Cardiac Arrhythmias
Sinus Bradycardia and Sinus Node Dysfunction
Vagally Mediated Sinus Arrest, Bradycardias, and Heart Block
Paroxysmal Supraventricular Tachycardia
Approach to Paroxysmal Supraventricular Tachycardia Therapy
Wolff-Parkinson-White Syndrome and Its Variants
Nonparoxysmal Atrioventricular Junctional Tachycardia, Paroxysmal Atrial Tachycardia with Block, and Automatic Atrioventricular Junctional Tachycardia
A BRIEF REVIEW OF ANTIARRHYTHMIC DRUGS
Metabolic Disturbances and Ischemia
Differential Diagnosis of Wide QRS Tachycardia
Approach to Ventricular Arrhythmias in the Critically Ill
CARDIAC ARREST AND ELECTRICAL STORM
CATHETER ABLATION OF CARDIAC ARRHYTHMIAS
PACEMAKERS AND IMPLANTABLE DEFIBRILLATORS
Critical care physicians deal with a plethora of medical and surgical problems in their patients. Arrhythmias may be the primary abnormality or may be secondary to myocardial ischemia, electrolyte imbalance, or toxic/metabolic disturbances. Optimal management of arrhythmias requires expertise in electrocardiography and clinical pharmacology as well as knowledge of arrhythmia precipitants, including proarrhythmia caused by antiarrhythmic drugs.1 The responsibility of the critical care physician is to facilitate transition from acute to chronic care by referring the patient with an arrhythmia to a cardiologist or an electrophysiologist. Therefore, the major emphasis of this chapter is on acute care of the patients with arrhythmias.
Bradycardias
Bradyarrhythmias usually present as either sinus node dysfunction (SND) or atrioventricular blockade (AV block). Bradyarrhythmias and indications and techniques for temporary cardiac pacing are reviewed extensively in Chapter 5. A brief overview is included here to highlight important issues for the intensivist.
Sinus Bradycardia and Sinus Node Dysfunction
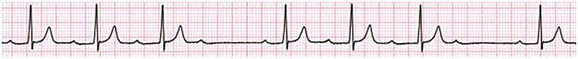
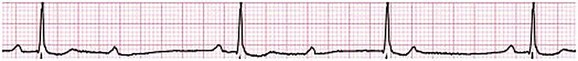
Sinus bradycardia is generally defined as periods of sinus rhythm with rates less than 60 beats per minute. In the absence of symptoms, it usually is benign and requires no treatment. Sinus bradycardia is common in young adults (particularly the physically fit). Nocturnal rates of 35 to 40 beats per minute and pauses during sleep of 2 seconds or longer are not uncommon. Sinus arrhythmia (Fig. 31.1) is a normal variant in which there are respirophasic changes in the RR interval on electrocardiogram (ECG) (prolongation of RR intervals during expiration).

Sinus bradycardia may also be a manifestation of certain pathologic conditions such as increased intracranial pressure, oculocardiac reflex after ophthalmologic surgery, cervical and mediastinal tumors, hypothyroidism, hypothermia, gram-negative sepsis, Chagas’ disease, depression, and anorexia nervosa. Sinus bradycardia is often seen after cardiac transplantation. Beta blockers, parasympathomimetic agents, calcium antagonists, amiodarone, and lithium commonly produce sinus bradycardia. Digoxin, in therapeutic doses, usually does not markedly affect the sinus node and is relatively safe to use in patients with SND.2 Sinus bradycardia complicates 10% to 15% of acute myocardial infarctions and is most common with inferior infarcts. It also may be seen after successful thrombolysis. In the absence of hemodynamic compromise, it is associated with a more favorable prognosis than sinus tachycardia.3
Sinus bradycardia (with or without AV block) may occur during periods of autonomic instability. Examples include carotid hypersensitivity and neurocardiogenic syncope. These syndromes have cardioinhibitory (bradycardic), vasodepressor (vasodilatory), and mixed forms. Permanent pacing (which must include the ability to pace the right ventricle for heart block) is a well-established therapy for cardioinhibitory carotid sinus hypersensitivity. Neurocardiogenic syncope generally is a benign condition that usually can be managed without permanent pacemaker therapy. However, treatment for patients with frequent and severe cardioinhibitory spells, especially those in whom asystolic periods exceeding 5 seconds can be demonstrated clinically or during head-up tilt table testing, may include palliative pacemaker therapy.4
Atrioventricular Block
Second-degree AV block is classified as Mobitz type I (Wenckebach), Mobitz type II, 2 : 1 AV block, or high-degree AV block. In Mobitz I (Wenckebach) block, PP intervals are constant, with gradual PR prolongation before failure of impulse conduction (nonconducted P wave) (Fig. 31.2). Although classic Wenckebach block involves simultaneous shortening of successive RR intervals before AV block, atypical alternations in RR intervals actually are more common. In younger people, Mobitz I AV block with normal QRS complexes generally is benign and does not progress to more advanced AV conduction disturbances. In older patients, the prognosis may be similar to that with Mobitz II block. Mobitz I second-degree AV block may accompany inferior myocardial infarction. The condition is benign, with a favorable prognosis, in the absence of hemodynamic compromise. The conduction disturbance usually is transient, and permanent pacing is not required.
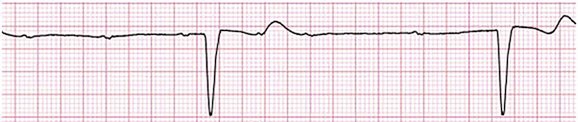
Mobitz II AV block is characterized by sudden failure of atrial impulse conduction without prior PR prolongation (Fig. 31.3). This form of AV block frequently heralds development of complete AV block and Adams-Stokes syncope. Mobitz II second-degree AV block in the setting of anterior infarction is associated with pump failure and high mortality rates. Survivors should receive permanent pacemakers.

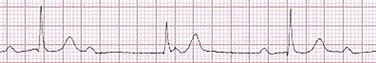
It is important to remember a few general rules to avoid common ECG misinterpretations of second-degree AV block. The 2 : 1 form of AV block may be nodal or infranodal. 2 : 1 block associated with narrow QRS complexes generally results from AV nodal block (Fig. 31.4). Wide QRS complexes are compatible with block in either the AV node or His-Purkinje system. If more than one P wave is not conducted to the ventricle, the term high-grade AV block is used (Fig. 31.5).
Complete AV block is diagnosed by the presence of independent atrial and ventricular activity on the ECG where the atrial rate is faster than the ventricular rate. When the atrial rhythm is sinus, AV dissociation is present, with the sinus rate exceeding the ventricular rate. The PP interval is constant. The RR interval is constant. The PR interval is variable in a random, nonrecurring pattern (Fig. 31.6). Complete AV block may also be present during all varieties of atrial tachycardia.
Acquired complete AV block most commonly occurs distal to the His bundle, usually is secondary to a trifasicular conduction disturbance, is potentially life-threatening, and generally is irreversible. A wide QRS escape rhythm with ventricular rates less than 40 beats per minute is the rule. An exception is seen in the setting of inferior infarction, in which recovery of complete (narrow QRS) AV nodal block occurs in greater than 90% of patients (time to recovery 30 minutes to 16 days).5
Drug toxicity, coronary artery disease, and degenerative disease of the conduction system are the most common causes of AV block in adults. Surgery, electrolyte disturbances (such as hyperkalemia), endocarditis, myocarditis (Lyme carditis), tumors, myxedema, rheumatoid nodules, Chagas’ disease, calcific aortic stenosis, polymyositis, amyloidosis, sarcoidosis, scleroderma, and vagotonic reflexes all may result in AV block. In truth, the number of factors and conditions that may result in AV block is nearly endless. “Hypervagal” responses (carotid hypersensitivity, neurocardiogenic syncope) may produce transient AV block (see later).3
Junctional Rhythm
When the sinus node does not depolarize the atrium for any reason (high vagal tone, SND), the cells near the AV junction (AV node, His bundle) may take over as the active pacemaker. Retrograde activation of the atrium, commonly manifest as negative P waves in the inferior leads (II, III, aVF), may be seen (Fig. 31.7). If the junctional rhythm goes faster than 60 beats per minute, it is termed an accelerated junctional rhythm. This is a common manifestation of digitalis toxicity.
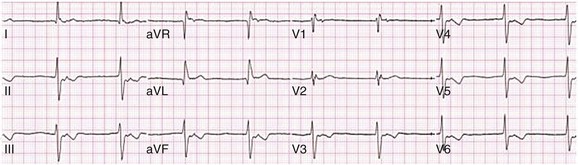
Vagally Mediated Sinus Arrest, Bradycardia, and Heart Block
The most common cause of nonconducted P waves during telemetry or Holter recordings is bradycardia-associated AV block. This manifests as sudden (usually nocturnal) block of one or more P waves with or without antecedent PR prolongation. This phenomenon is characterized by PP prolongation before AV block and is the result of transient increases in vagal tone. Vagally mediated sinus arrest, bradycardia, and heart block often occur in the intensive care unit (ICU) setting as a result of suctioning, gagging, femoral vessel compression (for hemostasis), and a variety of other triggers (Box 31.1). Vagal stimulation may lower blood pressure with or without significant bradycardia. Bradyarrhythmias and hypotension usually resolve when vagal stimulation ceases. Persistent bradycardia or hypotension mediated by vagal tone may require placing the patient in the Trendelenburg position, temporary saline infusion, or intravenous administration of atropine to fully resolve the episode.
Supraventricular Tachycardia
Overview
Tachyarrhythmias often occur in critically ill patients. Conditions such as hypoxemia, electrolyte imbalance, catecholamine excess (endogenous and exogenous), and other metabolic disturbances predispose patients (with or without preexisting arrhythmic substrates) to tachyarrhythmias. Intensivists must be prepared for acute management of supraventricular tachycardia. Knowledge of arrhythmia mechanisms, appropriate choices for acute pharmacotherapy, and indications for urgent or emergent direct current cardioversion are requisite.6 We will discuss the mechanisms of supraventricular tachyarrhythmias, give an ECG-guided approach to their diagnosis, and cover specific treatments for these dysrrhythmias (pharmacologic and catheter ablation).
Paroxysmal Supraventricular Tachycardia
AVNRT is by far the most common and in the past accounted for 50% to 60% of PSVTs evaluated at referral centers.7 The precise reentrant circuit is not defined; however, it is clear that the anterior and posterior AV nodal approaches and the perinodal atrial tissue are involved. In 76% to 90% of cases, antegrade conduction proceeds along the posterior (slow) AV nodal approach (pathway) and retrograde conduction along the anterior (fast) AV nodal pathway.7,8 This is slow-fast AVNRT. Because retrograde conduction is so rapid, atrial and ventricular activation are virtually simultaneous. P waves are usually not visible on the surface ECG or may appear in the terminal portion of the QRS complex (pseudo R′ in lead V1 or pseudo-S waves in the inferior leads). Atrial contraction on a closed AV valve may produce neck pounding.9 Less common (so-called “unusual”) variants (fast-slow, slow-slow, and slow–sort of slow) of AV nodal reentry also exist.8
AVNRT usually manifests after the age of 20 years8 and is more common in women than in men. The typical heart rate in AVNRT ranges from 150 to 250 beats per minute. Palpitations, lightheadedness, and near-syncope may accompany an episode. True syncope is unusual. Neck pounding (see previously) is virtually pathognomonic,8 but its absence does not exclude AVNRT.
Before catheter-based cures became routine, AVRT was the next most common (accounting for 30%) PSVT mechanism.7 AVRT (also commonly referred to as orthodromic tachycardia) manifests (on average) at a somewhat earlier age than that typical for AVNRT. The antegrade limb of the circuit proceeds down the normal AV nodal His-Purkinje system. The retrograde limb uses an accessory pathway that usually is located along the mitral or tricuspid valve annulus. Because the accessory pathway conducts in only retrograde fashion, it is concealed (not seen on surface ECG).
Because AVRT proceeds normally antegradely, the QRS complex is generally narrow. The AVRT reentry circuit travels antegradely through the AV node and His-Purkinje system to the ventricles before retrograde activation of the atria occurs via the bypass tract. The extra time taken to travel by way of the ventricles creates a longer RP interval during tachycardia compared with that seen in AVNRT. Because AVNRT and AVRT activate periannular atrial tissue first, P waves (if visible on surface ECG) will be negative in the inferior leads. Upright P waves in these leads indicate atrial (or sinus) tachycardia. AVRT tends to go faster than AVNRT and is more prone to manifest with QRS alternans or left bundle branch block (LBBB) aberrancy.10,11 A decrease in tachycardia rate on development of bundle branch block ipsilateral to the pathway is characteristic of AVRT. AV block is unusual during AVNRT and excludes the diagnosis of AVRT (which requires both atrial and ventricular participation). The presence of AV block strongly suggests the diagnosis of atrial tachycardia.
In the past, intra-atrial reentry, automatic atrial tachycardia, and sinus nodal reentry accounted for the remaining 8% to 10% of PSVTs.7 Sinus node reentry rarely occurs as an isolated phenomenon.12
Approximately 50% of patients with intra-atrial reentry have evidence of structural heart disease.13 This tachycardia is particularly prone to develop after surgery for congenital cardiac anomalies. Reentry occurs around structural barriers, such as suture lines. In patients without clear-cut structural disease, subtle changes such as scarring and fibrosis provide the substrate for reentry. Automatic atrial tachycardias occur along the crista terminalis, near the ostium of the coronary sinus, along the tricuspid and mitral annulus, in both atrial appendages, and within or in close proximity to the pulmonary veins. Automatic atrial tachycardias are exquisitely sensitive to catecholamines. Although these tachycardias may manifest in the absence of structural heart disease or obvious precipitants, they also are commonly associated with chronic lung disease, pneumonia, myocardial (atrial) infarction, and acute alcoholic binges. Amphetamine or cocaine abuse also may precipitate automatic atrial tachyarrhythmias.
Approach to Paroxysmal Supraventricular Tachycardia Therapy
Automatic atrial tachycardia is difficult to manage with pharmacotherapy. Precipitants should be treated or eliminated whenever possible. Beta blockers may slow atrial rate but rarely restore sinus rhythm. Adenosine may produce sinus rhythm; however, tachycardia may resume as soon as the drug is metabolized.14 Vagal maneuvers may produce AV block but do not terminate these arrhythmias. Clinical successes have been obtained with class IC agents and amiodarone. Flecainide should be avoided in patients with coronary artery disease or significant left ventricular dysfunction. Intravenous flecainide is not available in the United States (see later). Amiodarone is available for intravenous administration. Intravenous amiodarone may result in hypotension (vasodilation) but usually does not exacerbate heart failure or cause proarrhythmia in the setting of preexisting left ventricular dysfunction.
Wolff-Parkinson-White Syndrome and Its Variants
The ECG pattern of Wolff-Parkinson-White syndrome (see “Electrocardiographic Patterns Intensivists Should Recognize”), short PR interval with preexcitation (delta wave), has a reported prevalence of 0.1% to 0.3% in the general population. It is twice as common in men as in women.
Classic Wolff-Parkinson-White syndrome occurs when the accessory AV pathway is capable of bidirectional conduction (AV and ventriculoatrial). Symptomatic presentation usually is during the teenage years or early adulthood. Pregnancy may exacerbate symptoms. The most common tachycardia is AV reentry (down the AV node and His-Purkinje system, up the bypass tract), identical to AVRT involving a concealed bypass tract. Approximately 25% of patients with a Wolff-Parkinson-White ECG pattern are incapable of retrograde conduction via the accessory pathway (and therefore do not have orthodromic AVRT). Asymptomatic patients generally have a benign prognosis; however, the initial presentation may be ventricular fibrillation (see later).15
Accessory pathways generally have conduction properties similar to those of myocardium. Decremental conduction, (AV conduction delay or block) which is characteristic of the AV node, is uncommon. Pathways may therefore be capable of very rapid antegrade (AV) conduction. In these instances, atrial fibrillation may be associated with irregular wide QRS tachycardia and ventricular rates in excess of 300 beats per minute (Fig. 31.8). Syncope or SCD (degeneration to ventricular fibrillation) may ensue. Intravenous ibutilide (1 mg infused over 10 minutes; a second 1-mg dose may be given by infusion after a 10-minute wait, if necessary) also blocks antegrade accessory pathway conduction and is more likely to terminate acute episodes of atrial fibrillation or flutter (see later).16

Atriofascicular pathways (Mahaim fibers) connect the right atrium and the right bundle branch. During sinus rhythm, preexcitation is minimal or absent. Typical Mahaim reentry travels antegradely down the bypass tract and retrogradely through the normal conduction system (usually beginning with the right bundle branch). A regular wide QRS typical LBBB pattern is seen.17 These pathways conduct antegrade in a decremental fashion (retrograde accessory pathway conduction is absent) and occur much less frequently than typical AV accessory pathways. Patients with atriofascicular fibers frequently have multiple accessory pathways or AVNRT.15,18
Acute Management of Tachycardia Associated with Wolff-Parkinson-White Syndrome
Patients whose supine systolic blood pressure is greater than 90 mm Hg can be given intravenous adenosine. It is administered in the same manner as described previously. Tachycardia termination suggests a supraventricular mechanism that includes the AV node as a requisite part of the circuit. Transient AV block suggests an atrial tachycardia. No response to adenosine suggests a ventricular origin.19,20
Nonparoxysmal Atrioventricular Junctional Tachycardia, Paroxysmal Atrial Tachycardia with Block, and Automatic Atrioventricular Junctional Tachycardia
Digitalis toxicity also may precipitate atrial tachycardia (so-called paroxysmal atrial tachycardia with block) (Fig. 31.9). This tachycardia usually is managed by withholding digoxin and administering potassium. Lidocaine, phenytoin, and digoxin-specific antigen-binding fragments also may be used.
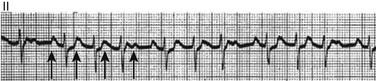
This tachyarrhythmia results in marked hemodynamic deterioration after corrective surgery for congenital heart disease. It generally appears within 12 hours postoperatively and terminates within a few days if the patient survives. Digitalis, beta blockers, and class IA antiarrhythmics are ineffective in children. Amiodarone (which may suppress tachycardia or control its rate) should be administered when rates less than 150 beats per minute cannot be achieved by other means.21 In adults, beta blockade may successfully control the rate. Adult automatic AV junctional tachycardia may be difficult to manage medically (Fig. 31.10). Recent reports suggest that catheter ablation can eliminate tachycardia while preserving AV conduction.22–24
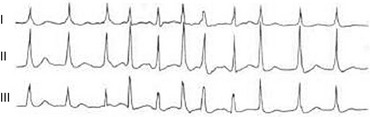
Multifocal Atrial Tachycardia
MAT is an automatic tachyarrhythmia. It is characterized by three or more morphologically distinct (nonsinus) P waves, atrial rates of 100 to 130 beats per minute, and variable AV block (Fig. 31.11).
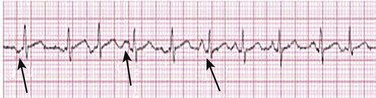
MAT is commonly associated with respiratory disease and CHF. It has been reported in patients with cancer, lactic acidosis, pulmonary emboli, renal disease, and infection. Hypoxemia frequently is present. MAT may be exacerbated by digitalis or theophylline toxicity, hypokalemia, hypomagnesemia, and hyponatremia. These precipitants usually do not result in MAT if respiratory decompensation is absent. Although MAT is (in general) an uncommon arrhythmia, it is relatively common in the critical care setting. Treatment of MAT usually is directed at elimination of the underlying precipitants. Metoprolol (used cautiously when bronchospasm is present) or verapamil may provide (atrial and ventricular) rate control and occasionally restore sinus rhythm.25,26 Potassium and magnesium supplements may help suppress MAT. Amiodarone has also been useful in restoring sinus rhythm. MAT may, superficially, resemble atrial fibrillation. Careful examination of a 12-lead ECG may be required to distinguish between these two entities. Differentiation is important for proper patient management. As noted, MAT does not respond to direct current cardioversion and is not amenable to catheter ablation.
Sinus Tachycardia
Sinus tachycardia usually is a normal reflex response to changes in physiologic, pharmacologic, or pathophysiologic stimuli such as exercise, emotional upset, fever, hemodynamic or respiratory compromise, anemia, thyrotoxicosis, poor physical conditioning, sympathomimetic or vagolytic agents, and abnormal hemoglobins.27 The resulting increase in cardiac output usually is beneficial. Heart rate generally does not exceed 180 beats per minute, except in young patients, who may achieve rates higher than 200 beats per minute during vigorous exercise.3 Tachycardia resolves when conditions return to baseline. The differential diagnosis for sinus tachycardia is presented in Table 31.1.
Atrial Flutter
Although the precise reentrant circuit is unknown, typical atrial flutter traverses (with either counterclockwise or clockwise rotation) through an isthmus formed by the inferior vena cava, tricuspid valve, eustachian ridge, and coronary sinus ostia. Counterclockwise rotation is more common and results in negative “flutter” waves in ECG leads II, III, aVF, and V6. Atrial activity in lead V1 is positively directed. Clockwise atrial flutter produces oppositely directed flutter waves in these leads. Atrial rates generally range between 250 and 350 beats per minute; however, slower rates may be seen in the presence of specific pharmacotherapy (which slows conduction within the circuit) or marked right atrial enlargement (presumably caused by a larger circuit).28 Atrial flutter usually manifests with 2 : 1 AV block and ventricular rates of approximately 150 beats per minute.
Atrial Fibrillation
Atrial fibrillation most often is associated with structural cardiac (diffuse atrial) disease. Unlike in typical atrial flutter, left atrial enlargement is more important than right atrial enlargement in the pathogenesis of atrial fibrillation.29 The chaotic ECG appearance of this arrhythmia usually is the result of shifting reentrant circuits (multiple wavelet hypotheses). Atrial fibrillation may have focal triggers (usually in one or more pulmonary veins).30 Causes of atrial fibrillation are listed in Box 31.2.
Acute Management of Atrial Fibrillation
Although atrial fibrillation is the most common sustained arrhythmia, there is no consensus on optimal atrial fibrillation management. In the critically ill, atrial fibrillation may be a “sign” (perhaps of disease severity) rather than an arrhythmic disease entity (as seen in noncritically ill patients with recurrent paroxysmal, persistent, and permanent atrial fibrillation). Critically ill patients are often in a hyperadrenergic state, which may increase ectopic triggers and shorten atrial refractoriness. This is likely the mechanism of atrial fibrillation in younger patients without chronic structural heart disease. In older patients with structural disease, increased adrenergic tone (and triggers) may precipitate atrial fibrillation when fibrosis has already created a suitable reentrant substrate.31
1. Is the diagnosis of atrial fibrillation correct?
2. Are there causes/precipitants (see Box 31.2) that can be eliminated or corrected?
3. Is it necessary to restore and maintain sinus rhythm?
4. Is atrial fibrillation causing hemodynamic impairment (angina, heart failure, hypotension)?
5. What are the potential adverse effects of the various therapeutic options?31
It is important to carefully analyze a 12-lead ECG because, as previously noted, MAT may be misinterpreted as atrial fibrillation on a rhythm strip. The treatment of choice for MAT remains correction of precipitants such as hypoxemia, digitalis or theophylline toxicity, hypokalemia, or hypomagnesemia. Atrial fibrillation may, likewise, terminate (and be less likely to recur) when precipitants are removed or corrected.31
Atrial fibrillation that does not compromise the patient may not require aggressive therapy. Rate control strategies may suffice. Spontaneous atrial fibrillation termination may be difficult to distinguish from a clear-cut benefit of specific antiarrhythmic pharmacotherapy. Hemodynamic impairment should be treated with urgent or emergent direct current cardioversion.31
Serial direct current shocks are not appropriate for recurrent (within hours or days) paroxysms (self-terminating episodes) of atrial fibrillation. This scenario is relatively common in ICUs or after cardiac surgery. It is also important to avoid repetitive, futile shocks or delivery of direct current to an inadequately sedated patient because both of these may heighten the hyperadrenergic state and create or exacerbate a downward clinical spiral.31–33 Restoration of sinus rhythm may be very difficult and impractical when a severe metabolic derangement or multisystem organ failure is present.32
Control of the ventricular rate (Table 31.2) is most frequently achieved using digoxin, beta blockers, calcium channel blockers (verapamil or diltiazem), or combinations of these agents. Verapamil should be administered cautiously to patients with significant left ventricular dysfunction. Although digoxin is effective in controlling rates at rest, exercise rate control is not often achieved. Digoxin remains appropriate therapy for patients with concomitant left ventricular dysfunction and CHF. Intravenous diltiazem is effective and well tolerated. Diltiazem may be administered by continuous intravenous infusion. The combination of efficacy, ease of parenteral delivery, and tolerance makes this agent an attractive option in the critical care setting.
Table 31.2
Intravenous Drugs for Atrial Fibrillation
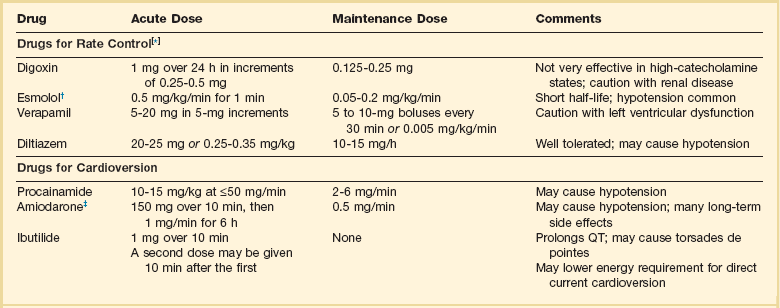
†Metoprolol and propranolol also may be used.
‡Amiodarone also is effective for rate control.
Adapted from Falk RH: Control of the ventricular rate in atrial fibrillation. In Falk RH, Podrid PJ (eds): Atrial Fibrillation: Mechanisms and Management. New York, Raven Press, 1992.
It has become relatively common to treat acute episodes with intravenous ibutilide. This unique class III agent prolongs action potential by blocking the rapid component of the delayed rectifier current.34,35 This increase results in QT interval prolongation. Patients receiving intravenous ibutilide should be carefully monitored34 (on telemetry for 4 to 8 hours) for development of torsades de pointes. Direct comparisons of intravenous procainamide and ibutilide have demonstrated clear superiority of ibutilide in conversion of atrial fibrillation and atrial flutter. Restoration of sinus rhythm with ibutilide occurred in 32% to 51% (atrial fibrillation) and 64% to 76% (atrial flutter) patients, compared with 0% to 5% (atrial fibrillation) and 0% to 14% (atrial flutter) after intravenous procainamide.34,36,37 As a result of this data, use of procainamide for this indication has become passé. Ibutilide is suitable for acute cardioversion; however, prolonged intravenous or oral dosing is not available to prevent arrhythmia recurrence. Ibutilide may be administered safely to patients on concomitant antiarrhythmic agents.38
Intravenous amiodarone is (initially) primarily a calcium channel and beta blocker. It may be effective for rate control when other agents fail. The temptation to use intravenous amiodarone to restore sinus rhythm should be tempered by knowledge of its acute electrophysiologic effects. Its class I and, particularly, class III effects take time to occur, making this a poor choice for rapid conversion. Bolus treatment with intravenous amiodarone has been very disappointing (4% conversion) for acute conversion of atrial fibrillation. By contrast, in approximately 20% to 50% of patients with persistent atrial fibrillation (lasting longer than 24 to 48 hours), reversion to sinus rhythm is achieved with sustained administration (loading periods of up to 4 weeks) of oral amiodarone.39 Intravenous amiodarone may result in less hypotension when used for rate control in the ICU than diltiazem.40,41
Intravenous class IC agents (such as flecainide and propafenone) are the most effective drugs for converting atrial fibrillation of recent onset. Unfortunately, they are not available in the United States. Ibutilide is more effective than intravenous class IC agents for restoration of sinus rhythm in atrial flutter.39 Electrical cardioversion remains the most effective way of restoring sinus rhythm in patients with atrial fibrillation. As noted, urgent electrical cardioversion should be contemplated for sustained tachycardias that precipitate angina, heart failure, or hypotension.
There are many pitfalls associated with acute atrial fibrillation management. Intravenous beta and calcium channel blockers may result in bradycardia, hypotension, and heart failure. Beta blockers may also aggravate or precipitate bronchospasm. Ibutilide may be proarrhythmic (torsades de pointes). Amiodarone is unlikely to be proarrhythmic in the absence of electrolyte abnormalities (hypokalemia, hypomagnesemia) or other drugs that have already prolonged the rate-corrected QT interval (QTc).31,34,41
In the absence of clear-cut evidence from randomized controlled trials, appropriate management should include treatment (or elimination) of potential precipitants and beta blockade (given the hyperadrenergic state of many ICU patients) as the initial pharmacotherapy of choice. If atrial fibrillation recurrence needs to be prevented and sinus rhythm maintained, institution of specific antiarrhythmic therapy (intravenous procainamide, intravenous or oral amiodarone, oral dofetilide, or sotalol) may be effective. We favor adding intravenous amiodarone. This recommendation is based on 100% bioavailability of the intravenous preparation (critically ill patients may not absorb oral drugs), amiodarone’s noncompetitive β-antagonistic effects, its benefit in the perioperative period of cardiac surgery42 (a time when endogenous and exogenous catecholamine levels are often high), and its efficacy in preventing atrial fibrillation recurrence.31,41
A recent meta-analysis of perioperative prophylactic amiodarone demonstrated decreased incidence of atrial fibrillation and flutter, ventricular tachyarrhythmias, stroke, and reduced length of stay after cardiac surgery.42 Not all studies included used beta blockade, and the course of therapy was inconsistent among trials. The Prophylactic Oral Amiodarone for the Prevention of Arrhythmias That Begin Early after Revascularization, Valve Replacement, or Repair (PAPABEAR), a large randomized controlled trial, compared perioperative amiodarone with placebo and showed significant reduction in postoperative atrial tachyarrhythmias.43 Toxicity risks were reduced because amiodarone was used for a short duration. Neither study demonstrated reduction in mortality rates. The data for perioperative amiodarone in cardiac surgery are compelling; however, incremental benefit beyond beta blockade alone remains unclear. It may still be reasonable to reserve amiodarone for postoperative atrial fibrillation in patients already receiving beta blockers and to limit use of amiodarone to 6 to 12 weeks postoperatively to prevent side effects.
Special Considerations for the Intensivist
The intensivist must balance complicated issues before undertaking direct current cardioversion. Strong effort should be focused on avoidance of low-yield attempts. Repeated doses of anesthesia and multiple shocks will ultimately result in further deterioration of critically ill patients. Optimal management of precipitants, careful choices, and monitoring of antiarrhythmic therapy, as well as a solid understanding of cardioversion and defibrillation techniques (see later), will maximize success. In some cases of atrial fibrillation, rapid ventricular rate cannot be controlled by pharmacotherapy. Radiofrequency ablation of the AV junction and permanent pacing may be required when medical therapy is ineffective.44
Anticoagulants for Stroke Prevention in Atrial Fibrillation.
Prevention of embolic strokes remains the most important goal of therapy for atrial fibrillation. Anticoagulation plays a pivotal role in minimizing the risk of emboli (and strokes) during elective cardioversion of atrial fibrillation.45 Classic recommendations for management of atrial fibrillation of more than 48 hours’ duration include 3 weeks of therapeutic warfarin (to achieve a prothrombin time [PT]/international normalized ratio [INR] of 2.0-3.0) before direct current shock administration and (at least) 4 more weeks of warfarin after the procedure. Although emboli may be less frequent with atrial flutter,45 it is clear that they occur,46 and the recommendations are the same as for atrial fibrillation. Novel oral anticoagulants have recently been introduced for stroke prophylaxis in atrial fibrillation (see later).
Anticoagulation in the ICU.
Short-term therapeutic anticoagulation with heparin before cardioversion (followed by warfarin in the usual manner) combined with transesophageal echocardiography (TEE) has gained acceptance as an alternative approach.47 Data from the ACUTE trial suggested similar embolic rates (0.5% versus 0.8%) comparing conventional and TEE-guided approaches.48
TEE is useful for detecting left atrial thrombi. It provides an excellent, minimally invasive view of the left atrial appendage. Patients with obvious thrombi should be anticoagulated for up to 8 weeks and have demonstrable resolution of clot before cardioversion is attempted.47
There are well-described risk factors that help the clinician balance the risk of anticoagulation and the risk of stroke from atrial fibrillation. The CHADS2 and CHA2DS2-VASc (Congestive heart failure, Hypertension, Age ≥ 75 years [doubled risk weight], Diabetes mellitus, previous Stroke/transient ischemic attack [doubled risk weight], Vascular disease, Age 65 to 74 years, female Sex) risk scores as well as a novel bleeding risk score, HAS-BLED (Hypertension, Abnormal renal/liver function, Stroke history, Bleeding history or predisposition, Labile international normalized ratio, Elderly [≥65 years], Drugs/alcohol concomitantly) aid the clinician in balancing a patient’s embolic stroke risk with the risk for bleeding.49 Patients who are younger than age 65 years with normal hearts and “lone” atrial fibrillation (i.e., with none of the aforementioned risk factors) can be anticoagulated with aspirin 325 mg daily or perhaps not at all. Patients with 1 point should have individualized treatment, and patients with 2 points should be anticoagulated. Patients with rheumatic mitral stenosis or the presence of a prosthetic heart valve are among the highest risk for stroke and should be anticoagulated regardless of the CHADS2 score.50
Recently, two classes of drugs, the direct thrombin (factor IIa) inhibitors and the factor Xa inhibitors (collectively termed “novel anticoagulants”) have emerged as options for prophylaxis against stroke in patients with nonvalvular atrial fibrillation. As a majority of patients in atrial fibrillation are willing to consider switching to these medications, intensivists will undoubtedly encounter patients on these drugs and deal with issues that arise from their use.51
Dabigatran, a direct thombin inhibitor, is an oral anticoagulant dosed twice a day. The drug was studied in comparision to warfarin for reduction of strokes in patients with atrial fibrillation. The RE-LY trial found a decreased incidence of stroke with the 150 mg dose of dabigatran and similar risk of bleeding.52 Dabigatran’s half-life is 14 to 18 hours and it is recommended that it be stopped 2 to 3 days prior to an elective surgery (4 to 5 days seems more prudent). There is no specific antidote to this drug. Local measures may suffice for minor bleeding. Dialysis and dabigatran’s relatively short half-life usually allow discontinuation of the drug to reverse the bleeding diathesis. The only current reversal option for dabigatran is emergency dialysis. Performing dialysis rapidly in unstable patients with bleeding or in those with large intracranial hemorrhage will present a very great challenge, even at level 1 trauma centers.53
In addition to thrombin (factor IIa), the coagulation factor Xa is a target for the novel anticoagulants. In the United States rivaroxaban and apixaban are approved for the treatment of atrial fibrillation, and additional drugs are being studied. The ROCKET-AF trial demonstrated that the once-daily drug rivaroxaban was noninferior to warfarin in reducing stokes with similar bleeding rates.54 The AVERROES trial demonstrated apixaban was superior to aspirin in patients in whom warfarin was unsuitable.55 Later, the ARISTOTLE trial demonstrated that apixiban was also superior to warfarin and caused less bleeding.56 As with the direct thrombin inhibitors, there is no specific reversal agent for factor Xa inhibitors.
The factor Xa inhibitors have a shorter half-life and they are generally stopped the day prior to a surgery.57 As mentioned, there has been little evidence to support any specific strategy of reversal of anticoagulation, and the rare nature of this problem makes randomized trials on the problem difficult to perform. Preliminary evidence suggests that prothrombin complex concentrate (PCC) immediately and completely reverses the anticoagulant effect of rivaroxaban in healthy subjects.58 PCC improved laboratory parameters but did not reverse apixaban-induced bleeding in a rabbit model.59
Direct Current Cardioversion
Deep Sedation for Cardioversion
Direct current shocks should never be administered to a conscious patient. A high-energy shock delivered to an awake patient may result in lifelong emotional trauma and has been appropriately termed a calamity.60
Our preferred drug for deep sedation before cardioversion is propofol.61 Dosing must be individualized. A bolus of 0.6 mg/kg usually is effective for routine elective cardioversion but may be excessive in a critically ill patient.62 Propofol’s adverse effects include apnea, bradycardia, hypotension, nausea, and pain and burning at the intravenous injection site that can be minimized by giving local lidocaine at the site. Overdose is treated with ventilation and oxygen, elevation of the legs, increasing flow rates of intravenous fluids, and administration of pressor agents and anticholinergic agents.
Regardless of who administers sedation, expert ability to manage the patient’s airway must be immediately available. Electrophysiology laboratory nurses are often quite capable of administering deep sedation.63 We recommend that an anesthesiologist be present for high-risk patients.
Technical Aspects
When the capacitors of a defibrillator charge, the device becomes capable of energy delivery (measured in watt-seconds or joules [J]). The energy is composed of voltage and current. Transthoracic current flow is partially determined by electrode placement. A variety of configurations has been employed. We have favored an anteroposterior (parasternal and left infrascapular) pathway for cardioverting atrial fibrillation and flutter and other atrial arrhythmias. This configuration provides the best vectors for energy delivery to the atria64 (Fig. 31.12). We also have found it to be optimal for patients with an implantable cardioverter-defibrillator (ICD) and epicardial patches.65
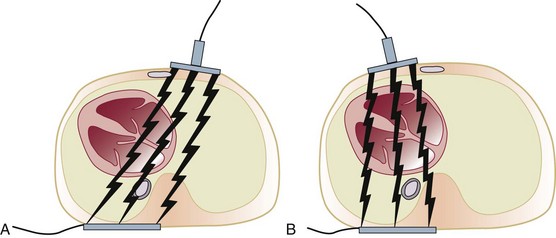
Although it was common to recommend an initial monophasic energy of 100 J for atrial fibrillation (with initial success rates of 50%), we agree with Ewy and begin with 200 J.64 An initial monophasic energy of 360 J for atrial fibrillation lasting longer than 48 hours also has been suggested.64,66 A similar recommendation of 200 J also applies to biphasic waveforms, particularly for cardioversion in patients with atrial fibrillation of long duration.47 Optimal monophasic energy delivery for cardioversion of atrial flutter was 100 J.67 We generally use 100 J for biphasic cardioversion of atrial flutter as well.
Determinants of Short- and Long-Term Success of Cardioversion
Transthoracic impedance influences current flow and procedural outcome. Current flow is inversely related to impedance. Impedance is influenced by a variety of factors. These factors include the phase of ventilation (impedance is lower with expiration than with inspiration), distance between electrodes, pressure on electrodes (air does not conduct well), effect of previous discharges (decreased impedance), time between discharges (waiting as long as 3 minutes may provide continued decreases in impedance), and patient body habitus (heavier weight or increased body mass index will decrease success).64,68
As noted, modern defibrillators have biphasic waveforms that are more effective per joule output than monophasic waveform defibrillators. Electrode size is also an important determinant of transthoracic impedance. Self-adhesive pads commonly are used in high-risk patients. They are easy to position precisely. Transthoracic impedance may be higher (70 to 100 ohms) with these pads compared with metal electrodes (50 ohms).64 Optimal paddle size ranges from 8 to 12 cm. A conductive gel or paste must be used between the metal electrodes and the chest skin.64 Smearing gel between paddles may deflect energy away from the heart.69 A switch from from self-adhesive pads to paddles with pressure is a simple method to increase current delivered per shock and to promote procedural success.
A Brief Review of Antiarrhythmic Drugs
Initiation of chronic oral antiarrhythmic drug therapy for tachyarrhythmias usually is in the realm of practice of general cardiologists and electrophysiologists. Nevertheless, intensivists should be familiar with these agents, as well as current philosophies for their use. The older class IA drugs, including quinidine, procainamide, and disopyramide, now are rarely used because of the risk of QT prolongation and torsades de pointes, low efficacy rates, and poor noncardiac side effect profiles resulting in high discontinuation rates.48,70 Antiarrythmic drugs in common use are described next.
Propafenone (class IC) is an equally effective drug for control of supraventricular tachycardia and atrial fibrillation. Newly formulated sustained-release propafenone has more convenient twice-daily dosing, starting at 225 mg orally twice daily, titrated up to 425 mg twice daily if necessary. Sustained-release propafenone was safe and effective in a large outpatient trial (RAFT).71 Side effects are similar to those of flecainide, with the addition of a “metallic taste in the mouth” (an unusual noncardiac side effect), which does not require drug discontinuation. Propafenone or flecainide can be beneficial on an outpatient basis in patients with paroxysmal atrial fibrillation who are in sinus rhythm when the drug is initiated.72 Propafenone has mild β-blocking properties (more prominent in slow metabolizers) but usually not enough to rely on for AV blockade in atrial fibrillation or atrial flutter. Like flecainide, propafenone should not be used in patients with significant structural heart disease.
Amiodarone is a class III antiarrhythmic with sodium, potassium, and calcium channel blocking properties. In addition, it is a noncompetitive beta blocker and inhibits peripheral conversion of thyroxine (T4) to triiodothyronine (T3). It is considered the best atrial fibrillation rhythm control drug and generally regarded as the most effective antiarrhythmic agent for all tachyarrhythmias.41 It is an excellent AV node–blocking drug. Even when the drug fails to achieve AF rhythm control, it usually will provide good rate control. Amiodarone has an extremely long half-life (a virtue for compliance, but a liability if side effects occur). Like dofetilide, it can be used in patients with low ejection fraction at risk of heart failure exacerbation. Amiodarone does not adversely affect survival and in some studies offered some protection against arrhythmia-related sudden death.65–67 It can cause significant bradycardia but is associated with a very low incidence of ventricular proarrhythmia (less than 1%), despite the fact that it prolongs the QT interval. The drug requires an initial loading dose (regimens vary). A maintenance dose in the range of 200 mg (or less) per day usually can be achieved and is helpful in minimizing side effects. Occasionally, maintenance doses of 400 mg per day are required for tachycardia control. Amiodarone’s long-term noncardiac side effects are significant and should limit its use in younger patients. Nevertheless, amiodarone is the most frequently prescribed specific antiarrhythmic drug in the United States. Side effects include pulmonary toxicity, polyneuropathy, photosensitivity, bradycardia, hepatic dysfunction, thyroid dysfunction, and ophthalmologic complications. Side effects are correlated with maintenance dose and duration of therapy. Baseline pulmonary function studies with diffusing capacity, chest radiograph, liver and thyroid function studies, and eye examination should be performed before or shortly after the drug is started. We recommend thyroid function tests, liver function tests, and a chest radiograph every 6 months to look for signs of toxicity. It is particularly disturbing that pulmonary toxicity (which may be fatal) may be missed even with careful surveillance.
Dronedarone is a derivative of amiodarone developed for the treatment of atrial fibrillation and atrial flutter. Like amiodarone, dronedarone is a potent blocker of multiple ion currents, including the rapidly activating delayed-rectifier potassium current, the slowly activating delayed-rectifier potassium current, the inward rectifier potassium current, the acetylcholine activated potassium current, peak sodium current, and the L-type calcium current. Dronedarone also exhibits antiadrenergic effects. Dronedarone has been studied for maintenance of sinus rhythm and control of ventricular response during episodes of atrial fibrillation. Dronedarone reduces mortality rate and morbidity in patients with high-risk atrial fibrillation but may be unsafe in patients with persistent or permanent atrial fibrillation as well as individuals with severe heart failure.73,74
The AFFIRM trial randomized over 4000 patients to receive rate or rhythm control treatment strategies (plus warfarin sodium in both treatment groups). Because AFFIRM demonstrated no significant stroke, quality of life, or mortality rate differences with rhythm versus rate control, physicians must consider the risk-benefit ratio of antiarrhythmics to maintain sinus rhythm.75–77 Patients with brief or minimally symptomatic recurrences of paroxysmal atrial fibrillation often do not require antiarrhythmic drugs. Patients with troublesome symptoms generally require suppressive antiarrhythmic therapy. Rate control and prevention of thromboembolism are appropriate in both situations.50
The 2011 ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation recommend that antiarrhythmic drugs for rhythm control of paroxysmal atrial fibrillation be chosen using an algorithm (Fig. 31.13).50 This algorithm takes into account whether or not the patient has structural heart disease, hypertension with or without “significant” left ventricular hypertrophy (echo wall thickness of 1.4 cm or greater), or coronary artery disease. If there is no structural heart disease, or minimal left ventricular hypertrophy only, the therapeutic agents of first choice are flecainide, propafenone, and sotalol. Second-line therapeutic options include amiodarone and dofetilide. If significant left ventricular hypertrophy is present, the treatment of first choice is amiodarone therapy. If coronary artery disease is present, the treatment of choice is sotalol (because of its β-blocking properties), with second-line therapies including dofetilide and amiodarone. If clinical heart failure is present, first-line agents are amiodarone and dofetilide.
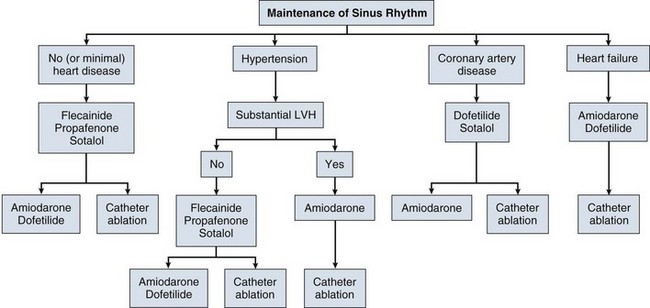
Catheter ablation is recommended for patients with symptomatic paroxysmal atrial fibrillation refractory to or intolerant of at least one antiarrythmic medication. Catheter ablation is considered reasonable for symptomatic paroxysmal atrial fibrillation prior to initation of antiarrythmic drug therapy or symptomatic persistent atrial fibrillation refractory to or intolerant of at least one antiarrythmic medication. Catheter ablation may be considered for symptomatic persistent or longstanding persistent atrial fibrillation prior to initation of antiarrythmic drug therapy and for longstanding persistent symptomatic atrial fibrillation refractory to or intolerant of at least one antiarrythmic medication.78
Ventricular Arrhythmias
The intensivist must be prepared to recognize and participate in the management of ventricular arrhythmias. However, long-term management usually is prescribed by cardiologists and electrophysiologists. Therefore this section concentrates on the intensive care and emergency treatment of ventricular arrhythmias and only briefly addresses chronic therapy. The emergency treatment of cardiac arrest is covered in Chapter 1.
Arrhythmogenesis
Triggered activity occurs when oscillations in membrane potential (called afterdepolarizations) reach the threshold for action potential formation, resulting in abnormal impulse formation. Early afterdepolarizations (EADs) result from delayed inactivation of inward ion currents during the plateau phase of the action potential. EADs appear to be important in the genesis of torsades de pointes (see later). Delayed afterdepolarizations (DADs) result from increased intracellular calcium. Digitalis toxicity is the classic example of DAD-induced triggered activity. Digitalis inhibits the Na+-K+ pump, thereby increasing intracellular Na+, which in turn increases intracellular Ca2+ via the Na+-Ca2+ exchange current.79 Idiopathic outflow tract ventricular tachycardia may also result from DADs (cyclic AMP-mediated triggered activity).80
Metabolic Disturbances and Ischemia
Further treatment of hyperkalemia involves removal of potassium from the body. The most common method is administration of the cation-exchange resin sodium polystyrene sulfonate (Kayexalate). The usual dose is 50 g given two or three times daily. The most effective means of reducing body potassium is dialysis. Care must be taken to avoid precipitous reduction, especially in patients taking digitalis glycosides.81 ECG patterns associated with electrolyte and metabolic disturbances are discussed in more detail later in this chapter.
Myocardial ischemia, from coronary artery disease, hemodynamic deterioration, or hypoxemia, can produce a variety of electrophysiologic effects. Acid-base disturbances and exogenous or endogenous catecholamines also can predispose affected patients to ventricular (often polymorphic) arrhythmias. The role of the sympathetic nervous system in arrhythmogenesis and electrical storm cannot be overemphasized.79,82,83 Reentry, enhanced automaticity, and triggered activity may all be provoked.
Differential Diagnosis of Wide QRS Tachycardia
Wide complex tachycardia (WCT) may be supraventricular or ventricular in origin. Evaluation of WCT remains a common dilemma for clinicians. Numerous algorithms aid in arriving at the correct diagnosis. Unfortunately they are difficult to remember and an overreliance on algorithms may prevent clinicians from understanding the underlying arrhythmic mechanisms. Presence of AV dissociation is diagnostic of ventricular tachycardia. In general, supraventricular tachycardia with aberrancy will manifest with a typical bundle branch block pattern, whereas ventricular tachycardia will have more “bizarre” QRS complexes. Preexcited supraventricular tachycardias may be impossible to distinguish from ventricular tachycardia using ECG criteria alone. Algorithms (with good sensitivity and specificity) to help distinguish supraventricular tachycardia from ventricular tachycardia are summarized in Table 31.3.83
Table 31.3

*Ventricular activation-velocity ratio (vi and vt = initial and terminal 40 ms, respectively, of QRS complex).
From Neiger JS, Trohman RG: Differential diagnosis of tachycardia with a typical left bundle branch block morphology. World J Cardiol 2011;3:127-134.
Approach to Ventricular Arrhythmias in the Critically Ill
A comprehensive discussion of advanced cardiovascular life support (ACLS) is beyond the scope of this chapter. Current recommendations for management of adult cardiac arrest are discussed in Chapter 1 of this book and are summarized in recently published ACLS guidelines.84
Specific Ventricular Arrhythmias
The approach to NSVT that persists after recovery from acute illness depends on the underlying cardiac substrate. Asymptomatic persons without structural disease require no therapy. Patients with reduced left ventricular function (less than or equal to 35%) should be evaluated for ICD therapy.85–87 Amiodarone, which has a neutral effect on mortality rates, may be used to suppress symptomatic NSVT in patients with significant structural heart disease.85
Acute management of sustained monomorphic ventricular tachycardia depends primarily on its hemodynamic stability. Unstable tachycardia should be treated promptly with direct current cardioversion. Stable arrhythmias may be treated with intravenous amiodarone or beta blockers. Although lidocaine remains an option, the International Cardiopulmonary Resuscitation (CPR) guidelines no longer recommend lidocaine as a first-line agent. Antitachycardia pacing may be considered in patients with transvenous or epicardial pacing leads. If these modalities are unsuccessful (or arrhythmia acceleration occurs), direct current cardioversion can be used to restore sinus rhythm. Electrical storm is defined as ventricular tachycardia or ventricular fibrillation occurring more than twice in 24 hours, usually requiring electrical cardioversion or defibrillation.88 Although data are limited, beta blockade in conjunction with amiodarone appears to be the most effective therapy for electrical storm.82,88
Because the arrhythmic substrate usually is fixed (most commonly, coronary artery disease and prior myocardial infarction), recurrence rates may remain high even after elimination of “reversible” causes.89,90 Although no definite guidelines exist in the presence of reversible causes, an ICD should be strongly considered in patients with left ventricular dysfunction (if comorbidity is not prohibitive). Electrophysiologic testing may be useful in patients with coronary artery disease but is much less reliable in patients with nonischemic cardiomyopathy.
Bundle branch reentry has been reported to account for up to 6% of the cases of monomorphic ventricular tachycardia.91 This percentage may rise to 40% to 50% in patients with idiopathic dilated cardiomyopathy.92 It has been our clinical experience that this arrhythmia is far less frequent. Nevertheless, bundle branch reentry should be considered in patients with marked left ventricular dysfunction (especially nonischemic), intraventricular conduction defects, and wide QRS tachycardia. It may appear after valve replacement surgery. The tachycardia circuit typically uses the right bundle branch as its antegrade limb and the left bundle branch as its retrograde limb. Tachycardia therefore manifests with classic LBBB morphology. Diagnosis and treatment may be accomplished during a single invasive electrophysiology session. The right bundle branch is easily ablated during sinus rhythm, permitting tachycardia cure without detailed mapping during hemodynamically unstable arrhythmias.93,94 Although the postablation prognosis has been said to be favorable for patients with isolated bundle branch reentry, patients with residual inducible or spontaneous ventricular tachycardia should be offered ICD therapy. Patients with significant residual infranodal conduction delay (His-ventricular rates longer than 90 ms) after ablation should be considered for permanent pacing (usually with an ICD). It is appropriate to implant an ICD after ablation in patients with heart failure and ejection fractions less than 35%.
Optimal long-term management of patients with structural heart disease and sustained monomorphic ventricular tachycardia is to implant an ICD.95,96 Hemodynamic stability does not predict a better long-term outcome.95 Ablation generally is regarded as palliative and is used primarily to reduce shock frequency in patients with recurrent ventricular tachycardias.97 However, substrate-based catheter ablation may reduce the ICD therapies in post-myocardial infarction patients who received ICDs for secondary prevention.98
The risk of recurrent polymorphic ventricular tachycardia/ventricular fibrillation is high, and the long-term prognosis is poor. Most patients, particularly those with left ventricular dysfunction, should have an ICD implanted if no contraindications exist. Idiopathic ventricular fibrillation may respond to catheter ablation if a single PVC focus (usually from the Purkinje system or the right ventricular outflow tract) is the consistent trigger.99,100 Coronary artery spasm may result in ventricular fibrillation caused by myocardial ischemia. Recognition of this uncommon cause of cardiac arrest is critical. Ideal treatment for patients with coronary spasm and associated ventricular arrhythmias remains controversial. Titration of calcium channel blocker dose to prevent ergonovine-induced spasm eliminated arrhythmia in one small series.101 Despite optimal medical therapy (nitrates and calcium channel blockers), this patient subgroup falls into a high-risk category of vasospastic angina and appears to be at greater risk for sudden death. Concomitant ICD implantation has been advocated to reduce this risk.102
Less Common Substrates
Idiopathic Ventricular Tachycardia
Idiopathic ventricular tachycardias tend to originate in a “line of fire” from the right ventricular outflow tract (90%), left ventricular outflow tract, aortic cusps, and mitral annulus.103 They often are facilitated by catecholamine infusion. The most common forms (right ventricular outflow tract tachycardia) have a typical, easily recognizable ECG pattern of LBBB with an inferior frontal lead axis (tall R waves in leads II, III, and aVF). These arrhythmias occur in the absence of apparent structural heart disease. Abnormalities may be detected using magnetic resonance imaging; however they do not definitely correlate with sites of arrhythmogenesis.104,105 Very frequent episodes may result in a tachycardia-mediated cardiomyopathy.106
More than 90% of idiopathic ventricular tachycardias can be cured by catheter ablation (Figs. 31.14 and 31.15).106,107 The most reliable method for localizing the site of origin is pace mapping. The 12-lead ECG will exactly match the spontaneous ventricular tachycardia QRS morphology at the site of origin of the ventricular tachycardia. If a perfect 12/12-lead ECG pace map match can be obtained, the site is ablated. These tachycardias are adenosine-sensitive and thought to be the result of cyclic AMP–mediated DADs.80 These arrhythmias may respond to treatment with adenosine and chronically to beta blockers or calcium channel blockers, which normally are ineffective in other ventricular tachycardias.
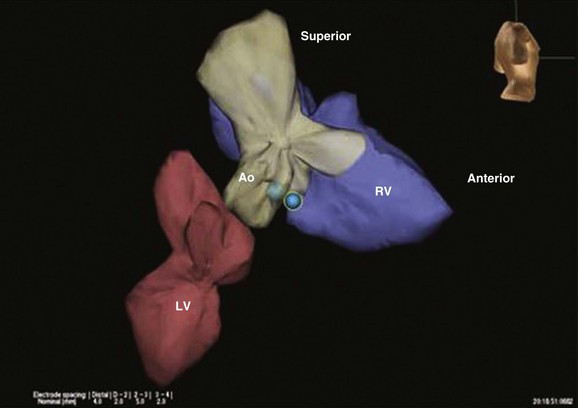
Another “idiopathic” left ventricular tachycardia manifests as a relatively narrow right bundle branch block (RBBB), left axis deviation tachycardia. The ECG and rhythm strip should be examined carefully for P waves. If the PP interval is slower and the P waves are dissociated, the diagnosis of ventricular tachycardia, rather than supraventricular tachycardia with aberrancy, is confirmed. This arrhythmia is verapamil-sensitive and can easily be ablated if necessary. The arrhythmia is due to macroentry in the terminal Purkinje fibers in the left distal third of the apical septum (Fig. 31.16). To ablate it, the lower third of the septum is mapped, looking for the sharpest, earliest Purkinje potential during ventricular tachycardia.108 A second technique (also using Purkinje potentials) is equally effective.109 This reentrant tachycardia is referred to by several different names, including fascicular ventricular tachycardia, verapamil-sensitive ventricular tachycardia, and Belhassen’s ventricular tachycardia.
In patients with ventricular arrhythmias and obvious significant right ventricular disease, the diagnosis of arrhythmogenic right ventricular dysplasia (ARVD)/cardiomyopathy can be made. The left ventricle generally has milder abnormalities. ARVD typically occurs in young patients (80% of patients are diagnosed before the age of 40 years) and is an important cause of SCD in this population. It should, however, be emphasized that the overall risk of SCD is low (2% to 2.5% per year).110
Males are predominantly affected. ARVD is transmitted in an autosomal dominant pattern with variable penetrance (abnormal loci have been mapped to chromosomal regions 14q23, 1q42, 14q12, 2q32, 17q21, and 3p23).110 Immunohistochemical analysis of endomyocardial biopsy samples revealing a diffusely reduced plakoglobin signal level appears to be a highly sensitive and specific test for ARVD.111
Ventricular arrhythmias in ARVD may be catecholamine dependent and are exacerbated during exercise tolerance testing in 50% of patients. Sotalol and amiodarone seem to be effective in ARVD. Catheter ablation has a palliative, complementary role. Arrhythmia recurrences at new foci may occur after apparent ablative success. Experience with ICDs in ARVD is limited. Patients resuscitated from cardiac arrest or those poorly responsive to (or intolerant of) antiarrhythmic drugs appear to be good candidates.112
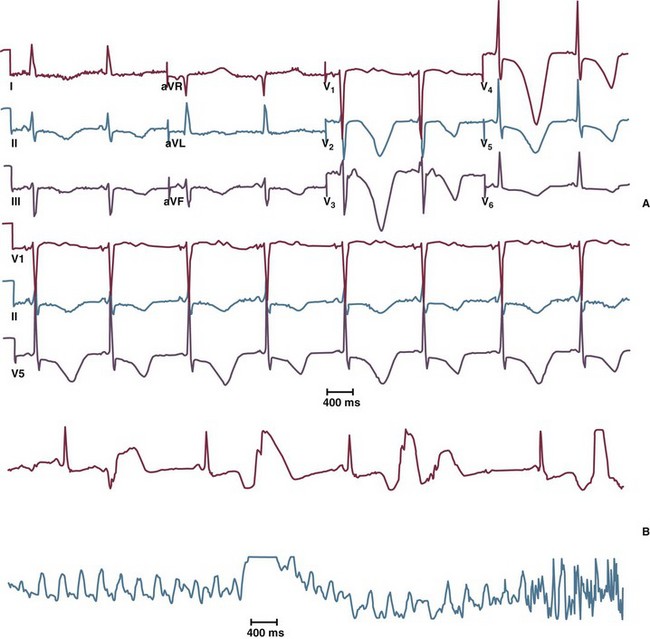
The congenital long QT syndrome is a manifestation of a variety of ion channel mutations that result in prolonged ventricular repolarization. The three main features of congenital long QT syndrome are (1) prolongation of the rate-corrected QT interval (QTc); (2) cardiac arrest secondary to torsades de pointes (Fig. 31.17); and (3) QT prolongation, syncope, or sudden death in family members. Syncope often occurs in association with physical activity, emotional reactions, or acute arousal with auditory stimuli (the specific trigger in a variant of long QT syndrome, LQT2).113,114 Beta blockers are the mainstay of treatment in patients with long QT syndrome. Permanent pacing (ideally via an ICD) may be beneficial for patients in whom beta blockade is not effective or in whom excessive bradycardia develops. Limited experience has been reported with left cervicothoracic sympathetic ganglionectomy in patients with drug-refractory long QT syndrome and surgical expertise is available in only a few centers. ICDs are recommended for high-risk patients, including those with recurrent syncope on beta blockers, aborted SCD, a strong family history of sudden death, and the Jervell and Lange-Nielsen syndrome (homozygotes or compound heterozygotes with mutations in KCNQ1 and KCNE1, resulting in abnormal Iks ion current long QT syndrome and hereditary deafness).115
Although the short-term effects of gene-specific therapy (e.g., mexiletine or flecainide in patients with sodium channel abnormalities, potassium plus spironolactone in potassium channel defects) on the QT interval are encouraging,116 long-term data are lacking regarding their ability to prevent arrhythmias in long QT syndrome. A trial of flecainide for another variant of the syndrome, LQT3, is ongoing.
QT prolongation may be acquired (most commonly caused by drug effects or toxic substances, electrolyte abnormalities, hypothermia, and central nervous system injury). Drug-induced QT prolongation usually is the result of Ikr ion current blockade.35 Intensivists need to be particularly aware of the pharmacologic causes of QT prolongation (Box 31.3). Drug-induced torsades de pointes is managed initially with intravenous magnesium sulfate. Isoproterenol and temporary pacing increase ventricular rates, shorten QT intervals, and help prevent recurrent arrhythmias until the effects of the offending agent diminish.
The Brugada syndrome (first described in 1992) is characterized by RBBB with ST-segment elevation in leads V1 to V3, polymorphic ventricular tachycardia, and ventricular fibrillation.117 Intensivists need to be particularly aware that febrile illnesses may trigger arrhythmic events.118 Brugada syndrome has been linked to mutation in the sodium channel gene SCN5A. This mutation decreases sodium channel activity. It is inherited in an autosomal dominant pattern with variable penetrance. Males are more likely to be affected and have an increased risk of sudden death, probably related to a more prominent transient outward potassium current. The ECG abnormality originally was thought to be persistent; however, transient forms (in which the ECG may be normal for periods of time) have been described.119 The electrocardiographic abnormalities may be unmasked by procainamide, flecainide, or ajmaline.
The cellular mechanism responsible for the ST-segment elevation is early repolarization of the ventricular epicardium as a result of rebalancing of currents at the end of phase I of the action potential. The transient outward potassium current (Ito) overwhelms inward currents. The action potential “dome” is abolished at some sites but not others. Propagation of the dome to sites where it is absent may result in so-called phase II reentry, the mechanism of arrhythmogenesis.120 In this instance, diminished sodium channel activity facilitates loss of the action potential dome as a result of a negative shift in the voltage at which phase I begins. Different mutations in SCN5A appear to account for LQT3; however, a recent report suggests a genetic (and perhaps clinical) link between the Brugada syndrome and LQT3.121 As with long QT syndrome, it appears that genetic heterogenicity exists in the Brugada syndrome.122 In Japan and Southeast Asia, the Brugada syndrome may account for 40% to 60% of cases of idiopathic ventricular fibrillation. The ICD is the only effective therapeutic intervention against SCD.
Catecholaminergic polymorphic ventricular tachycardia (CMPVT) typically manifests in childhood as syncope or aborted cardiac arrest. Young boys have the worst prognosis (perhaps they are more sensitive to adrenergic stimulation). Beta blockers are the cornerstone of therapy, and dosing may be titrated according to exercise response. In 40% of patients, arrhythmia control will remain inadequate despite dose optimization during repeat exercise testing. ICDs are the therapeutic option of choice in these patients.123
Short-coupled torsades de pointes (SC-TdP) occurs in patients with structurally normal hearts and unremarkable ECG tracings (normal QT intervals). The coupling interval of the initiating beat is invariably less than 300 ms. It is a rare, potentially fatal disorder whose pathophysiology is unknown. The prognosis is poor, and effective pharmacologic therapy has not been identified. ICDs may be the best option. SC-TdP shares features with idiopathic ventricular tachycardia, and speculation that it may respond to catheter ablation if a single PVC focus (usually from the Purkinje system) is the consistent trigger is not unreasonable.124,125
Short QT syndrome (SQTS) is a heritable primary electrical disease characterized by an abnormally short QT interval (less than 300 ms) and a propensity to atrial fibrillation or SCD, or both. As in the long QT syndrome, more than one relevant genetic mutation has been identified. Shortening of effective refractory periods combined with increased dispersion of repolarization is the likely substrate for reentry and life-threatening tachyarrhythmias. The best form of treatment is still unknown, but prevention of atrial fibrillation has been accomplished with propafenone. Implantation of an ICD is recommended for prevention of SCD.126,127
Patients who experience electrical shock (including lightning strikes) sustain a wide spectrum of injuries with unique pathophysiologic characteristics that require special management. Patients with serious burns admitted to the ICU are trauma patients and should be treated accordingly. Initial prediction of outcome for patients who have experienced electrical shock is difficult, because the full degree of injury often is not apparent. SCD due to ventricular fibrillation is more common with low-voltage alternating current, whereas asystole is more frequent with electric shocks from direct current or high-voltage alternating current. Potentially fatal arrhythmias are more likely to be caused by horizontal current flow (hand to hand); current passing in a vertical fashion (from head to foot) more commonly causes myocardial tissue damage. Lightning strike is unique because it causes cardiac and respiratory arrest, resulting in a 25% to 30% mortality rate.128
Aggressive and prolonged CPR in patients who have experienced electrical shock is indicated for several reasons.129 First, cardiac arrhythmias and prolonged respiratory arrest may be the only clinical problem, especially in patients struck by lightning. Second, as mentioned, patients who experience electrical shock commonly are young and have few or no comorbid conditions. These young patients may survive prolonged CPR with no or minor sequelae. It is important to remember that keraunoparalysis leading to autonomic dysfunction may masquerade as irreversible neurologic injury in patients who have been electrocuted. For practical purposes, guidelines for CPR as issued by the American Heart Association84 still apply. The algorithm for asystole acknowledges that “atypical clinical features” need to be considered in deciding whether CPR should be continued after initial unsuccessful attempts.
If more than one person has been electrocuted at a scene of injury, standard triage practices need to be modified, especially in those struck by lightning. Most patients who do not experience cardiac or respiratory arrest will survive.130 Thus, the usual triage principles should be reversed: First responders should focus initially on patients who appear clinically dead before patients who show signs of life are treated.
Cardiac Arrest and Electrical Storm
It is estimated that out of hospital sudden cardiac arrest (SCA) tragically ends over 300,000 lives in the United States131,132 and is responsible for 3 million deaths worldwide each year.133 Although the number of age-adjusted cardiovascular deaths has declined during the last 50 years, the proportion that are sudden has remained relatively constant (~50%).134,135 SCA claims more lives each year than stroke, lung cancer, breast cancer, and AIDS combined.136–138
SCD is defined as natural death due to cardiac causes, foreshadowed by abrupt loss of consciousness, occurring within 1 hour of an acute change in cardiovascular status.133,133 Unfortunately, SCA and SCD are nearly synonymous. Worldwide, survival after SCA is dismal (<1%). In the United States, mortality rate after SCA is about 95%.134
Merchant and colleagues139 estimated the annual incidence of in-hospital cardiac arrest in the United States. Their calculation of approximately 200,000 annual victims provides pivotal perspective on the magnitude of this problem. Despite the similarity in annual incidence, out-of-hospital SCA is far more familiar to the medical community.140
Coronary artery disease is the primary cause (80%) of out-of-hospital SCA. Nonischemic cardiomyopathies account for 10% to 15%. The remaining 5% are related to valvular disease, inherited ion channel or receptor defects (long QT syndrome, Brugada syndrome, CMPVT, etc.), congenital heart disease, and other causes. Sadly, over 60% of SCA occurs as an initial clinical event or in patients with clinical disease characteristics suggesting relatively low risk.133 Our risk stratification repertoire remains woefully inadequate for the vast majority of out-of-hospital SCA victims.131,140,141
In contrast, most in-hospital cardiac arrests result from preexisting conditions and are not due to sudden-onset cardiac arrhythmias. Progressive respiratory failure and shock are common precipitants. It has been suggested that this information be used to tailor better in-hospital cardiac arrest protocols.142
Ventricular fibrillation is the most frequently documented initial rhythm at the time of resuscitation from out-of-hospital cardiac arrest. In over 75% of cases the underlying cause is a ventricular tachyarrhythmia (ventricular fibrillation or pulseless ventricular tachycardia). Primary bradyarrhythmias (e.g., asystole) and pulseless electrical activity are less common.143
Interestingly, over 70% of in-hospital cardiac arrests present with asystole or pulseless electrical activity. Ventricular tachyarrhythmias are a far less common presentation (14%-24%).142,144,145
Out-of-hospital survival is linked directly to time between SCA onset and defibrillation or bystander CPR. Time to restoration of spontaneous circulation is also closely correlated with neurologic recovery. Brain damage begins in 4 to 6 minutes.146 Studies of outcome as a function of initial rhythm recorded at the scene of out-of-hospital cardiac arrest demonstrate that bradyarrhythmias and asystole have the worst prognosis.147,148
Likewise, survival to hospital discharge is substantially increased when the first documented in-hospital cardiac arrest rhythm is “shockable” (pulseless ventricular tachycardia or ventricular fibrillation).142 Shocks must take priority over CPR. Delayed defibrillation has been associated with a significantly lower probability of survival to hospital discharge.149,150 Asystole often occurs late in an arrest (after energy stores required to generate ventricular fibrillation have been exhausted) and can be a surrogate for prolonged downtime.140,148 Data exist suggesting that pulseless electrical activity portends a slightly better outcome compared to asystole.140,145
Children suffering in-hospital cardiac arrest have better survival to hospital discharge compared to adults. This is true (due to increased survival after pulseless electrical activity or asystole) despite a significantly lower prevalence of ventricular tachycardia/ventricular fibrillation as the initial rhythm in children suffering cardiac arrest.146 For in-hospital cardiac arrest, adult survival rates range from roughly 15% to 21%.142,144,145 In children, a survival rate of 27% has been reported.142 Aggressive, prolonged CPR should be considered in young patients (particularly those with few or no comorbid conditions). Young patients may survive prolonged CPR with no or only minor sequelae.128,140
Electrical storm is defined as ventricular tachycardia or ventricular fibrillation occurring two times or more in a 24-hour period, usually requiring electrical cardioversion or defibrillation.151 Small nonrandomized trials demonstrated amiodarone’s safety and efficacy for recurrent drug-refractory sustained ventricular arrhythmias.151,152 Intravenous amiodarone is more effective than lidocaine for out-of-hospital ventricular fibrillation resistant to shocks and epinephrine. More amiodarone-treated patients survive to hospital admission.153 Fogel and associates demonstrated 80% 1-year survival rate in patients with recurrent hemodynamically unstable ventricular arrhythmias initially treated with intravenous amiodarone who were receiving oral amiodarone at discharge.154 Patients with electrical storm following myocardial infarction treated with sympathetic blockade followed by oral amiodarone had significantly better short-term mortality rates compared with conventional antiarrhythmic drugs. Patients who received a combination of oral amiodarone and a beta blocker had the best outcomes.82 Although limited data exist, beta blockade in conjunction with amiodarone appears to be the most effective therapy for electrical storm.41
Key objectives in post–cardiac arrest care include (1) optimizing cardiopulmonary function and vital organ perfusion; (2) transportation to a hospital or critical-care unit with a comprehensive post–cardiac arrest treatment system of care; (3) identification and intervention for acute coronary syndromes; (4) temperature control to optimize neurologic recovery; and (5) anticipation, treatment, and prevention of multiple organ dysfunction.84
The most important manifestations of the post–cardiac arrest syndrome are often neurologic. About 80% of patients remain comatose for longer than 1 hour after resuscitation, and less than 50% of admitted patients have a good neurologic recovery. Targeted temperature management, or therapeutic hypothermia, is an intervention intended to limit neurologic injury after resuscitation from cardiac arrest.84 An Australian trial and a European multicenter trial suggested improved neurologic recovery and decreased mortality rate at 6 months after arrest.155–157 A 2008 statement from the International Liaison Committee on Resuscitation (ILCOR) incorporated targeted temperature management into the comprehensive treatment bundle of therapy for the post–cardiac arrest syndrome.158
Several different methods are available for cooling in therapeutic hypothermia. Many techniques use commercially developed equipment specifically designed for targeted temperature management. The optimal regimen is still a matter of debate.155
Targeted temperature management has been used almost exclusively in patients with an out-of-hospital cardiac arrest due to ventricular fibrillation or pulseless ventricular tachycardia. There are no data from studies of sufficient quality to recommend this therapy in an adult who has had a cardiac arrest that is not due to a hemodynamically unstable ventricular tachyarrhythmia. Indications and contraindications for targeted temperature management after cardiac arrest are summarized in Box 31.4.155
Catheter Ablation of Cardiac Arrhythmias
As previously noted, catheter ablation may be considered as first-line treatment for some patients with atrial fibrillation. The 2012 HRS/EHRA/ECAS Expert Consensus Statement on Catheter and Surgical Ablation of Atrial Fibrillation suggests that first-line ablation is reasonable for paroxysmal atrial fibrillation and may be considered for persistent or even longstanding persistent atrial fibrillation.78
Other operators have targeted complex fractionated electrograms recorded during atrial fibrillation. The theory behind this technique is that “rotors” or wavelets of reentry (the substrate for atrial fibrillation) can be eliminated. One report has suggested that attacking complex fractionated electrograms adds little to pulmonary vein isolation for persistent atrial fibrillation.159 A recent report that utilizes a special computer program to choose target sites has rekindled interest in this technique.160
Atrial fibrillation ablation success is highly dependent on careful patient selection. Patients with persistent or permanent atrial fibrillation, very large left atria, or age 75 years or older (similar success rates, complications more likely) are less likely to have an optimal outcome. Ideal candidates for catheter ablation of atrial fibrillation have symptomatic episodes of paroxysmal or persistent atrial fibrillation, have not responded to antiarrhythmic drugs, do not have severe comorbid conditions or significant structural heart disease, are younger than 65 to 70 years, have a left atrial diameter less than 50 to 55 mm, and have had persistent atrial fibrillation for less than 5 years.161
Although atrial fibrillation ablation is generally considered safe and effective, devastating complications may occur. The incidence of death associated with atrial fibrillation ablation is 0.1%. This is similar to the risk of death associated with ablation of regular supraventricular tachycardias. Cardiac tamponade is the most frequent fatal complication. Delayed development of an atrioesophageal fistula (10-16 days after procedure) is the second most frequent cause of death.162 This complication is unique to ablation in the posterior left atrium.
Pacemakers and Implantable Defibrillators
The classic indication for permanent pacing is symptomatic bradycardia that is not due to a transient cause and is, therefore, unlikely to reverse. Most commonly, this occurs because of SND or AV blockade. Permanent pacing at rates greater than 70 beats per minute and rate-smoothing algorithms are useful to prevent drug-induced torsades de pointes. Pacing at 80 beats per minute may help prevent torsades de pointes and torsades de pointes “storm” in patients with congenital long QT syndrome.4,163
The ICD has emerged as accepted therapy for primary and secondary prevention of sudden death.164 Evidence-based results have demonstrated the clinical benefits of ICD therapy and indications for implantation have expanded rapidly over the past 10 years (Box 31.5).165,166 Over 100,000 ICDs are implanted in the United States annually.167
Nevertheless, ICD therapy has associated risks including infection, inappropriate shocks, proarrhythmic potential, device malfunction, highly publicized manufacturer advisories, and procedural complications, which may adversely affect morbidity and quality of life.166 Defibrillator implantation is not always psychologically benign. ICD specific adjustment disorders are not uncommon. Syndromes described include anxiety with secondary panic reaction; defibrillator dependence, abuse, or withdrawal; negative body image; and imaginary (phantom) shocks.168 Recent problems with the Medtronic Sprint Fidelis and the St. Jude Riata and Riata ST transvenous high-voltage leads serve to emphasize the limitations of ICD therapy.169,170
Our increasing risk stratification repertoire remains inadequate for the vast majority of SCA victims.141 Some evidence suggests that only 35% to 51% of patients who are guideline eligible for an ICD receive one.171,172 In contrast, a recent analysis of ICD recipients in the National Cardiovascular Data Registry—Implantable Cardiac Defibrillators Registry suggested that 22.5% did not meet evidence-based implantation criteria.173 Patients are often referred for ICD replacement with little or no evidence of a well-informed discussion of the risks, expected benefits, and long-term goals of care.166
Investigators have raised important issues and asked pivotal questions such as the following: Who are we missing? Who are we overtreating? Who is best served by ICD therapy? Should all initial recipients receive replacement ICDs?167,174
Cardiac resynchronization therapy (CRT), or biventricular pacing, is an important therapeutic option with drug-refractory NYHA (New York Heart Association) class III or IV CHF, left ventricular ejection fraction at 35% or less, and a major left-sided conduction delay (QRS duration greater than 120 ms). Patients with RBBB and IVCD derive less benefit from CRT compared with those with a native LBBB. The underlying rhythm should be sinus or atrial fibrillation, with a slow enough ventricular response to allow continuous biventricular stimulation and capture. Biventricular pacing most frequently is used as part of an ICD system (CRT-D). Acute and chronic CHF contributes to the need for tachyarrhythmia treatment in ICD recipients. Although small trials (93 patients in total) have shown that biventricular pacing diminished ventricular arrhythmias, these results were not confirmed in larger trials.4
Electrocardiographic Patterns Intensivists Should Recognize
Electrolyte, Endocrine, and Metabolic Abnormalities
Hyperkalemia is frequent in the critically ill, in particular in patients with impaired renal function. As the serum potassium increases, the ECG follows a characteristic stepwise pattern of derangement. The first sign of hyperkalemia on ECG is peaked T waves. Although there is no universal definition of “peaked,” we agree with the definition of T waves greater than 10 mm in the precordium and 6 mm in the limb leads.175 In addition to being peaked, the T waves typically appear narrow. At a potassium level of greater than 6.0 mmol/L, T-wave peaking is pronounced, the P wave will begin to lose amplitude, and the QRS complex begins to widen. At potassium levels greater than 7 mmol/L the P wave is no longer visible and QRS widening progresses. The ensuing ECG pattern is known as sinoventricular rhythm (Fig. 31.18).176 Sine waves may become present as the widened QRS complex blends into the T wave (Fig. 31.19).177 An abnormal ECG may be the initial sign of dangerous hyperkalemia and therapy should begin immediately to eliminate the hyperkalemia and stabilize the heart (calcium gluconate infusion).
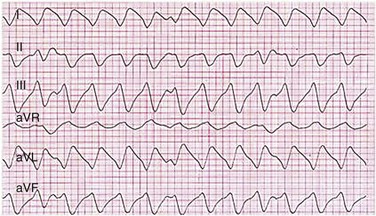
Figure 31.19 Sine waves seen in severe hyperkalemia. (From NEJM Image Challenge, http://www.nejm.org/action/showImageChallenge?ci=05032012, last accessed 6/13/12.)
Hypokalemia also results in characteristic ECG changes. The hallmark of hypokalemia is the U wave, an additional wave seen after the T wave (Fig. 31.20). As the hypokalemia becomes more severe, the U wave increases in amplitude, and T-wave amplitude will decrease. Eventually, the T wave and the U wave may fuse, giving the appearance of a widened T wave. This creates a prolonged QT interval, which may result in torsades de pointes. The risk of hypokalemia-induced arrhythmia is increased in patients receiving digitalis and during myocardial ischemia.178
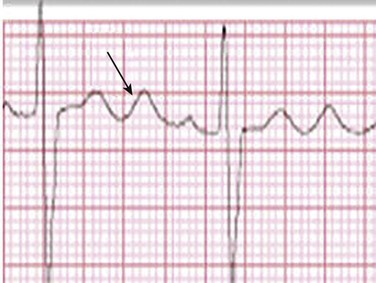
Hypomagnesemia is usually associated with potassium depletion. ECG abnormalities seen are produced by hypokalemia. Hypermagnesemia is uncommon and usually associated with renal failure. Observed ECG changes result from concomitant electrolyte disturbances (hyperkalemia and hypocalcemia).178
The principal electrocardiographic feature of hypercalcemia is a shortening of the QT interval (Fig. 31.21).176 This finding is not associated with arrhythmias. In contrast, patients with the rare SQTS have a QTc interval less than 360 ms (typically <300 ms) and a high risk of sudden death due to ventricular fibrillation (Fig. 31.22).179 Patients often have permanent or paroxysmal atrial fibrillation and occasionally manifest depression of the PR interval. Tall, peaked T waves without flat ST segments and impaired rate-dependent QT shortening has been recorded.180 SQTS appears to be inherited in an autosomal dominant pattern.
Figure 31.22 Patient with congenital short QT syndrome. (From http://www.shortqtsyndrome.org/dr_diagnosis.htm, last accessed 6/13/12.)
The principal finding in hypocalcemia is a prolongation of the QT interval. It should be noted that, unlike in hypokalemia, the T wave is normal sized and QT interval prolongation results from lengthening of the ST segment, not a broadening of the T wave due to fusion with a U wave. In severe hypocalcemia, PR and QRS intervals are frequently prolonged (Fig. 31.23).176 Second-degree or third-degree AV block has been reported. J waves (see later) have occasionally been reported.178
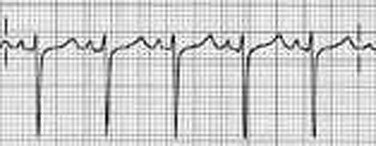
In addition to electrolyte abnormalities, metabolic derangements can cause characteristic ECG findings. Hypothermia, both from accidental exposure as well as therapeutic cooling after cardiac arrest, has classic findings on ECG (Fig. 31.24).181 As the core body temperature drops below 32° C, conduction becomes slower with slower sinus rates, prolonged PR and QT intervals, as well as junctional rhythms.182 The finding of an Osborn or J wave is a hallmark of hypothermia. This wave appears as an upward deflection at the terminal portion of the R wave, before the J point. Additionally, the ECG baseline may have artifact or oscillations from shivering. Though classically associated with hypothermia, the Osborn wave may also be a sign of other conditions such as sepsis and diabetic ketoacidosis (which can be hypothermic conditions) as well as normothermic conditions such as neurologic insults and hypercalcemia.183
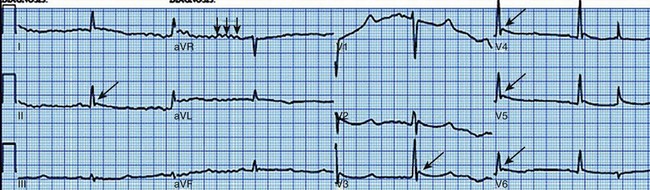
Brugada Pattern
As mentioned earlier, the Brugada syndrome is characterized by an inherited disorder of sodium channels. Brugada pattern is characterized on the ECG by RBBB (complete or incomplete) morphology and abnormal repolarization characterized by ST-segment elevation in leads V1 to V3. Three distinct patterns have been identified. Type 1 features a “coved” type ST segment (more than 2 mm) elevation and a negative T wave. Type 2 features a “saddleback” ST-T configuration, recognized by two notches in the T wave. Type 3 can have a coved or saddleback morphology. This configuration is less specific for Brugada syndrome (Fig. 31.25).184 A patient with any of the Brugada patterns and a history of cardiac arrest, syncope, or ventricular tachycardia should have an ICD implanted, as there is no effective drug therapy for this disease. It is not recommended that asymptomatic patients with the Brugada pattern on ECG receive a defibrillator, and clinical monitoring is advised instead. Should a patient suffer from electrical storm, isoproterenol is considered useful.132
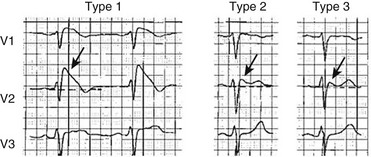
Long QT Syndromes
As mentioned previously, the congenital long QT syndrome is associated with SCD due to torsades de pointes. The three most common inherited forms (LQT1, LQT2, and LQT3) represent 75% of the inherited long QT syndrome. Each one of those subtypes has a specific mutation and an activity in which the arrhythmia (and hence syncope or cardiac arrest) is triggered. LQT1 is associated with events during exertion, LQT2 is associated with events occurring after hearing a sudden loud noise, and LQT3 is associated with events at rest or sleep. There are also ECG patterns associated with each of those three variants, though it should be noted that the clinical scenario will often correlate better than the ECG pattern to the specific gene mutation. The LQT1 has a broad T wave, the LQT2 has a bifid T wave, and the LQT3 has a long isoelectric segment and a narrow T wave. The LQT3 pattern appears somewhat similar to the ECG seen in hypocalcemia (Fig. 31.26).185
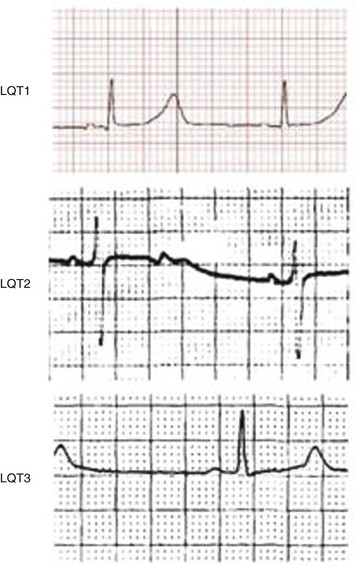
More commonly, however, intensivists will see patients with transient prolongations in their QT interval due to their illness, ingestion of medications, or antiarrhythmic drugs. These patients need to be monitored carefully for the development of torsades de pointes. In addition, an alternation in T wave amplitude can be seen. This is referred to as T wave alternans and represents a harbinger of worsening arrhythmias (Fig. 31.27). Dramatic QT prolongation and T wave stroke may occur after cardiovascular accident.
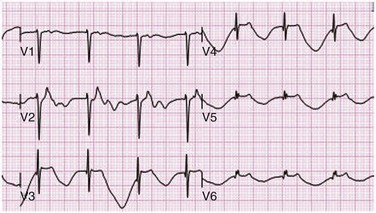
Torsades de Pointes
The term torsades or torsades de pointes refers to a polymorphic ventricular tachycardia that appears to rotate around a horizontal axis, hence the name (from the French “twisting of the points”) (Figs. 31.28 and 31.29).34 The term torsades de pointes should be used only in the setting of the ventricular tachycardia seen with a prolonged QT interval (either acquired or congenital). It is important to differentiate this rhythm from other types of polymorphic ventricular tachycardia (as seen in ischemia) because the management is different. Urgent management of acquired long QT prolongation and torsades de pointes is typically intravenous magnesium sulfate and defibrillation for hemodynamic instability. As mentioned previously, isoproterenol and temporary pacing increase ventricular rates, shorten QT intervals, and help prevent recurrent arrhythmias until the effects of the offending agent diminish. Torsades de pointes is often initiated by a short-long-short sequence of a premature ventricular beat (short segment) followed by a compensatory pause (long segment). The long delay from the compensatory pause delays repolarization. As a result, the QT segment is prolonged after the compensatory pause, and it is more likely that the next depolarization will occur when the ventricle is vulnerable (R on T phenomenon).
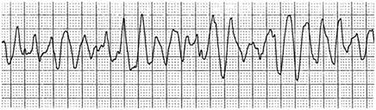
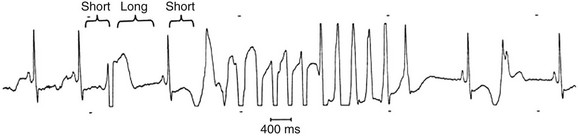
Wolf-Parkinson-White Pattern
As described previously, patients with Wolff-Parkinson-White syndrome can present in sinus rhythm or during an arrhythmia. There are three classic ECG patterns in this disorder depending on the patient’s presenting rhythm. Patients in sinus rhythm with activation of the ventricle by both the normal AV node and His-Purkinje system as well as by the accessory pathway will have a fusion beat on their ECG. This beat manifests as a delta wave and short PR interval as part of the ventricle is activated (preexcited) by the accessory pathway (Fig. 31.30). Patients with Wolff-Parkinson-White pattern in sinus rhythm are asymptomatic and usually come to attention when presenting for other reasons, after an arrhythmia has broken, or on a routine ECG that incidentally detects the abnormality. T-wave inversion may develop after accessory pathway ablation (Fig. 31.31). This is a transient, reversible, repolariztion phenomenon and does not reflect myocardial ischemia.
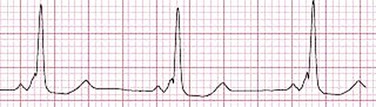
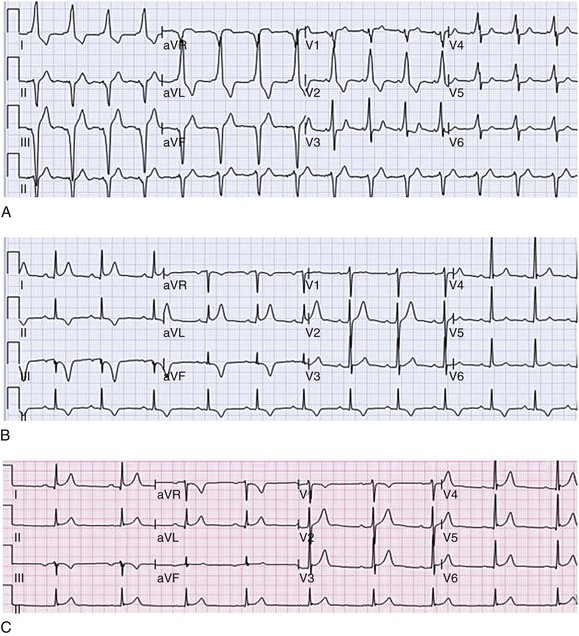
Arrhythmogenic Right Ventricular Dysplasia
In patients with ARVD, inverted T waves may be noted in the right precordial leads (V1, V2, and V3). In addition, an upward deflection, called an epsilon, may be seen just after the QRS complex and is due to late right ventricular activation (Fig 31.32).186 In addition, the QRS duration will be prolonged, often with a complete or incomplete RBBB pattern. The ECG may be the initial clue to this disease. Ventricular tachycardia with a LBBB-like morphology (right ventricular origin) is often precipitated by catecholamines (e.g., exercise).
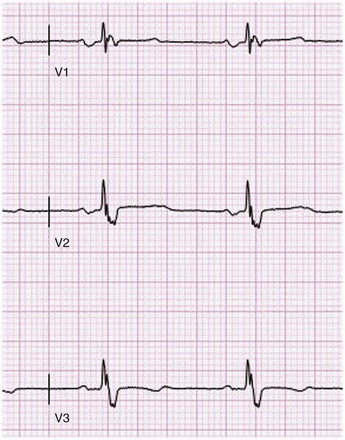
References
1. Trohman, RG, Parrillo, JE. Arrhythmias: Perspectives for the critical care physician. Crit Care Med. 2000; 28:N115.
2. Rubenstein, JJ, Schulman, CL, Yurchak, PM, DeSanctis, RW. Clinical spectrum of the sick sinus syndrome. Circulation. 1972; 46:5–13.
3. Zipes, DP. Specific arrhythmias: Diagnosis and treatment. In: Zipes DP, ed. Heart Disease. Philadelphia: WB Saunders, 1997.
4. Trohman, RG, Kim, MH, Pinski, SL. Cardiac pacing: The state of the art. Lancet. 2004; 364:1701–1719.
5. DeGuzman, M, Rahimtoola, SH. What is the role of pacemakers in patients with coronary artery disease and conduction abnormalities? Cardiovasc Clin. 1983; 13:191–207.
6. Trohman, RG. Supraventricular tachycardia: Implications for the intensivist. Crit Care Med. 2000; 28:N129–N135.
7. Tchou, PJ, Trohman, RG. Supraventricular tachycardias. In: Dale DC, Federman DD, eds. Scientific American Medicine. New York: WebMD, 2003.
8. Utomo, K, Wang, Z, Lazzara, R, Jackman, WM. Atrioventricular nodal reentrant tachycardia: Electrophysiologic characteristics of four forms and implications for the reentrant circuit. In: Zipes DP, Jalife J, eds. Cardiac Electrophysiology: From Cell to Bedside. 3rd ed. Philadelphia: WB Saunders; 2000:504–521.
9. Gursoy, S, Steurer, G, Brugada, J, et al. Brief report: The hemodynamic mechanism of pounding in the neck in atrioventricular nodal reentrant tachycardia. N Engl J Med. 1992; 327:772–774.
10. Knight, BP, Ebinger, M, Oral, H, et al. Diagnostic value of tachycardia features and pacing maneuvers during paroxysmal supraventricular tachycardia. J Am Coll Cardiol. 2000; 36:574–582.
11. Morady, F, DiCarlo, LA, Jr., Baerman, JM, et al. Determinants of QRS alternans during narrow QRS tachycardia. J Am Coll Cardiol. 1987; 9:489–499.
12. Sanders, WE, Jr., Sorrentino, RA, Greenfield, RA, et al. Catheter ablation of sinoatrial node reentrant tachycardia. J Am Coll Cardiol. 1994; 23:926–934.
13. Swerdlow, CD, Liem, LB. Atrial and junctional tachycardias. In: Zipes DP, Jalife J, eds. Cardiac Electrophysiology: From Cell to Bedside. 3rd ed. Philadelphia: WB Saunders; 2000:742–755.
14. Engelstein, ED, Lippman, N, Stein, KM, Lerman, BB. Mechanism-specific effects of adenosine on atrial tachycardia. Circulation. 1994; 89:2645–2654.
15. Blomstrom-Lundqvist, C, Scheinman, MM, Aliot, EM, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for the management of patients with supraventricular arrhythmias) developed in collaboration with NASPE-Heart Rhythm Society. J Am Coll Cardiol. 2003; 42:1493–1531.
16. Glatter, KA, Dorostkar, PC, Yang, Y, et al. Electrophysiological effects of ibutilide in patients with accessory pathways. Circulation. 2001; 104:1933–1939.
17. Neiger, JS, Trohman, RG. Differential diagnosis of tachycardia with a typical left bundle branch block morphology. World J Cardiol. 2011; 3(5):127–134.
18. Prystowsky, EN, Yee, R, Klein, GJ. Wolff-Parkinson-White syndrome. In Zipes DP, Jalife J, eds. : Cardiac Electrophysiology: From Cell to Bedside, 4th ed, Philadelphia: WB Saunders, 2004.
19. Trohman, RG. Adenosine for diagnosis of wide QRS tachycardia: Rapid infusion for an easier conclusion. Crit Care Med. 2009; 37:2651–2652.
20. Marill, KA, Wolfram, S, deSouza, IS, et al. Adenosine for wide-complex tachycardia: Efficacy and safety. Crit Care Med. 2009; 37:2512–2518.
21. Villain, E, Vetter, VL, Garcia, JM, et al. Evolving concepts in the management of congenital junctional ectopic tachycardia. A multicenter study. Circulation. 1990; 81:1544–1549.
22. Hamdan, MH, Badhwar, N, Scheinman, MM. Role of invasive electrophysiologic testing in the evaluation and management of adult patients with focal junctional tachycardia. Card Electrophysiol Rev. 2002; 6:431–435.
23. Scheinman, MM, Gonzalez, RP, Cooper, MW, et al. Clinical and electrophysiologic features and role of catheter ablation techniques in adult patients with automatic atrioventricular junctional tachycardia. Am J Cardiol. 1994; 74:565–572.
24. Trohman, RG, Haery, C, Pinski, SL. Focal radiofrequency catheter ablation of an irregularly irregular supraventricular tachycardia. Pacing Clin Electrophysiol. 1999; 22:360–362.
25. Hanau, SP, Solar, M, Ansura, EL. Metoprolol in the treatment of multifocal atrial tachycardia. Cardiovasc Rev Rep. 1984; 1182.
26. Levine, JH, Michael, JR, Guarnieri, T. Treatment of multifocal atrial tachycardia with verapamil. N Engl J Med. 1985; 312:21–25.
27. Dougherty, AH, Schroth, G, Ilkiw, RL. Episodic tachycardia in a 12-year-old girl. Circulation. 1995; 92:268–273.
28. Krishnan, K, Gupta, A, Halleran, SM, et al. Isthmus dependent atrial flutter cycle length correlates with right atrial cross-sectional area. Indian Pacing Electrophysiol J. 2009; 9(3):167–173.
29. Stevenson, JP, Pinski, SL, McLaughlin, VV, et al. Right atrial enlargement: An innocent bystander in the pathogenesis of atrial fibrillation. Circulation. 1999; 100:I-212.
30. Haissaguerre, M, Jais, P, Shah, DC, Garrigue, S, et al. Electrophysiological end point for catheter ablation of atrial fibrillation initiated from multiple pulmonary venous foci. Circulation. 2000; 101:1409–1417.
31. Trohman, RG. Atrial fibrillation in the critically ill: Common sense for a common problem. Crit Care Med. 2008; 36(5):1681–1682.
32. Trohman, RG, Parrillo, JE. Direct current cardioversion: Indications, techniques, and recent advances. Crit Care Med. 2000; 28(10 Suppl):N170–173.
33. Kowey, PR. The calamity of cardioversion of conscious patients. Am J Cardiol. 1988; 61:1106–1107.
34. Gupta, A, Lawrence, AT, Krishnan, K, et al. Current concepts in the mechanisms and management of drug-induced QT prolongation and torsades de pointes. Am Heart J. 2007; 153:891–899.
35. Roden, DM. Ibutilide and the treatment of atrial arrhythmias. A new drug—almost unheralded—is now available to U. S. physicians. Circulation. 1996; 94:1499–1502.
36. Stambler, BS, Wood, MA, Ellenbogen, KA. Antiarrhythmic actions of intravenous ibutilide compared with procainamide during human atrial flutter and fibrillation: Electrophysiological determinants of enhanced conversion efficacy. Circulation. 1997; 96:4298–4306.
37. Murray, KT. Ibutilide. Circulation. 1998; 97:493.
38. Volgman, AS, Carberry, PA, Stambler, B, et al. Conversion efficacy and safety of intravenous ibutilide compared with intravenous procainamide in patients with atrial flutter or fibrillation. J Am Coll Cardiol. 1998; 31:1414–1419.
39. Van, G, Tuinenburg, AE, Schoonderwoerd, BS, et al. Pharmacologic versus direct-current electrical cardioversion of atrial flutter and fibrillation. Am J Cardiol. 1999; 84:147R–151R.
40. Delle Karth, G, Geppert, A, Neunteufl, T, et al. Amiodarone versus diltiazem for rate control in critically ill patients with atrial tachyarrhythmias. Crit Care Med. 2001; 29:1149–1153.
41. Vassallo, P, Trohman, RG. Prescribing amiodarone: An evidence-based review of clinical indications. JAMA. 2007; 298:1312–1322.
42. Aasbo, JD, Lawrence, AT, Krishnan, K, et al. Amiodarone prophylaxis reduces major cardiovascular morbidity and length of stay after cardiac surgery: A meta-analysis. Ann Intern Med. 2005; 143:327–336.
43. Mitchell, LB, Exner, DV, Wyse, DG, et al. Prophylactic oral Amiodarone for the Prevention of Arrhythmias that Begin Early After Revascularization, valve replacement, or repair: PAPABEAR: A randomized controlled trial. JAMA. 2005; 294:3093–3100.
44. Trohman, RG, Simmons, TW, Moore, SL, et al. Catheter ablation of the atrioventricular junction using radiofrequency energy and a bilateral cardiac approach. Am J Cardiol. 1992; 70(18):1438–1443.
45. Arnold, AZ, Mick, MJ, Mazurek, RP, et al. Role of prophylactic anticoagulation for direct current cardioversion in patients with atrial fibrillation or atrial flutter. J Am Coll Cardiol. 1992; 19:851–855.
46. Lanzarotti, CJ, Olshansky, B. Thromboembolism in chronic atrial flutter: Is the risk underestimated? J Am Coll Cardiol. 1997; 30:1506–1511.
47. Fuster, V, Ryden, LE, Cannom, DS, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: Full text: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (writing committee to revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006; 8:651–745.
48. Asher, CR, Klein, AL. Transesophageal echocardiography to guide cardioversion in patients with atrial fibrillation: ACUTE trial update. Card Electrophysiol Rev. 2003; 7:387–391.
49. Parikh, MG, Aziz, Z, Krishnan, K, et al. Usefulness of transesophageal echocardiography to confirm clinical utility of CHA2DS2-VASc and CHADS2 scores in atrial flutter. Am J Cardiol. 2012; 109(4):550–555.
50. Wann, LS, Curtis, AB, January, CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (updating the 2006 guideline): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. writing on behalf of the 2006 ACC/AHA/ESC Guidelines for the Management of Patients With Atrial Fibrillation Writing Committee. J Am Coll Cardiol. 2011; 57:223–242.
51. Attaya, S, Bornstein, T, Ronquillo, N, et al. Study of warfarin patients investigating attitudes toward therapy change (SWITCH Survey). Am J Ther. 2012; 19(6):432–435.
52. Connolly, SJ, Ezekowitz, MD, Yusuf, S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009; 361:1139–1151.
53. Cotton, BA, McCarthy, JJ, Holcomb, JB. Acutely injured patients on dabigatran. N Engl J Med. 2011; 365:2039–2040.
54. Patel, MR, Mahaffey, KW, Garg, J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011; 365:883–891.
55. Connolly, SJ, Eikelboom, J, Joyner, C, et al. Apixaban in patients with atrial fibrillation. N Engl J Med. 2011; 364:806–817.
56. Granger, CB, Alexander, JH, McMurray, JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011; 365:981–992.
57. Viles-Gonzalez, JF, Fuster, V, Halperin, JL. New anticoagulants for prevention of stroke in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2011; 22:948–955.
58. Eerenberg, ES, Kamphuisen, PW, Sijpkens, MK, et al. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: A randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011; 124:1573–1579.
59. Anne-éline, M, Le-Bonniec Bernard, LB, Lecompte, T, et al. Evaluation of recombinant activated factor VII, prothrombin complex concentrate and fibrinogen concentrate to reverse apixaban in a rabbit model. J Am Coll Cardiol. 2012; 59(13):E573.
60. Kowey, PR. The calamity of cardioversion of conscious patients. Am J Cardiol. 1988; 61:1106–1107.
61. Bryson, HM, Fulton, BR, Faulds, D. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs. 1995; 50:513–559.
62. Schuten, C, May, C, Krishnan, K, Trohman, RG. Optimal propofol bolus dosing for rapid deep sedation. Heart Rhythm. 2009; 6(5):S2.
63. Tobin, MG, Pinski, SL, Tchou, PJ, et al. Cost effectiveness of administration of intravenous anesthetics for direct-current cardioversion by nonanesthesiologists. Am J Cardiol. 1997; 79(5):686–688.
64. Ewy, GA. The optimal technique for electrical cardioversion of atrial fibrillation. Clin Cardiol. 1994; 17:79–84.
65. Pinski, SL, Arnold, AZ, Mick, M, et al. Safety of external cardioversion/defibrillation in patients with internal defibrillation patches and no device. Pacing Clin Electrophysiol. 1991; 14:7–12.
66. Joglar, JA, Hamdan, MH, Ramaswamy, K, et al. Initial energy for elective external cardioversion of persistent atrial fibrillation. Am J Cardiol. 2000; 86:348–350.
67. Pinski, SL, Sgarbossa, EB, Ching, E, Trohman, RG. A comparison of 50-J versus 100-J shocks for direct-current cardioversion of atrial flutter. Am Heart J. 1999; 137:439–442.
68. Saliba, W, Juratli, N, Chung, MK, et al. Higher energy synchronized external direct current cardioversion for refractory atrial fibrillation. J Am Coll Cardiol. 1999; 34:2031–2034.
69. Caterine, MR, Yoerger, DM, Spencer, KT, et al. Effect of electrode position and gel-application technique on predicted transcardiac current during transthroacic defibrillation. Ann Emerg Med. 1997; 29:588–595.
70. Coplen, SE, Antman, EM, Berlin, JA, et al. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis of randomized control trials. Circulation. 1990; 82:1106–1116.
71. Pritchett, EL, Page, RL, Carson, M, for the Rythmol Atrial Fibrillation Trial (RAFT) Investigators. Efficacy and safety of sustained-release propafenone (propafenone SR) for patients with atrial fibrillation. Am J Cardiol. 2003; 92:941–946.
72. Wann, LS, Curtis, AB, January, CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (updating the 2006 guideline): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2006 Writing Committee Members, Fuster V, Rydén LE, Cannom DS, et al; ACCF/AHA Task Force Members, Jacobs AK, Anderson JL, Albert N, et al. Heart Rhythm. 2011; 8(1):157–176.
73. Patel, C, Yan, GX, Kowey, PR. Dronedarone. Circulation. 2009; 120:636–644.
74. Connolly, SJ, Camm, AJ, Halperin, JL, et al. Dronedarone in high-risk permanent atrial fibrillation. PALLAS Investigators. N Engl J Med. 2011; 365(24):2268–2276.
75. Wyse, DG, Waldo, AL, DiMarco, JP, et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med. 2002; 347:1825–1833.
76. Jenkins, LS, Brodsky, M, Schron, E, et al. Quality of life in atrial fibrillation: The Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) study. Am Heart J. 2005; 149:112–120.
77. Sherman, DG, Kim, SG, Boop, BS, et al. Occurrence and characteristics of stroke events in the Atrial Fibrillation Follow-up Investigation of Sinus Rhythm Management (AFFIRM) study. Arch Intern Med. 2005; 165:1185–1191.
78. Calkins, H, Kuck, KH, Cappato, R, et al. 2012 HRS/EHRA/ECAS expert consensus statement on catheter and surgical ablation of atrial fibrillation: Recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, end points, and research trial design: A report of the Heart Rhythm Society (HRS) Task Force on Catheter and Surgical Ablation of Atrial Fibrillation. Developed in partnership with the European Heart Rhythm Association (EHRA), a registered branch of the European Society of Cardiology (ESC) and the European Cardiac Arrhythmia Society (ECAS); and in collaboration with the American College of Cardiology (ACC), American Heart Association (AHA), the Asia Pacific Heart Rhythm Society (APHRS), and the Society of Thoracic Surgeons (STS). Endorsed by the governing bodies of the American College of Cardiology Foundation, the American Heart Association, the European Cardiac Arrhythmia Society, the European Heart Rhythm Association, the Society of Thoracic Surgeons, the Asia Pacific Heart Rhythm Society, and the Heart Rhythm Society. Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation. Heart Rhythm. 2012; 9(4):632–696.
79. Ramaswamy, K, Hamdan, MH. Ischemia, metabolic disturbances, and arrhythmogenesis: Mechanisms and management. Crit Care Med. 2000; 28:N151–N157.
80. Lerman, BB, Stein, K, Engelstein, ED, et al. Mechanism of repetitive monomorphic ventricular tachycardia. Circulation. 1995; 92(3):421–429.
81. Pastay, SO, Braunwald, E. Renal disorders and heart disease. In: Braunwald E, ed. Heart Disease. Philadelphia: WB Saunders, 1988.
82. Nademanee, K, Taylor, R, Bailey, WE, et al. Treating electrical storm: Sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000; 102:742–747.
83. Brugada, P, Brugada, J, Mont, L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991; 83:1649–1659.
84. Field, JM, Hazinski, MF, Sayre, MR, et al. Part 1: Executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010; 122:S640–656.
85. Bardy, GH, Lee, KL, Mark, KL, et al. Amiodarone or an implantable cardioverter defibrillator for congestive heart failure (Sed Heft Trial). N Engl J Med. 2005; 352:225–237.
86. Moss, AJ, Hall, WJ, Cannom, DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996; 335:1933–1940.
87. Moss, AJ, Zareba, W, Hall, WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002; 346:877–883.
87a. Wellens, HJ, Bär, FW, Lie, KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med. 1978; 64:27–33.
87b. Kindwall, KE, Brown, J, Josephson, ME. Electrocardiographic criteria for ventricular tachycardia in wide complex left bundle branch block morphology tachycardias. Am J Cardiol. 1988; 61:1279–1283.
87c. Akhtar, M, Shenasa, M, Jazayeri, M, et al. Wide QRS complex tachycardia. Reappraisal of a common clinical problem. Ann Intern Med. 1988; 109:905–912.
87d. Brugada, P, Brugada, J, Mont, L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991; 83:1649–1659.
87e. Vereckei, A, Duray, G, Szénási, G, et al. New algorithm using only lead aVR for differential diagnosis of wide QRS complex tachycardia. Heart Rhythm. 2008; 5:89–98.
88. Credner, SC, Klingenheben, T, Mauss, O, et al. Electrical storm in patients with transvenous implantable cardioverter-defibrillators: Incidence, management and prognostic implications. J Am Coll Cardiol. 1998; 32:1909–1915.
89. Kim, SG, Hallstrom, A, Love, JC, et al. Comparison of clinical characteristics and frequency of implantable defibrillator use between randomized patients in the Antiarrhythmics vs Implantable Defibrillators (AVID) trial and nonrandomized registry patients. Am J Cardiol. 1997; 80:454–457.
90. Pinski, SL, Yao, Q, Epstein, AE, et al. Determinants of outcome in patients with sustained ventricular tachyarrhythmias: The Antiarrhythmics versus Implantable Defibrillators (AVID) study registry. Am Heart J. 2000; 139:804–813.
91. Caceres, J, Jazayeri, M, McKinnie, J, et al. Sustained bundle branch reentry as a mechanism of clinical tachycardia. Circulation. 1989; 79:256–270.
92. Tchou, P, Jazayeri, M, Caceres, JA. Bundle branch reentrant ventricular tachycardia. Am Heart J. 1988; 116:1647–1648.
93. Tchou, P, Jazayeri, M, Denker, S, et al. Transcatheter electrical ablation of right bundle branch. A method of treating macroreentrant ventricular tachycardia attributed to bundle branch reentry. Circulation. 1988; 78:246–257.
94. Langberg, JJ, Desai, J, Dullet, N, Scheinman, MM. Treatment of macroreentrant ventricular tachycardia with radiofrequency ablation of the right bundle branch. Am J Cardiol. 1989; 63:1010–1013.
95. Raitt, MH, Renfroe, EG, Epstein, AE, et al. “Stable” ventricular tachycardia is not a benign rhythm: Insights from the Antiarrhythmics versus Implantable Defibrillators (AVID) registry. Circulation. 2001; 103:244–252.
96. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. N Engl J Med. 1997; 337:1576.
97. Strickberger, SA, Man, KC, Daoud, EG, et al. A prospective evaluation of catheter ablation of ventricular tachycardia as adjuvant therapy in patients with coronary artery disease and an implantable cardioverter-defibrillator. Circulation. 1997; 96:1525–1531.
98. Reddy, VY, Reynolds, MR, Neuzil, P, et al. Prophylactic catheter ablation for the prevention of defibrillator therapy. N Engl J Med. 2007; 357:2657–2665.
99. Haissaguerre, M, Shoda, M, Jais, P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002; 106:962–967.
100. Haissaguerre, M, Shah, DC, Jais, P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet. 2002; 359:677–678.
101. Myerburg, RJ, Kessler, KM, Mallon, SM, et al. Life-threatening ventricular arrhythmias in patients with silent myocardial ischemia due to coronary artery spasm. N Engl J Med. 1992; 326:1451–1455.
102. Parikh, MG, Kakodkar, SA, Neiger, JS, et al. Variant ventricular tachycardia. Pacing Clin Electrophysiol. 2012; 35:612–615.
103. Chen, Q, Kirsch, GE, Zhang, D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998; 392:293.
104. Carlson, MD, White, RD, Trohman, RG, et al. Right ventricular outflow tract ventricular tachycardia: Detection of previously unrecognized anatomical abormalities using cine magnetic imaging. J Am Coll Cardiol. 1994; 24:720.
105. Markowitz, SM, Litvak, BL, Ramirez de Arellano, EAM, et al. Adenosine-sensitive ventricular tachycardia: Right ventricular abnormalities delineated by magnetic resonance imaging. Circulation. 1997; 96:1192.
106. Kakodkar, S, Krishnan, K, Awad, S, et al. Reversible cardiomyopathy in an adolescent with idiopathic aortic cusp ventricular tachycardia. Pediatr Cardiol. 2010; 31(1):147–150.
107. Varma, N, Josephson, ME. Therapy of “idiopathic” ventricular tachycardia. J Cardiovasc Electrophysiol. 1997; 8(1):104–116.
108. Nakagawa, H, Beckman, KJ, McClelland, JH, et al. Radiofrequency catheter ablation of idiopathic left ventricular tachycardia guided by a Purkinje potential. Circulation. 1993; 88(6):2607–2617.
109. Nishimura, S, Yamauchi, Y, Aonuma, K, et al. Demonstration of diastolic and presystolic purkinje potentials as critical potentials in a macroreentry circuit of verapamil-sensitive idiopathic left ventricular tachycardia. J Am Coll Cardiol. 2000; 36:811–823.
110. Trohman, RG, Sahu, J. Arrhythmogenic right ventricular dysplasia. Curr Treat Opt Cardiovasc Med. 1999; 1:259.
111. Asimaki, A, Tandri, H, Huang, H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009; 360:1075–1084.
112. Marcus, FI, Fontaine, G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: A review. Pacing Clin Electrophysiol. 1995; 18:1298.
113. Wilde, AA, Jongbloed, RJ, Doevendans, PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J Am Coll Cardiol. 1999; 33:327–332.
114. Schwartz, PJ, Priori, SG, Spazzolini, C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001; 103:89–95.
115. Zareba, W, Moss, AJ, Daubert, JP, et al. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003; 14:337–341.
116. Windle, JR, Geletka, RC, Moss, AJ, et al. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A: DeltaKPQ mutation. Ann Noninvasive Electrocardiol. 2001; 6:153–158.
117. Brugada, P, Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992; 20:1391–1396.
118. Pasquie, JL, Sanders, P, Hocini, M, et al. Fever as a precipitant of idiopathic ventricular fibrillation in patients with normal hearts. J Cardiovasc Electrophysiol. 2004; 15:1271–1276.
119. Brugada, J, Brugada, P. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol. 1992; 20(6):1391–1396.
120. Gussak, I, Antzelevitch, C, Bjerregaard, P, et al. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999; 33:5–15.
121. Priori, SG, Napolitano, C, Schwartz, PJ, et al. The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation. 2000; 102:945–947.
122. Priori, SG, Napolitano, C, Gasparini, M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000; 102:2509–2515.
123. Napolitano, C, Priori, SG. Catecholaminergic polymorphic ventricular tachycardia and short-coupled torsades de pointes. In Zipes D, Jalife J, eds. : Cardiac Electrophysiology: From Cell to Bedside, 4th ed, Philadelphia: WB Saunders, 2004.
124. Haissaguerre, M, Shoda, M, Jais, P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002; 106:962–967.
125. Haissaguerre, M, Shah, DC, Jais, P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet. 2002; 359:677–678.
126. Bjerregaard, P, Gussak, I. Short QT syndrome. Ann Noninvasive Electrocardiol. 2005; 10:436–440.
127. Bjerregaard, P, Gussak, I. Short QT syndrome: Mechanisms, diagnosis and treatment. Nat Clin Pract Cardiovasc Med. 2005; 2:84–87.
128. Spies, C, Trohman, RG. Narrative review: Electrocution and life-threatening electrical injuries. Ann Intern Med. 2006; 145:531–537.
129. Kobernick, M. Electrical injuries: Pathophysiology and emergency management. Ann Emerg Med. 1982; 11:633–638.
130. Cooper, MA. Lightning injuries: Prognostic signs for death. Ann Emerg Med. 1980; 9:134.
131. Zipes, DP, Wellens, HJ. Sudden cardiac death. Circulation. 1998; 98(21):2334–2351.
132. European Heart Rhythm Association; Heart Rhythm SocietyZipes, DP, Camm, AJ, Borggrefe, M, et al. American College of Cardiology; American Heart Association Task Force; European Society of Cardiology Committee for Practice Guidelines. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death). J Am Coll Cardiol. 2006; 48(5):e247–346.
133. Myerberg, RJ, Castellanos, A. Cardiac arrest and sudden cardiac death. In: Braunwald E, ed. Heart Disease: A Textbook of Cardiovascular Medicine. 5th ed. New York: WB Saunders; 1997:742–779.
134. Myerburg, RJ. Sudden cardiac death: Exploring the limits of our knowledge. J Cardiovasc Electrophysiol. 2001; 12(3):369–381.
135. Huikuri, HV, Castellanos, A, Myerburg, RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001; 345(20):1473–1482.
136. Jemal, A, Thun, MJ, Ries, LAG, et al. Annual report to the nation on the status of cancer, 1975-2005, featuring trends in lung cancer, tobacco use and tobacco control. J Natl Cancer Inst. 2008; 100(23):1672–1694.
137. Centers for Disease Control. HIV/AIDS surveillance report-cases of HIV infection and AIDS in the United States and dependent areas. www.cdc.gov/hiv/topics/surveillance/resources/reports/2006report/pdf/2006SurveillanceReport.pdf, 2006. [(last accessed 12/23/08) 18:1-55. Available at].
138. American Cancer Society. Breast cancer facts and figures 2007-2008. Available at www.cancer.org/downloads/STT/BCFF-Final.pdf. [(last accessed 12/24/08)].
139. Merchant, RM, Yang, L, Becker, LB, et al. Incidence of treated cardiac arrest in hospitalized patients in the United States. Crit Care Med. 2011; 39:2401–2406.
140. Trohman, RG, Trohman, SD. Cardiac arrest: Unveiling the differences within. Crit Care Med. 2011; 39(11):2556–2557.
141. Goldberger, JJ, Cain, ME, Hohnloser, SH, et al. American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society Scientific Statement on Noninvasive Risk Stratification Techniques for Identifying Patients at Risk for Sudden Cardiac Death. A scientific statement from the American Heart Association Council on Clinical Cardiology Committee on Electrocardiography and Arrhythmias and Council on Epidemiology and Prevention. J Am Coll Cardiol. 2008; 52:1179–1199.
142. Nadkarni, VM, Larkin, GL, Peberdy, MA, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006; 295:50–57.
143. Brooks, R, McGovern, BA, Garan, H, et al. Current treatment of patients surviving out-of-hospital cardiac arrest. JAMA. 1991; 265:762–768.
144. Peberdy, MA, Ornato, JP, Larkin, GL, et al. Survival from in-hospital cardiac arrest during nights and weekends. JAMA. 2008; 299:785–792.
145. Meaney, PA, Nadkarni, VM, Kern, KB, et al. Rhythms and outcomes of adult in-hospital cardiac arrest. Crit Care Med. 2010; 38:101–108.
146. Sudden Deaths from Cardiac Arrest—Statistics. Available at http://www.senatormoore.com/news/archive/2010/06/AHA_Cardiac_Arrest.pdf, 2011. [Accessed June 7].
147. Myerburg, RJ, Estes, D, Zaman, L, et al. Outcome of resuscitation from bradyarrhythmic or asystolic prehospital cardiac arrest. J Am Coll Cardiol. 1984; 4:1118–1122.
148. Peberdy, MA, Kaye, W, Ornato, JP, et al. Cardiopulmonary resuscitation of adults in the hospital: A report of 14,720 cardiac arrests from the National Registry of Cardiopulmonary Resuscitation. Resuscitation. 2003; 58:297–308.
149. Skogvoll, E, Nordseth, T. The early minutes of in-hospital cardiac arrest: Shock or CPR? A population based prospective study. Scand J Trauma Resuscitation Emerg Med. 2008; 16:11.
150. Chan, PS, Krumholz, HM, Nichol, G, et al. Delayed time to defibrillation after in-hospital cardiac arrest. N Engl J Med. 2008; 358:9–17.
151. Credner, SC, Klingenheben, T, Mauss, O, et al. Electrical storm in patients with transvenous implantable cardioverter-defibrillators: Incidence, management and prognostic implications. J Am Coll Cardiol. 1998; 32(7):1909–1915.
152. Helmy, I, Herre, JM, Gee, G, et al. Use of intravenous amiodarone for emergency treatment of life-threatening ventricular arrhythmias. J AmColl Cardiol. 1988; 12(4):1015–1022.
153. Dorian, P, Cass, D, Schwartz, B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002; 346(12):884–890.
154. Fogel, RI, Herre, JM, Kopelman, HA, et al. Longterm follow-up of patients requiring intravenous amiodarone to suppress hemodynamically destabilizing ventricular arrhythmias. Am Heart J. 2000; 139(4):690–695.
155. Holzer, M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med. 2010; 363:1256–1264.
156. Bernard, SA, Gray, TW, Buist, MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002; 346:557–563.
157. The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002; 346:549–556.
158. Nolan, JP, Neumar, RW, Adrie, C, et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication: A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008; 79:350–379.
159. Oral, H, Chugh, A, Yoshida, K, et al. A randomized assessment of the incremental role of ablation of complex fractionated atrial electrograms after pulmonary vein isolation for long-lasting persistent atrial fibrillation. J Am Coll Cardiol. 2009; 53:782–789.
160. Narayan, SM, Sehra, R, Hopper, K, et al. Targeted ablation at sources alone (Focal Impulse and Rotor Modulation, FIRM) acutely terminates or slows human atrial fibrillation. Available at http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=147be3dc-ad59-43f7-89b1-09eb88e90ad7&cKey=9693d226-3477-4009-b5e7-3e4981dcdb84&mKey=%7bBAEF2DB4-7615-4F2C-851A-E5D7461EBD4E%7d. [Last accessed 5/22/12].
161. Oral, H, Morady, F. How to select patients for atrial fibrillation ablation. Heart Rhythm. 2006; 3:615–618.
162. Cappato, R, Calkins, H, Chen, S-A, et al. Prevalence and causes of fatal outcome in catheter ablation of atrial fibrillation. J Am Coll Cardiol. 2009; 53:1798–1803.
163. Pinski, SL, Eguia, LE, Trohman, RG. What is the minimal pacing rate that prevents torsades de pointes? Insights from patients with permanent pacemakers. Pacing Clin Electrophysiol. 2002; 25:1612–1615.
164. Myerburg, RJ, Reddy, V, Castellanos, A. Indications for implantable cardioverter-defibrillators based on evidence and judgment. J Am Coll Cardiol. 2009; 54:747–763.
165. Epstein, AE. Benefits of the implantable cardioverter-defibrillator. J Am Coll Cardiol. 2008; 52(14):1122–1127.
166. Tung, R, Zimetbaum, P, Josephson, ME. A critical appraisal of implantable cardioverter-defibrillator therapy for the prevention of sudden cardiac death. J Am Coll Cardiol. 2008; 52(14):1111–1121.
167. Kramer, DB, Buxton, AE, Zimetbaum, PJ. Time for a change—A new approach to ICD replacement. N Engl J Med.. 2012; 366(4):291–293.
168. Pinski, SL, Trohman, RG. Implantable cardioverter-defibrillators: Implications for the nonelectrophysiologist. Ann Intern Med. 1995; 122(10):770–777.
169. Hauser, RG, Maisel, WH, Friedman, PA, et al. Longevity of sprint fidelis implantable cardioverter-defibrillator leads and risk factors for failure: Implications for patient management. Circulation. 2011; 123:358–363.
170. Hauser, RG. Here we go again—Failure of postmarketing device surveillance. N Engl J Med. 2012; 366(10):873–875.
171. Redberg, RF. Disparities in use of implantable cardioverter-defibrillators. Moving beyond process measures to outcomes data. JAMA. 2007; 298:1564–1566.
172. Fonarow, GC, Yancy, CW, Albert, NM, et al. Heart failure care in the outpatient cardiology practice setting: Findings from IMPROVE HF. Circ Heart Fail. 2008; 1:98–106.
173. Al-Khatib, SM, Hellkamp, A, Curtis, J, et al. Non-evidence-based ICD implantations in the United States. JAMA. 2011; 305:43–49.
174. Epstein, AE. Subject of the year: Who are we missing, who are we overtreating, and who is best served? Refining the prescription of implantable cardioverter-defibrillator therapy. J Intervention Card Electrophysiol. 2009; 26(2):91–94.
175. O’Keefe, J, Jr., Hammil, SC, Freed, MS, Pogwizd, SM. The Complete Guide to ECGs, 3rd ed. Boston: Jones & Bartlett Publishers; 2008.
176. Baltazar, RF. Basic and Bedside Electrocardiography. Philadelphia: Lippincott Williams & Wilkins; 2009.
177. Image Challenge. Available at. http://www.nejm.org/action/showImageChallenge?ci=05032012 [Last accessed 6/1/12].
178. Van Mieghem, C, Sabbe, M, Knockaert, D. The clinical value of the ECG in noncardiac conditions. Chest. 2004; 125:1561–1576.
179. Short QT Syndrome. Available at. http://www.shortqtsyndrome.org/dr_diagnosis.htm [Last accessed 6/12/12].
180. Morita, H, Wu, J, Zipes, DP. The QT syndromes: Long and short. Lancet. 2008; 372(9640):750–763.
181. Sepehrdad, R, Paulsen, J, Amesterdan, EA. The ECG that came in from the cold. Am J Med. 2012; 125(3):246–248.
182. Alsafwah, S. Electrocardiographic changes in hypothermia heart and lung. J Acute Crit Care. 2001; 30(2):161–163.
183. Sheikh, AM, Hurst, JW. Osborn waves in the electrocardiogram, hypothermia not due to exposure, and death due to diabetic ketoacidosis. Clin Cardiol. 2003; 26:555–560.
184. Koonlawee Nademanee, Silvia G. Priori, SG, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation. 2002; 106:2514–2519.
185. Roden, DM. Long-QT Syndrome. N Engl J Med. 2008; 358:169–176.
186. Brugada, P, Geelen, P. Some electrocardiographic patterns predicting sudden cardiac death that every doctor should recognize. Acta Cardiol. 1997; 52(6):473–484.