (ACEI), angiotensin receptor blocker (ARB), beta-blocker, or calcium channel blocker. Most patients require at least two medications, each from different drug classifications, to normalize their blood pressure.13 Hypertensive emergencies with acute organ damage are discussed in the last section of this chapter.
Cigarette Smoking
The greater the number of cigarettes smoked per day, the greater the risk of developing CAD, acute MI, and stroke.14,16 In the United States, the prevalence of smoking has always been lower among women than men.1 Cigarette smoking unfavorably alters serum lipid levels, decreases the HDL cholesterol level, and increases LDL cholesterol and triglyceride levels. In the United States, smoking remains common, with an overall prevalence of 20.6% of the adult population. Smoking increases the risk of coronary heart disease at all levels.1 Smokers are two to four times more likely to develop CAD than nonsmokers.1 Passive, secondhand smoke exposure also increases cardiovascular risk; 34.7% of nonsmoking adults are exposed to environmental tobacco smoke at home or at work.15–17 Nicotine is addictive, and smoking cessation is difficult. People need tremendous support to be able to “kick the habit.” Chapter 25 provides patient education guidelines on how to stop smoking.
Diabetes Mellitus
Individuals with diabetes mellitus (types 1 and 2) have a higher incidence of coronary heart disease compared with the general population. Data from the Framingham Heart Study indicates a doubling in the incidence of diabetes over the past 30 years and most dramatically during the 1990s.1 Elevated blood glucose level is a known risk factor for development of vascular inflammation associated with atherosclerosis. The normoglycemia range is 70 to 100 mg/dL. It is recommended that patients in critical care have blood glucose levels maintained close to the normal range18 while avoiding hypoglycemic episodes.
A fasting blood glucose concentration between 100 and 125 mg/dL represents a prediabetic state and is a risk factor for the development of diabetes and CAD (Table 12-3). A fasting blood glucose above 126 mg/dL is indicative of diabetes. Patients with diabetes have an increased risk of developing CAD and have worse clinical outcomes after acute coronary syndrome events.19 In a multinational study of patients who were seen at hospitals with symptoms of acute coronary syndrome, almost one in four had a known history of diabetes.20
TABLE 12-3
FASTING BLOOD GLUCOSE AND RISK FOR CORONARY ARTERY DISEASE
| BLOOD GLUCOSE LEVEL | FASTING PLASMA GLUCOSE LEVEL* (mg/dL) |
| Normal | 70-100 |
| Prediabetic | 100-125 |
| Diabetic | 126 or higher |
*Values greater than normal increase the risk for CAD and kidney failure.
Chronic Kidney Disease
Chronic kidney disease is considered a risk equivalent for CAD.2,3,21 This means patients with chronic kidney disease have as much risk of experiencing a coronary event as if they already had CAD.2,3,22 The risk of death for the patient with acute MI rises as the serum creatinine level increases.22 In one study, the in-hospital mortality rates for patients with an acute MI were 2% for patients with normal kidney function, 6% for those with mild kidney failure, 14% for those with moderate kidney failure, 21% for those with severe kidney failure, and 30% for patients with end-stage kidney disease.22 Mortality following cardiac surgery is also higher for individuals with chronic kidney disease.23
Metabolic Syndrome
Metabolic syndrome refers to the clustering of risk factors associated with cardiovascular disease and type 2 diabetes.1 The prevalence of metabolic syndrome in 2006 for adults in the United States was 34%.1 Metabolic syndrome is diagnosed when three or more of the following risk factors are present:24,25
2 Serum triglyceride is level greater than 150 mg/dL (≥1.7 mmol/L).
4 Blood pressure is 130/85 mm Hg or higher, which is diagnostic for prehypertension or hypertension.
It is perhaps obvious that individuals with the signs of metabolic syndrome are at increased risk for CAD; even so, it is surprising to what degree this holds true. In the Framingham epidemiological study, presence of the factors associated with metabolic syndrome predicted 25% of all new-onset CAD and almost 50% of new-onset diabetes.23
Women and Heart Disease: Premenopause and Postmenopause
Serious CAD symptoms occur approximately 5 years later in women than in men.25 The average age for the first acute MI in men is 65.8 years and in women is 70.4 years.1 Incidence of CAD is two to three times higher among postmenopausal women than women who are premenopausal.1 In the past, it seemed logical to prescribe hormone replacement therapy (HRT) to treat the symptoms of menopause. However, well-designed research trials discovered an increase in cardiovascular events in the first year of HRT (estrogen plus progestin), although cardiac events declined after the first year.25,26 Subsequent studies have confirmed this result.24 In 2004 the estrogen-only HRT trial was stopped by the National Institutes of Health (NIH) because of an increased risk of stroke among the women taking estrogen. For these reasons, HRT is no longer recommended for prevention of atherosclerotic cardiovascular disease.27 Data from the Framingham Heart Study indicate the lifetime risk for cardiovascular disease is more than one in two for women at age 40.1
Cardiovascular disease kills more than one-half million women annually in the United States. To emphasize the magnitude of the problem, this represents more deaths than the next seven fatal diseases for women combined.28 Mortality rates for women after an acute MI are higher than for men: 38% and 25%, respectively. Risk factors more strongly associated with acute MI in women compared to men include hypertension, diabetes mellitus, alcohol intake, and physical inactivity.26 Many reasons contribute to women having higher mortality from acute MI, including waiting longer to seek medical care, having smaller coronary arteries, being older when symptoms occur, and experiencing very different symptoms from those of men of the same age.29 The 2007 evidence-based guidelines for cardiovascular disease prevention in women recommended a plan for a general approach to the female patient that classifies her as high risk, at risk, or at optimal risk.28 In 2007 50% of women surveyed by the American Heart Association (AHA) recognized that heart disease was the leading cause of death among women.
Vascular Inflammation
The link between vascular inflammation and atherosclerotic disease is well established.31 Measurement of this link has been more controversial, however.30 Many researchers think the development of atherosclerotic plaque occurs in response to inflammation. Noxious inflammatory agents circulate in the bloodstream and stimulate chemical mediators that directly modify the arterial wall. The initial inflammatory stimuli include increased blood glucose, elevated blood lipids, nicotine, and hypertension. However, other clinical conditions, such as connective tissue disorders and systemic infection, also produce an inflammatory state, and it is not clear what the impact of inflammation produced by these stimuli is on the vessels.30 Research to identify prognostic inflammatory markers is ongoing.
C-Reactive Protein
The inflammatory marker most frequently cited is C-reactive protein (CRP). It is measured as high-sensitivity C-reactive protein (hs-CRP).30 The higher the hs-CRP value, the greater the risk of a coronary event, especially if all other potential causes of systemic inflammation such as infection can be ruled out. If other systemic inflammatory conditions such as bronchitis or a urinary tract infection are present, the hs-CRP test loses all predictive value.31 CRP and other inflammatory markers are used to estimate the probability of future acute coronary events.30,31 During acute coronary syndrome events, there is widespread activation of neutrophils in the cardiac circulation (measured from the coronary sinus), which suggests that inflammation is not limited to one unstable plaque.32 Debate continues about whether CRP is simply a marker of vascular inflammation or also contributes to the proinflammatory state.9
Multifactorial Risk and Risk Equivalents
CAD has multifactorial causation; the greater the number of risk factors, the greater the risk of developing CAD.1,3,24,33 The best time for an individual to make lifestyle changes is before symptoms of CAD occur. Patients with two or more risk factors or with one or more of the CAD risk-equivalent diseases have the greatest potential to benefit from risk-factor reduction and lifestyle change.3
Certain medical conditions are considered risk equivalents of CAD. A risk equivalent means the person has the same risk of having an acute MI as if they had coronary heart disease already. Two noncardiac medical conditions are considered risk equivalents for CAD: diabetes mellitus and chronic kidney disease. Peripheral arterial disease and cerebral vascular disease are atherosclerotic conditions that are also considered CAD risk equivalents.
Primary Versus Secondary Prevention of Coronary Artery Disease
If a person has symptoms of CAD or has previously had an acute coronary syndrome event, the goal of any lifestyle change or medication is called secondary prevention, or preventing another heart attack. If an individual matches the risk profile described previously but does not have symptoms of CAD or has not had an acute MI, the treatment plan is described as primary prevention. The constellation of cardiac risk factors is well established and can predict development of CAD for most populations in the developed industrial world.
Pathophysiology of Coronary Artery Disease
Coronary heart disease is a progressive atherosclerotic disorder of the coronary arteries that results in narrowing or complete occlusion. Atherosclerosis affects the medium-size arteries that perfuse the heart and other major organs. Normal arterial walls are composed of three layers: the intima (inner lining), the media (middle muscular layer), and the adventitia (outer coat).
Development of Atherosclerosis
Atherosclerosis is a chronic inflammatory disorder that is characterized by an accumulation of macrophages and T lymphocytes in the arterial intimal wall. A high LDL cholesterol concentration is one of the triggers of vascular inflammation. The inflammation injures the wall, allowing the LDL cholesterol to move into the vessel wall below the endothelial surface.9 Blood monocytes adhere to endothelial cells and migrate into the vessel wall. Within the artery wall, some monocytes differentiate into macrophages that unite with and then internalize LDL cholesterol. The foam cells that result are the marker cells of atherosclerosis.9
Elevated LDL cholesterol levels promote low-level endothelial inflammation that allows lipoproteins to infiltrate the intimal vessel wall. After it has infiltrated under the endothelium, LDL cholesterol tends to stay within the vessel wall rather than return to the circulation.9 This contrasts with the actions of HDL cholesterol, which enters the vessel wall, helps efflux cholesterol from cells, and then returns to the circulation.9 The actions of HDL cholesterol may help minimize the number of foam cells in the artery wall.9
Atherosclerotic Plaque Rupture
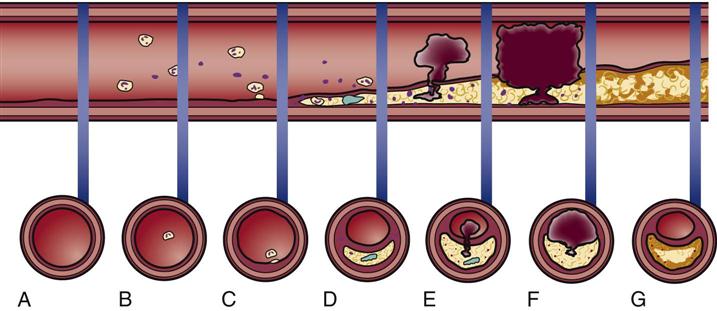
When a mature atherosclerotic plaque develops, it is not uniform in composition. It has a lipid liquid center filled with procoagulant factors. A connective tissue fibrous cap covers the top of the fluid lipid center.30–32 The abrupt rupture of this cap allows procoagulant lipids to flood into the vessel lumen and rapidly form a coronary thrombosis, as shown in Table 12-4. As the enlarging clot blocks blood flow through the coronary artery, a “heart attack” will occur unless there is adequate collateral circulation from other coronary vessels. Symptoms and suggested cardiac interventions at appropriate stages in development of CAD are listed in Table 12-4.
TABLE 12-4
TIMELINE OF ATHEROGENESIS DEVELOPMENT DEPICTED BY LONGITUDINAL SECTION OF AN ARTERY
| ATHEROGENESIS/THROMBOGENESIS | ASSOCIATED SYMPTOMS | CARDIAC INTERVENTION |
| A. Normal artery, normal vessel wall. | No symptoms | Primary prevention of CAD recommended: consume a low-fat diet, take regular physical exercise, avoid smoking, and achieve normal BMI |
| B. Lipids in bloodstream. | No symptoms | |
| C. Extracellular lipid accumulates in the intima of the artery (atheroma). | No symptoms | |
| D. Lipid accumulation evolves to become a fatty-fibrous (atherosclerotic) lesion. Some lesions contain a lipid interior covered by a fibrous cap. | Chest pain with exercise that is relieved by rest or NTG (stable angina) or possibly no symptoms until the lesion fills more than 75% of the vessel lumen | PCI if stable angina is present and CAD is diagnosed by cardiac catheterization |
| E. Rupture of the cap allows lipid in the center to be released into the bloodstream, stimulating clot formation (thrombogenesis). | Chest pain not relieved by rest or NTG (ACS – unstable angina) | Call 911 for immediate transport to a hospital, preferably one with experience treating ACS |
| F. Fresh clot blocks the vessel; spasm of the artery may occur near the thrombus. | Chest pain unrelieved by rest or NTG – severity, location of angina, and associated symptoms vary greatly among individuals (ACS – acute MI) | Emergency intervention to open the artery: fibrinolytic or catheter-based procedure (PCI) |
| G. Vessel is open, but the atherosclerotic lesion remains. | No symptoms | Secondary prevention of CAD to prevent repeat MI; beta-blockers to prevent arrhythmias; ACE-1 drugs to prevent ventricular remodeling and heart failure; elective PCI |
 |
||
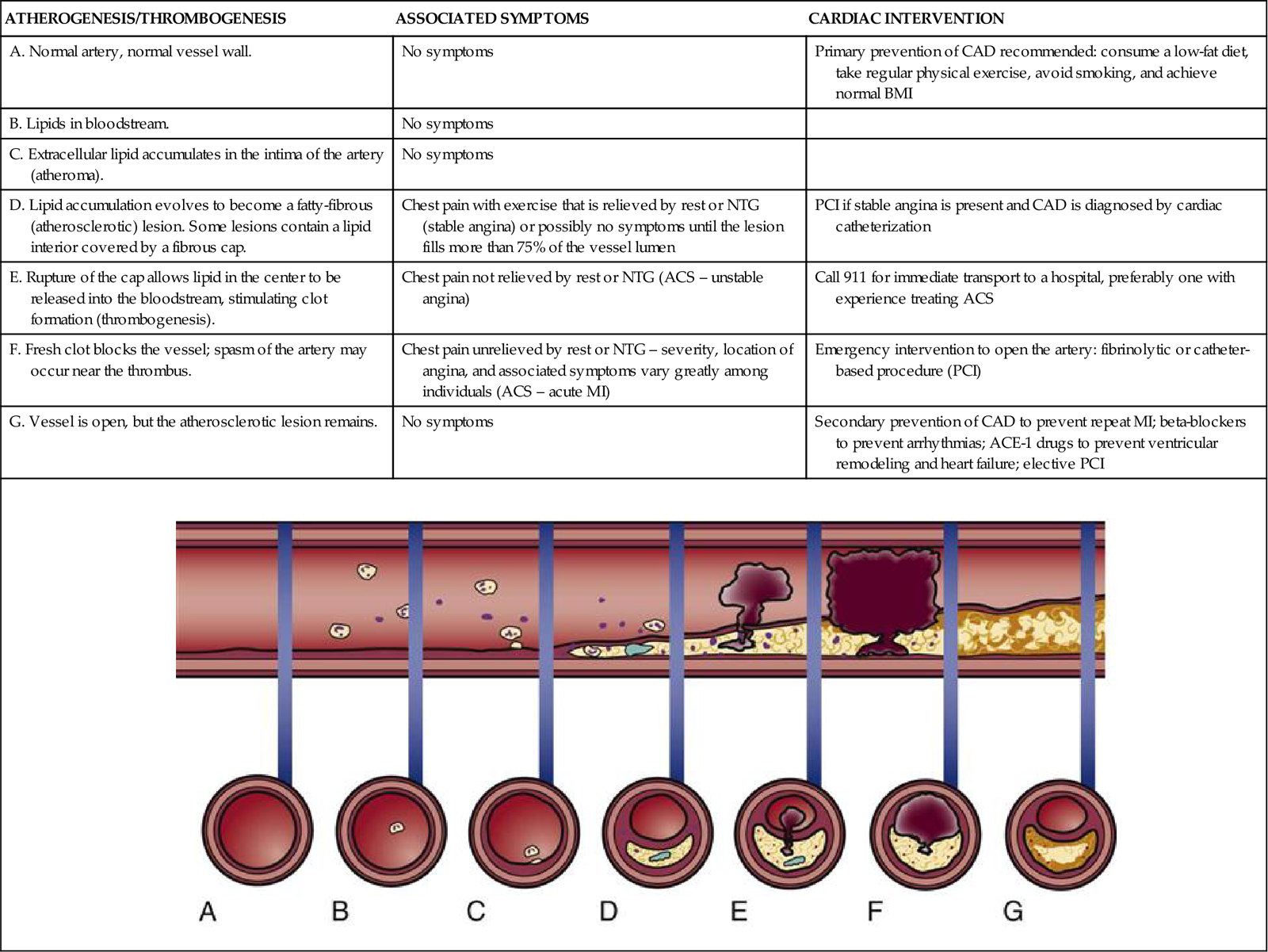
(Modified from Antman EM, et al: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary, Circulation 110(5):588, 2004.)
Plaques that are likely to rupture are saturated with macrophages and other inflammatory cells. These vulnerable plaques are usually not obstructive and are situated at bends or branch points in the arterial tree.3 It is not known what factors cause the fibrous cap to rupture or erode. As deep fissures in the cap expose the procoagulant factors to the blood plasma, an unstoppable cycle is put into motion. When platelets in the bloodstream are exposed to collagen, necrotic debris, von Willebrand factor, and thromboxane, a clot is formed that can occlude the coronary artery. Highly fibrotic plaques do not rupture. The type of atherosclerotic plaque that is prone to rupture has a weak fibrous cap and a large amount of liquid cholesterol within the core (see Table 12-4).32
Plaque Regression
A reduction in blood cholesterol decreases atherosclerotic plaque size by decreasing the amount of liquid cholesterol within the plaque core.7 Lowering cholesterol levels does not change the dimensions of the fibrous or calcified portions of the plaque. However, lower cholesterol levels reduce vascular inflammation and make vulnerable plaque less likely to rupture.
If diet is not effective in lowering blood cholesterol, lipid-lowering drugs are prescribed to lower the LDL cholesterol level below 100 mg/dL for patients at risk for CAD and to aim for an LDL level below 70 mg/dL for individuals with the highest-risk profile.7 Drugs, diet, and exercise are used to lower the triglyceride levels to less than 150 mg/dL, and to raise HDL cholesterol levels above 40 mg/dL for men and above 50 mg/dL for women (see Table 12-1).3,7,8,33
Acute Coronary Syndromes
The term acute coronary syndrome (ACS) is used to describe the array of clinical presentations of CAD that range from unstable angina to acute MI (see Table 12-4).1,3,5 The general public and media describe an acute MI as a “heart attack.” The following section discusses stable manifestations of CAD (stable angina) and acute manifestations described as an acute coronary syndrome (unstable angina and acute MI).
Angina
Angina pectoris, or chest pain, caused by myocardial ischemia is not a separate disease, but rather a symptom of CAD. It is caused by a blockage or spasm of a coronary artery, leading to diminished myocardial blood supply. The lack of oxygen causes myocardial ischemia, which is felt as chest discomfort, pressure, or pain. Angina may occur anywhere in the chest, neck, arms, or back, but the most commonly described location is pain or pressure behind the sternum. The pain often radiates to the left arm but can also radiate down both arms and to the back, the shoulder, the jaw, or the neck (Figure 12-1). Angina symptoms are not the same for all individuals, many patients may describe pressure or discomfort rather than pain and presenting symptoms can be highly individualized, as described in Box 12-3. Patients and families must be taught that angina does not always present in the dramatic heart attack scenario seen on television and in movies, in which the person clutches the throat or chest and exhibits extreme distress.3
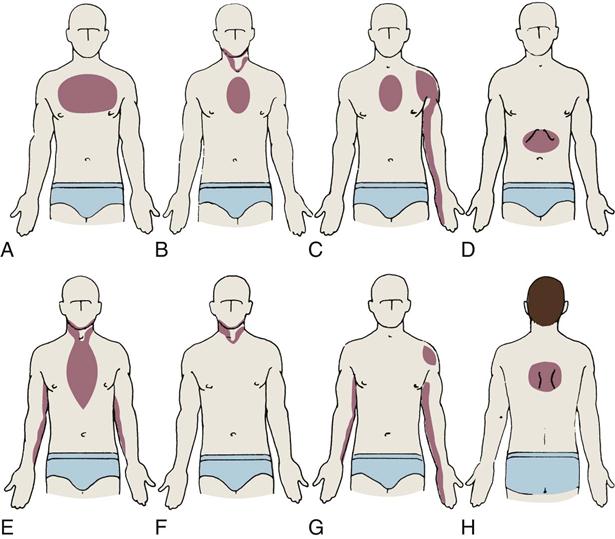
A, Upper part of chest. B, Beneath sternum, radiating to neck and jaw. C, Beneath sternum, radiating down left arm. D, Epigastric. E, Epigastric, radiating to neck, jaw, and arms. F, Neck and jaw. G, Left shoulder. H, Intrascapular.
Women and Angina
Many women experience a variety of different symptoms before an acute MI and during the acute event, as shown in Box 12-4.34 The recognition and publicity about the fact that many women do not experience “crushing chest pain” is important if women’s symptoms are not to be trivialized by clinicians.35 It is important that all patients are made aware of angina symptom equivalents, such as unexpected shortness of breath, breaking out in a cold sweat, or sudden fatigue, nausea, or lightheadedness.3 More women die every year in the United States of cardiovascular disease than men, a fact that is largely unknown.28
Box 12-4
Cardiovascular Symptoms Experienced by Women before Acute Myocardial Infarction
| SYMPTOMS 1 MONTH BEFORE ACUTE MI | SYMPTOMS DURING ACUTE MI |

From McSweeney JC, et al: Women’s early warning symptoms of acute myocardial infarction, Circulation 108(21):2619, 2003.
Stable Angina
Stable angina is predictable and caused by similar precipitating factors each time; typically, it is exercise induced. Patients become used to the pattern of this type of angina and may describe it as “my usual chest pain.” Pain control should be achieved within 5 minutes by rest and by taking sublingual nitroglycerin. Stable angina is the result of fixed lesions (blockages) of more than 75% of the coronary artery lumen. Ischemia and chest pain occur when myocardial demand from exertion exceeds the fixed blood oxygen supply.6 Additional information on CAD and stable angina is provided in the box Evidence-Based Collaborative Practice: Coronary Artery Disease and Stable Angina.
Unstable Angina
Unstable angina is defined as a change in a previously established stable pattern of angina. It is part of the continuum of ACS. Unstable angina usually is more intense than stable angina, may awaken the person from sleep, or may necessitate more than nitrates for pain relief. A change in the level or frequency of symptoms requires immediate medical evaluation. Severe angina that persists for more than 5 minutes, is worsening in intensity, and is not relieved by one nitroglycerin tablet is a medical emergency, and the patient or a family member must call 911 immediately.3 The 911 (Emergency Medical Services [EMS]) system is available to 90% of the population of the United States.3 In one study, patients with an acute MI who used 911 and were transported to the hospital by ambulance had significantly faster receipt of initial reperfusion therapies.36,37 Family and friends are discouraged from driving a person experiencing unstable angina to the hospital and instead are encouraged to call 911. Patients should be instructed never to drive themselves but to contact the EMS by calling 911.
Unstable angina is an indication of atherosclerotic plaque instability. It can signal atherosclerotic plaque rupture and thrombus formation that can lead to MI. The patient who comes to the emergency department with recent onset of unstable angina but who has nonspecific or nonelevated ST-segment changes on the 12-lead electrocardiogram (ECG) may be admitted to the critical care unit to rule out MI. If the symptoms are typical of MI, it is important to treat the patient according to the latest published guidelines, because not all patients who experience an MI have ST-segment elevation on the 12-lead ECG.5
Medical Management
Accurate assessment of chest pain symptoms is essential if unstable angina is to be recognized and treated effectively. An important reason to ask questions about the chest pain is to differentiate between stable and unstable angina. The change from stable to unstable angina is potentially life threatening for the patient. If the ST segments are elevated or there is a newly documented left bundle branch block on the 12-lead ECG, the patient will be treated for acute MI.3 However, if these classic ECG signs are missing and the chest pain continues, the current pharmacological treatments of choice are aspirin, vasodilation by nitroglycerin, intravenous antiplatelet agents such as the glycoprotein (GP) IIb/IIIa inhibitors, and intravenous unfractionated heparin (UFH).5 Low-molecular-weight heparin (LMWH) combined with fibrinolysis is an alternative to heparin fibrinolysis for patients younger than 75 years with a serum creatinine level below 2.5 mg/dL for men and below 2 mg/dL for women.3
Another option is to transport the patient directly to the cardiac catheterization laboratory for direct visualization of the coronary arteries by the cardiologist. Recanalization of the coronary arteries is recommended, provided the institution performs more than 200 procedures annually or the individual physician performs more than 75 interventional procedures annually.3,5
Nursing Management
Nursing management of the patient with CAD and angina incorporates a variety of nursing diagnoses (Nursing Diagnosis Priorities box on Coronary Artery Disease and Angina). Nursing priorities focus on (1) recognizing myocardial ischemia, (2) controlling chest pain, (3) maintaining a calm environment, and (4) providing patient education.
Recognizing Myocardial Ischemia
Complaints of chest discomfort (angina) must be evaluated quickly, because angina is an indicator of myocardial ischemia. The patient is asked to rate the intensity of the chest discomfort on a scale of 0 to 10. Pain levels must be assessed with sensitivity to differences in cultural manifestations of pain. The words “chest pain” are not to be used exclusively, because some patients describe their angina as “pressure” or “heaviness.” It is important to document the characteristics of the pain and the patient’s heart rate and rhythm, blood pressure, respirations, temperature, skin color, peripheral pulses, urine output, mentation, and overall tissue perfusion. A 12-lead ECG is used to identify the area of ischemic myocardium. The major concern is that the chest pain may represent preinfarction angina, and early identification is essential so that the patient can be immediately treated. A range of treatments are available that include fibrinolytic infusion immediately, or transfer to the cardiac catheterization laboratory for a coronary arteriogram and opening of a blocked artery. If the hospital does not have a cardiac catheterization laboratory, GP IIb/IIIa receptor blockers may be infused to prevent the evolution of the acute MI before transfer.3,5 Figure 12-2 presents additional information about transfer between hospitals in this circumstance.
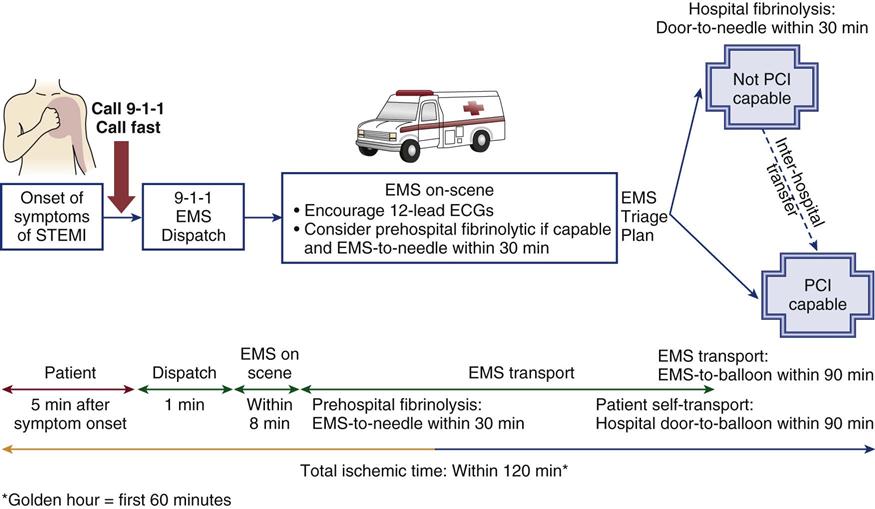
STEMI, ST elevation myocardial infarction; EMS, emergency medical services; PCI, percutaneous catheter intervention. (Illustration modified from Antman EM, et al: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction), Circulation 110(9):e82).
Relieving Chest Pain
In the critical care unit, control of angina is achieved by a combination of supplemental oxygen, nitrates, analgesia, and surveillance of the angina and of the effects of pharmacological therapy.
• Oxygen: All patients with acute ischemic pain are administered supplemental oxygen to increase myocardial oxygenation. Use of pulse oximetry is recommended to guide therapy and maintain oxygen saturation above 90%.3 Patients who develop symptoms of acute heart failure may require emergency intubation and mechanical ventilation to correct significant hypoxemia.3,5
• Nitrates: A combination of intravenous and sublingual nitroglycerin is used to vasodilate the coronary arteries and decrease pain. After nitrate administration, the critical care nurse closely observes the patient for relief of chest pain, for return of the ST segment to baseline, and for the potential development of unwanted side effects such as hypotension and headache. Administration of a nitrate is avoided if the SBP is below 90 mm Hg. Drug interactions with nitrates are another potential cause for concern. The phosphodiesterase inhibitor medication sidenafil (Viagra) is prescribed for several conditions including pulmonary hypertension and erectile dysfunction. Sidenafil and nitrates in combination may contribute to a precipitous fall in blood pressure.3,5
• Analgesia: Morphine (2 to 4 mg given intravenously) is the analgesic opiate of choice for preinfarction angina. It relieves pain and decreases fear and anxiety. After administration, the critical care nurse assesses the patient for pain relief and the development of unwanted side effects such as hypotension and respiratory depression.3,5
• Aspirin: Chewing an oral nonenteric-coated aspirin (162 to 325 mg) at the beginning of chest pain has been shown to reduce mortality. The nonenteric formulation is preferred because it increases absorption in the mouth when chewed, not swallowed.3,5
Maintaining a Calm Environment
Patients admitted to a critical care unit with unstable angina experience extreme anxiety and fear of death. The critical care nurse is faced with the challenge of ensuring that the elements of a calm environment that can alleviate the patient’s fear and anxiety are maintained, while being ready at all times to respond to an acute emergency, such as a cardiac arrest, or to assist with emergency intubation or insertion of hemodynamic monitoring catheters.
Providing Patient Education
In the critical care unit, the patient’s ability to retain educational information is severely affected by stress and pain. Initial patient education stresses the importance of alerting the nurse to any symptoms of chest pain or discomfort, and avoiding the Valsalva maneuver, which is defined as forced expiration against a closed glottis. This can be explained to the patient as “bearing down” when going to the bathroom or breath-holding when repositioning in bed. The Valsalva maneuver causes an increase in intrathoracic pressure that decreases venous return to the right side of the heart and is associated with low blood pressure and symptomatic bradycardia.
After the anginal pain is controlled, longer-term patient and family education can begin. Points to cover include risk factor modification, signs and symptoms of angina, when to call the physician, medications, and dealing with emotions and stress. However, because the acute hospital length of stay for uncomplicated angina is usually less than three days, referral to a cardiac rehabilitation program for a controlled exercise program and risk-factor modification after discharge may be the most helpful teaching intervention a critical care nurse can provide. Clinical practice guidelines for the management of CAD and stable angina are listed in the box Evidence-Based Collaborative Practice: Coronary Artery Disease and Stable Angina.
Myocardial Infarction
Description and Etiology
Myocardial infarction (MI) is the term used to describe irreversible myocardial necrosis (cell death) that results from an abrupt decrease or total cessation of coronary blood flow to a specific area of the myocardium.1 In the hospital, this is often referred to as an acute MI, indicating the sudden onset and the life-threatening nature of the event. Increasingly, an acute MI is described in relation to whether there was ST-segment elevation on the diagnostic 12-lead ECG. It may be labeled an acute non-ST-segment elevation MI (NSTEMI)5 or an acute ST-segment elevation MI (STEMI).3
Three mechanisms can block the coronary artery and are responsible for the acute reduction in oxygen delivery to the myocardium:
Myocardial tissue can best be salvaged within the first 2 hours (120 minutes) after the onset of anginal symptoms, as illustrated in Figure 12-2.3 The earlier the myocardium is revascularized, the better the survival.5 Unfortunately, many persons do not seek treatment until the acute phase has passed.3
Pathophysiology
Myocardial Ischemia
The outer region of the infarcted myocardial area is the zone of ischemia, as illustrated in Figure 12-3. It is composed of viable cells. Priority interventions are targeted to save this viable muscle. Repolarization in this zone is temporarily impaired but eventually will be restored to normal. Electrical conduction from areas of viable myocardium produces a normal QRS configuration as shown in Figure 12-4, A. Repolarization of ischemic myocardial cells manifests as T-wave inversion (see Figure 12-4, B).
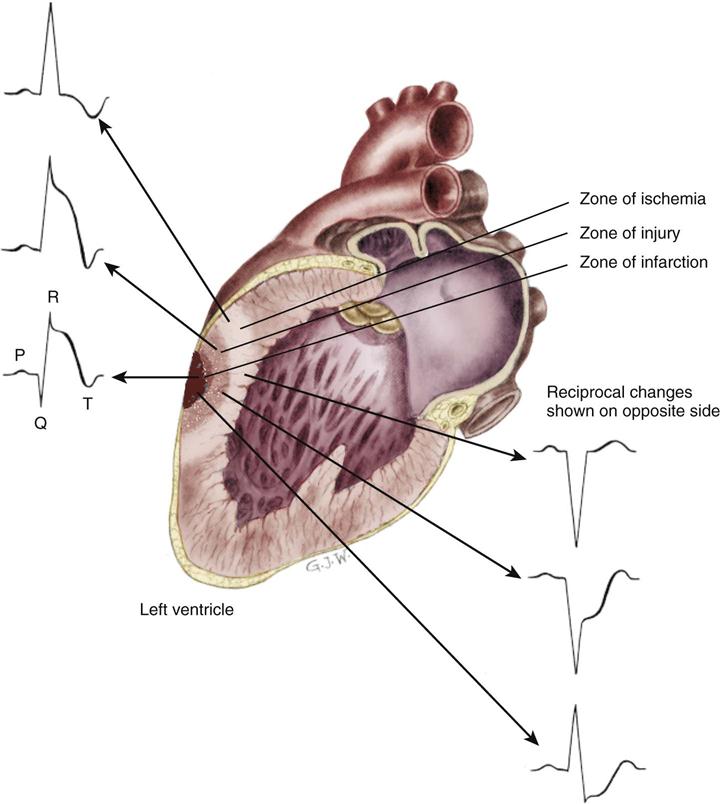
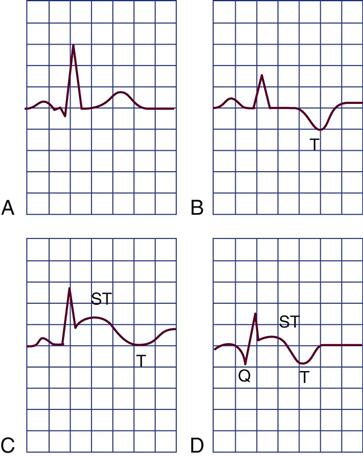
A, Normal ECG. B, Ischemia indicated by inversion of the T wave. C, Ischemia and current of injury indicated by T-wave inversion and ST-segment elevation. The ST segment may be elevated above or depressed below the baseline, depending on whether the tracing is from a lead facing toward or away from the infarcted area and depending on whether epicardial or endocardial injury occurs. Epicardial injury causes ST-segment elevation in leads facing the epicardium. D, Ischemia, injury, and myocardial necrosis. The Q wave indicates necrosis of the myocardium.
Myocardial Injury
The infarcted zone is surrounded by injured but still potentially viable tissue in an area known as the zone of injury (see Figure 12-3). Cells in this area do not fully repolarize because of the deficient blood supply. This is recorded on the ECG as elevation of the ST segment (see Figure 12-4, C).
Myocardial Infarction
The area of dead muscle (necrosis) in the myocardium is known as the zone of infarction (see Figure 12-3). On the ECG, evidence of this zone is seen by new pathological Q waves, which reflect a lack of depolarization from the cardiac surface involved in the MI (see Figure 12-4, D). As healing takes place, the cells in this area are replaced by scar tissue.
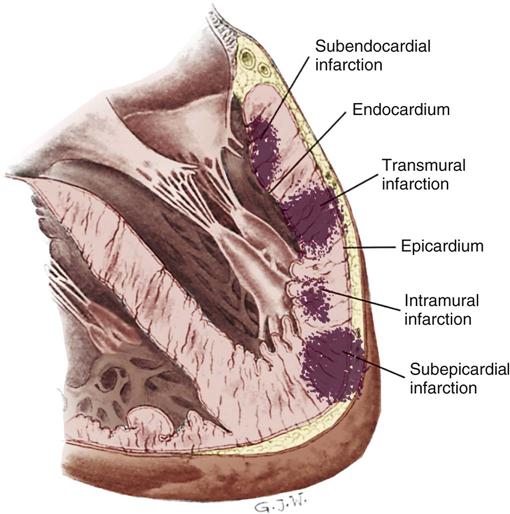
MIs are classified according to the location on the myocardial surface and the muscle layers affected. Not all infarctions cause necrosis in all layers, as shown in Figure 12-5. A transmural MI involves all three cardiac layers—the endocardium, the myocardium, and the epicardium. A transmural (full-thickness) MI usually provokes significant ECG changes (see Figure 12-4). This is also described as a Q-wave MI. Not every acute MI produces a recognizable series of Q waves on the 12-lead ECG. Some patients who had a demonstrated Q wave on a 12-lead ECG as a result of an acute MI lose the Q wave months or years later. The reasons for this are unknown, but it may represent the development of collateral circulation.
12-Lead Electrocardiographic Changes
The ECG changes produced by a transmural infarction demonstrate alteration in myocardial depolarization (QRS complex) and repolarization (ST segment). The changes in repolarization are seen by the presence of new Q waves. These new, pathological Q waves are deeper and wider than tiny Q waves found on the normal 12-lead ECG.3
Myocardial Infarction Location
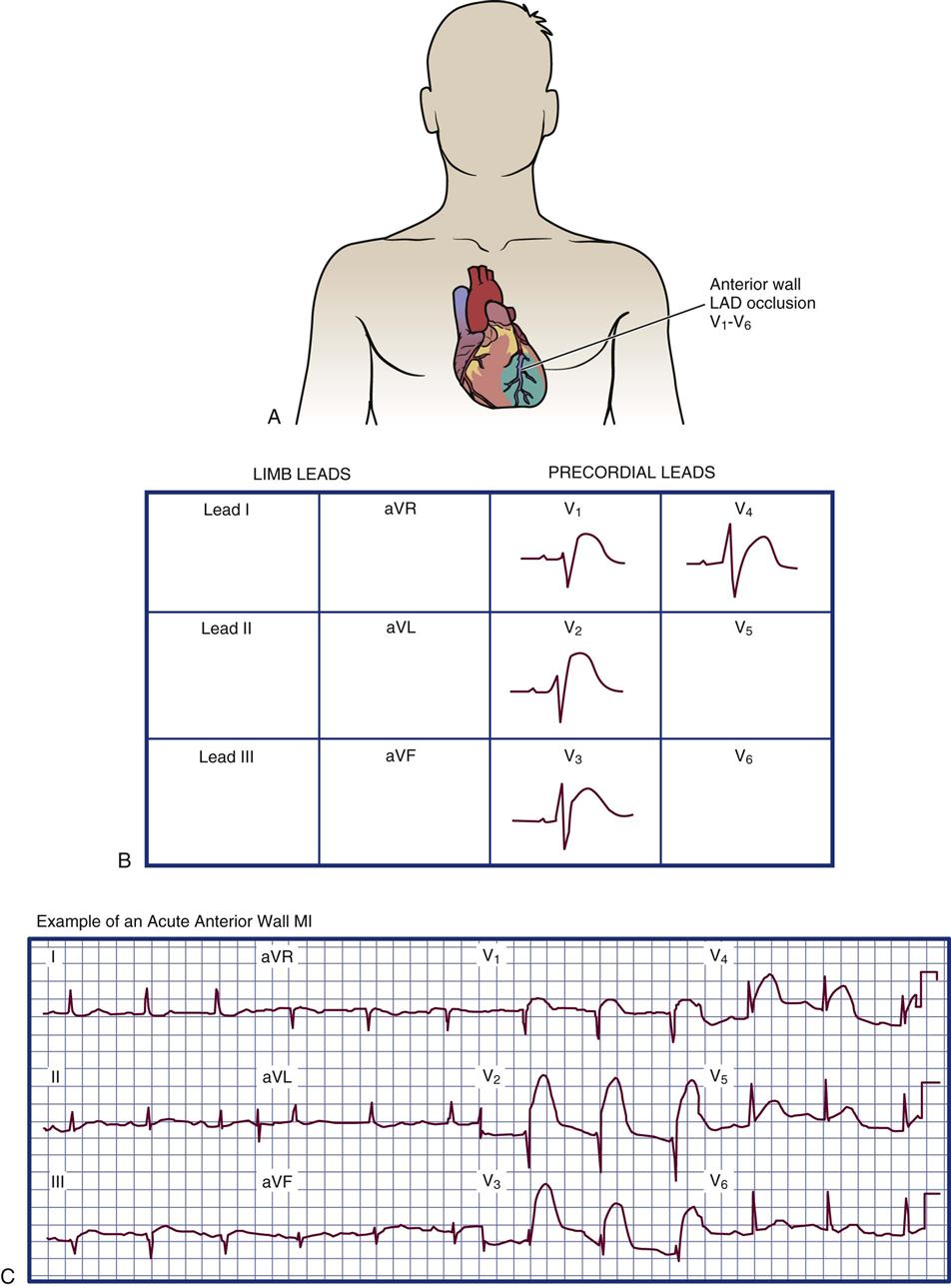
The location of infarction is determined by correlating the ECG leads with Q waves and the ST-segment T-wave abnormalities (Table 12-5). Infarction most commonly affects the left ventricle and the interventricular septum; however, the right ventricle can be infarcted, and many patients who sustain an inferior MI have some right ventricular damage. The ECG manifestations that are used to diagnose an MI and pinpoint the area of damaged ventricle include inverted T waves, ST-segment elevation, and pathological Q waves in specific lead groupings as described subsequently.
TABLE 12-5
CORRELATIONS AMONG VENTRICULAR SURFACES, ELECTROCARDIOGRAPHIC LEADS, AND CORONARY ARTERIES
| SURFACE OF LEFT VENTRICLE | ELECTROCARDIOGRAPHIC LEADS | CORONARY ARTERY USUALLY INVOLVED |
| Inferior | II, III, aVF | Right coronary artery |
| Lateral | V5-V6, I, aVL | Circumflex |
| Anterior | V2-V4 | Left anterior descending |
| Anterior lateral | V1-V6, I, aVL | Left main coronary artery |
| Septal | V1-V2 | Left anterior descending |
| Posterior | V1-V2 | Circumflex or right coronary artery (reciprocal changes) |
| V7-V9 (direct) |
Anterior Wall Infarction
Anterior wall infarction results from occlusion of the proximal left anterior descending artery (see Table 12-5). ST-segment elevation is expected in leads V1 through V4 on the 12-lead ECG, as shown in Figure 12-6. If the left main coronary artery is occluded, the ECG manifestations will involve almost all of the precordial leads V1 through V6 and leads I and aVL (augmented voltage left) as listed in Table 12-5. The specific groups of ECG changes that help to locate the part of the heart that is infarcting are called indicative changes. A large anterior wall MI may be associated with left ventricular pump failure, cardiogenic shock, or death.3
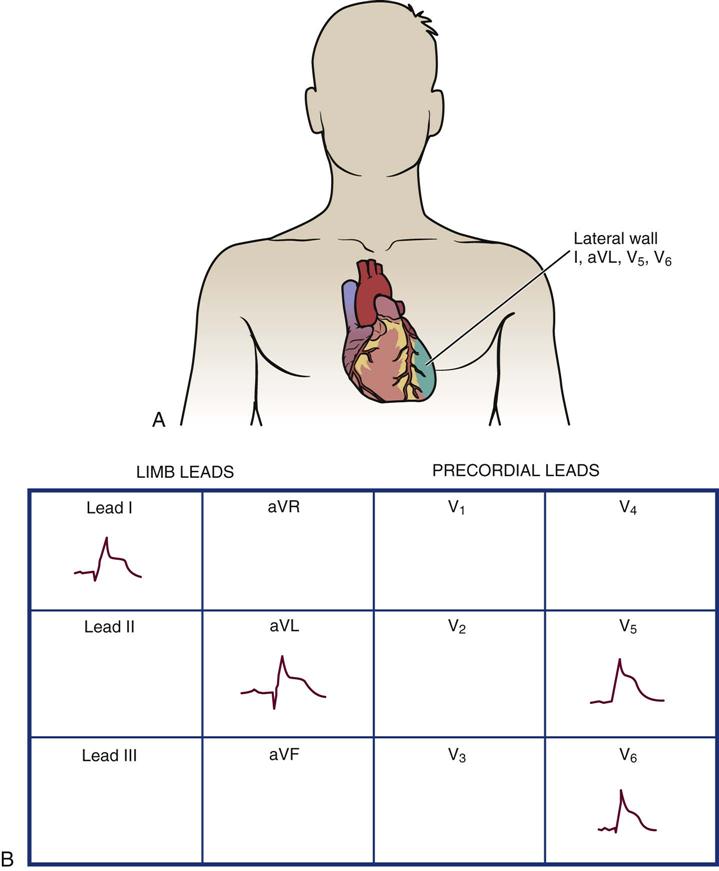
Left Lateral Wall Infarction
Left lateral wall infarction occurs as a result of occlusion of the circumflex coronary artery. On a 12-lead ECG, new Q waves and ST-segment T-wave changes are seen in leads I, aVL, V5, and V6 (Figure 12-7). In reality, few patients present with only lateral wall ECG changes, and some anterior wall leads (V3 and V4) may show evidence of injury or infarction.
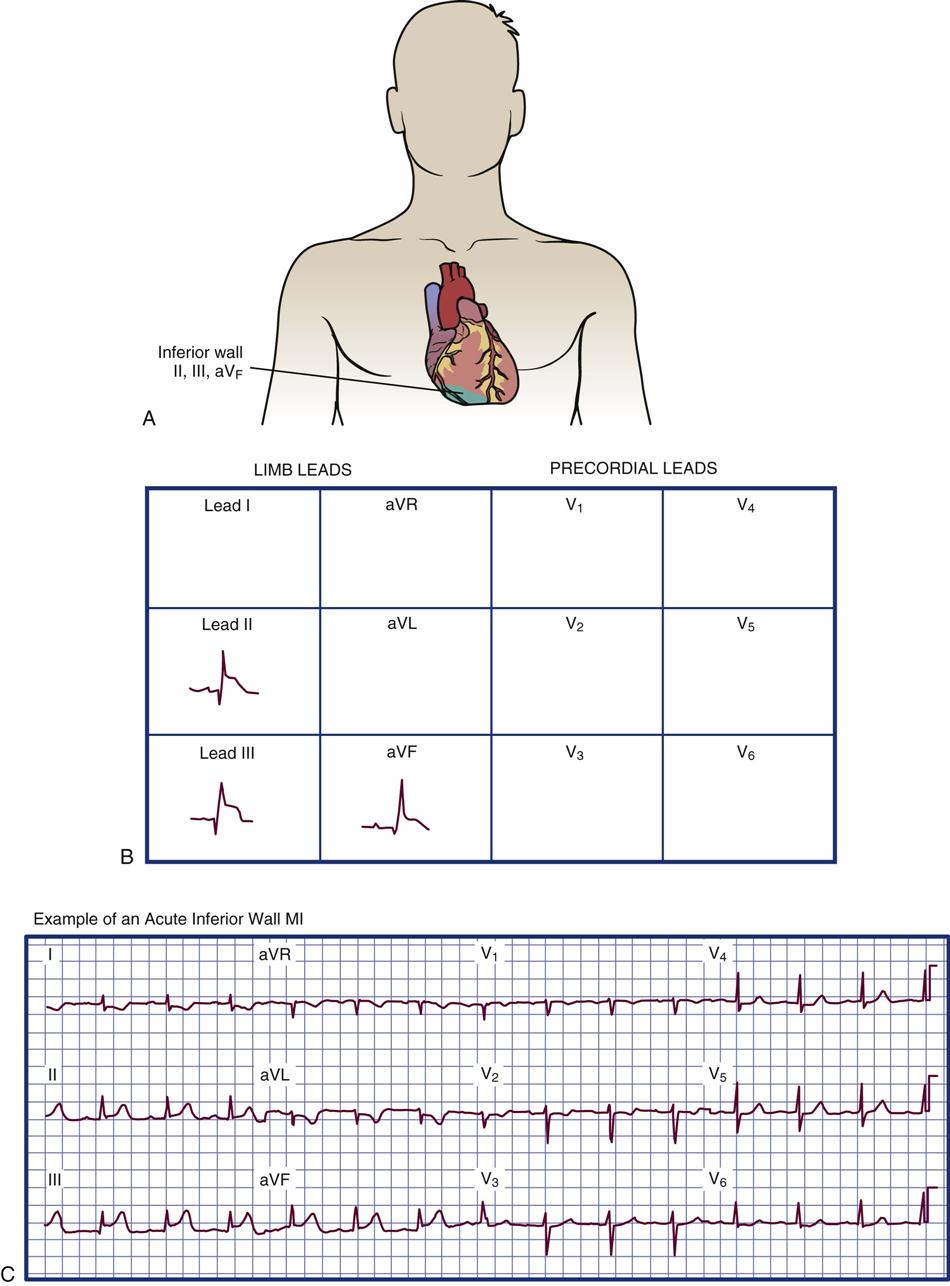
Inferior Wall Infarction
Inferior wall infarction occurs with occlusion of the right coronary artery. This infarction manifests by ECG changes in leads II, III, and aVF (Figure 12-8). Conduction disturbances are expected with an inferior wall MI and are related to the anatomy of the coronary arterial supply. Because the right coronary artery perfuses the sinoatrial (SA) node in slightly more than half of the population and supplies the proximal bundle of His and atrioventricular (AV) node in more than 90% of individuals, heart block and other conduction disturbances should be anticipated. Inferior wall MI carries a mortality rate of about 6%. If the right ventricle is involved, the mortality rate increases to 25% to 30%.3
Right Ventricular Infarction
Right ventricular infarction occurs when there is a blockage in a proximal section of the right coronary artery. This places all of the right ventricle and the inferior wall at risk. Right ventricular ischemia can be demonstrated in up to one half of inferior wall STEMIs, although only 10% to 15% show the hemodynamic abnormalities associated with classic infarction of the right ventricle.3 If massive infarction occurs, the patient can suffer cardiogenic shock, which carries a mortality rate of more than 50% in this population.37,38
Posterior Wall Infarction
Posterior wall infarction can occur because of a blockage in the right coronary artery or in the circumflex artery. This occurs because both arteries supply this section of the heart, although the right coronary artery is generally the dominant vessel. A posterior wall MI is difficult to detect but may be identified by specific leads placed in the left scapular area or by very tall R waves in leads V1 and V2.
Non-ST-Segment Elevation Myocardial Infarction
The 12-lead ECG is a highly useful diagnostic tool. For many years, it was considered the gold standard when diagnosing an acute MI. However, the ST segment is not elevated in every acute MI. One reason for the lack of ST-segment elevation may be that the infarction and subsequent necrosis are not full-thickness lesions. Because some of the muscle in the area can still be depolarized, ST-segment elevation may not occur. This type of MI is also less likely to develop Q waves on a subsequent 12-lead ECG after the acute phase has passed. This situation is diagnostically known as an NSTEMI.5 This condition has previously been described by several names, including nontransmural MI, non-Q-wave MI, and subendocardial MI. Because patients who sustain an NSTEMI do have CAD, it is important that they be treated aggressively to minimize the size of the infarcted area. Without the visual clue of the ST-segment elevation on the 12-lead ECG, the patients cannot receive immediate intravenous fibrinolytic agents, but they can be appropriately managed in an interventional catheterization laboratory and receive GP IIb/IIIa inhibitor therapy, as illustrated in the timeline in Figure 12-2. The 12-lead ECG plays a vital role in identifying the treatment plan for an ACS. ST-segment elevation is helpful when present, but it would be a mistake to believe the patient is not in danger of MI if the ST segment is not elevated. In this case, the definitive diagnosis may be made in the cardiac catheterization laboratory or by elevation of specific cardiac biomarkers.
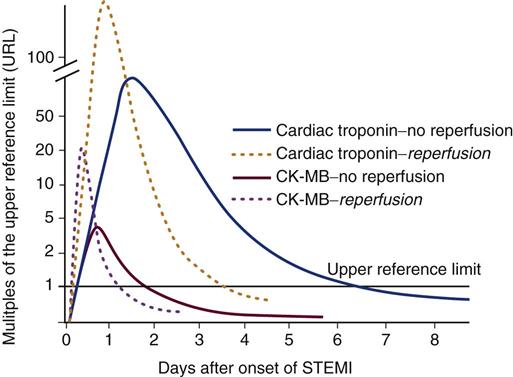
Cardiac Biomarkers During Myocardial Infarction
In the presence of damaged or necrotic myocardial muscle cells, cardiac biomarkers are released. These biomarkers are also called cardiac enzymes. To confirm the diagnosis of acute MI, the serum biomarkers creatine kinase-muscle/brain (CK-MB) and troponin I or troponin T are measured. If the coronary artery is opened by fibrinolytic therapy or a percutaneous catheter intervention (PCI), the biomarkers exhibit a more rapid rise and dramatic fall (Figure 12-9). Information on biomarkers is also shown in Table 11-14 in Chapter 11.
Complications of Acute Myocardial Infarction
Many patients experience complications occurring early or late in the postinfarction course. These complications may result from electrical dysfunction or from a cardiac contractility problem. The presence of a new murmur in a patient with an acute MI warrants special attention because it may indicate rupture of the papillary muscle. Electrical dysfunctions include bradycardia, bundle branch blocks, and various degrees of heart block. Pumping complications cause heart failure, pulmonary edema, and cardiogenic shock.3 The murmur can be indicative of severe damage and impending complications such as heart failure and pulmonary edema.
Sinus Bradycardia and Sinus Tachycardia
Sinus bradycardia (heart rate less than 60 beats/min) occurs in 30% to 40% of patients who sustain an acute MI.2 It is more prevalent with an inferior wall infarction in the first hour after STEMI.2 Symptomatic bradycardia with hypotension and low cardiac output is treated with atropine (0.5 to 1.0 mg by intravenous push), repeated every 3 to 5 minutes to a maximum dose of 0.03 mg/kg (e.g., 2 mg for a person who weighs 70 kg) per Advanced Cardiac Life Support (ACLS) Guidelines.
Sinus Tachycardia
Sinus tachycardia (heart rate more than 100 beats/min) most often occurs with an anterior wall MI. Anterior infarctions impair left ventricular pumping ability, thereby reducing the ejection fraction and the stroke volume. In an attempt to maintain cardiac output, the heart rate increases. Sinus tachycardia must be corrected, because it greatly increases myocardial oxygen consumption, leading to further ischemia.
Atrial Dysrhythmias
Premature atrial contractions (PACs) occur frequently in patients who sustain an acute MI. Atrial fibrillation is also common and may occur spontaneously or may be preceded by PACs. With onset of atrial fibrillation, the loss of organized atrial contraction decreases cardiac output by up to 20%. A global registry of patients with ACS found that almost 8% have preexisting atrial fibrillation, and about 6% develop new-onset atrial fibrillation during their hospitalization for ACS.39 Patients with new-onset or preexisting atrial fibrillation have a higher morbidity rate than patients without atrial fibrillation during an ACS event. ACS patients with new-onset atrial fibrillation experience a greater number of in-hospital adverse events such as reinfarction, shock, pulmonary edema, bleeding, and stroke.39 Atrial fibrillation during the hospitalization significantly affects risk of death in the setting of an acute MI; it increases in-hospital mortality by 20% and long-term mortality by 34%.2
Ventricular Dysrhythmias
Premature ventricular contractions (PVCs) are seen in almost all patients within the first few hours after an MI. They are initially controlled by administering oxygen to reduce myocardial hypoxia and by correcting acid-base or electrolyte imbalances. In the setting of an acute MI, PVCs are pharmacologically treated if they have the following characteristics: frequent, closely coupled (R-on-T phenomenon), multiform shapes, and occurrence in bursts of three or more, increasing the risk of sustained ventricular tachycardia (VT). Ventricular fibrillation (VF) is a life-threatening dysrhythmia associated with high mortality in acute MI. Beta-blockers are often prescribed after an acute MI to decrease mortality from ventricular dysrhythmias.3
Atrioventricular Heart Block During Myocardial Infarction
Heart block occurs in 6% to 14% of patients with STEMI, and those patients have increased mortality rates.2 In STEMI, AV block most often occurs after an inferior wall infarction. Because the right coronary artery supplies the AV node in 90% of the population, right coronary artery occlusion leads to ischemia and infarction of the AV node cells. The development of sudden heart block has become much less common because most patients receive fibrinolysis or undergo PCI to open the occluded vessel. In most cases, transcutaneous pacing is the primary intervention; transvenous pacemakers are used less frequently.2
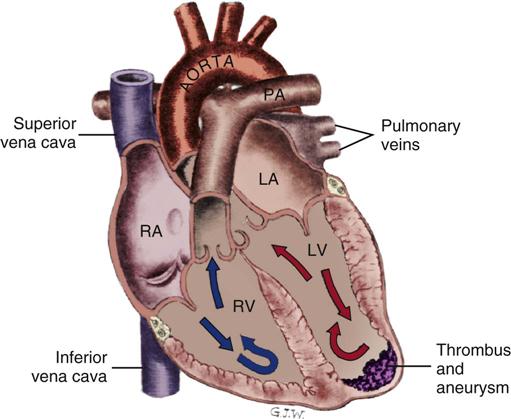
Ventricular Aneurysm After Myocardial Infarction
A ventricular aneurysm (Figure 12-10) is a noncontractile, thinned left ventricular wall that results from an acute transmural infarction. It most often occurs in the setting of an acute left anterior descending artery occlusion with a wide area of infarcted myocardium.2 The most effective prevention is early reperfusion of the myocardium, accomplished by opening the thrombosed coronary artery. In one study, the rate of ventricular aneurysm was reduced from 19% in untreated patients to 7% when the coronary artery was opened by fibrinolysis.2 The most common complications of a ventricular aneurysm are acute heart failure, systemic emboli, angina, and VT. Treatment is directed toward management of these complications and surgical repair by left ventricular aneurysmectomy. The affected area may be described as hypokinetic (contracts poorly), akinetic (noncontractile scar tissue), or dyskinetic (scar tissue that moves in the opposite direction of the normal contractile myocardium). The prognosis depends on the size of the aneurysm, the level of overall left ventricular dysfunction, and the severity of coexisting CAD.
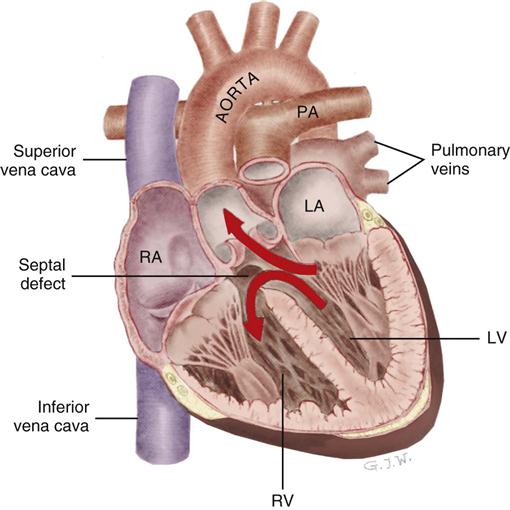
Ventricular Septal Rupture After Myocardial Infarction
Postinfarction rupture of the ventricular septal wall is a rare but potentially lethal complication of an acute anterior wall MI (Figure 12-11). Ventricular septal rupture, also known as an acquired ventricular septal defect (VSD), is an abnormal communication between the right and left ventricle. This complication occurs in less than 1% of all MIs, and the incidence has declined because most STEMI patients have the blocked coronary artery opened.2 Nevertheless, rupture of the ventricular septum carries an extremely high mortality rate.40 Mortality rates between 35% and 73% are typical.41,42 Most patients with septal rupture also have signs and symptoms of cardiogenic shock. Ventricular septal rupture manifests as severe chest pain, syncope, hypotension, and sudden hemodynamic deterioration caused by shunting of blood from the high-pressure left ventricle into the low-pressure right ventricle through the new septal opening. A holosystolic murmur (often accompanied by a thrill) can be auscultated and is best heard along the left sternal border. A diagnosis of postinfarction ventricular septal rupture can be made at the bedside with use of oxygen saturation assessment by means of a pulmonary artery catheter or by transesophageal echocardiography (TEE). Rupture of the septum is a medical and surgical emergency. The patient’s condition is stabilized with vasodilators and an intraaortic balloon pump (IABP) to decrease afterload.42 The goal of afterload reduction in this patient population is to decrease the amount of blood being shunted to the right side of the heart and consequently to increase the flow of blood to the systemic circulation. If the septal rupture is very small, and the patient’s condition is sufficiently stable to wait for scar tissue to form before surgical repair, survival improves. Unfortunately, when the septal opening is large, the massive left-to-right shunt across the septum makes the chances of survival dismal with or without surgery.41,42
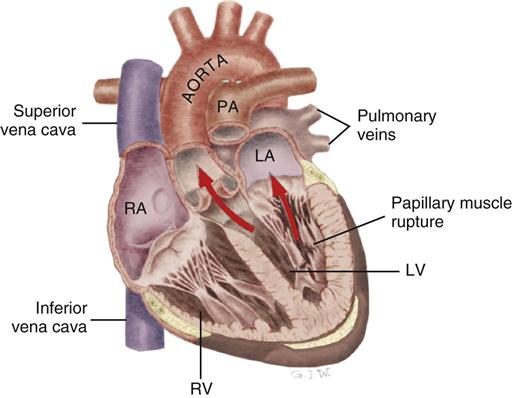
Papillary Muscle Rupture After Myocardial Infarction
Papillary muscle rupture can occur when the infarct involves the area around one of the papillary muscles that support the mitral valve. Infarction of the papillary muscles results in ineffective mitral valve closure, and blood is forced back into the low-pressure left atrium during ventricular systole. The rupture may be partial or complete. Complete rupture is catastrophic and precipitates severe acute mitral regurgitation, cardiogenic shock, and high risk of death.
Partial rupture of the valve structures (Figure 12-12) also results in mitral regurgitation, but the condition can be stabilized with aggressive medical management using the IABP and vasodilators. Urgent surgical intervention is required to replace the mitral valve.43 As with other structural complications during acute MI, the incidence is decreased in patients who have their myocardium reperfused early.2 Of the patients with acute MI who are admitted to the critical care unit in cardiogenic shock, 10% have papillary muscle rupture with acute mitral regurgitation. The mortality rate is 71% with medical treatment and 40% with surgical intervention to replace or repair the mitral valve.2
Cardiac Wall Rupture After Myocardial Infarction
Of the deaths that occur after MI, 1% to 6% can be attributed to cardiac rupture.2 The incidence of cardiac wall rupture has two peak times. The first occurs within the first 24 hours and the second between the third and fifth postinfarction days when leukocyte scavenger cells are removing necrotic debris, thinning the myocardial wall.2 The onset is sudden and usually catastrophic. Bleeding into the pericardial sac results in cardiac tamponade, cardiogenic shock, pulseless electrical activity (PEA), and death. Survival is rare. If rupture occurs in the hospital, emergency pericardiocentesis is required to relieve the tamponade until a surgical repair can be attempted. The best prevention is early reperfusion of the myocardium.2
Pericarditis After Myocardial Infarction
Pericarditis is inflammation of the pericardial sac. It can occur during a transmural MI or after an acute MI. Pericarditis may occur in 5% to 20% of transmural infarctions, but it is treated only if it is clinically significant.44 The damaged epicardium becomes rough and inflamed and irritates the pericardium lying adjacent to it, precipitating pericarditis. Pain is the most common symptom of pericarditis, and a pericardial friction rub is the most common initial sign. The friction rub is best auscultated with a stethoscope at the sternal border and is described as a grating, scraping, or leathery scratching. Pericarditis frequently produces a pericardial effusion (fluid).44 After the effusion occurs, the friction rub may disappear. On the 12-lead ECG, pericarditis may manifest as elevation of the ST segment in all of the typically upright leads.45 Pericarditis is treated with nonsteroidal antiinflammatory drugs. Pericarditis that occurs as a late complication of acute MI is also known as Dressler’s syndrome.44,46
Heart Failure and Acute Myocardial Infarction
Almost 20% of patients with acute STEMI also have acute heart failure on admission to the hospital. These patients have often waited longer to come to the hospital and are older and more likely to be female. Compared with acute MI patients without heart failure, these patients have a higher risk of adverse in-hospital events and have longer lengths of stay and higher in-hospital mortality rates.47 More detailed information about heart failure is presented later in this chapter.
Medical Management
Quality-outcomes research shows that compliance with the guidelines developed by the American College of Cardiology and the American Heart Association (ACC/AHA) decreases in-hospital mortality after acute MI.48,49 Patients admitted to hospitals who adhere to the AHA/ACC guidelines for treatment of STEMI or NSTEMI have an 8.3% in-hospital mortality rate, compared with a 15.3% mortality rate for patients managed at hospitals where the most recent guidelines are not fully used.48,49 The guidelines are research-based and are designed to improve the outcome of patients admitted to the hospital with an acute MI. Clinical guidelines address the issues of interventions to open the coronary artery, anticoagulation, prevention of dysrhythmias, tight glucose control, and prevention of ventricular remodeling after STEMI.3
Recanalization of the Coronary Artery
The essential immediate interventions are fibrinolytic therapy or PCI to open the occluded artery for the patient with an acute STEMI.2 All clinical guidelines emphasize the need for patients with symptoms of ACS to be rapidly triaged and treated.2
Anticoagulation
In the acute phase after STEMI, heparin is administered in combination with fibrinolytic therapy to recanalize (open) the coronary artery.2 For patients who will receive fibrinolytic therapy, an initial heparin bolus of 60 units/kg (maximum 4000 units) is given intravenously, followed by a continuous heparin drip at 12 units/kg/hr (maximum 1000 units/hr) to maintain an activated partial thromboplastin time (aPTT) between 50 and 70 seconds (1.5 to 2.0 times control). Alternatively, LMWH may be used at doses that provide full anticoagulation.2
It is also prudent to administer intravenous UFH or subcutaneous LMWH if the person is at risk for thrombus development.2 For patients with known heparin-induced thrombocytopenia (HIT),50 a third class of antithrombotic drugs is available as an alternative to LMWH or UFH. These are direct antithrombotic agents (e.g., hirudin, bivalirudin, argatroban, fondaparinux). Use of newer antithrombotic drugs remains low in clinical practice, however.52 Patients at risk for thrombotic emboli include those with an anterior wall infarction, atrial fibrillation, previous embolus, cardiomyopathy, or cardiogenic shock.
When the risk of systemic embolic complications remains high, especially in atrial fibrillation, the patient should be anticoagulated with warfarin (Coumadin).2
Dysrhythmia Prevention
The antidysrhythmic with the best safety record after STEMI is amiodarone. The reduction in death related to decreased dysrhythmias after MI is 13%.2 Beta-blockers are another class of antidysrhythmics that are recommended for all patients after STEMI. Beta-blockers prevent ventricular dysrhythmias, lower blood pressure, and prevent reinfarction, especially in patients with left ventricular dysfunction.2
Tight Glucose Control
Achievement of normal blood glucose levels during the acute phase and after MI improves survival.2
Prevention of Ventricular Remodeling
Many patients are at risk of developing heart failure after STEMI. Vasodilating drugs known as ACEIs or ARBs can stop or limit the ventricular remodeling that leads to heart failure. An ACEI is used, or if it is not tolerated, an ARB is indicated for all patients after STEMI.2 Information about the clinical effects of heart failure is provided later in this chapter.
Nursing Management
Nursing management of the patient with an acute MI incorporates a variety of nursing diagnoses (Nursing Diagnoses Priorities box on Myocardial Infarction). Nursing priorities focus on (1) balancing myocardial oxygen supply and demand, (2) preventing complications, and (3) providing patient education.
Balancing Myocardial Oxygen Supply and Demand
In the acute period, if severe heart muscle damage has occurred, myocardial oxygen supply is increased by the administration of supplemental oxygen to prevent tissue hypoxia. Drugs play an increasingly important role in balancing tissue oxygen supply and demand, and it is the critical care nurse who administers and monitors the effectiveness of these agents. For the patient with a low cardiac output, positive inotropic drugs such as dobutamine, dopamine, and milrinone are prescribed. These inotropic agents are used to increase cardiac contractility in the healthy areas of the heart (increasing oxygen supply), while avoiding damage to the recently infarcted areas. Myocardial oxygen supply can be further enhanced by the use of coronary artery vasodilators. Nitroglycerin is recommended for the first 48 hours to increase vasodilation and prevent myocardial ischemia.2 Research evidence supports the administration of early beta-blockade therapy to decrease myocardial workload and to prevent dysrhythmias. Administration of beta-blockers reduces mortality by 14% in the first 7 days after an MI and by 23% in the long-term.2 However, if the patient is in cardiogenic shock, beta-blockers are withheld until the cardiac output has improved.2 Other interventions to decrease cardiac work and myocardial oxygen consumption include bed rest with bedside commode privileges when the patient is clinically stable.
Preventing Complications
A thorough grasp of the range of potential complications that can occur after STEMI is essential. Cardiac monitoring for early detection of ventricular dysrhythmias is ongoing. Assessment for signs of continued ischemic pain is important, because angina is a warning sign of myocardium at risk. In response to angina, a 12-lead ECG is obtained to determine if there is an extension of the infarct, nitroglycerin is administered, and the physician is notified immediately so that interventions may be initiated to limit the size of the MI. Heart failure is a serious complication after STEMI. When the patient’s blood pressure is stable, treatment with ACEI is initiated. These vasodilators are used to prevent the left ventricular remodeling and dilation that occurs in many patients after an acute MI. Hypotension is a potential complication of ACEI, especially with the first dose. It is an important nursing responsibility to monitor blood pressure and patient symptoms after taking this medication. Surveillance to detect obvious and subtle signs of bleeding is also a priority because so many acute MI patients receive antiplatelet, anticoagulant, and fibrinolytic medications.2
In the first 24 hours, stable patients with acute MI may be given only a light diet, because their appetite is often poor. It is no longer considered necessary to restrict iced fluids or caffeine. While the patient is in bed, an upright position is preferred to foster better lung expansion. Deep breathing decreases the risk of atelectasis. An upright position also decreases venous return, lowers preload, and decreases cardiac work. The patient is taught to avoid increasing intraabdominal pressure (Valsalva maneuver). Stool softeners are given to the patient to lessen the risk of constipation from analgesics and bed rest and to decrease the risk of straining. The nurse controls the critical care unit environment by decreasing noise, diminishing sensory overload, and allowing adequate rest periods. (Evidence-Based Collaborative Practice box on Acute Coronary Syndrome and Acute Myocardial Infarction (Non-STEMI and STEMI).)
Providing Patient Education
It is important to discuss the risk of depression following acute MI. The incidence of depression after an MI ranges from 17% to 27%. Key symptoms of depression mentioned frequently by cardiac patients are fatigue, change in appetite, and sleep disturbance. Depression is an independent risk factor for increased morbidity and mortality in those with coronary heart disease, and it is a treatable condition.
After the acute phase has passed, education for the patient and family focuses on risk-factor reduction, manifestations of angina, when to call a physician or emergency services, medications, and resumption of physical and sexual activity. If possible, a referral is made to a cardiac rehabilitation program so that this education can be reinforced outside the acute care hospital environment.51
Cardiac Arrest and Sudden Cardiac Death
Description
Between 400,000 and 460,000 people die suddenly of cardiac causes each year.1 This number represents 60% of all cardiac deaths.1 These statistics represent deaths that occur outside the hospital with symptoms that last less than 1 hour or occur in a hospital emergency department.52 Sudden cardiac death represents about 5% of the total annual mortality rate for all causes.53
When the onset of symptoms is rapid, the most likely mechanism of death is VT, which degenerates into VF. This syndrome is called sudden cardiac death (SCD). Despite aggressive cardiopulmonary resuscitation (CPR) initiated outside the hospital, few who sustain an out-of-hospital cardiac arrest survive to hospital discharge. Strategies that have been shown to improve the resuscitation chain of survival involve huge community-wide programs to teach laypersons CPR and how to use an automated external defibrillator (AED). With such programs in place and with accessible AED units and rapid EMS support, survival to hospital discharge has been shown to improve.54
Etiology
Most SCD incidents occur in patients with preexisting ventricular dysfunction resulting from underlying heart disease that is either acquired or has a genetic component as listed in Box 12-5. Specific SCD risk factors include extensive coronary atherosclerosis with or without a history of an acute MI; dilated or hypertrophic cardiomyopathy; valvular heart disease; autonomic nervous system abnormalities; electrical system abnormalities, such as AV block, Wolff-Parkinson-White (WPW) syndrome, prolonged QT syndrome; and taking medications that prolong the QT interval.55–57 An ejection fraction of less than 30% and a history of ventricular dysrhythmias are powerful predictors of SCD. Unfortunately, many individuals are unaware of their risk. In one longitudinal analysis, 48% of the SCD victims had not previously been diagnosed with cardiac disease.52 Fifty percent of men and 64% of women who die suddenly of cardiovascular disease have no previous symptoms of the disease. Men account for 70% to 89% of SCD, an annual incidence that is three to four times higher for men than women.1
Therapeutic Hypothermia
Depending on the length of time the patient was unconscious following the cardiac arrest, cognitive defects can occur, caused by lack of cerebral blood flow and resultant hypoxia to the brain. For comatose patients at high risk for hypoxic brain injury after cardiac arrest, therapeutic hypothermia to about 33° centigrade is initiated for several hours to preserve brain function.58,59 Only one tenth to one third of people who are resuscitated after cardiac arrest are discharged from the hospital and able to lead an independent life.59 Post-arrest hypothermia treatment increases the likelihood that survivors of cardiac arrest will leave the hospital without major brain damage.59
Survivors receive antidysrhythmic agents and may also have an implantable cardioverter defibrillator (ICD) unit inserted.59–62 Prevention focuses on identification and treatment of high-risk cardiac patients (see “Implantable Cardioverter Defibrillator” and “Antidysrhythmic Drugs” in Chapter 13).
Heart Failure
Description and Etiology
The number of patients with heart failure is increasing in the United States. More than five million Americans have a diagnosis of heart failure, and about 550,000 new cases are diagnosed each year.63 This represents about 2.5% of the adult population. More than 282,000 people die of heart failure each year.1 Heart failure is primarily a disease of older persons; approximately 80% of patients hospitalized with heart failure are older than 65 years.63 Total inpatient and outpatient costs for heart failure treatment exceed $27.9 billion each year,63 with $2.9 billion required for heart failure drugs alone.63
Pathophysiology
Heart failure is a response to cardiac dysfunction, a condition in which the heart cannot pump blood at a volume required to meet the body’s needs. Any condition that impairs the ability of the ventricles to fill or eject blood can cause heart failure. CAD with resultant necrotic damage to the left ventricle is the underlying cause of heart failure in most patients. Other major conditions that lead to heart failure include valvular dysfunction, infection (myocarditis or endocarditis), cardiomyopathy, and uncontrolled hypertension.63 Hypertension is a precursor of heart failure in 75% of heart failure cases.1
Assessment and Diagnosis
Heart failure is typically classified using the New York Heart Association (NYHA) criteria. Patients are assigned into four groups, I through IV, depending on the degree of symptoms and the amount of patient effort required to elicit symptoms (Table 12-6). Research-based clinical guidelines suggest adding a second level of classification that emphasizes the progressive nature of heart failure through stages identified by increasing symptom distress and intensified clinical interventions (Figure 12-13).63 Heart failure can manifest in many different ways, depending on how far ventricular remodeling and dysfunction have advanced. Heart failure may be discovered because of a known clinical syndrome such as acute MI, or because of decreased exercise tolerance, fluid retention, or admission to the critical care unit for an unrelated condition.63 The clinical assessment signs diagnostic of fluid volume overload are jugular venous distention (JVD) and a third heart sound.63 The assessment procedures used to estimate JVD and auscultate a third heart sound are described in Chapter 11 (Box 11-1, Box 11-2; Figure 11-2, Figure 11-3). These clinical diagnostic skills, are also used to make the diagnosis of heart failure.63
TABLE 12-6
NEW YORK HEART ASSOCIATION FUNCTIONAL CLASSIFICATION OF HEART FAILURE
| CLASS | DEFINITION |
| I | Normal daily activity does not initiate symptoms. |
| II | Normal daily activities initiate onset of symptoms, but symptoms subside with rest. |
| III | Minimal activity initiates symptoms; patients are usually symptom-free at rest. |
| IV | Any type of activity initiates symptoms, and symptoms are present at rest. |
The first step in the diagnosis is to determine the underlying structural abnormality creating the ventricular dysfunction and symptoms. Various imaging tests are available to visualize cardiac anatomy, and laboratory tests are used to evaluate the impact of hormonal or electrolyte imbalance. The results of these tests permit the cardiology team to design a treatment plan to control symptoms and possibly correct the underlying cause. All patients do not have the same type of heart failure.
Left Ventricular Failure
Failure of the left ventricle is defined as a disturbance of the contractile function of the left ventricle, resulting in a low cardiac output state. This leads to vasoconstriction of the arterial bed that raises systemic vascular resistance (SVR), a condition also described as “high afterload,” and creates congestion and edema in the pulmonary circulation and alveoli. Clinical manifestations include decreased peripheral perfusion with weak or diminished pulses; cool, pale extremities; and in later stages, peripheral cyanosis (Table 12-7). Over time, with progression of the disease state the fluid accumulation behind the dysfunctional left ventricle elevates pulmonary pressures, contributes to pulmonary congestion and edema, and produces dysfunction of the right ventricle, resulting in failure of the right side of the heart.
TABLE 12-7
CLINICAL MANIFESTATIONS OF RIGHT- AND LEFT-SIDED HEART FAILURE
| LEFT VENTRICULAR FAILURE | RIGHT VENTRICULAR FAILURE | ||
| SIGNS | SYMPTOMS | SIGNS | SYMPTOMS |
| Tachypnea | Fatigue | Peripheral edema | Weakness |
| Tachycardia | Dyspnea | Hepatomegaly | Anorexia |
| Cough | Orthopnea | Splenomegaly | Indigestion |
| Bibasilar crackles | Paroxysmal nocturnal dyspnea | Hepatojugular reflux | Weight gain |
| Gallop rhythms (S3 and S4) | Nocturia | Ascites | Mental changes |
| Increased pulmonary artery pressures | Jugular venous distention | ||
| Hemoptysis | Increased central venous pressure | ||
| Cyanosis | Pulmonary hypertension | ||
| Pulmonary edema | |||
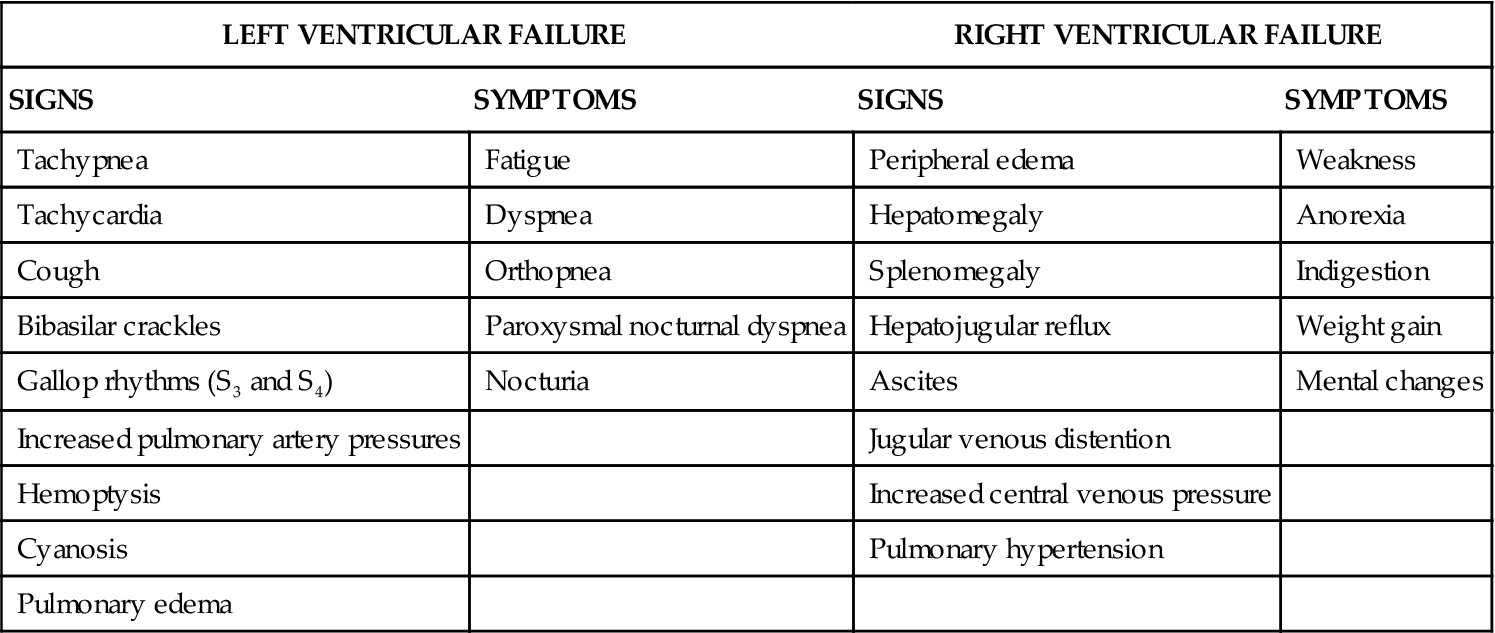
Right Ventricular Failure
Failure of the right side of the heart is defined as ineffective right ventricular contractile function. Pure failure of the right ventricle may result from an acute condition such as a pulmonary embolus or a right ventricular infarction, but it is most commonly caused by failure of the left side of the heart. The common manifestations of right ventricular failure are jugular venous distention, elevated central venous pressure (CVP), weakness, peripheral or sacral edema, hepatomegaly (enlarged liver), jaundice, and liver tenderness. Gastrointestinal symptoms include poor appetite, anorexia, nausea, and an uncomfortable feeling of fullness (see Table 12-7).
Heart Failure with Systolic Dysfunction
Systolic dysfunction describes an abnormality of the heart muscle that markedly decreases contractility during systole (ejection) and lessens the quantity of blood that can be pumped out of the heart. Patients with a diagnosis of systolic heart failure have signs and symptoms of heart failure combined with a below-normal ejection fraction. Left ventricular systolic dysfunction is the classic picture that most clinicians consider when thinking about heart failure. In addition to the signs and symptoms of left heart failure (described earlier), the patient has a low ejection fraction. There is some debate about how low the ejection fraction has to be to qualify as systolic heart failure, but the value is usually below 50%;64 some clinicians cite numbers below 45% or 40%.65,66 Symptoms of heart failure with systolic dysfunction include dyspnea, exercise intolerance, and fluid volume overload.
Heart Failure with Diastolic Dysfunction
Diastolic dysfunction describes an abnormality of the heart muscle that makes it unable to relax, stretch, or fill during diastole. The ejection fraction can be normal or abnormal (low), and the patient may be symptomatic or symptom-free.64,67,68 Half of patients with heart failure have diastolic muscle dysfunction.69 The principal causes are similar to systolic heart failure: CAD, myocardial ischemia, uncontrolled hypertension, and left ventricular hypertrophy.67 Some conditions that are known to markedly alter diastolic function include hypertrophic cardiomyopathy, restrictive cardiomyopathy, and infiltrative diseases such as amyloidosis and neoplastic infiltrate.
Systolic Versus Diastolic Dysfunction Heart Failure
It is impossible to determine whether a patient has systolic or diastolic heart failure from clinical assessment alone.64 Both types of heart failure produce similar signs and symptoms, and in most patients systolic and diastolic dysfunction coexist.63 The level of symptoms and quality of life varies between individuals, even when the ejection fraction and presumed cardiac dysfunction are the same.70 This may occur because most symptoms occur as a result of neurohormonal compensatory mechanisms (described later) rather than changes in cardiac output.
Doppler Echocardiography
The definitive diagnosis of the type of heart failure is often made using Doppler echocardiography. An echocardiogram performed at rest and during exercise (stress echo) permits visualization of heart wall movement during systole and diastole. Calculation of the ejection fraction can be determined using Doppler echocardiography or during a cardiac catheterization. These diagnostic tests also show when combined systolic and diastolic dysfunction coexist.64
The annual mortality rate for diastolic heart failure is 5% to 8%, markedly less than the 10% to 15% annual mortality for patients with systolic heart failure. To put this in perspective, age-matched controls without any heart failure have an annual mortality rate of just 1%.64,67
It is not possible to distinguish whether a patient has systolic or diastolic heart failure simply by looking at the medications he or she is prescribed. The same drugs are used to treat the two types of heart failure, although the underlying rationales may be different.65 For example, beta-blockers are used in diastolic heart failure to slow the heart rate, to prolong diastole to give more time for ventricular filling, and to modify the ventricular response to exercise, especially for patients who have a preserved ejection fraction.65 When beta-blockers are prescribed for treatment of systolic heart failure, the intent is to preserve long-term inotropic (contractile) function and prevent ventricular remodeling.65 Diuretics are used to treat both types of heart failure, although a smaller dosage is generally needed in diastolic heart failure.65 ACEIs and ARBs are also used to treat both types of heart failure. The medications used to treat heart failure are further discussed in Chapter 13 (see Table 13-19).
Acute Versus Chronic Heart Failure
Acute or chronic heart failure is determined by the rapidity with which the syndrome develops, the presence and activation of compensatory mechanisms, and the presence or absence of fluid accumulation in the interstitial space (see Figure 12-13). In clinical practice guidelines, the terms acute and chronic have replaced the older name of congestive heart failure (CHF), because not all heart failure involves pulmonary congestion.63 However, the descriptor CHF remains in common parlance in clinical practice.
Acute heart failure has a sudden onset, with no compensatory mechanisms. The patient may experience acute pulmonary edema, low cardiac output, or even cardiogenic shock. Patients with chronic heart failure are hypervolemic, have sodium and water retention, and have structural heart chamber changes such as dilation or hypertrophy.63
Chronic heart failure is ongoing, with symptoms that may be made tolerable by medication, diet, and a reduced activity level. The deterioration into acute heart failure can be precipitated by the onset of dysrhythmias, acute ischemia, sudden illness, or cessation of medications. This may necessitate admission to a critical care unit. Hypertension is the primary precursor of heart failure in women, whereas CAD, specifically acute MI, is the primary cause of heart failure in men.63
Neurohormonal Compensatory Mechanisms in Heart Failure
When the heart begins to fail and the cardiac output is no longer sufficient to meet the metabolic needs of the tissues, the body activates several major compensatory mechanisms: the sympathetic nervous system, the renin-angiotensin-aldosterone system (RAAS), and if hypertension is present, the development of ventricular hypertrophy (see Figure 12-13). This process ultimately reshapes the ventricle in a process described as ventricular remodeling. These pathophysiological processes and the pharmacological measures taken to limit ventricular remodeling are described in this section.
The sympathetic nervous system compensates for low cardiac output by increasing heart rate and blood pressure. As a result, levels of circulating catecholamines are increased, resulting in peripheral vasoconstriction. In addition to raising blood pressure and heart rate, catecholamines cause shunting of blood from nonvital organs, such as the skin, to vital organs, such as the heart and brain. This mechanism, although initially helpful, may become a negative factor if elevation of heart rate increases myocardial oxygen demand while shortening the amount of time for diastolic filling and coronary artery perfusion.
Activation of RAAS in heart failure promotes fluid retention.63,71 The RAAS is activated by low cardiac output that causes the hormone renin to be secreted by the kidneys. A physiological chain of events is then set in motion that leads to volume overload. The renin acts on angiotensinogen in the bloodstream and converts it to angiotensin I; when angiotensin passes through the lung tissues, it is activated by ACE, an enzyme that converts the angiotensin I to angiotensin II, a powerful vasoconstrictor that increases SVR, raises blood pressure, and increases the workload of the left ventricle; the increased SVR further lowers cardiac output. The mineralocorticoid hormone aldosterone is released from the adrenal glands and stimulates sodium retention by means of the distal tubules of the kidney. In response to the low cardiac output, the renal arterioles constrict, decrease glomerular filtration, and increase reabsorption of sodium from the proximal and distal tubules. To break the RAAS cycle of fluid retention in heart failure, two types of drugs are prescribed to interrupt the steps. To inhibit the conversion of angiotensin I to angiotensin II, an ACEI is prescribed (see Table 13-19 in Chapter 13). These agents prevent arterial vasoconstriction, decrease blood pressure and SVR, and decrease the amount of ventricular remodeling that often occurs with heart failure. A drug that inhibits angiotensin II directly may be prescribed instead. The drugs in this category are ARBs.74 Aldactone (spironolactone) is a drug from a different category that is also prescribed to break the RAAS cycle. Aldactone is a mineralocorticoid receptor antagonist that inhibits (blocks) the retention of sodium from the distal tubules of the kidney.71–73 Figure 12-14 shows the mechanism of action by which these drugs act on the RAAS.
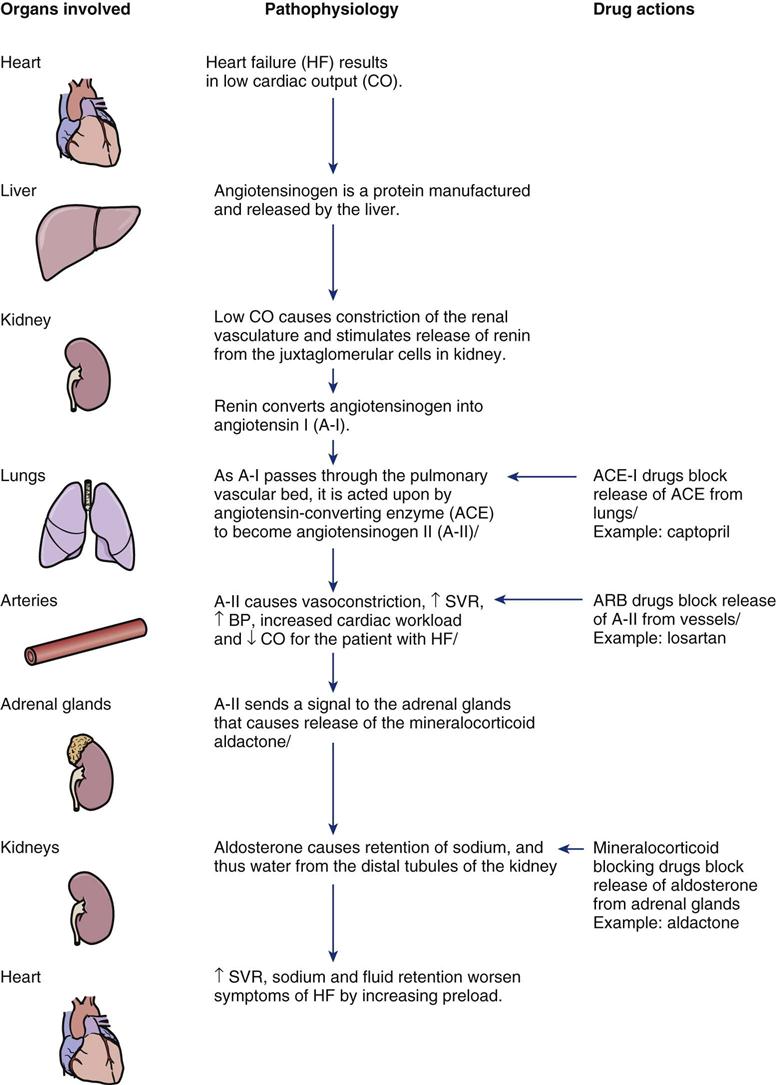
Ventricular hypertrophy is the final compensatory mechanism. It is also strongly associated with preexisting hypertension. Because myocardial hypertrophy increases the force of contraction, hypertrophy helps the ventricle overcome an increase in afterload. When this mechanism is no longer efficient for the ventricle, it will remodel by dilation.
Ventricular remodeling occurs as a result of the previously described mechanisms. The shape of the ventricle is changed, or is remodeled, to resemble a round bowl. A dilated ventricle has poor contractility and is enlarged without hypertrophy. Research trial evidence indicates that synergistic use of drugs from different categories—ACEI or ARB, Aldactone, plus beta-blockade—can halt or reduce the progression of heart failure remodeling.72,74,75
Pulmonary Complications of Heart Failure
The respiratory manifestations of acute heart failure result from tissue hypoperfusion and organ congestion and are progressive. The severity of respiratory manifestations progresses as heart failure worsens. Initially, symptoms of breathlessness appear only with exertion, but eventually symptoms also occur at rest.63
Shortness of Breath in Heart Failure
The patient experiences the feeling of shortness of breath first with exertion, but as heart failure worsens, symptoms are also present at rest. A diagnostic blood test is available to assist clinicians in differentiating whether a patient’s shortness of breath is caused by cardiac failure or by pulmonary complications.
BNP and NT-proBNP: Natriuretic peptides are released from the cardiac ventricles in response to increased wall tension. Heart failure increases left ventricular wall tension because of the excess preload in the ventricles causing increased wall stretch. The peptides are measured as brain natriuretic peptide (BNP) and N-terminal proB type natriuretic peptide (NT-proBNP).63
When the BNP blood level is greater than 100 pg/mL, the dyspnea is more likely to be related to cardiac rather than pulmonary failure.76–78 A BNP result greater than 400 is considered indicative of heart failure. The more severe the heart failure, the higher the BNP test result.79 If the patient has concomitant kidney failure, the BNP clinical diagnostic cut-point to diagnose heart failure rises to greater than 200 pg/mL.80 See Chapter 11, Figure 11-48 for more information on using BNP to diagnose heart failure.
Breathlessness in heart failure is described by the following terms:
Pulmonary Edema in Heart Failure
The alveolus is the primary site of gas exchange (Figure 12-15, A). Pulmonary edema, or protein-laden fluid in the alveoli, inhibits gas exchange by impairing the diffusion pathway between the alveolus and the capillary. It is caused by increased left atrial and ventricular pressures and results in an excessive accumulation of serous or serosanguineous fluid in the interstitial spaces and alveoli of the lungs. The formation of pulmonary edema has two stages. The first stage is not as severe and is characterized by interstitial edema, engorgement of the perivascular and peribronchial spaces, and increased lymphatic flow (see Figure 12-15, B). The later stage is characterized by alveolar edema resulting from fluid moving into the alveoli from the interstitium (see Figure 12-15, C). Eventually, blood plasma moves into the alveoli faster than the lymphatic system can clear it, interfering with diffusion of oxygen, depressing the arterial partial pressure of oxygen (PaO2), and leading to tissue hypoxia (see Figure 12-15, D).
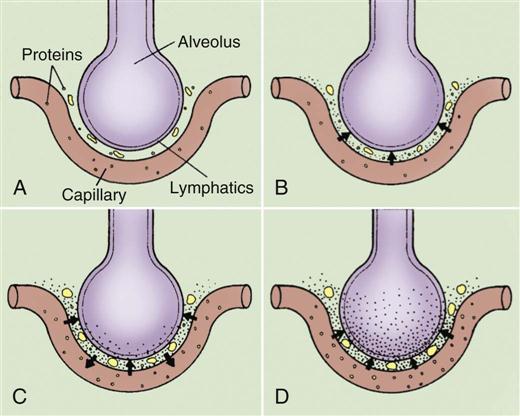
A, Normal relationship. B, Increased pulmonary capillary hydrostatic pressure causes fluid to move from the vascular space into the pulmonary interstitial space. C, Lymphatic flow increases in an attempt to pull fluid back into the vascular or lymphatic space. D, Failure of lymphatic flow and worsening of left-sided heart failure results in further movement of fluid into the interstitial space and the alveoli.
Heart failure patients in pulmonary edema are extremely breathless and anxious and have a sensation of suffocation. They expectorate pink, frothy liquid and feel as if they are drowning. They may sit bolt upright, gasp for breath, or thrash about. The respiratory rate is elevated, and accessory muscles of ventilation are used, with nasal flaring and bulging neck muscles. Respirations are characterized by loud inspiratory and expiratory gurgling sounds. Diaphoresis is profuse, and the skin is cold, ashen, and sometimes cyanotic, reflecting low cardiac output, increased sympathetic stimulation, peripheral vasoconstriction, and desaturation of arterial blood.
Arterial Blood Gases in Pulmonary Edema
Arterial blood gas values are variable. In the early stage of pulmonary edema, respiratory alkalosis may be present because of hyperventilation, which eliminates carbon dioxide. As the pulmonary edema progresses and gas exchange becomes impaired, acidosis (pH <7.35) and hypoxemia ensue. A chest radiograph usually confirms an enlarged cardiac silhouette, pulmonary venous congestion, and interstitial edema.
Dysrhythmias and Heart Failure
A ventricular ejection fraction below 30% and the presence of NYHA class III or IV heart failure are strongly associated with ventricular dysrhythmias and an increased risk of death.63,81–84 Because sustained VT or VF initiates sudden cardiac death, high-risk patients with severe heart failure are prescribed antidysrhythmic drugs and have an ICD inserted.82–84
Many patients with heart failure also have atrial fibrillation. Digoxin is frequently prescribed in atrial fibrillation to control ventricular heart rate. Digoxin does not prolong life but decreases symptoms and hospitalizations associated with heart failure. Digoxin may also work synergistically with specific beta-blockers (e.g., carvedilol) and make the symptoms more tolerable for patients with severe heart failure.83,85
Medical Management
The goals of the medical management of heart failure are to relieve heart failure symptoms, enhance cardiac performance, and correct known precipitating causes of acute heart failure.
Relief of Symptoms and Enhancement of Cardiac Performance
In the acute phase of advanced heart failure, the patient may have a pulmonary artery catheter in place so that left ventricular function can be followed closely. Control of symptoms involves management of fluid overload and improvement of cardiac output by decreasing SVR and increasing contractility. Diuretics are administered to decrease preload and to eliminate excess fluid from the body.73 If pulmonary edema develops, additional diuretics are used. Morphine is given to facilitate peripheral dilation and decrease anxiety. Afterload is decreased by vasodilators, such as sodium nitroprusside (Nipride) and nitroglycerin. Nitrates are used to decrease preload and vasodilate the coronary arteries if CAD is an underlying cause of the acute heart failure. For some patients, an intraaortic balloon pump (IABP) is temporarily required.86 Contractility is initially increased by continuous infusion of positive inotropic drugs (dopamine) or by combination inodilators such as dobutamine or milrinone. Nesiritide (Natrecor) or intravenous BNP is indicated for the relief of patients with acutely decompensated heart failure who have dyspnea at rest. Nesiritide lowers pulmonary artery pressures and wedge pressure, which decreases symptoms of dyspnea.87,88
After the acute heart failure is controlled, the patient is weaned off intravenous medications, which are gradually replaced by oral agents. Before the transition out of the critical care unit, the heart failure patient will receive ACEIs to inhibit left ventricular chamber remodeling and to slow left ventricular dilation.63,71,74 If the patient does not tolerate ACEIs, ARBs may be substituted.74 Low dosage beta-blockers such as carvedilol may also be prescribed, although strict surveillance is required to anticipate and avoid untoward negative inotropic effects.83,89 Digoxin may be added to the regimen, especially if the person has concomitant atrial fibrillation.85
Nonpharmacological interventions that are increasingly used include cardiac resynchronization therapy (CRT).90 CRT is biventricular pacing where the right and left ventricles each have a pacing lead in contact with the myocardium. The right ventricular lead is inside the right ventricle, and the left ventricular lead is positioned into a left wall tributary of the coronary sinus vein.90,91 In newer permanent pacemaker models, the right and left ventricular leads are paced to synchronize the ventricles and improve heart failure symptoms.
Correction of Precipitating Causes
After symptoms of heart failure are controlled, diagnostic studies such as cardiac catheterization, echocardiography, and thallium scanning are undertaken to uncover the cause of the heart failure and tailor long-term management to treat the cause. Some structural problems such as valvular disease may be amenable to surgical correction.
Palliative Care for End-Stage Heart Failure
In 2004 a consensus statement on palliative and supportive care in advanced heart failure was published.92 Because heart failure is a progressive disease, some patients will not recover.63 At some point, many NYHA class-IV heart failure patients will become candidates for palliative care.92 The primary aim of palliative care is symptom management. Symptom-management strategies are deployed to emphasize relief from suffering. Fundamental to all symptom-management strategies for heart failure is the optimization of medications according to current guidelines. The most common symptoms of advanced heart failure are dyspnea, pain, and fatigue.92
Nursing Management
Nursing management of the patient with heart failure incorporates a variety of nursing diagnoses (see Nursing Diagnosis Priorities box on Acute Heart Failure). Nursing priorities focus on (1) optimizing cardiopulmonary function, (2) promoting comfort and emotional support, (3) monitoring the effectiveness of pharmacological therapy, (4) providing adequate nutritional intake, and (5) providing patient education.
Optimizing Cardiopulmonary Function
The patient’s ECG is evaluated for any dysrhythmias that may be present or may develop as a result of drug toxicity or electrolyte imbalance. Patients with heart failure are prone to digoxin toxicity because of decreased renal perfusion; they are also prone to electrolyte imbalances. Breath sounds are auscultated frequently to determine the adequacy of respiratory effort and to assess for onset or worsening of pulmonary congestion. Oxygen through a nasal cannula is administered to relieve dyspnea. Diuretics or vasodilators are used to decrease excessive preload and afterload.75,88 If the patient is not hypotensive, morphine may be administered to decrease hyperventilation and anxiety. If the patient’s ventilatory status worsens, the nurse must be prepared for endotracheal intubation and mechanical ventilation. Obtaining daily weights is important until the weight stabilizes at a “dry” weight. Generally, the daily weight is used in fluid management and a weekly weight is optimally used for tracking body weight (e.g., muscle, fat).
Promoting Comfort and Emotional Support
During periods of breathlessness, activity must be restricted. Bed rest is usually prescribed for the patient, who is positioned with the head of the bed elevated to allow for maximal lung expansion. The arms can be supported on pillows so that no undue stress is placed on the shoulder muscles. The legs may be placed in a dependent position to encourage venous pooling, thereby decreasing venous return. Rest periods must be carefully planned and adhered to while independence within the patient’s activity prescription is fostered. Vital signs are recorded before an activity is begun and after it is completed. Signs of activity intolerance, such as dyspnea, fatigue, sustained increase in pulse, and onset of dysrhythmias, are documented and reported to the physician. Activity is gradually increased according to the patient’s tolerance. Skin breakdown is a risk because of the combination of bed rest, inadequate nutrition, peripheral edema, and decreased perfusion to the skin and subcutaneous tissue. Frequent position changes and mobilization can help to provide comfort and prevent this complication.
Monitoring the Effects of Pharmacological Therapy
Patients experiencing acute heart failure require aggressive pharmacological therapy.82,88,93,94 The nurse must know the action, side effects, therapeutic levels, and toxic effects of the diuretics and vasodilators used to decrease preload, the positive inotropic agents used to increase ventricular contractility, the vasodilators used to decrease afterload, and any antidysrhythmics used to control heart rate and prevent dysrhythmias. The patient’s hemodynamic response to these agents is closely monitored. Fluid intake and output balances are tabulated daily or even hourly in the critical care unit.
Providing Adequate Nutritional Intake
Patients experiencing heart failure often have decreased appetite and nausea, and small, frequent meals may be more appropriate than the standard three large meals. Food must be as tasty as possible; favorite foods and food from home may be incorporated into the diet as long as the foods are compatible with nutritional restrictions such as low levels of sodium to decrease the risk of fluid retention. Each patient must be assessed for nutritional imbalance individually. Some people with heart failure are well nourished, some are obese, and some are malnourished before they enter the hospital.
Providing Patient Education
The nurse assesses the patient’s and family’s understanding of the pathophysiology and individual risk-factor profile for heart failure.95 Primary topics of education include the importance of a low-salt diet, daily weight, fluid restrictions, and written information about the multiple medications used to control the symptoms of heart failure.63,96 Many patients with a diagnosis of heart failure also require education about lifestyle changes such as smoking cessation, weight loss, energy conservation, and how to incorporate exercise and sodium restriction into their daily lives.74,97,98 Achieving the optimal outcomes for the patient with heart failure requires contributions from a team of educated health care clinicians.63,74,97,98 Collaborative multidisciplinary goals, developed from clinical practice guidelines for management of the patient with symptoms of heart failure, are listed in the Evidence-Based Collaborative Practice box on Heart Failure.
Cardiomyopathy
Description and Etiology
Cardiomyopathy is a disease of the heart muscle: cardio (heart), myo (muscle), and pathy (pathology). Cardiomyopathies are classified on the basis of structural abnormalities and, if known, genotype. The cardiomyopathic categories are hypertrophic, restrictive, and dilated, as illustrated in Figure 12-16.
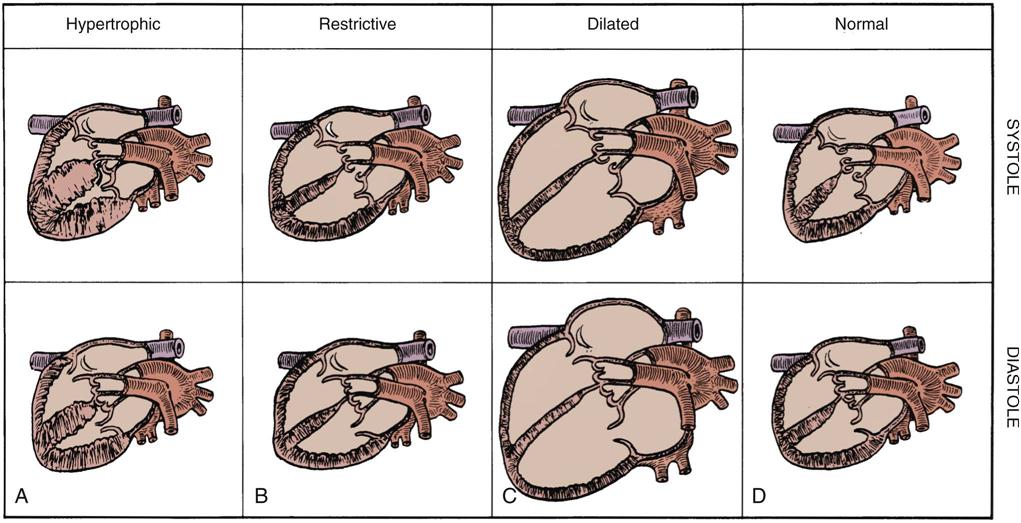
A, Hypertrophic. B, Restrictive. C, Dilated. D, Normal.
Hypertrophic Obstructive Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a genetically inherited disease that affects the myocardial sarcomere.99–102 As HCM progresses, the left ventricle becomes stiff, noncompliant, and hypertrophied, sometimes in an asymmetric fashion.99 HCM occurs in two forms. A well-known, but less frequent, manifestation is a stiff, noncompliant myocardial muscle with left ventricular hypertrophy and bizarre cellular hypertrophy of the upper ventricular septum. This left ventricular septal hypertrophy obstructs outflow through the aortic valve, especially during exercise (see Figure 12-16, A). It also pulls the papillary muscle out of alignment, causing mitral regurgitation. This form of HCM was previously known as idiopathic hypertrophic subaortic stenosis (IHSS); however, because IHSS does not describe all patients with hypertrophied hearts, the more general term of HCM is now used.99 Other patients with HCM have generalized left ventricular hypertrophy, but the septum is not more enlarged than the rest of the myocardium.99 HCM causes significant diastolic dysfunction because the muscle-bound, stiff, noncompliant heart muscle cannot fill adequately during diastole.
Two advances in diagnostic medicine have propelled understanding of the differences between these two forms of HCM. Two-dimensional transthoracic echocardiography (TTE) is often useful as the first diagnostic test to identify HCM.99 TTE enables visualization of the septal anatomy, septal movement, and ventricular wall thickness and motion. The second advance is diagnostic genetics. Genetic testing for HCM usually is performed at a center with expertise in this area.99 HCM is inherited as an autosomal dominant trait, and the clinical expression is caused by mutations in any of one of 10 genes. Each different gene encodes different protein components of the myocardial sarcomere.99 Genetic analysis is an expanding research area that will help clarify diagnosis and treatment options for this cardiomyopathy.101
Symptoms are similar to those seen with heart failure plus the symptoms of myocardial ischemia, supraventricular tachycardia (SVT), VT syncope, and stroke. Symptoms usually are more intense with physical exercise, especially in the obstructive form of HCM, in which the aortic outflow tract is obstructed by the enlarged left ventricular septum. Because there is a known association between HCM and SCD, limitation of physical activity may be recommended. Causes of SCD are thought to stem from ventricular dysrhythmias and atrial fibrillation.99 Episodes of paroxysmal atrial fibrillation occur in 20% to 25% of HCM patients.99 The atrial dysrhythmias are related to increased age and atrial enlargement.99 Pharmacological management includes beta-blockers to decrease left ventricular workload, medications to control and prevent atrial and ventricular dysrhythmias, anticoagulation if atrial fibrillation or left ventricular thrombi are present, and drugs to manage heart failure. Interventional procedures include insertion of an ICD to decrease the risk of SCD, and percutaneous alcohol ablation of the intraventricular septum to decrease the size of the septal wall.99,102 Surgical procedures such as septal myectomy and mitral valve replacement are options used less frequently than in the past.99
Dilated Cardiomyopathy
Dilated cardiomyopathy is characterized by gross dilation of both ventricles without muscle hypertrophy (see Figure 12-16, C). There are several distinct causes of dilated cardiomyopathy. It is estimated that 30% of patients with dilated cardiomyopathy may have a genetic cause.63
Ischemic Dilated Cardiomyopathy
Ischemic dilated cardiomyopathy results from repeated myocardial injury or infarction caused by the sequelae of CAD. It is the most common cause of dilated cardiomyopathy in the United States. Signs and symptoms of systolic heart failure and a low ejection fraction are present.
Familial Dilated Cardiomyopathy
When the cause of the dilated cardiomyopathy is unknown, it is called idiopathic. In some cases, the occurrence is linked to genetic inheritance. Scientific advances in molecular genetics permit detailed studies of families with a high incidence of dilated cardiomyopathy. It is estimated that 10% to 50% of familial idiopathic dilated cardiomyopathy cases reflect genetic transmission.103–105 In affected families, various genetic mutations occur in the gene that codes for the sarcomere contractile protein in the heart. The heritable trait can be expressed as an autosomal dominant or recessive inheritance pattern.103–105 Preliminary research indicates that the genetic picture is highly individual for different family groups, even if the clinical picture appears similar.103–105
Other Causes of Dilated Cardiomyopathy
There are many other nonischemic, nongenetic known causes of dilated cardiomyopathy. Injury can be caused by valvular heart dysfunction that has placed extreme pressure or volume on the chambers, and viral or bacterial infections such as myocarditis can lead to inflammatory changes that permanently remodel the heart.106 Other noncardiac causes include infiltration by systemic collagens as in amyloidosis or sarcoidosis.107
In dilated cardiomyopathy, the myocardial muscle fibers contract poorly, resulting in global left ventricular dysfunction, low cardiac output, atrial and ventricular dysrhythmias, blood pooling that leads to ventricular thrombi and embolic episodes, refractory heart failure, and premature death. The goals of the medical management of dilated cardiomyopathy are similar to those for systolic heart failure: improvement of pump function, removal of excess fluid, control of heart failure symptoms, anticipation and management of complications, and prevention of SCD.
Restrictive Cardiomyopathy
Restrictive cardiomyopathy is the least commonly encountered cardiomyopathy in industrialized societies (see Figure 12-16, B). As with the other cardiomyopathies, this form can be idiopathic or can have a known cause.108,109 Restrictive cardiomyopathy results in ventricular wall rigidity as a consequence of myocardial fibrosis. The overall effect is the diastolic inhibition of ventricular filling. Diastolic heart failure, low cardiac output, dyspnea, orthopnea, and liver engorgement are the most common clinical manifestations of restrictive cardiomyopathy. Medical management includes beta-blockers to slow the heart rate and allow more time for ventricular filling, diuretics to remove excess fluid, and a low-sodium diet.
Nursing Management
Nursing management of the patient with cardiomyopathy incorporates a variety of nursing diagnoses related to the symptoms of heart failure. These nursing diagnoses are reviewed in the Nursing Diagnosis Priorities box on Cardiomyopathy. Nursing priorities are individualized according to the type of cardiomyopathy and heart failure and focus on (1) achieving a stable fluid balance, (2) monitoring the effects of pharmacological therapy, (3) safely increasing mobility, and (4) providing patient and family education. As with heart failure, a collaborative team of compassionate, knowledgeable professionals is required to provide effective care and education for these challenging patients.96
Valvular Heart Disease
Description and Etiology
Valvular heart disease describes structural and functional abnormalities of single or multiple cardiac valves. The result is an alteration in blood flow across the valve. The two types of valvular lesions are stenotic and regurgitant. These are described with reference to the specific cardiac valves involved.
Usually, if a person is admitted to the critical care unit with valve disease, he or she is experiencing acute heart failure or is being admitted for cardiac surgical valvular replacement. In the past in the United States, most valvular lesions were rheumatic in origin, and damage was a direct result of group A β-hemolytic streptococcal pharyngitis. As a result of aggressive treatment of “strep throat,” this has become a rare problem, and older patients now are more likely to be seen with symptoms of heart failure and degenerative valve changes. These changes may be described as myxomatous leaflet degeneration or annular calcification.110,111
Pathophysiology
Mitral Valve Stenosis
Mitral valve stenosis describes a progressive narrowing of the mitral valve orifice. Primary cause is rheumatic endocarditis with rare occurrences related to congenital malformations. Mitral stenosis occurs in twice as many women as men.111 Symptoms occur when the normal valve size is reduced to 2 cm2 or less. Symptoms occur at rest when the valve area is reduced below 1 cm2.111 Narrowing is caused by aging valve tissue or by acute rheumatic valvulitis (Table 12-8, A). The diffuse valve leaflets fibrose and fuse, reducing mobility and thickening the chordae tendineae. As a result, the mitral valve can no longer open or close passively in response to left atrial and ventricular pressure changes. Blood flow across the valve is impeded. Mitral stenosis increases the risk of developing atrial fibrillation because of the high pressures in the left atrium that will stimulate left atrial remodeling and enlargement. Development of atrial fibrillation will significantly increase symptoms and may increase the need for surgical replacement of the valve.
TABLE 12-8
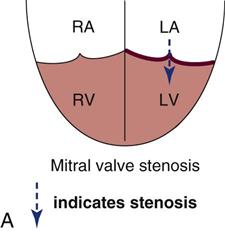
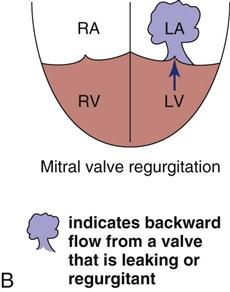
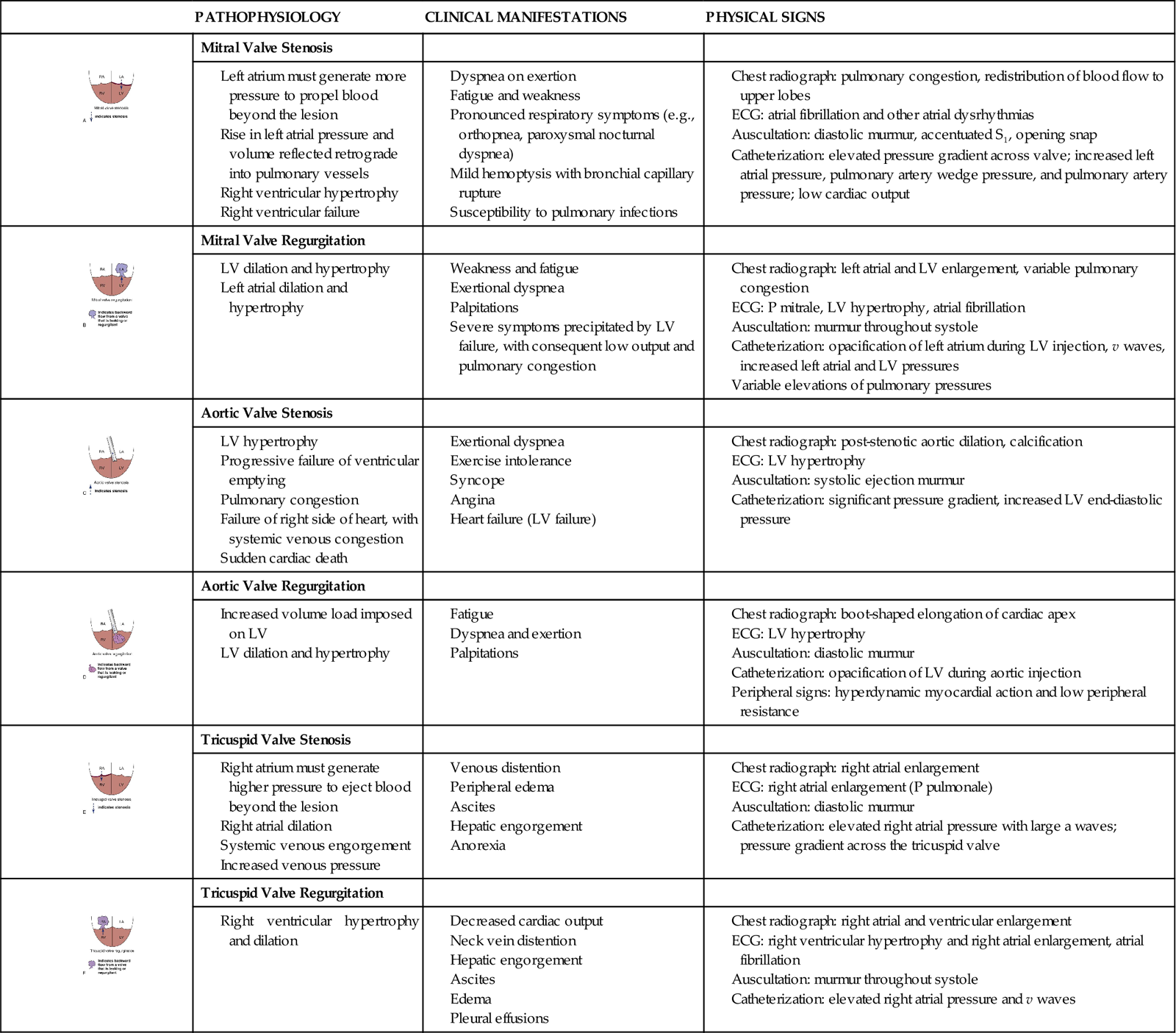
Mitral Valve Regurgitation
Mitral valve regurgitation may result from rheumatic disease, aging of the valve, endocarditis, collagen vascular disease, or papillary muscle dysfunction111 (see Table 12-8, B). In mitral regurgitation, the valve annulus, leaflets, chordae tendineae, and papillary muscles may all be dysfunctional, or the dysfunction may be isolated to just one component of the valve. Mitral valve regurgitation results in retrograde flow of blood into the left atrium with each ventricular contraction. It is always described as chronic or acute because of the very different impact on the left-sided chambers.
With chronic mitral valve regurgitation, the left atrium has dilated to accommodate the additional regurgitant volume, whereas the left ventricle has hypertrophied (increased muscle) to maintain an adequate stroke volume and cardiac output. In contrast, acute mitral valve regurgitation is precipitated by chordae tendineae or papillary muscle rupture resulting from an acute MI or infectious endocarditis.111 This is a medical emergency. The left atrium cannot accommodate the sudden increase in volume and pressure, and use of an IABP and inotropic drug support is often required to increase forward output and reduce pulmonary congestion. After the patient’s condition has stabilized, surgical replacement or repair of the incompetent valve is performed.111
Aortic Valve Stenosis
Aortic valve stenosis describes a narrowing of the aortic valve area. It can result from aging, rheumatic valvulitis, or deterioration of a congenital bicuspid valve111 (see Table 12-8, C). The pathological hallmarks are inflammation, fibrous valvular thickening, and tissue calcification. When the aortic valvular opening is reduced to less than 1.5 cm2, the condition is classified as mild. Cardiac catheterization or Doppler echocardiography can identify a gradient of less than 25 mm Hg across the valve.112 The gradient represents the difference in systolic pressure between the left ventricle and the aorta. A significant pressure difference is a diagnostic hallmark of valvular stenosis. If the valve orifice has narrowed to 1 cm2 or less, the gradient will be greater than 40 mm Hg, and the diagnosis will be upgraded to severe aortic valve stenosis.111 The impedance of left ventricular ejection into the aorta results in increased left ventricular systolic pressure, left ventricular hypertrophy, and eventually left ventricular dilation. When symptoms such as angina, dyspnea, syncope, and other indicators of heart failure develop, it is critical to intervene to prevent further damage to the left ventricle. Aortic valve replacement is usually indicated. Balloon valvotomy (dilation) may be an option for carefully selected patients with aortic stenosis without calcification, or for patients whose mortality risk during surgical aortic valve replacement is unacceptably high.111,112
Aortic Valve Regurgitation
Aortic regurgitation, also known as aortic insufficiency, can occur as a result of rheumatic fever, systemic hypertension, Marfan syndrome, syphilis, rheumatoid arthritis, aging valve tissue, or discrete subaortic stenosis (see Table 12-8, D). Aortic valve incompetence results in a reflux of blood back into the left ventricle during ventricular diastole. To accommodate this extra volume, the left ventricle initially dilates and then hypertrophies in an attempt to empty more completely and to meet the needs of the peripheral circulation. Aortic valve replacement is recommended for symptomatic patients with well-preserved or moderate left ventricular dysfunction.111
Tricuspid Valve Stenosis
Tricuspid valve stenosis is rarely an isolated lesion (see Table 12-8, E). It often occurs in conjunction with mitral or aortic disease. Its origin most often is rheumatic fever or a complication of intravenous drug abuse and resultant endocarditis.111 Tricuspid valve stenosis increases the pressure work of the usually low-pressure right atrium, resulting in right atrial hypertrophy. The right atrium dilates in an attempt to accommodate the residual right atrial volume and the incoming venous return. As a result, systemic venous congestion occurs—the consequences of which include jugular venous congestion, liver failure, hepatomegaly, ascites, and peripheral edema.
Tricuspid Valve Regurgitation
Tricuspid valve regurgitation usually results from advanced failure of the left side of the heart that eventually affects the right side of the heart, severe pulmonary hypertension, or as a complication of infectious endocarditis111 (see Table 12-8, F).
Pulmonic Valve Disease
Pulmonary valve disease is not a common disorder in adults. It is most often related to congenital anomalies and produces failure of the right side of the heart.
Mixed Valvular Lesions
Many persons have mixed valvular lesions as an element of stenosis and regurgitation. Mixed lesions can accentuate the severity of a condition. For example, when combined, aortic stenosis and aortic regurgitation increase left ventricular volume and pressure and thereby multiply the degree of left ventricular work.
Medical Management
Management of valvular disorders includes pharmacological therapy to control symptoms of heart failure and then cardiac surgical repair or replacement of the affected valve.111
Nursing Management
Nursing management of the patient with valvular disease incorporates a variety of nursing diagnoses (see the Nursing Diagnosis Priorities box on Valvular Heart Disease). Nursing priorities are focused on (1) maintaining adequate cardiac output, (2) optimizing fluid balance, and (3) providing patient education.
Maintaining Cardiac Output
Low cardiac output is a common finding in patients with valvular heart disease. It can occur because of decreased forward flow through a stenotic valve, because of bidirectional flow across an incompetent valve, or because of associated heart failure. Vital signs and the effect of positive inotropic and afterload-reducing agents are assessed and documented. If the patient has hemodynamic catheters inserted, cardiac output and hemodynamic parameters are measured and evaluated. Patient care activities are carefully planned to provide adequate rest periods to prevent fatigue.
Optimizing Fluid Balance
Fluid status is evaluated by auscultation of breath sounds for crackles, heart sounds for presence of an S3, daily weights to trend a “sudden weight gain,” and presence of peripheral edema. The appearance of pulmonary crackles or an S3 heart sound confirms volume overload. The jugular vein is assessed for signs of increased distention. Diuretics and vasodilators are administered to counteract excess fluid retention. The patient is weighed daily, and fluid intake and output are monitored and recorded.
Providing Patient Education
Education for the patient with acute or chronic heart failure caused by valvular dysfunction includes: (1) information related to diet, (2) fluid restrictions, (3) the actions and side effects of heart failure medications, (4) the need for prophylactic antibiotics before undergoing any invasive procedures such as dental work, and (5) when to call the health care provider to report a negative change in cardiac symptoms. Many patients also require information about valvular heart surgery. Achieving the optimal outcomes for the patient with valve disease requires contributions from a team of educated health care clinicians. Collaborative multidisciplinary priorities are listed in the box Evidence-Based Collaborative Practice: Valvular Heart Disease. The heart valve replacement section in Chapter 13 provides more information on surgical management.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
Objectives
• Describe the etiology and pathophysiology of atherosclerotic coronary artery disease.
• Identify the pathophysiology and clinical manifestations of acute heart failure.
• Discuss the nursing priorities for managing a patient with an acute cardiovascular disorder.
Be sure to check out the bonus material, including free self-assessment exercises, on the Evolve web site at http://evolve.elsevier.com/Urden/priorities/.
Cardiovascular disease remains the leading cause of mortality in the United States. The estimated direct and indirect national cost of cardiovascular disease in 2010 was $503.2 billion.1 Cardiovascular diseases are the leading cause of death for women and men. There are almost 80 million adults in the United States living with cardiovascular disease. Cardiovascular disease accounts for 1 of every 2.9 deaths in the United States.1 In terms of disability, the number of disability-adjusted life-years (DALYs) attributable to cardiovascular disease on a global basis is projected to double by the year 2020. An understanding of the pathology of cardiovascular disease processes and clinical management allows the critical care nurse to accurately anticipate and plan interventions. This chapter focuses on cardiac disorders commonly seen in the critical care environment.
Coronary Artery Disease
Description and Etiology
The biggest contributor to cardiovascular-related morbidity and mortality is coronary artery disease (CAD). Atherosclerosis is a progressive disease that affects arteries throughout the body. In the heart, atherosclerotic changes are clinically known as CAD. This disease process is also known by the term coronary heart disease (CHD) because other heart structures ultimately become involved in the disease process. The atherosclerotic vascular changes that lead to CAD may begin in childhood.2 Research and epidemiological data collected during the past 50 years have demonstrated a strong association between specific risk factors and the development of CAD.1 These risk factors are further delineated as nonmodifiable and modifiable coronary risk factors (Box 12-1).
Risk Factors for Coronary Artery Disease
Age, Gender, and Race
The severe effects of CAD occur as a person ages. In general, CAD symptoms are seen in middle and old age.1 Traditionally, CAD has been regarded as a male disease, but it is increasingly obvious that in modern society it affects both genders.1 The average age for a person having a first heart attack is 64.5 years for men and 70.3 years for women.1 Starting at age 75 years, the prevalence of cardiovascular disease is higher among women than men.2,3 CAD rates for postmenopausal women are two to three times higher than those for premenopausal women of the same age.1 People of color and multiracial populations of both genders have higher CAD mortality rates than do white populations of similar socioeconomic status.1
Family History
A positive family history is one in which a close blood relative has had a myocardial infarction (MI) or stroke before the age of 60. This family history suggests a genetic or lifestyle predisposition to the development of CAD. Individuals with a family history had a 50% greater risk of having an acute MI in the INTERHEART study.4 This was a large, international, standardized case-control study of similar cohorts in 52 countries that was designed to examine the importance of risk factors for CAD on a worldwide basis.4
Hyperlipidemia
Hyperlipidemia is a leading factor responsible for severe atherosclerosis and the development of CAD. Determining total serum cholesterol and triglyceride levels is a helpful start in the evaluation process.3,5 A lipid panel blood test can measure the following values:
• High-density lipoprotein (HDL) cholesterol
• Low-density lipoprotein (LDL) cholesterol
Treatment of hyperlipidemia has advanced beyond the concept of lowering total cholesterol to treatment of specific lipoprotein abnormalities.3,6–8 The target levels for specific serum lipids are listed in Table 12-1.
TABLE 12-1
LIPID GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE
| LIPID | TARGET VALUE* (mg/dl) |
| Total cholesterol | <200 |
| HDL cholesterol | |
| Men | >40 |
| Women | >50 |
| LDL cholesterol | |
| Very high risk | <70 |
| High risk | <100 |
| Low risk | <130 |
| VLDL cholesterol | <30 |
| Triglycerides | <150 |
HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.
*Values outside the target range increase the risk for coronary artery disease.
Total Cholesterol
The total cholesterol is the sum of the HDL, LDL, and VLDL cholesterol in the bloodstream. It is used as a starting point for lipid testing. A total cholesterol level higher than 200 mg/dL is an indication to investigate the lipid profile and other risk factors for CAD. More than 102 million American adults (46.8% of the adult population) have a total blood cholesterol level at or above 200 mg/dL.1
High-Density Lipoprotein Cholesterol
HDL cholesterol is frequently described as the “good cholesterol” because higher serum levels exert a protective effect against acute atherosclerotic events. All the reasons are not completely understood, but one recognized physiological effect is the ability of HDL to promote the efflux of cholesterol from cells. This process may minimize the accumulation of foam cells in the artery wall and decrease the risk of developing atherosclerosis.9 High HDL cholesterol levels confer antiinflammatory and antioxidant benefits on the arterial wall.9 In contrast, a low HDL cholesterol level is an independent risk factor for the development of CAD and other atherosclerotic conditions. HDL cholesterol is generally higher in women, and levels can be raised by physical exercise and by smoking cessation. In patients with low HDL cholesterol levels, when lifestyle changes are ineffective, the HDL level can be raised by drugs such as extended-release nicotinic acid (niacin) and fibrates. In 2006 16.2% of the adult U.S. population had HDL cholesterol levels below 40 mg/dL.1
Low-Density Lipoprotein Cholesterol
LDL cholesterol is usually described as the “bad cholesterol” because high levels are associated with an increased risk of acute coronary syndrome, stroke, and peripheral arterial disease (PAD). High LDL levels initiate the atherosclerotic process by infiltrating the vessel wall and binding to the matrix of cells beneath the endothelium.9 LDL cholesterol also exerts an inflammatory effect on the arterial vessel wall.9 A high LDL cholesterol level is initially managed by nonpharmacological lifestyle changes such as weight loss, smoking cessation, low-fat diet, physical exercise, and attainment of a normal body size as measured by the body mass index (BMI). If lifestyle changes are insufficient to reduce the LDL cholesterol level in the bloodstream, the drug category of choice is a statin. Numerous research studies have conclusively demonstrated that lowering the LDL cholesterol with statins for primary or secondary prevention is highly effective in lowering mortality due to CAD.1,3 Therapy should be tailored to treat the individual cardiovascular risk profile. The target LDL cholesterol is determined according to the individual’s risk profile as described in Table 12-1. Patients at highest risk are advised to maintain LDL cholesterol below 70 mg/dL; those at high risk, a level below 100 mg/dL; and those at low risk, LDL below 130 mg/dL. Controversy exists about whether the tiered LDL goals in the current guidelines are sufficiently low to prevent coronary atherosclerosis developing in persons without CAD. Some cardiologists advocate a reduction of the LDL goal to a 50 to 70 mg/dL range for everyone, not just those with a known cardiovascular disease.10 In 2006 32.6% of the adult U.S. population had LDL cholesterol levels of 130 mg/dL or higher.1
Very-Low-Density Lipoprotein Cholesterol
VLDL cholesterol is not usually measured, although a normal value is about 30 mg/dL.6 The value can be estimated by subtracting the sum of the HDL and LDL from the total cholesterol. When triglyceride levels are elevated, the VLDL cholesterol level also is high.
Triglycerides
Triglycerides are serum lipids that constitute an additional atherogenic risk factor. Triglycerides are carried by VLDL cholesterol in the bloodstream.7 An optimal triglyceride level is below 150 mg/dL, and the more elevated the triglyceride serum level, the higher the risk of developing CAD. A value between 150 and 199 mg/dL is borderline high; a value between 200 and 500 mg/dL is high; and a value above 500 mg/dL signals a high risk of atherogenic complications and a strong risk for the presence or development of type 2 diabetes.
Lipoprotein(a)
LDL cholesterol can be further analyzed by the category of lipid particles that make up the total LDL value. Researchers have investigated the function of several lipid particles to determine their role in the development of premature atherosclerotic CAD. One particle that has been extensively studied is lipoprotein(a), which is abbreviated Lp(a) and described verbally as “LP little a.” Lp(a) is manufactured in the liver and circulates in the bloodstream bound to a large glycoprotein called apolipoprotein(a), abbreviated as apo(a).11 The Lp(a)-apo(a) lipid particle concentration is elevated in the presence of inflammation, and it stimulates atheroma and clot formation in inflamed arteries.11 This effect is thought to occur because the apo(a) is structurally similar to plasminogen, a protein essential for clot formation.
Lp(a) levels are 90% genetically determined. Elevated Lp(a) plasma levels constitute the most frequently encountered genetic lipid disorder in families with premature CAD.11 Testing for Lp(a) is reserved for high-risk patient populations such as those with a strong family history of premature atherosclerotic disease and for patients with premature CAD who do not exhibit the expected cardiac risk factors. Reduction of Lp(a) levels to below 30 mg/dL is the therapeutic goal. This is usually achieved by ingesting high doses (1 to 2 grams/day) of extended-release nicotinic acid (niacin). The Lp(a) level is not reduced by statins, drugs that traditionally lower LDL levels, or by physical exercise, a low-fat diet, weight loss, or tight control of blood glucose levels.11 Lifestyle changes are recommended, and research is ongoing, but a cure is not yet discernible.
High-Fat Diet
A diet rich in saturated fats leads to elevated cholesterol levels in the blood. The first line of treatment to lower elevated serum cholesterol is a low-fat, high-fiber diet and increased physical exercise.7,8 If these measures are not effective, lipid-lowering drugs are indicated.1 This approach sounds simple, but less than one half of the people who qualify for lipid reduction therapy are taking their medications; only one third of treated patients reach their LDL target; and fewer than 20% of patients with CAD are at their LDL goal.1 Although lipid-lowering drugs are very helpful for some, they are not a panacea for everyone.
Obesity
Obesity is a disease of modern times. Global estimates are more than 1 billion overweight adults, and at least 300 million of these people are obese. Obesity is often associated with a sedentary lifestyle. A high risk of coronary heart disease is among the well-established adverse health effects associated with excess weight. Hypertension, hypercholesterolemia, and diabetes are among the clinical conditions that are important mediators of this association.12 In the United States in 2001, 122 million adults were overweight or obese.13 In 2006 144 million adults, or 66% of the U.S. adult population, were overweight or obese.1 Obesity is defined using the body mass index (BMI).
The BMI is calculated as the weight in kilograms divided by the square of the height in meters (kg/m2); it assesses body weight relative to height. BMI is used to evaluate the threat of excess pounds as a risk factor for CAD and permits comparisons of people of different gender, age, height, and body type.1 BMI is calculated as the weight in kilograms divided by the square of the height in meters (kg/m2). The BMI calculation in metric units is shown in Box 12-2. A normal BMI is between 18.5 and 25 kg/m2. A BMI between 25 and 30 kg/m2 indicates the person is overweight. A BMI greater than 30 kg/m2 is the definition of obesity.1
The distribution pattern of fat on the body is a CAD risk factor. The more weight carried in the abdominal area, producing a large waist, the greater the risk of CAD. Excess abdominal adiposity (apple body shape) indicates additional fat around the abdominal organs compared with individuals who have a smaller waist and larger hips (pear body shape). A waist size greater than 40 inches in men and 35 inches in women increases their risk for CAD. Physical exercise assists with weight reduction, lowers the risk for CAD, and decreases the risk of developing type 2 diabetes.
Physical Inactivity
Regular vigorous physical activity using large muscle groups promotes physiological adaptation to aerobic exercise that can prevent the development of CAD and reduce symptoms in patients with established cardiovascular disease.14 Exercise also reduces the incidence of many other diseases, including type 2 diabetes, osteoporosis, obesity, depression, and cancers of the colon and breast.14 Many research trials have demonstrated the positive effects of physical activity on the other major cardiac risk factors.14 Exercise alters the lipid profile by decreasing LDL cholesterol and triglyceride levels and increasing HDL cholesterol levels.14 Exercise reduces insulin resistance at the cellular level, lowering the risk for developing type 2 diabetes, especially if combined with a weight-loss program.14 Epidemiological studies indicate that physical athletics as a young person do not confer protection in later years. A sedentary lifestyle has negative effects, regardless of age, gender, BMI, smoking status, presence or absence of hypertension, or abnormal lipoprotein profile. Lifelong physical activity is necessary to prevent atherosclerotic CAD and stroke.14 In 2005 the prevalence of adults not engaging in any physical activity during leisure or work time was 10.3% in the United States.1 In affluent countries, only one third of the population exercises moderately for the recommended 30 minutes, five times per week.
Hypertension
Normal blood pressure is described as a systolic blood pressure (SBP) below 120 mm Hg and a diastolic blood pressure (DBP) below 80 mm Hg. Hypertension is defined as an SBP greater than 140 mm Hg or DBP higher than 90 mm Hg. Controlled hypertension describes a situation in which administration of antihypertensive medications maintains the patient’s blood pressure within the normal range.
Hypertension is a cardiac risk factor because the high SBP damages the arterial endothelium, leading to vascular inflammation that encourages formation of plaque. Hypertension is a complex, multifactorial disease process. Hypertension is divided into stages for the purposes of treatment, as shown in Table 12-2.
TABLE 12-2
BLOOD PRESSURE GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE
| CATEGORY | SYSTOLIC BP* (mm Hg) | DIASTOLIC BP* (mm Hg) |
| Normal (optimal)* | <120 | <80 |
| Prehypertension | 120-139 | 80-89 |
| Stage 1 hypertension | 140-159 | 90-99 |
| Stage 2 hypertension | ≥160 | ≥100 |
BP, blood pressure; CAD, coronary artery disease.
*Values greater than normal increase the risk for CAD and heart failure.
Prehypertension is an SBP of 120 to 139 mm Hg or DBP above 85 mm Hg.1,13 In the United States, one in five adults is prehypertensive. Hypertension is diagnosed when the blood pressure is above 140/90 mm Hg. Hypertension affects another one in four adults in the United States. After the blood pressure is above 140/80 mm Hg, hypertension is described as stage 1 or stage 2 depending on the severity (see Table 12-2).
Hypertension is often described as the “silent killer,” because 30% of those affected are unaware they have seriously elevated blood pressure.1 A higher percentage of men than women have hypertension until age 45, but from the ages of 45 to 54 years, the percentage of men and women with hypertension is similar.1 It is essential that patients understand that sustained elevation of blood pressure leads inexorably toward atherosclerosis, heart failure, kidney failure, stroke, and heart attack.13 So widespread is hypertension in industrialized societies that even a normotensive person at age 55 has a 90% lifetime risk of developing hypertension. This implies that even normotensive persons should adopt interventions to maintain a normal blood pressure.13 The estimated direct and indirect cost of hypertension in the United States for 2010 is $76.6 billion.1
According to recent guidelines, the goal of treatment for the hypertensive person without other risk factors is to achieve a blood pressure below 140/80 mm Hg. For the hypertensive person who already has diabetes or kidney disease, the target blood pressure is below 130/80 mm Hg. A normal blood pressure is below 120/80 mm Hg.13
Lifestyle interventions that can normalize blood pressure include physical exercise, a low-salt diet, limiting alcohol intake, and achieving normal body weight. Most patients are started on a diuretic, and if this is insufficient, they may be placed on an angiotensin-converting enzyme inhibitor (ACEI), angiotensin receptor blocker (ARB), beta-blocker, or calcium channel blocker. Most patients require at least two medications, each from different drug classifications, to normalize their blood pressure.13 Hypertensive emergencies with acute organ damage are discussed in the last section of this chapter.
Cigarette Smoking
The greater the number of cigarettes smoked per day, the greater the risk of developing CAD, acute MI, and stroke.14,16 In the United States, the prevalence of smoking has always been lower among women than men.1 Cigarette smoking unfavorably alters serum lipid levels, decreases the HDL cholesterol level, and increases LDL cholesterol and triglyceride levels. In the United States, smoking remains common, with an overall prevalence of 20.6% of the adult population. Smoking increases the risk of coronary heart disease at all levels.1 Smokers are two to four times more likely to develop CAD than nonsmokers.1 Passive, secondhand smoke exposure also increases cardiovascular risk; 34.7% of nonsmoking adults are exposed to environmental tobacco smoke at home or at work.15–17 Nicotine is addictive, and smoking cessation is difficult. People need tremendous support to be able to “kick the habit.” Chapter 25 provides patient education guidelines on how to stop smoking.
Diabetes Mellitus
Individuals with diabetes mellitus (types 1 and 2) have a higher incidence of coronary heart disease compared with the general population. Data from the Framingham Heart Study indicates a doubling in the incidence of diabetes over the past 30 years and most dramatically during the 1990s.1 Elevated blood glucose level is a known risk factor for development of vascular inflammation associated with atherosclerosis. The normoglycemia range is 70 to 100 mg/dL. It is recommended that patients in critical care have blood glucose levels maintained close to the normal range18 while avoiding hypoglycemic episodes.
A fasting blood glucose concentration between 100 and 125 mg/dL represents a prediabetic state and is a risk factor for the development of diabetes and CAD (Table 12-3). A fasting blood glucose above 126 mg/dL is indicative of diabetes. Patients with diabetes have an increased risk of developing CAD and have worse clinical outcomes after acute coronary syndrome events.19 In a multinational study of patients who were seen at hospitals with symptoms of acute coronary syndrome, almost one in four had a known history of diabetes.20
TABLE 12-3
FASTING BLOOD GLUCOSE AND RISK FOR CORONARY ARTERY DISEASE
| BLOOD GLUCOSE LEVEL | FASTING PLASMA GLUCOSE LEVEL* (mg/dL) |
| Normal | 70-100 |
| Prediabetic | 100-125 |
| Diabetic | 126 or higher |
*Values greater than normal increase the risk for CAD and kidney failure.
Chronic Kidney Disease
Chronic kidney disease is considered a risk equivalent for CAD.2,3,21 This means patients with chronic kidney disease have as much risk of experiencing a coronary event as if they already had CAD.2,3,22 The risk of death for the patient with acute MI rises as the serum creatinine level increases.22 In one study, the in-hospital mortality rates for patients with an acute MI were 2% for patients with normal kidney function, 6% for those with mild kidney failure, 14% for those with moderate kidney failure, 21% for those with severe kidney failure, and 30% for patients with end-stage kidney disease.22 Mortality following cardiac surgery is also higher for individuals with chronic kidney disease.23
Metabolic Syndrome
Metabolic syndrome refers to the clustering of risk factors associated with cardiovascular disease and type 2 diabetes.1 The prevalence of metabolic syndrome in 2006 for adults in the United States was 34%.1 Metabolic syndrome is diagnosed when three or more of the following risk factors are present:24,25
2 Serum triglyceride is level greater than 150 mg/dL (≥1.7 mmol/L).
4 Blood pressure is 130/85 mm Hg or higher, which is diagnostic for prehypertension or hypertension.
It is perhaps obvious that individuals with the signs of metabolic syndrome are at increased risk for CAD; even so, it is surprising to what degree this holds true. In the Framingham epidemiological study, presence of the factors associated with metabolic syndrome predicted 25% of all new-onset CAD and almost 50% of new-onset diabetes.23
Women and Heart Disease: Premenopause and Postmenopause
Serious CAD symptoms occur approximately 5 years later in women than in men.25 The average age for the first acute MI in men is 65.8 years and in women is 70.4 years.1 Incidence of CAD is two to three times higher among postmenopausal women than women who are premenopausal.1 In the past, it seemed logical to prescribe hormone replacement therapy (HRT) to treat the symptoms of menopause. However, well-designed research trials discovered an increase in cardiovascular events in the first year of HRT (estrogen plus progestin), although cardiac events declined after the first year.25,26 Subsequent studies have confirmed this result.24 In 2004 the estrogen-only HRT trial was stopped by the National Institutes of Health (NIH) because of an increased risk of stroke among the women taking estrogen. For these reasons, HRT is no longer recommended for prevention of atherosclerotic cardiovascular disease.27 Data from the Framingham Heart Study indicate the lifetime risk for cardiovascular disease is more than one in two for women at age 40.1
Cardiovascular disease kills more than one-half million women annually in the United States. To emphasize the magnitude of the problem, this represents more deaths than the next seven fatal diseases for women combined.28 Mortality rates for women after an acute MI are higher than for men: 38% and 25%, respectively. Risk factors more strongly associated with acute MI in women compared to men include hypertension, diabetes mellitus, alcohol intake, and physical inactivity.26 Many reasons contribute to women having higher mortality from acute MI, including waiting longer to seek medical care, having smaller coronary arteries, being older when symptoms occur, and experiencing very different symptoms from those of men of the same age.29 The 2007 evidence-based guidelines for cardiovascular disease prevention in women recommended a plan for a general approach to the female patient that classifies her as high risk, at risk, or at optimal risk.28 In 2007 50% of women surveyed by the American Heart Association (AHA) recognized that heart disease was the leading cause of death among women.
Vascular Inflammation
The link between vascular inflammation and atherosclerotic disease is well established.31 Measurement of this link has been more controversial, however.30 Many researchers think the development of atherosclerotic plaque occurs in response to inflammation. Noxious inflammatory agents circulate in the bloodstream and stimulate chemical mediators that directly modify the arterial wall. The initial inflammatory stimuli include increased blood glucose, elevated blood lipids, nicotine, and hypertension. However, other clinical conditions, such as connective tissue disorders and systemic infection, also produce an inflammatory state, and it is not clear what the impact of inflammation produced by these stimuli is on the vessels.30 Research to identify prognostic inflammatory markers is ongoing.
C-Reactive Protein
The inflammatory marker most frequently cited is C-reactive protein (CRP). It is measured as high-sensitivity C-reactive protein (hs-CRP).30 The higher the hs-CRP value, the greater the risk of a coronary event, especially if all other potential causes of systemic inflammation such as infection can be ruled out. If other systemic inflammatory conditions such as bronchitis or a urinary tract infection are present, the hs-CRP test loses all predictive value.31 CRP and other inflammatory markers are used to estimate the probability of future acute coronary events.30,31 During acute coronary syndrome events, there is widespread activation of neutrophils in the cardiac circulation (measured from the coronary sinus), which suggests that inflammation is not limited to one unstable plaque.32 Debate continues about whether CRP is simply a marker of vascular inflammation or also contributes to the proinflammatory state.9
Multifactorial Risk and Risk Equivalents
CAD has multifactorial causation; the greater the number of risk factors, the greater the risk of developing CAD.1,3,24,33 The best time for an individual to make lifestyle changes is before symptoms of CAD occur. Patients with two or more risk factors or with one or more of the CAD risk-equivalent diseases have the greatest potential to benefit from risk-factor reduction and lifestyle change.3
Certain medical conditions are considered risk equivalents of CAD. A risk equivalent means the person has the same risk of having an acute MI as if they had coronary heart disease already. Two noncardiac medical conditions are considered risk equivalents for CAD: diabetes mellitus and chronic kidney disease. Peripheral arterial disease and cerebral vascular disease are atherosclerotic conditions that are also considered CAD risk equivalents.
Primary Versus Secondary Prevention of Coronary Artery Disease
If a person has symptoms of CAD or has previously had an acute coronary syndrome event, the goal of any lifestyle change or medication is called secondary prevention, or preventing another heart attack. If an individual matches the risk profile described previously but does not have symptoms of CAD or has not had an acute MI, the treatment plan is described as primary prevention. The constellation of cardiac risk factors is well established and can predict development of CAD for most populations in the developed industrial world.
Pathophysiology of Coronary Artery Disease
Coronary heart disease is a progressive atherosclerotic disorder of the coronary arteries that results in narrowing or complete occlusion. Atherosclerosis affects the medium-size arteries that perfuse the heart and other major organs. Normal arterial walls are composed of three layers: the intima (inner lining), the media (middle muscular layer), and the adventitia (outer coat).
Development of Atherosclerosis
Atherosclerosis is a chronic inflammatory disorder that is characterized by an accumulation of macrophages and T lymphocytes in the arterial intimal wall. A high LDL cholesterol concentration is one of the triggers of vascular inflammation. The inflammation injures the wall, allowing the LDL cholesterol to move into the vessel wall below the endothelial surface.9 Blood monocytes adhere to endothelial cells and migrate into the vessel wall. Within the artery wall, some monocytes differentiate into macrophages that unite with and then internalize LDL cholesterol. The foam cells that result are the marker cells of atherosclerosis.9
Elevated LDL cholesterol levels promote low-level endothelial inflammation that allows lipoproteins to infiltrate the intimal vessel wall. After it has infiltrated under the endothelium, LDL cholesterol tends to stay within the vessel wall rather than return to the circulation.9 This contrasts with the actions of HDL cholesterol, which enters the vessel wall, helps efflux cholesterol from cells, and then returns to the circulation.9 The actions of HDL cholesterol may help minimize the number of foam cells in the artery wall.9
Atherosclerotic Plaque Rupture
When a mature atherosclerotic plaque develops, it is not uniform in composition. It has a lipid liquid center filled with procoagulant factors. A connective tissue fibrous cap covers the top of the fluid lipid center.30–32 The abrupt rupture of this cap allows procoagulant lipids to flood into the vessel lumen and rapidly form a coronary thrombosis, as shown in Table 12-4. As the enlarging clot blocks blood flow through the coronary artery, a “heart attack” will occur unless there is adequate collateral circulation from other coronary vessels. Symptoms and suggested cardiac interventions at appropriate stages in development of CAD are listed in Table 12-4.
TABLE 12-4
TIMELINE OF ATHEROGENESIS DEVELOPMENT DEPICTED BY LONGITUDINAL SECTION OF AN ARTERY
| ATHEROGENESIS/THROMBOGENESIS | ASSOCIATED SYMPTOMS | CARDIAC INTERVENTION |
| A. Normal artery, normal vessel wall. | No symptoms | Primary prevention of CAD recommended: consume a low-fat diet, take regular physical exercise, avoid smoking, and achieve normal BMI |
| B. Lipids in bloodstream. | No symptoms | |
| C. Extracellular lipid accumulates in the intima of the artery (atheroma). | No symptoms | |
| D. Lipid accumulation evolves to become a fatty-fibrous (atherosclerotic) lesion. Some lesions contain a lipid interior covered by a fibrous cap. | Chest pain with exercise that is relieved by rest or NTG (stable angina) or possibly no symptoms until the lesion fills more than 75% of the vessel lumen | PCI if stable angina is present and CAD is diagnosed by cardiac catheterization |
| E. Rupture of the cap allows lipid in the center to be released into the bloodstream, stimulating clot formation (thrombogenesis). | Chest pain not relieved by rest or NTG (ACS – unstable angina) | Call 911 for immediate transport to a hospital, preferably one with experience treating ACS |
| F. Fresh clot blocks the vessel; spasm of the artery may occur near the thrombus. | Chest pain unrelieved by rest or NTG – severity, location of angina, and associated symptoms vary greatly among individuals (ACS – acute MI) | Emergency intervention to open the artery: fibrinolytic or catheter-based procedure (PCI) |
| G. Vessel is open, but the atherosclerotic lesion remains. | No symptoms | Secondary prevention of CAD to prevent repeat MI; beta-blockers to prevent arrhythmias; ACE-1 drugs to prevent ventricular remodeling and heart failure; elective PCI |
|
|
||
(Modified from Antman EM, et al: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary, Circulation 110(5):588, 2004.)
Plaques that are likely to rupture are saturated with macrophages and other inflammatory cells. These vulnerable plaques are usually not obstructive and are situated at bends or branch points in the arterial tree.3 It is not known what factors cause the fibrous cap to rupture or erode. As deep fissures in the cap expose the procoagulant factors to the blood plasma, an unstoppable cycle is put into motion. When platelets in the bloodstream are exposed to collagen, necrotic debris, von Willebrand factor, and thromboxane, a clot is formed that can occlude the coronary artery. Highly fibrotic plaques do not rupture. The type of atherosclerotic plaque that is prone to rupture has a weak fibrous cap and a large amount of liquid cholesterol within the core (see Table 12-4).32
Plaque Regression
A reduction in blood cholesterol decreases atherosclerotic plaque size by decreasing the amount of liquid cholesterol within the plaque core.7 Lowering cholesterol levels does not change the dimensions of the fibrous or calcified portions of the plaque. However, lower cholesterol levels reduce vascular inflammation and make vulnerable plaque less likely to rupture.
If diet is not effective in lowering blood cholesterol, lipid-lowering drugs are prescribed to lower the LDL cholesterol level below 100 mg/dL for patients at risk for CAD and to aim for an LDL level below 70 mg/dL for individuals with the highest-risk profile.7 Drugs, diet, and exercise are used to lower the triglyceride levels to less than 150 mg/dL, and to raise HDL cholesterol levels above 40 mg/dL for men and above 50 mg/dL for women (see Table 12-1).3,7,8,33
Acute Coronary Syndromes
The term acute coronary syndrome (ACS) is used to describe the array of clinical presentations of CAD that range from unstable angina to acute MI (see Table 12-4).1,3,5 The general public and media describe an acute MI as a “heart attack.” The following section discusses stable manifestations of CAD (stable angina) and acute manifestations described as an acute coronary syndrome (unstable angina and acute MI).
Angina
Angina pectoris, or chest pain, caused by myocardial ischemia is not a separate disease, but rather a symptom of CAD. It is caused by a blockage or spasm of a coronary artery, leading to diminished myocardial blood supply. The lack of oxygen causes myocardial ischemia, which is felt as chest discomfort, pressure, or pain. Angina may occur anywhere in the chest, neck, arms, or back, but the most commonly described location is pain or pressure behind the sternum. The pain often radiates to the left arm but can also radiate down both arms and to the back, the shoulder, the jaw, or the neck (Figure 12-1). Angina symptoms are not the same for all individuals, many patients may describe pressure or discomfort rather than pain and presenting symptoms can be highly individualized, as described in Box 12-3. Patients and families must be taught that angina does not always present in the dramatic heart attack scenario seen on television and in movies, in which the person clutches the throat or chest and exhibits extreme distress.3
A, Upper part of chest. B, Beneath sternum, radiating to neck and jaw. C, Beneath sternum, radiating down left arm. D, Epigastric. E, Epigastric, radiating to neck, jaw, and arms. F, Neck and jaw. G, Left shoulder. H, Intrascapular.
Women and Angina
Many women experience a variety of different symptoms before an acute MI and during the acute event, as shown in Box 12-4.34 The recognition and publicity about the fact that many women do not experience “crushing chest pain” is important if women’s symptoms are not to be trivialized by clinicians.35 It is important that all patients are made aware of angina symptom equivalents, such as unexpected shortness of breath, breaking out in a cold sweat, or sudden fatigue, nausea, or lightheadedness.3 More women die every year in the United States of cardiovascular disease than men, a fact that is largely unknown.28
Box 12-4
Cardiovascular Symptoms Experienced by Women before Acute Myocardial Infarction
| SYMPTOMS 1 MONTH BEFORE ACUTE MI | SYMPTOMS DURING ACUTE MI |
From McSweeney JC, et al: Women’s early warning symptoms of acute myocardial infarction, Circulation 108(21):2619, 2003.
Stable Angina
Stable angina is predictable and caused by similar precipitating factors each time; typically, it is exercise induced. Patients become used to the pattern of this type of angina and may describe it as “my usual chest pain.” Pain control should be achieved within 5 minutes by rest and by taking sublingual nitroglycerin. Stable angina is the result of fixed lesions (blockages) of more than 75% of the coronary artery lumen. Ischemia and chest pain occur when myocardial demand from exertion exceeds the fixed blood oxygen supply.6 Additional information on CAD and stable angina is provided in the box Evidence-Based Collaborative Practice: Coronary Artery Disease and Stable Angina.
Unstable Angina
Unstable angina is defined as a change in a previously established stable pattern of angina. It is part of the continuum of ACS. Unstable angina usually is more intense than stable angina, may awaken the person from sleep, or may necessitate more than nitrates for pain relief. A change in the level or frequency of symptoms requires immediate medical evaluation. Severe angina that persists for more than 5 minutes, is worsening in intensity, and is not relieved by one nitroglycerin tablet is a medical emergency, and the patient or a family member must call 911 immediately.3 The 911 (Emergency Medical Services [EMS]) system is available to 90% of the population of the United States.3 In one study, patients with an acute MI who used 911 and were transported to the hospital by ambulance had significantly faster receipt of initial reperfusion therapies.36,37 Family and friends are discouraged from driving a person experiencing unstable angina to the hospital and instead are encouraged to call 911. Patients should be instructed never to drive themselves but to contact the EMS by calling 911.
Unstable angina is an indication of atherosclerotic plaque instability. It can signal atherosclerotic plaque rupture and thrombus formation that can lead to MI. The patient who comes to the emergency department with recent onset of unstable angina but who has nonspecific or nonelevated ST-segment changes on the 12-lead electrocardiogram (ECG) may be admitted to the critical care unit to rule out MI. If the symptoms are typical of MI, it is important to treat the patient according to the latest published guidelines, because not all patients who experience an MI have ST-segment elevation on the 12-lead ECG.5
Medical Management
Accurate assessment of chest pain symptoms is essential if unstable angina is to be recognized and treated effectively. An important reason to ask questions about the chest pain is to differentiate between stable and unstable angina. The change from stable to unstable angina is potentially life threatening for the patient. If the ST segments are elevated or there is a newly documented left bundle branch block on the 12-lead ECG, the patient will be treated for acute MI.3 However, if these classic ECG signs are missing and the chest pain continues, the current pharmacological treatments of choice are aspirin, vasodilation by nitroglycerin, intravenous antiplatelet agents such as the glycoprotein (GP) IIb/IIIa inhibitors, and intravenous unfractionated heparin (UFH).5 Low-molecular-weight heparin (LMWH) combined with fibrinolysis is an alternative to heparin fibrinolysis for patients younger than 75 years with a serum creatinine level below 2.5 mg/dL for men and below 2 mg/dL for women.3
Another option is to transport the patient directly to the cardiac catheterization laboratory for direct visualization of the coronary arteries by the cardiologist. Recanalization of the coronary arteries is recommended, provided the institution performs more than 200 procedures annually or the individual physician performs more than 75 interventional procedures annually.3,5
Nursing Management
Nursing management of the patient with CAD and angina incorporates a variety of nursing diagnoses (Nursing Diagnosis Priorities box on Coronary Artery Disease and Angina). Nursing priorities focus on (1) recognizing myocardial ischemia, (2) controlling chest pain, (3) maintaining a calm environment, and (4) providing patient education.
Recognizing Myocardial Ischemia
Complaints of chest discomfort (angina) must be evaluated quickly, because angina is an indicator of myocardial ischemia. The patient is asked to rate the intensity of the chest discomfort on a scale of 0 to 10. Pain levels must be assessed with sensitivity to differences in cultural manifestations of pain. The words “chest pain” are not to be used exclusively, because some patients describe their angina as “pressure” or “heaviness.” It is important to document the characteristics of the pain and the patient’s heart rate and rhythm, blood pressure, respirations, temperature, skin color, peripheral pulses, urine output, mentation, and overall tissue perfusion. A 12-lead ECG is used to identify the area of ischemic myocardium. The major concern is that the chest pain may represent preinfarction angina, and early identification is essential so that the patient can be immediately treated. A range of treatments are available that include fibrinolytic infusion immediately, or transfer to the cardiac catheterization laboratory for a coronary arteriogram and opening of a blocked artery. If the hospital does not have a cardiac catheterization laboratory, GP IIb/IIIa receptor blockers may be infused to prevent the evolution of the acute MI before transfer.3,5 Figure 12-2 presents additional information about transfer between hospitals in this circumstance.
STEMI, ST elevation myocardial infarction; EMS, emergency medical services; PCI, percutaneous catheter intervention. (Illustration modified from Antman EM, et al: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction), Circulation 110(9):e82).
Relieving Chest Pain
In the critical care unit, control of angina is achieved by a combination of supplemental oxygen, nitrates, analgesia, and surveillance of the angina and of the effects of pharmacological therapy.
• Oxygen: All patients with acute ischemic pain are administered supplemental oxygen to increase myocardial oxygenation. Use of pulse oximetry is recommended to guide therapy and maintain oxygen saturation above 90%.3 Patients who develop symptoms of acute heart failure may require emergency intubation and mechanical ventilation to correct significant hypoxemia.3,5
• Nitrates: A combination of intravenous and sublingual nitroglycerin is used to vasodilate the coronary arteries and decrease pain. After nitrate administration, the critical care nurse closely observes the patient for relief of chest pain, for return of the ST segment to baseline, and for the potential development of unwanted side effects such as hypotension and headache. Administration of a nitrate is avoided if the SBP is below 90 mm Hg. Drug interactions with nitrates are another potential cause for concern. The phosphodiesterase inhibitor medication sidenafil (Viagra) is prescribed for several conditions including pulmonary hypertension and erectile dysfunction. Sidenafil and nitrates in combination may contribute to a precipitous fall in blood pressure.3,5
• Analgesia: Morphine (2 to 4 mg given intravenously) is the analgesic opiate of choice for preinfarction angina. It relieves pain and decreases fear and anxiety. After administration, the critical care nurse assesses the patient for pain relief and the development of unwanted side effects such as hypotension and respiratory depression.3,5
• Aspirin: Chewing an oral nonenteric-coated aspirin (162 to 325 mg) at the beginning of chest pain has been shown to reduce mortality. The nonenteric formulation is preferred because it increases absorption in the mouth when chewed, not swallowed.3,5
Maintaining a Calm Environment
Patients admitted to a critical care unit with unstable angina experience extreme anxiety and fear of death. The critical care nurse is faced with the challenge of ensuring that the elements of a calm environment that can alleviate the patient’s fear and anxiety are maintained, while being ready at all times to respond to an acute emergency, such as a cardiac arrest, or to assist with emergency intubation or insertion of hemodynamic monitoring catheters.
Providing Patient Education
In the critical care unit, the patient’s ability to retain educational information is severely affected by stress and pain. Initial patient education stresses the importance of alerting the nurse to any symptoms of chest pain or discomfort, and avoiding the Valsalva maneuver, which is defined as forced expiration against a closed glottis. This can be explained to the patient as “bearing down” when going to the bathroom or breath-holding when repositioning in bed. The Valsalva maneuver causes an increase in intrathoracic pressure that decreases venous return to the right side of the heart and is associated with low blood pressure and symptomatic bradycardia.
After the anginal pain is controlled, longer-term patient and family education can begin. Points to cover include risk factor modification, signs and symptoms of angina, when to call the physician, medications, and dealing with emotions and stress. However, because the acute hospital length of stay for uncomplicated angina is usually less than three days, referral to a cardiac rehabilitation program for a controlled exercise program and risk-factor modification after discharge may be the most helpful teaching intervention a critical care nurse can provide. Clinical practice guidelines for the management of CAD and stable angina are listed in the box Evidence-Based Collaborative Practice: Coronary Artery Disease and Stable Angina.
Myocardial Infarction
Description and Etiology
Myocardial infarction (MI) is the term used to describe irreversible myocardial necrosis (cell death) that results from an abrupt decrease or total cessation of coronary blood flow to a specific area of the myocardium.1 In the hospital, this is often referred to as an acute MI, indicating the sudden onset and the life-threatening nature of the event. Increasingly, an acute MI is described in relation to whether there was ST-segment elevation on the diagnostic 12-lead ECG. It may be labeled an acute non-ST-segment elevation MI (NSTEMI)5 or an acute ST-segment elevation MI (STEMI).3
Three mechanisms can block the coronary artery and are responsible for the acute reduction in oxygen delivery to the myocardium:
Myocardial tissue can best be salvaged within the first 2 hours (120 minutes) after the onset of anginal symptoms, as illustrated in Figure 12-2.3 The earlier the myocardium is revascularized, the better the survival.5 Unfortunately, many persons do not seek treatment until the acute phase has passed.3
Pathophysiology
Myocardial Ischemia
The outer region of the infarcted myocardial area is the zone of ischemia, as illustrated in Figure 12-3. It is composed of viable cells. Priority interventions are targeted to save this viable muscle. Repolarization in this zone is temporarily impaired but eventually will be restored to normal. Electrical conduction from areas of viable myocardium produces a normal QRS configuration as shown in Figure 12-4, A. Repolarization of ischemic myocardial cells manifests as T-wave inversion (see Figure 12-4, B).
A, Normal ECG. B, Ischemia indicated by inversion of the T wave. C, Ischemia and current of injury indicated by T-wave inversion and ST-segment elevation. The ST segment may be elevated above or depressed below the baseline, depending on whether the tracing is from a lead facing toward or away from the infarcted area and depending on whether epicardial or endocardial injury occurs. Epicardial injury causes ST-segment elevation in leads facing the epicardium. D, Ischemia, injury, and myocardial necrosis. The Q wave indicates necrosis of the myocardium.
Myocardial Injury
The infarcted zone is surrounded by injured but still potentially viable tissue in an area known as the zone of injury (see Figure 12-3). Cells in this area do not fully repolarize because of the deficient blood supply. This is recorded on the ECG as elevation of the ST segment (see Figure 12-4, C).
Myocardial Infarction
The area of dead muscle (necrosis) in the myocardium is known as the zone of infarction (see Figure 12-3). On the ECG, evidence of this zone is seen by new pathological Q waves, which reflect a lack of depolarization from the cardiac surface involved in the MI (see Figure 12-4, D). As healing takes place, the cells in this area are replaced by scar tissue.
MIs are classified according to the location on the myocardial surface and the muscle layers affected. Not all infarctions cause necrosis in all layers, as shown in Figure 12-5. A transmural MI involves all three cardiac layers—the endocardium, the myocardium, and the epicardium. A transmural (full-thickness) MI usually provokes significant ECG changes (see Figure 12-4). This is also described as a Q-wave MI. Not every acute MI produces a recognizable series of Q waves on the 12-lead ECG. Some patients who had a demonstrated Q wave on a 12-lead ECG as a result of an acute MI lose the Q wave months or years later. The reasons for this are unknown, but it may represent the development of collateral circulation.
12-Lead Electrocardiographic Changes
The ECG changes produced by a transmural infarction demonstrate alteration in myocardial depolarization (QRS complex) and repolarization (ST segment). The changes in repolarization are seen by the presence of new Q waves. These new, pathological Q waves are deeper and wider than tiny Q waves found on the normal 12-lead ECG.3
Myocardial Infarction Location
The location of infarction is determined by correlating the ECG leads with Q waves and the ST-segment T-wave abnormalities (Table 12-5). Infarction most commonly affects the left ventricle and the interventricular septum; however, the right ventricle can be infarcted, and many patients who sustain an inferior MI have some right ventricular damage. The ECG manifestations that are used to diagnose an MI and pinpoint the area of damaged ventricle include inverted T waves, ST-segment elevation, and pathological Q waves in specific lead groupings as described subsequently.
TABLE 12-5
CORRELATIONS AMONG VENTRICULAR SURFACES, ELECTROCARDIOGRAPHIC LEADS, AND CORONARY ARTERIES
| SURFACE OF LEFT VENTRICLE | ELECTROCARDIOGRAPHIC LEADS | CORONARY ARTERY USUALLY INVOLVED |
| Inferior | II, III, aVF | Right coronary artery |
| Lateral | V5-V6, I, aVL | Circumflex |
| Anterior | V2-V4 | Left anterior descending |
| Anterior lateral | V1-V6, I, aVL | Left main coronary artery |
| Septal | V1-V2 | Left anterior descending |
| Posterior | V1-V2 | Circumflex or right coronary artery (reciprocal changes) |
| V7-V9 (direct) |
Anterior Wall Infarction
Anterior wall infarction results from occlusion of the proximal left anterior descending artery (see Table 12-5). ST-segment elevation is expected in leads V1 through V4 on the 12-lead ECG, as shown in Figure 12-6. If the left main coronary artery is occluded, the ECG manifestations will involve almost all of the precordial leads V1 through V6
[/not-level-membership-for-critical-care-medicine-category]
