Chapter 414 Cardiac Development
414.1 Early Cardiac Morphogenesis
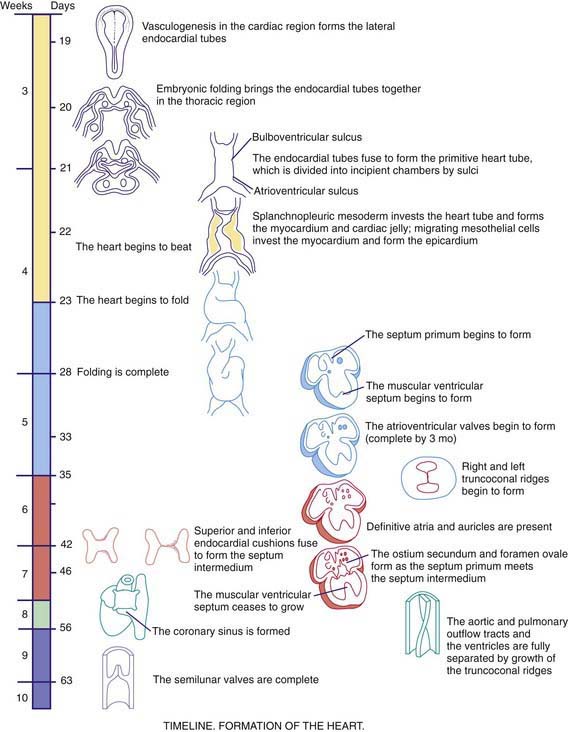
As early as 20-22 days, before cardiac looping, the embryonic heart begins to contract and exhibit phases of the cardiac cycle that are surprisingly similar to those in the mature heart. Morphologists initially identified segments of the heart tube that were believed to correspond to structures in the mature heart (Web Fig. 414-1): the sinus venosus and atrium (right and left atria), the primitive ventricle (left ventricle), the bulbus cordis (right ventricle), and the truncus arteriosus (aorta and pulmonary artery). However, this model is oversimplified. Only the trabecular (most heavily muscularized) portions of the left ventricular myocardium are present in the early cardiac tube; the cells that will become the inlet portion of the left ventricle migrate into the cardiac tube at a later stage (after looping is initiated). Even later to appear are the primordial cells that give rise to the great arteries (truncus arteriosus), including cells derived from the neural crest, which are not present until after cardiac looping is complete. Chamber-specific transcription factors participate in the differentiation of the right and left ventricles. The basic helix-loop-helix (bHLH) transcription factor dHAND is expressed in the developing right ventricle; disruption of this gene or of other transcriptional factors such as myocyte enhancer factors 2C (MEF2C) in mice leads to hypoplasia of the right ventricle. The transcription factor eHAND is expressed in the developing left ventricle and conotruncus and is also critical to their development.
414.2 Cardiac Looping
At ≈22-24 days, the heart tube begins to bend ventrally and toward the right (see Web Fig. 414-1). The heart is the 1st organ to escape from the bilateral symmetry of the early embryo. Looping brings the future left ventricle leftward and in continuity with the sinus venosus (future left and right atria), whereas the future right ventricle is shifted rightward and in continuity with the truncus arteriosus (future aorta and pulmonary artery). This pattern of development explains the relatively common occurrence of the cardiac anomalies double-outlet right ventricle and double-inlet left ventricle and the extreme rarity of double-outlet left ventricle and double-inlet right ventricle (Chapter 424.5). When cardiac looping is abnormal (situs inversus, heterotaxia), the incidence of serious cardiac malformations is high and there are usually associated abnormalities in the L-R patterning of the lungs and abdominal viscera.
414.3 Cardiac Septation
The heart tube now consists of several layers of myocardium and a single layer of endocardium separated by cardiac jelly, an acellular extracellular matrix secreted by the myocardium. Septation of the heart begins at approximately day 26 with the ingrowth of large tissue masses, the endocardial cushions, at both the atrioventricular and conotruncal junctions (see Web Fig. 414-1). These cushions consist of protrusions of cardiac jelly, which, in addition to their role in development, also serve a physiologic function as primitive heart valves. Endocardial cells dedifferentiate and migrate into the cardiac jelly in the region of the endocardial cushions, eventually becoming mesenchymal cells that will form part of the atrioventricular valves.
Complete septation of the atrioventricular canal occurs with fusion of the endocardial cushions. Most of the atrioventricular valve tissue is derived from the ventricular myocardium in a process involving undermining of the ventricular walls. Because this process occurs asymmetrically, the tricuspid valve annulus sits closer to the apex of the heart than the mitral valve annulus does. Physical separation of these 2 valves produces the atrioventricular septum, the absence of which is the primary common defect in patients with atrioventricular canal defects (Chapter 420.5). If the process of undermining is incomplete, one of the atrioventricular valves may not separate normally from the ventricular myocardium, a possible cause of Ebstein anomaly (Chapter 424.7).
Septation of the atria begins at ≈30 days with growth of the septum primum downward toward the endocardial cushions (see Web Fig. 414-1). The orifice that remains is the ostium primum. The endocardial cushions then fuse and, together with the completed septum primum, divide the atrioventricular canal into right and left segments. A 2nd opening appears in the posterior portion of the septum primum, the ostium secundum, and it allows a portion of the fetal venous return to the right atrium to pass across to the left atrium. Finally, the septum secundum grows downward, just to the right of the septum primum. Together with a flap of the septum primum, the ostium secundum forms the foramen ovale, through which fetal blood passes from the inferior vena cava to the left atrium (Chapter 415).
Septation of the ventricles begins at about embryonic day 25 with protrusions of endocardium in both the inlet (primitive ventricle) and outlet (bulbus cordis) segments of the heart. The inlet protrusions fuse into the bulboventricular septum and extend posteriorly toward the inferior endocardial cushion, where they give rise to the inlet and trabecular portions of the interventricular septum. Ventricular septal defects can occur in any portion of the developing interventricular septum (Chapter 420.6). The outlet or conotruncal septum develops from ridges of cardiac jelly, similar to the atrioventricular cushions. These ridges fuse to form a spiral septum that brings the future pulmonary artery into communication with the anterior and rightward right ventricle and the future aorta into communication with the posterior and leftward left ventricle. Differences in cell growth of the outlet septum lead to lengthening of the segment of smooth muscle beneath the pulmonary valve (conus), a process that separates the tricuspid and pulmonary valves. In contrast, disappearance of the segment beneath the aortic valve leads to fibrous continuity of the mitral and aortic valves. Defects in these processes are responsible for conotruncal and aortic arch defects (truncus arteriosus, tetralogy of Fallot, pulmonary atresia, double-outlet right ventricle, interrupted aortic arch), a group of cardiac anomalies often associated with deletions of the DiGeorge critical region of chromosome 22q11 (Chapters 417 and 418). The transcription factor Tbx1 has been implicated as a candidate gene, which may be responsible for DiGeorge syndrome. Several genes have been implicated in valve formation, including Ptpn11, which encodes the tyrosine phosphatase Shp-2, and when present in a mutated from leads to Noonan syndrome, associated with pulmonary valve stenosis; and NOTCH1, a regulator of cell differentiation that has been associated with aortic valve disease.
414.4 Aortic Arch Development
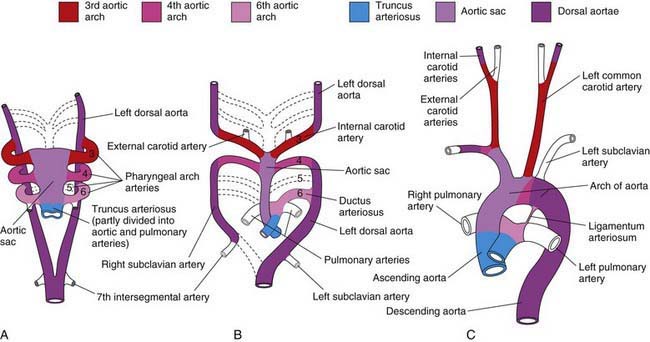
The aortic arch, head and neck vessels, proximal pulmonary arteries, and ductus arteriosus develop from the aortic sac, arterial arches, and dorsal aortae. When the straight heart tube develops, the distal outflow portion bifurcates into the right and left 1st aortic arches, which join the paired dorsal aortae (Fig. 414-1). The dorsal aortae will fuse to form the descending aorta. The proximal aorta from the aortic valve to the left carotid artery arises from the aortic sac. The 1st and 2nd arches largely regress by about 22 days, with the 1st aortic arch giving rise to the maxillary artery and the 2nd to the stapedial and hyoid arteries. The 3rd arches participate in the formation of the innominate artery and the common and internal carotid arteries. The right 4th arch gives rise to the innominate and right subclavian arteries, and the left 4th arch participates in formation of the segment of the aortic arch between the left carotid artery and the ductus arteriosus. The 5th arch does not persist as a major structure in the mature circulation. The 6th arches join the more distal pulmonary arteries, with the right 6th arch giving rise to a portion of the proximal right pulmonary artery and the left 6th arch giving rise to the ductus arteriosus. The aortic arch between the ductus arteriosus and the left subclavian artery is derived from the left-sided dorsal aorta, whereas the aortic arch distal to the left subclavian artery is derived from the fused right and left dorsal aortae. Abnormalities in development of the paired aortic arches are responsible for right aortic arch, double aortic arch, and vascular rings (Chapter 426.1).
414.5 Cardiac Differentiation
The process by which the totipotential cells of the early embryo become committed to specific cell lineages is differentiation. Precardiac mesodermal cells differentiate into mature cardiac muscle cells with an appropriate complement of cardiac-specific contractile elements, regulatory proteins, receptors, and ion channels. Expression of the contractile protein myosin occurs at an early stage of cardiac development, even before fusion of the bilateral heart primordia. Differentiation in these early mesodermal cells is regulated by signals from the anterior endoderm, a process known as induction. Several putative early signaling molecules include fibroblast growth factor, activin, and insulin. Signaling molecules interact with receptors on the cell surface; these receptors activate 2nd messengers, which, in turn, activate specific nuclear transcription factors (GATA-4, MEF2, Nkx, bHLH, and the retinoic acid receptor family) that induce the expression of specific gene products to regulate cardiac differentiation. Some of the primary disorders of cardiac muscle, the cardiomyopathies, may be related to defects in some of these signaling molecules (Chapter 433).
Developmental processes are chamber specific. Early in development, ventricular myocytes express both ventricular and atrial isoforms of several proteins, such as atrial natriuretic peptide (ANP) and myosin light chain (MLC). Mature ventricular myocytes do not express ANP and express only a ventricular-specific MLC 2v isoform, whereas mature atrial myocytes express ANP and an atrial-specific MLC 2a isoform. Heart failure (Chapter 436), volume overload (Chapters 420 and 422), and pressure overload hypertrophy (Chapter 421) are associated with a recapitulation of fetal cell phenotypes in which mature myocytes re-express fetal proteins. Because different isoforms have different contractile behavior (fast vs slow activation, high vs low adenosine triphosphatase activity), expression of different isoforms may have important functional consequences.
414.6 Developmental Changes in Cardiac Function
The sarcoplasmic reticulum (SR), a series of tubules surrounding the myofibrils, controls the intracellular calcium concentration. A series of pumps regulate calcium release to the myofibrils for initiation of contraction (ryanodine-sensitive calcium channel) and calcium uptake for initiation of relaxation (adenosine triphosphate–dependent SR calcium pump). In immature hearts, this SR calcium transport system is less well developed, and such hearts consequently have an increased dependence on transport of calcium from outside the cell for contraction. In a mature heart, the majority of the calcium to activate contraction comes from the SR. This developmental phenomenon may explain the sensitivity of the infant heart to sarcolemmal calcium channel blockers such as verapamil, which often results in a marked depression in contractility and cardiac arrest (Chapter 429).
Changes in myocardial structure and myocyte biochemistry result in easily quantifiable differences in cardiac function with development. Fetal cardiac function is less responsive to changes in both preload (filling volume) and afterload (systemic resistance). The most effective means of increasing ventricular function in a fetus is through increasing the heart rate. After birth and with further maturation, preload and afterload play an increasing role in regulating cardiac function. The rate of cardiac relaxation is also developmentally regulated. The decreased ability of the immature SR calcium pump to remove calcium from the contractile apparatus is manifested as a decreased ability of the fetal heart to enhance relaxation in response to sympathetic stimulation. This decreased ability of the immature myocardium to use preload effectively may partly explain the difficulty that most premature infants have in compensating for the left-to-right shunt through a patent ductus arteriosus Chapters 95.3 and 420.8).
Brand T. Heart development: molecular insights into cardiac specification and early morphogenesis. Dev Biol. 2003;258:1-19.
Breckpot J, Thienpont B, Peeters H, et al. Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J Pediatr. 2010;156:810-817.
Chen JN, Fishman MC. Genetics of heart development. Trends Genet. 2000;16:383-388.
Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: the scientific foundations of cardiac repair. J Clin Invest. 2005;115:572-583.
Epstein JA. Cardiac development and implication for heart disease. N Engl J Med. 2010;363:1638-1646.
Epstein JA, Buck CA. Transcriptional regulation of cardiac development: implications for congenital heart disease and DiGeorge syndrome. Pediatr Res. 2000;48:717-724.
Erdogan F, Larsen LA, Zhang L, et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridization in patients with isolated congenital heart disease. J Med Genet. 2008;45:704-709.
Gittenberger-De Groot AC, Bartelings MM, Deruiter MC, et al. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169-176.
Kathiriya IS, Srivistava D. Left-right asymmetry and cardiac looping: implications for cardiac development and congenital heart disease. Am J Med Genet. 2000;97:271-279.
Okoromah CAN. Prevalence, profile and predictors of malnutrition in children with congenital heart defects: a case-control observational study. Arch Dis Child. 2011;96:354-360.
Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922-1927.
Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287-335.
Richards AA, Santos LJ, Nichols HA, et al. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr Res. 2008;64:358-363.
Srivastava D. Genetic regulation of cardiogenesis and congenital heart disease. Annu Rev Pathol. 2006;1:199-213.
Srivastava D. HAND proteins: molecular mediators of cardiac development and congenital heart disease. Trends Cardiovasc Med. 1999;9:11-18.
Stevens KN, Hakonarson H, Kim CE, et al. Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. PLoS One. 2010;5(5):e10855.
Towbin JA, Belmont J. Molecular determinants of left and right outflow tract obstruction. Am J Med Genet. 2000;97:297-303.
Wright EMMB, Kerr B. RAS-MAPK pathway disorders: important causes of congenital heart disease, feeding difficulties, developmental delay and short stature. Arch Dis Child. 2010;95:724-730.
Zarchi O, Attias J, Raveh E, et al. A comparative study of hearing loss in two microdeletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. J Pediatr. 2011;158:301-306.



