Chapter 6 Cardiology
Long Case
Cardiac disease
This chapter deals with problems that are likely to be discussion areas in the long-case section. Cardiac long-case patients may have complex cyanotic heart disease, or heart disease and other medical problems either causally related, as in Noonan syndrome or congenital rubella, or as a complication of their heart disease or its treatment (e.g. hemiparesis with cyanotic heart disease).
There have been many advances on several fronts in cardiology recently:
• Subacute bacterial endocarditis prophylaxis guidelines have been modified to reduce the number of cardiac conditions for which antibiotics are recommended with dental procedures (see below).
• Advances have been made in understanding the genetics and clinical aspects of the long QT syndrome, with risk stratification for experiencing a sudden life-threatening cardiac event (see below).
• Medical therapy for Marfan syndrome with angiotensin II receptor blockers (ARBs) has been shown to decrease the risk of aortic root dilatation, after a specific metabolic defect of the aortic wall was discovered that improved with ARBs (see below).
• Echocardiography has benefited from recent advances in computer technology, signal processing and high-frequency transducer design, to encompass new modalities: three-dimensional echocardiography (3DE), tissue Doppler imaging (TDI) and speckle tracking echocardiography (STE) are used to evaluate parameters including ventricular volume, myocardial velocity, left-ventricular twist/torsion (during systole) (STE), regional strain and strain rate (TDI and STE) and mechanical dyssynchrony (TDI, SDE, 3DE).
• Modification of cardiac risk factors is now actively occurring on children. In 2008, both the American Academy of Pediatrics and the American Heart Association reviewed obesity and the metabolic syndrome with insulin resistance, hypertension and early coronary artery disease, and now diet and drug therapy (including statins) are being recommended for children as young as 10 years.
History
Diagnosis
When made (birth, days, weeks or months); where; symptoms at diagnosis (e.g. cyanosis, tachypnoea, poor feeding, failure to thrive, recurrent infection); how (initial investigations: chest X-ray, electrocardiography, echocardiography, angiography).
Past history
1. Aetiology (e.g. rheumatic fever, pregnancy complicated by maternal rubella, smoking, alcoholism, or drugs such as lithium, warfarin or phenytoin).
2. Indications for, and number of, previous hospital admissions.
3. Pattern and timing of change in condition (e.g. when cyanosis or heart failure developed, when and how it was controlled).
4. Episodes of infective endocarditis.
5. Other complications of disease (e.g. cerebral thrombosis or cerebral abscess with cyanotic heart disease).
Treatment
1. Past surgery, complications thereof, plans for further operative procedures.
2. Past interventional catheter procedures.
3. Past ablation therapy, pacemaker or defibrillator placement.
4. Medications, past and present, the side effects of these, monitoring levels (e.g. digoxin), treatment plans for future.
5. Exercise restrictions (may be inappropriately applied by parents).
6. Recommendations for antibiotic prophylaxis for dental procedures; maintenance of dental hygiene.
8. Any need for identification bracelet.
9. Instructions for air travel and high altitude.
10. Any recent investigations monitoring treatment (e.g. Holter monitoring; oxygen saturation).
11. Any recent changes in treatment regimen, and indications for these.
Current state of health
• Symptoms of cardiac failure, such as fatigue and shortness of breath (in comparison with peers), cough, sweating, poor feeding, recurrent chest infections.
• Cyanosis, squatting with ‘Fallot turns’.
• Episodes suggestive of arrhythmias, such as syncope, alteration of consciousness, dizziness, shortness of breath, palpitations in older children (what was the rate; i.e. faster than when usually playing or excited), or a ‘funny feeling’ in the chest, sweating, nausea, vomiting, or the parents being unable to count the rapid pulse rate, suggesting a tachyarrhythmia.
• Chest pain (e.g. myocardial ischaemia versus musculoskeletal, pleural or pericardial causes) or headache (e.g. polycythaemia in cyanotic heart disease, severe hypertension, cerebral abscess).
• Recent change in condition. Note the temporal relationship between associated symptoms with any tachycardia or palpitation: they should coincide—functional chest pain can be followed by a reflex sinus tachycardia.
Other associated problems
These may either be related to the main diagnosis (e.g. Down syndrome, Marfan syndrome) or be more general problems (e.g. poor growth, developmental delay, especially involving gross motor abilities, exercise limitations and psychological effects).
Social history
1. Disease impact on child; for example, growth, development, schooling (academic performance, sports restrictions, teachers’ attitudes, peers’ attitudes, teasing, amount of school missed and whether schooling is appropriate).
2. Disease impact on parents; for example, financial situation, financial burden of disease so far, government allowances being received, marriage stability, restrictions on social life, plans for further children, genetic counselling, or at least an awareness of the risks of recurrence, and availability of fetal echocardiography.
3. Disease impact on siblings; for example, sibling rivalry, effect of family financial burden.
4. Social supports; for example, social worker, contact with other families of children with similar problems, break from managing child.
5. Coping: contingency plans (e.g. plan if child develops severe febrile illness); parents’ degree of understanding regarding cause, prognosis, exercise restriction, and antibiotic prophylaxis for operations and dental procedures.
6. Access to local doctor, paediatrician, cardiac outpatients clinic (where, how often), other clinics attended.
Management issues
1. General development, growth and nutrition
Chronic left-to-right shunts sufficient to cause cardiomegaly often cause growth retardation. This may be an indication for surgical correction even in the absence of other indications such as pulmonary hypertension. Marked hypoxia may be associated with growth retardation, but the hypoxia has to be severe to do so. Most patients who have Eisenmenger syndrome with cyanotic heart disease and pulmonary hypertension do not have increased energy requirements or inadequate caloric intake, and do not have growth retardation. It takes profound hypoxia to cause growth retardation.
2. Prophylaxis against subacute bacterial endocarditis (SBE) risk
Genitourinary and gastroenterological surgery
As enterococci are the usual problem, the recommended treatment should adequately cover these organisms; thus parenteral gentamicin and ampicillin is an appropriate choice.
4. Social issues
Impact of disease
Siblings receiving less parental attention than expected in a normal family is a commonly encountered problem.
Travel
All these children should carry a letter explaining the nature of their heart lesion, their usual medications, and the name of their doctor or usual treatment centre, and have access to a hospital if they have a disease prone to sudden deterioration (e.g. Fallot spells). An identification bracelet should also be worn. Those with pacemakers in particular should carry written details regarding their unit and its program. Patients with severe congestive cardiac failure, severe cyanosis or pulmonary hypertension must avoid high altitudes (e.g. above 1500 m) and require supplemental oxygen to be available during commercial air travel. Hot climates can be a problem in children with polycythaemia, and adequate hydration must be maintained. The child’s exercise tolerance must be considered when planning the trip itinerary.
5. Specific problems
Specific syndromes: cardiac involvement
Marfan syndrome
This is an autosomal dominant condition caused by defective fibrillin, a protein important to the integrity of connective tissue (see Figure 6.1). The relevant gene (FBN1) has been mapped to chromosome 15q21.1. The cardiac features are the most important and life-threatening aspects of Marfan syndrome, manifesting in childhood in 25% of those affected. The cardiac involvement is progressive in around one third of these children. Features include the major diagnostic criterion of dilatation of the ascending aorta with or without aortic regurgitation, and involving at least the sinuses of Valsalva or dissection of the ascending aorta. Minor diagnostic criteria for Marfan syndrome include mitral valve prolapse with or without mitral valve regurgitation, dilatation of the main pulmonary artery in the absence of another anatomic cause (before age 40), calcification of the mitral annulus (before age 40), and dilatation or dissection of the descending thoracic or abdominal aorta (before age 50).
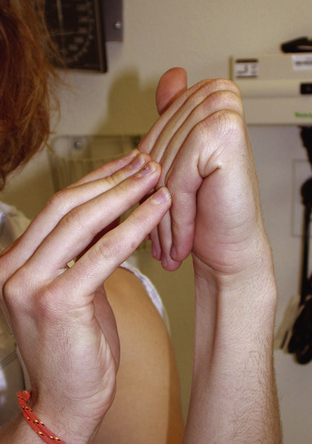
Figure 6.1 Marfan syndrome—joint hypermobility.
Jones, Kenneth. 2005. Smith’s Recognizable Patterns of Human Malformation, 6th edition, p. 550.
Parental education regarding the importance of avoiding strenuous exercise and competitive or contact sports is important, and should begin before preschool, placing less emphasis on the importance of sporting activities. Non-strenuous activities should be encouraged (e.g. walking, fishing, golf). The symptoms of aortic dissection must be discussed, including chest pain and syncope.
Dilatation or dissection of aorta at level of sinuses of Valsalva
Displacement of lens (ectopia lentis)—directed upward; also early cataract formation
Dolichostenomelia: disproportionately long extremities; decreased upper to lower segment ratio (US:LS < 0.85 for older children/adults) or arm span to height ratio > 1.05; other diagnostic skeletal features—
Distinctive facial features (dolichocephaly, down-slanting palpebral fissures, deeply set eyes (enophthalmos), decreased malar prominence (malar hypoplasia), diminished jaw (retrognathia)
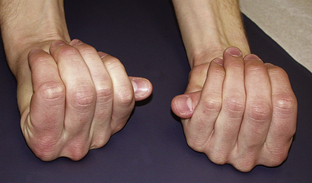
Figure 6.2 Steinberg thumb sign.
Jones, Kenneth. 2005. Smith’s Recognizable Patterns of Human Malformation, 6th edition, Figure 2B.
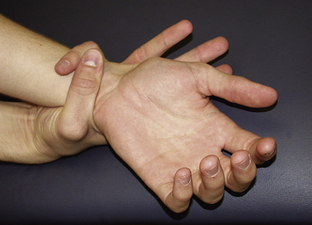
Figure 6.3 Walker–Murdoch wrist sign.
Jones, Kenneth. 2005. Smith’s Recognizable Patterns of Human Malformation, 6th edition, Figure 2C.
The acronym MARFANS also can be used as an (alternative) aide-mémoire:
M. Mitral prolapse ± Mitral regurgitation
A. Ascending aorta dilatation (involving sinuses of Valsalva)/dissection/Arched palate (high arched, with tooth crowding)/Acetabuli (protrusio acetabuli)
R. Regurgitation of aorta/mitral/Retinal detachment/Reduced US:LS ratio
F. Fibrillin defective/Facial gestalt (dolichocephaly, down-slanting palpebral fissures, deeply set eyes (enophthalmos), decreased malar prominence (malar hypoplasia), diminished jaw (retrognathia)/Flat cheek bones/Flat cornea/Flexible joints (joint hypermobility)
A. Apical blebs (on CXR)/Air leaks (spontaneous pneumothorax)/Arachnodactyly

S. Scoliosis/Sternal deformity/Skeletal dolichostenomelia/Sacral dural ectasia
Noonan syndrome (NS)
NS is an autosomal dominant condition, associated with four genes. In 50% of cases, the associated gene locus is at 12q24.1, with mutations in PTPN11, the gene encoding the non-receptor type protein, tyrosine phosphatase SHP-2. The other genes are KRAS, SOS and RAF1. Almost all patients with NS have some cardiac defect, particularly a dysplastic (and often stenotic) pulmonary valve, which is more common with a PTPN11 mutation, or hypertrophic cardiomyopathy (HCM), which affects around 20–30% of NS children, but is less common with a PTPN11 mutation. The ECG frequently shows left-axis deviation and a dominant S wave over the praecordial leads, even in NS patients with no known cardiac disease; the cause for this is not known. Phenotypic features of NS include dysmorphic facial features, short stature, webbed neck and skeletal anomalies (see the short case on dysmorphism).
NS patients with dysplastic pulmonary valves can have rapid progression of pulmonary valvular obstruction and may require review more frequently than for non-NS pulmonary valve lesions. Also, NS-associated valve obstruction is more likely to require surgical intervention. Balloon valvoplasty is usually unsuccessful in abolishing the obstruction, and simple valvotomy may be inadequate. Often complete excision of the valve, resection of the right-ventricular outflow muscle, and occasionally an outflow tract patch may be needed. Atrial septal defects (ASDs) (Figure 6.10) and pulmonary artery branch stenoses may coexist with valvular pulmonary stenosis (Figure 6.4). Other infrequent findings with NS include ventricular septal defects (VSDs) (Figure 6.12) and tetralogy of Fallot (Figure 6.17 and Figure 6.16).
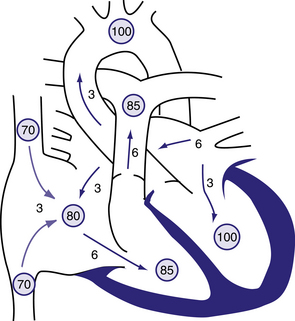
Figure 6.10 Physiology of atrial septal defect (ASD).
Circled numbers represent oxygen saturation values. The numbers next to the arrows represent volumes of blood flow (in L/min/m2). This illustration shows a hypothetical patient with a pulmonary-to-systemic blood flow ratio (Qp:Qs) of 2:1. Desaturated blood enters the right atrium from the vena cavae at a volume of 3 L/min/m2 and mixes with an additional 3 L of fully saturated blood shunting left to right across the ASD; the result is an increase in oxygen saturation in the right atrium. Six litres of blood flows through the tricuspid valve and causes a mid-diastolic flow rumble. Oxygen saturation may be slightly higher in the right ventricle because of incomplete mixing at the atrial level. The full 6 L flows across the right-ventricular outflow tract and causes a systolic ejection flow murmur. Six litres returns to the left atrium, with 3 L shunting left to right across the defect and 3 L crossing the mitral valve to be ejected by the left ventricle into the ascending aorta (normal cardiac output). Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 419.1.
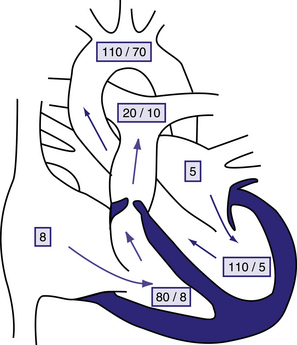
Figure 6.4 Physiology of valvular pulmonary stenosis.
Boxed numbers represent pressure in mmHg. Because of the absence of right-to-left or left-to-right shunting, blood flow through all cardiac chambers is normal at 3 L/min/m2. The pulmonary-to-systemic blood flow ratio (Qp:Qs) is 1:1. Right atrial pressure is increased slightly as a result of decreased right ventricular compliance. The right-ventricle is hypertrophied, and systolic and diastolic pressure is increased. The pressure gradient across the thickened pulmonary valve is 60 mmHg. The main pulmonary artery pressure is slightly low, and poststenotic dilatation is present. Left heart pressure is normal. Unless right-to-left shunting is occurring through a foramen ovale, the patient’s systemic oxygen saturation will be normal. Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 420.1.
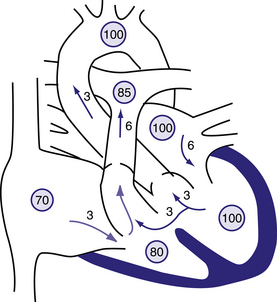
Figure 6.12 Physiology of a large ventricular septal defect (VSD).
Circled numbers represent oxygen saturation values. The numbers next to the arrows represent volumes of blood flow (in L/min/m2). This illustration shows a hypothetical patient with a pulmonary-to-systemic blood flow ratio (Qp:Qs) of 2:1. Desaturated blood enters the right atrium from the vena cava at a volume of 3 L/min/m2 and flows across the tricuspid valve. An additional 3 L of blood shunts left to right across the VSD, the result being an increase in oxygen saturation in the right ventricle. Six litres of blood is ejected into the lungs. Pulmonary arterial saturation may be further increased because of incomplete mixing at right-ventricular level. Six litres returns to the left atrium, crosses the mitral valve, and causes a middiastolic flow rumble. Three litres of this volume shunts left to right across the VSD, and 3 L is ejected into the ascending aorta (normal cardiac output). Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 419.5.
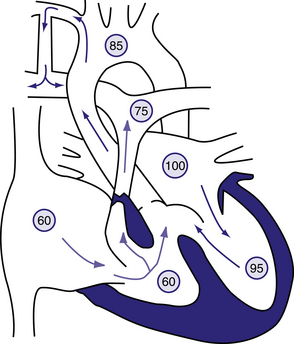
Figure 6.17 Blalock–Taussig shunt in the patient with tetralogy of Fallot.
Circled numbers represent oxygen saturation values. The intracardiac shunting pattern is as described for Figure 423.1. Blood shunting left to right across the shunt from the right subclavian artery to the right pulmonary artery increases total pulmonary blood flow and results in a higher oxygen saturation than would exist without the shunt (see Fig. 423.1). Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 423.5.
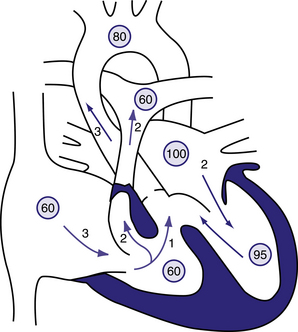
Figure 6.16 Physiology of the tetralogy of Fallot.
Circled numbers represent oxygen saturation values. The numbers next to the arrows represent volumes of blood flow (in L/min/m2). Atrial (mixed venous) oxygen saturation is decreased because of the systemic hypoxemia. A volume of 3 L/min/m2 of desaturated blood enters the right atrium and traverses the tricuspid valve. Two litres flows through the right-ventricular outflow tract into the lungs, whereas 1 L shunts right to left through the ventricular septal defect (VSD) into the ascending aorta. Thus, pulmonary blood flow is two thirds normal (Qp:Qs of 0.7:1). Blood returning to the left atrium is fully saturated. Only 2 L of blood flows across the mitral valve. Oxygen saturation in the left ventricle may be slightly decreased because of right-to-left shunting across the VSD. Two litres of saturated left-ventricular blood mixing with 1 L of desaturated right-ventricular blood is ejected into the ascending aorta. Aortic saturation is decreased, and cardiac output is normal. Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 423.1.
The acronym NOONANS can be used as an aide-mémoire:
 Neurodevelopmental problems: 25% learning disability, 10–15% special education, <30% mild intellectual impairment, 72% articulation problems, non-verbal abilities better than verbal, specific learning problems/Neuromuscular examination—hypotonia (floppy strong): joint hyperextensibility/Neurosurgical—Arnold–Chiari type I
Neurodevelopmental problems: 25% learning disability, 10–15% special education, <30% mild intellectual impairment, 72% articulation problems, non-verbal abilities better than verbal, specific learning problems/Neuromuscular examination—hypotonia (floppy strong): joint hyperextensibility/Neurosurgical—Arnold–Chiari type I
O. Obstructive heart lesions: RVOT (pulmonary valve); LVOT (HCM)/Other: ASD, VSD, tetralogy of Fallot, coarctation of aorta, branch pulmonary artery stenosis
O. Ocular anomalies in up to 95%: strabismus, refractive errors, amblyopia, nystagmus; hypertelorism, epicanthic folds, droopy eyelids, vivid blue or blue–green irises

A. Abnormal coagulation (33% have one or more defects)/Abnormal lymphatics: lymphoedema (hands/feet/scrotum/vulva), lymphangiectasia (lung, gut or testis)

S. Sternal deformity (superior carinatum, inferior excavatum)/Short stature (GH Rx)/Secondary sexual characteristics: delayed puberty, cryptorchidism in males/Skin findings: pigmented naevi (25%), café-au-lait patches (10%), lentigines (3%)
• Cardio-facial-cutaneous (CFC) syndrome (cardiac lesions—pulmonary valve stenosis; atrial septal defects); hyperkeratosis, haemangiomata, icthyosis. Four genes associated: BRAF (70–80%), MAPK1 and MAPK2 (10–15%), and KRAS (<5%).
• LEOPARD syndrome (Lentigines, ECG abnormalities [axis deviations, unilateral or bilateral hypertrophy, conduction abnormalities], Ocular hypertelorism, Pulmonary stenosis, Abnormalities of genitalia, Retardation of growth, and Deafness). Two genes associated: PTPN11 and RAF1; testing these finds mutations in 93% of affected people.
• Neurofibromatosis–Noonan syndrome; gene map locus 17q11.2; the clinical overlap between NFNS and NS is explained by the respective gene products, neurofibromin and SHP2, exerting their modulatory effects via a common pathway.
• Watson syndrome; gene map locus 17q11.2 (like NFNS, and NF1). Features: short stature, pulmonary valve stenosis, pigmented skin lesions (café-au-lait spots), intellectual development variable. Phenotype overlaps with NF1 as well as NS.
• Costello syndrome; gene map locus 11p15.5. Gene: the HRAS proto-oncogene; phenotypic overlap with NS and CFC. Features: short stature, pulmonary stenosis, HCM, hypertelorism, epicanthic folds, intellectual development variable.
22q11.2 deletion syndrome: neural crest—associated conotruncal defects
Note that similar phenotypic characteristics may occur in association with microdeletions on chromosomes 5, 10 and 17, as well as 22. The 22q11.2-deletion-associated cardiac defects include Truncus arteriosus, Tetralogy of fallot and Tricuspid atresia with d-malposition of the aorta (three Ts); Aortic arch interruption, Atrial septal defect and Aberrant right subclavian artery (three As); and Pulmonary atresia and ventricular septal defect, Pulmonary valve absence and Patent ductus arteriosus (three Ps). Deletions of 22q11.2 can also be seen in isolated heart disease (e.g. found in 30% of interrupted aortic arch, 20% of truncus arteriosus and 8% of tetralogy of Fallot).
The acronym CATCH-22 can be expanded to CATCHING, to include more features:
C. Cardiac: 74%; Conotruncal; 3 Ts, 3As, 3Ps (see text above); causes > 90% of all deaths
A. Abnormal face: long face, malar flatness, hypertelorism, hooded eyelids, ptosis, ear anomalies (overfolded helices, cupped, microtic, protruberant ears, preauricular pits), prominent nasal root, bulbous nasal tip, nasal dimple, asymmetric crying face)/Autoimmune diseases: juvenile idiopathic arthritis, Graves’ disease, vitiligo
T. Thymic hypoplasia, T-cell deficiency; 77% have immunodeficiency
C. Cleft palate, and other palatal anomalies: velopharyngeal incompetence (VPI), submucosal cleft palate (SMCP), bifid uvula, cleft lip; 69% have palatal problem/Craniosynostosis and other skeletal anomalies: hemivertebrae, polydactyly, extra ribs/Croup-like: laryngotrachealoesophageal anomalies—vascular ring, laryngeal web
H. Hypoparathyroidism/Hypocalcaemia/Hearing loss (conductive and sensorineural)
I. Intellectual issues: learning problems (non-verbal), 70–90%; mean full scale IQ 75–80; neuropsychological—ADHD, ASD; schizophrenia, bipolar, anxiety, depression/Impaired swallowing (dysmotile pharyngoesophageal area [derived from 3rd and 4th pharyngeal pouches]; nasopharygeal reflux, abnormal cricopharyngeal closure)

G. Growth hormone deficiency/Genitals: hypospadias, cryptorchidism, absent uterus/Gastrointestinal: atresias (oesophageal, jejunal, anal), malrotation, Hirschsprung/GP1BB mutation can cause coexistent Bernard–Soulier syndrome (BSS) (thrombocytopaenia and giant platelets); risk of bleeding significant
Williams syndrome (WS)
Williams syndrome is due to a deletion of a region of chromosome 7, termed the Williams–Beuren Syndrome Critical Region; it comprises 1.5–1.8 million base pairs and has 26–28 genes (see Figure 6.6). This area is predisposed to misalignment, during meiosis, of ‘duplicons’, which are low-copy-repeat blocks of homologous groups of genes and pseudogenes flanking the WBSCR. Hemizygosity for the elastin gene (ELN deletion) leads to the elastin arteriopathies supravalvular aortic stenosis (SVAS), peripheral pulmonary arterial stenosis (PPS) and other vascular stenoses. Hemizygosity for the gene LIM-kinase 1 leads to impaired visuospatial construction cognition. WS is a polyendocrine disorder that can involve all endocrine organs, and it is also a neurodevelopmental disorder. Children with this condition are friendly, outgoing and gregarious, are easily noticed and approach strangers readily. It is inherited as autosomal dominant. Cardiovascular conditions seen in WS include: SVAS, PPS, mitral valve prolapse, ventricular and atrial septal defects, renal artery stenosis with hypertension, and hypoplastic aorta. The mnemonic WILLIAMS HYPERCALCAEMIA has most of the salient features of WS:
W. Williams–Beuren Syndrome Critical Region (WBSCR); chromosome locus 7q11.2
I. Intelligence quotient (IQ) average 50–60/Impaired vision: reduced stereopsis
• Low tone (hypotonia)/Low pitched or hoarse voice, vocal cord paralysis
• Lax joints (joint hypermobility)/Loquacious, over-friendly, excessively empathic
I. Impaired feeding; tactile sensory defensiveness (difficulty with food textures); vomiting
A. ADHD symptomatology/Anxiety (somatisation can lead to abdominal pain)
M. Mitral valve prolapse (MVP)
S. Supravalvular aortic stenosis (SVAS)/Spine: scoliosis, kyphosis/Sternum: excavatum
HY. HYpercalcaemia, hypercalciuria, hypertension
P. Peripheral pulmonary arterial stenosis (PPW)/Puberty early (but not precocious)
E. Elastin arteriopathy (SVAS, PPS, aortic insufficiency, stenosis of mesenteric arteries)/Endocrine: hypothyroidism; IDDM in adults/Elfin face (see details below under A for appearance)
R. Renal anomalies (nephrocalcinosis, pelvic kidney)
C. Chronic otitis media (50%)/Characteristic personality: over-friendly, people-orientated
A. Audiological problems: high-frequency sensorineural hearing loss, hyperacusis (in 90%)
• Linear growth failure; postnatal growth rate 75% of normal/Loquacious personality
C. Cognitive: good verbal short-term memory, language; poor visuospatial construction
A. Appearance: broad brow, bitemporal narrowness, medial eyebrow flare, short palpebral fissures, epicanthic folds, blue stellate iris, short nose, full nasal tip, full cheeks, malar hypoplasia, long philtrum, full lips, wide mouth, small jaw, prominent earlobes
E. Eyes: hypotelorism, strabismus (50%), amblyopia, refractive errors (hyperopia in 50%)
M. Malocclusion, microdontia, enamel hypoplasia, widely spaced teeth, missing teeth
I. Intestinal problems: constipation, diverticulosis, coeliac disease
A. Abdominal pain: reflux oesophagitis; cholelithiasis; diverticulitis; ischaemic bowel disease
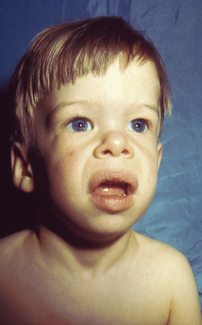
Figure 6.6 Williams syndrome.
Note the depressed nasal bridge, epicanthal folds, periorbital fullness, anteverted nares, long philtrum and prominent lips with large mouth. (A–C, From Jones KL, Smith DW: J Pediatr 86:718, 1975, with permission.) Jones, Kenneth. 2007. Smith’s recognizable patterns of human malformation, 3rd edition, p.122, Figure 1A.
Supraventricular tachycardia (SVT)
The commonest sustained tachyarrhythmia in children, SVT is caused mainly by an additional electrical connection between the atria and ventricle (accessory atrioventricular connection, AAVC) in those under 12; in teenagers, atrioventricular node re-entry tachycardia (AVNRT; has the functional equivalent of an extra connection within the AVN) may be a cause. Those with AAVCs often have ‘orthodromic’ tachycardia, with antegrade conduction down the atrioventricular (AV) node and retrograde conduction up the AAVC (the right way down the right path, the wrong way up the wrong path). Some patients have an AAVC that conducts in an antegrade fashion as well (giving ‘antidromic’ tachycardia, with retrograde conduction either up the AV node or the AAVC). Re-entry, the mechanism underlying these forms of SVT, requires two electrophysiologically distinct pathways around an insulated core, such as the AV valve annulus. Most cases of re-entrant SVT are sporadic.
Wolff–Parkinson–White (WPW) syndrome is a combination of pre-excitation on surface ECG and episodic SVT, whether orthodromic or antidromic. In WPW, the ECG shows a short PR interval, a delta wave (initial slurring of the QRS complex) and a wide QRS complex. During episodes of tachycardia the ECG develops either a narrow QRS tachycardia with a retrograde P wave after the QRS (orthodromic SVT) or a wide QRS (antidromic SVT). Children with WPW can develop atrial flutter, and also have a small risk of sudden cardiac death (SCD) from extremely fast atrial tachycardias (e.g. atrial flutter or atrial fibrillation being conducted down the AAVC, producing ventricular tachycardia [VT] or ventricular fibrillation [VF]).
Long QT syndrome (LQTS)
Children with arrhythmias (e.g. sick sinus syndrome, severe bradycardia, complete AV block), congestive cardiac failure, myocarditis or anthracycline cardiotoxicity, or those on certain medications (e.g. several antiarrhythmics [especially quinidine], some antibiotics, diuretics [by causing acute/chronic hypokalaemia], promotility agents, [cisapride]) have an increased risk of developing LQTS, and subsequent ‘torsades de pointes’ (TdP) (twisting of points) ventricular tachycardia, which can present with Sudden death, Syncope or Seizures (the three Ss). Recent molecular breakthroughs are unravelling the congenital forms of LQTS.
Romano–Ward syndrome (RWS)
This autosomal dominant cardiac electrophysiological disorder is characterised by LQTS and T-wave abnormalities on ECG, and by TdP; TdP is usually self-terminating and causes syncope particularly with exercise or emotional stress, with no warning—sometimes at rest, or in sleep. TdP can degenerate to ventricular fibrillation (VF) and causes sudden death, or aborted cardiac arrest if defibrillated successfully. Molecular genetic testing can identify the genes associated with RWS that code for potassium or sodium channels: KCNQ1 (chromosome 11p15.5), KCNH2 (chromosome 7q35–q36), SCN5A (chromosome 3p21), KCNE1 (chromosome 21q22.1–q22.2), KCNE2 (chromosome 21q22.1) and SCN4B (chromosome 3p21). The first five account for 70% known mutations; the last is more recently described, and the proportion attributable to this is unknown. Three clinical phenotypes are recognised: LQT1 is due to mutations in KCNQ1 and KCNE1 causing IKs potassium channel dysfunction, and cardiac events triggered by exercise and emotion; LQT2 is due to mutations in KNH2 and KCNE2 causing IKr potassium channel dysfunction, and cardiac events triggered by exercise, emotion and sleep; and LQT3 is due to mutations in SCN5A, the cardiac sodium channel gene, causing INa channel dysfunction, and cardiac events triggered by sleep. Management for the LQT1 and LQT2 phenotypes is beta blocker or pacemaker if beta blocker produces symptomatic bradycardia; recurrence of events while receiving beta blockers is usually the result of inadequate dosing; for the LQT3 phenotype, implantable cardioverter–defibrillator (ICD); ICD may also be required for the LQT1 and LQT2 if resistant to beta blockers or there is a past history of cardiac arrest. Automatic external defibrillators should be readily available at home, at school and at work. It is very important these patients avoid drugs that increase QT: see www.azcert.org for an updated list (this stands for Arizona Centre for Education and Research on Therapeutics).
Jervell and Lange–Nielsen syndrome (JLNS)
Abnormal cardiac depolarisation and repolarisation can lead to prolonged QT interval and tachyarrhythmias, including VT, TdP and VF, which can cause syncope or sudden death: 50% of patients will have cardiac events before 3 years of age, triggered by emotions and exercise. Corrected QT prolongation in JLNS is associated with increased risk of sudden infant death syndrome (SIDS), and if untreated, more than half of the children with JLNS will die prior to 15 years of age. Up to 95% of people with JLNS have a cardiac event before adulthood. Although the sex ratio among patients with JLNS is equal, females have lower risk of SCD. Heterozygotes have normal hearing; in some heterozygotes, there is no QT prolongation; in others, there can be QT prolongation with fainting and risk of SCD, which is RWS as above. Only the genes KCNQ1 and KCNE1 are involved in both RWS and JLNS.
LQTS is also associated with other cardiac channelopathies: Brugada syndrome (see below), Andersen–Tawil syndrome (LQTS, episodic flaccid muscle paralysis, and dysmorphic features; potassium channel gene KCNJ2) and Timothy syndrome (LQTS with syndactyly, dysmorphic facial features and neurological disorders; average age of death from VT 2.5 years; calcium channel gene CACA1C; the recommended treatment is ICD). Acquired causes for LQTS include electrolyte abnormalities (hypokalaemia, hypocalcaemia and hypomagnesaemia), malnutrition, myocardial disease (cardiomyopathy, myocarditis) and drugs, including some vasodilators, tricyclic antidepressants, antihistamines and phenothiazines; the full list is at www.azcert.org, as mentioned above.
Brugada syndrome
Described in 1992, this is an inherited cardiac disease causing ventricular tachyarrhythmias in patients with structurally normal hearts, presenting with syncope or cardiac arrest, and a strong family history of syncope or sudden death. It is inherited as autosomal dominant. There are eight types of Brugada syndrome listed in OMIM. For Brugada syndrome 1, the gene known to be associated is SCN5A, located at 3p21, which encodes the sodium channel protein type 5 subunit alpha in cardiac myocytes; there are over 160 known mutations in SCN5A. Mutations in SCN5A are found in 20–25% of patients with Brugada syndrome. It has a characteristic ECG pattern: right bundle branch block (RBBB) and ST elevation in V1 to V3; some patients with normal resting ECGs can have the classic changes induced by giving ajmaline, an antiarrhythmic medication. Implantable cardioverter–defibrillators (ICDs) can be placed in patients with a history of syncope or cardiac arrest, to prevent life-threatening arrhythmias; this is the only therapy known to be effective in these patients. Isoproterenol infusion can be used for electrical storms. Quinidine also has been used to prevent symptoms and resolve ECG features.
Myocardial disease
Dilated cardiomyopathy (DCM)—familial dilated cardiomyopathy (FDC) and idiopathic dilated cardiomyopathy (IDC)
The most common form of cardiomyopathy, DCM, is characterised by left-ventricular enlargement, systolic dysfunction and reduced myocardial contraction force. It can present with Dysrhythmia, Congestive failure and Mural thrombus, leading to thromboembolic disease. The term ‘DCM’ can have a generic meaning (describing two features: left-ventricular enlargement and systolic dysfunction), and can be familial dilated cardiomyopathy (FDC), idiopathic dilated cardiomyopathy (IDC), which may in fact be familial in 20–50% of cases, or it may be secondary to therapeutic toxins (anthracyclines), radiation, inflammatory conditions, myocarditis, long-standing severe hypertension or thyroid disease. DCM (FDC) has been researched at the molecular level, identifying over 20 genes for autosomal dominant FDC, the commonest ones encoding the structural proteins of cardiac muscle: lamin-A/C, gene LMNA (7–8%); myosin 7, gene MYH7 (5–8%); sodium channel protein type 5 subunit alpha, gene SCN5A (2–4%), at least two genes for X-linked DCM, these genes encoding the proteins—dystrophin, gene DMD, and tafazzin, gene TAZ, and one gene for autosomal recessive inheritance DCM, encoding troponin I, cardiac muscle, gene TNNI3. Other genes are involved in metabolic causes, such as deficiencies of enzymes needed for myocardial fatty acid oxidation. First-degree relative screening should include the medical history, physical examination, an echocardiogram and ECG. Symptomatic DCM indicates late disease. Symptoms may be those of CCF (oedema, orthopnoea, paroxysmal dyspnoea), palpitations, chest pain, or exercise intolerance or syncope; signs include hypotension, weak peripheral pulses and hepatomegaly, with investigations showing cardiomegaly (CXR), arrhythmias (ECG), dilatation of the left ventricle and left atrium (echocardiography).
Familial hypertrophic cardiomyopathy (HCM): also called hypertrophic obstructive cardiomyopathy (HOCM) and idiopathic hypertrophic subaortic stenosis (IHSS)
Symptoms include failure to thrive, CCF, cyanosis, shortness of breath on exertion, dyspnoea, fatigue with exercise, chest pain, presyncope, syncope and palpitations. Signs include prominent left-ventricular apical impulse or lift, systolic murmur (increased by exercise, standing, straining; decreased by squatting), extra heart sounds (S3 and S4) and mid-diastolic rumble (mitral flow murmur with severe mitral regurgitation, with systolic anterior motion of the mitral valve). LVOT or intracavity obstruction may need provocative manoeuvres to detect their presence, such as the Valsalva manoeuvre, standing from squatting, and exercise. Investigations show cardiomegaly (CXR), right- and left-ventricular hypertrophy (RVH and LVH) in infants, LVH and abnormal Q waves in older children, LQTS in infants or older children, or arrhythmias (ECG), asymmetric septal hypertrophy, and concentric and apical hypertrophy (echocardiography or cardiac MRI). Other tests may include gated technetium-99m labelled blood pool scan (assess ejection fraction), thallium perfusion scan (regional perfusion abnormalities) and positron emission tomography (regional metabolic abnormalities).
Congestive cardiac failure (CCF)
Principles/aspects of managing CCF can be listed as follows (mnemonic ASPECTS):
A. Afterload reduction: ACEIs, ARBs, brain natriuretic peptide (BNP), milrinone, nitrates
S. Sympathetic inhibition: beta blockers, BNP, digoxin
P. Preload reduction: BNP, diuretics
E. Enhanced contractility (inotropy): digoxin
C. Cardiac remodelling prevention: mineralocorticoid inhibitors (spironolactone)
T. Timely surgical repair of structural congenital heart disease
S. Systemic disease recognition and treatment (of pathology underlying CCF)
Angiotensin-converting enzyme (ACE) inhibitors (e.g. captopril) are very useful in lowering afterload and have been shown to decrease mortality in adults (but must be avoided in HCM, as above); they are usually started in hospital due to the risk of initial dose hypotension and the worsening of any unrecognised renovascular pathology. Diuretics (e.g. combined low-dose loop and thiazide diuretics) give rapid symptomatic relief; diuretics reduce preload, which prevents high cardiac filling pressures (which could lead to pulmonary oedema). Mineralocorticoid inhibitors (e.g. spironolactone) can help prevention of maladaptive cardiac remodelling and interstitial fibrosis. Digoxin is still widely used; it acts as an inotrope, but its use remains controversial and in adults it does not increase survival in CCF—indeed, there are no studies demonstrating its efficacy. Beta blockers (e.g. metoprolol, carvedilol) also are used increasingly in carefully graduated doses. Growth hormone has been used in a small number of patients (e.g. pre-transplant) and has been associated with improved indices on echocardiography, increased exercise capacity and decreased myocardial oxygen consumption. Nesiritide, which is a recombinant form of BNP, causes both diuresis and vasodilation, decreases both preload and afterload, inhibits the sympathetic nervous system, promotes cardiac myocyte survival and inhibits cardiac fibroblast activation; it shows promise as a third-line therapy, but studies in the paediatric age group are lacking at this stage.
Cardiac transplantation
Life-saving: (CC,UU,RR): CCF with symptomatic ventricular dysfunction secondary to myocardial disease or palliated CHD; Complex CHD with failed surgical correction; Unresectable tumours; Unresectable ventricular diverticula; Rhythm disturbances: life-threatening (arrhythmias), resistant to therapy; Retransplantation (graft vasculopathy, ventricular dysfunction).
Life-enhancing: (CC,FF,II): CCF associated with pulmonary hypertension; Cardiomyopathy—restrictive, with poor survival overall; Fontan circulation with protein-losing enteropathy resistant to medical therapy; Fontan failing with decreased exercise tolerance, and declining quality of life; Inoperable AV valve or aortic regurgitation; Inoperable CHD with severe oxygen desaturation.
There are some fetal indications for listing on the transplant waiting list in North America. These include: hypoplastic left-heart syndrome, where transplantation is primary therapy; intractable arrhythmias; unresectable cardiac tumours; cardiomyopathies with poor ventricular function; right-atrial isomerism syndromes; and single-ventricle anatomy with risk factors for surgical palliation (severe atrioventricular valve regurgitation, decreased function). Candidates are listed from 35 weeks gestation and weight over 2.5 kg; if a donor heart becomes available, then patients are delivered by caesarean section, followed by immediate transplantation.
The commonest long-term side effects are related to immunosuppression:
• Coronary artery disease in up to 75% examined by intravascular ultrasound 5 years after transplant. Symptoms can include presyncope, syncope, exercise intolerance and chest pain (rare due to denervation of the transplanted heart). This form of chronic rejection is an accelerated graft vasculopathy that presents at a median of 6 years. To assess risk, coronary angiography can be done 1–2 yearly and, if positive, infers severe disease. Other imaging studies can be used: myocardial perfusion scanning, dobutamine stress echocardiography and MRI studies. Several agents are being investigated for prevention of vasculopathy, including calcium antagonists, ACEIs, vitamin E, statins, aspirin, MMF and rapamycin. Coronary artery disease may lead to need for retransplantation.
• Hypertension in up to 60% at 5 years; more in those on steroids and CSA; less with CSA alone; least with TRL alone. Aggressive treatment is required, usually with an ACE inhibitor or the calcium blocker diltiazem. For those with relevant congenital heart pathology, evaluation for residual pathology (e.g. residual coarctation) is required; check renal function yearly; nuclear scans may be needed to calculate the glomerular filtration rate (GFR).
• Neoplasia in 20%, most commonly lymphoproliferative diseases, at 6 months to 6 years after transplantation.
• Abnormal renal function in up to 25% at 5 years. With longer survival, a small number of patients are developing end-stage renal disease (ESRD) and then have a renal transplant; increased use of MMF and sirolimus (no renal toxicity) may allow decreased use of CSA and TRL and thus less nephrotoxicity.
• Osteoporosis probably occurs in 100%: steroids and calcineurin inhibitors decrease bone formation and increase bone destruction; steroids decrease calcium absorption. Supplemental calcium and vitamin D are recommended; protocols have been developed that include biphosphates, calcitonin and hormone replacement, although paediatric data is scant. Yearly bone density (DEXA) scanning is useful.
Telemedicine
Advances in telecommunications technology, with the widespread introduction of integrated services digital network (ISDN) lines, allows transmission of echocardiogram images from peripheral to tertiary centres, with a paediatric cardiologist interpreting the transmitted images and guiding the performance of the scan to ensure that the best possible views are obtained. This approach has been used successfully in many countries. The defects that must be correctly diagnosed early include those causing cyanosis, obstructive lesions of the left and right heart, and total anomalous pulmonary venous drainage (TAPVD), the latter providing the greatest diagnostic difficulty. The other two lesions that can be difficult to diagnose are coarctation of the aorta (obtaining good aortic arch views can be challenging) and patent ductus arteriosus (and, if present, ascertaining how much it contributes clinically).
Short Case
The cardiovascular system
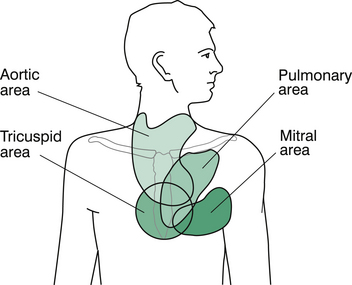
Now, turn your attention to the chest. If not already done, check for scars and asymmetry carefully. At this point, lie the child down on the examination bed. Look for the apex beat and then palpate for it. Describe the location (make a show of counting down the intercostal spaces) and the quality of the impulse. Beware of dextrocardia if the apex appears elusive. After the apex, feel the parasternal border and substernal region for heaves, and the suprasternal and supraclavicular regions for thrills, and feel over the pulmonary area for palpable closure of the pulmonary valve (i.e. ‘palpable S2’).
Additional manoeuvres may be needed to clarify suspected diagnoses. The Valsalva manoeuvre is useful in identifying hypertrophic cardiomyopathy (HCM), as it increases the intensity of the murmur (via increased intrathoracic pressure, decreased venous return and hence decreased intracardiac volume and more severe LVOTO), and in mitral valve prolapse, where the murmur is also increased and the systolic click is heard earlier. Innocent systolic outflow tract murmurs decrease in intensity in response to the Valsalva manoeuvre.
Exercising the child is especially useful in bringing out a tricuspid diastolic murmur in an atrial septal defect (ASD; see Figure 6.10); this is most easily achieved by having the child do several sit-ups. In patients with ventricular septal defects (VSDs) and ASDs, the appearance of a mid-diastolic murmur suggests a pulmonary blood flow at least twice that of the systemic circulation, and in patients with mitral or tricuspid incompetence, such a murmur suggests at least a moderate degree of regurgitation.
Figure 6.8 shows the major points as outlined above. A more comprehensive listing of possible findings on cardiovascular examination is given in Table 6.1 (see also Figure 6.9 and Figure 6.7).
Table 6.1 Additional information: possible findings on cardiovascular examination
| General observations |
| Height |
• Rate, amplitude, character (lift arm to detect hyperdynamic pulsation of Al) and rhythm
• Radial (both sides, for radioradial delay)
• Brachial and femoral together (for brachiofemoral delay in older adolescents)
• Femorals (both sides): reduction of one femoral only is not due to coarctation, but to local trauma (cardiac catheter)
Chest X-ray and electrocardiography
Chest X-ray (CXR)
Increased pulmonary vascularity occurs in the following:
1. Truncus arteriosus (Figure 6.5).
2. Total anomalous pulmonary venous drainage (TAPVD).
3. One condition of ‘textbook interest’, rarely seen outside the neonatal period nowadays, which is transposition of the great arteries (TGA).
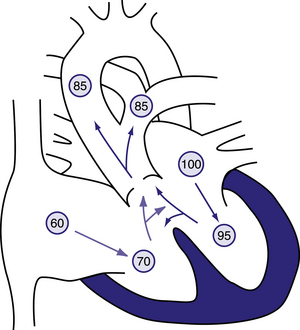
Figure 6.5 Physiology of truncus arteriosus.
Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. Desaturated blood enters the right atrium, flows through the tricuspid valve into the right ventricle, and is ejected into the truncus. Saturated blood returning from the left atrium enters the left ventricle and is also ejected into the truncus. The common aortopulmonary trunk gives rise to the ascending aorta and to the main or branch pulmonary arteries. Oxygen saturation in the aorta and pulmonary arteries is usually the same (definition of a total mixing lesion). As pulmonary vascular resistance decreases over the first few weeks of life, pulmonary blood flow increases dramatically and mild cyanosis and congestive heart failure result. Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 424.6.
Decreased pulmonary vascularity occurs in the following:
1. Pulmonary atresia with MAPCs (Figure 6.14).
2. Tricuspid atresia (Figure 6.15).
3. Ebstein’s anomaly (Figure 6.13).
4. Tetralogy of Fallot: 25% of these have a right-sided aortic arch.
5. Another cause is critical pulmonary stenosis associated with a right-to-left shunt in the neonatal period, but this again is more of ‘textbook interest’.
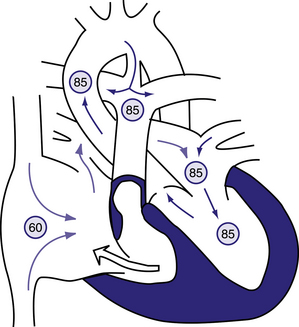
Figure 6.14 Physiology of pulmonary atresia with an intact ventricular septum.
Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. A small amount of the blood entering the right atrium may cross the tricuspid valve, which is often stenotic as well. The right-ventricular cavity is hypertrophied and may be hypoplastic. No outlet from the right ventricle exists because of the atretic pulmonary valve; thus, any blood entering the right ventricle returns to the right atrium via tricuspid regurgitation. Most of the desaturated blood shunts right to left via the foramen ovale into the left atrium, where it mixes with fully saturated blood returning from the lungs. The only source of pulmonary blood flow is via the patent ductus arteriosus. Aortic and pulmonary arterial oxygen saturation will be identical (definition of a total mixing lesion). Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 423.6.
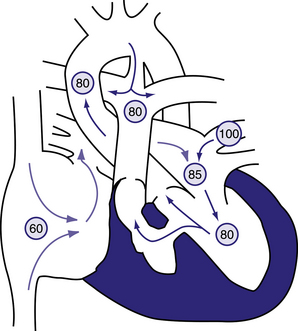
Figure 6.15 Physiology of tricuspid atresia with normally related great vessels.
Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. The tricuspid valve is non-patent, and the right ventricle may manifest varying degrees of hypoplasia. The only outlet from the right atrium involves shunting right to left across an atrial septal defect or patent foramen ovale to the left atrium. There, desaturated blood mixes with saturated pulmonary venous return. Blood enters the left ventricle and is ejected either through the aorta or via a ventricular septal defect (VSD) into the right ventricle. In this example, some pulmonary blood flow is derived from the right ventricle, the rest from a patent ductus arteriosus (PDA). In patients with tricuspid atresia, the PDA may close or the VSD may grow smaller and result in a marked decrease in systemic oxygen saturation. Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 423.7.
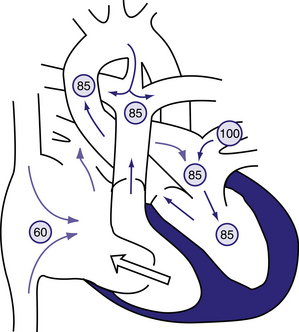
Figure 6.13 The physiology of the Ebstein anomaly of the tricuspid valve.
Circled numbers represent oxygen saturation values. Inferior displacement of the tricuspid valve leaflets into the right ventricle has resulted in a thin-walled, low-pressure ‘atrialized’ segment of right ventricle. The tricuspid valve is grossly insufficient (clear arrow). Right atrial blood flow is shunted right to left across an atrial septal defect or patent foramen ovale into the left atrium. Some blood may cross the right-ventricular outflow tract and enter the pulmonary artery; however, in severe cases, the right ventricle may generate insufficient force to open the pulmonary valve, and ‘functional pulmonary atresia’ results. In the left atrium, desaturated blood mixes with saturated pulmonary venous return. Blood enters the left ventricle and is ejected via the aorta. In this example, some pulmonary blood flow is derived from the right ventricle, the rest from a patent ductus arteriosus (PDA). Severe cyanosis will develop in neonates with a severe Ebstein anomaly when the PDA closes. Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 423.10.
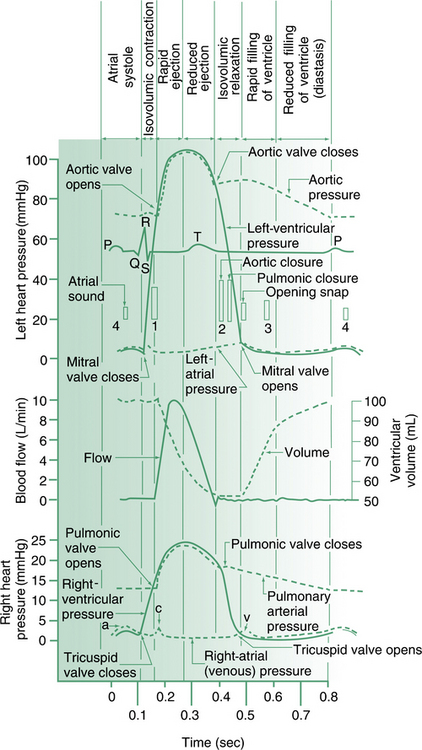
Electrocardiography
Axis
Right-axis deviation (RAD)—that is, an axis at +90° to +180° (after infancy)—is often associated with right-ventricular hypertrophy, whereas left-axis deviation (LAD) has numerous causes, including atrioventricular canal (Figure 6.11), tricuspid atresia and conduction anomalies.
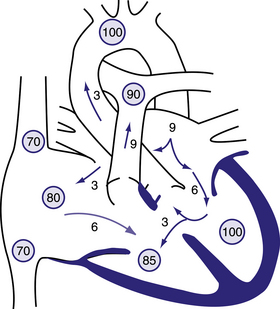
Figure 6.11 Physiology of atrioventricular septal defect (AVSD).
Circled numbers represent oxygen saturation values. The numbers next to the arrows represent volumes of blood flow (in L/min/m2). This illustration shows a hypothetical patient with a pulmonary-to-systemic blood flow ratio (Qp:Qs) of 3:1. Desaturated blood enters the right atrium from the vena cavae at a volume of 3 L/min/m2 and mixes with 3 L of fully saturated blood shunting left to right across the atrial septal defect; the result is an increase in oxygen saturation in the right atrium. Six litres of blood flows through the right side of the common AV valve, joined by an additional 3 L of saturated blood shunting left to right at the ventricular level, further increasing oxygen saturation in the right ventricle. The full 9 L flows across the right-ventricular outflow tract into the lungs. Nine litres returns to the left atrium, with 3 L shunting left to right across the defect and 6 L crossing the left side of the common AV valve and causing a mid-diastolic flow rumble. Three litres of this volume shunts left to right across the VSD, and 3 L is ejected into the ascending aorta (normal cardiac output). Behrman et al, 2007. Nelson Textbook of Paediatrics, 17th edition, Figure 419.2.
Causes of atrial hypertrophy
QRS and Q wave abnormalities
Prolonged QRS is seen in the following:
Q waves occur in the following circumstances. They are normal in leads II, III, aVf, V5, V6, and abnormal in other leads. Think of:
Specific diagnosis by ECG
Although a pathophysiological approach is, of course, preferable to a mnemonic or list in cardiology, ECGs do lend themselves to the latter. Table 6.2 is one such list, but rather than rote-learning this, the candidate should endeavour to understand the pathophysiology and always consider the ECG in the context of the clinical findings.
Table 6.2 Electrocardiographic findings and associated pathologies
| If the finding is | Think of |
|---|---|
| Prolonged P–R interval | Endocardial cushion defect |
| Ebstein anomaly | |
| Acute rheumatic fever | |
| Congenital block (maternal SLE) | |
| Partial RBBB | |
| with LAD | Ostium primum ASD |
| with RAD | Ostium secundum ASD |
| with RAH, delta waves | Ebstein anomaly |
| Complete RBBB | Post-ventriculotomy |
| LAD | Endocardial cushion defect |
| Tricuspid atresia | |
| Hypertrophic cardiomyopathy | |
| Inlet VSD | |
| RAH | Ebstein anomaly |
| without RVH | Tricuspid atresia (LAD) |
| Pulmonary atresia with intact septum | |
| with axis over 90° | Truncus arteriosus |
| Tetralogy with large VSD | |
| Deep Q waves | Hypertrophic cardiomyopathy |
| Transposition of great arteries | |
| Anomalous left coronary artery |
ASD = atrial septal defect; LAD = left axis deviation; RAD = right axis deviation; RAH = right atrial hypertrophy; RVH = right-ventricular hypertrophy; VSD = ventricular septal defect.
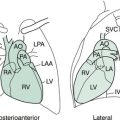
Selected diagrams
The following pages contain useful illustrations outlining the physiology of some of the more common congentital heart conditions: ASD, atrioventricular canal, VSD, Ebstein anomaly, pulmonary atresia with intact ventricular septum, tricuspid atresia, tetralogy of Fallot, and Blalock-Taussig shunt in the patient with tetralogy of Fallot.