[level-membership-for-cardiovascular-category]
Chapter 2 Cardiac Ion Channels
Ion channels are pore-forming membrane proteins that regulate the flow of ions passively down their electrochemical gradient across the membrane. Ion channels are present on all membranes of cells (plasma membrane) and intracellular organelles (nucleus, mitochondria, endoplasmic reticulum). There are more than 300 types of ion channels in a living cell. The channels are not randomly distributed in the membrane, but tend to cluster at the intercalated disc in association with modulatory subunits.1
Importantly, channel opening and closing are not instantaneous but usually take time. The transition from the resting (closed) state to the open state is called activation. Once opened, channels do not remain in the open state, but instead they undergo conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening and must undergo a recovery or reactivation process back to the resting state to regain their ability to open. Inactivation curves of the various voltage-gated ion channel types differ in their slopes and midpoints of inactivation and can overlap, in which case a steady-state or noninactivating current flows.1
Ion channels differ with respect to the number of subunits of which they are composed and other aspects of structure. Many ion channels function as part of macromolecular complexes in which many components are assembled at specific sites within the membrane. For most ion channels, the pore-forming subunit is called the α subunit, whereas the auxiliary subunits are denoted β, gamma, and so on. Most ion channels have a single pore; however, some have two.1
Sodium Channels
Structure and Physiology
The cardiac Na+ channel complex is composed of a primary α and multiple ancillary β subunits. The approximately 2000-amino-acid α subunit contains the channel’s ion-conducting pore and controls the channel selectivity for Na+ ions and voltage-dependent gating machinery. This subunit contains all the drug and toxin interaction sites identified to date. The α subunit (Nav1.5), encoded by the SCN5A gene, consists of four internally homologous domains (I to IV) that are connected to each other by cytoplasmic linkers (Fig. 2-1). Each domain consists of six membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops. The four domains are arranged in a fourfold circular symmetry to form the channel. The extracellular loops between S5 and S6 (termed the P segments) have a unique primary structure in each domain (Fig. 2-2). The P segments curve back into the membrane to form an ion-conducting central pore whose structural constituents determine the selectivity and conductance properties of the Na+ channel.2
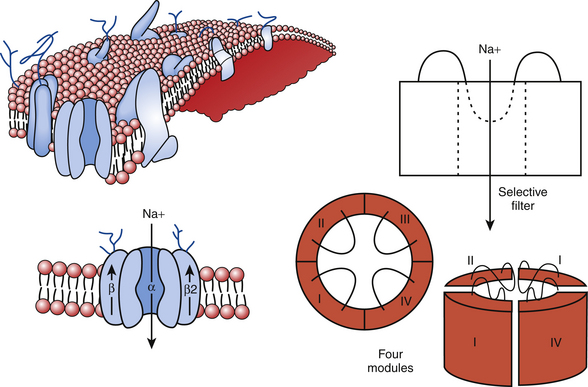
FIGURE 2-1 The sodium channel macromolecular complex. See text for discussion.
(From Boussy T, Paparella G, de Asmundis C, et al: Genetic basis of ventricular arrhythmias. Heart Fail Clin 6:249-266, 2010.)
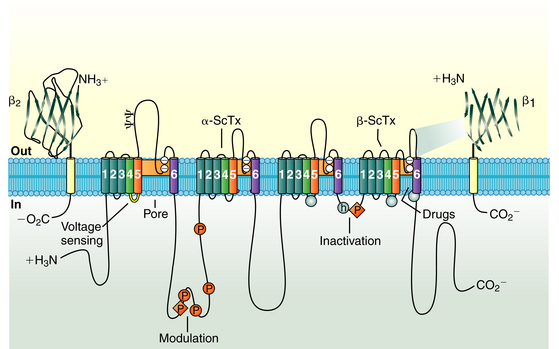
Four auxiliary β subunits (Navβ1 to Navβ4, encoded by the genes SCN1B to SCN4B, respectively) have been identified; each is a glycoprotein with a single membrane-spanning segment. The β1 subunit likely plays a role in modulation of the gating properties and level of expression of the Na+ channel.2
Na+ channels are the typical example of voltage-gated ion channels. Na+ channels switch among three functional states: deactivated (closed), activated (open), and inactivated (closed), depending on the membrane potential (Em). These channel states control Na+ ion permeability through the channel into the cardiomyocyte. Na+ channel activation allows Na+ ion influx into the cell, and inactivation blocks entry of Na+ ions.2
On excitation of the cardiomyocyte by electrical stimuli from adjacent cells, its resting Em (approximately −85 mV) depolarizes. The positively charged S4 segment of each domain of the α subunit functions as the sensor of the transmembrane voltage; these segments are believed to undergo rapid structural conformational changes in response to membrane depolarization, thus leading to channel opening (activation) from its resting (closed) state and enabling a large and rapid influx of Na+ (inward Na+ current [INa]) during the rapid upstroke (phase 0) of the action potential in atrial, ventricular, and Purkinje cardiomyocytes.3
Normally, activation of Na+ channels is transient; fast inactivation (closing of the pore) starts simultaneously with activation, but because inactivation is slightly delayed relative to activation, the channels remain transiently open to conduct INa during phase 0 of the action potential before it closes. Each Na+ channel opens very briefly (less than 1 millisecond) during phase 0 of the action potential; collectively, activation of the channel lasts a few milliseconds and is followed by fast inactivation.
Na+ channel inactivation comprises different conformational states, including fast, intermediate, and slow inactivation. Fast inactivation is at least partly mediated by rapid occlusion of the inner mouth of the pore by the cytoplasmic interdomain linker between domains III and IV of the α subunit, which has a triplet of hydrophobic residues that likely functions as a hinged “latch” that limits or restricts Na+ ion pass through the pore. The carboxyl terminus (C-terminus) also plays an important role in the control of Na+ channel inactivation and stabilizing the channels in the inactivated state by interacting with the loop linking domains III and IV. Importantly, although most Na+ channels open before inactivating, some actually inactivate without ever opening (a process known as closed-state inactivation).1,4
In addition to these rapid gating transitions, Na+ channels are also susceptible to slower inactivating processes (slow inactivation) if the membrane remains depolarized for a longer time. These slower events can contribute to the availability of active channels under various physiological conditions. Whereas fast-inactivated Na+ channels recover rapidly (within 10 milliseconds) during the hyperpolarized interval between stimuli, slow inactivation requires much longer recovery times (ranging from hundreds of milliseconds to many seconds). The molecular movements leading to slow inactivation are less well understood. The P segments seem to play a key role in slow inactivation.4
Some Na+ channels occasionally show alternative gating modes consisting of isolated brief openings occurring after variable and prolonged latencies and bursts of openings during which the channel opens repetitively for hundreds of milliseconds. The isolated brief openings are the result of the occasional return from the inactivated state. The bursts of openings are the result of occasional failure of inactivation.1 Prolonged opening or reopening of some Na+ channels during phases 2 and 3 can result in a small late INa (INaL). Despite its minor contribution in healthy hearts, INaL can potentially play an important role in diseased hearts.3
Function
The cardiac Na+ channel also plays a crucial role in the propagation of action potentials throughout the atrium, HPS, and ventricles. The opening of Na+ channels in the atria underlies the P wave on the ECG, and in the ventricles INa underlies the QRS complex and enables a synchronous ventricular contraction. Because the upstroke of the electrical potential primarily determines the speed of conduction between adjacent cells, Na+ channels are present in abundance in tissues where speed is of importance. Cardiac Purkinje cells contain up to 1 million Na+ channels, a finding that illustrates the importance of rapid conductance in the heart.2
Na+ channels also make a contribution in the plateau phase (phase 2) and help determine the duration of the action potential. After phase 0 of the action potential, INa decreases to less than 1% of its peak value over the next several milliseconds because of voltage-dependent inactivation. This persistent or “late” inward INa (INaL), along with the L-type Ca2+ current (ICaL), helps maintain the action potential plateau.5
Regulation
The regulatory proteins interacting with Nav1.5 may be classified as follows: (1) anchoring-adaptor proteins (e.g., ankyrin-G, syntrophin proteins, multicopy suppressor of gsp1 [MOG1]), which play roles only in trafficking and targeting the channel protein in specific membrane compartments; (2) enzymes interacting with and modifying the channel structure (post-translational modifications), such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 on binding (e.g., caveolin-3, calmodulin, glycerol 3-phosphate dehydrogenase 1–like [G3PD1L], telethonin, Plakophilin-2).5 Coexpression of Nav1.5 with its β subunits induces acceleration in the recovery from inactivation and enhancement of INa amplitude.
The cardiac Na+ channels are subject to phosphorylation and dephosphorylation by kinases or phosphatases. The intracellular linker between domains I and II contains eight consensus sites for cyclic adenosine monophosphate (cAMP)–dependent protein kinase A (PKA) phosphorylation. cAMP-dependent PKA and G protein stimulatory α subunit (Gsα) modulate the function of expressed cardiac Na+ channels on β-adrenergic stimulation and enhance INa.1
In contrast, activation of α-adrenergic stimulating protein kinase C (PKC) results in the reduction of INa. The effect of PKC is largely attributable to phosphorylation of a highly conserved serine in the linker between domains III and IV. PKC reduces the maximal conductance of the channels and alters gating. Na+ channels exhibit a hyperpolarizing shift in the steady-state availability curve, suggesting an enhancement of inactivation from closed states.
All subunits of the Na+ channel are modified by glycosylation. The β1 and β2 subunits are heavily glycosylated, with up to 40% of the mass being carbohydrate. In contrast, the cardiac α subunit is only 5% sugar by weight. Sialic acid is a prominent component of the N-linked carbohydrate of the Na+ channel. The addition of such a highly charged carbohydrate has predictable effects on the voltage dependence of gating through alteration of the surface charge of the channel protein.1
Pharmacology
Na+ channels are the targets for the action of class I antiarrhythmic drugs. Na+ channel blockers bind to a specific receptor within the channel’s pore. The binding blocks ion movement through the pore and stabilizes the inactivated state of Na+ channels. Blockade of Na+ channels tends to decrease tissue excitability and conduction velocity (by attenuating peak INa) and can shorten action potential duration (by attenuating late INa).1,6
One important component in the action of antiarrhythmic drugs is a voltage-dependent change in the affinity of the drug-binding site (i.e., the channel is a modulated receptor). Additionally, restricted access to binding sites can contribute to drug action, a phenomenon that has been called the guarded receptor model. Open and inactivated channels are more susceptible to block than resting channels, likely because of a difference in binding affinity or state-dependent access to the binding site. Consequently, binding of antiarrhythmic drug occurs primarily during the action potential (known as use-dependent block), and the block dissipates after repolarization (i.e., in the interval between action potentials). When the time interval between depolarizations is insufficient for block to recover before the next depolarization occurs (secondary to either abbreviation of the interval between action potentials during fast heart rates or slow kinetics of the unbinding of the Na+ channel blocker), block of Na+ channels accumulates (resulting in an increased number of blocked channels and enhanced blockade).6 A drug with rapid kinetics produces less channel block with the subsequent depolarization than does a drug with slower recovery. Use-dependent block is important for the action of antiarrhythmic drugs because it allows strong drug effects during fast heart rates associated with tachyarrhythmias but limits Na+ channel block during normal heart rates. Importantly, drug recovery kinetics can potentially be slowed by pathophysiological conditions such as membrane depolarization, ischemia, and acidosis.1 This property is known as use-dependence and is seen most frequently with the class IC agents, less frequently with the class IA drugs, and rarely with the class IB agents.
Inherited Channelopathies
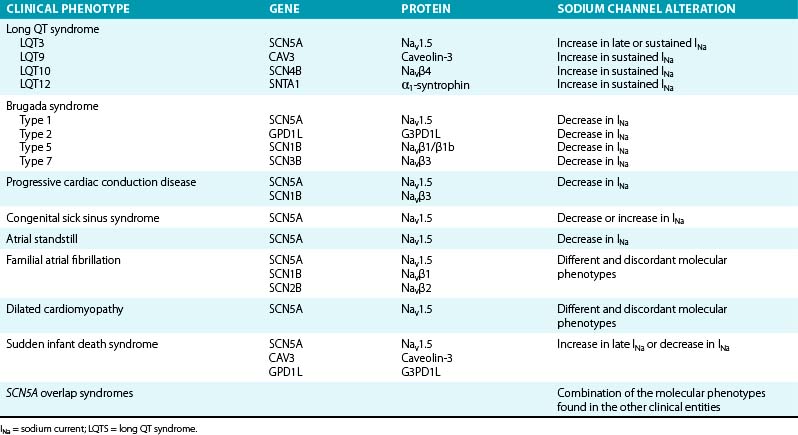
Mutations in genes that encode various subunits of the cardiac Na+ channel or proteins involved in regulation of the inward INa have been linked to several types of electrical disorders (Table 2-1). Depending on the mutation, the consequence is either a gain of channel function (with consequent prolongation of action potential duration because more positive ions accumulate in the cell) or an overall loss of channel function that influences the initial depolarizing phase of the action potential (with consequent decrease in cardiac excitability and electrical conduction velocity). It is noteworthy that a single mutation can cause different phenotypes or combinations thereof.2
Long QT Syndrome
In contrast to most long QT syndrome (LQTS) phenotypes, which are based on mutations that modify the cardiac K+ currents, type 3 congenital LQTS (LQT3), which accounts for approximately 8% of congenital LQTS cases, is caused by gain-of-function mutations on the Na+ channel gene, SCN5A. More than 80 mutations have been identified in the SCN5A gene, with most being missense mutations mainly clustered in Nav1.5 regions that are involved in fast inactivation (i.e., S4 segment of domain IV, the domain III–domain IV linker, and the cytoplasmic loops between the S4 and S5 segments of domain III and domain IV), or in regions that stabilize fast inactivation (e.g., the C-terminus).2,4,7
Several mechanisms have been identified to underlie ionic effects of SCN5A mutations in LQT3. Most SCN5A mutations cause a gain of function through disruption of fast inactivation, thus allowing repeated reopening during sustained depolarization and resulting in an abnormal, small, but functionally important sustained (or persistent) noninactivating Na+ current (Isus) during action potential plateau. Because the general membrane conductance is small during the action potential plateau, the presence of a persistent inward INa, even of small amplitude, can potentially have a major impact on the plateau duration and can be sufficient to prolong repolarization and QT interval. QT prolongation and the risk of developing arrhythmia are more pronounced at slow heart rates, when the action potential duration is longer, thereby allowing more INa to enter the cell.2,4,7
Other less common mechanisms of SCN5A mutations to cause LQT3 include increased window current, which results from delayed inactivation of mutant Na+ channels, occurring at more positive potentials and widening the voltage range during which the Na+ channel may reactivate without inactivation. Additionally, some mutations cause slower inactivation, which allows longer channel openings and causes a slowly inactivating INa. This current is INaL and is to be distinguished from Isus (which does not inactivate). Comparable to Isus, both the window current and INaL exert their effects during phases 2 and 3 of the action potential, in which normally no or very small INa is present. Other mutations induce prolonged action potential duration by enhancing recovery from inactivation, an effect that leads to larger peak INa by increasing the fraction of channels available for activation (because of faster recovery) during subsequent depolarizations. Finally, some mutations can cause increased expression of mutant Nav1.5 through enhanced mRNA translation or protein trafficking to the sarcolemma, decreased protein degradation, or altered modulation by β subunits and regulatory proteins. These effects lead to larger INa density during phase 0 of the action potential. Importantly, one single SCN5A mutation can potentially cause several changes in the expression and/or gating properties of the resulting Na+ channels.2
Regardless of the mechanism, increased Na+ current (Isus, window current, INaL, or peak INa) upsets the balance between depolarizing and repolarizing currents in favor of depolarization. The resulting delay in the repolarization process triggers early afterdepolarizations (EADs; i.e., reactivation of the L-type Ca2+ channel during phase 2 or 3 of the action potential), especially in Purkinje fiber myocytes, in which action potential durations are intrinsically longer.2
LQT9 is caused by gain-of-function mutations on the CAV3 gene, which encodes caveolin-3, a plasma membrane scaffolding protein that interacts with Nav1.5 and plays a role in compartmentalization and regulation of channel function. Mutations in caveolin-3 induce kinetic alterations of the Nav1.5 current that result in persistent Na+ current (Isus) and have been reported in cases of sudden infant death syndrome (SIDS).2,8
LQT10 is caused by loss-of-function mutations on the SCN4B gene, which encodes the β subunit (Navβ4) of the Nav1.5 channel. To date, only a single mutation in one patient has been described. This mutation caused a shift in the inactivation of the INa toward more positive potentials, but it did not change the activation. This resulted in increased window currents at an Em corresponding to the phase 3 of the action potential.2,8
LQT12 is caused by mutations on the SNTA1 gene, which encodes α1 syntrophin, a cytoplasmic adaptor protein that enables the interaction among Nav1.5, nitric oxide synthase, and the sarcolemmal Ca2+ adenosine triphosphatase (ATPase) complex that appears to regulate ion channel function. By disrupting the interaction between Nav1.5 and the sarcolemmal Ca2+ ATPase complex, SNTA1 mutations cause increased Nav1.5 nitrosylation with consequent reduction of channel inactivation and enhanced Isus densities.2,9
Brugada Syndrome
The Brugada syndrome is an autosomal dominant inherited channelopathy characterized by an elevated ST segment or J wave appearing in the right precordial leads. This syndrome is associated with a high incidence of sudden cardiac death (SCD) secondary to a rapid polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF). Approximately 65% of mutations identified in the SCN5A gene are associated with the Brugada syndrome phenotype (Brugada syndrome type 1), and they account for approximately 18% to 30% of cases of Brugada syndrome. So far, more than 200 Brugada syndrome–associated loss-of-function (i.e., reduced peak INa) mutations have been described in SCN5A. Some of these mutations result in loss of function secondary to impaired channel trafficking to the cell membrane (i.e., reduced expression of functional Na+ channels), disrupted ion conductance (i.e., expression of nonfunctional Na+ channels), or altered gating function. Altered gating properties comprise delayed activation (i.e., activation at more positive potentials), earlier inactivation (i.e., inactivation at more negative potentials), faster inactivation, and enhanced slow inactivation.1–3,10
Most of the mutations are missense mutations, whereby a single amino acid is replaced by a different amino acid. Missense mutations commonly alter the gating properties of mutant channels. Because virtually all reported SCN5A mutation carriers are heterozygous, mutant channels with altered gating may cause up to 50% reduction of INa. Different SCN5A mutations can cause different degrees of INa reduction and therefore different degrees of severity of the clinical phenotype of Brugada syndrome.10–12
In addition to SCN5A alterations, mutations in the GPD1L gene, which encodes the glycerol 3-phosphate dehydrogenase 1–like protein (G3PD1L), affect the trafficking of the cardiac Na+ channel to the cell surface and result in reduction of INa and Brugada syndrome type 2.10 Brugada syndrome associated with GPD1L gene mutations is characterized by progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis.2,13
Furthermore, reduction in INa can be caused by mutations in the SCN1B gene (encoding the β1 and β1b subunits of the Na+ channel) and the SCN3B gene (encoding the β3 subunit of the Na+ channel), resulting in Brugada syndrome type 5 and type 7, respectively.10,13
Familial Progressive Cardiac Conduction Disease
Loss-of-function SCN5A mutations have been linked to familial forms of progressive cardiac conduction disease (referred to as hereditary Lenègre disease, primary cardiac conduction system disease, and familial atrioventricular [AV] block). This disease is characterized by slowing of electrical conduction through the atria, AVN, His bundle, Purkinje fibers, and ventricles, accompanied by an age-related degenerative process and fibrosis of the cardiac conduction system, in the absence of structural or systemic disease. It is often reflected by varying degrees of AV block and bundle branch block. Whether the age-dependent fibrosis of the conduction system is a primary degenerative process in progressive cardiac conduction disease or a physiological process that is accelerated by INa reduction remains to be investigated. A single loss-of-function SCN5A mutation can cause isolated progressive cardiac conduction disease or can be combined with the Brugada syndrome (overlap syndrome). Loss-of-function mutations in SCN1B also have been identified in patients with progressive cardiac conduction disease who carried no mutation in SCN5A.2
Congenital Sick Sinus Syndrome
Although INa does not play a prominent role in sinus node activity, mutations in SCN5A have been linked to sick sinus syndrome, manifesting as sinus bradycardia, sinus arrest, sinoatrial block, or a combination of these conditions, which can progress to atrial inexcitability (atrial standstill). Loss-of-function SCN5A mutations result in reduced peak INa density, hyperpolarizing shifts in the voltage dependence of steady-state channel availability, and slow recovery from inactivation. These effects likely cause reduced automaticity, decreased excitability, and conduction slowing or block of impulses generated in the sinus node to the surrounding atrial tissue. Sinus node dysfunction can also manifest concomitantly with other phenotypes that are linked to SCN5A loss-of-function mutations such as Brugada syndrome and progressive cardiac conduction disorders.2,14
Familial Atrial Fibrillation
Loss-of-function mutations, gain-of-function mutations, and common polymorphisms on the SCN5A gene have been identified in some cases of atrial fibrillation (AF) occurring in young patients with structurally normal hearts. It is speculated that INa reduction may predispose to AF by slowing the electrical conduction velocity and thereby facilitating reentry. On the other hand, gain-of-function mutations can potentially predispose to AF by increasing atrial excitability. AF can occur in patients with other phenotypes of Na+ channelopathies, including LQT3, Brugada syndrome, dilated cardiomyopathy, and sinus node dysfunction. Furthermore, mutations in the SCN1B gene (encoding the β1 subunit of the Na+ channel) and the SCN2B gene (encoding the β2 subunit of the Na+ channel) have been identified in patients with AF, many of whom displayed ECG patterns suggestive of the Brugada syndrome.1,2
Dilated Cardiomyopathy
Some cases of familial dilated cardiomyopathy have been linked to SCN5A mutations. Dilated cardiomyopathy–linked SCN5A mutations cause diverse loss-of-function and gain-of-function changes in the gating properties, but how such changes evoke contractile dysfunction is not understood. It is speculated that SCN5A mutations disrupt the interactions between the mutant Na+ channels and intracellular (or extracellular) proteins that are essential for normal cardiomyocyte structure and architecture. Notably, dilated cardiomyopathy with SCN5A mutations display atrial or ventricular arrhythmias (including AF, VT, and VF), or both, as well as sinus node dysfunction, AV block, and intraventricular conduction delay.2
Acquired Diseases
In heart failure, peak INa is reduced (likely secondary to reduced SCN5A expression), whereas INaL is increased (likely because of increased phosphorylation of Na+ channels). Nav1.5 expression is reduced in the surviving myocytes in the border zone of the myocardial infarct (MI). Importantly, Na+ channel blockers can increase the risk for SCD in patients with ischemic heart disease, possibly by facilitating the initiation of reentrant excitation waves. Additionally, INaL increases during myocardial ischemia, explaining why INaL inhibition may be an effective therapy for chronic stable angina. Nav1.5 expression is reduced in response to persistent atrial tachyarrhythmias as part of the “electrical remodeling” process, leading to attenuation of INa.3
Potassium Channels
Structure and Physiology
Cardiac K+ channels are membrane-spanning proteins that allow the passive movement of K+ ions across the cell membrane along its electrochemical gradient. The ion-conducting or pore-forming subunit is generally referred to as the α subunit. The tripeptide sequence glycine-tyrosine-glycine GYG is common to the pore of all K+ channels and constitutes the signature motif for determining K+ ion selectivity. A gating mechanism controls switching between open-conducting and closed-nonconducting states.1,15
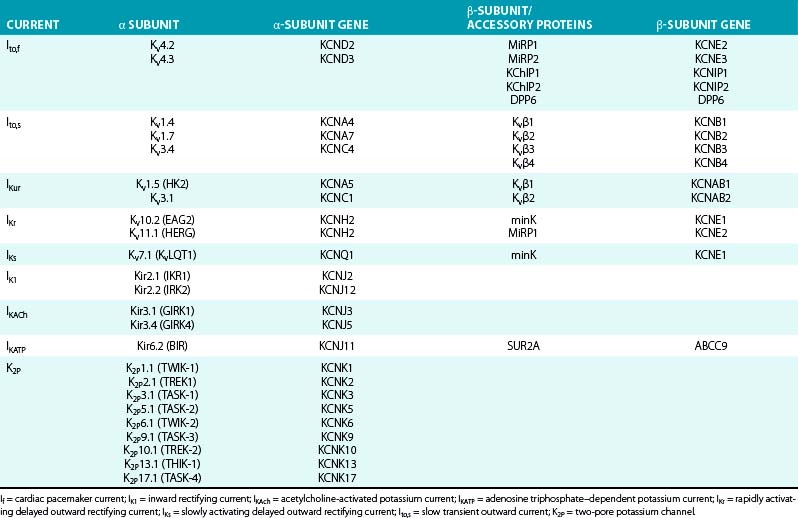
K+ channels represent the most diverse class of cardiac ion channels (Fig. 2-3). Cardiac K+ currents can be categorized as voltage-gated (Kv) and ligand-gated channels. In Kv channels, pore opening is coupled to the movement of a voltage sensor within the membrane electric field, and they include the rapidly activating and inactivating transient outward current (Ito); the ultrarapid (IKur), rapid (IKr), and slow (IKs) components of the delayed rectifier current; and the inward rectifier current (IK1). In contrast, pore opening in ligand-gated channels is coupled to the binding of an organic molecule, including channels activated by a decrease in the intracellular concentration of adenosine triphosphate (KATP) or activated by acetylcholine (KACh). Other classes of K+ channels respond to different stimuli, including changes in intracellular Ca2+ concentration and G proteins.15
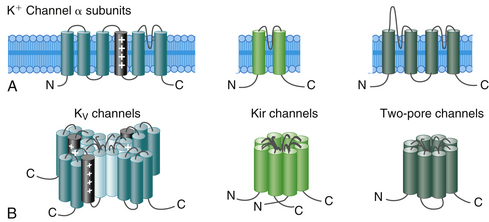
On the basis of the primary amino acid sequence of the α subunit, K+ channels have been classified into three major families (Table 2-2):
1. Channels containing six transmembrane segments and a single pore. This architecture is typical of Kv channels.
2. Channels containing two transmembrane segments (M1 and M2) and a pore. This architecture is typical of inward rectifier K+ (Kir) channels, including K1, KATP, and KACh channels. They conduct K+ currents more in the inward direction than the outward and play an important role in setting the resting potential close to the equilibrium potential for K+ and in repolarization. Kir channels form either homotetramers or heterotetramers.
3. Channels containing four transmembrane segments and two pores (K2P). These channels exist as homodimers or heterodimers. K2P currents display little time or voltage dependence. There are four classes of cardiac K2P: TASK, TWIK, TREK, and THIK.15,16
Each voltage-gated K+ channel (Kv family) is formed by the coassembly of 4 identical (homotetramers) or a combination of 4 different (from the same subfamily, heterotetramers) α subunits. A total of 38 genes has been cloned and assigned to 12 subfamilies of voltage-gated K channels (Kv1 to Kv12) on the basis of sequence similarities. Each α subunit contains one domain consisting of 6 membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops (similar to 1 of the 4 domains of voltage-gated Na+ and Ca2+ channels), with both the amino terminus (N-terminus) and the C-terminus located on the intracellular side of the membrane. The central ion-conducting pore region is formed by the S5 and S6 segments and the S5-S6 linker (P segment); the S5-S6 linker is responsible for K+ ion selectivity. The S4 segment serves as the voltage sensor.1,15
The α subunits of Kv channels can generate voltage dependent K+ current when expressed in heterologous systems. However, the assembly of a functional tetramer can occur only in the presence of multiple auxiliary units (see Table 2-2). In many cases, auxiliary subunits coassociate with the α subunits and likely modulate cell surface expression, gating kinetics, and drug sensitivity of the α subunit complex. Most K+ channel β subunits assemble with α subunits and give rise to an α4β4 complex. K+ channel β subunits represent a diverse molecular group, which includes cytoplasmic proteins (Kvβ1 to Kvβ3, KChIP, and KChAP) that interact with the intracellular domains of Kv channels; single transmembrane spanning proteins (e.g., minK and minK-related proteins [MiRPs]) encoded by the KCNE gene family; and large ATP-binding cassette (ABC) transport-related proteins (e.g., the sulfonylurea receptors [SURs]).1,16
As with voltage-dependent Na+ and Ca2+ channels, Kv channels typically fluctuate among distinct conformational states because of molecular movements in response to voltage changes across the cell membrane (voltage-dependent gating). The Kv channel activates (opens) on membrane depolarization, thus allowing the rapid passage of K+ ions across the sarcolemma. After opening, the channel undergoes conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening, even if the transmembrane voltage is favorable, unless they “recover” from inactivation (i.e., enter the closed state) on membrane repolarization. Closed channels are nonconducting but can be activated on membrane depolarization.17
Three mechanistically distinct types of Kv channel inactivation that are associated with distinct molecular domains have been identified: N-type, C-type, and U-type. N-type (“ball and chain”) inactivation involves physical occlusion of the intracellular mouth of the channel pore through binding of a small group of amino acids (“inactivation ball tethered to a chain”) at the extreme N-terminus. In contrast, C-type inactivation involves conformational changes in the external mouth of the pore. C-type inactivation exists in almost all K+ channels and may reflect a slow constriction of the pore. This inactivation process is thought to be voltage independent, coupled to channel opening, and is usually slower than N-type inactivation. Recovery from C-type inactivation is relatively slow and weakly voltage dependent. Importantly, the rate of C-type inactivation and recovery can be strongly influenced by other factors, such as N-type inactivation, drug binding, and changes in extracellular K+ concentration. These interactions render C-type inactivation an important biophysical process in regulating repetitive electrical activity and determining certain physiological properties such as refractoriness, drug binding, and sensitivity to extracellular K+.17
In addition to N-type and C-type inactivation, some Kv channels also show another type of inactivation (U-type), which exhibits a U-shaped voltage dependence with prolonged stimulation rates. Those channels appear to exhibit preferential inactivation from partially activated closed states, rapid and strongly voltage-dependent recovery from inactivation and, in some channel types, accelerated inactivation with elevation of extracellular K+. The exact conformational changes underlying U-type inactivation remain unclear. Importantly, there is extreme diversity in the kinetic and potentially molecular properties of Kv channel inactivation, particularly of C-type inactivation.17
Function
K+ channels are a diverse and ubiquitous group of membrane proteins that regulate K+ ion flow across the cell membrane on the electrochemical gradient and regulate the resting Em, the frequency of pacemaker cells, and the shape and duration of the cardiac action potential. Because the concentration of K+ ions outside the cell membrane is approximately 25-fold lower than that in the intracellular fluid, on activation, the opening of K+ channels generates an outward current resulting from the efflux of positively charged ions that offers a mechanism to counteract, dampen, or restrict the depolarization front (phases 1 through 4 of the action potential) triggered by an influx of cations (Na+ and Ca2+).1,3,15,16
The variation in the level of expression of K+ channels that participate in the genesis of the cardiac action potential explains the regional differences of the configuration and duration of cardiac action potentials from sinus node and atrial to ventricular myocytes and across the myocardial wall (endocardium, midmyocardium, and epicardium). Moreover, the expression and properties of K+ channels are not static; heart rate, neurohumoral state, pharmacological agents, cardiovascular diseases (cardiac hypertrophy and failure, MI) and arrhythmias (e.g., AF) can influence those properties, and they underlie the change in action potential configuration in response to variation in heart rate and various physiological and pathological conditions.1,15,16
Transient Outward Potassium Current (Ito)
Structure and Physiology
Cardiac Ito channels are macromolecular protein complexes, comprising four pore-forming Kv α subunits and a variety of Kv channel accessory (β) subunits (see Fig. 2-3). Ito is the sum of a voltage-dependent, Ca2+-independent K+ current (Ito1) and a Ca2+-activated Cl− or K+ current (Ito2). In human atrial and ventricular myocytes, the presence of Ito2 has not been clearly demonstrated.
Kv channels mediating Ito,s are formed by the coassembly of four α subunits from the Kv1.x subfamily (primarily Kv1.4, and possibly Kv1.7 and Kv3.4), whereas those mediating Ito,f are formed by the coassembly of four α subunits from the Kv4.x subfamily (primarily Kv4.3, and possibly Kv4.2) (see Table 2-2). Among the various accessory subunits identified, a crucial role has been definitively demonstrated only for KChIP2, and potentially for MiRP2.1,16,18
Function
The density of Ito varies across the myocardial wall and in different regions of the heart. In human ventricles, Ito densities are much higher in the epicardium and midmyocardium than in the endocardium. Furthermore, Ito,f and Ito,s are differentially expressed in the myocardium, thus contributing to regional heterogeneities in action potential waveforms. Ito,f is the principal subtype expressed in human atrium. The markedly higher densities of Ito,f, together with the expression of the ultrarapid delayed rectifier current, accelerate the early phase of repolarization and lead to lower plateau potentials and shorter action potentials in atrial as compared with ventricular cells.17,18
Although both Ito,f and Ito,s are expressed in the ventricle, Ito,f is more prominent in the epicardium and midmyocardium (putative M cells) than in the endocardium. These regional differences are responsible for the shorter duration and the prominent phase 1 notch and the “spike-and-dome” morphology of epicardial and midmyocardial compared with endocardial action potentials. A prominent Ito-mediated action potential notch in ventricular epicardium but not endocardium produces a transmural voltage gradient during early ventricular repolarization that registers as a J wave or J point elevation on the ECG.19 Ito densities are also reportedly higher in right than in left (midmyocardial and epicardial) ventricular myocytes, consistent with the more pronounced spike-and-dome morphology of right, compared with left, ventricular action potentials.1,3,17,18
Furthermore, variations in cardiac repolarization associated with Ito regional differences strongly influence intracellular Ca2+ transient by modulating Ca2+ entry via ICaL and Na+-Ca2+ exchange, thereby regulating excitation-contraction coupling and regional modulation of myocardial contractility and hence synchronizing the timing of force generation between different ventricular regions and enhancing mechanical efficiency.17
Regulation
Transient outward channels are subject to α- and β-adrenergic regulation. α-Adrenergic stimulation reduces Ito; concomitant β-adrenergic stimulation appears to counteract the α-adrenergic effect, at least in part. The effects of α- and β-adrenergic stimulation are exerted by phosphorylation of the Kv1.4, Kv4.2, and Kv4.3 α-subunits by PKA as well as PKC. Calmodulin-dependent kinase II, on the other hand, has been shown to be involved in enhancement of Ito. Adrenergic stimulation is also an important determinant of transient outward channel downregulation in cardiac disease. Chronic α-adrenergic stimulation and angiotensin II reduce Ito channel expression.20
KChIP2, when coexpressed with Kv4.3, increases surface channel density and current amplitude, slows channel inactivation, and markedly accelerates the recovery from inactivation. In the ventricle, KChIP2 mRNA is 25-fold more abundant in the epicardium than in the endocardium. This gradient parallels the gradient in Ito expression, whereas Kv4.3 mRNA is expressed at equal levels across the ventricular wall. Thus, transcriptional regulation of the KChIP2 gene (KChIP2) is the primary determinant of Ito expression in the ventricular wall.1,16
Observations suggest that MiRP2 is required for the physiological functioning of human Ito,f channels and that gain-of-function mutations in MiRP2 predispose to Brugada syndrome through augmentation of Ito,f.18
Ito is strongly rate dependent. Ito fails to recover from previous inactivation at very fast heart rates, which can be manifest as a decrease in the magnitude of the J wave on the surface ECG. Hence, abrupt changes in rate and pauses have important consequences for the early repolarization of the membrane.19
Ito can be enhanced by aging, low sympathetic activity, high parasympathetic activity, bradycardia, hypothermia, estrogen reduction, and drugs. Estrogen suppresses the expression of the Kv4.3 channel and results in reduced Ito and a shallow phase 1 notch.13
Phase 1 notch of the action potential modulates the kinetics of slower activating ion currents and consequently the later phases of the action potential. Initial enhancement of phase 1 notch promotes phase 2 dome and delays repolarization, presumably by delaying the peak of ICaL. However, further enhancement of phase 1 notch prevents the rising of phase 2 dome and abbreviates action potential duration, presumably by deactivation or voltage modulation that reduces ICaL. Thus, progressive deepening of phase 1 notch can cause initial enhancement followed by sudden disappearance of phase 2 dome and corresponding prolongation followed by abbreviation of action potential duration. On the other hand, modulators that decrease Ito lead to a shift of the plateau phase into the positive range of potentials, thus increasing the activation of the delayed rectifier currents, promoting faster repolarization, and reducing the electrochemical driving force for Ca2+ and hence ICaL. Phase 1 notch also affects the function of the Na+-Ca2+ exchanger and subsequently intracellular Ca2+ handling and Na+ channel function.1,16
Pharmacology
Quinidine, 4-aminopyridine, flecainide, and propafenone produce an open channel blockade and accelerate Ito inactivation. Quinidine, but not flecainide or propafenone, produces a frequency-dependent block of Ito that results from a slow rate of drug dissociation from the channel. Quinidine has relatively strong Ito blocking effect, whereas flecainide mildly blocks Ito.18,20
Ito blockers can potentially prolong the action potential duration in the atrial and in ischemic ventricular myocardium. However, because the net effects of Ito blockade on repolarization depend on secondary changes in other currents, the reduction of Ito density can result in a shortening of the ventricular action potential. Moreover, heterogeneous ventricular distribution of Ito can cause marked dispersion of repolarization across the ventricular wall that, when accompanied by prominent conduction delays related to Na+ channel blockade, results in extrasystolic activity through a phase 2 reentrant mechanisms.18,20
Currently, a cardioselective and channel-specific Ito opener or blocker is not available for clinical use. Development of an Ito-selective drug is expected to be beneficial in patients with primary abnormality in the Ito or in other channels, such as the Brugada syndrome, in which heterogeneity in the expression of Ito between epicardium and endocardium in the right ventricle (RV) results in the substrate responsible for reentry and ventricular arrhythmias.18,20
Inherited Channelopathies
To date, only mutations in KCNE3 (MiRP2) are linked to inherited arrhythmia. KCNE3 mutations were identified in five related patients with Brugada syndrome. When expressed with Kv4.3, the mutation increased Ito,f density.3
Another KCNE3 mutation was identified in one patient with familial AF. The mutation was found to increase Ito,f and was postulated to cause AF by shortening action potential duration and facilitating atrial reentrant excitation waves.3
A genome wide haplotype-sharing study associated a haplotype on chromosome 7, harboring DPP6, with idiopathic VF in three distantly related families. Overexpression of DPP6, which encodes dipeptidyl-peptidase 6, a putative component of the Ito channel complex, was proposed as the likely pathogenetic mechanism. DPP6 significantly alters the inactivation kinetics of both Kv4.2 and Kv4.3 and promotes expression of these α subunits in the cell membrane.3,21
Importantly, the normally functioning Ito channels play an important role in the electrophysiological consequences of the ionic current abnormalities in the Brugada and J wave syndromes. Heterogeneity in the distribution of Ito channels across the myocardial wall, being more prominent in ventricular epicardium than endocardium, and particularly in the RV, results in the shorter duration and the prominent phase 1 notch and the spike-and-dome morphology of the epicardial action potential as compared with the endocardium. The resultant transmural voltage gradient during the early phases (phases 1 and 2) of the action potential is thought to be responsible for the inscription of the J wave on the surface ECG.18,19 An increase in net repolarizing current, secondary to either a decrease in the inward currents (INa and ICaL) or an increase in the outward K+ currents (Ito, IKr, IKs, IKAch, IKATP), or both, can accentuate the action potential notch and lead to augmentation of the J wave or the appearance of ST segment elevation on the surface ECG. An outward shift of currents that extends beyond the action potential notch not only can accentuate the J wave but also can lead to partial or complete loss of the dome of the action potential, thus leading to a protracted transmural voltage gradient that manifests as greater ST segment elevation and gives rise to J wave syndromes. The type of the ion current affected and its regional distribution in the ventricles determine the particular phenotype (including the Brugada syndrome, early repolarization syndrome, hypothermia-induced ST segment elevation, and MI-induced ST segment elevation).18,19,22 The degree of accentuation of the action potential notch leading to loss of the dome depends on the magnitude of Ito. These changes are more prominent in regions of the myocardium exhibiting a relatively large Ito, such as the RV epicardium; this explains the appearance of coved ST segment elevation, characteristic of Brugada syndrome, in the right precordial ECG leads.19,20
In this context, factors that influence the kinetics of Ito or the other repolarization currents can modify the manifestation of the J wave on the ECG. Na+ channel blockers (procainamide, pilsicainide, propafenone, flecainide, and disopyramide), which reduce the inward INa, can accentuate the J wave and ST segment elevation in patients with concealed J wave syndromes. Quinidine, which inhibits both Ito and INa, reduces the magnitude of the J wave and normalizes ST segment elevation. Additionally, acceleration of the heart rate, which is associated with reduction of Ito (because of slow recovery of Ito from inactivation), results in a decrease in the magnitude of the J wave. Male predominance can potentially result from larger epicardial Ito density versus Ito in women.19,22
The increased transmural heterogeneity of ventricular repolarization (i.e., dispersion of repolarization between epicardium and endocardium), which is responsible for J point elevation and early repolarization pattern on the surface ECG, is also responsible for the increased vulnerability to ventricular tachyarrhythmias. A significant outward shift in current can cause partial or complete loss of the dome of the action potential in regions where Ito is prominent (epicardium), with the consequent loss of activation of ICaL. The dome of the action potential then can propagate from regions where it is preserved (midmyocardium endocardium) to regions where it is lost (epicardium), thus giving rise to phase 2 reentry, which can generate premature ventricular complexes that in turn can initiate polymorphic VT or VF.19,20,22,23
Acquired Diseases
An alteration in the expression and distribution of Ito is observed in various pathophysiological conditions. Adrenergic effects seem to be involved in at least some of these Ito-regulating processes during heart disease.20
In general, myocardial ischemia, MI, dilated cardiomyopathy, and end-stage heart failure cause downregulation of Ito. In fact, Ito downregulation is the most consistent ionic current change in the failing heart. The reduction in Ito results in attenuation of early repolarization (phase 1) and affects the level of plateau (phase 2) of the action potential and other currents involved in delayed repolarization (phase 3), with resulting prolongation and increased heterogeneity of action potential duration. The prominent epicardial Ito contributes to the selective electrical depression of the epicardium. This process leads to the development of a marked dispersion of repolarization between normal and abnormal epicardium and between epicardium and endocardium, which provides the substrate for reentrant arrhythmias and may underlie the increased predisposition to ventricular arrhythmias and SCD in patients with heart failure and ischemic heart disease.18,20,24 Additionally, downregulation of Ito in advanced heart failure likely slows the time course of force generation, thereby contributing to reduced myocardial performance.
On the other hand, compensatory ventricular hypertrophy preceding heart failure is associated with an upregulation of Ito. The prolongation of the action potential, concomitant with an increase in Ito, presumably results from the more negative level of the plateau with less ICaL inactivation and probably less delayed rectifier activation. In contrast, progression of hypertrophy to heart failure is associated with a clear reduction in Ito.20
Chronic AF reduces Ito density and Kv4.3 mRNA levels. Hypothyroidism reduces the expression of KCND2 (Kv4.2) genes. Additionally, Ito may also be reduced and contribute to QT interval prolongation in diabetes. Importantly, with some delay, insulin therapy partially restores Ito, maybe by enhancing Kv4.3 expression.3
Ultrarapidly Activating Delayed Outward Rectifying Current (IKur)
Structure and Physiology
The ion-conducting pore of IKur channels is formed by four Kv1.5 α subunits, whereas the ancillary β subunits Kvβ1.2, Kvβ1.3, and Kvβ2.1 control channel trafficking and plasma membrane integration as well as activation and inactivation kinetics.3,25
IKur activates rapidly on depolarization in the plateau range and displays outward rectification, but it inactivates very slowly during the time course of the action potential. Inactivation accelerates when Kv1.5 is coexpressed with its β subunits.3
Function
IKur is detected only in human atria and not in the ventricles, so that it is the predominant delayed rectifier current responsible for human atrial repolarization and is a basis for the much shorter duration of the action potential in the atrium.1,3
Regulation
β-Adrenergic stimulation enhances IKur, whereas α-adrenergic stimulation inhibits it, effects likely mediated by PKA and PKC, respectively. Membrane depolarization and elevated extracellular K+ concentrations reduce Kv1.5 expression. Additionally, cAMP, mechanical stretch, hyperthyroidism, and dexamethasone increase Kv1.5 expression, whereas extracellular acidosis, phenylephrine, and hypothyroidism decrease it.1,3
Pharmacology
IKur is relatively insensitive to class III antiarrhythmics of the methane-sulfonanilide group, but it is highly sensitive to 4-aminopyridine. Selective inhibition of IKur by 4-aminopyridine prolongs the human atrial action potential duration.16
IKur is a promising target for the development of new, safer antiarrhythmic drugs to prevent AF or atrial flutter, or both, without a risk of ventricular proarrhythmia. Because IKur is atrium specific, a drug specifically targeting Kv1.5 channels would be expected to terminate AF by preventing reentry through atrial action potential prolongation. The drug vernakalant is an IKur/INa channel blocker and is undergoing review by the U.S. Food and Drug Administration for the acute termination of AF. However, because Kv1.5 is downregulated in AF, the beneficial effect of IKur block becomes less certain. Furthermore, because Kv1.5 is also expressed in other organs (e.g., brain), discovery of drugs that selectively inhibit atrial Kv1.5 channels remains necessary.1,3,16
Physiologically, rapid activation of IKur in the positive potential range following the action potential upstroke can offset depolarizing ICaL and hence lead to the less positive plateau phase in atrial compared with ventricular cardiomyocytes. Conversely, block of IKur produces a more pronounced spike-and-dome configuration and therefore shifts the potential into a more positive range in which ICaL activation enhances systolic Ca2+ influx during a free-running action potential. Such an indirect effect on ICaL should be shared by all IKur blockers and is expected to result in a positive atrial inotropic effect.25
Inherited Channelopathies
KCNA5 nonsense mutations have been reported in individuals with familial AF. Heterologous expression of these mutations revealed complete IKur loss of function. Absence of IKur can excessively prolong atrial action potential duration with an enhanced risk of EADs that can trigger or maintain AF.3,25
Acquired Diseases
A prolonged period of rapid activation of the atria as occurs during AF leads to a decrease in IKur. Additionally, IKur may be affected in myocardial ischemia. Decreased Kv1.5 mRNA levels were reported for the epicardial border zone of infarcted hearts. Moreover, ischemic damage disrupted the normal location of Kv1.5 in the intercalated discs.1,3,26
Rapidly Activating Delayed Outward Rectifying Current (IKr)
Structure and Physiology
IKr is formed by coassembly of four pore-forming α subunits (Kv11.1, encoded by the KCNH2 gene, also called the human-ether-a-go-go-related gene, HERG, so-named because the mutation in the Drosophila fruit fly caused it to shake like a go-go dancer) and β subunits (MiRP1, encoded by the KCNE2 gene). The current generated by HERG channels shows unusual voltage dependence. In contrast to IKs, IKr activates relatively rapidly (on the order of 10s of milliseconds) on membrane depolarization. Activation of IKr occurs with steep voltage dependence and reaches half-maximum activation at membrane voltage of approximately –20 mV. The magnitude of IKr increases as a function of Em up to approximately 0 mV, but it declines with stronger depolarization (higher than 0 mV), resulting in a negative slope conductance of the current-voltage relationship. During repolarization of the action potential, IKr rapidly recovers from inactivation, thus causing the current to peak at –40 mV. The amplitude of the tail current on repolarization exceeds that of the current during the depolarizing pulse.3,16,27,28
The unusual voltage dependence of IKr results from a fast, voltage-dependent C-type inactivation process, which limits outward K+ flow at positive voltages. The large tail current on repolarization from positive voltages results from the rapid recovery of inactivated channels into a conducting state.27 On repolarization, HERG channels deactivate (close) via a slow, voltage-independent process (in contrast to the voltage-dependent inactivation process).3
Unlike most Kv channels, HERG channels exhibit inward rectification. Rectification describes the property of an ion channel to allow currents preferentially to flow in one direction or limit currents from flowing in the other direction. In other words, conductivity of channels carrying such currents is not constant but is altered at a different Em. A channel that is inwardly rectifying is one that passes current (positive charge) more easily into the cell. This property is critical for limiting outward K+ conductance during the plateau phase of the cardiac action potential. Unlike typical Kir channels, in which rectification derives from blockade of the channel pore by intracellular polyamines (see later discussion), the mechanism of HERG inward rectification is a very rapid inactivation that develops at far more negative potentials (–85 mV) than channel activation (–20 mV).28
Function
IKr activates relatively fast on membrane depolarization and allows outward diffusion of K+ ions in accordance with its electrochemical gradient, but voltage-dependent inactivation thereafter is very fast; hence, only limited numbers of channels remain in the open state, whereas a considerable fraction resides in the nonconducting inactivated state. The fast voltage-dependent inactivation limits outward current through the channel at positive voltages and thus helps maintain the action potential plateau phase that controls contraction and prevents premature excitation. However, as the voltage becomes less positive at the end of the plateau phase of repolarization, the channels recover rapidly from inactivation, thus leading to a progressive increase in IKr amplitudes during action potential phases 2 and 3, with maximal outward current occurring before the final rapid declining phase of the action potential. Next, the channel deactivates (closes) slowly. The resulting large and transient outward current adds considerably to the ongoing repolarization and makes HERG especially suitable for robust control of the repolarization phase.3,16,27
Regulation
β-Adrenergic stimulation and elevation of intracellular cAMP levels enhance IKr amplitude both through PKA-mediated effects and by direct interaction with the protein. α-Adrenergic stimulation is inhibitory. Coexpression of HERG with its β subunit (KCNE1 or KCNE2) accentuates the cAMP-induced voltage shift. The net result of these effects is a reduction in IKr.3,16,27
Extracellular Na+ potently inhibits IKr by binding to an outer pore site, and it also speeds recovery from inactivation. The inhibitory effect of Na+ is potently relieved by physiological levels of extracellular K+. Competition with external K+ for a binding site near the external pore explains the finding that elevation of extracellular K+ concentration paradoxically enhances IKr despite the decrease in the electrochemical driving force. Hypokalemia causes prolongation of the action potential duration as a result of reduced K+ conductance. Low extracellular K+ levels accelerate fast inactivation of the HERG channel and further decrease IKr.17
Pharmacology
IKr is the target of class III antiarrhythmic drugs of the methanesulfonanilide group (almokalant, dofetilide, d-sotalol, E-4031, ibutilide, and MK-499). These drugs produce a voltage- and use-dependent block, shorten open times in a manner consistent with open channel block, and exhibit low affinity for closed and inactivated states. IKr blockers prolong atrial and ventricular action potential duration (and the QT interval) and refractoriness in the absence of significant changes in conduction velocity (A-H, H-V, and PR intervals do not prolong). Although selective IKr blockers exhibit antiarrhythmic properties against reentrant arrhythmias, they are probably not effective against triggered activity or increased automaticity.16
Selective IKr blockers have several disadvantages. These drugs tend to prolong the action potential duration in the Purkinje and midmyocardial cells more than in the subepicardial or subendocardial cells, thus resulting in increased dispersion of repolarization across the ventricular wall and, as a consequence, increased arrhythmogenesis. Moreover, the effects of these drugs increase with decreasing heart rate. This reverse frequency-dependent nature of IKr blockers can potentially result in excessive prolongation of the QT interval during bradycardia, potentially precipitating torsades de pointes, whereas this prolongation is much less marked or even absent following β-adrenergic stimulation or during sustained tachycardia. This phenomenon limits the efficacy of these drugs in terminating tachyarrhythmias, while maximizing the risk of torsades de pointes during slow heart rates, such as during sinus rhythm after termination of AF. Reverse use-dependence has been attributed, at least in part, to the incomplete deactivation (accumulation) of IKs during fast heart rates that leads to a progressive increase in current amplitude, which counteracts the action potential prolongation effects of IKr blockers.3,16,27
Furthermore, HERG channels display an unusual susceptibility to blockade by a variety of drugs compared with other voltage-gated K+ channels. Increasing numbers of drugs with diverse chemical structures (including some antihistaminics, antipsychotics, and antibiotics) decrease IKr by depressing HERG channel gating, delay ventricular repolarization, prolong the QT interval (acquired LQTS), and induce torsades de pointes. In fact, almost all drugs that cause acquired LQTS target HERG channels, likely because of unique structural properties rendering this channel unusually susceptible to a wide range of different drugs. Compared with other cardiac K+ channels, the HERG channel has a large, funnel-like vestibule that allows many small molecules to enter and block the channel. The more spacious inner cavity results from a lack of the S6 helix bending Pro-X-Pro sequence, which presumably facilitates access of drugs to the pore region from the intracellular side of the channel to block the channel current. Additionally, the HERG channel contains two aromatic residues located in the S6 domain facing the channel vestibule (not present in most other K+ channels) that provide high-affinity binding sites for a wide range of structurally diverse compounds. The accessory β subunit (MiRP1, KCNE2) also determines the drug sensitivity. Interaction of these compounds with the channel’s pore causes functional alteration of its biophysical properties or occlusion of the permeation pathway, or both.8,16,29
One novel mechanism for acquired LQTS involves compounds interfering with HERG trafficking (i.e., moving the HERG protein from the endoplasmic reticulum to the cell membrane), rather than direct pore blocking. These compounds include arsenic trioxide, pentamidine, probucol (a cholesterol-lowering therapeutic compound), and cardiac glycosides.16,29,30
Some drugs (almokalant, norpropoxyphene, azimilide, candesartan, and E3174, the active metabolite of losartan) can enhance IKr. Flufenamic acid and niflumic acid also increase IKr by accelerating channel opening. These observations open the possibility of developing new IKr openers for the treatment of patients with congenital (LQT2) or drug-induced LQTS.16
Inherited Channelopathies
Long QT Syndrome
The LQTS variants in which IKr is dysfunctional include LQT2 (caused by KCNH2 [HERG] loss-of-function mutations) and LQT6 (caused by KCNE2 [MiRP1] mutations); most are LQT2, which is the second most prevalent type of LQTS. More than 200 putative disease-causing mutations have been identified for KCNH2; most appear to disrupt the maturation and trafficking of IKr α subunit (Kv11.1) to the sarcolemma, thereby reducing the number of functional ion channels at the cell surface membrane. Mutations involving the pore region of the HERG channel are associated with a significantly more severe clinical course than nonpore mutations; most pore mutations are missense mutations with a dominant negative effect. Attenuation of IKr results in prolongation of the action potential and the QT interval and can potentially generate EADs and torsades de pointes.3,7,27,30
The trafficking of some mutant channels into the sarcolemma can be restored by HERG channel blockers (e.g., cisapride, terfenadine, astemizole, E-4031), even when fexofenadine rescues mutant HERG channels at concentrations that do not cause channel block. However, because IKr blockers failed to rescue other trafficking-defective mutants, it is evident that multiple mechanisms may exist for pharmacological rescue of LQT2 mutations.7,16
Proarrhythmia induced by conditions associated with reduction of IKr (acquired or congenital LQTS) is related to excessive prolongation of action potential duration near plateau voltages, especially those that favor the development of EADs. It is also related to a more marked prolongation of the action potential duration in midmyocardial than in subepicardial or subendocardial ventricular cells possibly because of the relative scarcity of IKs and hence less “repolarization reserve” in the midmyocardial cells. Thus, triggered focal activity and ventricular reentry associated with an increased heterogeneity of repolarization across the ventricular wall would lead to the development of torsades de pointes.16
Short QT Syndrome
Short QT syndrome (SQTS) is a rare disease associated with short QT intervals and increased risk for AF and VF. A gain-of-function mutation in KCNH2 (HERG) is linked to SQTS type 1 (SQT1). A gain-of-function mutation on KCNH2 causes a shift of voltage dependence of inactivation of IKr by +90 mV out of the range of the action potential leading to a significant increase of IKr during the action potential plateau. The resulting IKr increase achieved by altered gating hastens repolarization, thereby shortening action potential duration and facilitating reentrant excitation waves to induce atrial or ventricular arrhythmia, or both. Additionally, gain-of-function mutations in KCNE2 (MiRP1) have been found in two families with AF.3,27,30
Acquired Diseases
MI can result in reduction in Kv11.1 mRNA levels and IKr with consequent prolongation of the action potential duration. Conversely, IKr density increases in subendocardial Purkinje cells in the infarcted heart at 48 hours, which can potentially increase the proarrhythmic effects of IKr blockers in patients with MI. Additionally, during acute ischemia, IKr is increased and action potential duration is shortened. Such changes can be arrhythmogenic. IKr is unchanged in patients with chronic AF and is homogeneously distributed in failing canine hearts.3,16
ATP, derived from either glycolysis or oxidative phosphorylation, is critical for HERG channel function. Both hyperglycemia and hypoglycemia depress IKr and can cause QT prolongation and ventricular arrhythmias. In diabetes, Kv11.1 levels are downregulated, leading to reduction in IKr and contributing to QT interval prolongation. Importantly, insulin therapy restores IKr function and shortens QT intervals.3,16
Unlike with most other K+ currents, IKr amplitude increases on elevation of extracellular K+ concentrations and decreases after removal of extracellular K+. Elevation of extracellular K+ concentration reduces C-type inactivation and increases the single channel conductance of HERG channels. This explains why the action potential durations are shorter at higher extracellular K+ concentrations and longer at low concentrations, and it clarifies the associations among hypokalemia, action potential duration prolongation, and induction of torsades de pointes in patients treated with IKr blockers. In contrast, modest elevations of extracellular K+ concentrations using K+ supplements and spironolactone in patients given IKr blockers or with LQT2 significantly shorten the QT interval and may prevent torsades de pointes. Moreover, the antiarrhythmic actions of IKr blockers can be reversed during ischemia, which is frequently accompanied by elevations of the extracellular K+ concentrations in the narrow intercellular spaces and by catecholamine surges that occur with exercise or other activities associated with fast heart rates.16
Slowly Activating Delayed Outward Rectifying Current (IKs)
Structure and Physiology
IKs is formed by coassembly of four pore-forming α subunits (Kv7.1, encoded by the KCNQ1 gene, also known as KvLQT1) and β subunits (minK, encoded by the KCNE1 gene). IKs is a K+-selective current that activates very slowly in response to membrane depolarization to potentials greater than –30 mV and reaches half-maximum activation close to +20 mV. IKs has a linear current-voltage relationship, its time course of activation is extremely slow, slower than any other known K+ current, and steady-state amplitude is achieved only with extremely long membrane depolarization.3,16,27,28
Inactivation of KCNQ1 channels is half maximal at –18 mV and, at its maximum, inactivation reduces fully activated current by approximately 35%. In addition, unlike inactivation of other Kv channels, the onset of IKs inactivation occurs after a delay (a delay of approximately 75 milliseconds at +40 mV). In contrast, when inactivation is induced after transient recovery of channels to open states, the onset of inactivation is 10 times faster. The molecular mechanism of KCNQ1 channel inactivation is unknown, but in contrast to a classical C-type inactivation, KCNQ1 inactivation is independent of extracellular K+ concentration.28
Function
IKs contributes to human atrial and ventricular repolarization, particularly during action potentials of long duration. IKs gradually increases during the plateau phase of the action potential because its activation is delayed and very slow. As a consequence, the contribution of IKs to the net repolarizing current is greatest late in the cardiac action potential plateau phase. IKs is expressed in all cell types, but it is reduced in midmyocardial cells. The midmyocardial cells have the longest action potential duration across the myocardial wall.28
IKs plays an important role in determining the rate-dependent shortening of the cardiac action potential. As heart rate increases, IKs increases because channel deactivation is slow and incomplete during the shortened diastole. This allows IKs channels to accumulate in the open state during rapid heart rates and contribute to the faster rate of repolarization.16,28
Importantly, IKs is functionally upregulated when other repolarizing currents (e.g., IKr) are reduced, potentially serving as a safeguard against loss of repolarizing power. As such, several redundant mechanisms contribute to repolarization constituting the repolarization reserve, in which IKs plays an important role.8,28
Regulation
IKs is markedly enhanced by β-adrenergic stimulation through channel phosphorylation by PKA (requiring A-kinase anchoring protein 9 [AKAP9, also known as Yotiao]) and PKC (requiring minK). This produces a rate-dependent shortening of the action potential duration such as seen during exercise-induced sinus tachycardia. IKs is also modulated by α-adrenergic receptors through the PKC pathway. Lowering extracellular K+ and Ca2+ concentrations increases IKs.16,27
Coexpression of KCNQ1 α subunits with minK regulates the α-subunit trafficking and behavior and results in a seven-fold increase in IKs magnitude, marked slowing of the time course of activation, and removal (or significant slowing) of inactivation of KCNQ1 channels.16
As noted, IKr and IKs are functionally linked; when IKr is reduced, the action potential is prolonged, causing IKs activation to increase to prevent excess repolarization delay. Hence, the duration of the action potential is very tightly tuned via IKs and IKr regulation.27
Pharmacology
IKs is resistant to methanesulfonanilides (almokalant, dofetilide, d-sotalol, E-4031, ibutilide, and MK-499), but it is selectively blocked by chromanols, indapamide, thiopentone, propofol, and benzodiazepines. IKs is also blocked, although nonselectively, by amiodarone, dronedarone, and azimilide. KCNE1 modulates the effects of IKs blockers and agonists. In fact, KCNQ1/KCNE1 channels have 6- to 100-fold higher affinity for some IKs blockers than KCNQ1 channels.16
Selective IKs blockers prolong the cardiac action potential duration and QT interval and suppress electrically induced ventricular tachyarrhythmias in animals with acute coronary ischemia and exercise superimposed on a healed MI.16
IKs blockade seems to have less proarrhythmic potency as compared with IKr blockade, likely the result of less drug-induced dispersion in repolarization. Additionally, because IKs accumulates at fast driving rates because of its slow deactivation, IKs blockers can be expected to be more effective in prolonging action potential duration and refractoriness at fast rates. Furthermore, because IKs activation occurs at approximately 0 mV and this voltage is more positive than the Purkinje fiber action potential plateau voltage, IKs blockade should not be expected to prolong the action potential duration at this level. Conversely, in ventricular muscle, the plateau voltage is more positive (approximately +20 mV), thus allowing IKs to be substantially more activated, so that IKs blockade would be expected to markedly increase action potential duration.16
β-Adrenergic agonists increase IKs density and produce a rate-dependent shortening of the action potential duration and can also decrease the antiarrhythmic effects of IKs blockers. Additionally, in the presence of IKs blockade, isoproterenol seems to abbreviate the action potential duration of epicardial and endocardial, but not midmyocardial, cells, an effect that can accentuate transmural dispersion of repolarization and precipitate torsades de pointes. These observations may explain the therapeutic actions of β-blockers in patients with LQTS syndromes linked to attenuation of IKs and the increased risk of fatal cardiac arrhythmias under physical activity or stressful situations that increase sympathetic activity in these patients.16
Inherited Channelopathies
KCNQ1 and KCNE1 mutations can lead to a defective protein and several forms of inherited arrhythmias, including LQTS (comprising the autosomal dominant Romano-Ward syndrome and the autosomal recessive Jervell and Lange-Nielsen syndrome), SQTS, and familial AF.3,31
Long QT Syndrome
The most common type of LQTS, LQT1, is caused by autosomal dominant loss-of-function mutations on the KCNQ1 gene (KvLQT1). More than 170 mutations of this gene have been reported. They comprise many Romano-Ward (autosomal dominant) syndromes and account for approximately 45% of all genotyped LQTS families.7 Individuals with the less prevalent LQTS type 5 (LQT5) carry loss-of-function autosomal dominant mutations in KCNE1 and display a phenotype similar to that seen in patients with LQT1.8
Loss-of-function mutations in both alleles of KCNQ1 or KCNE1 (i.e., inherited from both parents, autosomal recessive) cause the very rare Jervell and Lange-Nielsen syndrome type 1 or 2, respectively. Jervell and Lange-Nielsen syndrome encompasses 1% to 7% of all genotyped patients with LQTS and is characterized by severe QT interval prolongation, high risk of sudden death, and congenital deafness; the deafness results from deficient endolymph secretion (KCNQ1 and KCNE1 are also expressed in the inner ear, where they enable endolymph secretion).3,8,9,31,32
LQT11 is caused by loss-of-function mutations on the AKAP9 gene, which encodes an A-kinase anchoring protein (Yotiao), shown to be an integral part of the IKs macromolecular complex. The presence of Yotiao is necessary for the physiological response of the IKs channel to β-adrenergic stimulation.9 A mutation in AKAP9 (Yotiao) in the IKs channel (Kv7.1) binding domain reduces the interaction between the IKs channel and Yotiao. This, in turn, reduces the cAMP-induced phosphorylation of the channel and prevents the functional response of the IKs channel to cAMP and adrenergic stimulation (i.e., prevents the increase in magnitude of IKs and the shortening of action potential duration in response to sympathetic stimulation). The final result is an attenuation of IKs, resulting in a delay in ventricular repolarization and QT interval prolongation.3,8
Mutations in LQT1, LQT5, and LQT11 result in attenuation of IKs, which causes prolongation of repolarization, action potential duration, and QT interval, which may be especially notable during periods of increased sympathetic activity, such as exercise, when IKs becomes the predominant repolarization current rather than IKr. In LQT1, ventricular arrhythmias are usually triggered by emotional or physical stress, probably because mutant IKs does not increase sufficiently (i.e., has less repolarization reserve) during β-adrenergic stimulation. Accordingly, β-adrenergic blocking drugs suppress arrhythmic events in LQT1.3,8,33
Short QT Syndrome
SQT2 is caused by mutations on the KCNQ1 gene (KvLQT1). A gain-of-function mutation on KCNQ1 causes a shift of voltage dependence of activation of IKs by −20 mV and acceleration of activation kinetics, leading to enhancement of IKs and shortening of the action potential duration and QT interval. KCNQ1 gain-of-function mutations likely predispose to AF and VF by shortening refractoriness and facilitating reentry.3,8
Acquired Diseases
Heart failure reduces IKs in atrial, ventricular, and sinus node myocytes. Given that IKr is unchanged, IKs reduction may largely account for the prolonged action potential duration in heart failure.3
IKs density and KCNQ1/KCNE1 mRNA levels are reduced in myocytes from infarcted border zones 2 days after MI. However, KCNQ1 expression is restored 5 days after MI, whereas KCNE1 expression remains decreased.3
Inward Rectifying Current (IK1)
Structure and Physiology
The Kir channels are formed by the coassembly of four α subunits (see Fig. 2-3). The α subunit (Kir2.1) of IK1 is encoded by KCNJ2 and consists of two transmembrane domains (M1 and M2) connected by a pore-forming P loop (H5) along with the cytoplasmic N- and C-termini. The tetrameric Kir channel complex can be formed by identical (homotetramers) or different (heterotetramers) α subunits. Several IK1 channels with different conductances are recorded in human atrial myocytes. Similarly, different gene families (Kir2.1 to Kir2.3) have been found in human heart encoding IK1.16,34
Kir channels exhibit a strong inward rectification property because conductance to K+ ions alters at a different Em. As noted, rectification describes the property of an ion channel to allow currents preferentially to flow in one direction or limit currents from flowing in the other direction. A channel that is inwardly rectifying is one that passes current (positive charge) more easily into the cell. In the case of Kir channels, inward rectification is a strongly voltage-dependent decline of K+ efflux (i.e., reduction of outward current) on membrane depolarization that produces a characteristic region of so-called negative slope conductance. As such, IK1 is a strong rectifier that passes K+ currents over a limited range of Em; at a negative Em, IK1 conductance is much larger than that of any other current, and so it clamps the resting Em close to the reversal potential for K+ (EK). On depolarization, IK1 channels close almost immediately and thus limit K+ efflux at potentials more positive than the EK, remain closed throughout the plateau, and open again at potentials negative to –20 mV. Nevertheless, IK1 channels also conduct a substantial outward current at an Em between −40 and −90 mV. Within this voltage range, outward IK1 is larger at more negative potentials. Thus, IK1 also contributes to terminal phase 3 of repolarization. Because an Em negative to EK is not reached in cardiomyocytes, only the outward IK1 plays a role in action potential formation.16,34
The phenomenon of inward rectification of IK1 channels results from high-affinity and strongly voltage-dependent blockade of the inner channel pore by cytosolic magnesium (Mg2+), Ca2+, and polyamines (spermine, spermidine, putrescine), which plug the channel pore at depolarized potentials, resulting in a decline in outward currents, but are displaced by incoming K+ ions at hyperpolarized potentials. This voltage-dependent block by polyamines causes currents to be conducted well only in the inward direction. As such, IK1 channels are voltage regulated despite the lack of the classic voltage-sensing mechanism of Kv channels.16,17,34
Function
IK1 channels close on depolarization. The strong inward rectification of the IK1 limits the outward current during the positive phase of the action potential (phases 0, 1, and 2), thus allowing membrane depolarization following Na+ channel activation, slowing membrane repolarization, and helping maintain a more prolonged cardiac action potential. This also confers energetic efficiency in the generation of the action potential.16,34
IK1 density is much higher in ventricular than in atrial myocytes, a finding that explains the steep repolarization phase in the ventricles (where more abundant IK1 plays a larger role in accelerating the terminal portion of repolarization) and the more shallow phase in the atria. The higher IK1 channel expression in the ventricle protects the ventricular cell from pacemaker activity. By contrast, IK1 is almost absent in sinus node and AVN cells, thus allowing for relatively more depolarized resting diastolic potentials compared with atrial and ventricular myocytes.16,34
A unique property of Kir currents is the unusual dependence of rectification on extracellular K+ concentration. Specifically, on increase in extracellular K+, the IK1 current-voltage relationship shifts nearly in parallel with the EK and leads to a crossover phenomenon. One important consequence of such behavior is that at potentials positive to the crossover, K+ conductance increases rather than decreases, against an expectation based on a reduced driving force for K+ ions in response to elevated extracellular K+ concentration.34
Fast heart rates increase the K+ concentration in the narrow intercellular space to several millimolars and the IK1 density, which results in a shortening of the action potential duration that may offset the ability of IKr blockers to prolong the action potential duration under these conditions.16,34
Regulation
β-Adrenergic stimulation inhibits IK1 in ventricular myocytes via PKA-mediated phosphorylation of the channel. In atrial myocytes, α1-adrenergic stimulation reduces IK1 via PKC-dependent pathways.16,34
Kir2.1 overexpression increases IK1 density, shortens the action potential duration, and hyperpolarizes the resting Em. In contrast, suppression of IK1 prolongs the action potential duration and disrupts effective clamping of the resting Em, thus precipitating spontaneous pacemaker activity in otherwise nonpacemaking atrial and ventricular cardiomyocytes.16
Pharmacology
Barium (Ba2+) is a potent IK1 blocker. Blocking IK1 by extracellular Ba2+ results in depolarization of the resting potential and mild action potential prolongation.3,34
IK1 blockers prolong atrial and ventricular action potential duration and are effective against various types of experimental reentrant VTs. Moreover, IK1 blockers produce membrane depolarization, an effect that slows conduction velocity as a result of voltage-dependent inactivation of Na+ channels, and prolongs the QT interval; both actions are proarrhythmic.16,34
Inherited Channelopathies
Long QT Syndrome
More than 33 loss-of-function mutations of KCNJ2 gene encoding Kir2 result in dominant negative effects on the current and have been linked to Andersen-Tawil syndrome (LQT7), a rare autosomal dominant disorder characterized by the triad of skeletal developmental abnormalities, periodic paralysis, and usually ventricular arrhythmias, often associated with prominent U waves and mild QT interval prolongation (Kir2.1 channels are expressed primarily in skeletal muscle, heart, and brain).3,16,34 The arrhythmias displayed by affected patients are more benign compared with other LQTS and rarely degenerate into hemodynamically compromising rhythms such as torsades de pointes, as ultimately evidenced by the lack of SCD cases so far.8,35,36
Disruption of the IK1 function can potentially lead to prolongation of the terminal repolarization phase and QT interval, which can predispose to the generation of EADs and delayed afterdepolarizations (DADs) that cause ventricular arrhythmias. However, unlike other types of LQTS in which the afterdepolarizations arise from reactivation of L-type Ca2+ channels, the EADs and DADs generated in LQT7 are likely secondary to Na+-Ca2+ exchanger-driven depolarization. It is believed that the differential origin of the triggering beat is responsible for the observed discrepancy in arrhythmogenesis and the clinical features compared with other types of LQTS. Additionally, it is likely that prolongation of the action potential duration in LQT7 is somewhat homogeneous across the ventricular wall (i.e., transmural dispersion of repolarization is less prominent than in other types of LQTS), and this can potentially explain the low frequency of torsades de pointes.8,35,36
Catecholaminergic Polymorphic Ventricular Tachycardia
Three novel loss-of-function mutations of KCNJ2 have been found in patients with catecholaminergic polymorphic VT (CPVT). These patients had prominent U waves, ventricular ectopy, and polymorphic VT, but no dysmorphic features or skeletal muscle abnormalities.34
IK1 reduction can trigger arrhythmia by allowing inward currents, which are no longer counterbalanced by the strong outward IK1, to depolarize the Em gradually during phase 4. Membrane depolarization during phase 4 induces arrhythmia by facilitating spontaneous excitability.3
Short QT Syndrome
A gain-of-function mutation of KCNJ2 has been identified and linked to SQTS type 3 (SQT3). The mutation causes a significant increase in the outward IK1 at potentials between −75 mV and −45 mV, thus leading to acceleration of the terminal phase of repolarization and, as a consequence, shortening the action potential duration and QT interval and asymmetrical T waves with a rapid terminal phase.37
Acquired Diseases
IK1 is downregulated in patients with severe heart failure and cardiomyopathy. The downregulation of IK1 produces membrane depolarization and prolongation of the action potential duration, and it can facilitate spontaneous excitability and trigger arrhythmia (both EADs and DADs). The ventricular myocytes from patients with idiopathic dilated cardiomyopathy exhibit decreased channel activity, longer action potential duration, and a lower resting Em than those from patients with ischemic cardiomyopathy. Upregulation of IK1 can be observed in ventricular hypertrophy.3,34
Atrial IK1 is upregulated in patients with chronic AF, resulting in more negative resting potentials and, together with reduced ICaL, accounting for action potential shortening in AF.3
Acetylcholine-Activated Potassium Current (IKACh)
Structure and Physiology
IKACh results from a heterotetrameric complex of two Kir3.1 (encoded by KCNJ3) and two Kir3.4 (encoded by KCNJ5) α subunits. IKACh is a receptor-activated Kir channel; it has large cytoplasmic domains that harbor specific binding sites for cytosolic effectors (G proteins). The channel conducts IKACh in response to the stimulation of G protein−coupled muscarinic (M2) and adenosine (A1) receptors.16
Cardiac IK1 and IKACh are the major K+ currents displaying classical strong inward rectification with membrane depolarization, a unique property that is critical for their roles in cardiac excitability.34
Function
IKACh has generally an opposite distribution to that of IK1. IKACh is more prominent in atrial tissue, as well as in the sinus node and AVN, and is largely absent in the ventricles. The regional distribution of IKACh is also heterogeneous within and between the atria.16,34
IKACh mediates vagal influences on sinus rate and atrial repolarization, as well as AVN conduction. Activation IKACh by acetylcholine hyperpolarizes the Em and shortens action potential duration. These effects result in slowing of phase 4 depolarization, reduction in the spontaneous firing rate of the pacemaker cells of the sinus node and AVN, and slowing of AVN conduction. These effects explain why vagal maneuvers or intravenous adenosine can terminate reentrant supraventricular tachycardias using the AVN.16,34
Regulation
Vagal stimulation produces a nonuniform shortening of the atrial action potential duration and refractoriness mediated by activation of IKACh, an effect that can contribute to the perpetuation of AF.16
Atrial IKACh is inhibited by membrane stretch, possibly serving as a mechanoelectrical feedback pathway, a property conferred by the Kir3.4 subunit.16
Pharmacology
IKACh activity can be stimulated by intracellular ATP, the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) and ETA endothelin, A opioid, and α2-adrenergic agonists. α1-Adenosine receptor agonists stimulate IKACh, whereas methylxanthines, such as theophylline and aminophylline, antagonize the effects of adenosine. Dipyridamole prolongs the action of adenosine by disturbing the action of the cell membrane transporter of adenosine.1,38
IKACh is inhibited by several antiarrhythmic drugs, including amiodarone, dronedarone, disopyramide, procainamide, flecainide, and propafenone. Disopyramide and procainamide mainly block the muscarinic receptors, whereas flecainide and propafenone act as open channel blockers. Blockade of IKACh by dronedarone is approximately 100 times more potent than that of amiodarone.16
A potent IKACh blocking property is of additional therapeutic value especially for treatment of AF, because IKACh plays a prominent role in vagally induced AF and has been shown to be constitutively active in chronic AF. In fact, IKACh is a promising target for AF therapy, and series of chemical compounds have been tested as antiarrhythmic agents. Inhibition of IKACh in the setting of AF can potentially produce proportionally greater action potential duration prolongation than under control conditions and can even terminate experimental atrial tachyarrhythmias and AF without ventricular side effects.26
Inherited Channelopathies
LQT13 is caused by loss-of-function mutations on the KCNJ5 gene. The KCNJ5 mutation is the most recently identified LQTS-associated gene, and it exerts dominant-negative effects on Kir3.1-Kir3.4 channel complexes by disrupting membrane targeting and stability of Kir3.4.39
Acquired Diseases
IKACh is downregulated during chronic AF, possibly to counteract the AF-induced nonuniform shortening of the atrial refractoriness.16 However, IKACh channels can develop constitutive activity during human AF (i.e., these channels become activated despite the absence of stimulating acetylcholine). This increase in functionally uncoupled IKACh in human AF is possibly the result of increased phosphorylation of Kir3 channels by PKC or a reduction in inhibitory Gαi-3 subunits. Constitutively active IKACh can hyperpolarize the membrane and, hence, contribute to AF-related electrical remodeling and to the persistence of AF by stabilization of rotors. Therefore, selectively targeting constitutively active IKACh channels only may preserve physiological stimulation by vagal nerves and could serve as a promising remodeling-related drug target.26,40
ATP-Dependent Potassium Current (IKATP)
Structure and Physiology
Cardiac ATP-sensitive K+ (KATP) channels (also termed the adenosine diphosphate [ADP]–activated K+ channel) are formed by the unique combination of two dissimilar proteins: four pore-forming α subunits (Kir6.2, encoded by KCNJ4) and four regulatory ATP-binding cassette proteins (sulfonylurea receptor subunits [SUR2A] encoded by ABCC9). The Kir6.2 subunits have two transmembrane spans and form the channel’s pore and large cytoplasmic domains that provide the binding sites for ATP. The SUR2A subunits have three transmembrane domains and contain two nucleotide-binding domains on the cytoplasmic side. These allow for nucleotide-mediated regulation of KATP and are critical in its role as a sensor of metabolic status. The SUR2A subunits are also sensitive to sulfonylureas, Mg-ATP, Mg-ADP, and some other pharmacological channel openers. The SUR2A subunit also harbors an ATPase for ATP hydrolysis, which gates the K+ permeation through the Kir6.2 α subunit.16,41–43
KATP is a receptor-activated weak inward rectifier channel, regulated by intracellular ATP and ADP concentrations. An increase in the ratio of ATP to ADP closes the channel, and a decrease opens it, linking the metabolic state to the cellular Em.16,41–43
Function
IKATP is inhibited by intracellular ATP and activated by Mg-ADP, so that the channel activity is regulated by the ATP/ADP ratio, coupling cell metabolism to the Em. In responding to cytoplasmic nucleotide levels, KATP channel activity provides a unique link between cellular energetics and electrical excitability and hence contractility. Under normal metabolic conditions, sarcolemmal KATP channels are predominantly closed (inhibited by intracellular ATP), and they do not significantly contribute to the cardiac action potential, resting Em, or cell excitability. However, when exposed to a severe metabolic stress such as anoxia, metabolic inhibition, or ischemia, KATP channels become activated (secondary to reduced intracellular ATP levels) and conduct an outward repolarizing K+ current (IKATP), which results in abbreviation of the action potential duration and reduction of Ca2+ influx through L-type Ca2+ channels. By reducing Ca2+ entry, KATP channels depress muscle contractility, thereby conserving scarce energy resources, and prevent the damaging effects of intracellular Ca2+ overload.42,43
Accordingly, cardiac KATP channels act as membrane-based metabolic sensors that receive energetic signals of cellular distress and provide adaptive response to acute stress capable of controlling cardiac action potential duration and associated cellular functions and adjusting cellular excitability to match demand.43
Additionally, activation of IKATP plays an important role in ischemic preconditioning; brief periods of myocardial ischemia confer protection against subsequent prolonged ischemia, reducing MI size, severity of stunning, and incidence of cardiac arrhythmias. However, the role of sarcolemmal KATP channels in ischemic preconditioning versus that of mitochondrial KATP channels (which appear to be pharmacologically distinct from sarcolemmal KATP) has been debated.42,43
On the other hand, activation of IKATP also results in shortening of the action potential duration, accumulation of extracellular K+, membrane depolarization, and slowed conduction velocity, effects that render the ischemic heart vulnerable to reentrant arrhythmias.42
KATP channels have been further implicated in the adaptive cardiac response to chronic pathophysiological hemodynamic load. KATP channel deficiency affects structural remodeling, renders the heart vulnerable to Ca2+-dependent maladaptation, and predisposes to heart failure.43
Regulation
ATP (with or without Mg2+) inhibits IKATP by stabilizing the closed state of the channel by interacting directly with Kir6.2. In addition, in the presence of Mg2+, ATP and ADP can activate the channel through interaction with the SUR2A subunit. Inhibition by ATP binding to Kir6.2 and activation by Mg nucleotides is the primary physiological regulatory mechanism.16,41–43
KATP channel’s sensitivity to ATP is not fixed, and it can be modulated by other cellular factors. Nucleotide diphosphates, lactate, oxygen-derived free radicals, and adenosine α1 receptor stimulation desensitize KATP to inhibition by intracellular ATP. Additionally, the phospholipid PIP2 directly interacts with Kir6.2 subunit stabilizing the open state of the channel and antagonizes ATP inhibition of IKATP.42
Pharmacology
K+ channel openers (pinacidil, cromakalim, rimakalim, and nicorandil) bind at two distinct regions of SUR2A subunits and can exert cardioprotective effects in patients with acute MI. However, K+ channel openers also activate vascular KATP (Kir6.1/SUR2B) and produce hypotensive effects that limit their use in the treatment of myocardial ischemia. Moreover, because IKATP density is larger in the epicardium, K+ channel openers produce a more marked shortening of action potential duration in epicardial cells, thus leading to a marked dispersion of repolarization and to the development of extrasystolic activity via a mechanism of phase 2 reentry. On the other hand, K+ channel openers shorten the action potential duration (and QT interval), reduce transmural dispersion of repolarization, and suppress EADs and DADs induced in patients with LQT1. Thus, K+ channel openers may prevent spontaneous torsades de pointes when congenital or acquired LQTS is secondary to reduced IKr or IKs.16,42
On the other hand, cardioselective IKATP blockers (clamikalant, HMR 1098) inhibited hypoxia-induced shortening of the action potential duration and prevented VF induced by coronary artery occlusion in postinfarcted conscious dogs at doses that had no effect on insulin release, blood pressure, or coronary blood flow. Thus, these drugs may represent a new therapeutic approach to the treatment of ventricular arrhythmias in patients with coronary heart disease.16,42
It is still unclear whether opening of KATP channels has completely proarrhythmic or antiarrhythmic effects. Increased K+ conductance should stabilize the Em during ischemic insults and reduce the extent of infarct and ectopic pacemaker activity. On the other hand, K+ channel opening accelerates repolarization of the action potential, possibly inducing arrhythmic reentry.16,42
Inherited Channelopathies
Mutations in ABCC9 resulting in reduced intrinsic channel ATPase activity, metabolic sensing deficit, and dysfunctional KATP channels have been found in patients with idiopathic dilated cardiomyopathy and rhythm disturbances. These mutations confer susceptibility to Ca2+-dependent maladaptive remodeling, progressing to cardiomyopathy and congestive heart failure. Additionally, a loss-of-function ABCC9 mutation has been linked to predisposition to adrenergic AF. KATP channel-pore polymorphisms have also been linked to SCD.43
Many mutations in the Kir6.2 subunit have now been identified as causal in human neonatal diabetes mellitus, a very severe form of diabetes that typically occurs within the first days or weeks of life. All the identified Kir6.2 mutations result in a reduced channel sensitivity to ATP inhibition, leading to channel activation at elevated glucose, maintained hyperpolarization of pancreatic islet α cells, and electrical inexcitability, with consequent inhibition of insulin secretion. Although cardiac KATP channels (Kir6.2/SUR2A) have the same Kir6.2 subunit as that in pancreatic KATP channels (Kir6.2/SUR1), there are no reports of any cardiac abnormalities in patients with neonatal diabetes mellitus; this finding suggests that factors other than nucleotide sensitivity play an important role.42
Acquired Diseases
Metabolic dysregulation of IKATP created by disease-induced structural remodeling appears to contribute to the dysfunction of heart failure.26,43
The effects of Kir6.2 modulation in AF have not extensively been studied. However, evidence indicates a reduction of IKATP in patients with chronic AF, a finding suggesting that regulation of this current may not contribute importantly to AF-related ionic remodeling.26,43
Two-Pore Potassium Channels (K2P)
Structure and Physiology
K2P channels are composed of four transmembrane domains and two pore-forming P loops arranged in tandem, one between the first and second transmembrane domains and the other between the third and fourth domains (see Fig. 2-3). The proteins mainly form functional homodimers, although heterodimers combining different K2P subunits have been reported. Several subfamilies of K2P channels have been identified, including the TWIK-related acid-sensitive K+ (TASK) channels and TWIK-related K+ (TREK) channels.44
TREK channels, which comprise TREK-1 (KCNK2), TREK-2 (KCNK10), and TRAAK (KCNK4), display low basal activity, but are stimulated by stretch of the cell membrane, lysophospholipids, and arachidonic acid and are inactivated by hypo osmolarity and phosphorylation by PKA and PKC.44
Several members of the K2P channel family are expressed in the heart and in the systemic or pulmonary circulations, and some contribute to background K+ currents and the control of Em in vascular smooth muscle cells. The K+ selectivity, voltage-independent gating, and rectification of K2P currents are characteristics that make them strong candidates for mediating background K+ currents. Importantly, the sensitivity of K2P channels to numerous chemical and physical physiological stimuli (e.g., pH, oxygen, phospholipids, neurotransmitters, G protein–coupled receptors, and stretch) allow these channels to play a role in regulating the Em and excitability in various cell types under a range of physiological and pathological situations.44,45
Function
There is clear evidence for TREK-1 and TASK-1 in the heart and these channels are likely to regulate cardiac action potential duration through their regulation by stretch, polyunsaturated fatty acids, pH, and neurotransmitters. TREK-1 may also have a critical role in mediating the vasodilator response of resistance arteries to polyunsaturated fatty acids, thus contributing to their protective effect on the cardiovascular system. TASK-1, on the other hand, is a strong candidate for a role in hypoxic vasoconstriction of pulmonary arteries.44
In working atrial and ventricular myocytes, background K+ currents are crucial for stabilizing the Em at a hyperpolarized value toward the K+ equilibrium potential and regulating action potential duration in various physiological and pathological conditions. The background current is mainly carried by inward rectifier channels (including IK1, IKACh, and IKATP). Several K2P channels have been proposed to contribute to the cardiac background or “leak” K+ channels (i.e., channels with properties similar to the steady-state noninactivating K+ current [Iss] that is well characterized in rodent myocytes). Among them, TREK-1 and TASK-1 have been the most extensively studied.44
TREK-1, as an outwardly rectifying current, can potentially participate in balancing the Em and action potential duration. Indeed, on a beat-to-beat basis, it could be involved in a negative feedback loop, hyperpolarizing the Em in response to a stretch stimulus following the stretch activation of nonselective cation channels. The expression of TREK-1 appears to be nonuniform in the heart, with stronger TREK-1 mRNA expression in endocardial cells compared with epicardial cells. This finding possibly reflects different amounts of stretch experienced by muscle cells in different parts of the ventricular wall, leading to differential mechanoelectrical feedback and thereby reducing action potential repolarization in areas of the myocardium where conduction velocity is slower. Mechanoelectric feedback following an increase in atrial volume may be arrhythmogenic, changing the shape of the action potential. Physiological evidence of the direct involvement of TREK-1 current in mechanoelectric feedback in the heart has still to be provided.44
Acquired Diseases
TREK-1 activity may have some importance in pathological conditions such as ischemia, when released purinergic agonists such as ADP and ATP lead to arachidonic acid production. Activation of TREK-1 by ATP during ischemia may contribute to electrophysiological disturbances in the ventricular wall. As a stretch-activated K+ channel in atrial cells, TREK-1 could additionally be involved in regulating the release of atrial natriuretic peptide, which is released by a stretch-induced increase in intracellular Ca2+ concentration. Further work will be necessary to clarify the possible role of TREK or other stretch-dependent channels in the pathological heart.44
L-Type Calcium Current (ICaL)
Structure and Physiology
In cardiac muscle, two types of voltage-dependent Ca2+ channels, the L-type and the T-type, transport Ca2+ into the cells. The L-type channel (L for long-lasting, because of its slow kinetics of current decay as compared with Na+ channels) is found in all cardiac cell types. The T-type channel (T for tiny and transient) is found principally in pacemaker, atrial, and Purkinje cells. The term Ca2+ channels is used to refer to the L-type channel.46–48
Cardiac L-type Ca2+ channels are composed of four polypeptide subunits (α1C, β, α2, delta) and form a heterotetrameric complex. The α1C subunit (Cav1.2, encoded by the CACNAIC gene) has a structure similar to that of the Na+ channel: four homologous domains (I to IV), each consisting of six transmembrane segments (S1 to S6). The S5 and S6 segments and the membrane-associated pore loop (P loop) between them form the central pore through which ions flow down their electrochemical gradient. The P loop contains four negatively charged glutamate residues (EEEE) that are required for the Ca2+ selectivity of the channel. S4 in each homologous domain contains a highly conserved positively charged residue (arginine or lysine) at every third or fourth position. This segment serves as the voltage sensor for gating.46,47,49
The α1C subunit is the main and largest subunit, and it determines most of the channel characteristics because it harbors the ion-selective pore, voltage sensor, gating apparatus, and binding sites for channel-modulating drugs and is autoregulatory. To form a functional L-type Ca2+ channel, the α1C subunit coassembles with auxiliary subunits in a 1:1:1 ratio: the β subunit, the α2 subunit, and the delta subunit.46,47,49
The β subunit (Cavβ, encoded by the CACNB gene) is entirely intracellular and is tightly bound to a highly conserved motif in the cytoplasmic linker between domains I and II of the α1C subunit. Coexpression of β subunits modulates the biophysical properties of the α1C subunit. The β subunit has a prominent role in channel expression, trafficking, regulation, and facilitation. Sites possible for phosphorylation by various protein kinases (PKA, PKC, protein kinase G [PKG]) have been identified in these subunits. The β subunits are also involved in channel regulation by β-adrenergic stimulation and in response to the changes of the pH of the cell. In addition, the β subunit increases Ca2+ current amplitude, accelerates the kinetics of Ca2+ channel activation, and alters pharmacological properties of the channel.46,47
The α2 and delta subunits are encoded by the same gene (CACNA2D); the mature forms of these subunits are derived by post-translational proteolytic cleavage, but they remain associated through a disulfide bond. The α2 subunit is completely extracellular, whereas the delta subunit has a single membrane-spanning segment with a very short intracellular part that anchors the α2-delta subunit complex to the α1C subunit. The α2-delta subunit complex has less influence on channel function than the β subunit. The α2-delta subunit slightly increases Ca2+ current amplitude and accelerates channel inactivation and can change the properties of Ca2+ channel activation. It can also affect channel density trafficking.46,47
Although ICaL is normally activated during phase 0 by the regenerative depolarization caused by the fast INa, ICaL is much smaller than the peak INa. In addition, the amplitude of ICaL is not maximal near the action potential peak because of the time-dependent nature of ICaL activation, as well as the low driving force (Em – reversal potential of a cardiac Ca2+ channel [ECa]) for ICaL.50
The decay of ICaL during depolarization (i.e., time-dependent inactivation) is very slow and depends on two mechanisms: voltage-dependent inactivation and Ca2+-dependent inactivation. These two mechanisms control Ca2+ influx into cardiomyocytes and hence regulate signal transduction to sarcoplasmic reticulum Ca2+ channels (ryanodine receptor 2 [RyR2]) and ensure normal contraction and relaxation of the heart.46–48
Fast Ca2+-dependent inactivation serves as a negative feedback for Ca2+ to limit further Ca2+ entry via L-type Ca2+ channels. The slow voltage-dependent inactivation (induced by membrane depolarization) prevents a premature rise in ICaL when intracellular Ca2+ concentration decreases and Ca2+-dependent inactivation terminates during maintained depolarization. Although still under dispute, the relative contribution of Ca2+-dependent inactivation to total inactivation of ICaL appears to be greater at negative potentials when voltage-dependent inactivation, which typically exhibits a U-shaped availability curve, is weak. After β-adrenergic stimulation, Ca2+-dependent inactivation becomes the main inactivation mechanism as a result of a slowing down of voltage-dependent inactivation.46–48
Voltage steady-state activation and inactivation are sigmoidal, with an activation range over –40 to +10 mV (with a half-activation potential near –15 mV) and a half-inactivation potential near –35 mV. However, a relief of inactivation for voltages positive to 0 mV leads to a U-shaped voltage curve for steady-state inactivation. Overlap of the steady-state voltage-dependent inactivation and activation relations defines a window current near the action potential plateau, within which transitions from closed and open states can occur that may participate in action potential repolarization and may play a major role in the initiation of EADs.47,48
After inactivation, the transition of Ca2+ channels from the inactivated to the closed resting state (i.e., recovery from inactivation [reactivation, restoration, or repriming]) is also Ca2+ and voltage dependent. Reduction of intracellular Ca2+ concentration in the immediate vicinity of the channel allows recovery from Ca2+-dependent inactivation. Acceleration of Ca2+ channel reactivation, as may occur secondary to reuptake of Ca2+ by the sarcoplasmic reticulum during prolonged depolarization, can result in the recovery from Ca2+-dependent inactivation and enable secondary depolarization. This leads to instability of the cell Em during repolarization and may be the basis for the EADs that are capable of initiating torsades de pointes.46,48
In contrast, at normal resting potentials, recovery of ICaL from inactivation is fast, and ICaL may increase progressively during repetitive stimulation. This positive staircase or rate-dependent potentiation of contractility is Ca2+ dependent and likely is the result of diminished Ca2+-dependent inactivation at frequencies with less sarcoplasmic reticulum Ca2+ release. Additionally, similar to Ca2+-dependent inactivation, Ca2+-dependent facilitation requires high-affinity binding of calmodulin to the C-terminal tail of the Cav1.2 channel and may be facilitated by calmodulin kinase II–dependent phosphorylation. Calmodulin kinase II is a Ca2+/calmodulin-dependent serine/threonine kinase that is activated by low intracellular Ca2+ concentration. The facilitatory effect of Ca2+ entry on subsequent ICaL is distinct from, but coexistent with, Ca2+-dependent inactivation.46–48
Function
ICaL is activated by membrane depolarization. It is largely responsible for the action potential plateau and is a major determinant of the duration of the plateau phase and hence of action potential duration and refractoriness. ICaL also links membrane depolarization to myocardial contraction and constitutes the dominant factor in mediating positive inotropy in all types of cardiac tissue. Additionally, ICaL is responsible for the upstroke (phase 0) of slow response action potentials (in pacemaking cardiomyocytes and regions of depressed resting Em) and contributes to physiological frequency regulation in the sinus node.46–48
L-type Ca2+ channels are the principal portal of entry of Ca2+ into the cells during depolarization. Ca2+ influx during the action potential plateau triggers more massive Ca2+ release (Ca2+ transients) from the sarcoplasmic reticulum into the cytosol via activation of Ca2+-release channels (e.g., RyR2). This amplifying process, termed Ca2+-induced Ca2+ release (CICR), causes a rapid increase in intracellular Ca2+ concentration (from approximately 100 nM to approximately 1 µM) to a level required for optimal binding of Ca2+ to troponin C and induction of contraction. Most of the L-type Ca2+ channels in the adult myocyte are localized in the transverse tubules (T tubules) facing the sarcoplasmic reticulum junction and the RyR2, organized as a “complex” that ensures coordinated Ca2+ release during excitation-contraction coupling.46,49
Cytosolic Ca2+ concentration decreases during diastole: contraction is followed by Ca2+ release from troponin C and its reuptake by the sarcoplasmic reticulum via activation of the sarcoplasmic reticulum Ca2+-ATPase Ca2+ pump, in addition to extrusion across the sarcolemma via the Na+-Ca2+ exchanger. Intracellular Ca2+-dependent inactivation limits Ca2+ influx during action potential.46,47,49
For maintenance of intracellular Ca2+ homeostasis and balanced cardiac activity, Ca2+ influx into cytoplasm via L-type Ca2+ channels has to be terminated. This is achieved by Ca2+-dependent inactivation of L-type Ca2+ channels. This inactivation serves as a negative feedback mechanism for regulating Ca2+ entry into the cell and as a physiological safety mechanism against a harmful Ca2+ overload in the cell, which can cause both arrhythmias and cell death. Ca2+-dependent inactivation is also a major determinant of action potential duration, and it ensures that contraction and relaxation cycles of the heart muscle fiber are coordinated. A failure to deactivate ICaL completely may possibly be an essential mechanism underlying EADs caused by suppression of the K+ delayed rectifier currents.50
Inhibition of α1C subunit binding to calmodulin eliminates Ca2+-dependent inactivation, thus promoting Ca2+-dependent facilitation, which contributes to a force-frequency relationship in the heart.46,47,49
Regulation
Phosphorylation of the pore-forming α1C subunits by different kinases is one of the most important pathways to change the activity of the L-type Ca2+ channel. Phosphorylation by PKA is the main mechanism of Ca2+ channel activation, because it increases the probability and duration of the open state of the channels and consequently increases ICaL.46,47,49
Several different agonists (e.g., catecholamines, glucagon, histamine, serotonin) can activate PKA-mediated phosphorylation and activation of the L-type Ca2+ channel via an intracellular signaling cascade. Once one of these agonists binds to its receptor, receptor stimulation activates guanosine triphosphate (GTP)–binding protein (Gs), which activates adenylyl cyclase, which, in turn, mediates the conversion of ATP into cAMP. The increased cAMP levels stimulate cAMP-dependent PKA phosphorylation of the α1C subunit of L-type Ca2+ channels and result in an increase in ICaL amplitude and a shift in activation to a more negative Em. cAMP is degraded by cAMP phosphodiesterases, and the signaling cascade is then suppressed limiting cAMP-dependent phosphorylation; in addition, the signaling cascade is terminated by serine/threonine phosphatases that remove a phosphate group from kinase-phosphorylated proteins.46,47
The suppression of adenylyl cyclase activity is one of the most common pathways to interrupt PKA-dependent Ca2+ channel stimulation. Adenylyl cyclase is usually suppressed (and cAMP synthesis is blocked) by activation of Gi proteins. Stimulation of various Gi protein–coupled receptors (e.g., M2 muscarinic receptors, adenosine A1 receptors, opiates, and atrial natriuretic peptides) does not change basal ICaL in most cases, but reduces ICaL increased via stimulation of β-adrenergic receptors. Activation of phosphodiesterases is another way to reduce PKA-dependent channel phosphorylation. Phosphodiesterases hydrolyze cAMP and cyclic guanosine monophosphate (cGMP) and decrease their intracellular concentrations.46,47,49
The physiological functions of cardiac L-type Ca2+ channels are under control of catecholamines of circulating and neurohumoral origin. The effects of adrenergic stimulation are exerted by phosphorylation of the L-type Ca2+ channel subunits by PKA, PKC, and PKG. β1-Adrenergic receptors couple exclusively to the Gs protein, thus producing a widespread increase in cAMP levels in the cell, whereas β2-adrenergic receptors couple to both Gs and Gi, thus producing a more localized activation of L-type Ca2+ channels.46,49
The effect of PKC-mediated phosphorylation on ICaL can be highly diverse. PKC can either increase or decrease ICaL. Activation of Gq subunits by Gq protein–coupled receptors (e.g., α-adrenergic receptors, endothelin, angiotensin II, and muscarinic receptors) stimulates phospholipase C, which hydrolyzes PIP2 to inositol 1,4,5-triphosphate (InsP3) and diacylglycerol (DAG). DAG activates PKC, which, in turn, phosphorylates L-type Ca2+ channels. The mechanism of the effect of PKC on the activity of cardiac L-type Ca2+ channels is not exactly known. PKC phosphorylates the N-terminus of the α1C subunit, and the effect on the channel can be either stimulating or suppressive.46,47
Activation of soluble guanylate cyclase (primarily by nitric oxide) results in the conversion of GTP into cGMP. cGMP activates PKG, which phosphorylates the α1C subunit of the L-type Ca2+ channel, with a resulting inhibition of ICaL. Besides direct phosphorylation of the L-type Ca2+ channel, it is also possible that PKG activates a protein phosphatase, which dephosphorylates the channel, or that cGMP activates phosphodiesterase 2, which reduces cAMP levels. Thus, stimulation of ICaL by PKA is inhibited. However, besides an inhibition of ICaL, stimulatory effects of the PKG pathway have been shown.46,47,49
ICaL is blocked by several cations (e.g., Mg2+, nickel [Ni2+], zinc [Zn2+]) and drugs (dihydropyridines, phenylalkylamines, benzothiazepines). In addition, coexpression of the β-subunit increases ICaL amplitude, accelerates the kinetics of Ca2+ channel activation, and alters pharmacological properties of the channel.47,49
Pharmacology
Cardiac L-type Ca2+ channels are the targets for the interaction with class IV antiarrhythmic drugs. The three classes of organic Ca2+ channel blockers include dihydropyridines (e.g., nifedipine, nicardipine, amlodipine, felodipine), phenylalkylamines (verapamil), and benzothiazepines (diltiazem). Each drug type binds specifically to separate binding sites on the channel’s α1C subunit. The combined use of Ca2+ channel blockers can enhance or weaken the block effect because, at least in part, of the different binding sites for those drugs. It is noteworthy that increased extracellular Ca2+ concentrations inhibit the binding of phenylalkylamines and dihydropyridines to their receptors on the Ca2+ channel.
Verapamil and diltiazem preferentially block open and inactivated states of the channel. The more frequently the Ca2+ channel opens, the better is the penetration of the drug to the binding site; hence, these drugs cause use-dependent block of conduction in cells with Ca2+-dependent action potentials such as those in the sinus node and AVN. This explains their preferential effect on nodal tissue in paroxysmal supraventricular tachycardia.46,47,49
The dihydropyridines block open Ca2+ channels. However, the lack of use-dependence and the presence of voltage sensitivity of the dihydropyridines with regard to their binding explain their vascular selectivity. The kinetics of recovery from block is sufficiently fast that these drugs produce no significant cardiac effect but effectively block the smooth muscle Ca2+ channel because of its low resting potential.46,47,49
Inherited Channelopathies
Long QT Syndrome
Gain-of-function mutations of the CACNA1C gene encoding the α1C subunit (Cav1.2) result in nearly complete elimination of voltage-dependent inactivation of Cav1.2 channels, thus leading to inappropriate continuation of the depolarizing ICaL and lengthening the plateau phase. The resultant sustained Ca2+ influx, action potential (and QT interval) prolongation, and Ca2+ overload promote EADs and DADs. These mutations have been linked to Timothy syndrome, a rare disease with QT interval prolongation (LQT8). Because the Ca2+ channel Cav1.2 is abundant in many tissues, patients with Timothy syndrome have many clinical manifestations including congenital heart disease, autism, syndactyly, and immune deficiency.3,47,49
Brugada Syndrome
Approximately 12% of cases of the Brugada syndrome are attributable to loss-of-function mutations in the cardiac Ca2+ channel resulting in a reduction of the depolarizing ICaL. Brugada syndrome type 3 is caused by mutations in the CACNA1C gene, which encodes the pore-forming α1 subunit (Cav1.2). Brugada syndrome type 4 is caused by mutations in the CACNB2 gene, which encodes for the regulatory β2 subunit (Cavβ2), which modifies gating of ICaL. The mechanism of Brugada syndrome type 3 and type 4 involves a reduction of the depolarizing ICaL. Mutations in the α and β subunits of the Ca2+ channel can also lead to a shorter than normal QT interval, creating a new clinical entity consisting of a combined Brugada/short QT syndrome.10
Short QT Syndrome
SQT4 is caused by mutations on the CACNA1C gene (encoding the α1C subunit, Cav1.2) and SQT5 is caused by mutations on the CACNB2 gene (encoding the β2b subunit). Loss-of-function mutations on those genes result in major attenuation in ICaL amplitude, leading to shortening of the action potential duration, and are associated with asymmetrical T waves, an attenuated QT–heart rate relationship, and AF. The three patients reported to harbor these mutations had a Brugada type 1 phenotype.1,49
Acquired Diseases
Abnormalities in Ca2+ currents or intracellular Ca2+ transients, or both, in acquired diseases may induce both arrhythmia and contractile dysfunction. In AF, Cav1.2 mRNA and protein levels are downregulated, resulting in ICaL reduction, which contributes to action potential shortening.3
In heart failure, the membrane density of ICaL channels is reduced. However, channel phosphorylation is increased, leading to reduced response to phosphorylating interventions and causing increased single channel open probability that compensates for the reductions in channel density. Despite unchanged ICaL, sarcoplasmic reticulum Ca2+ transients are smaller and slower in heart failure, and they cause contractile dysfunction.3,24,49
T-Type Calcium Current (ICaT)
Structure and Physiology
The cardiac T-type Ca2+ channel (originally called low-voltage–activated channels) is composed of a single α subunit. Cav3.1 (α1G subunit encoded by CACNA1G) and Cav3.2 (α1H subunit encoded by CACNA1H) isoforms are major candidates for the cardiac T-type Ca2+ channel. It is not yet clear whether auxiliary subunits exist for native T-type Ca2+ channels. The structure of the α1H and α1G subunits is similar to that involved in the L-type Ca2+ channels.46,48
T-type Ca2+ channels can be distinguished from L-type Ca2+ channels on the basis of their distinctive gating and conductance properties. Compared with L-type Ca2+ channels, T-type Ca2+ channels have a smaller conductance and transient openings, and they open at the significantly more negative Em that overlaps the pacemaker potentials of sinus node cells. The threshold for activation of ICaT is –70 to –60 mV, and ICaT is fully activated at –30 to –10 mV at physiological Ca2+ concentration. Membrane depolarization also causes inactivation of ICaT. The inactivation threshold is near –90 mV, with half-maximal inactivation of –60 mV. In contrast to L-type Ca2+ channels, T-type Ca2+ channels do not inactivate in a Ca2+-dependent manner. The activation and steady-state inactivation overlap near the activation threshold (–60 to –30 mV), thus providing a constant inward current (a window current). This window component may help in facilitating the slow diastolic depolarization in sinus node cells and contribute to automaticity. Unlike L-type channels, T-type Ca2+ channels are relatively insensitive to dihydropyridines.46,48
Function
Cav3 channels conduct the T-type Ca2+ current (ICaT), which is important in a wide variety of physiological functions, including neuronal firing, hormone secretion, smooth muscle contraction, cell proliferation of some cardiac tissues, and myoblast fusion. In the heart, T-type channels are abundant in sinus node pacemaker cells and Purkinje fibers of many species and are important for maintenance of pacemaker activity by setting the frequency of action potential firing.46,48
Because T-type Ca2+ channels are most prevalent in the conduction system in the adult heart and the activation range of ICaT overlaps the pacemaker potential, it has been suggested that T-type Ca2+ channels play a role in generating pacemaker depolarization and contribute to automaticity. However, experimental evidence indicates that ICaT is not a primary pacemaker current, but it can modify depolarization frequency only slightly. Although organic T-type Ca2+ channel blockers (mibefradil) result in marked decrease in firing frequency of sinus node cells in clinical studies, the possibility that these blockers affect other ionic currents, including ICaL, cannot be entirely excluded. In fact, it is has not yet been determined whether ICaT exists functionally in atrial, ventricular, and sinoatrial node cells in the human heart. Further studies are necessary to clarify whether T-type Ca2+ channels contribute to the automaticity of the human heart.48,51
Acquired Diseases
Interestingly, the T- type Ca2+ channels are re-expressed in atrial and ventricular myocytes in animal models under various pathological conditions such as cardiac hypertrophy, MI, and heart failure. These findings reflect a reversion to a fetal or neonatal pattern of gene expression, and ICaT contributes to abnormal electrical activity and excitation-contraction coupling. It is possible that similar remodeling occurs in the hypertrophied human heart; however, to date, T-type Ca2+ channels have not been detected in normal or diseased human myocardial cells.46,48,51
Furthermore, experimental evidence suggests that T-type Ca2+ channels may be of functional importance in arrhythmogenesis in cardiomyocytes in pulmonary veins, which can initiate paroxysmal AF. ICaT may directly and indirectly participate in pacemaker depolarization in sinoatrial and other regions of the heart, and this mechanism may become more important in failing hearts.46,48
Cardiac Pacemaker Current (If)
Structure and Physiology
If is a mixed Na+-K+ current, with a threefold higher selectivity for Na+ than for K+. Despite the GYG amino acid motif, HCN channels are more permeable Na+ than K+ ions. Unlike most voltage-gated channels, which are activated on membrane depolarization, HCN channels are activated on hyperpolarization. The HCN channel activates slowly on hyperpolarization (at voltages lower than approximately −40 to −45 mV) and inactivate slowly in a voltage-independent manner on depolarization. The speed of channel opening is strongly dependent on Em and is faster at more negative potentials. If conducts an inward current during phases 3 and 4 of the action potential and may underlie slow membrane depolarization in cells with pacemaker activity (i.e., cells with If and little or no IK1).52
Function
The If channels are deactivated during the action potential upstroke and the initial plateau phase of repolarization, but they begin to activate at the end of the action potential as repolarization brings the Em to levels more negative than approximately −40 to −50 mV, and they are fully activated at approximately −100 mV. Once activated, If depolarizes the membrane back toward a level at which the Ca2+ current activates to initiate the action potential.1 In its range of activation, which quite properly comprises the voltage range of diastolic depolarization in sinus node cells (approximately −40 to −65 mV), the current is inward, and its reversal occurs at approximately −10 to −20 mV. At the end of the repolarization phase of an action potential, because If activation occurs in the background of a decaying outward (K+ time-dependent) current, current flow quickly shifts from outward to inward, giving rise to a sudden reversal of voltage change (from repolarizing to depolarizing) at the maximum diastolic potential. Hence, If first opposes and then stops the repolarization process (at the maximum diastolic potential) and finally initiates the diastolic depolarization.52–55
The If contribution terminates when, in the late part of diastolic depolarization, Ca2+-dependent processes take over, and the threshold for L-type Ca2+ current activation and action potential firing is reached. Although deactivation of If at depolarized voltages is rapid, complete switch off of the current occurs only during the very early fraction of the action potential, which provides a brief time interval during which If carries an outward current at positive voltages.52
If is not only involved in principal rhythm generation but also plays a key role in heart rate regulation. The degree of activation of If determines, at the end of an action potential, the steepness of phase 4 depolarization and hence the frequency of action potential firing. Additionally, If represents a basic physiological mechanism mediating autonomic regulation of heart rate. If is regulated by intracellular cAMP and is thus activated and inhibited by β-adrenergic and muscarinic M2 receptor stimulation, respectively.52,56
However, given the complexity of the cellular processes involved in rhythmic activity, exact quantification of the extent to which If and other mechanisms contribute to pacemaking is still a debated issue.52
Regulation
The voltage dependence of If activation is regulated by cAMP direct binding to the cyclic nucleotide-binding domain in the HCN channel and not via phosphorylation-dependent activation mechanisms. Direct interaction of cAMP to the channel shifts the activation curve to more depolarized voltages and strongly accelerates channel activation kinetics. Sympathetic stimulation activates If and hence accelerates heart rate via β-adrenoceptor-triggered cAMP production, whereas low-level vagal stimulation lowers heart rate via inhibition of cAMP synthesis and an ensuing inhibition of If activity. High vagal tone most likely lowers the heart rate mainly via the activation of IKACh.56
Pharmacology
Given the key role of HCN channels in cardiac pacemaking, If has become a pharmacological target for the development of novel and more specific heart rate–reducing agents in patients with ischemic heart disease. Whereas current heart rate–lowering drugs adversely affect cardiac contractility, selective If inhibition is believed to lower heart rate without impairing contractility. In the past, several agents inhibiting cardiac If were developed. Early drugs identified as pure bradycardic agents include zatebradine and cilobradine, which are derived from the L-type Ca2+ channel blocker verapamil. More recently, ivabradine was introduced into clinical use as the first therapeutic If blocker for the treatment of chronic stable angina. The principal action of all these substances is to reduce the frequency of pacemaker potentials in the sinus node by inducing a reduction of the diastolic depolarization slope. Ivabradine blocks HCN4 and HCN1 channels by accessing the channels from their intracellular side and by exerting a use- and current-dependent block. Interestingly, ivabradine acts as open channel blocker in HCN4 (as in sinus nodal If), whereas block of HCN1 requires channels either to be closed or in a transitional state between an open and closed configuration.52,56 With the exception of ivabradine, other HCN channel blockers are not specific enough for sinus nodal (mainly HCN4-mediated) If; they also block neuronal HCN channels (Ih current) in several regions of the nervous system, and this has prevented their clinical utility.56
Clonidine, an α2-adrenergic agonist, was shown to block sinus nodal If. Clonidine produces a shift in the voltage dependence of the channel by 10 to 20 mV to more hyperpolarizing potentials.56
Sarcoplasmic Reticulum Calcium Release Channels (Ryanodine Receptor 2)
Structure and Physiology
The Ca2+ release channel is a macromolecular complex, formed by the cardiac ryanodine receptor isoform (RyR2, encoded by the RYR2 gene) homotetramer and certain proteins localized on both the cytosolic and the luminal side of the sarcoplasmic reticulum membrane. The cardiac RyR2, by far the largest protein of the complex, operates as a Ca2+-conducting channel. RyR2 channels are approximately 10 times larger than voltage-gated Ca2+ and Na+ channels.57,58
Each RyR2 monomer contains a transmembrane domain, the pore-forming region that is composed of an even, but still undetermined number (likely six to eight) of transmembrane segments. This domain encompasses only approximately 10% of the protein clustered at the C-terminus, but it has a critical functional role because it contains sequences that control RyR2 localization and oligomerization and is sufficient to form a functional Ca2+ release channel. The remaining 90% of the protein at the N-terminus comprises an enormous cytoplasmic domain that serves as a cytosolic scaffold that interacts with regulatory molecules (including Ca2+, ATP) and proteins (including FKBP12.6, calmodulin). On the luminal (sarcoplasmic reticulum) side, RyR2 forms a part of a large quaternary complex with calsequestrin (CASQ2), triadin, and junctin. Together these four proteins form the core of the Ca2+ release channel complex.57–60
Cardiac RyR2 functions as a ligand-activated ion channel that activates (opens) on Ca2+ binding. However, the exact structural determinants of RyR gating are as yet unknown. RyR2 is normally closed at low cytosolic diastolic Ca2+ concentrations (approximately 100 to 200 nM). At submicromolar cytosolic Ca2+ concentrations, Ca2+ binds to high-affinity binding sites on RyR2 and thus increases the open probability of the channel (two Ca2+ ions are required to open the RyR2 channel) and allows Ca2+ release from the sarcoplasmic reticulum into the cytosol.50,59,60
The precise juxtaposition of the sarcolemmal specialized invaginations (known as T tubules) and sarcoplasmic reticulum forms specific junctional microdomains, creating a 10- to 12-nM gap, known as the dyadic cleft. RyR2s are assembled in a paracrystalline lattice in each dyad, containing 80 to 260 channels, where the RyR2 cytoplasmic region resides, and its transmembrane region spans the sarcoplasmic reticulum membrane to immerse the luminal portion into the sarcoplasmic reticulum Ca2+ store. Each array of RyR2s is faced by 10 to 25 L-type Ca2+ channels in the sarcolemmal T tubule. Hence, each dyad constitutes a local Ca2+ signaling complex, or couplon, whereby these proteins are coordinately regulated via the changing concentrations of Ca2+, Na+, and K+ within the dyadic cleft.50,59,60
After approximately 10 milliseconds of RyR2 channel opening, Ca2+ release from the sarcoplasmic reticulum terminates, and the Ca2+ spark signal starts to decay, mostly owing to diffusion of Ca2+ away from its source. RyR2 channel activity is maximal at cytosolic Ca2+ concentrations of approximately 10 µM. Elevating cytosolic Ca2+ concentrations beyond this point leads to a reduction in the open probability of the channel, possibly because of Ca2+ binding to low-affinity inhibitory binding sites on the RyR2 channel.60
Function
The RyR2 channels are an essential component of the excitation-contraction coupling and act as sentinels to the large sarcoplasmic reticulum Ca2+ store. Excitation-contraction coupling describes the physiological process of converting an electrical stimulus (action potential) to a mechanical response (muscle contraction). The contraction of a cardiac myocyte is governed primarily by intracellular Ca2+ concentration (see Fig. 1-3). Ca2+ enters the cell during the plateau phase of the action potential through L-type Ca2+ channels that line the sarcolemmal T tubules. However, the rise in intracellular Ca2+ is small and not sufficient to induce contraction. Nonetheless, the small amount of Ca2+ entering the cell via ICaL triggers a rapid mobilization of Ca2+ from the sarcoplasmic reticulum into the cytosol by opening the RyR2 channels in the CICR process. Approximately 75% of Ca2+ present in the cytoplasm during contraction is released from the sarcoplasmic reticulum. The close proximity of the RyR2 to the T tubule enables each L-type Ca2+ channel to activate 4 to 6 RyR2s and generate a Ca2+ spark. Ca2+ influx via ICaL simultaneously activates approximately 10,000 to 20,000 couplons in each ventricular myocyte with every action potential. Such sophisticated coordination in opening and closing is required to ensure that Ca2+ release occurs during the systolic phase of the cardiac cycle and functional silence during diastole.50,59,60
Luminal Ca2+-dependent deactivation of RyR2 is a process by which the decline in sarcoplasmic reticulum Ca2+ that follows sarcoplasmic reticulum Ca2+ release renders RyR2s functionally inactive. This results in termination of CICR and induction of a refractory state that suppresses Ca2+ release during the diastolic phase, which is an important determinant of the mechanical refractoriness required for efficient relaxation and refilling of the heart.50,58
Regulation
Many proteins interact directly and indirectly with the N-terminal cytoplasmic domain of RyR2 including FK506-binding protein (calstabin2 or FKBP12.6), PKA, Ca2+-calmodulin-dependent kinase II (CaMKII), phosphodiesterase 4D3, calmodulin, protein phosphatases 1 and 2A, and sorcin. CASQ2, junctin, and triadin bind with the luminal (sarcoplasmic reticulum) C-terminus of RyR2.58–60
Calsequestrin
CASQ2 is a low-affinity, high-capacity Ca2+-binding protein, which serves as a Ca2+ storage reservoir that is able to bind luminal Ca2+ (40 to 50 Ca2+ ions per molecule) during diastole, thus buffering Ca2+ within the sarcoplasmic reticulum (i.e., preventing Ca2+ precipitation and lowering luminal free Ca2+ concentration) and preventing diastolic Ca2+ release via RyR2 to cytosol. CASQ2 presumably serves as a luminal Ca2+ sensor that modulates the responsiveness of the RyR2 to luminal Ca2+. At low luminal Ca2+ concentrations, CASQ2 interacts with RyR2 via binding to triadin and junctin and inhibits the activity of the RyR2. When sarcoplasmic reticulum Ca2+ levels increase, Ca2+ binds to CASQ2, resulting in weakened interactions or complete dissociation of CASQ2 from triadin. This process relieves the inhibitory action of CASQ2 on the RyR2 complex, allows Ca2+ release to the cytosol, and hence normalizes sarcoplasmic reticulum Ca2+ load. Triadin and junctin not only mediate functional interactions of CASQ2 and RyR2 but also modulate RyR2 function by themselves by increasing the activity of the RyR2 channel. This Ca2+-dependent modulation of RyR2s by triadin, junctin, and CASQ2 may contribute to deactivation and refractoriness of RyR2 channels after sarcoplasmic reticulum Ca2+ release.58,60,61
FK506-Binding Protein
FKBP12.6 (the 12.6-kDa cytosolic FK506-binding protein), also known as calstabin2 (Ca2+ channel-stabilizing binding protein), stabilizes the closed conformational state of the RyR2 channel, thus enabling the channel to close completely during diastole (at low intracellular Ca2+ concentrations), preventing aberrant Ca2+ leakage from the sarcoplasmic reticulum, and ensuring muscle relaxation. PKA phosphorylation of RyR2 decreases the binding affinity of FKBP12.6 to RyR2 and thereby increases the probability of an open state and amplifies the response to Ca2+-dependent activation.50,59,60,62
Calmodulin
Calmodulin is a Ca2+-binding protein containing four Ca2+-binding sites and four sites that bind to RyR2 monomers. Calmodulin preferentially inhibits RyR2 at Ca2+ concentrations lower than 10 µM by binding to a region on RyR2. Calmodulin may function to assist closing RyR2 following sarcoplasmic reticulum Ca2+ release in excitation-contraction coupling.60
Protein Kinase A
Stimulation of β-adrenergic receptors results in an increase in cAMP and consequent PKA activation. PKA interacts with the RyR2 channel via binding to the muscle A kinase anchoring protein (mAKAP). PKA phosphorylation of RyR2 activates the channel, at least in part by increasing the sensitivity of RyR2 to cytosolic Ca2+ and a transient decrease in the binding affinity of calstabin2. This allows for increased sarcoplasmic reticulum Ca2+ release on Ca2+ influx via ICaL as a part of the fight-or-flight mechanism. By contrast, chronic PKA hyperphosphorylation of RyR2 can result in incomplete channel closure and a Ca2+ leak during diastole, which causes depletion of the sarcoplasmic reticulum Ca2+ store and reduced Ca2+ release on receptor activation.50,60,62
Ca2+-Calmodulin-Dependent Protein Kinase II
CaMKII is a dodecameric holoenzyme activated by Ca2+-bound calmodulin, possibly at high cellular Ca2+ loads. CaMKII phosphorylation increases RyR2 channel open probability, but to a smaller extent than PKA phosphorylation. CaMKII activity increases at faster heart rates (typically mediated by β-adrenergic stimulation and PKA activation and associated with increased cytosolic Ca2+) and phosphorylates RyR2 to enhance sarcoplasmic reticulum Ca2+ release, which helps maintain the positive force-frequency relationship (i.e., cardiac contractility increases as a function of heart rate). Increased CaMKII activity also phosphorylates phospholamban to help accelerate diastolic filling of the ventricles at higher heart rates. Physiologically, sympathetic activation stimulates both PKA and CaMKII, which thus function synergistically. CaMKII is typically considered to be downstream of PKA and elevated Ca2+ transients.50,60,62
Pharmacology
Among class I antiarrhythmic drugs, only flecainide and propafenone were found to inhibit RyR2 channels (by inducing brief closures of open RyR2 to subconductance states), suppress arrhythmogenic Ca2+ sparks, and prevent CPVT in experimental studies. The potency of RyR2 channel inhibition rather than Na+ channel blockade appears to determine the efficacy of class I agents for the prevention of CPVT. Flecainide has been demonstrated to prevent lethal ventricular arrhythmias in patients with familial CPVT.63,64
Inherited Channelopathies
CPVT is caused by mutations in genes that encode for key Ca2+ regulatory proteins. Two genetic variants of CPVT have been described: an autosomal-dominant trait (CPVT1; most common) caused by mutations in the cardiac RyR2 gene, and a recessive form (CPVT2; rare) associated with homozygous mutations in the CASQ2 gene (CASQ2).
Approximately 50% to 70% of patients with CPVT harbor RyR2 mutations. More than 70 RyR2 mutations linked to CPVT have been identified. CPVT mutant RyR2 typically shows gain-of-function defects following channel activation by PKA phosphorylation (in response to β-adrenergic stimulation or caffeine), resulting in uncontrolled Ca2+ release from the sarcoplasmic reticulum during electrical diastole. The exaggerated spontaneous Ca2+ release from the sarcoplasmic reticulum facilitates the development of DADs and triggered arrhythmias.58,59
The molecular mechanisms by which RyR2 mutations alter the physiological properties and function of RyR2 are not completely defined. It has been suggested that CPVT mutations in RyR2 reduce the binding affinity of RyR2 for the regulatory protein FKBP12.6 (calstabin2) that stabilizes the closed conformational state of the RyR2 channel, thus enabling the channel to close completely during diastole (at low intracellular Ca2+ concentrations), preventing aberrant Ca2+ leakage from the sarcoplasmic reticulum, and ensuring muscle relaxation. PKA phosphorylation (induced by β-adrenergic stimulation) of the mutant channels results in further worsening of the binding affinity of FKBP12.6 to the mutant RyR2 and increases the probability of an open state at diastolic Ca2+ concentrations. As a consequence, the mutant RyR2 channel fails to close completely during diastole, with a resulting diastolic Ca2+ leak from the sarcoplasmic reticulum during stress or exercise.58–60
An alternative hypothesis is that CPVT mutations in RyR2 sensitize the channel to luminal (sarcoplasmic reticulum) Ca2+ such that under baseline conditions, when sarcoplasmic reticulum load is normal, there is no Ca2+ leak. Under β-adrenergic (sympathetic) stimulation, sarcoplasmic reticulum Ca2+ concentration becomes elevated above the reduced threshold, causing Ca2+ to leak out of the sarcoplasmic reticulum. A third hypothesis for RyR2-related CPVT is that mutations in RyR2 impair the intermolecular interactions between discrete RyR2 domains necessary for proper folding of the channel and self-regulation of channel gating.58,60
So far, seven CASQ2 mutations linked to CPVT have been reported. Although some of these mutations are thought to compromise CASQ2 synthesis and result in reduced expression or complete absence of CASQ2 in the heart, other mutations seem to cause expression of defective CASQ2 proteins with abnormal regulation of cellular Ca2+ homeostasis. CASQ2 mutations result in disruption of the control of RyR2s by luminal Ca2+ required for effective termination of sarcoplasmic reticulum Ca2+ release and prevention of spontaneous Ca2+ release during diastole, thus leading to diminished Ca2+ signaling refractoriness and generation of arrhythmogenic spontaneous Ca2+ releases.58,59,65
Importantly, in the setting of digitalis poisoning, the abnormal RyR2 behavior leading to spontaneous Ca2+ release and DADs is secondary to the elevation of the sarcoplasmic reticulum Ca2+ content (SOICR). In CPVT, on the other hand, spontaneous Ca2+ release and DADs can occur without Ca2+ overload. Mutations in RyR2 or CASQ2 lead to defective Ca2+ signaling lowering of the sarcoplasmic reticulum Ca2+ threshold for spontaneous Ca2+ release to less than the normal baseline level (perceived Ca2+ overload).58
Missense mutations in RyR2 also been linked to a form of arrhythmogenic cardiomyopathy (ARVD-2) characterized by exercise-induced polymorphic VT that does not appear to have a reentrant mechanism and occurs in the absence of significant structural abnormalities. Patients do not develop characteristic features of ARVD on the 12-lead ECG or signal-averaged ECG, and global RV function remains unaffected. ARVD-2 shows a closer resemblance to familial CPVT in both etiology and phenotype; its inclusion under the umbrella term of ARVD remains controversial.66–68
Acquired Diseases
RyR2 dysfunction is a key factor leading to arrhythmias in heart failure. Chronic β-adrenergic stimulation results in PKA hyperphosphorylation of RyR2. This process causes the dissociation of the channel-stabilizing protein calstabin and leads to diastolic Ca2+ leak from the sarcoplasmic reticulum and the generation of spontaneous Ca2+ waves, which can be maintained despite a reduced Ca2+ gradient, thus underlying DAD-induced triggered arrhythmias in heart failure.24,69,70
Cardiac Gap Junctions
Structure and Physiology
Cardiomyocytes make contact with each other via multiple intercalated discs, which mediate the transmission of force, electrical continuity, and chemical communication between adjacent cells. Three types of specialized junctions exist in the intercalated disc: (1) the fascia adherens, (2) the macula adherens (desmosome), and (3) the gap junction (nexus). The fascia adherens is an anchoring site for myofibrils, facilitating the transmission of mechanical energy between neighboring cells. The desmosomes link to the cytoskeleton of adjacent cells to provide strong localized adhesion sites that resist shearing forces generated during contraction. Gap junctions are assemblies of intercellular channels that provide electrical continuity and chemical communication between adjacent cells.71
In addition to the end-to-end and side-to-side gap junctions localized at the intercalated discs, lateral (side-to-side) gap junctions can exist in nondisc lateral membranes of cardiomyocytes, but they are much less common, occurring more in atrial than ventricular myocardium.71
Each gap junction channel is constructed of two hemichannels (connexons) aligned head-to-head in mirror symmetry across a narrow extracellular gap, one provided by each of the adjoining cells. Each connexon is composed of six integral membrane proteins called connexins (Cx) hexagonally arranged around the pore. Each connexin consists of four membrane-spanning domains (M1 to M4), two extracellular loops (E1, E2), one intracellular loop, and cytoplasmic N- and C-termini. The extracellular loops mediate the docking of the two hemichannels.71,72
Up to 24 different connexin types have been identified. They are named after their theoretical molecular weight in daltons. In the heart, Cx40, Cx43, and Cx45 are most important for action potential propagation. Although each connexin exhibits a distinct tissue distribution, most cardiomyocytes express more than one connexin isoform. Cx43 is by far the most abundant and is expressed between atrial and ventricular myocytes and distal parts of the Purkinje system. Cx40 is mainly expressed in the atrial myocytes, AVN, and HPS. Cx45 appears to be primarily expressed in nodal tissue (the sinus and compact AVNs), and more weakly in the atrium, His bundle, bundle branches, and Purkinje fibers.71
Connexons can be composed by the oligomerization of a single connexin type (homomeric) or of different types (heteromeric). In addition, the gap junction channel as a whole may be formed of two matching hemichannels (homotypic) or nonmatching hemichannels (heterotypic).71
The individual gap junction channels allow exchange of nutrients, metabolites, ions (e.g., Na+, Cl−, K+, Ca2+) and small molecules (e.g., cAMP, cGMP, inositol triphosphate [IP3]) with molecular weights up to approximately 1000 Da.71
Function
Gap junctions maintain direct cell-to-cell communication in the heart by providing biochemical and low-resistance electrical coupling between adjacent cardiomyocytes. Thus, gap junctions are responsible for myocardial electrical current flow propagation from one cardiac cell to another and are crucial in myocardial synchronization and heart function. Gap junctions also provide biochemical coupling, which allows intercellular movement of second-messenger substances (e.g., ATP, cyclic nucleotides, and IP3) and hence enables coordinated responses of the myocardial syncytium to physiological stimuli.
Under physiological conditions, a given cardiomyocyte in the adult working myocardium is electrically coupled to an average of approximately 11 adjacent cells, with gap junctions predominantly localized at the intercalated discs at the ends of the rod-shaped cells. Lateral (side-to-side) gap junctions in nondisc lateral membranes of cardiomyocytes are much less abundant and occur more often in atrial than ventricular tissues.71 Thus, intercellular current flow occurs primarily at the cell termini, although propagation can occur both longitudinally and transversely. This particular subcellular distribution of gap junctions underlies uniform anisotropic impulse propagation throughout the myocardium, whereby conduction in the direction parallel to the long axis of the myocardial fiber bundles is approximately 3 to 5 times more rapid than that in the transverse direction. This property is attributable principally to the lower resistivity of myocardium provided by the gap junctions in the longitudinal versus the transverse direction.73–75
Regulation
Gap junction channels have a voltage-dependent gating mechanism, depending primarily on transjunctional voltage (i.e., the potential difference between the cytoplasm of the two adjacent cells). At rest, when the junctional voltage is zero, the channels usually are open. During the course of a propagated action potential, the channels tend to close in a voltage- and time-dependent manner. Gap junction channel gating can also be altered by specific changes in intracellular ions and by post-translational modifications (“loop” gating). Cytosolic Na+ and Ca2+ overload, acidosis, and reduced ATP levels decrease gap junction channel function. Unlike the voltage gate, which closes rapidly and incompletely, the chemical gate closes slowly and completely.76
The regulation of gap junction trafficking, assembly and disassembly, and degradation is likely to be critical in the control of intercellular communication. Phosphorylation also appears to play a key role in channel gating that determines channel conductance and has been implicated in the regulation of the connexin “life cycle” at several stages. Gap junction coupling also is regulated by certain endogenous mediators (e.g., acetylcholine, norepinephrine, and angiotensin), likely via phosphorylation-mediated mechanisms. Importantly, channels composed of different connexins possess different properties and are susceptible to different regulation.71,76
Pharmacology
Antimalarial drugs, particularly quinine and quinine derivatives such as mefloquine, can reduce gap junction channel conductance, and their effects seem to be connexin subtype specific. In addition, it has been suggested that the cardiac glycosides strophanthidin, ouabain, and digitoxin decrease intercellular coupling.76
Experimental evidence suggests that increasing gap junction conductance can potentially confer an antiarrhythmic effect. Some compounds, including antiarrhythmic peptides and their derivatives (e.g., AAP10, ZP123, rotigaptide) can upregulate Cx43 via modulation either synthesis or degradation and enhance gap junctional communication and were found to reduce conduction slowing and prevent AF and ischemic reentrant VT in various cell and animal models. These peptides presumably act by modulating Cx43 phosphorylation.77,78
Acquired Diseases
Modification of cell-to-cell coupling occurs in numerous pathophysiological settings (e.g., myocardial ischemia, ventricular hypertrophy, cardiomyopathy) as a consequence of acute changes in the average conductance of gap junctions secondary to ischemia, hypoxia, acidification, or an increase in intracellular Ca2+, or it can be produced by changes in expression or cellular distribution patterns of gap junctions.74,79–81
Remodeling includes a decrease in the number of gap junction channels resulting from the interruption of communication between cells by fibrosis and downregulation of Cx43 formation or of trafficking to the intercalated disc. Additionally, gap junctions can become more prominent along lateral membranes of myocytes (so-called structural remodeling). Influences of Cx43 lateralization on impulse propagation have not yet been well defined.82
Alterations in distribution and function of cardiac gap junctions are associated with conduction delay or block. Inactivation of gap junctions decreases transverse conduction velocity to a greater degree than longitudinal conduction, thus resulting in exaggeration of anisotropy and providing a substrate for reentrant activity and increased susceptibility to arrhythmias.74
AF is associated with abnormal expression and distribution of atrial Cx40, which can potentially lead to inhomogeneous electrical coupling and abnormal impulse formation and conduction and thereby provide the substrate for atrial arrhythmias. Furthermore, a rare single nucleotide polymorphism in the atrial-specific Cx40 gene has been found to increase the risk of idiopathic AF.83
1. Grant A.O. Cardiac ion channels. Circ Arrhythm Electrophysiol. 2009;2:185-194.
2. Amin A.S., Asghari-Roodsari A., Tan H.L. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223-237.
3. Amin A.S., Tan H.L., Wilde A.A. Cardiac ion channels in health and disease. Heart Rhythm. 2010;7:117-126.
4. Viswanathan P.C., Balser J.R. Biophysics of normal and abnormal cardiac sodium channel function. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:93-104.
5. Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2-11.
6. January C.T., Makielski J.C. Pharmacology of the cardiac sodium channel. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:169-174.
7. Moss A.J., Goldenberg I. Importance of knowing the genotype and the specific mutation when managing patients with long QT syndrome. Circ Arrhythm Electrophysiol. 2008;1:213-226.
8. Saenen J.B., Vrints C.J. Molecular aspects of the congenital and acquired long QT syndrome: clinical implications. J Mol Cell Cardiol. 2008;44:633-646.
9. Lu J.T., Kass R.S. Recent progress in congenital long QT syndrome. Curr Opin Cardiol. May 2010;25:216-221. 3
10. Campuzano O., Brugada R., Iglesias A. Genetics of Brugada syndrome. Curr Opin Cardiol. May 2010;25:210-215. 3
11. Gehi A.K., Duong T.D., Metz L.D., et al. Risk stratification of individuals with the Brugada electrocardiogram: a meta-analysis. J Cardiovasc Electrophysiol. 2006;17:577-583.
12. Antzelevitch C., Brugada P., Borggrefe M., et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670.
13. Morita H., Zipes D.P., Wu J. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 2009;6(Suppl):S34-S43.
14. Lei M., Huang C.L., Zhang Y. Genetic Na+ channelopathies and sinus node dysfunction. Prog Biophys Mol Biol. 2008;98:171-178.
15. Katz A.M. Cardiac ion channels. In: Katz A.M., editor. Physiology of the heart. ed 5. Philadelphia: Lippincott Williams & Wilkins; 2011:3433-3468.
16. Tamargo J., Caballero R., Gomez R., et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9-33.
17. Oudit G.Y., Backx P.H. Voltage-regulated potassium channels. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:29-42.
18. Niwa N., Nerbonne J.M. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12-25.
19. Antzelevitch C., Yan G.X. J wave syndromes. Heart Rhythm. 2010;7:549-558.
20. van der Heyden M.A., Wijnhoven T.J., Opthof T. Molecular aspects of adrenergic modulation of the transient outward current. Cardiovasc Res. 2006;71:430-442.
21. Alders M., Koopmann T.T., Christiaans I., et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468-476.
22. Benito B., Guasch E., Rivard L., Nattel S. Clinical and mechanistic issues in early repolarization of normal variants and lethal arrhythmia syndromes. J Am Coll Cardiol. 2010;56:1177-1186.
23. Tikkanen J.T., Anttonen O., Junttila M.J., et al. Long-term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361:2529-2537.
24. Aiba T., Tomaselli G.F. Electrical remodeling in the failing heart. Curr Opin Cardiol. 2010;25:29-36.
25. Ravens U., Wettwer E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc Res. 2011;89:776-785.
26. Ehrlich J.R. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J Cardiovasc Pharmacol. 2008;52:129-135.
27. Charpentier F., Merot J., Loussouarn G., Baro I. Delayed rectifier K(+) currents and cardiac repolarization. J Mol Cell Cardiol. 2010;48:37-44.
28. Tristani-Firouzi M., Sanguinetti M.C. Structural determinants and biophysical properties of HERG and KCNQ1 channel gating. J Mol Cell Cardiol. 2003;35:27-35.
29. Smyth J.W., Shaw R.M. Forward trafficking of ion channels: what the clinician needs to know. Heart Rhythm. 2010;7:1135-1140.
30. Ruan Y., Liu N., Napolitano C., Priori S.G. Therapeutic strategies for long QT syndrome: does the molecular substrate matter? Circ Arrhythm Electrophysiol. 2008;1:290-297.
31. Roden D.M. Clinical practice: long QT syndrome. N Engl J Med. 2008;358:169-176.
32. Schwartz P.J., Spazzolini C., Crotti L., et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783-790.
33. Sy R.W., Chattha I.S., Klein G.J., et al. Repolarization dynamics during exercise discriminate between LQT1 and LQT2 genotypes. J Cardiovasc Electrophysiol. 2010;21:1242-1246.
34. Anumonwo J.M., Lopatin A.N. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010;48:45-54.
35. Lu C.W., Lin J.H., Rajawat Y.S., et al. Functional and clinical characterization of a mutation in KCNJ2 associated with Andersen-Tawil syndrome. J Med Genet. 2006;43:653-659.
36. Tsuboi M., Antzelevitch C. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm. 2006;3:328-335.
37. Schimpf R., Borggrefe M., Wolpert C. Clinical and molecular genetics of the short QT syndrome. Curr Opin Cardiol. 2008;23:192-198.
38. Callans D.J. Patients with hemodynamically tolerated ventricular tachycardia require implantable cardioverter defibrillators. Circulation. 2007;116:1196-1203.
39. Yang Y., Yang Y., Liang B., et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010;86:872-880.
40. Ravens U., Cerbai E. Role of potassium currents in cardiac arrhythmias. Europace. 2008;10:1133-1137.
41. Nichols C.G. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470-476.
42. Zhang H., Flagg T.P., Nichols C.G. Cardiac sarcolemmal K(ATP) channels: latest twists in a questing tale!. J Mol Cell Cardiol. 2010;48:71-75.
43. Kane G.C., Liu X.K., Yamada S., et al. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38:937-943.
44. Gurney A., Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38:305-318.
45. Enyedi P., Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559-605.
46. Bodi I., Mikala G., Koch S.E., et al. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306-3317.
47. Benitah J.P., Alvarez J.L., Gomez A.M. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010;48:26-36.
48. Ono K., Iijima T. Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol. 2010;48:65-70.
49. van der Heyden M.A., Wijnhoven T.J., Opthof T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc Res. 2005;65:28-39.
50. Bers D.M. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23-49.
51. Nerbonne J.M., Kass R.S. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205-1253.
52. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434-446.
53. Mangoni M.E., Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008;88:919-982.
54. Lakatta E.G. A paradigm shift for the heart’s pacemaker. Heart Rhythm. 2010;7:559-564.
55. Venetucci L.A., Trafford A.W., O’Neill S.C., Eisner D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008 January 15;77:285-292.
56. Biel M., Wahl-Schott C., Michalakis S., Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847-885.
57. Zalk R., Lehnart S.E., Marks A.R. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367-385.
58. Gyorke S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2009;6:123-129.
59. Mohamed U., Napolitano C., Priori S.G. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2007;18:791-797.
60. Kushnir A., Marks A.R. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol. 2010;59:1-30.
61. Ter Keurs H.E., Boyden P.A. Calcium and arrhythmogenesis. Physiol Rev. 2007;87:457-506.
62. Blayney L.M., Lai F.A. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther. 2009;123:151-177.
63. Cerrone M., Napolitano C., Priori S.G. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6:1652-1659.
64. Hwang H.S., Hasdemir C., Laver D., et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4:128-135.
65. Napolitano C., Priori S.G. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:675-678.
66. Otten E., Asimaki A., Maass A., et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010;7:1058-1064.
67. Maass K. Arrhythmogenic right ventricular cardiomyopathy and desmin: another gene fits the shoe. Heart Rhythm. 2010;7:1065-1066.
68. Hamilton R.M., Fidler L. Right ventricular cardiomyopathy in the young: an emerging challenge. Heart Rhythm. 2009;6:571-575.
69. Jin H., Lyon A.R., Akar F.G. Arrhythmia mechanisms in the failing heart. Pacing Clin Electrophysiol. 2008;31:1048-1056.
70. Laurita K.R., Rosenbaum D.S. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J Mol Cell Cardiol. 2008;44:31-43.
71. Jansen J.A., van Veen T.A., de Bakker J.M., van Rijen H.V. Cardiac connexins and impulse propagation. J Mol Cell Cardiol. 2010;48:76-82.
72. Yeager M., Harris A.L. Gap junction channel structure in the early 21st century: facts and fantasies. Curr Opin Cell Biol. 2007;19:521-528.
73. Valderrabano M. Influence of anisotropic conduction properties in the propagation of the cardiac action potential. Prog Biophys Mol Biol. 2007;94:144-168.
74. Kleber A.G., Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431-488.
75. Saffitz J.E., Lerner D.L. Gap junction distribution and regulation in the heart. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 4. Philadelphia: Saunders; 2004:181-191.
76. Lewandowski R., Petersen J.S., Delmar M. Connexins as potential targets for cardiovascular pharmacology. In: Zipes D.P., Jalife J., editors. Cardiac electrophysiology: from cell to bedside. ed 5. Philadelphia: Saunders; 2009:205-213.
77. Wit A.L., Duffy H.S. Drug development for treatment of cardiac arrhythmias: targeting the gap junctions. Am J Physiol. 2008;294:H16-H18.
78. Lin X., Zemlin C., Hennan J.K., et al. Enhancement of ventricular gap-junction coupling by rotigaptide. Cardiovasc Res. 2008;79:416-426.
79. van Rijen H.V., van Veen T.A., Gros D., et al. Connexins and cardiac arrhythmias. Adv Cardiol. 2006;42:150-160.
80. Lee P.J., Pogwizd S.M. Micropatterns of propagation. Adv Cardiol. 2006;42:86-106.
81. Cascio W.E., Yang H., Muller-Borer B.J., Johnson T.A. Ischemia-induced arrhythmia: the role of connexins, gap junctions, and attendant changes in impulse propagation. J Electrocardiol. 2005;38(Suppl):55-59.
82. Cabo C., Yao J., Boyden P.A., et al. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72:241-249.
83. Chaldoupi S.M., Loh P., Hauer R.N., et al. The role of connexin40 in atrial fibrillation. Cardiovasc Res. 2009;84:15-23.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 2 Cardiac Ion Channels
Ion channels are pore-forming membrane proteins that regulate the flow of ions passively down their electrochemical gradient across the membrane. Ion channels are present on all membranes of cells (plasma membrane) and intracellular organelles (nucleus, mitochondria, endoplasmic reticulum). There are more than 300 types of ion channels in a living cell. The channels are not randomly distributed in the membrane, but tend to cluster at the intercalated disc in association with modulatory subunits.1
Importantly, channel opening and closing are not instantaneous but usually take time. The transition from the resting (closed) state to the open state is called activation. Once opened, channels do not remain in the open state, but instead they undergo conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening and must undergo a recovery or reactivation process back to the resting state to regain their ability to open. Inactivation curves of the various voltage-gated ion channel types differ in their slopes and midpoints of inactivation and can overlap, in which case a steady-state or noninactivating current flows.1
Ion channels differ with respect to the number of subunits of which they are composed and other aspects of structure. Many ion channels function as part of macromolecular complexes in which many components are assembled at specific sites within the membrane. For most ion channels, the pore-forming subunit is called the α subunit, whereas the auxiliary subunits are denoted β, gamma, and so on. Most ion channels have a single pore; however, some have two.1
Sodium Channels
Structure and Physiology
The cardiac Na+ channel complex is composed of a primary α and multiple ancillary β subunits. The approximately 2000-amino-acid α subunit contains the channel’s ion-conducting pore and controls the channel selectivity for Na+ ions and voltage-dependent gating machinery. This subunit contains all the drug and toxin interaction sites identified to date. The α subunit (Nav1.5), encoded by the SCN5A gene, consists of four internally homologous domains (I to IV) that are connected to each other by cytoplasmic linkers (Fig. 2-1). Each domain consists of six membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops. The four domains are arranged in a fourfold circular symmetry to form the channel. The extracellular loops between S5 and S6 (termed the P segments) have a unique primary structure in each domain (Fig. 2-2). The P segments curve back into the membrane to form an ion-conducting central pore whose structural constituents determine the selectivity and conductance properties of the Na+ channel.2
FIGURE 2-1 The sodium channel macromolecular complex. See text for discussion.
(From Boussy T, Paparella G, de Asmundis C, et al: Genetic basis of ventricular arrhythmias. Heart Fail Clin 6:249-266, 2010.)
Four auxiliary β subunits (Navβ1 to Navβ4, encoded by the genes SCN1B to SCN4B, respectively) have been identified; each is a glycoprotein with a single membrane-spanning segment. The β1 subunit likely plays a role in modulation of the gating properties and level of expression of the Na+ channel.2
Na+ channels are the typical example of voltage-gated ion channels. Na+ channels switch among three functional states: deactivated (closed), activated (open), and inactivated (closed), depending on the membrane potential (Em). These channel states control Na+ ion permeability through the channel into the cardiomyocyte. Na+ channel activation allows Na+ ion influx into the cell, and inactivation blocks entry of Na+ ions.2
On excitation of the cardiomyocyte by electrical stimuli from adjacent cells, its resting Em (approximately −85 mV) depolarizes. The positively charged S4 segment of each domain of the α subunit functions as the sensor of the transmembrane voltage; these segments are believed to undergo rapid structural conformational changes in response to membrane depolarization, thus leading to channel opening (activation) from its resting (closed) state and enabling a large and rapid influx of Na+ (inward Na+ current [INa]) during the rapid upstroke (phase 0) of the action potential in atrial, ventricular, and Purkinje cardiomyocytes.3
Normally, activation of Na+ channels is transient; fast inactivation (closing of the pore) starts simultaneously with activation, but because inactivation is slightly delayed relative to activation, the channels remain transiently open to conduct INa during phase 0 of the action potential before it closes. Each Na+ channel opens very briefly (less than 1 millisecond) during phase 0 of the action potential; collectively, activation of the channel lasts a few milliseconds and is followed by fast inactivation.
Na+ channel inactivation comprises different conformational states, including fast, intermediate, and slow inactivation. Fast inactivation is at least partly mediated by rapid occlusion of the inner mouth of the pore by the cytoplasmic interdomain linker between domains III and IV of the α subunit, which has a triplet of hydrophobic residues that likely functions as a hinged “latch” that limits or restricts Na+ ion pass through the pore. The carboxyl terminus (C-terminus) also plays an important role in the control of Na+ channel inactivation and stabilizing the channels in the inactivated state by interacting with the loop linking domains III and IV. Importantly, although most Na+ channels open before inactivating, some actually inactivate without ever opening (a process known as closed-state inactivation).1,4
In addition to these rapid gating transitions, Na+ channels are also susceptible to slower inactivating processes (slow inactivation) if the membrane remains depolarized for a longer time. These slower events can contribute to the availability of active channels under various physiological conditions. Whereas fast-inactivated Na+ channels recover rapidly (within 10 milliseconds) during the hyperpolarized interval between stimuli, slow inactivation requires much longer recovery times (ranging from hundreds of milliseconds to many seconds). The molecular movements leading to slow inactivation are less well understood. The P segments seem to play a key role in slow inactivation.4
Some Na+ channels occasionally show alternative gating modes consisting of isolated brief openings occurring after variable and prolonged latencies and bursts of openings during which the channel opens repetitively for hundreds of milliseconds. The isolated brief openings are the result of the occasional return from the inactivated state. The bursts of openings are the result of occasional failure of inactivation.1 Prolonged opening or reopening of some Na+ channels during phases 2 and 3 can result in a small late INa (INaL). Despite its minor contribution in healthy hearts, INaL can potentially play an important role in diseased hearts.3
Function
The cardiac Na+ channel also plays a crucial role in the propagation of action potentials throughout the atrium, HPS, and ventricles. The opening of Na+ channels in the atria underlies the P wave on the ECG, and in the ventricles INa underlies the QRS complex and enables a synchronous ventricular contraction. Because the upstroke of the electrical potential primarily determines the speed of conduction between adjacent cells, Na+ channels are present in abundance in tissues where speed is of importance. Cardiac Purkinje cells contain up to 1 million Na+ channels, a finding that illustrates the importance of rapid conductance in the heart.2
Na+ channels also make a contribution in the plateau phase (phase 2) and help determine the duration of the action potential. After phase 0 of the action potential, INa decreases to less than 1% of its peak value over the next several milliseconds because of voltage-dependent inactivation. This persistent or “late” inward INa (INaL), along with the L-type Ca2+ current (ICaL), helps maintain the action potential plateau.5
Regulation
The regulatory proteins interacting with Nav1.5 may be classified as follows: (1) anchoring-adaptor proteins (e.g., ankyrin-G, syntrophin proteins, multicopy suppressor of gsp1 [MOG1]), which play roles only in trafficking and targeting the channel protein in specific membrane compartments; (2) enzymes interacting with and modifying the channel structure (post-translational modifications), such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 on binding (e.g., caveolin-3, calmodulin, glycerol 3-phosphate dehydrogenase 1–like [G3PD1L], telethonin, Plakophilin-2).5 Coexpression of Nav1.5 with its β subunits induces acceleration in the recovery from inactivation and enhancement of INa amplitude.
The cardiac Na+ channels are subject to phosphorylation and dephosphorylation by kinases or phosphatases. The intracellular linker between domains I and II contains eight consensus sites for cyclic adenosine monophosphate (cAMP)–dependent protein kinase A (PKA) phosphorylation. cAMP-dependent PKA and G protein stimulatory α subunit (Gsα) modulate the function of expressed cardiac Na+ channels on β-adrenergic stimulation and enhance INa.1
In contrast, activation of α-adrenergic stimulating protein kinase C (PKC) results in the reduction of INa. The effect of PKC is largely attributable to phosphorylation of a highly conserved serine in the linker between domains III and IV. PKC reduces the maximal conductance of the channels and alters gating. Na+ channels exhibit a hyperpolarizing shift in the steady-state availability curve, suggesting an enhancement of inactivation from closed states.
All subunits of the Na+ channel are modified by glycosylation. The β1 and β2 subunits are heavily glycosylated, with up to 40% of the mass being carbohydrate. In contrast, the cardiac α subunit is only 5% sugar by weight. Sialic acid is a prominent component of the N-linked carbohydrate of the Na+ channel. The addition of such a highly charged carbohydrate has predictable effects on the voltage dependence of gating through alteration of the surface charge of the channel protein.1
Pharmacology
Na+ channels are the targets for the action of class I antiarrhythmic drugs. Na+ channel blockers bind to a specific receptor within the channel’s pore. The binding blocks ion movement through the pore and stabilizes the inactivated state of Na+ channels. Blockade of Na+ channels tends to decrease tissue excitability and conduction velocity (by attenuating peak INa) and can shorten action potential duration (by attenuating late INa).1,6
One important component in the action of antiarrhythmic drugs is a voltage-dependent change in the affinity of the drug-binding site (i.e., the channel is a modulated receptor). Additionally, restricted access to binding sites can contribute to drug action, a phenomenon that has been called the guarded receptor model. Open and inactivated channels are more susceptible to block than resting channels, likely because of a difference in binding affinity or state-dependent access to the binding site. Consequently, binding of antiarrhythmic drug occurs primarily during the action potential (known as use-dependent block), and the block dissipates after repolarization (i.e., in the interval between action potentials). When the time interval between depolarizations is insufficient for block to recover before the next depolarization occurs (secondary to either abbreviation of the interval between action potentials during fast heart rates or slow kinetics of the unbinding of the Na+ channel blocker), block of Na+ channels accumulates (resulting in an increased number of blocked channels and enhanced blockade).6 A drug with rapid kinetics produces less channel block with the subsequent depolarization than does a drug with slower recovery. Use-dependent block is important for the action of antiarrhythmic drugs because it allows strong drug effects during fast heart rates associated with tachyarrhythmias but limits Na+ channel block during normal heart rates. Importantly, drug recovery kinetics can potentially be slowed by pathophysiological conditions such as membrane depolarization, ischemia, and acidosis.1 This property is known as use-dependence and is seen most frequently with the class IC agents, less frequently with the class IA drugs, and rarely with the class IB agents.
Inherited Channelopathies
Mutations in genes that encode various subunits of the cardiac Na+ channel or proteins involved in regulation of the inward INa have been linked to several types of electrical disorders (Table 2-1). Depending on the mutation, the consequence is either a gain of channel function (with consequent prolongation of action potential duration because more positive ions accumulate in the cell) or an overall loss of channel function that influences the initial depolarizing phase of the action potential (with consequent decrease in cardiac excitability and electrical conduction velocity). It is noteworthy that a single mutation can cause different phenotypes or combinations thereof.2
Long QT Syndrome
In contrast to most long QT syndrome (LQTS) phenotypes, which are based on mutations that modify the cardiac K+ currents, type 3 congenital LQTS (LQT3), which accounts for approximately 8% of congenital LQTS cases, is caused by gain-of-function mutations on the Na+ channel gene, SCN5A. More than 80 mutations have been identified in the SCN5A gene, with most being missense mutations mainly clustered in Nav1.5 regions that are involved in fast inactivation (i.e., S4 segment of domain IV, the domain III–domain IV linker, and the cytoplasmic loops between the S4 and S5 segments of domain III and domain IV), or in regions that stabilize fast inactivation (e.g., the C-terminus).2,4,7
Several mechanisms have been identified to underlie ionic effects of SCN5A mutations in LQT3. Most SCN5A mutations cause a gain of function through disruption of fast inactivation, thus allowing repeated reopening during sustained depolarization and resulting in an abnormal, small, but functionally important sustained (or persistent) noninactivating Na+ current (Isus) during action potential plateau. Because the general membrane conductance is small during the action potential plateau, the presence of a persistent inward INa, even of small amplitude, can potentially have a major impact on the plateau duration and can be sufficient to prolong repolarization and QT interval. QT prolongation and the risk of developing arrhythmia are more pronounced at slow heart rates, when the action potential duration is longer, thereby allowing more INa to enter the cell.2,4,7
Other less common mechanisms of SCN5A mutations to cause LQT3 include increased window current, which results from delayed inactivation of mutant Na+ channels, occurring at more positive potentials and widening the voltage range during which the Na+ channel may reactivate without inactivation. Additionally, some mutations cause slower inactivation, which allows longer channel openings and causes a slowly inactivating INa. This current is INaL and is to be distinguished from Isus (which does not inactivate). Comparable to Isus, both the window current and INaL exert their effects during phases 2 and 3 of the action potential, in which normally no or very small INa is present. Other mutations induce prolonged action potential duration by enhancing recovery from inactivation, an effect that leads to larger peak INa by increasing the fraction of channels available for activation (because of faster recovery) during subsequent depolarizations. Finally, some mutations can cause increased expression of mutant Nav1.5 through enhanced mRNA translation or protein trafficking to the sarcolemma, decreased protein degradation, or altered modulation by β subunits and regulatory proteins. These effects lead to larger INa density during phase 0 of the action potential. Importantly, one single SCN5A mutation can potentially cause several changes in the expression and/or gating properties of the resulting Na+ channels.2
Regardless of the mechanism, increased Na+ current (Isus, window current, INaL, or peak INa) upsets the balance between depolarizing and repolarizing currents in favor of depolarization. The resulting delay in the repolarization process triggers early afterdepolarizations (EADs; i.e., reactivation of the L-type Ca2+ channel during phase 2 or 3 of the action potential), especially in Purkinje fiber myocytes, in which action potential durations are intrinsically longer.2
LQT9 is caused by gain-of-function mutations on the CAV3 gene, which encodes caveolin-3, a plasma membrane scaffolding protein that interacts with Nav1.5 and plays a role in compartmentalization and regulation of channel function. Mutations in caveolin-3 induce kinetic alterations of the Nav1.5 current that result in persistent Na+ current (Isus) and have been reported in cases of sudden infant death syndrome (SIDS).2,8
LQT10 is caused by loss-of-function mutations on the SCN4B gene, which encodes the β subunit (Navβ4) of the Nav1.5 channel. To date, only a single mutation in one patient has been described. This mutation caused a shift in the inactivation of the INa toward more positive potentials, but it did not change the activation. This resulted in increased window currents at an Em corresponding to the phase 3 of the action potential.2,8
LQT12 is caused by mutations on the SNTA1 gene, which encodes α1 syntrophin, a cytoplasmic adaptor protein that enables the interaction among Nav1.5, nitric oxide synthase, and the sarcolemmal Ca2+ adenosine triphosphatase (ATPase) complex that appears to regulate ion channel function. By disrupting the interaction between Nav1.5 and the sarcolemmal Ca2+ ATPase complex, SNTA1 mutations cause increased Nav1.5 nitrosylation with consequent reduction of channel inactivation and enhanced Isus densities.2,9
Brugada Syndrome
The Brugada syndrome is an autosomal dominant inherited channelopathy characterized by an elevated ST segment or J wave appearing in the right precordial leads. This syndrome is associated with a high incidence of sudden cardiac death (SCD) secondary to a rapid polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF). Approximately 65% of mutations identified in the SCN5A gene are associated with the Brugada syndrome phenotype (Brugada syndrome type 1), and they account for approximately 18% to 30% of cases of Brugada syndrome. So far, more than 200 Brugada syndrome–associated loss-of-function (i.e., reduced peak INa) mutations have been described in SCN5A. Some of these mutations result in loss of function secondary to impaired channel trafficking to the cell membrane (i.e., reduced expression of functional Na+ channels), disrupted ion conductance (i.e., expression of nonfunctional Na+ channels), or altered gating function. Altered gating properties comprise delayed activation (i.e., activation at more positive potentials), earlier inactivation (i.e., inactivation at more negative potentials), faster inactivation, and enhanced slow inactivation.1–3,10
Most of the mutations are missense mutations, whereby a single amino acid is replaced by a different amino acid. Missense mutations commonly alter the gating properties of mutant channels. Because virtually all reported SCN5A mutation carriers are heterozygous, mutant channels with altered gating may cause up to 50% reduction of INa. Different SCN5A mutations can cause different degrees of INa reduction and therefore different degrees of severity of the clinical phenotype of Brugada syndrome.10–12
In addition to SCN5A alterations, mutations in the GPD1L gene, which encodes the glycerol 3-phosphate dehydrogenase 1–like protein (G3PD1L), affect the trafficking of the cardiac Na+ channel to the cell surface and result in reduction of INa and Brugada syndrome type 2.10 Brugada syndrome associated with GPD1L gene mutations is characterized by progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis.2,13
Furthermore, reduction in INa can be caused by mutations in the SCN1B gene (encoding the β1 and β1b subunits of the Na+ channel) and the SCN3B gene (encoding the β3 subunit of the Na+ channel), resulting in Brugada syndrome type 5 and type 7, respectively.10,13
Familial Progressive Cardiac Conduction Disease
Loss-of-function SCN5A mutations have been linked to familial forms of progressive cardiac conduction disease (referred to as hereditary Lenègre disease, primary cardiac conduction system disease, and familial atrioventricular [AV] block). This disease is characterized by slowing of electrical conduction through the atria, AVN, His bundle, Purkinje fibers, and ventricles, accompanied by an age-related degenerative process and fibrosis of the cardiac conduction system, in the absence of structural or systemic disease. It is often reflected by varying degrees of AV block and bundle branch block. Whether the age-dependent fibrosis of the conduction system is a primary degenerative process in progressive cardiac conduction disease or a physiological process that is accelerated by INa reduction remains to be investigated. A single loss-of-function SCN5A mutation can cause isolated progressive cardiac conduction disease or can be combined with the Brugada syndrome (overlap syndrome). Loss-of-function mutations in SCN1B also have been identified in patients with progressive cardiac conduction disease who carried no mutation in SCN5A.2
Congenital Sick Sinus Syndrome
Although INa does not play a prominent role in sinus node activity, mutations in SCN5A have been linked to sick sinus syndrome, manifesting as sinus bradycardia, sinus arrest, sinoatrial block, or a combination of these conditions, which can progress to atrial inexcitability (atrial standstill). Loss-of-function SCN5A mutations result in reduced peak INa density, hyperpolarizing shifts in the voltage dependence of steady-state channel availability, and slow recovery from inactivation. These effects likely cause reduced automaticity, decreased excitability, and conduction slowing or block of impulses generated in the sinus node to the surrounding atrial tissue. Sinus node dysfunction can also manifest concomitantly with other phenotypes that are linked to SCN5A loss-of-function mutations such as Brugada syndrome and progressive cardiac conduction disorders.2,14
Familial Atrial Fibrillation
Loss-of-function mutations, gain-of-function mutations, and common polymorphisms on the SCN5A gene have been identified in some cases of atrial fibrillation (AF) occurring in young patients with structurally normal hearts. It is speculated that INa reduction may predispose to AF by slowing the electrical conduction velocity and thereby facilitating reentry. On the other hand, gain-of-function mutations can potentially predispose to AF by increasing atrial excitability. AF can occur in patients with other phenotypes of Na+ channelopathies, including LQT3, Brugada syndrome, dilated cardiomyopathy, and sinus node dysfunction. Furthermore, mutations in the SCN1B gene (encoding the β1 subunit of the Na+ channel) and the SCN2B gene (encoding the β2 subunit of the Na+ channel) have been identified in patients with AF, many of whom displayed ECG patterns suggestive of the Brugada syndrome.1,2
Dilated Cardiomyopathy
Some cases of familial dilated cardiomyopathy have been linked to SCN5A mutations. Dilated cardiomyopathy–linked SCN5A mutations cause diverse loss-of-function and gain-of-function changes in the gating properties, but how such changes evoke contractile dysfunction is not understood. It is speculated that SCN5A mutations disrupt the interactions between the mutant Na+ channels and intracellular (or extracellular) proteins that are essential for normal cardiomyocyte structure and architecture. Notably, dilated cardiomyopathy with SCN5A mutations display atrial or ventricular arrhythmias (including AF, VT, and VF), or both, as well as sinus node dysfunction, AV block, and intraventricular conduction delay.2
Acquired Diseases
In heart failure, peak INa is reduced (likely secondary to reduced SCN5A expression), whereas INaL is increased (likely because of increased phosphorylation of Na+ channels). Nav1.5 expression is reduced in the surviving myocytes in the border zone of the myocardial infarct (MI). Importantly, Na+ channel blockers can increase the risk for SCD in patients with ischemic heart disease, possibly by facilitating the initiation of reentrant excitation waves. Additionally, INaL increases during myocardial ischemia, explaining why INaL inhibition may be an effective therapy for chronic stable angina. Nav1.5 expression is reduced in response to persistent atrial tachyarrhythmias as part of the “electrical remodeling” process, leading to attenuation of INa.3
Potassium Channels
Structure and Physiology
Cardiac K+ channels are membrane-spanning proteins that allow the passive movement of K+ ions across the cell membrane along its electrochemical gradient. The ion-conducting or pore-forming subunit is generally referred to as the α subunit. The tripeptide sequence glycine-tyrosine-glycine GYG is common to the pore of all K+ channels and constitutes the signature motif for determining K+ ion selectivity. A gating mechanism controls switching between open-conducting and closed-nonconducting states.1,15
K+ channels represent the most diverse class of cardiac ion channels (Fig. 2-3). Cardiac K+ currents can be categorized as voltage-gated (Kv) and ligand-gated channels. In Kv channels, pore opening is coupled to the movement of a voltage sensor within the membrane electric field, and they include the rapidly activating and inactivating transient outward current (Ito); the ultrarapid (IKur), rapid (IKr), and slow (IKs) components of the delayed rectifier current; and the inward rectifier current (IK1). In contrast, pore opening in ligand-gated channels is coupled to the binding of an organic molecule, including channels activated by a decrease in the intracellular concentration of adenosine triphosphate (KATP) or activated by acetylcholine (KACh). Other classes of K+ channels respond to different stimuli, including changes in intracellular Ca2+ concentration and G proteins.15
On the basis of the primary amino acid sequence of the α subunit, K+ channels have been classified into three major families (Table 2-2):
1. Channels containing six transmembrane segments and a single pore. This architecture is typical of Kv channels.
2. Channels containing two transmembrane segments (M1 and M2) and a pore. This architecture is typical of inward rectifier K+ (Kir) channels, including K1, KATP, and KACh channels. They conduct K+ currents more in the inward direction than the outward and play an important role in setting the resting potential close to the equilibrium potential for K+ and in repolarization. Kir channels form either homotetramers or heterotetramers.
3. Channels containing four transmembrane segments and two pores (K2P). These channels exist as homodimers or heterodimers. K2P currents display little time or voltage dependence. There are four classes of cardiac K2P: TASK, TWIK, TREK, and THIK.15,16
Each voltage-gated K+ channel (Kv family) is formed by the coassembly of 4 identical (homotetramers) or a combination of 4 different (from the same subfamily, heterotetramers) α subunits. A total of 38 genes has been cloned and assigned to 12 subfamilies of voltage-gated K channels (Kv1 to Kv12) on the basis of sequence similarities. Each α subunit contains one domain consisting of 6 membrane-spanning segments (S1 to S6), connected to each other by alternating intracellular and extracellular peptide loops (similar to 1 of the 4 domains of voltage-gated Na+ and Ca2+ channels), with both the amino terminus (N-terminus) and the C-terminus located on the intracellular side of the membrane. The central ion-conducting pore region is formed by the S5 and S6 segments and the S5-S6 linker (P segment); the S5-S6 linker is responsible for K+ ion selectivity. The S4 segment serves as the voltage sensor.1,15
The α subunits of Kv channels can generate voltage dependent K+ current when expressed in heterologous systems. However, the assembly of a functional tetramer can occur only in the presence of multiple auxiliary units (see Table 2-2). In many cases, auxiliary subunits coassociate with the α subunits and likely modulate cell surface expression, gating kinetics, and drug sensitivity of the α subunit complex. Most K+ channel β subunits assemble with α subunits and give rise to an α4β4 complex. K+ channel β subunits represent a diverse molecular group, which includes cytoplasmic proteins (Kvβ1 to Kvβ3, KChIP, and KChAP) that interact with the intracellular domains of Kv channels; single transmembrane spanning proteins (e.g., minK and minK-related proteins [MiRPs]) encoded by the KCNE gene family; and large ATP-binding cassette (ABC) transport-related proteins (e.g., the sulfonylurea receptors [SURs]).1,16
As with voltage-dependent Na+ and Ca2+ channels, Kv channels typically fluctuate among distinct conformational states because of molecular movements in response to voltage changes across the cell membrane (voltage-dependent gating). The Kv channel activates (opens) on membrane depolarization, thus allowing the rapid passage of K+ ions across the sarcolemma. After opening, the channel undergoes conformational transition in a time-dependent manner to a stable nonconducting (inactivated) state. Inactivated channels are incapable of reopening, even if the transmembrane voltage is favorable, unless they “recover” from inactivation (i.e., enter the closed state) on membrane repolarization. Closed channels are nonconducting but can be activated on membrane depolarization.17
Three mechanistically distinct types of Kv channel inactivation that are associated with distinct molecular domains have been identified: N-type, C-type, and U-type. N-type (“ball and chain”) inactivation involves physical occlusion of the intracellular mouth of the channel pore through binding of a small group of amino acids (“inactivation ball tethered to a chain”) at the extreme N-terminus. In contrast, C-type inactivation involves conformational changes in the external mouth of the pore. C-type inactivation exists in almost all K+ channels and may reflect a slow constriction of the pore. This inactivation process is thought to be voltage independent, coupled to channel opening, and is usually slower than N-type inactivation. Recovery from C-type inactivation is relatively slow and weakly voltage dependent. Importantly, the rate of C-type inactivation and recovery can be strongly influenced by other factors, such as N-type inactivation, drug binding, and changes in extracellular K+ concentration. These interactions render C-type inactivation an important biophysical process in regulating repetitive electrical activity and determining certain physiological properties such as refractoriness, drug binding, and sensitivity to extracellular K+.17
In addition to N-type and C-type inactivation, some Kv channels also show another type of inactivation (U-type), which exhibits a U-shaped voltage dependence with prolonged stimulation rates. Those channels appear to exhibit preferential inactivation from partially activated closed states, rapid and strongly voltage-dependent recovery from inactivation and, in some channel types, accelerated inactivation with elevation of extracellular K+. The exact conformational changes underlying U-type inactivation remain unclear. Importantly, there is extreme diversity in the kinetic and potentially molecular properties of Kv channel inactivation, particularly of C-type inactivation.17
Function
K+ channels are a diverse and ubiquitous group of membrane proteins that regulate K+ ion flow across the cell membrane on the electrochemical gradient and regulate the resting Em, the frequency of pacemaker cells, and the shape and duration of the cardiac action potential. Because the concentration of K+ ions outside the cell membrane is approximately 25-fold lower than that in the intracellular fluid, on activation, the opening of K+ channels generates an outward current resulting from the efflux of positively charged ions that offers a mechanism to counteract, dampen, or restrict the depolarization front (phases 1 through 4 of the action potential) triggered by an influx of cations (Na+ and Ca2+).1,3,15,16
The variation in the level of expression of K+ channels that participate in the genesis of the cardiac action potential explains the regional differences of the configuration and duration of cardiac action potentials from sinus node and atrial to ventricular myocytes and across the myocardial wall (endocardium, midmyocardium, and epicardium). Moreover, the expression and properties of K+ channels are not static; heart rate, neurohumoral state, pharmacological agents, cardiovascular diseases (cardiac hypertrophy and failure, MI) and arrhythmias (e.g., AF) can influence those properties, and they underlie the change in action potential configuration in response to variation in heart rate and various physiological and pathological conditions.1,15,16
Transient Outward Potassium Current (Ito)
Structure and Physiology
Cardiac Ito channels are macromolecular protein complexes, comprising four pore-forming Kv α subunits and a variety of Kv channel accessory (β) subunits (see Fig. 2-3). Ito is the sum of a voltage-dependent, Ca2+-independent K+ current (Ito1) and a Ca2+-activated Cl− or K+ current (Ito2). In human atrial and ventricular myocytes, the presence of Ito2 has not been clearly demonstrated.
Kv channels mediating Ito,s are formed by the coassembly of four α subunits from the Kv1.x subfamily (primarily Kv1.4, and possibly Kv1.7 and Kv3.4), whereas those mediating Ito,f are formed by the coassembly of four α subunits from the Kv4.x subfamily (primarily Kv4.3, and possibly Kv4.2) (see Table 2-2). Among the various accessory subunits identified, a crucial role has been definitively demonstrated only for KChIP2, and potentially for MiRP2.1,16,18
Function
The density of Ito varies across the myocardial wall and in different regions of the heart. In human ventricles, Ito densities are much higher in the epicardium and midmyocardium than in the endocardium. Furthermore, Ito,f and Ito,s are differentially expressed in the myocardium, thus contributing to regional heterogeneities in action potential waveforms. Ito,f is the principal subtype expressed in human atrium. The markedly higher densities of Ito,f, together with the expression of the ultrarapid delayed rectifier current, accelerate the early phase of repolarization and lead to lower plateau potentials and shorter action potentials in atrial as compared with ventricular cells.17,18
Although both Ito,f and Ito,s are expressed in the ventricle, Ito,f is more prominent in the epicardium and midmyocardium (putative M cells) than in the endocardium. These regional differences are responsible for the shorter duration and the prominent phase 1 notch and the “spike-and-dome” morphology of epicardial and midmyocardial compared with endocardial action potentials. A prominent Ito-mediated action potential notch in ventricular epicardium but not endocardium produces a transmural voltage gradient during early ventricular repolarization that registers as a J wave or J point elevation on the ECG.19 Ito densities are also reportedly higher in right than in left (midmyocardial and epicardial) ventricular myocytes, consistent with the more pronounced spike-and-dome morphology of right, compared with left, ventricular action potentials.1,3,17,18
Furthermore, variations in cardiac repolarization associated with Ito regional differences strongly influence intracellular Ca2+ transient by modulating Ca2+ entry via ICaL and Na+-Ca2+ exchange, thereby regulating excitation-contraction coupling and regional modulation of myocardial contractility and hence synchronizing the timing of force generation between different ventricular regions and enhancing mechanical efficiency.17
Regulation
Transient outward channels are subject to α- and β-adrenergic regulation. α-Adrenergic stimulation reduces Ito; concomitant β-adrenergic stimulation appears to counteract the α-adrenergic effect, at least in part. The effects of α- and β-adrenergic stimulation are exerted by phosphorylation of the Kv1.4, Kv4.2, and Kv4.3 α-subunits by PKA as well as PKC. Calmodulin-dependent kinase II, on the other hand, has been shown to be involved in enhancement of Ito. Adrenergic stimulation is also an important determinant of transient outward channel downregulation in cardiac disease. Chronic α-adrenergic stimulation and angiotensin II reduce Ito channel expression.20
KChIP2, when coexpressed with Kv4.3, increases surface channel density and current amplitude, slows channel inactivation, and markedly accelerates the recovery from inactivation. In the ventricle, KChIP2 mRNA is 25-fold more abundant in the epicardium than in the endocardium. This gradient parallels the gradient in Ito expression, whereas Kv4.3 mRNA is expressed at equal levels across the ventricular wall. Thus, transcriptional regulation of the KChIP2 gene (KChIP2) is the primary determinant of Ito expression in the ventricular wall.1,16
Observations suggest that MiRP2 is required for the physiological functioning of human Ito,f channels and that gain-of-function mutations in MiRP2 predispose to Brugada syndrome through augmentation of Ito,f.18
Ito is strongly rate dependent. Ito fails to recover from previous inactivation at very fast heart rates, which can be manifest as a decrease in the magnitude of the J wave on the surface ECG. Hence, abrupt changes in rate and pauses have important consequences for the early repolarization of the membrane.19
Ito can be enhanced by aging, low sympathetic activity, high parasympathetic activity, bradycardia, hypothermia, estrogen reduction, and drugs. Estrogen suppresses the expression of the Kv4.3 channel and results in reduced Ito and a shallow phase 1 notch.13
Phase 1 notch of the action potential modulates the kinetics of slower activating ion currents and consequently the later phases of the action potential. Initial enhancement of phase 1 notch promotes phase 2 dome and delays repolarization, presumably by delaying the peak of ICaL. However, further enhancement of phase 1 notch prevents the rising of phase 2 dome and abbreviates action potential duration, presumably by deactivation or voltage modulation that reduces ICaL. Thus, progressive deepening of phase 1 notch can cause initial enhancement followed by sudden disappearance of phase 2 dome and corresponding prolongation followed by abbreviation of action potential duration. On the other hand, modulators that decrease Ito lead to a shift of the plateau phase into the positive range of potentials, thus increasing the activation of the delayed rectifier currents, promoting faster repolarization, and reducing the electrochemical driving force for Ca2+ and hence ICaL. Phase 1 notch also affects the function of the Na+-Ca2+ exchanger and subsequently intracellular Ca2+ handling and Na+ channel function.1,16
Pharmacology
Quinidine, 4-aminopyridine, flecainide, and propafenone produce an open channel blockade and accelerate Ito inactivation. Quinidine, but not flecainide or propafenone, produces a frequency-dependent block of Ito that results from a slow rate of drug dissociation from the channel. Quinidine has relatively strong Ito blocking effect, whereas flecainide mildly blocks Ito.18,20
Ito blockers can potentially prolong the action potential duration in the atrial and in ischemic ventricular myocardium. However, because the net effects of Ito blockade on repolarization depend on secondary changes in other currents, the reduction of Ito density can result in a shortening of the ventricular action potential. Moreover, heterogeneous ventricular distribution of Ito can cause marked dispersion of repolarization across the ventricular wall that, when accompanied by prominent conduction delays related to Na+ channel blockade, results in extrasystolic activity through a phase 2 reentrant mechanisms.18,20
Currently, a cardioselective and channel-specific Ito opener or blocker is not available for clinical use. Development of an Ito-selective drug is expected to be beneficial in patients with primary abnormality in the Ito or in other channels, such as the Brugada syndrome, in which heterogeneity in the expression of Ito between epicardium and endocardium in the right ventricle (RV) results in the substrate responsible for reentry and ventricular arrhythmias.18,20
Inherited Channelopathies
To date, only mutations in KCNE3 (MiRP2) are linked to inherited arrhythmia. KCNE3 mutations were identified in five related patients with Brugada syndrome. When expressed with Kv4.3, the mutation increased Ito,f density.3
Another KCNE3 mutation was identified in one patient with familial AF. The mutation was found to increase Ito,f and was postulated to cause AF by shortening action potential duration and facilitating atrial reentrant excitation waves.3
A genome wide haplotype-sharing study associated a haplotype on chromosome 7, harboring DPP6, with idiopathic VF in three distantly related families. Overexpression of DPP6, which encodes dipeptidyl-peptidase 6, a putative component of the Ito channel complex, was proposed as the likely pathogenetic mechanism. DPP6 significantly alters the inactivation kinetics of both Kv4.2 and Kv4.3 and promotes expression of these α subunits in the cell membrane.3,21
Importantly, the normally functioning Ito channels play an important role in the electrophysiological consequences of the ionic current abnormalities in the Brugada and J wave syndromes. Heterogeneity in the distribution of Ito channels across the myocardial wall, being more prominent in ventricular epicardium than endocardium, and particularly in the RV, results in the shorter duration and the prominent phase 1 notch and the spike-and-dome morphology of the epicardial action potential as compared with the endocardium. The resultant transmural voltage gradient during the early phases (phases 1 and 2) of the action potential is thought to be responsible for the inscription of the J wave on the surface ECG.18,19 An increase in net repolarizing current, secondary to either a decrease in the inward currents (INa and ICaL) or an increase in the outward K+ currents (Ito, IKr, IKs, IKAch, IKATP), or both, can accentuate the action potential notch and lead to augmentation of the J wave or the appearance of ST segment elevation on the surface ECG. An outward shift of currents that extends beyond the action potential notch not only can accentuate the J wave but also can lead to partial or complete loss of the dome of the action potential, thus leading to a protracted transmural voltage gradient that manifests as greater ST segment elevation and gives rise to J wave syndromes. The type of the ion current affected and its regional distribution in the ventricles determine the particular phenotype (including the Brugada syndrome, early repolarization syndrome, hypothermia-induced ST segment elevation, and MI-induced ST segment elevation).18,19,22 The degree of accentuation of the action potential notch leading to loss of the dome depends on the magnitude of Ito. These changes are more prominent in regions of the myocardium exhibiting a relatively large Ito, such as the RV epicardium; this explains the appearance of coved ST segment elevation, characteristic of Brugada syndrome, in the right precordial ECG leads.19,20
In this context, factors that influence the kinetics of Ito or the other repolarization currents can modify the manifestation of the J wave on the ECG. Na+ channel blockers (procainamide, pilsicainide, propafenone, flecainide, and disopyramide), which reduce the inward INa, can accentuate the J wave and ST segment elevation in patients with concealed J wave syndromes. Quinidine, which inhibits both Ito and INa, reduces the magnitude of the J wave and normalizes ST segment elevation. Additionally, acceleration of the heart rate, which is associated with reduction of Ito (because of slow recovery of Ito from inactivation), results in a decrease in the magnitude of the J wave. Male predominance can potentially result from larger epicardial Ito density versus Ito in women.19,22
The increased transmural heterogeneity of ventricular repolarization (i.e., dispersion of repolarization between epicardium and endocardium), which is responsible for J point elevation and early repolarization pattern on the surface ECG, is also responsible for the increased vulnerability to ventricular tachyarrhythmias. A significant outward shift in current can cause partial or complete loss of the dome of the action potential in regions where Ito is prominent (epicardium), with the consequent loss of activation of ICaL. The dome of the action potential then can propagate from regions where it is preserved (midmyocardium endocardium) to regions where it is lost (epicardium), thus giving rise to phase 2 reentry, which can generate premature ventricular complexes that in turn can initiate polymorphic VT or VF.19,20,22,23
Acquired Diseases
An alteration in the expression and distribution of Ito is observed in various pathophysiological conditions. Adrenergic effects seem to be involved in at least some of these Ito-regulating processes during heart disease.20
In general, myocardial ischemia, MI, dilated cardiomyopathy, and end-stage heart failure cause downregulation of Ito. In fact, Ito downregulation is the most consistent ionic current change in the failing heart. The reduction in Ito results in attenuation of early repolarization (phase 1) and affects the level of plateau (phase 2) of the action potential and other currents involved in delayed repolarization (phase 3), with resulting prolongation and increased heterogeneity of action potential duration. The prominent epicardial Ito contributes to the selective electrical depression of the epicardium. This process leads to the development of a marked dispersion of repolarization between normal and abnormal epicardium and between epicardium and endocardium, which provides the substrate for reentrant arrhythmias and may underlie the increased predisposition to ventricular arrhythmias and SCD in patients with heart failure and ischemic heart disease.18,20,24 Additionally, downregulation of Ito in advanced heart failure likely slows the time course of force generation, thereby contributing to reduced myocardial performance.
On the other hand, compensatory ventricular hypertrophy preceding heart failure is associated with an upregulation of Ito. The prolongation of the action potential, concomitant with an increase in Ito, presumably results from the more negative level of the plateau with less ICaL inactivation and probably less delayed rectifier activation. In contrast, progression of hypertrophy to heart failure is associated with a clear reduction in Ito.20
Chronic AF reduces Ito density and Kv4.3 mRNA levels. Hypothyroidism reduces the expression of KCND2 (Kv4.2) genes. Additionally, Ito may also be reduced and contribute to QT interval prolongation in diabetes. Importantly, with some delay, insulin therapy partially restores Ito, maybe by enhancing Kv4.3 expression.3
Ultrarapidly Activating Delayed Outward Rectifying Current (IKur)
Structure and Physiology
The ion-conducting pore of IKur channels is formed by four Kv1.5 α subunits, whereas the ancillary β subunits Kvβ1.2, Kvβ1.3, and Kvβ2.1 control channel trafficking and plasma membrane integration as well as activation and inactivation kinetics.3,25
IKur activates rapidly on depolarization in the plateau range and displays outward rectification, but it inactivates very slowly during the time course of the action potential. Inactivation accelerates when Kv1.5 is coexpressed with its β subunits.3
Function
IKur is detected only in human atria and not in the ventricles, so that it is the predominant delayed rectifier current responsible for human atrial repolarization and is a basis for the much shorter duration of the action potential in the atrium.1,3
Regulation
β-Adrenergic stimulation enhances IKur, whereas α-adrenergic stimulation inhibits it, effects likely mediated by PKA and PKC, respectively. Membrane depolarization and elevated extracellular K+ concentrations reduce Kv1.5 expression. Additionally, cAMP, mechanical stretch, hyperthyroidism, and dexamethasone increase Kv1.5 expression, whereas extracellular acidosis, phenylephrine, and hypothyroidism decrease it.1,3
Pharmacology
IKur is relatively insensitive to class III antiarrhythmics of the methane-sulfonanilide group, but it is highly sensitive to 4-aminopyridine. Selective inhibition of IKur by 4-aminopyridine prolongs the human atrial action potential duration.16
IKur is a promising target for the development of new, safer antiarrhythmic drugs to prevent AF or atrial flutter, or both, without a risk of ventricular proarrhythmia. Because IKur is atrium specific, a drug specifically targeting Kv1.5 channels would be expected to terminate AF by preventing reentry through atrial action potential prolongation. The drug vernakalant is an IKur/INa channel blocker and is undergoing review by the U.S. Food and Drug Administration for the acute termination of AF. However, because Kv1.5 is downregulated in AF, the beneficial effect of IKur block becomes less certain. Furthermore, because Kv1.5 is also expressed in other organs (e.g., brain), discovery of drugs that selectively inhibit atrial Kv1.5 channels remains necessary.1,3,16
[/not-level-membership-for-cardiovascular-category]
