[level-membership-for-critical-care-medicine-category]22
Cardiogenic Shock
Cardiogenic shock is the syndrome that ensues when the heart is unable to deliver enough blood to maintain adequate tissue perfusion. Acute myocardial infarction (MI) is the leading cause, but other potential etiologic factors need to be considered.1,2 Without prompt diagnosis and appropriate management, morbidity and mortality rates are substantial, approaching 60% for all age groups.2,3 Rapid evaluation and prompt initiation of supportive measures and definitive therapy in patients with cardiogenic shock may improve both early and long-term outcomes.
Definition
The clinical definition of cardiogenic shock includes decreased cardiac output and evidence of tissue hypoxia in the presence of adequate intravascular volume. The diagnosis of circulatory shock (Box 22.1) is made at the bedside by the presence of hypotension along with a combination of clinical signs indicative of poor tissue perfusion, including oliguria, clouded sensorium, and cool, mottled extremities. Hemodynamic criteria include sustained hypotension (systolic blood pressure < 90 mm Hg for at least 30 minutes) and a reduced cardiac index (<2.2 L/min/m2) in the presence of elevated filling pressures (pulmonary capillary occlusion pressure > 15 mm Hg).4 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion or correction of factors such as hypovolemia, hypoxia, and acidosis.
Epidemiology
Pump failure due to cardiogenic shock has long been known to carry a high mortality rate. The seminal article outlining prognosis after MI was a single center series of 250 patients reported by Killip in 1967.5 Killip divided patients into four classes as follows:
Killip class I: no evidence of congestive heart failure
Killip class II: presence of an S3 gallop and/or bibasilar rales
Killip class III: pulmonary edema (rales greater than halfway up the lung fields)
Nineteen percent of the 250 patients were in class IV at presentation, and their mortality rate was 81%.5
With the advent of right-sided heart catheterization, Forrester and Swan defined hemodynamic subsets after MI analogous to the clinical subsets outlined by Killip.4 Subset I consisted of patients with normal pulmonary capillary wedge pressure (PCWP) and cardiac output, subset II consisted of patients with elevated PCWP and normal cardiac output, subset III consisted of patients with normal PCWP and decreased cardiac output, and subset IV consisted of patients with elevated PCWP and decreased cardiac output.4
Despite advances in management of heart failure and acute MI, the mortality rate of patients with cardiogenic shock has remained high.2,6–8 Data suggest an increase in survival in the 1990s, coincident with the use of reperfusion strategies.7–9 Cardiogenic shock, however, remains the most common cause of death in hospitalized patients with acute MI.
Incidence
Accurate determination of the precise incidence of cardiogenic shock is difficult because patients who die of MI prior to reaching the hospital generally do not receive this diagnosis.6,10–13 Nonetheless, estimates from a variety of sources have been fairly consistent. The Worcester Heart Attack Study,6 a community-wide analysis, found an incidence of cardiogenic shock of 7.5%, an incidence that remained fairly stable from 1975 to 1997.6,9 The incidence was similar in the randomized GUSTO (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) trial (7.2%),14 in other multicenter thrombolytic trials,10–12 and in patients with ST-segment elevation MI in the National Registry of Myocardial Infarction (NRMI) database from 1995 to 2004 (8.6%).7 More recently, however, the incidence of cardiogenic shock has fallen from about 8% to about 6% of MIs, with most of the change resulting from a decrease in cardiogenic shock developing after initial presentation, supporting the notion that early revascularization strategies are an important contributor to the decline.3
Etiology
The most common cause of cardiogenic shock is left ventricular failure in the setting of an extensive acute MI, although a smaller infarction in a patient with previously compromised left ventricular function may also precipitate shock. Cardiogenic shock can also be caused by mechanical complications such as acute mitral regurgitation, rupture of the interventricular septum, or rupture of the free wall—or by large right ventricular infarctions. In a report of the SHOCK (SHould we emergently revascularize Occluded Coronaries for shocK) trial registry of 1160 patients with cardiogenic shock,2 78.5% of patients had predominant left ventricular failure, 6.9% had acute mitral regurgitation, 3.9% had ventricular septal rupture, 2.8% had isolated right ventricular shock, 1.4% had tamponade or cardiac rupture, and 6.5% had shock resulting from other causes.
Other causes of cardiogenic shock include myocarditis, end-stage cardiomyopathy, myocardial contusion, septic shock with severe myocardial depression, myocardial dysfunction after prolonged cardiopulmonary bypass, valvular heart disease, and hypertrophic obstructive cardiomyopathy (Box 22.2). An important consideration is that some cardiogenic shock may have an iatrogenic component. Early diagnosis of impending shock or of patients at high risk for development of shock is essential, both to speed intervention and to avoid therapies that may worsen hemodynamics. In many cases of cardiogenic shock in the setting of MI, the diagnosis is not made until the patient has been triaged and admitted to an inpatient setting. Patients may have received early beta blockade or angiotensin-converting enzyme inhibition, therapies that may impact hemodynamics substantially.
Patients may have cardiogenic shock at initial presentation, but most do not; shock usually evolves over several hours,15,16 suggesting that early treatment may potentially prevent shock. In fact, some data indicate that early thrombolytic therapy may decrease the incidence of cardiogenic shock.17 In the SHOCK trial registry, 75% of patients developed cardiogenic shock within 24 hours after presentation, with a median delay of 7 hours.2 Results from the GUSTO trial were similar;13 among patients with shock, 11% were in shock on arrival and 89% developed shock after admission.
Risk factors for the development of cardiogenic shock in MI generally parallel those for left ventricular dysfunction and the severity of coronary artery disease. Shock is more likely to develop in patients who are elderly, are diabetic, and have anterior MI.5,15,18,19 Patients with cardiogenic shock are also more likely to have histories of previous infarction, peripheral vascular disease, and cerebrovascular disease.18,19 Decreased ejection fractions and larger infarctions (as evidenced by higher cardiac enzymes) are also predictors of the development of cardiogenic shock.18,19 Analysis from the GUSTO-3 trial has identified age, lower systolic blood pressure, heart rate, and Killip class as significant predictors of the risk for development of cardiogenic shock after presentation with acute MI.20 Use of a predictive scoring system derived from this study may be useful in identifying patients at high risk for the development of cardiogenic shock and targeting such patients for closer monitoring.20
Cardiogenic shock is most often associated with anterior MI. In the SHOCK trial registry, 55% of infarctions were anterior, 46% were inferior, 21% were posterior, and 50% were in multiple locations.2 These findings were consistent with those in other series.21 Angiographic evidence most often demonstrates multivessel coronary disease (left main occlusion in 20% of patients, three-vessel disease in 64%, two-vessel disease in 23%, and one-vessel disease in 13% of patients).22 The high prevalence of multivessel coronary artery disease is important because compensatory hyperkinesis normally develops in myocardial segments that are not involved in an acute MI, and this response helps maintain cardiac output. Failure to develop such a response, because of previous infarction or high-grade coronary stenoses, is an important risk factor for cardiogenic shock and death.16,23
Pathogenesis
Systemic Effects
Cardiac dysfunction in patients with cardiogenic shock is usually initiated by MI or ischemia. The myocardial dysfunction resulting from ischemia worsens that ischemia, creating a downward spiral24 (Fig. 22.1). When a critical mass of ischemic or necrotic left ventricular myocardium fails to pump, stroke volume and cardiac output decrease. Myocardial perfusion, which depends on the pressure gradient between the coronary arterial system and the left ventricle and on the duration of diastole, is compromised by hypotension and tachycardia, exacerbating ischemia. The increased ventricular diastolic pressures caused by pump failure reduce coronary perfusion pressure, and the additional wall stress elevates myocardial oxygen requirements, further worsening ischemia. Decreased cardiac output also compromises systemic perfusion.
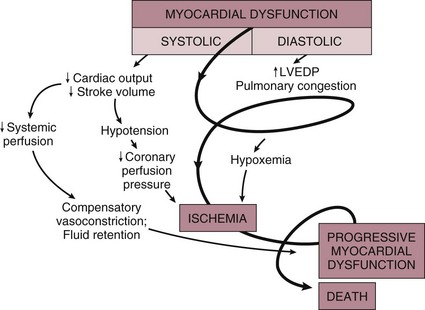
When myocardial function is depressed, several compensatory mechanisms are activated, including sympathetic stimulation to increase heart rate and contractility and renal fluid retention to increase preload. These compensatory mechanisms may become maladaptive and can actually worsen the situation when cardiogenic shock develops. Increased heart rate and contractility increase myocardial oxygen demand and exacerbate ischemia. Fluid retention and impaired diastolic filling caused by tachycardia and ischemia may result in pulmonary congestion and hypoxia. Vasoconstriction to maintain blood pressure increases myocardial afterload, further impairing cardiac performance and increasing myocardial oxygen demand. This increased demand, in the face of inadequate perfusion, worsens ischemia and begins a vicious cycle that will end in death if not interrupted (see Fig. 22.1). The interruption of this cycle of myocardial dysfunction and ischemia forms the basis for the therapeutic regimens for cardiogenic shock.
Not all patients fit into this classic paradigm. In the SHOCK trial, the average systemic vascular resistance (SVR) was not elevated, and the range of values was wide, suggesting that compensatory vasoconstriction is not universal. Some patients had fever and elevated white blood cell counts along with decreased SVR, suggesting a systemic inflammatory response syndrome.25 This has led to an expansion of the paradigm to include the possibility of the contribution of inflammatory responses to vasodilation and myocardial stunning, leading clinically to persistence of shock (Fig. 22.2).25 Supporting this notion is the fact that the mean ejection fraction in the SHOCK trial was only moderately decreased (30%), suggesting that mechanisms other than pump failure were operative.25 Immune activation appears to be common to a number of different forms of shock. Activation of inducible nitric oxide synthase (iNOS) with production of nitric oxide and peroxynitrate has been proposed as one potential mechanism.
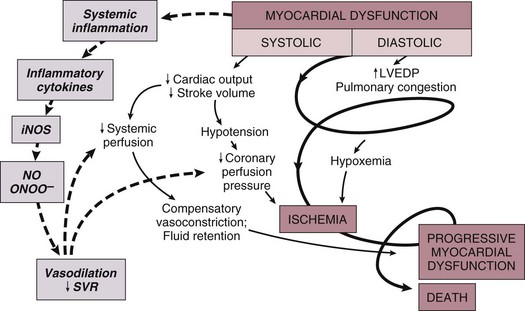
Figure 22.2 Expansion of the pathophysiologic paradigm of cardiogenic shock to include the potential contribution of inflammatory mediators. Inhibition of nitric oxide, however, has not been shown to be beneficial in patients with cardiogenic shock.149 iNOS, inducible nitric oxide synthase; LVEDP, left ventricular end-diastolic pressure; NO, nitric oxide; ONOO−, peroxynitrite; SVR, systemic vascular resistance. (Adapted from Hochman JS: Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation 2003;107:2999.)
Myocardial Pathology
Cardiogenic shock is characterized by both systolic and diastolic myocardial dysfunction.16,26 Progressive myocardial necrosis has been observed consistently in clinical and pathologic studies of patients with cardiogenic shock.16,27 Patients who develop shock after admission often have evidence of infarct extension, which can result from reocclusion of a transiently patent infarct artery, propagation of intracoronary thrombus, or a combination of decreased coronary perfusion pressure and increased myocardial oxygen demand.18,19 Myocytes at the border zone of an infarction are more susceptible to additional ischemic episodes; therefore, these adjacent segments are at particular risk.28 Mechanical infarct expansion, which is seen most dramatically after extensive anterior MI, can also contribute to late development of cardiogenic shock.18,29
Ischemia remote from the infarct zone may be particularly important in producing systolic dysfunction in patients with cardiogenic shock.23,30 Patients with cardiogenic shock usually have multivessel coronary artery disease,2,16 with limited vasodilator reserve, impaired autoregulation, and consequent pressure-dependent coronary flow in several perfusion territories.31 Hypotension and metabolic derangements thus have the potential to impair the contractility of noninfarcted myocardium in patients with shock.32 This can limit hyperkinesis of uninvolved segments, a compensatory mechanism typically seen early after MI.23,30
Myocardial diastolic function is also impaired in patients with cardiogenic shock. Myocardial ischemia causes decreased compliance, increasing the left ventricular filling pressure at a given end-diastolic volume.33,34 Compensatory increases in left ventricular volumes to maintain stroke volume further increase filling pressures. Elevation of left ventricular pressures can lead to pulmonary edema and hypoxemia (see Fig. 22.1).
Cellular Pathology
Tissue hypoperfusion and consequent cellular hypoxia lead to anaerobic glycolysis, with depletion of adenosine triphosphate and intracellular energy reserves. Anaerobic glycolysis also causes accumulation of lactic acid and resultant intracellular acidosis. Failure of energy-dependent ion transport pumps decreases transmembrane potential, causing intracellular accumulation of sodium and calcium and myocyte swelling.35 Cellular ischemia and intracellular calcium accumulation can activate intracellular proteases.36 If the ischemia is severe and prolonged enough, myocardial cellular injury can become irreversible, with the classic pattern of myonecrosis: mitochondrial swelling, accumulation of denatured proteins and chromatin in the cytoplasm, lysosomal breakdown, and fracture of the mitochondria, nuclear envelope, and plasma membrane.35,36
Accumulating evidence indicates that apoptosis (programmed cell death) may also contribute to myocyte loss in MI.28,36,37 Although myonecrosis clearly outweighs apoptosis in the core of an infarcted area, evidence for apoptosis has been found consistently in the border zone of infarcts after ischemia and reperfusion and sporadically in areas remote from the ischemia area.28,37 Activation of inflammatory cascades, oxidative stress, and stretching of myocytes have been proposed as mechanisms that activate the apoptotic pathways.36,37 Although the magnitude of apoptotic cell loss in MI remains uncertain, inhibitors of apoptosis have been found to attenuate myocardial injury in animal models of postischemic reperfusion; these inhibitors may also have therapeutic potential for myocyte salvage after large infarctions.37
Reversible Myocardial Dysfunction
A key to understanding the pathophysiology and treatment of cardiogenic shock is to realize that large areas of nonfunctional but viable myocardium can also cause or contribute to the development of cardiogenic shock in patients after MI (Fig. 22.3). This reversible dysfunction can be described in two main categories: stunning and hibernation.
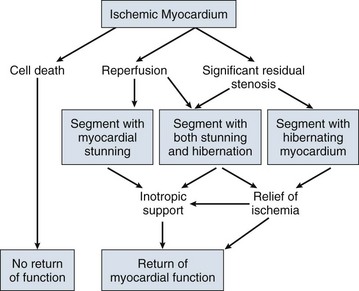
Myocardial stunning represents postischemic dysfunction that persists despite restoration of normal blood flow; eventually, however, myocardial performance recovers completely.38 Originally defined in animal models of ischemia and reperfusion,39 stunning has been recognized in the clinical arena.38,40 Direct evidence for myocardial stunning in humans has been found using positron emission tomography (PET) scanning in patients with persistent wall motion abnormalities after angioplasty for acute coronary syndromes; perfusion measured by 13N-ammonia was normal in the presence of persistent contractile dysfunction.41 The pathogenesis of stunning has not been conclusively established but appears to involve a combination of oxidative stress,42 perturbation of calcium homeostasis, and decreased myofilament responsiveness to calcium.38,43,44 In addition to these direct effects, data from studies in isolated cardiac myocytes suggest that circulating myocardial depressant substances may contribute to contractile dysfunction in myocardial stunning.45 The intensity of stunning is determined primarily by the severity of the antecedent ischemic insult.38
Myocardial hibernation comprises segments with persistently impaired function at rest due to severely reduced coronary blood flow; inherent in the definition of hibernating myocardium is the notion that function can be normalized by improving blood flow.46–48 Hibernation can be seen as an adaptive response to reduce contractile function of hypoperfused myocardium and restore equilibrium between flow and function, thereby minimizing the potential for ischemia or necrosis.49 Revascularization of hibernating myocardium can lead to improved myocardial function,50 and improved function appears to translate into improved prognosis.51,52
Although hibernation is conceptually and pathophysiologically different from myocardial stunning, the two conditions are difficult to distinguish in the clinical setting and may in fact coexist.38,52 Repetitive episodes of myocardial stunning can coexist with or mimic myocardial hibernation.38,46,53 Consideration of myocardial stunning and hibernation is vital in patients with cardiogenic shock because of their therapeutic implications. Hibernating myocardium improves with revascularization, and stunned myocardium retains inotropic reserve and can respond to inotropic stimulation.38 In addition, the fact that the severity of the antecedent ischemic insult determines the intensity of stunning38 provides one of the rationales for reestablishment of patency of occluded coronary arteries in patients with cardiogenic shock. Finally, the notion that some myocardial tissue may recover function emphasizes the importance of measures to support hemodynamics and thus minimize myocardial necrosis in patients with shock.
Clinical Assessment
Evaluation
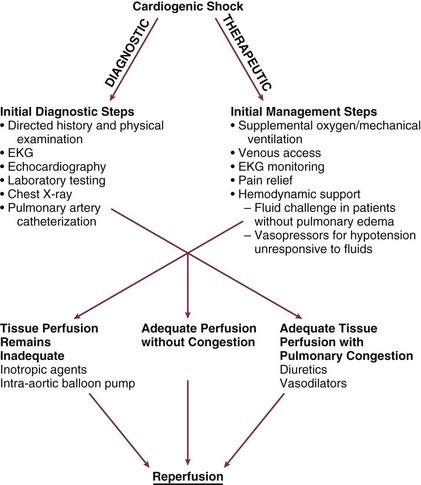
Cardiogenic shock is an emergency. The clinician must initiate therapy before shock irreversibly damages vital organs; at the same time, he or she must perform the clinical assessment required to understand the cause of shock and to target therapy to that cause. A practical approach is to make a rapid initial evaluation on the basis of a limited history, physical examination, and specific diagnostic procedures (Fig. 22.4).24 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion of alternative causes of hypotension such as hypovolemia, hemorrhage, sepsis, pulmonary embolism, tamponade, aortic dissection, and preexisting valvular disease.
Echocardiography is an excellent initial tool for confirming the diagnosis of cardiogenic shock and ruling out other causes of shock (Box 22.3); therefore, early echocardiography should be routine. Echocardiography provides information on overall and regional systolic function, and can rapidly diagnose mechanical causes of shock such as papillary muscle rupture and acute mitral regurgitation, acute ventricular septal defect, and free wall rupture and tamponade.54,55 Unsuspected severe mitral regurgitation is not uncommon. In some cases, echocardiography may reveal findings compatible with right ventricular infarction.
Invasive hemodynamic monitoring can be quite useful to exclude volume depletion, right ventricular infarction, and mechanical complications.16,35 The hemodynamic profile of cardiogenic shock includes a pulmonary capillary occlusion pressure greater than 15 mm Hg and a cardiac index less than 2.2 L/min/m2. It should be recognized that optimal filling pressures may be greater than 15 mm Hg in individual patients due to left ventricular diastolic dysfunction. Right-sided heart catheterization may reveal an oxygen step-up diagnostic of ventricular septal rupture or a large v wave that suggests severe mitral regurgitation. The hemodynamic profile of right ventricular infarction includes high right-sided filling pressures in the presence of normal or low occlusion pressures.56,57
Initial Management
Maintenance of adequate oxygenation and ventilation are critical; intubation and mechanical ventilation are often required, if only to reduce the work of breathing and facilitate sedation and stabilization before cardiac catheterization. Central venous and arterial access, bladder catheterization, and pulse oximetry are routine. Electrolyte abnormalities should be corrected. Hypokalemia and hypomagnesemia are predisposing factors to ventricular arrhythmias, and acidosis can decrease contractile function. Relief of pain and anxiety with morphine sulfate (or fentanyl if systolic pressure is compromised) can reduce excessive sympathetic activity and decrease oxygen demand, preload, and afterload. Arrhythmias and heart block may have major effects on cardiac output, and should be corrected promptly with antiarrhythmic drugs, cardioversion, or pacing. Cardiology consultation has been shown to be associated with improved outcomes in patients with MI and is strongly indicated in the setting of cardiogenic shock.58 In addition, measures proven to improve outcome after MI, such as nitrates, beta blockers, and angiotensin-converting enzyme inhibitors,59 have the potential to exacerbate hypotension in cardiogenic shock and should be stopped until the patient stabilizes.
Therapy
Following initial stabilization and restoration of adequate blood pressure, tissue perfusion should be assessed (see Fig. 22.4). If tissue perfusion remains inadequate, inotropic support or intra-aortic balloon pumping (IABP) should be initiated. If tissue perfusion is adequate but significant pulmonary congestion remains, diuretics may be employed. Vasodilators can be considered as well, depending on the blood pressure.
The initial approach to the hypotensive patient should include fluid resuscitation unless frank pulmonary edema is present. Patients are commonly diaphoretic and relative hypovolemia may be present. In the original description of hemodynamic subsets in MI, approximately 20% of patients had low cardiac index and low PCWP; most had reduced stroke volume and compensatory tachycardia.60 Some of these patients would be expected to respond to fluid infusion with an increase in stroke volume, although the magnitude of such a response depends on the degree of ischemia and cardiac reserve.
Fluid infusion is best initiated with predetermined boluses titrated to clinical end points of heart rate, urine output, and blood pressure.61 Ischemia produces diastolic as well as systolic dysfunction, and thus elevated filling pressures may be necessary to maintain stroke volume in patients with cardiogenic shock. Patients who do not respond rapidly to initial fluid boluses or those with poor physiologic reserve should be considered for invasive hemodynamic monitoring. Optimal filling pressures vary from patient to patient; hemodynamic monitoring can be used to construct a Starling curve at the bedside, identifying the filling pressure at which cardiac output is maximized. Maintenance of adequate preload is particularly important in patients with right ventricular infarction.
When arterial pressure remains inadequate, therapy with vasopressor agents, titrated not only to blood pressure but to clinical indices of perfusion and mixed venous oxygen saturation, may be required to maintain coronary perfusion pressure. Maintenance of adequate blood pressure is essential to break the vicious cycle of progressive hypotension with further myocardial ischemia. Norepinephrine is preferable to dopamine for hypotension in this situation. Dopamine acts as both an inotrope (particularly 3-10 µg/kg/minute) and a vasopressor (10-20 µg/kg/minute). Norepinephrine (0.02-1.0 µg/kg/minute) acts primarily as a vasoconstrictor, has a mild inotropic effect, and increases coronary flow. A recent randomized trial comparing norepinephrine and dopamine in 1678 patients with shock found no significant difference in 28-day mortality rate in the overall trial, but a prespecified subgroup analysis did find increased mortality rate with dopamine in the 280 patients with cardiogenic shock.62 Phenylephrine, a selective α1-adrenergic agonist, may be employed to support blood pressure when tachyarrhythmias limit therapy with other vasopressors, although it does not improve cardiac output. Vasopressin, which causes vasoconstriction, has a neutral or slightly depressant effect upon cardiac output, and increases vascular sensitivity to norepinephrine, may be added to catecholamines if needed.
In patients with inadequate tissue perfusion and adequate intravascular volume, cardiovascular support with inotropic agents should be initiated. Dobutamine, a selective β1-adrenergic receptor agonist, can improve myocardial contractility and increase cardiac output without markedly changing heart rate or SVR; it is the initial agent of choice in patients with systolic pressures greater than 90 mm Hg.63–65 Dobutamine may exacerbate hypotension in some patients and can precipitate tachyarrhythmias. Phosphodiesterase inhibitors such as milrinone increase intracellular cyclic adenosine monophosphate (cAMP) by mechanisms not involving adrenergic receptors, producing both positive inotropic and vasodilatory actions. Milrinone has fewer chronotropic and arrhythmogenic effects than catecholamines.66 In addition, because milrinone does not stimulate adrenergic receptors directly, its effects may be additive to those of the catecholamines.67 Milrinone, however, has the potential to cause hypotension and has a long half-life; in patients with tenuous clinical status, its use is often reserved for situations in which other agents have proved ineffective.16 Standard administration of milrinone calls for a loading dose followed by an infusion, but most clinicians eschew the loading dose in patients with marginal blood pressure.
Levosimendan, a calcium sensitizer, has both inotropic and vasodilatory properties and does not increase myocardial oxygen consumption. Levosimendan reduces the calcium-binding coefficient of troponin C by stabilizing the conformational shape, which enhances myocardial contraction with lower intracellular calcium concentrations.68 Several relatively small studies have shown hemodynamic benefits with levosimendan in cardiogenic shock after MI,69 one suggesting a better hemodynamic effect than dobutamine,70 but survival benefits with use of levosimendan have not been shown in either cardiogenic shock or acute heart failure.71 Levosimendan has the potential to cause hypotension and thus should be used with some caution in patients with cardiogenic shock. Levosimendan is not available in the United States.
Infusions of vasoactive agents need to be titrated carefully in patients with cardiogenic shock to maximize coronary perfusion pressure with the least possible increase in myocardial oxygen demand. Invasive hemodynamic monitoring can be extremely useful in allowing optimization of therapy in these unstable patients, because clinical estimates of filling pressure can be unreliable;72 in addition, changes in myocardial performance and compliance and therapeutic interventions can change cardiac output and filling pressures precipitously. Optimization of filling pressures and serial measurements of cardiac output (and other parameters, such as mixed venous oxygen saturation) allow for titration of the dosage of inotropic agents and vasopressors to the minimum dosage required to achieve the chosen therapeutic goals. This control minimizes the increases in myocardial oxygen demand and arrhythmogenic potential.61,73
Diuretics should be used to treat pulmonary congestion and enhance oxygenation. Vasodilators should be used with extreme caution in the acute setting owing to the risk of precipitating further hypotension and decreasing coronary blood flow. After blood pressure has been stabilized, however, vasodilator therapy can decrease both preload and afterload. Sodium nitroprusside is a balanced arterial and venous vasodilator that decreases filling pressures and can increase stroke volume in patients with heart failure by reducing afterload.74 Nitroglycerin is an effective venodilator that reduces the pulmonary capillary occlusion pressure and can decrease ischemia by reducing left ventricular filling pressure and redistributing coronary blood flow to the ischemic zone.75 Both agents may cause acute and rapid decreases in blood pressure and dosages must be titrated carefully; invasive hemodynamic monitoring can be useful in optimizing filling pressures when these agents are used.
Thrombolytic Therapy
Although it has been demonstrated convincingly that thrombolytic therapy reduces mortality rates in patients with acute MI,10,76–78 the benefits of this therapy in patients with cardiogenic shock are less certain. It is clear that thrombolytic therapy can reduce the likelihood of subsequent development of shock after initial presentation.14,76,77,79 This is important because most patients develop cardiogenic shock more than 6 hours after hospital presentation.2,14
Nonetheless, no trials have demonstrated that thrombolytic therapy reduces mortality rate in patients with established cardiogenic shock. The numbers of patients are small because most thrombolytic trials have excluded patients who have cardiogenic shock at presentation.80 In the GISSI (Gruppo Italiano per lo Studio della Streptochinasi Nell’Infarto Miocardico) trial,10,80 30-day mortality rates were 69.9% in 146 patients with cardiogenic shock who received streptokinase and 70.1% in 134 patients receiving placebo. The International Study Group reported a mortality rate of 65% in 93 patients with shock treated with streptokinase and a mortality rate of 78% in 80 patients treated with recombinant tissue plasminogen activator (rt-PA).12 In the GUSTO trial,13 315 patients had shock on arrival; mortality rate was 56% in patients treated with streptokinase and 59% in patients treated with rt-PA.14,81
The failure of thrombolytic therapy to improve survival in patients with cardiogenic shock may seem paradoxical in light of evidence that the absolute reduction in mortality rate with thrombolytics is greatest in those at highest risk at presentation. The meta-analysis performed by the Fibrinolytic Therapy Trialists (FTT) Collaborative Group demonstrated a reduction in mortality rate from 36.1% to 29.7% when thrombolytic therapy was used in patients with initial systolic blood pressures less than 100 mm Hg. In patients with initial heart rates greater than 100 beats per minute the mortality rate decreased from 23.8% to 18.9%.82 However, most patients in these subgroups did not meet criteria for cardiogenic shock.
Consideration of the efficacy of thrombolytic therapy once cardiogenic shock has been established makes the disappointing results in this subgroup of patients easier to understand. The degree of reperfusion correlates with outcome,79,83 and reperfusion has been shown to be less likely for patients in cardiogenic shock.21,83,84 When reperfusion is successful, mortality rate has been shown to be significantly reduced.21 The lower rates of reperfusion in patients with shock may explain some of the disappointing results in this subgroup in the thrombolytic trials.
The reasons for decreased thrombolytic efficacy in patients with cardiogenic shock include hemodynamic, mechanical, and metabolic factors. Decreased arterial pressure limits the penetration of thrombolytic agents into a thrombus.85 Passive collapse of the infarct artery in the setting of hypotension can also contribute to decreased thrombolytic efficacy, as can acidosis, which inhibits the conversion of plasminogen to plasmin.85 Two small studies support the notion that vasopressor therapy to increase aortic pressure improves thrombolytic efficacy.86,87
Intra-aortic Balloon Pumping
IABP reduces systolic afterload and augments diastolic perfusion pressure, increasing cardiac output and improving coronary blood flow.88,89 These beneficial effects, in contrast to those of inotropic or vasopressor agents, occur without an increase in oxygen demand. IABP is efficacious for initial stabilization of patients with cardiogenic shock.90,91 Small randomized trials in the prethrombolytic era, however, failed to show that IABP alone increases survival.92,93 IABP alone does not substantially improve blood flow distal to a critical coronary stenosis.94
IABP is probably not best used as an independent modality to treat cardiogenic shock. It may, however, be an essential support mechanism to allow definitive therapeutic measures to be undertaken. In the GUSTO trial, patients who presented with shock and had early IABP placement showed a trend toward lower mortality rates, even after exclusion of patients who underwent revascularization.13,95 A similar trend was seen in the SHOCK trial registry, although it did not persist after adjustment for age and catheterization.2 Several observational studies have also suggested that IABP can improve outcome in patients with shock, although revascularization procedures are a confounding factor in these studies.96–99 IABP has been shown to decrease reocclusion and cardiac events after emergency angioplasty for acute MI.100,101 The TACTICS trial randomized 57 patients with MI complicated by hypotension or cardiogenic shock to IABP or placebo in conjunction with fibrinolysis; the trial was terminated early due to difficulties with enrollment and was thus underpowered.102 Although there was no difference in the primary end point of 6-month mortality rate (34% versus 43%, p = 0.23), patients presenting in Killip class III or IV heart failure showed a trend toward benefit with IABP (39% versus 80%, p = 0.05).102
Revascularization
Direct Coronary Angioplasty
Reestablishment of brisk (TIMI [Thrombolysis in Myocardial Infarction] grade 3) flow in the infarct-related artery is an important determinant of left ventricular function and survival after MI.79 Direct percutaneous transluminal coronary angioplasty (PTCA) can achieve TIMI grade 3 flow in 80% to 90% of patients with MI103–105 compared with rates of 50% to 60% 90 minutes after thrombolytic therapy.79,106 In addition to improving wall motion in the infarct territory, increased perfusion of the infarct zone has been associated with augmented contraction of remote myocardium, possibly due to recruitment of collateral blood flow.23
Use of angioplasty in patients with cardiogenic shock grew out of its use as primary therapy in patients with MI.21,107–117 Observational studies from registries of randomized trials, most notably the GUSTO-1 trial, have also reported improved outcomes in patients with cardiogenic shock selected for revascularization,14,84,118 and these findings have also been confirmed in reports from NRMI.119
Randomized Studies
Prompt revascularization is the only intervention that has been shown consistently to reduce mortality rates in cardiogenic shock. In the landmark SHOCK trial, patients with shock caused by left ventricular failure complicating ST-segment elevation myocardial infarction (STEMI) were randomized to emergency revascularization (n = 152), accomplished by either coronary artery bypass grafting (CABG) or angioplasty, or initial medical stabilization (n = 150). IABPs were used in 86% of patients in both groups. The landmark “Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock” (SHOCK) study 120,121 was a randomized, multicenter international trial that assigned patients with cardiogenic shock to receive optimal medical management—including IABP and thrombolytic therapy—or to cardiac catheterization with revascularization using PTCA or CABG.120 The trial enrolled 302 patients and was powered to detect a 20% absolute decrease in 30-day all-cause mortality rates. Mortality rate at 30 days was 46.7% in patients treated with early intervention and 56% in patients treated with initial medical stabilization, but this difference did not quite reach statistical significance (p = 0.11).120 It is important to note that the control group (patients who received medical management) had a lower mortality rate than that reported in previous studies; this may reflect the aggressive use of thrombolytic therapy (64%) and balloon pumping (86%) in these control subjects. These data provide indirect evidence that the combination of thrombolysis and IABP may produce the best outcomes when cardiac catheterization is not immediately available. At 6 months, mortality rate in the SHOCK trial was reduced significantly (50.3% compared with 63.1%, p = 0.027),120 and this risk reduction was maintained at 12 months (mortality rate 53.3% versus 66.4%, p < 0.03) (Fig. 22.5).121 Encouragingly, this 13% absolute improvement in survival remained stable at both 3 and 6 years of follow-up.122 In addition, most survivors have good functional status.123
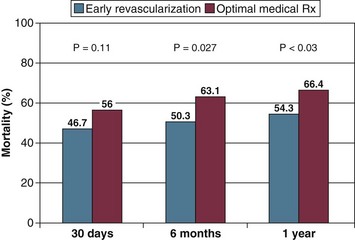
Subgroup analysis showed a substantial improvement in mortality rates in patients younger than 75 years of age at both 30 days (41.4% versus 56.8%, p = 0.01) and 6 months (44.9% versus 65.0%, p = 0.003).120 For patients older than 75, no benefit of revascularization was demonstrated in the SHOCK trial, although this was a small subgroup, and further analysis suggested baseline differences so that the elderly patients randomized to medical therapy appeared to have been a lower-risk group.124 In the SHOCK trial registry, elderly patients treated with early revascularization had better outcomes than those treated medically, suggesting that it is possible to select elderly patients who will benefit from aggressive treatment.125
The SMASH (Swiss Multicenter Trial of Angioplasty for Shock) trial was independently conceived and had a very similar design, although a more rigid definition of cardiogenic shock resulted in enrollment of sicker patients and a higher mortality rate.126 The trial was terminated early due to difficulties in patient recruitment for two different reasons: Early on, several centers declined to participate because it was felt that it would not be ethical to undertake early invasive evaluation in such extremely ill patients, and then, after publication of several encouraging studies documenting the superiority of percutaneous coronary intervention (PCI) over thrombolysis for acute MI, many centers felt that it had become unethical not to proceed to early evaluation and revascularization.127 In the SMASH trial, although the patient numbers were very small (55 patients in all), an absolute reduction in 30-day mortality rate similar to that seen in the SHOCK trial was observed (69% mortality rate in the invasive group versus 78% in the medically managed group, RR [relative risk] = 0.88, 95% CI [confidence interval] = 0.6-1.2, p = NS [not significant]).126 This benefit was also maintained at 1 year.
When the results of both the SHOCK and SMASH trials are put into perspective with results from other randomized, controlled trials of patients with acute MI, an important point emerges: Despite the moderate relative risk reduction (for the SHOCK trial, RR 0.72, CI 0.54-0.95; for the SMASH trial, RR 0.88, CI, 0.60-1.20) the absolute benefit is important, with 9 lives saved for 100 patients treated at 30 days in both trials, and 13.2 lives saved for 100 patients treated at 1 year in the SHOCK trial. This latter figure corresponds to a number needed to treat of 7.6, one of the lowest figures ever observed in a randomized, controlled trial of cardiovascular disease. In our judgment, these data strongly support the superiority of a strategy of early revascularization in most patients with cardiogenic shock (see Fig. 22.4). In the latest ACC/AHA guidelines for the management of acute MI, primary coronary intervention was given a class I indication for patients younger than 75 and a class IIa indication for patients older than 75.128
Coronary Artery Bypass Surgery
Analysis of the SHOCK trial helps to define the indications for CABG in the setting of cardiogenic shock. CABG should be the first line of therapy offered in cases of left main artery disease or triple-vessel disease as well as in cases in which the patient has sustained mechanical complications necessitating surgical repair. In patients with multivessel disease in the SHOCK trial, complete revascularization was achieved more frequently (87% versus 23%). Long-term mortality rates were similar in the CABG and PCI groups despite worse coronary anatomy and more diabetes in the surgical group.129
Other Causes of Cardiogenic Shock
Right Ventricular Infarction
Right ventricular infarction occurs in up to 30% of patients with inferior infarction and is clinically significant in 10%.130 Patients present with hypotension, elevated jugular venous pressure, and clear lung fields. The diagnosis is made by identifying ST-segment elevation in right precordial leads or by characteristic hemodynamic findings on right-sided heart catheterization (elevated right atrial and right ventricular end-diastolic pressures with normal to low pulmonary artery occlusion pressure and low cardiac output). Echocardiography can demonstrate depressed right ventricular contractility.57 Patients with cardiogenic shock on the basis of right ventricular infarction have a better prognosis than those with left-sided pump failure.130 This difference may be due in part to the fact that right ventricular function tends to return to normal over time with supportive therapy,131 although such therapy may need to be prolonged.
Supportive therapy for patients with right ventricular infarction begins with maintenance of right ventricular preload with fluid administration. In some cases, however, fluid resuscitation may increase pulmonary capillary occlusion pressure but may not increase cardiac output, and overdilation of the right ventricle can compromise left ventricular filling and cardiac output.131 Inotropic therapy with dobutamine may be more effective in increasing cardiac output in some patients, and monitoring with serial echocardiograms may also be useful to detect right ventricular overdistention.131 Maintenance of atrioventricular synchrony is also important in these patients to optimize right ventricular filling.57 For patients with continued hemodynamic instability, IABP may be useful, particularly because elevated right ventricular pressures and volumes increase wall stress and oxygen consumption and decrease right coronary perfusion pressure, exacerbating right ventricular ischemia.
Reperfusion of the occluded coronary artery is also crucial. Restoration of normal flow by direct angioplasty resulted in dramatic recovery of right ventricular function and a mortality rate of only 2%, whereas unsuccessful reperfusion was associated with persistent hemodynamic compromise and a mortality rate of 58%.132 Prompt revascularization of patients with right ventricular infarction is a class I recommendation in the American College of Cardiology/American Heart Association (ACC/AHA) guidelines for the treatment of acute MI.133
Acute Mitral Regurgitation
Ischemic mitral regurgitation is usually associated with inferior MI and ischemia or infarction of the posterior papillary muscle, which has a single blood supply, usually from the posterior descending branch of a dominant right coronary artery.134 Papillary muscle rupture typically occurs 2 to 7 days after acute MI and presents dramatically with pulmonary edema, hypotension, and cardiogenic shock. When a papillary muscle ruptures, the murmur of acute mitral regurgitation may be limited to early systole because of rapid equalization of pressures in the left atrium and left ventricle. More importantly, the murmur may be soft or inaudible, especially when cardiac output is low.135
Echocardiography is extremely useful in the differential diagnosis, which includes free wall rupture, ventricular septal rupture, and infarct extension with pump failure. Hemodynamic monitoring with pulmonary artery catheterization may also be helpful. Management includes afterload reduction with nitroprusside and IABP as temporizing measures. Inotropic or vasopressor therapy may also be needed to support cardiac output and blood pressure. Definitive therapy, however, is surgical valve repair or replacement, which should be undertaken as soon as possible because clinical deterioration can be sudden.135,136 Although mortality rate is 20% to 40%, survival and ventricular function are improved compared with medical therapy.137
Ventricular Septal Rupture
Rapid stabilization, using IABP and pharmacologic measures followed by operative repair, is the only viable option for long-term survival. Because perforations are exposed to shear forces, the rupture site can expand abruptly. Repair can be technically difficult owing to the need to suture in areas of necrosis. Surgical mortality rate is 20% to 50%, especially for serpiginous inferoposterior ruptures, which typically are less well circumscribed than anteroapical ruptures. Right ventricular function is an important determinant of outcome in this setting. Timing of surgery has been controversial, but guidelines now recommend that operative repair should be undertaken early, within 48 hours of the rupture.59 Placement of a septal occluding device may be helpful in selected patients.
Free Wall Rupture
Ventricular free wall rupture usually occurs during the first week after MI; the classic patient is elderly, female, and hypertensive. The early use of thrombolytic therapy reduces the incidence of cardiac rupture, but late use may increase the risk, particularly in older patients.138 Free wall rupture presents as a catastrophic event with a pulseless rhythm. Salvage is possible with prompt recognition, pericardiocentesis to relieve acute tamponade, and thoracotomy with repair.139
Myocardial Dysfunction After Cardiopulmonary Bypass
Transient depression of ventricular contractility is common after cardiopulmonary bypass, and can represent a significant clinical problem. The differential diagnosis includes inadequate operation, cardiac tamponade (which may be localized and difficult to detect), and increased right ventricular afterload, but most cases likely result from myocardial stunning. The heart is rendered globally ischemic during aortic cross-clamping and then reperfused, and because demonstrable myocardial necrosis is rare, stunning can be implicated. Stunning after bypass has been documented in a study in which an ultrasonic probe was left on the epicardial surface for 2 to 3 days in 31 patients following bypass surgery; left ventricular wall thickening fell after surgery, reached a nadir at 2 to 6 hours, and subsequently improved, usually returning to baseline by 24 to 48 hours.140
The degree of myocardial dysfunction after cardiac surgery is variable, and may depend on the cardioplegia solution, the method of administration (antegrade or retrograde), the mode of administration (continuous or intermittent), and the temperature of the solution and of the patient during surgery.38 In the clinical setting, transient depression of ventricular contractility is common and usually reversible within 24 to 48 hours. The depression of contractility can be severe to cause cardiogenic shock. In this event, therapy with inotropic agents, vasodilators, and IABP is necessary. Occasionally, even a left ventricular assist device may be employed.40 Better understanding of the mechanisms of post–cardiopulmonary bypass myocardial dysfunction may lead to better preventive and therapeutic approaches.
Myocarditis
Evidence exists that some patients with myocarditis will benefit from immunosuppressive therapy, but how to identify which patients should be treated remains controversial. A trial initiated at the National Institutes of Health randomized 102 patients with dilated cardiomyopathy, no significant coronary artery disease, and ejection fraction less than 35% to oral prednisone or placebo.141 The prospectively defined end point, an increase in radionuclide-measured ejection fraction of more than 5 percentage points, was observed in 53% of patients treated with prednisone compared to only 27% of control subjects at 3 months (p < 0.05), but the improvement did not persist at 9 months when patients were switched to alternate-day prednisone therapy.141 Another clinical trial of immunosuppressive therapy for myocarditis showed no improvement in mean ejection fraction with immunosuppression, although the admission criteria in this trial were quite restrictive, the therapeutic regimens heterogeneous, and the incidence of definitive myocarditis uncertain.142 We advocate consideration of corticosteroids in patients with myocarditis who do not respond to conventional heart failure therapies.
Although it might seem that patients with fulminant myocarditis might be the best candidates for immunosuppressive therapy, a recent series confounds this notion by reporting excellent long-term survival in patients with myocarditis and a fulminant course.143 Patients with acute myocarditis without a fulminant course had a much worse prognosis in this series,143 pointing up the need for further research to identify subgroups of patients with dilated cardiomyopathy who may benefit from adjunctive therapies.
Left Ventricular Assist Devices
In patients with potentially reversible causes of myocardial dysfunction, aggressive cardiovascular support with a combination of inotropic agents and intra-aortic balloon counterpulsation may be required for hours or days to allow sufficient time for recovery. If these measures fail, mechanical circulatory support with left ventricular assist devices (LVADs) can be considered.144 Mechanical support with LVADs can interrupt the downward spiral of myocardial dysfunction, hypoperfusion, and ischemia in cardiogenic shock, allowing time for recovery of stunned or hibernating myocardium.
Percutaneously implanted LVADs are used in situations of cardiogenic shock, during high-risk percutaneous interventions, in postcardiotomy shock, and in fulminant myocarditis. Two currently approved devices, the TandemHeart (Fig. 22.6) and the Impella (Fig. 22.7), can be placed in the cardiac catheterization laboratory. The TandemHeart device is a bypass system with inflow of oxygenated blood from the left atrium and outflow to the femoral artery using a centrifugal flow pump; it can provide blood flow up to 5 L/minute. The Impella device is inserted across the aortic valve and pumps blood from a distal port from within the ventricle out to the ascending aorta through a proximal port of the device; there are two versions, one capable of pumping 2.5 L/minute, and one with a larger diameter that can provide up to 5 L/minute, although the actual flow is dependent on afterload. The inflow cannula with its pump is under fluoroscopic guidance. The device sits across the aortic valve. These devices augment cardiac output and blood pressure while decreasing myocardial oxygen demand. Both devices offer the potential for near-complete cardiac support but do require adequate right ventricular function. Known complications of percutaneous LVAD use include limb ischemia and bleeding.
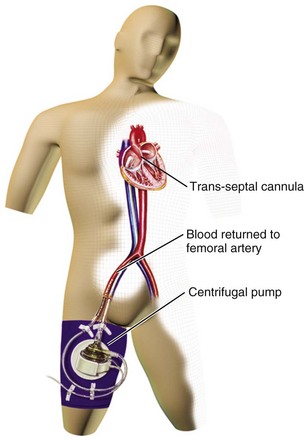
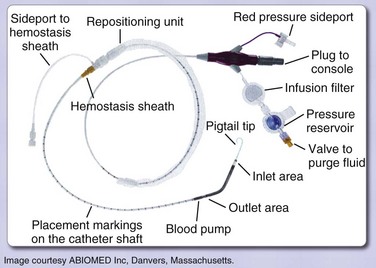
Two recent trials compared the use of IABP to the TandemHeart for patients with cardiogenic shock,145,146 and another compared use of the Impella to IABP therapy;147 the results of these trials were combined in a meta-analysis that included 100 patients.148 Hemodynamic benefits for the percutaneous LVADs compared with IABP were shown, with higher cardiac indices and mean arterial pressures as well as lower PCWPs. However, LVAD use showed no mortality rate benefit over IABP at 30 days.148
Conclusion
Cardiogenic shock remains a prevalent and dangerous syndrome that requires accurate and efficient diagnosis. Mortality rates in patients with cardiogenic shock have improved but remain frustratingly high. Its pathophysiology involves a downward spiral in which ischemia causes myocardial dysfunction, which in turn worsens ischemia. Areas of nonfunctional but viable myocardium can also cause or contribute to the development of cardiogenic shock. Expeditious coronary revascularization is crucial, and the randomized multicenter SHOCK trial120 has provided important data that help clarify the appropriate role and timing of revascularization in patients with cardiogenic shock. The potential for reversal of myocardial dysfunction with revascularization provides the rationale for supportive therapy to maintain coronary and tissue perfusion until more definitive revascularization measures can be undertaken. Application of a thorough understanding of the essentials of pathophysiology, diagnosis, and treatment of cardiogenic shock can allow for expeditious management and improved outcomes.
References
1. Hollenberg, SM. Recognition and treatment of cardiogenic shock. Semin Respir Crit Care Med. 2004; 25:661–671.
2. Hochman, JS, Boland, J, Sleeper, LA, et al. Current spectrum of cardiogenic shock and effect of early revascularization on mortality. Results of an International Registry. Circulation. 1995; 91:873–881.
3. Goldberg, RJ, Spencer, FA, Gore, JM, et al. Thirty-year trends (1975 to 2005) in the magnitude of, management of, and hospital death rates associated with cardiogenic shock in patients with acute myocardial infarction: A population-based perspective. Circulation. 2009; 119:1211–1219.
4. Forrester, JS, Diamond, G, Chatterjee, K, Swan, HJC. Medical therapy of acute myocardial infarction by application of hemodynamic subsets. I. N Engl J Med. 1976; 295:1356–1362.
5. Killip, T, Kimball, JT. Treatment of myocardial infarction in a coronary care unit. A two-year experience with 250 patients. Am J Cardiol. 1967; 20:457–464.
6. Goldberg, RJ, Gore, JM, Alpert, JS, et al. Cardiogenic shock after acute myocardial infarction. Incidence and mortality from a community-wide perspective, 1975 to 1988. N Engl J Med. 1991; 325:1117–1122.
7. Babaev, A, Frederick, PD, Pasta, DJ, et al. Trends in management and outcomes of patients with acute myocardial infarction complicated by cardiogenic shock. JAMA. 2005; 294:448–454.
8. Fang, J, Mensah, GA, Alderman, MH, Croft, JB. Trends in acute myocardial infarction complicated by cardiogenic shock, 1979-2003, United States. Am Heart J. 2006; 152:1035–1041.
9. Goldberg, RJ, Samad, NA, Yarzebski, J, et al. Temporal trends in cardiogenic shock complicating acute myocardial infarction. N Engl J Med. 1999; 340:1162–1168.
10. Gruppo Italiano per lo Studio Della Streptochinasi Nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet. 1986; 2:397–402.
11. ISIS-3 (Third International Study of Infarct Survival) Collaborative Group. ISIS-3: A randomised comparison of streptokinase vs tissue plasminogen activator vs anistreplase and of aspirin plus heparin vs aspirin alone among 41,299 cases of suspected acute myocardial infarction. Lancet. 1992; 339:753–770.
12. International Study Group. In-hospital mortality and clinical course of 20,891 patients with suspected acute myocardial infarction randomised between alteplase and streptokinase with or without heparin. Lancet. 1990; 336:71–75.
13. GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993; 329:673–682.
14. Holmes, DR, Jr., Bates, ER, Kleiman, NS, et al. Contemporary reperfusion therapy for cardiogenic shock: The GUSTO-I trial experience. The GUSTO-I Investigators. Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries. J Am Coll Cardiol. 1995; 26:668–674.
15. Scheidt, S, Ascheim, R, Killip, T. Shock after acute myocardial infarction. A clinical and hemodynamic profile. Am J Cardiol. 1970; 26:556–564.
16. Califf, RM, Bengtson, JR. Cardiogenic shock. N Engl J Med. 1994; 330:1724–1730.
17. Bonnefoy, E, Lapostolle, F, Leizorovicz, A, et al. Primary angioplasty versus prehospital fibrinolysis in acute myocardial infarction: A randomised study. Lancet. 2002; 360:825–829.
18. Hands, ME, Rutherford, JD, Muller, JE, et al. The in-hospital development of cardiogenic shock after myocardial infarction: Incidence, predictors of occurrence, outcome and prognostic factors. The MILIS Study Group. J Am Coll Cardiol. 1989; 14:40–46.
19. Leor, J, Goldbourt, U, Reicher-Reiss, H, et al. Cardiogenic shock complicating acute myocardial infarction in patients without heart failure on admission: Incidence, risk factors, and outcome. SPRINT Study Group. Am J Med. 1993; 94:265–273.
20. Hasdai, D, Califf, RM, Thompson, TD, et al. Predictors of cardiogenic shock after thrombolytic therapy for acute myocardial infarction. J Am Coll Cardiol. 2000; 35:136–143.
21. Bengtson, JR, Kaplan, AJ, Pieper, KS, et al. Prognosis in cardiogenic shock after acute myocardial infarction in the interventional era. J Am Coll Cardiol. 1992; 20:1482–1489.
22. Wong, SC, Sanborn, T, Sleeper, LA, et al. Angiographic findings and clinical correlates in patients with cardiogenic shock complicating acute myocardial infarction: A report from the SHOCK Trial Registry. SHould we emergently revascularize Occluded Coronaries for cardiogenic shocK? J Am Coll Cardiol. 2000; 36:1077–1083.
23. Grines, CL, Topol, EJ, Califf, RM, et al. Prognostic implications and predictors of enhanced regional wall motion of the noninfarct zone after thrombolysis and angioplasty therapy of acute myocardial infarction. The TAMI Study Groups. Circulation. 1989; 80:245–253.
24. Hollenberg, SM, Kavinsky, CJ, Parrillo, JE. Cardiogenic shock. Ann Intern Med. 1999; 131:47–59.
25. Hochman, JS. Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation. 2003; 107:2998–3002.
26. Greenberg, MA, Menegus, MA. Ischemia-induced diastolic dysfunction: New observations, new questions. J Am Coll Cardiol. 1989; 13:1071–1072.
27. Page, DL, Caulfield, JB, Kaster, JA, et al. Myocardial changes associated with cardiogenic shock. N Engl J Med. 1971; 285:133–137.
28. Olivetti, G, Quaini, F, Sala, R, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1994; 28:2005–2016.
29. Weisman, HF, Healy, B. Myocardial infarct expansion, infarct extension, and reinfarction: Pathophysiologic concepts. Prog Cardiovasc Dis. 1987; 30:73–110.
30. Widimsky, P, Gregor, P, Cervenka, V, et al. Severe diffuse hypokinesis of the remote myocardium—the main cause of cardiogenic shock? An echocardiographic study of 75 patients with extremely large myocardial infarctions. Cor Vasa. 1988; 30:27–34.
31. McGhie, AI, Golstein, RA. Pathogenesis and management of acute heart failure and cardiogenic shock: Role of inotropic therapy. Chest. 1992; 102:626S–632S.
32. Webb, JG. Interventional management of cardiogenic shock. Can J Cardiol. 1998; 14:233–244.
33. Harizi, RC, Bianco, JA, Alpert, JS. Diastolic function of the heart in clinical cardiology. Arch Intern Med. 1988; 148:99–109.
34. Oh, JK, Hatle, L, Tajik, AJ, Little, WC. Diastolic heart failure can be diagnosed by comprehensive two-dimensional and Doppler echocardiography. J Am Coll Cardiol. 2006; 47:500–506.
35. Hollenberg, SM, Parrillo, JE. Shock. In: Fauci AS, Braunwald E, Isselbacher KJ, et al, eds. Harrison’s Principles of Internal Medicine. 14th ed. New York: McGraw-Hill; 1997:214–222.
36. Okuda, M. A multidisciplinary overview of cardiogenic shock. Shock. 2006; 25:557–570.
37. Bartling, B, Holtz, J, Darmer, D. Contribution of myocyte apoptosis to myocardial infarction? Basic Res Cardiol. 1998; 93:71–84.
38. Bolli, R. Basic and clinical aspects of myocardial stunning. Prog Cardiovasc Dis. 1998; 40:477–516.
39. Arnold, JM, Braunwald, E, Sandor, T, Kloner, RA. Inotropic stimulation of reperfused myocardium with dopamine: Effects on infarct size and myocardial function. J Am Coll Cardiol. 1985; 6:1026–1034.
40. Ballantyne, CM, Verani, MS, Short, HD, et al. Delayed recovery of severely “stunned” myocardium with the support of a left ventricular assist device after coronary artery bypass graft surgery. J Am Coll Cardiol. 1987; 10:710–712.
41. Gerber, BL, Wijns, W, Vanoverschelde, JL, et al. Myocardial perfusion and oxygen consumption in reperfused noninfarcted dysfunctional myocardium after unstable angina: Direct evidence for myocardial stunning in humans. J Am Coll Cardiol. 1999; 34:1939–1946.
42. Jeroudi, MO, Hartley, CJ, Bolli, R. Myocardial reperfusion injury: Role of oxygen radicals and potential therapy with antioxidants. Am J Cardiol. 1994; 73:2B–7B.
43. Gao, WD, Liu, Y, Mellgren, R, Marban, E. Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium. A consequence of Ca2+-dependent proteolysis? Circ Res. 1996; 78:455–465.
44. Atar, D, Gao, WD, Marban, E. Alterations of excitation-contraction coupling in stunned myocardium and in failing myocardium. J Mol Cell Cardiol. 1995; 27:783–791.
45. Brar, R, Kumar, A, Schaer, GL, et al. Release of soluble myocardial depressant activity by reperfused myocardium (abstract). J Am Coll Cardiol. 1996; 27:386A.
46. Wijns, W, Vatner, SF, Camici, PG. Hibernating myocardium. N Engl J Med. 1998; 339:173–181.
47. Kloner, RA, Jennings, RB. Consequences of brief ischemia: Stunning, preconditioning, and their clinical implications: Part 1. Circulation. 2001; 104:2981–2989.
48. Bito, V, Heinzel, FR, Weidemann, F, et al. Cellular mechanisms of contractile dysfunction in hibernating myocardium. Circ Res. 2004; 94:794–801.
49. Marban, E. Myocardial stunning and hibernation. The physiology behind the colloquialisms. Circulation. 1991; 83:681–688.
50. Topol, EJ, Weiss, JL, Guzman, PA, et al. Immediate improvement of dysfunctional myocardial segments after coronary revascularization: Detection by intraoperative transesophageal echocardiography. J Am Coll Cardiol. 1984; 4:1123–1134.
51. Tillisch, J, Brunken, R, Marshall, R, et al. Reversibility of cardiac wall-motion abnormalities predicted by positron tomography. N Engl J Med. 1986; 314:884–888.
52. Wu, KC, Lima, JA. Noninvasive imaging of myocardial viability: Current techniques and future developments. Circ Res. 2003; 93:1146–1158.
53. Kim, SJ, Peppas, A, Hong, SK, et al. Persistent stunning induces myocardial hibernation and protection: Flow/function and metabolic mechanisms. Circ Res. 2003; 92:1233–1239.
54. Nishimura, RA, Tajik, AJ, Shub, C, et al. Role of two-dimensional echocardiography in the prediction of in-hospital complications after acute myocardial infarction. J Am Coll Cardiol. 1984; 4:1080–1087.
55. Berkowitz, MJ, Picard, MH, Harkness, S, et al. Echocardiographic and angiographic correlations in patients with cardiogenic shock secondary to acute myocardial infarction. Am J Cardiol. 2006; 98:1004–1008.
56. Kinch, JW, Ryan, TJ. Right ventricular infarction. N Engl J Med. 1994; 330:1211–1217.
57. Nedeljkovic, ZS, Ryan, TJ. Right ventricular infarction. In: Hollenberg SM, Bates ER, eds. Cardiogenic Shock. Armonk, NY: Futura Publishing Co. ; 2002:161–186.
58. Jollis, JG, DeLong, ER, Peterson, ED, et al. Outcome of acute myocardial infarction according to the specialty of the admitting physician. N Engl J Med. 1996; 335:1880–1887.
59. Antman, EM, Anbe, DT, Armstrong, PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2004; 110:588–636.
60. Forrester, JS, Diamond, G, Chatterjee, K, Swan, HJC. Medical therapy of acute myocardial infarction by application of hemodynamic subsets. II. N Engl J Med. 1976; 295:1404–1413.
61. Hollenberg, SM, Ahrens, TS, Annane, D, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 2004; 32:1928–1948.
62. De Backer, D, Biston, P, Devriendt, J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010; 362:779–789.
63. Tuttle, RR, Mills, J. Dobutamine: Development of a new catecholamine to selectively increase cardiac contractility. Circ Res. 1975; 36:185–196.
64. Gillespie, TA, Ambos, HD, Sobel, BE, Roberts, R. Effects of dobutamine in patients with acute myocardial infarction. Am J Cardiol. 1977; 39:588–594.
65. Keung, EC, Siskind, SJ, Sonneblick, EH, et al. Dobutamine therapy in acute myocardial infarction. JAMA. 1981; 245:144–146.
66. Benotti, JR, Grossman, W, Braunwald, E, Carabello, BA. Effects of amrinone on myocardial energy metabolism and hemodynamics in patients with severe congestive heart failure due to coronary artery disease. Circulation. 1980; 62:28–34.
67. Gage, J, Rutman, H, Lucido, D, LeJemtel, T. Additive effects of dobutamine and amrinone on myocardial contractility and ventricular performance in patients with severe heart failure. Circulation. 1986; 74:367–374.
68. Elahi, MM, Lam, J, Asopa, S, Matata, BM. Levosimendan versus an intra-aortic balloon pump in adult cardiac surgery patients with low cardiac output. J Cardiothorac Vasc Anesth. 2011; 25:1154–1162.
69. Russ, MA, Prondzinsky, R, Christoph, A, et al. Hemodynamic improvement following levosimendan treatment in patients with acute myocardial infarction and cardiogenic shock. Crit Care Med. 2007; 35:2732–2739.
70. Garcia-Gonzalez, MJ, Dominguez-Rodriguez, A, Ferrer-Hita, JJ, et al. Cardiogenic shock after primary percutaneous coronary intervention: Effects of levosimendan compared with dobutamine on haemodynamics. Eur J Heart Fail. 2006; 8:723–728.
71. Mebazaa, A, Nieminen, MS, Packer, M, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: The SURVIVE Randomized Trial. JAMA. 2007; 297:1883–1891.
72. Hansen, RM, Viquerat, CE, Matthay, MA, et al. Poor correlation between pulmonary arterial wedge pressure and left ventricular end-diastolic volume after coronary artery bypass graft surgery. Anesthesiology. 1986; 64:764–770.
73. Hollenberg, SM, Hoyt, JW. Pulmonary artery catheters in cardiovascular disease. New Horizons. 1997; 5:207–213.
74. Cohn, JN, Burke, LP. Nitroprusside. Ann Intern Med. 1979; 91:752–757.
75. Flaherty, JT, Becker, LC, Bulkley, BH, et al. A randomized trial of intravenous nitroglycerin in patients with acute myocardial infarction. Circulation. 1983; 68:576–588.
76. Wilcox, RG, von der Lippe, G, Olsson, CG, et al. Trial of tissue plasminogen activator for mortality reduction in acute myocardial infarction. Anglo-Scandinavian Study of Early Thrombolysis (ASSET). Lancet. 1988; 2:525–530.
77. AIMS Trial Study Group. Effect of intravenous APSAC on mortality after acute myocardial infarction: Preliminary report of a placebo-controlled clinical trial. Lancet. 1988; 1:545–549.
78. ISIS-2 Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet. 1988; 2:349–360.
79. GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med. 1993; 329:1615–1622.
80. Col, NF, Gurwitz, JH, Alpert, JS, Goldberg, RJ. Frequency of inclusion of patients with cardiogenic shock in trials of thrombolytic therapy. Am J Cardiol. 1994; 73:149–157.
81. Bates, ER, Moscucci, M. Post-myocardial infarction cardiogenic shock. In: Brown DL, ed. Cardiac Intensive Care. Philadelphia: WB Saunders; 1998:215–228.
82. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: Collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994; 343:311–322.
83. Kennedy, HL, Whitlock, JA, Sprague, MK, et al. Long-term follow-up of asymptomatic healthy subjects with frequent and complex ventricular ectopy. N Engl J Med. 1985; 312:193–197.
84. Berger, PB, Holmes, DR, Jr., Stebbins, AL, et al. Impact of an aggressive invasive catheterization and revascularization strategy on mortality in patients with cardiogenic shock in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO-I) trial. An observational study. Circulation. 1997; 96:122–127.
85. Becker, RC. Hemodynamic, mechanical, and metabolic determinants of thrombolytic efficacy: A theoretic framework for assessing the limitations of thrombolysis in patients with cardiogenic shock. Am Heart J. 1993; 125:919–929.
86. Garber, PJ, Mathieson, AL, Ducas, J, et al. Thrombolytic therapy in cardiogenic shock: Effect of increased aortic pressure and rapid tPA administration. Can J Cardiol. 1995; 11:30–36.
87. Prewitt, RM, Gu, S, Garber, PJ, Ducas, J. Marked systemic hypotension depresses coronary thrombolysis induced by intracoronary administration of recombinant tissue-type plasminogen activator. J Am Coll Cardiol. 1992; 20:1626–1633.
88. Mueller, H, Ayres, SM, Giannelli, SJ, et al. Effect of isoproterenol, l-norepinephrine, and intraaortic counterpulsation on hemodynamics and myocardial metabolism in shock following acute myocardial infarction. Circulation. 1972; 45:335–351.
89. Santa-Cruz, RA, Cohen, MG, Ohman, EM. Aortic counterpulsation: A review of the hemodynamic effects and indications for use. Cathet Cardiovasc Interv. 2006; 67:68–77.
90. Willerson, JT, Curry, GC, Watson, JT, et al. Intraaortic balloon counterpulsation in patients in cardiogenic shock, medically refractory left ventricular failure and/or recurrent ventricular tachycardia. Am J Med. 1975; 58:183–191.
91. Trost, JC, Hillis, LD. Intra-aortic balloon counterpulsation. Am J Cardiol. 2006; 97:1391–1398.
92. O’Rourke, MF, Norris, RM, Campbell, TJ, et al. Randomized controlled trial of intraaortic balloon counterpulsation in early myocardial infarction with acute heart failure. Am J Cardiol. 1981; 47:815–820.
93. Flaherty, JT, Becker, LC, Weiss, JL, et al. Results of a randomized prospective trial of intraaortic balloon counterpulsation and intravenous nitroglycerin in patients with acute myocardial infarction. J Am Coll Cardiol. 1985; 6:434–446.
94. Kern, MJ, Aguirre, F, Bach, R, et al. Augmentation of coronary blood flow by intra-aortic balloon pumping in patients after coronary angioplasty. Circulation. 1993; 87:500–511.
95. Anderson, RD, Ohman, EM, Holmes, DR, Jr., et al. Use of intraaortic balloon counterpulsation in patients presenting with cardiogenic shock: Observations from the GUSTO-I Study. Global Utilization of Streptokinase and TPA for Occluded Coronary Arteries. J Am Coll Cardiol. 1997; 30:708–715.
96. DeWood, MA, Notske, RN, Hensley, GR, et al. Intraaortic balloon counterpulsation with and without reperfusion for myocardial infarction shock. Circulation. 1980; 61:1105–1112.
97. Waksman, R, Weiss, AT, Gotsman, MS, Hasin, Y. Intra-aortic balloon counterpulsation improves survival in cardiogenic shock complicating acute myocardial infarction. Eur Heart J. 1993; 14:71–74.
98. Stomel, RJ, Rasak, M, Bates, ER. Treatment strategies for acute myocardial infarction complicated by cardiogenic shock in a community hospital. Chest. 1994; 105:997–1002.
99. Kovack, PJ, Rasak, MA, Bates, ER, et al. Thrombolysis plus aortic counterpulsation: Improved survival in patients who present to community hospitals with cardiogenic shock. J Am Coll Cardiol. 1997; 29:1454–1458.
100. Ishihara, M, Sato, H, Tateishi, H, et al. Intraaortic balloon pumping as the postangioplasty strategy in acute myocardial infarction. Am Heart J. 1991; 122:385–389.
101. Ohman, EM, George, BS, White, CJ, et al. Use of aortic counterpulsation to improve sustained coronary artery patency during acute myocardial infarction. Results of a randomized trial. Circulation. 1994; 90:792–799.
102. Ohman, EM, Nanas, J, Stomel, RJ, et al. Thrombolysis and counterpulsation to improve survival in myocardial infarction complicated by hypotension and suspected cardiogenic shock or heart failure: Results of the TACTICS Trial. J Thromb Thrombolysis. 2005; 19:33–39.
103. Grines, CL, Browne, KF, Marco, J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. N Engl J Med. 1993; 328:673–679.
104. Zijlstra, F, de Boer, MJ, Hoorntje, JC, et al. A comparison of immediate coronary angioplasty with intravenous streptokinase in acute myocardial infarction. N Engl J Med. 1993; 328:680–684.
105. Gibbons, RJ, Holmes, DR, Reeder, GS, et al. Immediate angioplasty compared with the administration of a thrombolytic agent followed by conservative treatment for myocardial infarction. N Engl J Med. 1993; 328:685–691.
106. TIMI Study Group. The thrombolysis in myocardial infarction (TIMI) trial. Phase I findings. N Engl J Med. 1985; 312:932–936.
107. O’Keefe, JH, Jr., Bailey, WL, Rutherford, BD, Hartzler, GO. Primary angioplasty for acute myocardial infarction in 1,000 consecutive patients. Results in an unselected population and high-risk subgroups. Am J Cardiol. 1993; 72:107G–115G.
108. O’Neill, W, Erbel, R, Laufer, N, et al. Coronary angioplasty therapy of cardiogenic shock complicating acute myocardial infarction (abstract). Circulation. 1985; 72(Suppl II):309.
109. Lee, L, Bates, ER, Pitt, B, et al. Percutaneous transluminal coronary angioplasty improves survival in acute myocardial infarction complicated by cardiogenic shock. Circulation. 1988; 78:1345–1351.
110. Lee, L, Erbel, R, Brown, TM, et al. Multicenter registry of angioplasty therapy of cardiogenic shock: Initial and long-term survival. J Am Coll Cardiol. 1991; 17:599–603.
111. Gacioch, GM, Ellis, SG, Lee, L, et al. Cardiogenic shock complicating acute myocardial infarction: The use of coronary angioplasty and the integration of the new support devices into patient management. J Am Coll Cardiol. 1992; 19:647–653.
112. Hibbard, MD, Holmes, DR, Jr., Bailey, KR, et al. Percutaneous transluminal coronary angioplasty in patients with cardiogenic shock. J Am Coll Cardiol. 1992; 19:639–646.
113. Moosvi, AR, Khaja, F, Villanueva, L, et al. Early revascularization improves survival in cardiogenic shock complicating acute myocardial infarction. J Am Coll Cardiol. 1992; 19:907–914.
114. Seydoux, C, Goy, JJ, Beuret, P, et al. Effectiveness of percutaneous transluminal coronary angioplasty in cardiogenic shock during acute myocardial infarction. Am J Cardiol. 1992; 69:968–969.
115. Eltchaninoff, H, Simpfendorfer, C, Franco, I, et al. Early and 1-year survival rates in acute myocardial infarction complicated by cardiogenic shock: A retrospective study comparing coronary angioplasty with medical treatment. Am Heart J. 1995; 130:459–464.
116. Vogt, A, Niederer, W, Pfafferott, C, et al. Direct percutaneous transluminal coronary angioplasty in acute myocardial infarction. Predictors of short-term outcome and the impact of coronary stenting. Study Group of The Arbeitsgemeinschaft Leitender Kardiologischer Krankenhausarzte (ALKK). Eur Heart J. 1998; 19:917–921.
117. Antoniucci, D, Santoro, GM, Bolognese, L, et al. A clinical trial comparing primary stenting of the infarct-related artery with optimal primary angioplasty for acute myocardial infarction: Results from the Florence Randomized Elective Stenting in Acute Coronary Occlusions (FRESCO) trial. J Am Coll Cardiol. 1998; 31:1234–1239.
118. Berger, PB, Tuttle, RH, Holmes, DR, Jr., et al. One-year survival among patients with acute myocardial infarction complicated by cardiogenic shock, and its relation to early revascularization: Results From the GUSTO-I Trial. Circulation. 1999; 99:873–878.
119. Rogers, WJ, Canto, JG, Lambrew, CT, et al. Temporal trends in the treatment of over 1. 5 million patients with myocardial infarction in the US from 1990 through 1999: The National Registry of Myocardial Infarction 1, 2 and 3. J Am Coll Cardiol. 2000; 36:2056–2063.
120. Hochman, JS, Sleeper, LA, Webb, JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. N Engl J Med. 1999; 341:625–634.
121. Hochman, JS, Sleeper, LA, White, HD, et al. One-year survival following early revascularization for cardiogenic shock. JAMA. 2001; 285:190–192.
122. Hochman, JS, Sleeper, LA, Webb, JG, et al. Early revascularization and long-term survival in cardiogenic shock complicating acute myocardial infarction. JAMA. 2006; 295:2511–2515.
123. Sleeper, LA, Ramanathan, K, Picard, MH, et al. Functional status and quality of life after emergency revascularization for cardiogenic shock complicating acute myocardial infarction. J Am Coll Cardiol. 2005; 46:266–273.
124. Dzavik, V, Sleeper, LA, Picard, MH, et al. Outcome of patients aged > or = 75 years in the SHould we emergently revascularize Occluded Coronaries in cardiogenic shocK (SHOCK) trial: Do elderly patients with acute myocardial infarction complicated by cardiogenic shock respond differently to emergent revascularization? Am Heart J. 2005; 149:1128–1134.
125. Dzavik, V, Sleeper, LA, Cocke, TP, et al. Early revascularization is associated with improved survival in elderly patients with acute myocardial infarction complicated by cardiogenic shock: A report from the SHOCK Trial Registry. Eur Heart J. 2003; 24:828–837.
126. Urban, P, Stauffer, JC, Bleed, D, et al. A randomized evaluation of early revascularization to treat shock complicating acute myocardial infarction. The (Swiss) Multicenter Trial of Angioplasty for Shock-(S)MASH. Eur Heart J. 1999; 20:1030–1038.
127. Urban, P, Stauffer, JC. Randomized trials of revascularization therapy for cardiogenic shock. In: Hollenberg SM, Bates ER, eds. Cardiogenic Shock. Armonk, NY: Futura Publishing Co. ; 2002:135–144.
128. Kushner, FG, Hand, M, Smith, SC, Jr., et al. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009; 120:2271–2330.
129. White, HD, Assmann, SF, Sanborn, TA, et al. Comparison of percutaneous coronary intervention and coronary artery bypass grafting after acute myocardial infarction complicated by cardiogenic shock: Results from the SHould we emergently revascularize Occluded Coronaries for cardiogenic shocK (SHOCK) trial. Circulation. 2005; 112:1992–2001.
130. Zehender, M, Kasper, W, Kauder, E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med. 1993; 328:981–988.
131. Dell’Italia, LJ, Starling, MR, Blumhardt, R, et al. Comparative effects of volume loading, dobutamine, and nitroprusside in patients with predominant right ventricular infarction. Circulation. 1985; 72:1327–1335.
132. Bowers, TR, O’Neill, WW, Grines, C, et al. Effect of reperfusion on biventricular function and survival after right ventricular infarction. N Engl J Med. 1998; 338:933–940.
133. Antman, EM, Hand, M, Armstrong, PW, et al. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2008; 51:210–247.
134. Voci, P, Bilotta, F, Caretta, Q, et al. Papillary muscle perfusion pattern. A hypothesis for ischemic papillary muscle dysfunction. Circulation. 1995; 91:1714–1718.
135. Khan, SS, Gray, RJ. Valvular emergencies. Cardiol Clin. 1991; 9:689–709.
136. Bolooki, H. Emergency cardiac procedures in patients in cardiogenic shock due to complications of coronary artery disease. Circulation. 1989; 79:1137–1148.
137. Thompson, CR, Buller, CE, Sleeper, LA, et al. Cardiogenic shock due to acute severe mitral regurgitation complicating acute myocardial infarction: A report from the SHOCK Trial Registry. SHould we use emergently revascularize Occluded Coronaries in cardiogenic shocK? J Am Coll Cardiol. 2000; 36:1104–1109.
138. Bueno, H, Martinez-Selles, M, Perez-David, E, et al. Effect of thrombolytic therapy on the risk of cardiac rupture and mortality in older patients with first acute myocardial infarction. Eur Heart J. 2005; 26:1705–1711.
139. Reardon, MJ, Carr, CL, Diamond, A, et al. Ischemic left ventricular free wall rupture: Prediction, diagnosis, and treatment. Ann Thorac Surg. 1997; 64:1509–1513.
140. Bolli, R, Hartley, CJ, Chelly, JE, et al. An accurate nontraumatic ultrasonic method to monitor myocardial wall thickening in patients undergoing cardial surgery. J Am Coll Cardiol. 1990; 15:1055–1065.
141. Parrillo, JE, Cunnion, RE, Epstein, SE, et al. A prospective, randomized, controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. 1989; 321:1061–1068.
142. Mason, JW, O’Connell, JB, Herskowitz, A, et al. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995; 333:269–275.
143. McCarthy, RE, 3rd., Boehmer, JP, Hruban, RH, et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med. 2000; 342:690–695.
144. Hunt, SA, Abraham, WT, Chin, MH, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult-Summary Article. J Am Coll Cardiol. 2005; 46:1116–1143.
145. Thiele, H, Sick, P, Boudriot, E, et al. Randomized comparison of intra-aortic balloon support with a percutaneous left ventricular assist device in patients with revascularized acute myocardial infarction complicated by cardiogenic shock. Eur Heart J. 2005; 26:1276–1283.
146. Burkhoff, D, Cohen, H, Brunckhorst, C, O’Neill, WW. A randomized multicenter clinical study to evaluate the safety and efficacy of the TandemHeart percutaneous ventricular assist device versus conventional therapy with intraaortic balloon pumping for treatment of cardiogenic shock. Am Heart J. 2006; 152:469.
147. Seyfarth, M, Sibbing, D, Bauer, I, et al. A randomized clinical trial to evaluate the safety and efficacy of a percutaneous left ventricular assist device versus intra-aortic balloon pumping for treatment of cardiogenic shock caused by myocardial infarction. J Am Coll Cardiol. 2008; 52:1584–1588.
148. Cheng, JM, den Uil, CA, Hoeks, SE, et al. Percutaneous left ventricular assist devices vs. intra-aortic balloon pump counterpulsation for treatment of cardiogenic shock: A meta-analysis of controlled trials. Eur Heart J. 2009; 30:2102–2108.
149. Alexander, JH, Reynolds, HR, Stebbins, AL, et al. Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: The TRIUMPH randomized controlled trial. JAMA. 2007; 297:1657–1666.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]22
Cardiogenic Shock
Cardiogenic shock is the syndrome that ensues when the heart is unable to deliver enough blood to maintain adequate tissue perfusion. Acute myocardial infarction (MI) is the leading cause, but other potential etiologic factors need to be considered.1,2 Without prompt diagnosis and appropriate management, morbidity and mortality rates are substantial, approaching 60% for all age groups.2,3 Rapid evaluation and prompt initiation of supportive measures and definitive therapy in patients with cardiogenic shock may improve both early and long-term outcomes.
Definition
The clinical definition of cardiogenic shock includes decreased cardiac output and evidence of tissue hypoxia in the presence of adequate intravascular volume. The diagnosis of circulatory shock (Box 22.1) is made at the bedside by the presence of hypotension along with a combination of clinical signs indicative of poor tissue perfusion, including oliguria, clouded sensorium, and cool, mottled extremities. Hemodynamic criteria include sustained hypotension (systolic blood pressure < 90 mm Hg for at least 30 minutes) and a reduced cardiac index (<2.2 L/min/m2) in the presence of elevated filling pressures (pulmonary capillary occlusion pressure > 15 mm Hg).4 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion or correction of factors such as hypovolemia, hypoxia, and acidosis.
Epidemiology
Pump failure due to cardiogenic shock has long been known to carry a high mortality rate. The seminal article outlining prognosis after MI was a single center series of 250 patients reported by Killip in 1967.5 Killip divided patients into four classes as follows:
Killip class I: no evidence of congestive heart failure
Killip class II: presence of an S3 gallop and/or bibasilar rales
Killip class III: pulmonary edema (rales greater than halfway up the lung fields)
Nineteen percent of the 250 patients were in class IV at presentation, and their mortality rate was 81%.5
With the advent of right-sided heart catheterization, Forrester and Swan defined hemodynamic subsets after MI analogous to the clinical subsets outlined by Killip.4 Subset I consisted of patients with normal pulmonary capillary wedge pressure (PCWP) and cardiac output, subset II consisted of patients with elevated PCWP and normal cardiac output, subset III consisted of patients with normal PCWP and decreased cardiac output, and subset IV consisted of patients with elevated PCWP and decreased cardiac output.4
Despite advances in management of heart failure and acute MI, the mortality rate of patients with cardiogenic shock has remained high.2,6–8 Data suggest an increase in survival in the 1990s, coincident with the use of reperfusion strategies.7–9 Cardiogenic shock, however, remains the most common cause of death in hospitalized patients with acute MI.
Incidence
Accurate determination of the precise incidence of cardiogenic shock is difficult because patients who die of MI prior to reaching the hospital generally do not receive this diagnosis.6,10–13 Nonetheless, estimates from a variety of sources have been fairly consistent. The Worcester Heart Attack Study,6 a community-wide analysis, found an incidence of cardiogenic shock of 7.5%, an incidence that remained fairly stable from 1975 to 1997.6,9 The incidence was similar in the randomized GUSTO (Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries) trial (7.2%),14 in other multicenter thrombolytic trials,10–12 and in patients with ST-segment elevation MI in the National Registry of Myocardial Infarction (NRMI) database from 1995 to 2004 (8.6%).7 More recently, however, the incidence of cardiogenic shock has fallen from about 8% to about 6% of MIs, with most of the change resulting from a decrease in cardiogenic shock developing after initial presentation, supporting the notion that early revascularization strategies are an important contributor to the decline.3
Etiology
The most common cause of cardiogenic shock is left ventricular failure in the setting of an extensive acute MI, although a smaller infarction in a patient with previously compromised left ventricular function may also precipitate shock. Cardiogenic shock can also be caused by mechanical complications such as acute mitral regurgitation, rupture of the interventricular septum, or rupture of the free wall—or by large right ventricular infarctions. In a report of the SHOCK (SHould we emergently revascularize Occluded Coronaries for shocK) trial registry of 1160 patients with cardiogenic shock,2 78.5% of patients had predominant left ventricular failure, 6.9% had acute mitral regurgitation, 3.9% had ventricular septal rupture, 2.8% had isolated right ventricular shock, 1.4% had tamponade or cardiac rupture, and 6.5% had shock resulting from other causes.
Other causes of cardiogenic shock include myocarditis, end-stage cardiomyopathy, myocardial contusion, septic shock with severe myocardial depression, myocardial dysfunction after prolonged cardiopulmonary bypass, valvular heart disease, and hypertrophic obstructive cardiomyopathy (Box 22.2). An important consideration is that some cardiogenic shock may have an iatrogenic component. Early diagnosis of impending shock or of patients at high risk for development of shock is essential, both to speed intervention and to avoid therapies that may worsen hemodynamics. In many cases of cardiogenic shock in the setting of MI, the diagnosis is not made until the patient has been triaged and admitted to an inpatient setting. Patients may have received early beta blockade or angiotensin-converting enzyme inhibition, therapies that may impact hemodynamics substantially.
Patients may have cardiogenic shock at initial presentation, but most do not; shock usually evolves over several hours,15,16 suggesting that early treatment may potentially prevent shock. In fact, some data indicate that early thrombolytic therapy may decrease the incidence of cardiogenic shock.17 In the SHOCK trial registry, 75% of patients developed cardiogenic shock within 24 hours after presentation, with a median delay of 7 hours.2 Results from the GUSTO trial were similar;13 among patients with shock, 11% were in shock on arrival and 89% developed shock after admission.
Risk factors for the development of cardiogenic shock in MI generally parallel those for left ventricular dysfunction and the severity of coronary artery disease. Shock is more likely to develop in patients who are elderly, are diabetic, and have anterior MI.5,15,18,19 Patients with cardiogenic shock are also more likely to have histories of previous infarction, peripheral vascular disease, and cerebrovascular disease.18,19 Decreased ejection fractions and larger infarctions (as evidenced by higher cardiac enzymes) are also predictors of the development of cardiogenic shock.18,19 Analysis from the GUSTO-3 trial has identified age, lower systolic blood pressure, heart rate, and Killip class as significant predictors of the risk for development of cardiogenic shock after presentation with acute MI.20 Use of a predictive scoring system derived from this study may be useful in identifying patients at high risk for the development of cardiogenic shock and targeting such patients for closer monitoring.20
Cardiogenic shock is most often associated with anterior MI. In the SHOCK trial registry, 55% of infarctions were anterior, 46% were inferior, 21% were posterior, and 50% were in multiple locations.2 These findings were consistent with those in other series.21 Angiographic evidence most often demonstrates multivessel coronary disease (left main occlusion in 20% of patients, three-vessel disease in 64%, two-vessel disease in 23%, and one-vessel disease in 13% of patients).22 The high prevalence of multivessel coronary artery disease is important because compensatory hyperkinesis normally develops in myocardial segments that are not involved in an acute MI, and this response helps maintain cardiac output. Failure to develop such a response, because of previous infarction or high-grade coronary stenoses, is an important risk factor for cardiogenic shock and death.16,23
Pathogenesis
Systemic Effects
Cardiac dysfunction in patients with cardiogenic shock is usually initiated by MI or ischemia. The myocardial dysfunction resulting from ischemia worsens that ischemia, creating a downward spiral24 (Fig. 22.1). When a critical mass of ischemic or necrotic left ventricular myocardium fails to pump, stroke volume and cardiac output decrease. Myocardial perfusion, which depends on the pressure gradient between the coronary arterial system and the left ventricle and on the duration of diastole, is compromised by hypotension and tachycardia, exacerbating ischemia. The increased ventricular diastolic pressures caused by pump failure reduce coronary perfusion pressure, and the additional wall stress elevates myocardial oxygen requirements, further worsening ischemia. Decreased cardiac output also compromises systemic perfusion.
When myocardial function is depressed, several compensatory mechanisms are activated, including sympathetic stimulation to increase heart rate and contractility and renal fluid retention to increase preload. These compensatory mechanisms may become maladaptive and can actually worsen the situation when cardiogenic shock develops. Increased heart rate and contractility increase myocardial oxygen demand and exacerbate ischemia. Fluid retention and impaired diastolic filling caused by tachycardia and ischemia may result in pulmonary congestion and hypoxia. Vasoconstriction to maintain blood pressure increases myocardial afterload, further impairing cardiac performance and increasing myocardial oxygen demand. This increased demand, in the face of inadequate perfusion, worsens ischemia and begins a vicious cycle that will end in death if not interrupted (see Fig. 22.1). The interruption of this cycle of myocardial dysfunction and ischemia forms the basis for the therapeutic regimens for cardiogenic shock.
Not all patients fit into this classic paradigm. In the SHOCK trial, the average systemic vascular resistance (SVR) was not elevated, and the range of values was wide, suggesting that compensatory vasoconstriction is not universal. Some patients had fever and elevated white blood cell counts along with decreased SVR, suggesting a systemic inflammatory response syndrome.25 This has led to an expansion of the paradigm to include the possibility of the contribution of inflammatory responses to vasodilation and myocardial stunning, leading clinically to persistence of shock (Fig. 22.2).25 Supporting this notion is the fact that the mean ejection fraction in the SHOCK trial was only moderately decreased (30%), suggesting that mechanisms other than pump failure were operative.25 Immune activation appears to be common to a number of different forms of shock. Activation of inducible nitric oxide synthase (iNOS) with production of nitric oxide and peroxynitrate has been proposed as one potential mechanism.
Figure 22.2 Expansion of the pathophysiologic paradigm of cardiogenic shock to include the potential contribution of inflammatory mediators. Inhibition of nitric oxide, however, has not been shown to be beneficial in patients with cardiogenic shock.149 iNOS, inducible nitric oxide synthase; LVEDP, left ventricular end-diastolic pressure; NO, nitric oxide; ONOO−, peroxynitrite; SVR, systemic vascular resistance. (Adapted from Hochman JS: Cardiogenic shock complicating acute myocardial infarction: Expanding the paradigm. Circulation 2003;107:2999.)
Myocardial Pathology
Cardiogenic shock is characterized by both systolic and diastolic myocardial dysfunction.16,26 Progressive myocardial necrosis has been observed consistently in clinical and pathologic studies of patients with cardiogenic shock.16,27 Patients who develop shock after admission often have evidence of infarct extension, which can result from reocclusion of a transiently patent infarct artery, propagation of intracoronary thrombus, or a combination of decreased coronary perfusion pressure and increased myocardial oxygen demand.18,19 Myocytes at the border zone of an infarction are more susceptible to additional ischemic episodes; therefore, these adjacent segments are at particular risk.28 Mechanical infarct expansion, which is seen most dramatically after extensive anterior MI, can also contribute to late development of cardiogenic shock.18,29
Ischemia remote from the infarct zone may be particularly important in producing systolic dysfunction in patients with cardiogenic shock.23,30 Patients with cardiogenic shock usually have multivessel coronary artery disease,2,16 with limited vasodilator reserve, impaired autoregulation, and consequent pressure-dependent coronary flow in several perfusion territories.31 Hypotension and metabolic derangements thus have the potential to impair the contractility of noninfarcted myocardium in patients with shock.32 This can limit hyperkinesis of uninvolved segments, a compensatory mechanism typically seen early after MI.23,30
Myocardial diastolic function is also impaired in patients with cardiogenic shock. Myocardial ischemia causes decreased compliance, increasing the left ventricular filling pressure at a given end-diastolic volume.33,34 Compensatory increases in left ventricular volumes to maintain stroke volume further increase filling pressures. Elevation of left ventricular pressures can lead to pulmonary edema and hypoxemia (see Fig. 22.1).
Cellular Pathology
Tissue hypoperfusion and consequent cellular hypoxia lead to anaerobic glycolysis, with depletion of adenosine triphosphate and intracellular energy reserves. Anaerobic glycolysis also causes accumulation of lactic acid and resultant intracellular acidosis. Failure of energy-dependent ion transport pumps decreases transmembrane potential, causing intracellular accumulation of sodium and calcium and myocyte swelling.35 Cellular ischemia and intracellular calcium accumulation can activate intracellular proteases.36 If the ischemia is severe and prolonged enough, myocardial cellular injury can become irreversible, with the classic pattern of myonecrosis: mitochondrial swelling, accumulation of denatured proteins and chromatin in the cytoplasm, lysosomal breakdown, and fracture of the mitochondria, nuclear envelope, and plasma membrane.35,36
Accumulating evidence indicates that apoptosis (programmed cell death) may also contribute to myocyte loss in MI.28,36,37 Although myonecrosis clearly outweighs apoptosis in the core of an infarcted area, evidence for apoptosis has been found consistently in the border zone of infarcts after ischemia and reperfusion and sporadically in areas remote from the ischemia area.28,37 Activation of inflammatory cascades, oxidative stress, and stretching of myocytes have been proposed as mechanisms that activate the apoptotic pathways.36,37 Although the magnitude of apoptotic cell loss in MI remains uncertain, inhibitors of apoptosis have been found to attenuate myocardial injury in animal models of postischemic reperfusion; these inhibitors may also have therapeutic potential for myocyte salvage after large infarctions.37
Reversible Myocardial Dysfunction
A key to understanding the pathophysiology and treatment of cardiogenic shock is to realize that large areas of nonfunctional but viable myocardium can also cause or contribute to the development of cardiogenic shock in patients after MI (Fig. 22.3). This reversible dysfunction can be described in two main categories: stunning and hibernation.
Myocardial stunning represents postischemic dysfunction that persists despite restoration of normal blood flow; eventually, however, myocardial performance recovers completely.38 Originally defined in animal models of ischemia and reperfusion,39 stunning has been recognized in the clinical arena.38,40 Direct evidence for myocardial stunning in humans has been found using positron emission tomography (PET) scanning in patients with persistent wall motion abnormalities after angioplasty for acute coronary syndromes; perfusion measured by 13N-ammonia was normal in the presence of persistent contractile dysfunction.41 The pathogenesis of stunning has not been conclusively established but appears to involve a combination of oxidative stress,42 perturbation of calcium homeostasis, and decreased myofilament responsiveness to calcium.38,43,44 In addition to these direct effects, data from studies in isolated cardiac myocytes suggest that circulating myocardial depressant substances may contribute to contractile dysfunction in myocardial stunning.45 The intensity of stunning is determined primarily by the severity of the antecedent ischemic insult.38
Myocardial hibernation comprises segments with persistently impaired function at rest due to severely reduced coronary blood flow; inherent in the definition of hibernating myocardium is the notion that function can be normalized by improving blood flow.46–48 Hibernation can be seen as an adaptive response to reduce contractile function of hypoperfused myocardium and restore equilibrium between flow and function, thereby minimizing the potential for ischemia or necrosis.49 Revascularization of hibernating myocardium can lead to improved myocardial function,50 and improved function appears to translate into improved prognosis.51,52
Although hibernation is conceptually and pathophysiologically different from myocardial stunning, the two conditions are difficult to distinguish in the clinical setting and may in fact coexist.38,52 Repetitive episodes of myocardial stunning can coexist with or mimic myocardial hibernation.38,46,53 Consideration of myocardial stunning and hibernation is vital in patients with cardiogenic shock because of their therapeutic implications. Hibernating myocardium improves with revascularization, and stunned myocardium retains inotropic reserve and can respond to inotropic stimulation.38 In addition, the fact that the severity of the antecedent ischemic insult determines the intensity of stunning38 provides one of the rationales for reestablishment of patency of occluded coronary arteries in patients with cardiogenic shock. Finally, the notion that some myocardial tissue may recover function emphasizes the importance of measures to support hemodynamics and thus minimize myocardial necrosis in patients with shock.
Clinical Assessment
Evaluation
Cardiogenic shock is an emergency. The clinician must initiate therapy before shock irreversibly damages vital organs; at the same time, he or she must perform the clinical assessment required to understand the cause of shock and to target therapy to that cause. A practical approach is to make a rapid initial evaluation on the basis of a limited history, physical examination, and specific diagnostic procedures (Fig. 22.4).24 Cardiogenic shock is diagnosed after documentation of myocardial dysfunction and exclusion of alternative causes of hypotension such as hypovolemia, hemorrhage, sepsis, pulmonary embolism, tamponade, aortic dissection, and preexisting valvular disease.
Echocardiography is an excellent initial tool for confirming the diagnosis of cardiogenic shock and ruling out other causes of shock (Box 22.3); therefore, early echocardiography should be routine. Echocardiography provides information on overall and regional systolic function, and can rapidly diagnose mechanical causes of shock such as papillary muscle rupture and acute mitral regurgitation, acute ventricular septal defect, and free wall rupture and tamponade.54,55 Unsuspected severe mitral regurgitation is not uncommon. In some cases, echocardiography may reveal findings compatible with right ventricular infarction.
Invasive hemodynamic monitoring can be quite useful to exclude volume depletion, right ventricular infarction, and mechanical complications.16,35 The hemodynamic profile of cardiogenic shock includes a pulmonary capillary occlusion pressure greater than 15 mm Hg and a cardiac index less than 2.2 L/min/m2. It should be recognized that optimal filling pressures may be greater than 15 mm Hg in individual patients due to left ventricular diastolic dysfunction. Right-sided heart catheterization may reveal an oxygen step-up diagnostic of ventricular septal rupture or a large v wave that suggests severe mitral regurgitation. The hemodynamic profile of right ventricular infarction includes high right-sided filling pressures in the presence of normal or low occlusion pressures.56,57
Initial Management
Maintenance of adequate oxygenation and ventilation are critical; intubation and mechanical ventilation are often required, if only to reduce the work of breathing and facilitate sedation and stabilization before cardiac catheterization. Central venous and arterial access, bladder catheterization, and pulse oximetry are routine. Electrolyte abnormalities should be corrected. Hypokalemia and hypomagnesemia are predisposing factors to ventricular arrhythmias, and acidosis can decrease contractile function. Relief of pain and anxiety with morphine sulfate (or fentanyl if systolic pressure is compromised) can reduce excessive sympathetic activity and decrease oxygen demand, preload, and afterload. Arrhythmias and heart block may have major effects on cardiac output, and should be corrected promptly with antiarrhythmic drugs, cardioversion, or pacing. Cardiology consultation has been shown to be associated with improved outcomes in patients with MI and is strongly indicated in the setting of cardiogenic shock.58 In addition, measures proven to improve outcome after MI, such as nitrates, beta blockers, and angiotensin-converting enzyme inhibitors,59 have the potential to exacerbate hypotension in cardiogenic shock and should be stopped until the patient stabilizes.
Therapy
Following initial stabilization and restoration of adequate blood pressure, tissue perfusion should be assessed (see Fig. 22.4). If tissue perfusion remains inadequate, inotropic support or intra-aortic balloon pumping (IABP) should be initiated. If tissue perfusion is adequate but significant pulmonary congestion remains, diuretics may be employed. Vasodilators can be considered as well, depending on the blood pressure.
The initial approach to the hypotensive patient should include fluid resuscitation unless frank pulmonary edema is present. Patients are commonly diaphoretic and relative hypovolemia may be present. In the original description of hemodynamic subsets in MI, approximately 20% of patients had low cardiac index and low PCWP; most had reduced stroke volume and compensatory tachycardia.60 Some of these patients would be expected to respond to fluid infusion with an increase in stroke volume, although the magnitude of such a response depends on the degree of ischemia and cardiac reserve.
[/not-level-membership-for-critical-care-medicine-category]
