[level-membership-for-neonatal-and-perinatal-medicine-category]
Carbohydrate, fat, and protein metabolism
Metabolism comes from the Greek word meaning “to change” and is the totality of chemical reactions within a living organism. Four principles that underlie and guide metabolic functions in humans are “(1) plasma glucose must be maintained within normal limits; (2) an optimal source of glycogen must be maintained as an emergency fuel; (3) an optimal supply of protein must be maintained for use in enzymatic mechanisms of metabolism as well as muscular mobility; excess protein is converted to fat and the nitrogen released is excreted in urine; and (4) protein must be conserved when it is scarce and stored fat used in time of caloric need.”47,p.2
Maternal physiologic adaptations
Metabolic adaptations during pregnancy are directed toward (1) ensuring satisfactory growth and development of the fetus; (2) providing the fetus with adequate stores of energy and substrates needed for transition to extrauterine life; (3) meeting maternal needs to cope with the increased physiologic demands of pregnancy; and (4) providing energy and substrate stores for the demands of pregnancy, labor, and lactation.6 The first two demands compete with the third and fourth demands. As a result, alterations in maternal metabolic processes can significantly affect maternal and fetal health status.
Pregnancy involves a “coordinated series of physiologic adjustments which act in concert to preserve maternal homeostasis while at the same time providing for fetal growth and development.”47 Pregnancy is primarily an anabolic state in which food intake and appetite are increased, activity is decreased, approximately 3.5 kg of fat is deposited, energy reserves of approximately 30,000 kcal (125,550 J) are established, and 900 g of new protein is synthesized by the mother, fetus, and placenta. The overall energy cost of reproduction is estimated at 75,000 to 85,000 kcal (313,875 to 355,725 J).6,80,82 Anabolic aspects are most prominent during the first two trimesters when, due to enhanced lipogenesis, accumulation of maternal fat and increased blood volume lead to maternal weight gain (Figure 16-1).19,78 Protein and glycogen synthesis increase in muscle with increased glycogenolysis (breakdown of glycogen to form glucose) and decreased glycolysis in the liver.78 Insulin increases in response to glucose with a normal or slight increase in peripheral insulin sensitivity and serum glucose levels. This results in uptake of nutrients and maternal fat accumulation. During the third trimester, the woman’s metabolic status becomes more catabolic as stored fat is used (see Figure 16-1). Gluconeogenesis (formation of glucose from amino acids and glycerol) in the liver decreases and intestinal dietary fat absorption increases.78 Lipolytic activity within adipose tissue is enhanced with increases in plasma free fatty acids and glycerol. Maternal ketone production is increased. Counterinsulin hormones increase, leading to insulin resistance. During this phase, maternal weight gain is primarily due to the growing fetus and placenta; the fetus gains 90% of its growth in the last half of pregnancy.12,19,57,80,82
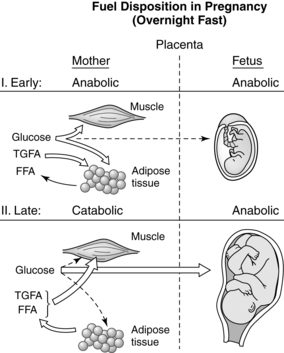
Antepartum period
Pregnancy is associated with major changes in metabolic processes and endocrine function. Pregnancy has been characterized as a metabolic “tug of war” between the competing needs of the mother and the fetus.47 The fetus and placenta influence maternal metabolic alterations in that these tissues become an additional site for metabolism of maternal hormones as well as new sites for hormonal biosynthesis. Many of the metabolic changes during pregnancy are aimed at providing substances (especially glucose, lipids, and amino acids) for the growth and development of the fetus. As the fetus and placenta grow, maternal fuel economy is altered to support this growth.38,109
Basal metabolic rate
The basal metabolic rate (BMR) increases during pregnancy (Figure 16-2). The rate of change varies with maternal prepregnant nutritional status and fetal growth. Significant variations are seen among individual women with up to an eightfold increase reported. Similar variations are reported for fat accretion.17,79,91 If a woman has low energy reserves at conception, there is less of an increase in the BMR and energy is conserved.
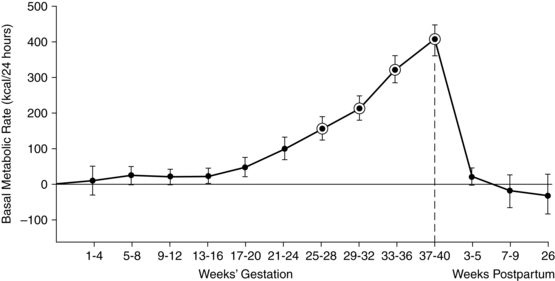
The total energy required for pregnancy can be divided into three parts: (1) obligatory energy needed for the fetus, placenta, uterus, and breasts (which is the smallest part); (2) energy for maternal fat storage; and (3) energy maintenance of these new tissues.79 If a woman has lower energy stores, less of the maternal energy intake is needed to maintain new tissues and energy is conserved for maternal basic needs and the fetal-placental unit.79,80,107,109 For example, in undernourished women, fetal weight accounts for 60% of the pregnancy weight gain, versus 25% in a well-nourished woman.109 This energy-sparing response may allow the woman to sustain the pregnancy but is often at the expense of fetal growth, with decreased birth weight and risk of fetal growth restriction.109 Prentice and Goldberg suggest that leptin might monitor a woman’s prepregnancy energy stores and adjust or coordinate maternal metabolic resources.109 Women with large-for-gestational-age (LGA) infants tend to have increased BMR with less maternal energy storage.79,111
The pregnant woman meets the energy demands of pregnancy by increasing her intake, decreasing her activity, or limiting fat storage.80,107 King and colleagues propose three examples of how women in different situations might alter their energy to sustain pregnancy.79 First, an underweight impoverished woman with limited food, poor fat stores, and need for physical work cannot increase food intake or limit physical activity during pregnancy. Her body responds by decreasing basal energy expenditures so that pregnant energy needs are similar to prepregnant needs. Second, a normal weight woman in a developed country with fat stores before pregnancy and adequate nutrition during pregnancy increases fat stores in pregnancy and increases her BMR slightly. Finally, an overweight woman in a developed country increases her BMR by 20% or more, perhaps to reduce additional fat deposition.79
Carbohydrate metabolism
Basal endogenous hepatic glucose production remains sensitive to insulin and increases up to 30% by the third trimester to meet fetal and placental needs.12,16,72,98 Endogenous glucose production increases with gestational age, paralleling fetal and maternal needs.72 Maternal glucose levels are generally 10% to 20% lower than in nonpregnant women. In addition, during the overnight fasting period, maternal plasma glucose values fall to levels 15 to 20 mg/dL (0.8 to 1.1 mmol/L) lower than in nonpregnant women.88 This decrease in glucose leads to lower insulin levels during the postabsorptive state (between meals and overnight) and a tendency toward hypoglycemia and ketosis. The decreased glucose level in the postabsorptive state is due to increased plasma volume in early pregnancy and later to placental transport of glucose to the fetus, which increases during gestation as fetal glucose needs increase.98,101
During the first two trimesters, the pregnant woman is in an anabolic state. Insulin secretion increases with increased peripheral glucose use without increases in insulin resistance.47 As pregnancy progresses, peripheral glucose use by the mother decreases because of increasing insulin resistance. This reduces maternal glucose utilization and makes glucose more readily available to the fetus. The mother compensates by using fat stores to meet her energy needs with breakdown of glycerol to glucose.57 Insulin resistance in the latter part of pregnancy is believed to be a result of a decrease in sensitivity of cell receptors that results from the insulin antagonism effects of hPL, progesterone, and cortisol.16,47 The insulin antagonism is partially modulated by pancreatic β-cell hyperplasia and hypertrophy with increased insulin availability after a meal.16 Pregnancy is also characterized by greater oscillations in insulin and glucagon levels.47 A reduction in the extraction of insulin by the liver may contribute to the peripheral hyperinsulinemia. Variations in hepatic insulin binding may lead to alterations in the ratio of insulin to glucagon.6
Progesterone augments insulin secretion, decreases peripheral insulin effectiveness, and increases insulin levels after a meal. Estrogen increases the level of plasma cortisol (an insulin antagonist), stimulates β-cell hyperplasia (and thus insulin production), and enhances peripheral glucose utilization. Increased levels of both bound and free cortisol decrease hepatic glycogen stores and increase hepatic glucose production. These changes further increase glucose availability for the fetus.88,98
hPL levels correlate with fetal and placental weight and are higher in multiple pregnancies.16 hPL increases synthesis and availability of lipids. Lipids can be used by the mother as an alternative fuel, enhancing availability and transfer of glucose and amino acids to the fetus. A mild form of the metabolic changes seen during pregnancy can be induced by giving hPL to nonpregnant women. However, there is no consistent relationship between hPL levels and insulin requirements in the pregnant diabetic woman.
Protein metabolism
Decreased serum amino acid and serum protein levels are found in pregnancy.6,42,72 This decrease is related to increased placental uptake, increased insulin levels, hepatic diversion of amino acids for gluconeogenesis, and transfer of amino acids to the fetus.6,42 The fetus uses some of these amino acids for glucose formation. Maternal plasma levels of glucogenic amino acids (e.g., those that can be converted into glucose), such as alanine, threonine, glutamate, and serine, are reduced due to placental transfer of these amino acids.31,72 Maternal plasma alanine levels, in particular, are lower because alanine is a key precursor for glucose formation (via gluconeogenesis) by the fetal liver.72
Alterations in protein metabolism during pregnancy have a biphasic pattern.72 During the first half of gestation, maternal protein storage increases, with a net retention of 1.3 g/day of nitrogen.80 Most of this is transported to the fetus, but some is retained in maternal tissues. During the second half, maternal protein use is more economic, with decreased urinary nitrogen excretion, thus conserving protein.86 These changes may be mediated by decreased activity of hepatic enzymes involved in amino acid deamination and urea synthesis.
Lipid metabolism
Pregnancy results in marked alterations in lipid metabolism with a markedly different lipoprotein profile, with marked increases in triglycerides, as well as increases in phospholipids and cholesterol. Basal oxidation of fatty acids increases 70% in pregnancy leading to a relative hyperlipidemia.27 Synthesis of very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL), which are contained in triglycerides, and LDL and HDL, which are found in cholesterol, also increase.59
Lipid metabolism in pregnancy is characterized by two phases and is analogous to the patterns of change in carbohydrate and protein metabolism.19,57 During the first two trimesters, triglyceride synthesis and fat storage increase. VLDL increases threefold in the second and third trimesters. LDL decreases slightly initially, followed by a progressive rise.100 HDL increases progressively to 24 weeks, then decreases to 32 weeks and stabilizes for the remainder of pregnancy.100 Triglycerides increase 40% by 18 weeks and 250% by term.87 Phospholipids and cholesterol levels also increase; the triglyceride-to-cholesterol level remains stable since cholesterol also increases.59,58 By late pregnancy, cholesterol levels are 50% higher than prior to pregnancy, regardless of maternal dietary intake.42 These changes enhance the availability of substrates for the fetus.57
Maternal fat storage is most prominent from 10 to 30 weeks, before the peak of fetal energy demands.79 Promotion of lipogenesis and suppression of lipolysis during this phase are mediated by progressive increases in insulin responsiveness and enhanced by progesterone, cortisol, leptin, and prolactin.6,16,30,42,59 Estrogens decrease lipoprotein lipase activity.18 During this period the pregnant woman experiences a physiologic ketosis with a twofold to threefold increase in baseline ketone body production, with an acute increase after fasting, suggesting enhanced fat utilization.116,130
Lipid metabolism in late pregnancy is illustrated in Figure 16-3. The third trimester is characterized by both lipogenesis and lipolysis, with increased breakdown of fat deposits.19,32,57,58 These changes are mediated by increased adipose tissue lipolytic activity and decreased lipoprotein lipase (LPL) activity (due to estrogens).19,58 LPL, which is present in the capillary endothelium of extrahepatic tissues, hydrolyzes circulating triglycerides, including VLDL, producing free fatty acids and glycerol.58 Adipose tissue is broken down into free fatty acids and glycerol, which circulate to the liver and are converted to active forms (acyl-CoA and glycerol-3-phosphate) and re-esterified into triglycerides (circulate as VLDL).19,58 Glycerol can also be used for glucose synthesis (gluconeogenesis). The increased lipolysis is due to the rise of hPL levels with its antiinsulinogenic and lipolytic effects, as well as the effects of cortisol, glucagon, and prolactin, resulting in increased lipolytic activity in adipose tissue.16,19,57,58,78 Enhanced ketogenesis in the liver is a consequence of increased oxidation of free fatty acids for energy and release of ketone bodies.58 The fat mobilization is associated with increased glucose and amino acid uptake by the fetus. Thus fats are used by the mother as an alternative energy substrate, allowing the mother to conserve glucose for the fetus and her central nervous system (CNS) during the second half of gestation.6,16,59,58 Fetal glucose and ketone uptake increases. Ketones are used by the fetus for oxidative metabolism, lipogenesis, triglyceride production, and as substrate for brain lipid synthesis.58,78,116,130
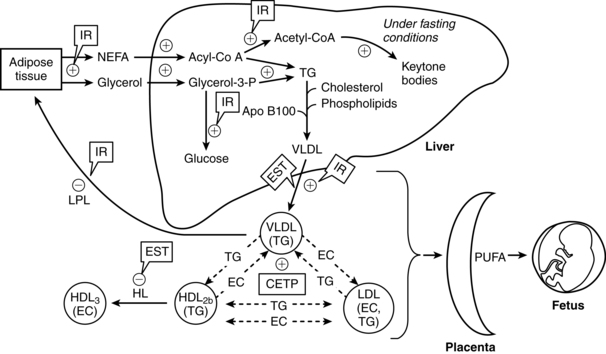
The changes in lipid metabolism during pregnancy are reflected in changes in maternal serum free fatty acid concentrations as well as plasma triglyceride, cholesterol, and phospholipid levels. These changes may be exaggerated in obese women.27 The minimal change in free fatty acids during early pregnancy is probably due to increased fat storage and augmented fat utilization. As maternal fat stores are mobilized, serum levels of triglycerides, free fatty acids, glycerol, and triglyceride-rich lipoproteins (VLDL) increase to peak near term.19,58,59 Because elevations in free fatty acids are due to catabolism of stored triglycerides (into free fatty acid and glycerol), changes in free fatty acids are mirrored by changes in glycerol.6 Hypertriglyceridemia during the third trimester is primarily due to increases in VLDLs and decreased peripheral clearance.16,19,58,59 The increase is due to reduced VLDL clearance secondary to decreased activity of LPL in the liver and adipose tissue, enhanced activity of cholesterol ester transferase, increased gastrointestinal absorption of lipids, and increased hepatic triglyceride production.16,32,58,59 The decrease in lipoprotein lipase is due to increases in estrogens in late pregnancy and the increasing insulin resistance.58,78 Near term, lipoprotein lipase activity increases in the mammary glands. This enhances availability of triglycerides for milk production.57
Changes in lipid metabolism are accompanied by functional and morphologic changes in the adipocytes. Hypertrophy of these cells accommodates the increased fat storage during the first two thirds of pregnancy. In the last trimester, maximal glucose transport, glucose oxidation, and lipogenesis within the adipocytes decrease.42 The number of insulin receptors on the adipocytes increases in the first part of pregnancy and returns to prepregnant levels by term.6,42 Because responsiveness of adipose tissue to insulin is not diminished as much as that of other tissues, these changes in the adipocytes facilitate fat storage.42 After a meal, maternal fat stores are replenished by increased glucose uptake, incorporation of glucose into glycerol, and esterification of fatty acids by adipocytes.6
Insulin
Insulin levels and responsiveness of tissues to insulin change dramatically during pregnancy, leading to peripheral insulin resistance. Peripheral insulin resistance is “the decreased ability of insulin to affect glucose uptake, primarily in skeletal muscle and to a lesser degree in adipose tissue.”98 Actions of insulin are summarized in Table 16-1.40 Insulin production and sensitivity during pregnancy differ in early compared to later pregnancy.16 During early pregnancy (up to 12 to 14 weeks), insulin responses are enhanced, sending glucose to the embryo and young fetus. The pregnant woman has a normal glucose tolerance, basal glucose production, and peripheral muscle sensitivity to insulin.16,110 Adipose tissue is more sensitive to insulin during this period, resulting in lipogenesis and fat storage.110
Table 16-1
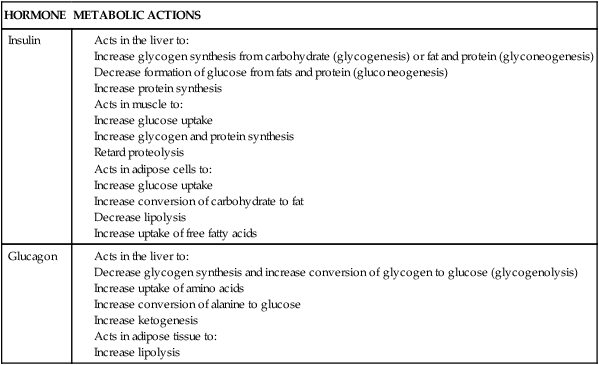
Compiled from Guyton, A.C., & Hall, J.E. (2010). Textbook of medical physiology (12th ed.). Philadelphia: Saunders Elsevier; Widmaier, E., Raff, H., & Strang, K.T. (2005). Vander’s Human physiology: The mechanism of body function (10th ed.). New York: McGraw-Hill.
In later pregnancy (from 20 weeks to term), insulin sensitivity decreases and insulin secretion and resistance increase with decreased glucose uptake by muscle and adipose tissue.16,131 Maternal insulin levels increase 2.5- to threefold by the third trimester.47,78,98 These changes are accompanied by maternal pancreatic hypertrophy and hyperplasia.110 Increased insulin secretion ensures adequate maternal protein synthesis in the face of increasing resistance of peripheral tissues to the effects of insulin. With increasing gestation, mean insulin sensitivity decreases up to 50% to 70%.78 Tissue resistance is most prominent in liver, adipose, and muscle cells, and is further altered in women with preeclampsia.119 The increasing insulin resistance promotes nutrient flux from the mother to the fetus and promotion of adipose tissue accumulation.78 Insulin resistance is often exaggerated in overweight and obese women.27
Insulin receptor binding does not change significantly during pregnancy, however, the postreceptor insulin signaling cascade, and thus handling of glucose by cells, is altered in skeletal muscle and adipose tissue.110 During pregnancy insulin receptor substrate-1 (IRS-1) is down-regulated, affecting insulin uptake and use by cells.98 The postreceptor handling of glucose during pregnancy is altered by decreased IRS-1 expression (especially in skeletal muscle), altered expression of tyrosine kinase activity, and decreased expression of GLUT-4 glucose transporter protein in adipose tissue, which normally promotes glucose uptake.98,110
Insulin resistance is mediated by the increasing levels of placental hormones, especially estrogens, progesterone, and hPL, as well as prolactin and cortisol, and is minimally affected by changes in blood glucose levels.98 Other factors that play an important role in the insulin resistance of pregnancy are tumor necrosis factor-α and other cytokines, circulating free fatty acids, leptin, and placental growth hormone, which stimulates insulin-like growth factor-I (IGF-I).15,78,197,124 In late pregnancy, although basal insulin levels are elevated, maternal blood glucose values are similar to prepregnant levels.6 Increased insulin secretion after a meal (in response to the higher blood glucose) offsets the contrainsulin effects of the placental hormones and facilitates movement of nutrients to the fetus. The insulin resistance of late pregnancy enhances maternal fat breakdown and increased gluconeogenesis and ketogenesis in the postabsorptive state, the state in which changes in insulin response are most apparent.16,47,58 If the pregnant woman is not able to elevate her insulin secretion to overcome the increasing pregnancy-induced insulin resistance, maternal and fetal hyperglycemia will result and metabolic abnormalities such as gestational diabetes may develop or existing metabolic problems such as diabetes will be aggravated.
Alterations in insulin production and responsiveness are critical in integrating changes in carbohydrate and fat metabolism throughout the course of pregnancy. Baird summarizes these interactions as follows: During early pregnancy, increased insulin in response to glucose, minimal changes in insulin sensitivity, and increased number of insulin receptors on the adipocytes result in normal or slightly enhanced carbohydrate tolerance. The increased hepatic synthesis and secretion of triglycerides during this period, along with a normal or slightly elevated removal of triglycerides from the circulation, lead to a net storage of fat. During late pregnancy, the elevated plasma insulin, decrease in numbers of adipocyte insulin receptors to prepregnant levels, and increasing insulin resistance result in reduced assimilation of glucose and triglycerides by maternal tissues, greater transfer of these substances to the fetus, and increased lipolysis. The net result is a decrease in maternal blood glucose, increased glucose turnover, and greater maternal reliance on lipid catabolism for energy.6 Changes in lipid, carbohydrate, and protein metabolism are summarized in Table 16-2.
Table 16-2
Maternal Metabolic Processes during Pregnancy: Relationship between Hormonal and Metabolic Changes
| HORMONAL CHANGE | EFFECT | METABOLIC CHANGE |
| Increased hPL | ||
| Increased prolactin | Insulin resistance | |
| Increased bound and free cortisol | Ensures glucose and amino acids to fetus | |
| Increased estrogen, progesterone, and insulin during early pregnancy | Anabolic fat storage during early pregnancy | |
| Increased hPL in late pregnancy | Lipolysis | Catabolic fat mobilization in late pregnancy |
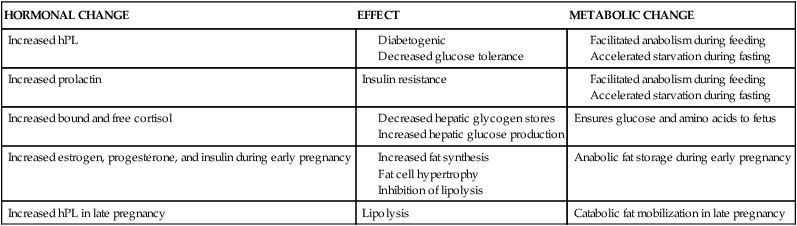
Adapted from Moore, T.R. (2004). Diabetes and pregnancy. In R.K. Creasy, R. Resnik, & J.D. Iams (Eds.), Maternal-fetal medicine: Principles and practice (5th ed.). Philadelphia: Saunders. hPL, human placental lactogen.
Absorptive versus postabsorptive states
In addition to phasic changes in metabolic processes in early versus late pregnancy, metabolism of amino acids, carbohydrates, and fats also varies on a daily basis depending on whether the mother is in the absorptive (fed) or postabsorptive state (Figures 16-4 and16-5). As a result of these changes, pregnancy has been characterized as a time of both “accelerated starvation” and “facilitated anabolism.”96
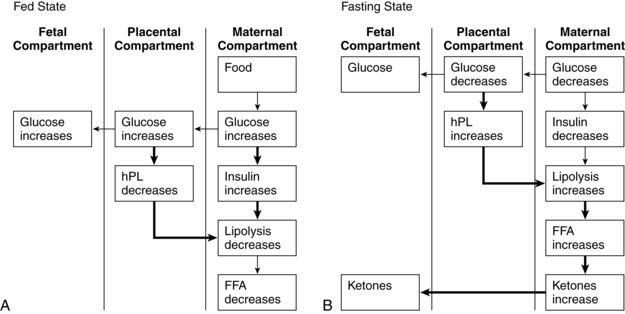
Absorptive state.
During the absorptive (fed) state, ingested nutrients (amino acid, glucose, triglyceride) are entering the blood from the gastrointestinal tract (see Chapter 12) and must be oxidized for energy, used for protein synthesis, or stored. The average meal takes 4 to 6 hours for complete absorption. In this state anabolism exceeds catabolism and glucose is the major energy source. Small amounts of amino acid and fat are converted into energy or used to resynthesize body proteins or for structural fat. Most of the amino acid and fat and any extra carbohydrate are transformed into adipose tissue; carbohydrate is also stored as glycogen.
Insulin has an anabolic and anticatabolic role during this state.78 Insulin secretion increases and plasma insulin levels rise. Insulin promotes glucose uptake by the hepatocytes and peripheral tissues, inhibits glycogen breakdown, and inhibits lipolysis in adipose tissue. Glucose is converted to glycogen for storage in the liver, cardiac muscle, and skeletal muscle. Muscle amino acid uptake is enhanced and proteolysis inhibited.78
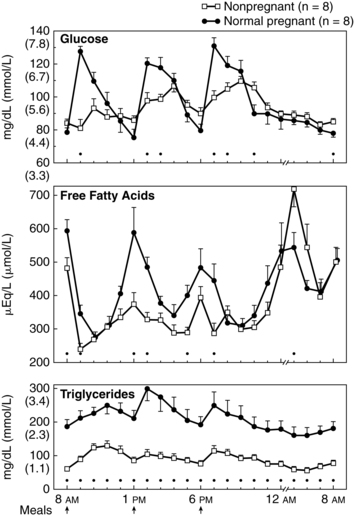
After a meal the pregnant woman has higher glucose, insulin, and triglyceride levels and suppression of glycogen compared to nonpregnant women. Thus the absorptive state during pregnancy (see Figure 16-4, A) is characterized by relative hyperinsulinemia (related to decreased insulin sensitivity), hyperglycemia (due to failure of liver glucose uptake), insulin resistance (especially in skeletal muscle), hypertriglyceridemia, and lipogenesis (more glucose is converted to triglyceride for storage).6,16,78,119,129 Pregnancy has been called a state of facilitated anabolism to describe these metabolic alterations that conserve energy, especially during early pregnancy.94 These changes increase glucose availability for transport to the fetus; increase availability of an alternate energy source (triglycerides) for maternal needs; and provide fewer stimuli for maternal gluconeogenesis, glycogenolysis, and ketogenesis.42,47
Maternal blood glucose levels may rise transiently to 130 to 140 mg/dL (7.2 to 7.8 mmol/L) (see Figure 16-5). Gluconeogenesis and circulating free fatty acids are decreased. Under the influence of placental hormones, resistance of the liver and peripheral tissues to insulin is increased by as much as 60% to 70%.119,129 The hyperinsulinemic response is most marked during the third trimester because of hypertrophy and hyperplasia of islet β cells. These cells become more responsive to alterations in blood glucose and amino acid levels. The increased insulin levels after eating overcome the insulin resistance to allow glucose uptake by muscles for storage as glycogen.119 Even with increased production of insulin, however, overall glucose levels are maintained, although at a relatively lower level than in nonpregnant women because of the counterbalancing effects of estrogen, progesterone, and hPL.
Postabsorptive and fasting state.
In the postabsorptive state (i.e., when nutrients are not entering the blood from the intestines, beginning 4 to 6 hours after a meal) and fasting state (12 or more hours after the last meal), energy must be supplied by body stores. Insulin levels are low. Fat and protein synthesis are decreased and catabolism exceeds anabolism. Gluconeogenesis (production of new glucose from lactate, amino acids [especially alanine], and glycerol) and catabolism of fat are the main sources of energy.78 Plasma glucose levels are maintained during the postabsorptive state by use of these alternate sources of glucose and glucose-sparing or fat-utilization reactions.16,42 The CNS continues to use glucose, while other organs and tissues become glucose sparing, depending on fat as the primary energy source.
During the fasting state (e.g., overnight between the evening meal and breakfast), plasma glucose levels decline. The magnitude of the decline is greater in pregnant women than in nonpregnant women, due to the continuous transfer of glucose to the fetus, and is associated with a more rapid conversion to fat metabolism. Fatty acids are liberated by breakdown of triglycerides. Lipolysis yields glycerol (converted to glucose by the liver) and free fatty acids, which are catabolized to ketone bodies (oxidized for energy). Under homeostatic conditions ketones do not accumulate in the body to produce ketoacidosis because excess ketones not needed for energy are rapidly cleared by the kidneys.78
This response is an exaggeration of the changes normally seen during the overnight fast in nonpregnant women. These changes would normally raise blood glucose levels, but in the pregnant woman the fasting glucose level tends to be lower because of the limited availability of substrate for gluconeogenesis. For example, as early as 15 weeks’ gestation, maternal glucose levels after a 12- to 14-hour overnight fast are 15 to 20 mg (0.8 to 1.1 mmol/L) lower than levels in nonpregnant women. The decrease in glucose during the overnight “fasting” period is especially prominent during the second and third trimesters.6,78
The exaggerated maternal responses during the fasting state are influenced by (1) continuous placental uptake of glucose and amino acids from the maternal circulation; (2) decreased peripheral utilization of glucose as plasma concentrations of ketones and free fatty acids increase; (3) decreased renal absorption of glucose; and (4) decreased hepatic glucose production. This response, seen primarily in late pregnancy, is characterized by lower fasting glucose and amino acid levels, increased blood glucose levels after eating, and increased plasma free fatty acids, triglycerides, ketones, and insulin secretion in response to glucose.12,42,78 Thus the postabsorptive fasting state in pregnancy is characterized by a relative hypoglycemia (due to the fetal siphon, increased renal losses, and decreased liver production), hyperketonuria (ketones used as an alternative energy source), hypoaminoacidemia (due to placental transfer for use in fetal glucose production), and hypoinsulinemia (see Figures 16-4, B, and 16-5).12,24,57,72,78,98 Hypoalaninemia also develops because maternal protein stores can provide only limited substrate, which is insufficient to meet both maternal and fetal amino acid needs.47 Increased insulin during the postabsorptive state enhances uptake of glucose into the maternal skeletal muscle and adipose tissue. In lean women, this leads to suppression of hepatic glucose production in late pregnancy. Less suppression is seen in obese women, therefore the alteration in insulin sensitivity is greater in obese than nonobese women.98
Levels of lipoprotein lipase are increased, enhancing triglyceride breakdown with release of free fatty acids and glycerol (which is broken down into glucose) and production of ketone bodies to provide energy when plasma glucose supply is low.16 Glycerol is a source for hepatic gluconeogenesis. The elevated free fatty acids prevent glucose uptake and oxidation by maternal cells, thus preserving glucose for the maternal CNS and the fetus.12 In the nonpregnant woman this switch to fat oxidation occurs after 14 to 18 hours of fasting; during pregnancy the switch occurs after 2 to 3 hours and has been termed accelerated starvation.12,19,42,96
Metabolic changes characteristic of this state are primarily due to hPL, which promotes lipolysis to increase free fatty acid levels and opposes insulin action, thus increasing glucose availability to the fetus. Other factors influencing this response include increased glucose utilization by the fetus (“the fetal siphon”) and mother, along with an increase in the volume of distribution for glucose (i.e., hemodilution).78 During the second half of pregnancy, effectiveness of insulin in translocating glucose into cells is reduced.78 Because insulin is the ultimate arbitrator of both the absorptive and postabsorptive states, alterations in insulin secretion alter substrate availability to the mother and fetus.47 The insulin antagonism in pregnancy is progressive, paralleling the growth of the fetoplacental unit, and disappears immediately after delivery. Placental hormones and other substances are major factors in producing this insulin antagonism.
Drainage of glucose and amino acids by the fetus may lead to increased maternal appetite and a feeling of faintness sometimes experienced in pregnancy. Pregnant women may experience more rapid development of ketosis and fasting hypoglycemia after food deprivation. With greater maternal reliance on fat utilization during pregnancy, production of ketone bodies and risks of abnormalities such as acidosis are increased. Dieting and caloric restriction during pregnancy should be considered potentially dangerous to both the mother and fetus.37
Effects of placental hormones and other substances
The phasic changes in carbohydrate, lipid, and protein metabolism during pregnancy are due to the interplay of placental hormones, especially estrogen, progesterone, hPL, and leptin.75,98 During the first half of pregnancy, metabolism is affected primarily by estrogens and progesterone. In late pregnancy the influences of increasing concentrations of hPL and leptin become more prominent. Maternal metabolic changes are also influenced by prolactin and cortisol.12
Estrogen stimulates islet β-cell hyperplasia and insulin secretion, enhances glucose utilization in peripheral tissues, and increases plasma cortisol, an insulin antagonist. As a result, particularly in the first half of gestation, estrogen decreases fasting glucose levels, improves glucose tolerance, and increases glycogen storage.78 Progesterone augments insulin secretion, increases fasting plasma insulin concentrations, and diminishes peripheral insulin effectiveness. Cortisol mediates these changes by inhibiting glucose uptake and oxidation, increasing liver glucose production, and possibly augmenting glucagon secretion.12 Cortisol increases to 2.5 times normal levels by late pregnancy.12,131 Prolactin increases fivefold to 10-fold and stimulates insulin production and, at least in animal models, increases the number of β-cell receptors.131
Human placental lactogen (hPL), also called human chorionic somatomammotropin (hCS), is a polypeptide hormone produced by the syncytiotrophoblast (see Chapter 3). hPL is the most potent insulin antagonist of the placental hormones. This hormone is secreted primarily into the maternal circulation although some is also secreted into fetal circulation after around 6 weeks.43 Levels of hPL increase markedly after 20 weeks. Because effects of hPL are similar to those of growth hormone, it has been called the “growth hormone” of the second half of pregnancy.12 hPL action increases availability of maternal glucose and amino acids to the fetus. Other effects of hPL include diminished tissue response to insulin; increased β-cell mass; lipolysis, which increases plasma free fatty acids; enhanced nitrogen retention; decreased urinary potassium excretion; and increased calcium excretion. The major effect is sparing of maternal carbohydrate (glucose) by providing alternative energy sources such as free fatty acids for the mother.16
Several adipokines, including leptin, adiponectin, and retinal binding protein 4 (RBP4), are also important for ensuring adequate substrate for fetal growth.58 Leptin, a protein product of the obese (ob) gene, was identified in 1994. It is produced and secreted by adipose tissue and other tissues, including the placenta during pregnancy. Leptin is involved in regulating appetite and food intake and enhancing energy expenditure, and may also be important in signaling readiness for sexual maturation at puberty.128 Leptin receptors are found in the hypothalamus, placenta, muscle, liver, lymphoid tissue, uterus, chorion, amnion, pancreas, ovary, and adipose tissue.78,124 Placental leptin is secreted into both maternal and fetal circulations.86
Leptin plays a role in maturation and regulation of reproduction and “may serve as a detector of long-term metabolic fuel availability signaling the presence of significant maternal fat stores to initiate reproduction.”16 Leptin modulates glucose metabolism, insulin sensitivity, and adipose tissue lipolysis during pregnancy, enhancing breakdown of maternal fat stores in the second half of pregnancy and availability of glucose and lipids for the fetus.58 Other roles of leptin in pregnancy may be to mediate changes in appetite, thermogenesis, and lipid metabolism.16,49 Leptin may also modulate fetal growth. Concentrations of leptin increase from 6 to 8 weeks’ gestation, rising further in the second and (especially) third trimesters.81 The increase is probably primarily due to increased placental leptin production. Levels in early pregnancy correlate with maternal weight and body mass index; this correlation is not found in later pregnancy and may reflect a form of leptin resistance (similar to that seen with obesity).16,81
Adiponectin is a protein, secreted by adipose tissue, that is involved in modulating glucose and lipid metabolism and enhancing insulin sensitivity.58 Changes in leptin and adiponectin have been reported in complicated pregnancies. For example, both leptin and adiponectin are increased in women with severe preeclampsia.7,84,86 Adiponectin is decreased in women with gestational diabetes mellitus, whereas tumor necrosis factor-α (an inflammatory marker) and cord blood leptin are increased.55,58,90,112 Low levels of adiponectin in women with gestational diabetes may play a role in the increased insulin resistance seen in these women.58 Low leptin levels are associated with spontaneous abortion.83 RBP4 may also have a role in glucose metabolism and insulin sensitivity. RBP4 increases with increasing gestational age (probably due to placental production) and is increased in pregnancies complicated by gestational diabetes.58
Intrapartum period
The processes of parturition are dependent on an available supply of glucose and triglycerides as energy sources. In addition, essential fatty acids are important as precursors of prostaglandins (arachidonic acid, from which prostaglandins are derived, is a derivative of essential fatty acid), which are critical to the onset of labor. These relationships are described in Chapter 4.
During labor and delivery, maternal glucose consumption increases markedly to produce the energy required by the uterus and skeletal muscles.47 As a result, maternal insulin requirements fall. Oxytocin may augment or supplant insulin during this period. In animals, oxytocin has been demonstrated to act similarly to insulin; that is, oxytocin stimulates glucose oxidation, lipogenesis, glycogen synthesis, and protein formation.47
Postpartum period
With removal of the placenta, concentrations of placental hormones such as hPL, estrogens, and progesterone fall rapidly within hours after delivery (see Chapter 5). The postpartum woman is in a state of relative hypopituitarism with blunted production of gonadotropins and growth hormone.47 This hypopituitarism may result from the feedback effects of elevated hPL and prolactin levels during pregnancy on the pituitary gland. hPL is similar to growth hormone, and its disappearance with removal of the placenta leaves the woman without its contrainsulin effects during a period of relative deficiency of growth hormone.47 Plasma leptin levels decrease by 24 hours after delivery.83
Fasting plasma glucose levels fall within a few days, then increase reaching late pregnancy fasting levels by 5 days.42 It is unclear when these levels return to prepregnant values.42 The insulin resistance of late pregnancy is reversed soon after delivery.98
Plasma free fatty acids fall to prepregnant levels by 3 days; triglycerides by 2 weeks.18,42 During the first week the decrease in triglycerides coincides with the fall in hPL. This fall is more rapid in women who are breastfeeding.18,58 In these women, free fatty acids rise to late pregnancy levels by 6 weeks, followed by a decrease to prepregnant levels by 3 to 6 months.6 The increasing levels of fatty acids from 1 to 6 weeks postpartum may reflect maternal use of other nutrients for milk production.6 Cholesterol levels slowly decrease to prepregnant levels over the first few weeks postpartum.42 Maternal plasma amino acid levels return to prepregnant values of approximately 4.3 mg/dL (0.043 g/L) (versus pregnant values of 3.5 mg/dL [0.35 g/L]) by several days after birth.1,35
Clinical implications for the pregnant woman and her fetus
The metabolic adaptations of pregnancy safeguard against variations in maternal caloric intake, changes in activity, increased metabolic efficiency, and changes in the metabolism of carbohydrates, fats, and proteins.6 These metabolic changes occur in a phasic pattern—probably programmed by placental hormones—that spreads the energy costs and protein requirements of pregnancy over the entire 9 months of gestation. In early pregnancy, energy is conserved (facilitated anabolism), followed by later redirection of energy (glucose) to the fetus (“accelerated starvation”), whereas throughout pregnancy the mother uses protein more economically to provide adequate amino acids for development of the fetal brain and other organs.6,47 Pregnancy has also been characterized as a diabetogenic state. This state is reflected in the elevated blood glucose levels in association with increasing insulin resistance. This state is described in this section along with the basis for alterations in the glucose tolerance test (GTT) and the effects of the normal metabolic changes of pregnancy on the diabetic woman and her fetus.
The diabetogenic effects of pregnancy are reflected by alterations in the GTT, with higher glucose values after a meal reflecting an acquired resistance to insulin. The alterations in carbohydrate metabolism are most evident during late pregnancy in the absorptive state (see Figure 16-4, A). When the woman is in this state and glucose is being added to the plasma from the gut, her blood glucose levels do not drop as rapidly as usual, even in the face of higher circulating insulin levels. This response results from decreased maternal sensitivity to insulin due to the action of hormones such as hPL, progesterone, and cortisol. Secretion of these hormones increases during the second half of pregnancy; therefore diabetogenic effects are most prominent during this period. Insulin resistance is somewhat compensated for by increased plasma insulin concentrations.78
Effects of metabolic changes on glucose tolerance tests
Two methods commonly used to evaluate glucose tolerance in pregnancy are (1) a one-step 75-g, 2-hour test (recommended by the World Health Organization); and (2) a two-step test with a 1-hour 50-g glucose challenge (screening test) followed by a 100-g, 3-hour oral glucose tolerance test (OGTT) if challenge levels are either 135 mg/dL (7.5 mmol/L) or greater (more women meet criteria for the 3-hour test, but a greater proportion of women with gestational diabetes mellitus [GDM] are identified) or 140 mg/dL (7.8 mmol/L).78,98 The one-step testing has a lower detection rate for GDM.97 The two-step test has been used most commonly in the United States, whereas the one-step test is used most commonly in other countries.95,98 The Fifth International Workshop Conference on Gestational Diabees Mellitus recommended the two-step test.95 However, following the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) recommended use of a 75-g glucose load followed by fasting, 12-, and 2-hour plasma glucose concentrations.64
During pregnancy, the initial fasting blood glucose value is lower than in nonpregnant individuals, because of decreased glucose utilization and increased fat utilization by the mother (making increased glucose available to the fetus) and the subsequent effects of the fetal siphon. Blood glucose levels tend to remain high after ingestion of carbohydrates for a longer period of time secondary to insulin antagonism and decreased insulin sensitivity. Normally the magnitude of the increase in blood glucose after a carbohydrate feeding is a reflection of failure in glucose uptake by the liver. During pregnancy the increased glucose response in the face of increased endogenous insulin confirms the relative insensitivity and resistance of the liver (as well as peripheral tissues such as muscle and adipose tissue) to insulin.78,98
Maternal-fetal relationships
Growth and development of the fetus is dependent upon the availability of a constant supply of glucose, amino acids, and lipids from the mother for energy, protein synthesis, and production of new tissues. The fetus must also develop adequate stores of these substances to meet the demands of the intrapartum period and transition to extrauterine life. Placental transfer of selected nutrients and hormones is summarized in Figure 16-6. Fetal requirements for substrates involved in carbohydrate, fat, and protein metabolism are discussed in the next section.
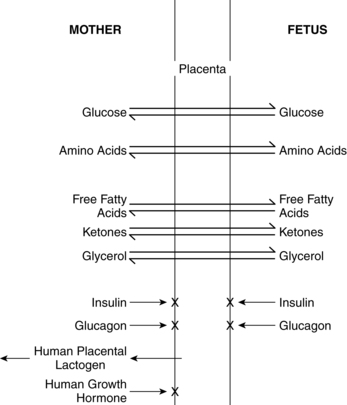
Alterations in maternal metabolic processes or in placental transfer of essential nutrients that increase the availability of specific substrates are usually an advantage to the fetus but can be a disadvantage in altered maternal metabolic states. For example, because fetal energy requirements are met almost exclusively by glucose, the metabolic changes in pregnancy increase availability of glucose in maternal plasma for placental transfer by reducing the efficiency of maternal glucose storage. If the usual metabolic changes of pregnancy or placental function are altered, however, variations in fetal growth—such as occur in the infant of a diabetic mother or the an infant with fetal growth restriction—may develop.122 Maternal glucose infusions during the intrapartum period can lead to a fetal hyperglycemia that stimulates insulin and inhibits glucagon secretion. This may delay gluconeogenesis after birth and increase the risk of neonatal hypoglycemia.
The placenta is a highly metabolic organ with its own substrate needs. Placental metabolic activities include glycolysis, gluconeogenesis, glycogenesis, oxidation, protein synthesis, amino acid interconversion, triglyceride synthesis, and alterations in the length of fatty acid chains (see Chapter 3). The placenta can modify nutrient uptake to meet fetal growth demands.115 The degree of glucose uptake by the placenta is similar to that of the brain. Cholesterol from the mother is essential for placental synthesis of estrogens and progesterone. Fatty acids are needed by the placenta for oxidation and membrane formation.16 Leptin may have a role in coordinating placental metabolism.81
Glucose is the nutrient that crosses the placenta in highest concentrations. The increased maternal reliance on fats as an alternative energy source, especially during the maternal postabsorptive fasting state, conserves maternal glucose for transfer to the fetus. The fetal-placental unit uses approximately 50% of the total maternal glucose needed for pregnancy.57 Glucose crosses via facilitated transport by insulin dependent glucose transporters.81 Amino acids are transferred via active transport because levels are higher in the fetus than in the mother. Amino acids are removed from maternal circulation and concentrated in the placental intercellular matrix. As fetal amino acid levels fall, these placental stores are transferred to the fetus. Mechanisms for amino acid transfer include direct transfer from mother to fetus without modification in the placenta, metabolism of maternally derived amino acids by the placenta to produce other amino acids (which are then transferred to the fetus), and production of new amino acids by the placenta for fetal transfer.57,61 Maternal lipoproteins do not cross the placenta directly, but are taken up by the placenta where placental lipoprotein lipase along with other lipases, enzymes, receptors, and fatty acid binding proteins facilitate transfer of fatty acids to the fetus.57,58,59,78 Essential fatty acids and long-chain polyunsaturated fatty acids needed for fetal growth and development cross the placenta.59 Free fatty acids are transferred to the fetus according to maternal-fetal concentration gradients mediated by specific fatty acid carriers. The pattern of essential and other fatty acids in the fetus reflect maternal concentrations.57,61 Ketones, especially acetoacetate and β-hydroxybutyrate, diffuse readily across the placenta; concentrations are similar in the mother and fetus.78 Inadequate maternal dietary fatty acids such as docosahexaenoic acid (DHA) increase the risk of altered neurologic and vision development in offspring.18,44
The pregnant diabetic woman
The classification system for diabetes proposed by the National Diabetes Data Group and adopted by the American Diabetic Association includes (1) type 1 diabetes (“β-cell destruction, usually leading to absolute insulin deficiency”), (2) type 2 diabetes (“ranging from predominantly insulin resistance with relative insulin deficiency to predominantly an insulin secretory defect with insulin resistance”), (3) other specific types of diabetes (including genetic defects, pancreas alterations, or endocrine, drug-induced, or infectious etiologies), and (4) gestational diabetes mellitus.3 Individuals with impaired fasting glucose and/or impaired glucose tolerance are referred to as having “pre-diabetes.”3
The metabolic changes during pregnancy contribute to alterations in insulin requirements in insulin-dependent pregnant diabetic women. Because the metabolic changes in pregnancy normally lead to increased insulin availability by the end of pregnancy, it is not surprising that pregnant diabetic women experience an increase in insulin requirements by this time.47,78,98
Management of the pregnant diabetic woman and the woman who develops gestational diabetes is complex. There are many controversies regarding screening, diagnosis, treatment, and outcomes of women with gestational diabetes.46,70,78,87 Guidelines for screening and diagnosis of gestational diabetes from various groups around the world were summarized by Leary et al. in a recent article.87 Some of the current controversies relate to the findings and subsequent recommendations from the HAPO study.41,45,46,87 The HAPO study was a prospective, randomized controlled multinational study of 25,000 pregnant women in 10 countries who did not have a diagnosis of diabetes on study entry.45,46 This study examined the relationship between maternal glucose concentrations lower than those diagnostic of diabetes and adverse perinatal outcomes.45,85 Findings demonstrated a linear association between findings on the 75-g OGTT and the following fetal/maternal outcomes: birth weight greater than 90th percentile, increased cord blood C-peptide level, and risks of neonatal hypoglycemia, fetal shoulder dystocia, neonatal hyperbilirubinemia, and maternal preeclampsia.45,46 In the HAPO study, both fasting and post 75-g OGTT levels were correlated to maternal and neonatal outcomes.26,41,45,85 In response to the HAPO study, an IADPSG consensus panel recently published new guidelines on screening and diagnosis of GDM at the first prenatal visit for all or high-risk women and screening all women at 24 to 28 weeks. With the IADPSG recommendations women would be diagnosed with GDM earlier on the basis of an initial fasting glucose of 92 to 126 mg/dL (5.1 to 7.0 mmol/L) or an abnormal fasting (92 mg/dL or 5.1 mmol/L), 1-hour (180 mg/dL or 10.0 mmol/L), or 2-hour (153 mg/dL or 8.5 mmol/L) 75-g OGTT with a single abnormal glucose considered adequate for the diagnosis.64 Recommendations are controversial and their effect on current practice and policies is unclear.41,87,132
GDM is defined as “carbohydrate intolerance of various degrees of severity with onset or first recognition during pregnancy.”98 GDM occurs in 3% to 9% of pregnancies in the United States.98 Women with GDM have a pronounced peripheral insulin resistance, decreased numbers of insulin receptors, and decreased binding of insulin to target cells, which results in a progressive alteration in glucose tolerance.16 Fasting, postprandial, and 24-hour glucose, and lipid and amino acid concentrations are altered.16 Possible etiologies include an autoimmune defect in the β cells, impaired β-cell function, increased insulin degradation, and decreased tissue sensitivity to insulin, either due to impaired insulin-receptor binding or intracellular insulin signaling.110 Two to 13% of women with GDM have specific antibodies against β-cell antigens.98 Most women with GDM have impaired β-cell function and adaptation during pregnancy with chronic insulin resistance, which has an additive effect in pregnancy.98 It is unclear if insulin resistance occurs prior to alterations in β-cell function or if these events occur together.98 A decrease in IRS-1 concentration and decreased ability of insulin receptor-B (found inside skeletal muscle cells) to be phosphorylated by tyrosine have been reported.98 These changes further alter insulin signaling and reduces glucose transport activity. In late pregnancy, the pregnant woman normally increases insulin secretion, but the woman with GDM, because of underlying chronic insulin resistance, has even greater insulin resistance, producing less insulin compared to the amount of resistance.98
During the postpartum period, women with GDM continue to show defects in insulin action and sensitivity, even after glucose tolerance tests have normalized.2 Women with GDM have an increased risk for later development of diabetes, primarily type 2.16 Approximately 10% develop diabetes in the first months postpartum, 50% by 5 years postpartum, and 70% by 10 years postpartum.2,16,17 The risk is greater with weight gain after pregnancy or GDM in a subsequent pregnancy.2,16 Several studies suggest that infant outcomes, including macrosomia and shoulder dystocia, could be reduced by treating women with mild hyperglycemia who did not reach diagnostic criteria for GDM.26,85
Pregnant women with type 1 diabetes may experience no change or decreased insulin requirements during the first trimester because of increased glucose siphoning by the fetus, which decreases maternal blood glucose levels. Maternal food intake may also decrease during this period because of the nausea and vomiting of pregnancy. Because circulating glucose levels are reduced, maternal insulin requirements are often lowered. As pregnancy progresses, the diabetogenic actions of increasing amounts of placental hormones and the increasing insulin insensitivity outweigh the effects of the fetal siphon. Women with type 1 diabetes appear to have similar alterations in insulin sensitivity during pregnancy to woman with normal glucose tolerance.98 Thus maternal insulin requirements usually increase during the second half of gestation to levels two to three times higher than prepregnancy values.47,78,98
The diabetic woman may have altered insulin requirements during labor, probably because of an increase in energy needs (and thus glucose utilization) and the presence of oxytocin, with its insulin-like effects. After delivery and removal of the placenta, levels of estrogens, progesterone, and hPL fall rapidly. This quickly reverses the insulin insensitivity of pregnancy. Maternal insulin requirements usually fall rapidly to prepregnancy levels or even below (due to a rebound phenomenon). Oxytocin may also contribute to these changes. As a result, the insulin-dependent diabetic woman may need little or no exogenous insulin the first few days after delivery. Insulin requirements generally return to prepregnancy levels by 4 to 6 weeks postpartum.37
Levels of glycosylated hemoglobin and other glycosylated proteins are useful in monitoring glucose concentrations over time and in genetic counseling and have been used to evaluate fetal and maternal risks for complications in a pregnancy complicated by maternal diabetes. Glycosylated hemoglobin is formed slowly over the lifespan of the red blood cell and represents an overall measure of glycemia. The parameters used most frequently are hemoglobin A1c (the most abundant component of hemoglobin A) and total amounts of hemoglobin A. Levels of glycosylated hemoglobin A reflect ambient glucose concentrations over the previous 4 to 6 weeks and have been used to monitor maternal glycemic control on a monthly basis. Hemoglobin A1c is higher in pregnant diabetic women than in other pregnant women, but lower than in nonpregnant diabetic women. Maternal levels of hemoglobin A1c correlate with development of fetal anomalies, with the lowest levels having the least risk.98,111 Hemoglobin A1c is not as useful as an independent measure of glycemic control in pregnancy, because these levels may not be a good predictor of capillary blood glucose levels in the woman.
Fetus of a diabetic mother
Maternal metabolic abnormalities, particularly hyperglycemia during the period of embryonic organogenesis (3 to 8 weeks’ gestation), have been associated with an increased risk of congenital anomalies.1,78,81,98 The most common anomalies involve the cardiovascular system (especially atrial septal defects, ventricular septal defects, and transposition of the great vessels) and central nervous system (neural tube defects), with urinary tract malformations, anal/rectal atresias, and caudal regression syndrome also seen with increased frequency.78 The risk is increased four- to eight-fold in women with overt diabetes prior to conception, with no increase in women who develop GDM after the first trimester or in offspring of diabetic fathers.98 Rigid glycemic control before conception and during early pregnancy has been associated with a reduction in the frequency of congenital anomalies, but the risk is still higher than in the nondiabetic woman.62,98 Preconceptional counseling and glycemic control are critical for improving pregnancy outcome in a diabetic mother. Using data from a meta-analysis, Ray and colleagues reported that mean fasting capillary glucose levels of 70 to 130 mg/dL (3.9 to 7.2 mmol/L) and a glycosylated hemoglobin level less than 4 standard deviations above normal during the periconceptional period minimized the risk of glycemia-related anomalies.111 The basis for the increase in anomalies is not completely understood. Possible etiologies include excessive formation of free oxygen radicals in the mitochondria; inhibition of prostacyclin formation resulting in an excess of thromboxane A2, as compared to prostacyclin (thromboxane A2 is a potent vasoconstrictor that alters vascularization of tissues); altered levels of arachidonic acid and myoinositol; lipid peroxidation; increased somatomedin inhibitors; accumulation of sorbitol and trace metals; and hyperglycemia-induced apoptosis with exaggerated programmed cell death (glucose alters the expression of regulating genes).78,98,104
Macrosomia is seen in up to 20% of infants of women with GDM and 35% of women with other forms of diabetes.116 This phenomenon is generally thought to arise from increased fetal production of insulin and other growth factors, especially IGF-I and leptin, in response to fetal hyperglycemia.66,98,105 This leads to excessive transfer of nutrients across the placenta. Because maternal insulin does not cross the placenta (see Figure 16-6), fetal hyperinsulinemia arises as a response to increased placental transfer of substrates, particularly glucose. Maternal hyperglycemia increases fetal insulin, IGFs, and leptin, which up-regulates glucose transporters (GLUT) that move glucose across the placenta and into fetal cells.41,58,62,103 Up-regulation of these glucose transporters in women with type 1 diabetes may lead to excessive transfer of glucose to the fetus even with good maternal glucose control.32 Hyperglycemia in pregnant diabetic women results in fetal hyperglycemia and subsequent hyperplasia of the fetal pancreatic islet cells, with increased production of insulin, enhanced glycogen synthesis, lipogenesis, and increased protein synthesis.98,129
Increased levels of other substances (“mixed nutrients”), particularly amino acids and fatty acids, are also thought to be important in the development of fetal macrosomia, because fetal overgrowth remains a problem even with strict glycemic control.58,67 Maternal diabetes alters lipid metabolism and increases transfer of fatty acids to the fetus and the amount of triglyceride stored in the placenta.58 These changes are secondary to alterations in several factors that influence fatty acid transfer, including maternal and fetal blood flow and concentrations of serum proteins and placental fatty acid–binding protein. Elevated levels of triglycerides in the pregnant diabetic woman also enhance lipid availability to the fetus.58 Maternal triglycerides and nonesterified fatty acid levels in women with well controlled GDM correlated with fat mass and weight in the newborn.117 Increased intrauterine fat deposition may also be due to altered fat metabolism in the fetus of women with GDM.103
Levels of endogenous insulin and IGF-I in the fetus are correlated with the development of macrosomia.67 Insulin and IGF-I are major mediators of fetal growth; therefore fetal hyperinsulinemia leads to increased body fat and organ size. The major organs affected are the heart, lungs, liver, spleen, thymus, and adrenal gland. The brain and kidney are not significantly affected. The organomegaly probably arises from increased protein synthesis. Even short-term fetal hyperinsulinemia promotes storage of excess nutrients. Stringent maternal glucose control, especially during the third trimester, when fetal growth peaks, reduces the risk of macrosomia, primarily be reducing fetal adipose tissue mass.105 However, the accelerated growth velocity seen in these infants may continue into childhood and adulthood.
The placenta is also affected, especially in women whose diabetes is poorly controlled, with increased peripheral and capillary surface area and intervillous space volume. These changes may result from the increased glucose load and abnormal metabolic environment with fetal hyperinsulinemia or as a compensatory mechanism to increase oxygen delivery. The fetus of a diabetic mother shows increases in metabolic rate and oxygen consumption due to metabolism of excessive glucose and other substrates.6 The risk of later development of type 1 diabetes in infants whose mother has type 1 diabetes is 1.3%. The risk of later development of type 2 diabetes is 15% if one parent (up to 60% if both parents) has type 2 diabetes.78 Additional morbidities in infants of diabetic women are discussed on pp. 583-584
Summary
Maternal adaptations during pregnancy alter the woman’s metabolic processes. These changes are critical for protection of the mother and promote her ability to adapt to pregnancy. Maternal adaptations are essential to ensure that the fetus obtains an adequate supply of nutrients to support growth and development. Alterations in metabolic processes in the mother also interact with the course of disorders such as diabetes mellitus. An understanding of the normal metabolic changes during pregnancy increases understanding of the alterations seen in the pregnant diabetic woman, the fetus, and the newborn. Implications for clinical practice are summarized in Table 16-3.
Table 16-3
Recognize the usual changes in carbohydrate, protein, and fat metabolism during pregnancy (pp. 560-567 and Table 16-2).
Assess and monitor maternal nutrition in terms of carbohydrate, protein, and fat intake (pp. 560-567 and Chapter 12).
Counsel women regarding nutrient and energy requirements to meet maternal and fetal needs during pregnancy (pp. 560-567, 569-570).
Monitor maternal glucose and ketone status (pp. 562, 569-570).
Monitor fetal growth (pp. 569-570, 572-573).
Understand the implications of changes in the absorptive and postabsorptive states for the pregnant woman and her fetus (pp. 565-567 and Figures 16-4 and 16-5).
Monitor maternal energy status during the intrapartum period (p. 568).
Counsel women regarding changes in appetite and weight during pregnancy (pp. 560-562 and Chapter 12).
Know how glucose tolerance test parameters are altered during pregnancy (p. 569).
Recognize the effects of metabolic changes on insulin requirements of diabetic women during the prenatal, intrapartum, and postpartum periods (pp. 570-572).
Evaluate and monitor metabolic and insulin status in the pregnant diabetic woman (pp. 570-572).
Counsel diabetic women regarding the effects of diabetes on pregnancy and the fetus and of pregnancy on diabetes (pp. 570-572 and Figure 16-11).
Counsel diabetic women regarding strategies prior to pregnancy to optimize maternal and fetal outcomes (pp. 570-572).
Recognize the potential effects of diabetes on the fetus and newborn (pp. 570-572 and Figure 16-11).
Development of carbohydrate, fat, and protein metabolism in the fetus
The placenta and fetal liver function as a “coordinated multiorgan system for the exchange of nutrients and for ensuring the production of nutrients sufficient to meet fetal requirements.”9 Fetal metabolic processes are dominated by anabolism and governed primarily by glucose with little oxidation of fat. The fetus must produce energy and maintain oxidative phosphorylation in the face of a low-oxygen environment. Although energy is produced in the fetus under aerobic conditions, the fetus has a greater capacity for anaerobic metabolism and is efficient in using lactate. Oxygen consumption in the fetus is 8 mL/kg/min. Glucose contributes more than half of the substrate for oxygen consumption.9,108
The fetal caloric requirement has been estimated to average 90 to 100 kcal/kg/day. Almost all of the fetal fuel requirements are met by metabolism of glucose; lactate; and amino acids such as alanine, glutamate, and serine.9,51 The fetus also uses these substrates as major precursors for storage of fuels (e.g., fatty acids, glycogen). The stored fuels are critical energy sources during the intrapartum period and transition to extrauterine life. By term, the fetus has increased its weight 175-fold, protein content 400-fold, and fat content 5000-fold. Fetal nutrient uptake and growth is influenced by maternal nutrition and health; uterine blood flow; placental nutrient uptake, metabolism, and transfer; umbilical blood flow; and the fetal endocrine system.38
Substances are transported across the placental syncytiotrophoblast from maternal to fetal circulations. The syncytiotrophoblast is “the transporting epithelium of the human placenta,”32 consisting of two polarized plasma membranes. The microvillous membrane (MVM) faces maternal blood in the intervillous space; the basal membrane (BM) faces the fetal capillary epithelium.32
Insulin-like growth factors (IGF-I and IGF-II) are the dominant endocrine regulators of fetal growth and cord blood levels correlated with birth weight.14,20 IGF-I and IGF-II levels increase longitudinally to about 33 weeks, then increase twofold to threefold more to term. IGF-I is thought to be the primary controller of fetal size in the second half of pregnancy. IGF-I (but not IGF-II) is significantly lower in infants with fetal growth restriction.20 Cord blood IGF-I levels are also lower in women who smoke.20
Fetal leptin, adiponectin, and other adipokines (resistin, visfatin, and apelin) are involved in controlling fetal growth.15, Leptin may act by modulating growth hormone secretion and may also have a role in hematopoiesis and angiogenesis. Leptin has been found in immature subcutaneous fat cells by 6 to 10 weeks’ gestation.4 Circulating levels of leptin, an adipostatic hormone, increase after 32 to 34 weeks’ gestation, around the time of increasing body fat mass.102,105 Levels of leptin decrease rapidly after birth and may help limit energy expenditure and conserve the infant’s nutrient reserves for later growth and development.55 Leptin levels at birth have been reported to correlate with intrauterine growth and be a predictor of neonatal bone mass.20,69 Concentrations of leptin are higher in large-for-gestational-age (LGA) infants than in infants of appropriate size for gestational age; concentrations are also higher in LGA infants than in small-for-gestational-age (SGA) infants.
The nutritional, metabolic, and hormonal status during fetal and early postbirth life can alter organ development including the hypothalamus and other endocrine structures (see Chapter 19).18,99 The mechanisms by which the fetal environment influences later status are thought to be related to placental adaptive responses to the intrauterine environment. After birth these adaptations may no longer be appropriate for the extrauterine environment and lead to altered glucose-insulin metabolism, lipid metabolism, and endocrine programming. Alterations in fetal and early neonatal nutrition “result in neuroendocrine, pancreatic, skeletal muscle, and adipose tissue dysfunction, and increased food intake and decreased energy expenditure. This leads to increased adiposity and adult disease.”18 These adult onset disorders include insulin resistance and type 2 diabetes, obesity, hypertensive disorders, coronary artery disease, and osteoporosis.18,28,33,36,39,99
Carbohydrate metabolism
The fetus has been described as a “glucose-dependent parasite” and uses glucose from the mother as the major substrate for energy production.82 Eighty percent of fetal energy comes from carbohydrate (glucose) oxidation.71,114 Fetal glucose utilization rates (averaging 5 mg/kg/min) are higher than in adults (2 to 3 mg/kg/min).51,71 Even with fetal growth restriction, maternal glucose is the major energy substrate for the fetus, although the placenta can produce alternate substrates such as lactate and ketone bodies for use as energy and for glycogen synthesis.71
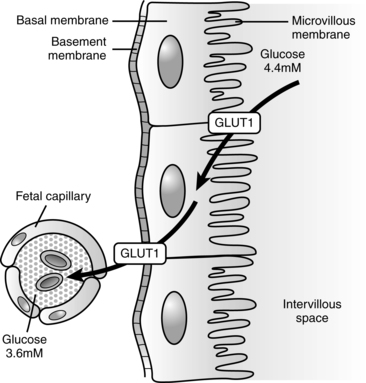
Glucose is transported across the placenta via sodium-independent carrier-mediated facilitated diffusion.68,116 The placenta has a high facility for glucose uptake and transport via a family of membrane transport proteins (GLUTs, which is the gene symbol for facilitated glucose transporter) on the microvillous membrane facing maternal blood and fetal-facing basal membrane of the placenta.62,68,108 Various GLUT isoforms are expressed in the syncytiotrophoblast, including GLUT-1, -3, -4, and -12. GLUT-4 and -12 are sensitive to insulin.68 GLUT-1 and GLUT-2 are expressed early in development and are found on both the trophoblast and blastocyst.104 GLUT-1 is the major fetal glucose transporter; fetal GLUT-2 levels remain low.118 GLUT-1 is expressed on almost all fetal tissues, enhancing cellular glucose uptake.121 In animal models maternal hyperglycemia during early gestation down-regulates these receptors, increasing the risk of apoptosis and neural tube and limb defects. There is a fivefold greater increase in GLUT-1 on the microvillous membrane facing maternal blood than on the fetal-facing basal membrane during the first trimester.62,68,108 This increases the movement of maternal glucose into the placenta, where approximately 30% to 40% is used by the placenta for oxidation or converted to glycogen and lactate to meet its energy needs.68
Basal membrane glucose transport is the rate-limiting step in fetal glucose transfer.8,68 In the second half of pregnancy, basal membrane GLUT-1 receptors increase at least twofold and their activity increases 50% to meet increasing demands with fetal growth in late pregnancy.62,108 If the uteroplacental nutrient supply is diminished, the fetus consumes nutrients and oxidizes them at the usual rate, but the placenta reduces consumption of both nutrients and oxygen. Increased placental GLUT-1 receptors with increased glucose transport to the fetus have been found in diabetic women with hyperglycemia.120 Placental glucose transport is summarized in Figure 16-7.
During the first few days after fertilization, the zygote has a limited ability to metabolize glucose. After the embryonic genome is activated, glucose metabolism increases.29 Both glucose and pyruvate uptake increase initially; glucose uptake remains high throughout pregnancy, while pyruvate uptake falls.
The major regulators of fetal growth are IGF-I and IGF-II, which stimulate cell proliferation, differentiation, and metabolism.38 IGFs act via cell membrane receptors and are modulated by a group of binding proteins. The liver is the main source of IGFs, which have autocrine, paracrine, and endocrine functions. IGF-I can be detected in fetal tissue by 9 weeks’ gestation and in fetal circulation by 15 weeks’ gestation.34 Both IGF-I and IGF-II increase with gestational age. In early gestation, IGF-II activity, which is not significantly affected by nutritional factors, is predominant. In late pregnancy, IGF-I, which is regulated by nutrient availability, is predominant. Fetal growth in later gestation is regulated by the interaction of glucose, insulin, and IGF-I.38 Glucose transfer across the placenta stimulates fetal insulin release. Insulin in turn stimulates lipogenesis and IGF, which increases fetal anabolism and placental uptake of glucose and other nutrients for fetal (versus placental) use.
Lower levels of IGF-I are seen in preterm and growth-restricted infants.89 Levels are also reduced in infants of mothers who smoke.113 Even though glucose and insulin concentrations are lower with fetal growth restriction, the fetus maintains glucose uptake and utilization due to increased insulin sensitivity, enhancing movement of glucose into cells. This is mediated by increased expression of GLUT and insulin-responsive glucose transporters.116 With chronic substrate deprivation, the growth restricted fetus will develop systems for glucose production, initially by glycogenolysis (breakdown of glycogen to glucose) and later by gluconeogenesis (production of new glucose from lactate, amino acids [especially alanine], and glycerol) along with the reduction in fetal growth.116 Poor fetal nutrition may alter the ability to produce and respond to insulin in adulthood, increasing the risk of type 2 diabetes, or may alter fetal low-density lipoprotein metabolism, increasing the risk of later coronary artery disease.18,36,89,74,131
Under basal, nonstressed conditions, the fetal glucose pool is in equilibrium with the maternal pool.132 Enzymes for fetal gluconeogenesis are present by 3 months’ gestation, but fetal glucose production is minimal and maternal glucose remains the source of fetal glucose.52,71,116 Ambient fetal glucose levels are generally 20 to 40 mg/dL (1.1 to 2.2 mmol/L) less than maternal levels (or 70% to 80% of maternal values) and increase slightly toward the end of gestation.71,78 This gradient is regulated by the placenta and favors transfer of glucose across the placenta from the mother through carrier-mediated facilitated diffusion (GLUTs) (Figure 16-7).
The usual lower limit for fetal glucose is 54 mg/dL (3 mmol/L), especially after 30 weeks.51 If fetal glucose supply decreases, then placental glucose uptake increases, and vice versa, as seen with the infant of a diabetic mother.68,71 Extra glucose is stored by the fetus as glycogen and triglycerides.73 The levels at which glucose carriers become saturated are significantly above the usual maternal blood glucose level, which promotes a constant supply of glucose to the fetus.82 There is no net transfer of insulin or glucagon to the fetus (see Figure 16-6). With adequate maternal nutrition, gluconeogenesis and ketogenesis are not seen in the fetus.93,116 If the placental supply of glucose is inadequate, the fetus can use ketone bodies and other substrates as alternative energy sources.116 As noted above, with prolonged glucose deprivation, the fetus can produce some glucose initially by glycogenolysis and later by gluconeogenesis, probably mediated by cortisol.51,116
Fetal glucose utilization is independent of maternal glucose availability. The linear relationship between maternal and fetal glucose levels is maintained during maternal euglycemia, hypoglycemia, and hyperglycemia.73 The mother meets this demand by an increasing reliance on fat metabolism for her own fuel needs. If the maternal system is not able to meet the fetal demand for gluconeogenic precursors, hypoglycemia can result. Because fetal blood glucose levels are 70% to 80% of maternal values, maternal hypoglycemia leads to even lower fetal blood glucose levels.78 Placental glucose transfer increases with increasing gestational age not only by increases in GLUT, but also by increases in insulin receptors on fetal tissues particularly adipose tissue and skeletal muscle. Glucose is also needed by the fetus for protein synthesis, as a precursor for fat synthesis, for conversion to glycogen for storage, and as the primary substrate for oxidative metabolism. Most of the transferred glucose is oxidized to carbon dioxide and water with release of energy by the tricarboxylic acid cycle and oxidative phosphorylation.
The fetus has an active capacity for anaerobic metabolism, which has a greater role in fetal metabolic processes than it does in adults. The fetus has increased amounts of glycolytic isoenzymes such as hexokinase, glucose-6-phosphate dehydrogenase (G6PD), and pyruvate dehydrogenase, which favor anaerobic glycolysis.73 Fetal and especially placental tissues actively metabolize glucose to lactate. The lactate generated by the placenta serves as an important fuel for the fetus.51 Lactate is a major precursor of fetal hepatic glycogen and fatty acid synthesis.73 Under aerobic conditions the fetus is a net consumer of lactate. The placenta produces large amounts of lactate and ammonia, which may help in regulating metabolic activities in the fetal hepatocytes.51 The fetus can also use ketone bodies produced from β-oxidation of fatty acids as another alternative fuel source.116
The fetal liver contains enzymes needed to both synthesize and catabolize glycogen to store and release glucose. The fetus can also synthesize glycogen from pyruvate, acetate, and alanine.73 Glycogen synthesis is greater than glycogenolysis in the fetus. Glycogen synthetase and other gluconeogenic enzymes can be found in the liver from the eighth week and increase to term, with a rapid increase seen after 36 weeks’ gestation.73,93 Deposition of hepatic glycogen during the perinatal period is regulated by glucocorticoids and insulin. Glucocorticoids may induce glycogen synthetase, which is then activated by insulin. Fetal cells have increased insulin receptors, greater receptor affinity for glucose, and delayed maturation of hepatic glucagon receptors. These changes promote storage of glucose as glycogen and fat. Glycogen can be synthesized from lactate, pyruvate, alanine, and glycerol as well as glucose.108
Glycogen is stored in fetal tissues from 9 weeks’ gestation on and increases slowly from 15 to 20 weeks, then more rapidly during the third trimester.71 Until 20 to 24 weeks, the fetal liver is the main glycogen storehouse; after that time, glycogen is stored in cardiac and skeletal muscle, and to a lesser extent in the kidneys, intestines, and brain (primarily in the astrocytes).107,108 Liver and skeletal muscle glycogen concentrations peak at term.108 Compared with adults, the term fetus has significantly more liver, skeletal muscle, and cardiac muscle glycogen stores. Liver glycogen synthesis is regulated by fetal insulin, the hypothalamic-pituitary axis, and thyroid hormones.108
Immunoreactive insulin is present in fetal plasma and islet tissue as early as 8 weeks’ gestation.71,120 Insulin levels are dependent upon fetal glucose levels.51 Insulin production is stimulated by increasing glucose and amino acid concentrations, especially after 20 weeks.71 Fetal glucose metabolism is relatively independent of the insulin-glucagon regulatory mechanisms seen after birth, however. Acute changes in glucose concentrations leading to hypoglycemia or hyperglycemia do not significantly alter fetal insulin or glucagon secretion.118 However, secretion of these hormones is markedly altered by chronic changes such as long-term hyperglycemia in a diabetic woman, which augments insulin secretion by β-cell hyperplasia and suppresses glucagon, or by chronic maternal malnutrition, which depresses insulin and stimulates release of fetal glucagon.71,132 Because insulin is a fetal growth hormone, fetal hyperinsulinemic states are associated with fetal and neonatal macrosomia.57,71
Glucagon is found in fetal plasma by 15 weeks’ gestation and reaches peak concentrations at 24 to 26 weeks. In comparison with adults, the number of fetal hepatic glucagon receptors is decreased and insulin receptors are increased. Fetal liver cells, erythrocytes, monocytes, and lung cells have an increased affinity for insulin. These attributes promote insulin-mediated anabolic processes such as glycogen formation and decrease glucagon-mediated catabolism.132
The fetal liver receives the highest net flux of maternal glucose because it is the first organ system encountered by blood returning from the placenta (see Chapter 9). Fetal hepatic enzymes for glycogenesis (carbohydrate to glycogen) are increased, whereas enzymes for glycolysis (carbohydrate to pyruvate and lactate) and gluconeogenesis (fat and protein to glucose) are present but decreased. For example, glucose-6-phosphatase, an enzyme involved in gluconeogenesis and inhibited by glucose and amino acids, is at 20% to 50% of adult levels at midgestation. These relationships are maintained until birth, when decreased glucose availability and onset of high-fat feedings stimulate decreases in glycogenolytic enzymes.
Lipid metabolism
Lipids are a critical component of brain development, of retinal development, and for structural and functional integrity of neuronal and glial membranes, and are the main component of the myelin sheath.32,58,63,127 Fetal fat content increases during gestation from 0.5% of body weight in early gestation to approximately 3.5% by 28 weeks and 16% by term or by approximately 3.4 g/kg/day in the third trimester.127 This increase is due to transfer of fatty acids from the mother and active lipogenesis in the fetal liver and other tissues. Lipogenesis is primarily through the fatty acid synthetase pathway, which is highly active in the fetus. The placenta modulates the fatty acid supply for its use and for fetal use.19 Placental leptin stimulates lipolysis and is excreted to both maternal and fetal circulations. As a result the placenta may have a role in modulating fatty acid supply based on fetal demands.44
Long-chain polyunsaturated fatty acids in the mother are primarily found in the form of esterified fatty acids such as triglycerides, phospholipids, and esterified cholesterol. Maternal lipoproteins do not cross the placenta but are taken up by the placenta where they are broken down and their products used for energy or steroid hormone production or released to the fetus.59 The placenta contains various tissue receptors, enzymes, and fatty acid binding proteins to upload these lipoproteins and to transport polyunsaturated fatty acids and nonesterified fatty acids to the fetus.32,68 Free fatty acids cross by diffusion in limited amounts, with a net flux of unesterified fatty acids to the fetus.58,59,125 These fatty acids are derived primarily from maternal circulating free fatty acids or cleavage of maternal triglycerides by lipoprotein lipase.56 The maternal diet is reflected in the fatty acid content of fetal tissues.59 Ketone bodies and glycerol also cross the placenta.59 Placental lipid transport is illustrated in Figure 16-8. Because transport of fatty acids is partly controlled by maternal concentrations, increased maternal levels are associated with increased transfer and fetal storage.16,19,62,123,125 Other factors influencing free fatty acid transfer include serum albumin level, fatty acid chain length, lipid solubility, uteroplacental and umbilical blood flow, α-fetoprotein, placental proteins that bind fatty acids, and binding affinity.58,71,125
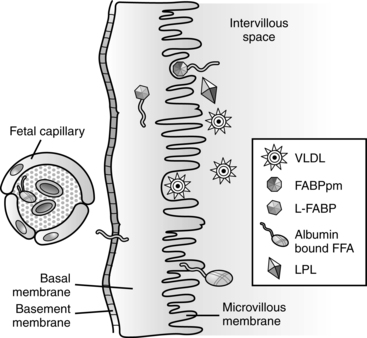
Essential fatty acids (EFA), such as linoleic and α-linolenic acids, cannot be synthesized by the fetus and must be transported across the placenta.32,58,59,68 These substances can then be desaturated by the fetus to form other fatty acids.58 Transfer of essential fatty acids increases in late pregnancy, when demand for brown and white adipose tissue development and for vascular and neural growth increases; however, some early transfer is needed for uteroplacental vascular development.125 Long-chain polyunsaturated fatty acids (LCPUFA) from the mother are critical for development of the brain and retinal and brain growth.44,58 The fetus has limited capacity to synthesize LCPUFA due to low levels of desaturating enzymes.44,58 In the third trimester when fetal neuronal and vascular growth is high, selective transport of LCPUFA derivatives such as arachidonic acid and docosahexaenoic acid (DHA) increases.58,59,126 The preterm infant thus needs a diet that includes these critical substances.44 Linoleic acid is converted to arachidonic acid, the main precursor for prostaglandins, thromboxanes, and leukotrienes; α-linolenic acid is converted to DHA (needed for fetal growth and development) and eicosapentaenoic acid (precursor of prostaglandins that inhibit platelet aggregation).19
Increased fat deposition during the third trimester is associated with increasing fetal weight and decreased serum triglycerides because these are used in fat deposition.23 Most (80%) of the fetal fat accretion during this period is due to de novo synthesis from acetyl coenzyme A (CoA) with formation of palmitic acid, especially in the brain and liver.56,125
Fetal lipid metabolism is characterized by early development of mechanisms for cholesterol metabolism and lipogenesis with decreased lipolytic activity throughout gestation, except in the liver.101 Fetal fatty acid synthesis occurs through lipogenesis and desaturation of essential fatty acids. Lipogenesis is dependent upon substrate availability. Lipogenic precursors include glucose, lactate, and ketone bodies; the latter two are the most important.56,109
The rate of synthesis is primarily controlled by the ratio of plasma insulin-to-glucagon levels. Insulin stimulates and glucagon inhibits fatty acid synthesis. The action of glucagon is mediated by cyclic adenosine monophosphate (cAMP), which inhibits acetyl-CoA enzymes.56 The high insulin-to-glucagon ratio in the fetus promotes fatty acid synthesis. Lipogenesis is increased by glucose, fatty acids, and T4 (see Chapter 19) and reduced by catecholamines.125 The fetal liver and brain contain enzymes for ketone oxidation as an alternative energy substrate; the fetal brain can use ketones for energy by 10 to 12 weeks’ gestation.59,108 Lipolytic activity becomes active after delivery, when the infant can no longer rely on a constant glucose supply from the mother and must cope with a relatively high fat intake.
By 15 weeks the fetus has developed enzymes to convert acetate or citrate to fatty acid and thus the potential to use fat as an alternative source of energy. Blood lipid and free fatty acid levels are relatively stable after about 26 weeks but remain low until after delivery. The free fatty acids that cross the placenta are used by the fetus in organ development; synthesis of pulmonary surfactant, other phospholipids, bile, and serum lipoprotein; formation of cell membranes; precursors for prostaglandins; and as second messenger precursors. Fatty acids needed by developing neuronal and glial cells and in formation of the myelin sheath are synthesized within the brain.56
The fetus needs cholesterol for cell membranes, bile acid synthesis, embryogenesis (cell proliferation and differentiation), synaptogenesis, production of steroid hormones, and cell-cycle regulation.58 The fetus obtains cholesterol from endogenous biosynthesis and placental transport.59 Fetal cholesterol levels are high in early pregnancy, decrease, then rise, peaking in the third trimester.59,101 Fetal and maternal cholesterol levels are not correlated.59 Activity of the pentose phosphate intermediary metabolic pathway is also high in the fetus. Activity of this pathway is associated with cell proliferation and an increased requirement for ribose phosphate precursors of nucleic acids and provision of nicotinamide-adenine dinucleotide (NADH) for synthesis of long-chain fatty acids.
Protein metabolism
At least 10 amino acids are essential for the fetus, including those essential for adults plus cysteine and histidine, with a dependency on arginine and tyrosine. Arginine and leucine are insulin secretogogues; arginine may also enhance vascular development. Taurine helps regulate metabolism and is needed in development of the heart, eye, and brain. Because of the relative inactivity of hepatic enzymes such as cystathionase and phenylalanine hydroxylase, the fetus cannot synthesize tyrosine and cysteine from phenylalanine and methionine. The fetus uses amino acids for protein synthesis or oxidation because organ development involves continuous remodeling (breakdown and resynthesis) of tissue. The availability of glucose and other substances influences fetal amino acid catabolism and protein accretion.54
There is a net flux of most amino acids from the mother to the fetus.82 Because concentrations of most amino acids are higher in fetal than maternal blood, amino acids are actively transported from maternal blood across the placental microvillous membrane.68 Amino acid levels in the syncytiotrophoblast are higher than in either maternal or fetal circulations. Facilitated diffusion moves substances across the syncytiotrophoblast basal membrane to fetal blood.68 There are at least 15 different amino acid transporters, each mediating uptake of several different amino acids, and each amino acid may use multiple transport systems.68
Placental amino acid transport is illustrated in Figure 16-9. Fetal-to-maternal amino acid nitrogen ratios average 1.03 to 3.0, with a net active transfer of nitrogen to the fetus of 54 nmol/day and a total accumulation of about 400 g of protein by term.6,54,82 Amino acid transport is not significantly affected by fluctuations in uterine or placental blood flow.54 Transport may be down-regulated in the growth restricted fetus.68
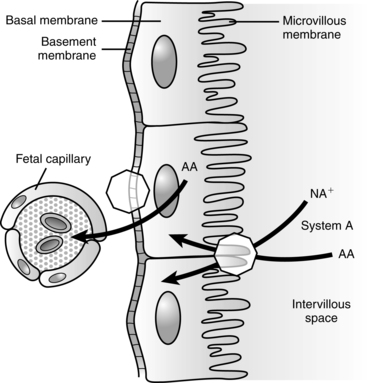
Amino acids are supplied to the fetus in greater amounts than are needed for nitrogen accretion. The fetus uses the carbon from excess amino acids for oxidation and to make nonessential amino acids.54 Some critical amino acids are not transferred across the placenta directly but are, instead, produced in the placenta. For example, glutamate, which is needed for neurotransmitters and brain development, is not transferred from mother to fetus. Instead glutamine is transferred from the mother to the placenta, where it is used to produce glutamate. Similarly asparagine is used by the placenta to produce aspartate, another neurotransmitter. Both glutamate and aspartate are toxic, so by transferring precursors, the placenta can produce only the amounts that are needed by the fetus. The placenta also produces ammonia, which is used by the fetal liver for additional protein synthesis.
Neonatal physiology
Transitional events
Metabolic transition is characterized by a shift from the anabolic-dominant fetal state to the catabolic state of the neonate. This transition is influenced by genetic, environmental, and endocrine factors as well as by major alterations in energy metabolism within the mitochondria. Changes in metabolism with birth are regulated by the expression of specific genes and gene products that alter the activity of various enzymes.23 Shortly prior to term birth, induction of hepatic glucose production begins and is enhanced postbirth by increased glucocorticoids and glucagon.116 The ability of the fetus to use glucose anaerobically and to readily metabolize lactate may be important in maintaining homeostasis during the stresses of labor and delivery. The fetus prepares for this transition during the last weeks of gestation by increasing fuel storage in the form of glycogen and lipids. Glycogen is critical in order to maintain glucose homeostasis immediately after birth, whereas the fat stores, through lipolysis of fatty acids and ketone bodies, serve as an alternate energy source. Postnatal changes in metabolism involve the transition from the almost exclusive reliance on glucose for energy production in the fetus to markedly increased use of fatty acid oxidation and ketone body use for energy production in the neonate. An increase in epinephrine, norepinephrine, and glucagon and a decrease in insulin at birth promote mobilization of fatty acids and glycogen metabolism.73
Carbohydrate metabolism
Birth results in the loss of the maternal glucose source. As a result, neonatal blood glucose normally falls after birth, reaching a nadir at 30 to 90 (generally around 60) minutes after birth (Figure 16-10).71 This fall in glucose is thought to be necessary for activating postnatal glucose production processes.116 Glucose values then rise and stabilize by 2 to 3 hours after birth.52,71 The glucose nadir and timing of the nadir are influenced by maternal glucose infusion during the intrapartum period.71 Steady-state hepatic release of glucose at 4 to 6 mg/kg/min is seen by 2 to 3 hours in term infants.40,107 Mean glucose levels in term healthy infants in the first week average 80 mg/dL.71 The basis for the fall in blood glucose is summarized in Table 16-4.
Table 16-4
Basis for Changes in Blood Glucose Levels Immediately after Birth
| CHARACTERISTIC | BASIS |
| Immature liver enzyme systems | Promotes glucose storage rather than release |
| Larger brain in proportion to body size | Obligatory glucose user |
| Increased red blood cell volume | Obligatory glucose user |
| Decreased liver response to glucagons | Limits release of glucose from glycogen stores |
| Increased energy needs | Increased metabolic and motor activity with birth |
The newborn responds to the decrease in blood glucose in several ways. The rapid glycogenolysis (liberation of glucose from glycogen) after birth is stimulated by a fall in the insulin-to-glucagon ratio and sluggish insulin secretion (because of the decreased blood glucose with removal of the placenta), increased serum glucagon, stimulation of the sympathetic nervous system with catecholamine release, increased thyroid stimulating hormone, and increases in hepatic cAMP.23,107,118 Because insulin promotes transfer of glucose out of the blood into cells, the lowered levels decrease transfer and elevate blood glucose levels. Glucagon stimulates conversion of glycogen into glucose, also raising blood glucose levels.
Hepatic glycogen stores decrease markedly during this period. In the term infant, glycogen stores only last an estimated 10 hours after birth before glucose must be produced by gluconeogenesis (production of new glucose from lactate, amino acids [especially alanine], and glycerol).40 As glycogen falls, the newborn responds by mobilizing fat stores with release of free fatty acids. This response is stimulated by the catecholamine release associated with cooling at birth (see Chapter 20), which rapidly increases free fatty acids.121 Increased secretion of catecholamines at birth increases glucagon secretion (together these activate hepatic glycogen phosphorylase and glycogenolysis), suppresses insulin, reverses the fetal insulin-to-glucagon ratio, increases lipolysis to provide an alternate energy source, activates enzymes needed for gluconeogenesis, and augments growth hormone secretion.116,121,132 Healthy infants are able to readily mobilize free fatty acids and oxidize ketones to maintain their blood glucose levels.
Breastfed infants have lower blood glucose levels and higher ketone bodies than bottle-fed infants.52,116 The elevated ketone bodies may provide an alternate fuel during the period of lower nutrient intake as breastfeeding is being established. This may be especially important for the brain, which can readily use ketones as an alternate energy source.23,52,116,121 Production of glucose from amino acids, especially alanine, increases and accounts for 4% to 10% of the glucose used for energy in term infants.
Gluconeogenesis also contributes to systemic glucose production after birth. Substrates metabolized by the liver to produce glucose include glycerol, lactate, alanine, and pyruvate.40,71,73,121 Lactate may account for up to 30% of hepatic glucose production, with 5% to 10% from alanine and glycerol via gluconeogenesis.94 Gluconeogenic enzyme activity increases after birth. Both glycogenolysis and gluconeogenesis pathways are dependent on the hepatic microsomal glucose-6-phosphatase (G6PD) enzyme system, which has only about 10% of adult activity at birth.60 The cortisol surge with birth stimulates G6PD activity in the liver and hepatic glucose release.116 Adult values are reached by about 3 days in term infants, but take longer in preterm infants.60 By 12 to 24 hours after birth, both gluconeogenesis and ketogenesis are active.93,121 From late fetal life to 3 days after birth, blood glucose regulation is glucose dominant. As a result, gluconeogenesis is diminished during this period. After this period, blood glucose regulation becomes insulin dominant (the adult pattern). The increased gluconeogenesis after birth is regulated by changes in the serum insulin-to-glucagon ratio, catecholamine secretion, fatty acid oxidation, and activation of hepatic enzyme systems.121 The very low–birth weight (VLBW) infant is able to produce glucose to meet basal metabolic needs with appropriate responses to exogenous glucose and amino acid infusions, although hepatic glucose production may be sluggish or incomplete. In the first day after birth in term infants, about 50% of glucose is produced via glucogenolysis and 30% to 40% by gluconeogenesis (primarily from glycerol, but also from alanine and lactate).52
After birth, GLUT-1 glucose transporters decrease, whereas GLUT-2 (involved in uptake of glucose by liver, pancreatic β cells, and intestinal and renal epithelium) and GLUT-3 (involved in uptake by neurons, especially in the cerebellum, skeletal muscle, and other tissues) increase rapidly.35,52,120,122 Activity of GLUT-4 (involved in uptake by muscle and adipose tissue) and other GLUTs increases more slowly.120,122 GLUT-1, although lower in the neonate than in the fetus, remains active and needed for transfer of glucose into red blood cells and across the blood-brain barrier.126 Both GLUT-1 and GLUT-3 are important in regulating brain glucose supply and are prominent in the blood-brain barrier epithelial cells, astrocytes, oligodendrocytes, choroid plexus, and cerebral cortex.52 Major alternative fuels for the brain are pyruvate, lactate, and ketones. These are transported across the blood-brain barrier by monocarboxylate transporters.52
Increased glucagon concentrations and norepinephrine are important in subsequent activation of the hepatic gluconeogenic enzymes.71,73,77,122 The predominant enzymes activated during this period are hepatic glycogen phosphorylase, glucose-6-phosphate dehydrogenase (G6PD), and phosphoenolpyruvate carboxykinase (PEP-CK). Hepatic glycogen phosphorylase is activated by norepinephrine and glucagon and stimulates glycogenolysis.40,116 G6PD activity increases markedly after birth, increasing hepatic release of glucose.116 This is probably due to surges of glucagon and cAMP, which help shift the activity of the liver from glycogen storage to glucose production. PEP-CK, which is inhibited by insulin, is the rate-limiting enzyme for gluconeogenesis.40,116 With changes in the insulin-to-glucagon ratio after birth, this enzyme increases about 20-fold, reaching adult levels by 24 hours after birth and thus increasing gluconeogenesis.40,121
The resumption of a carbohydrate source (i.e., feeding) generally stabilizes the infant’s glucose concentration, and most neonates achieve a steady-state in their glucose concentrations by about 5 days. The first enteral feeding immediately increases blood glucose levels. This increase is accompanied by an increase in plasma insulin levels in term infants and development of cyclic changes in insulin and blood glucose levels. In preterm infants the initial feeding is not accompanied by similar hormonal changes and the cyclic responses in insulin and blood glucose take 2 to 3 days to develop (and longer in VLBW infants or infants who are not fed). Enteral feeding stimulates production of digestive hormones and secretion of peptides critical for induction of gastrointestinal tract maturation and development of the enteroinsular axis (see Chapter 12). These changes lead to additional modifications of hepatic metabolism.
Basal glucose production in newborns is 4 to 6 mg/kg/min versus 2 to 3 mg/kg/min in adults and approximately 6 to 8 mg/kg/min in preterm infants and infants with symmetric growth restriction.40,51,52,116 The high glucose needs in neonates reflect the increased brain-to-body mass, in that the brain has a high obligatory glucose use.62,73 For infants weighing less than 1000 g, glucose is the major energy source for most of the neonatal period.35 Newborns are able to use ketone bodies and lactate for brain energy metabolism if inadequate glucose is available, although excess levels can be damaging. This mechanism is present in term, large preterm, and term small-for-gestational-age (SGA) infants but is absent in VLBW infants and other intrauterine growth–restricted infants who are at risk for hypoglycemia.50 Glycogen stores in astrocytes may provide another alternative source of glucose for the neurons during periods of low glucose.50
Lipid metabolism
The inability of the fetus to readily oxidize fatty acids is rapidly reversed at birth because of changes in the functional ability of enzymes such as carnitine palmitoyltransferase. During this period the levels of glucose and free fatty acids are mirror images of each other. Lipolysis, with enhanced oxidation of free fatty acids and ketogenesis, increases quickly after birth, reaching a maximum within a few hours.36,56,76,107 This increase is reflected in changes in plasma free fatty acid levels, which rise rapidly beginning 4 to 6 hours after birth and reach adult levels by 24 hours. By this time, 60% to 70% of the infant’s energy is produced from oxidation of fat.114
Fat is the major form of stored calories in the newborn, and the preferred energy source for tissues such as the heart and adrenal cortex, which have high energy demands. Fat stores are significantly reduced in low–birth weight infants. After birth, mobilization of fatty acids from these stores is reflected in the rise in serum levels over the first few hours. This process is initiated by the increase in catecholamines and glucagon, resulting in increased cAMP followed by an increase in protein kinases, phosphorylation, and activation of adipose tissue lipase with release of fatty acids.36,56
The transition from glucose to fatty acid oxidation after birth is reflected in the fall of the respiratory quotient from 0.9 to less than 0.8 by 2 hours.76,114 This indicates that the infant has moved from obtaining nearly two thirds of its energy from oxidation of glycogen immediately after birth, to deriving most of its energy from fat metabolism, conserving glucose to ensure an adequate glucose supply for the central nervous system.40,71,107 This transition reflects increasing dependence on oxidative metabolism and is associated with an increase in the number of mitochondria and enzymes of the Krebs cycle and changes in serum free fatty acid levels. This shift to fat metabolism is delayed in infants of diabetic mothers with hyperinsulinemia.71,73 The neonate’s brain may use free fatty acids, along with branched-chain amino acids and ketones, as additional energy sources.40
Serum cholesterol levels in cord blood are 50 to 75 mg/dL (1.3 to 1.9 mmol/L), about one-half adult values, and serum triglycerides average 30 to 50 mg/dL (0.3 to 0.6 mmol/L).101 Cholesterol in preterm infants may be as high as 100 mg/dL (2.6 mmol/L) if they are born during the time of the normal fetal cholesterol peak in the third trimester.101 Low-density lipoprotein cholesterol concentrations decrease with increasing gestational age and are 25 to 30 mg/dL (0.5 to 0.8 mmol/L, about one fifth adult values) at term; high-density lipoprotein cholesterol is 25 mg/dL (0.6 mmol/L) at term (about one-half adult values).101 Very-low-density lipoprotein (VLDL) transports less than one third of the total serum triglycerides in the term infant, versus that in the adult in whom VLDL is the major transporter of triglycerides.101 Serum cholesterol and lipoprotein increase rapidly after birth, especially in breastfed infants.101
Protein metabolism
Serum amino acid levels are higher during the first few weeks of life.92 Urinary amino acids are elevated immediately after birth, with excretion of 8.8 mg/day in preterm and 7.6 mg/day in term infants versus 2.5 mg/day in older children. The average body nitrogen content at birth is 2%.92
Clinical implications for neonatal care
The newborn’s transitional state in relation to glucose homeostasis can result in problems even for healthy newborns as they attempt to provide adequate energy for maintenance and growth. Alterations in metabolic processes in the newborn can result in clinical problems, most notably hypoglycemia. The status of metabolic function in the newborn also influences nutritional needs (see Chapter 12).
Neonatal hypoglycemia
The neonate may develop hypoglycemia if glycogen stores are insufficient to provide fuel during transition until production of energy by fat oxidation is adequate, or if the infant fails to adequately mobilize available glycogen stores. The physiologically optimal range for plasma glucose is given as 70 to 100 mg/dL (3.9 to 5.6 mmol/L).77,122 Plasma glucose levels (whole blood glucose levels are 10% to 15% lower than plasma levels) below 40 mg/dL (2.2 mmol/L) are uncommon in the first few hours after birth in healthy infants with early feeding, and the lowest optimal level of glucose is probably around 60 mg/dL (3.3 mmol/L).24,71,77,122 Arterial samples are slightly higher than venous samples (by 10% to 15%) with capillary samples intermediate.10 Elevated hematocrits can lead to lower glucose levels due to the high glucose utilization by the red blood cells. Newer technologies for continuous interstitial glucose monitoring, currently being investigated for use in the neonate, have identified more episodes of intermittent blood glucose levels than previously recognized, however the physiologic significance and long-term significance of these episodes are unknown.48,53
Four approaches have been used to define hypoglycemia.23,25 These include approaches based on appearance of clinical manifestations, measured glucose value ranges, acute metabolic changes and endocrine responses, and long-term neurologic outcomes.23 None have been satisfactory. Aynsley-Green and Hawdon note that “hypoglycemia is a continuum, no single blood glucose concentration reflecting functional changes in every infant at that level.”5 Similarly, an expert panel concluded that the “rational definition of hypoglycemia is clearly not a specific value but a continuum of falling blood glucose values, creating thresholds for neurologic dysfunction, which may vary from one cause of hypoglycemia or clinical circumstance to another.”22
Focus should be on promoting normoglycemia with prompt intervention for less optimal values.22,104 The optimal glucose value and risk for neurologic sequelae probably varies from infant to infant depending on their brain maturity, glycogen stores, presence of hypoxia or ischemia, activity of gluconeogenic pathways, glucose transport status, and brain glucose demand.24 Most sources cite levels below 40 to 45 mg/dL (2.2 to 2.5 mmol/L) as cutoff values for neonatal hypoglycemia.48,52,71,77,116 There is no evidence that the preterm infant is able to tolerate low glucose better than term infants.5,122 Infants who are very immature or ill (hypoxia, ischemia, sepsis) may have greater glucose needs and be more vulnerable to the effects of hypoglycemia.24 The American Academy of Pediatrics Committee on Fetus and Newborn recently published a practice guideline and algorithm for screening and management of hypoglycemia in late preterm and term newborns.21 This guideline suggests that for these infants, a “reasonable (although arbitrary) cutoff for treating symptomatic infants is 40 mg/dL. This value is higher than the physiologic nadir and higher than concentrations usually associated with clinical signs . . . . A reasonable goal is to maintain plasma glucose concentrations in symptomatic infants between 40 and 50 mg/dL.”21,p.678
Clinical signs of hypoglycemia include tremors, jitteriness, irregular respiration, hypotonia, apnea, cyanosis, poor feeding, high-pitched cry, lethargy, irritability, hypothermia, and seizures. Because hypoglycemic infants may be symptomatic or asymptomatic and signs of hypoglycemia are often nonspecific, careful monitoring of infants at risk for development of hypoglycemia is critical. Multiple factors influence the outcome of infants with hypoglycemia, including severity and duration of the episode, cerebral blood flow and central nervous system (CNS) glucose levels, rates of glucose uptake, maturity, availability of alternative substrates, response to intervention, and type of clinical manifestation.22,24,50,93,116
Glucose is critical for the brain and hypoglycemia increases the risk of brain injury. Decreased glucose availability increases release of glutamate, free radicals, and other toxic metabolites that can lead to mitochondrial damage, altered ATP-dependent ion transport across neuronal membranes, changes in cell membranes, cellular edema, and neuronal necrosis (see Chapter 15). Unfortunately many studies on the outcome of infants with hypoglycemia have significant methodologic limitations and there have been no controlled prospective studies.13,107,116 Infants with persistent and recurrent hypoglycemia have the poorest neurologic outcomes.116 Hypoglycemia and hypoxic ischemic events may have an additive effect on neuronal injury.116 Brain neuroprotective responses to hypoglycemia include increased epinephrine, increased cerebral blood flow, use of alternative fuels such as ketone bodies and lactate, and degradation of glycogen stored in astrocytes.107,115 This may contribute to the lack of clinical signs even with low blood glucose levels.40 Kalahan and Devaskar concluded that “transient asymptomatic hypoglycemia in an otherwise healthy neonate has been associated with a good prognosis. Several studies of small groups of subjects have suggested that symptomatic hypoglycemia in the infant results in long-term neurologic damage. However, these data should be interpreted with caution because of a number of confounding variables.”71
Neonatal hypoglycemia can arise from an inadequate supply of glucose, alterations in endocrine regulation, or increased glucose regulation.24,25,71,77 Preterm and SGA infants tend to develop hypoglycemia because of insufficient glycogen and fat stores and a decreased rate of gluconeogenesis. Normally with hypoglycemia, brain glucose utilization decreases by up to 50%, with increased reliance on ketones and lactate for energy. Preterm infants may be limited in their ability to mobilize these responses and thus more vulnerable to the effects of hypoglycemia.35 Infants of diabetic mothers usually have sufficient stores; however, glycogenolysis is prevented by their high insulin levels and inability to secrete glucagon despite falling blood glucose levels. Infants at risk for neonatal hypoglycemia and associated mechanisms are summarized in Table 16-5.
Table 16-5
Causes and Time Course of Neonatal Hypoglycemia
| MECHANISM | CLINICAL SETTING | EXPECTED DURATION |
| Decreased substrate availability |
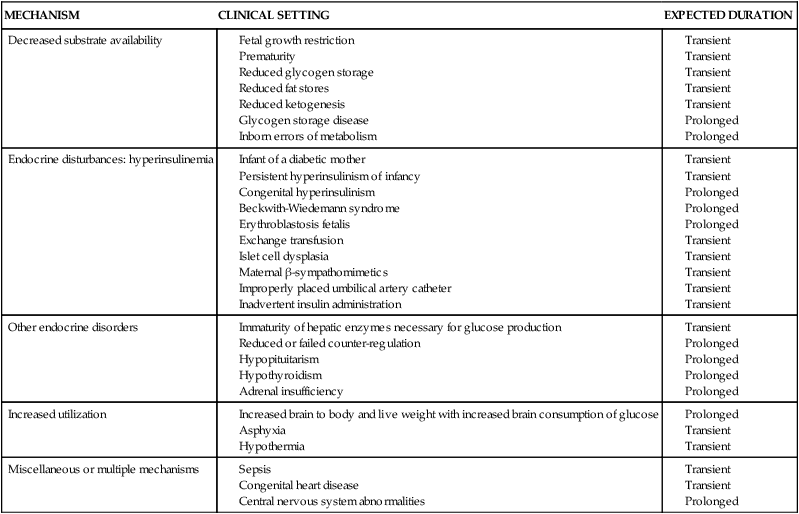
From McGowan, J.E., et al. (2011). Glucose homeostasis. In S.L. Gardner, et al. (Eds.), Merenstein & Gardner’s Handbook of neonatal intensive care (7th ed.). St. Louis: Mosby Elsevier, p. 360.
The preterm infant
Hypoglycemia is a common problem of preterm infants. Preterm infants usually develop hypoglycemia secondary to inadequate intake or decreased hepatic glucose production. These infants have decreased glycogen and fat reserves (accumulation of these stores occurs during the third trimester), and immature hepatic function, with low levels of gluconeogenic and glycogenolytic enzymes, especially glucose-6-phosphatase, which is important in glycogenolysis and gluconeogenesis.60,71 Their initial hormonal response to low glucose levels may be limited.60 Preterm infants are less able to produce alternate substrates such as ketone bodies. These infants may also have altered metabolic demands from tachypnea, respiratory distress syndrome, hypoxia, hypothermia, or other events that increase glucose use. Infants of mothers treated long term with β-adrenergic agonists for preterm labor (these agents are now being used less frequently) may develop hypoglycemia secondary to hyperinsulinemia.104 These agents rapidly cross the placenta and stimulate β-cell receptors on the fetal pancreas, with subsequent insulin release and altered glucose homeostasis in the fetus and newborn. Maternal treatment with benzothiazide diuretics can also stimulate fetal β-cell receptors and increase maternal and thus fetal glucose levels.71 Maternal propranolol, which can interfere with the catecholamine surge at birth, may also increase the risk of hypoglycemia.71
The growth restricted infant
Growth restricted infants are at risk for hypoglycemia primarily because of alterations in hepatic glucose production and increased glucose utilization.71,77 These infants may have reduced glycogen stores due to altered placental transport of substrates during fetal life, delayed maturation of gluconeogenesis, and a tendency toward hyperinsulinemia.93 Insulin sensitivity is decreased in the liver and increased in the peripheral cells. Growth restricted infants have increased energy demands due to their greater brain-to-body mass size, increased metabolic rate, and tendency toward polycythemia, but smaller energy stores. Because the brain and red blood cells are obligatory glucose users, these factors can markedly increase glucose needs even in nonstressed infants. Glucose utilization may be further increased by chronic or acute perinatal hypoxia. Secretion of hepatic gluconeogenic enzymes (especially PEP-CK) is impaired in these infants, further limiting their ability to increase glucose production to meet metabolic demands.116 growth restricted infants are at risk for later obesity and other long-term health risks (see Chapter 12).11,74,131
The infant of a diabetic mother
Although improved preconceptional care and careful metabolic control of pregnant diabetic women have reduced the incidence of significant macrosomia and improved perinatal mortality, infants of a diabetic mothers (IDMs) continue to be at risk for these as well as other health problems.130 The cause of these problems relates to fetal and neonatal responses to maternal metabolic alterations and consequences to the neonate of cessation of placental transfer of substrates after birth. Many IDMs are LGA, lethargic, poor feeders, and at risk for the problems summarized in Figure 16-11 (see also pp. 572-573 for a discussion of the proposed basis for macrosomia and risk of congenital anomalies in IDMs). Infants born to diabetic mothers with significant vascular involvement are often SGA, but with more mature liver enzyme systems and lungs due to the effects of intrauterine stress. This group of infants is at particular risk for problems associated with chronic hypoxia such as asphyxia and polycythemia.
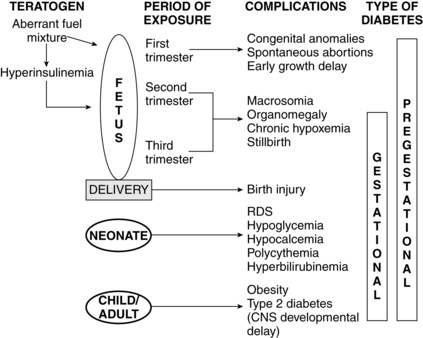
A prominent problem seen in IDMs is hypoglycemia. Hyperinsulinemia and a blunted glucagon response, aggravated by decreased hepatic responsiveness to glucose, are responsible for the hypoglycemia.118 Levels of epinephrine and norepinephrine are also elevated, suggesting that hypoglycemia in these infants also may be related to adrenal medullary exhaustion.25, In addition exposure in utero to marked increases in glucose (due to higher maternal glucose levels) up-regulates insulin secretion and alters glucose disposal. This leads to rapid insulin secretion and rebound hypoglycemia after an intravenous glucose bolus in the newborn.116
Large-for-gestational-age (LGA) infants.
LGA infants (weight greater than 90th percentile or greater than 2 standard deviations for their gestational age) who are not offspring of diabetic women are also at higher risk for developing hypoglycemia and hyperinsulinemia.107,116 The exact mechanism is unclear but may be related to increased insulin sensitivity or due to borderline maternal glucose tolerance.71,107,116
The infant with asphyxia
After intrauterine stress, levels of IGF-I are low with increased insulin-like growth factor binding protein-1 and interleukin-6. This leads to decreases in peripheral glucose consumption, conserving glucose for the central nervous system.40 Hypoxia and asphyxia alter glucose production and utilization with an increase in glycogenolysis to meet the increased metabolic and energy demands. Because oxygen availability is compromised, the infant switches from aerobic to anaerobic glycolysis. Anaerobic metabolism is less efficient than aerobic metabolism, producing a net increase of only two molecules of adenosine triphosphate (ATP) per molecule of glucose oxidized (versus 36 molecules of ATP per glucose molecule under aerobic conditions). These changes rapidly deplete glucose (glycogen) reserves, with decreased energy production that may be inadequate to maintain normal cell biologic processes and lead to accumulation of lactic acid.25 Hypoxic-ischemic damage to the liver may further impair glucose production and delay the postnatal increase in gluconeogenesis. These infants may also develop a transient hyperinsulinemia.25,93
Neonatal hyperglycemia
Neonatal hyperglycemia is a plasma glucose level greater than 180 mg/dL.71 Hyperglycemia occurs less frequently than hypoglycemia but has the potential for significant alterations in neurodevelopmental outcome.22,35,71,77 Hyperglycemia is seen predominantly in preterm infants, especially those weighing less than 1000 g and receiving parenteral glucose infusions.25,35 Hyperglycemia is negatively correlated with birth weight and positively correlated with the rate of glucose infusion. Markedly increased blood glucose levels can lead to osmotic changes and fluid shifts within the CNS (with a risk of intraventricular hemorrhage) and to glycosuria (with increased fluid and electrolyte losses and subsequent dehydration).
Hyperglycemia in low–birth weight infants “is most likely related to the secretion of glucose counter regulatory hormones as a result of stress, or to the release of cytokines in infected infants.”71 Other mechanisms that have been suggested for neonatal hyperglycemia include an inability of the immature infant to suppress endogenous glucose production while receiving a glucose infusion.25,35 This inability may be due to a decrease in expression of GLUT-2 on hepatocytes, an increased ratio of GLUT-1/GLUT-2 transporters, decreased sensitivity and response to glucose and insulin with hyperglycemia, and decreased ability of the pancreatic β cells to adequately increase insulin in response to increased glucose.35,98 Stressed infants seem to be at particular risk for hyperglycemia because of the simultaneous increase in catecholamine release (as a stress response), which further increases glucose levels by inhibiting insulin release and glucose utilization. Other infants at risk for hyperglycemia include infants treated with methylxanthines for apnea or with lipid infusions given at rates greater than 0.25 g/kg/hr, infants with sepsis or after surgery, and term newborns with severe growth restriction who develop transient neonatal diabetes. Chronic glucose deficiency in the growth restricted fetus leads to fewer pancreatic β cells with a decreased ability to secrete insulin. As a result these infant may develop hepatic insulin resistance with increased PEP-CK and increased hepatic glucose production rate, which can lead to a persistent hyperglycemia.116 Hyperglycemia can also occur, rarely, in infants with very early onset diabetes mellitus.110
Maturational changes during infancy and childhood
Energy and calorie requirements per unit of body weight remain higher in children than in adults because of their higher metabolic rate and growth needs. The relative requirement for carbohydrates is similar for children and adults. For infants, generally not more than 40% of the total calories should be carbohydrates; for children and adults, this value is 40% to 60%.92 Cerebral glucose utilization is increased until childhood.
The fasting response pattern is similar in infants and children as in adults; however, it proceeds more rapidly due to the greater brain mass–to–body weight ratio and increased glucose utilization rates. Therefore infants and young children develop fasting hyperketonemia more rapidly (within 24 hours) than adults (36 to 48 hours).122
IGF-I levels along with insulin continue as important growth regulators during infancy and have an inverse relationship with body weight.106 Catch-up growth is associated with IGF-I elevations during early childhood.106 Growth hormone receptors increase in the first 2 years with a gradual transition to growth hormone regulation of growth.106
Serum amino acid levels decrease and urinary excretion increases until early childhood. Total body protein increases and reaches adult proportions (3%) by 4 years. Retention of nitrogen decreases during this time to adult values (11 mg/kg/day).92 Diet alters the fatty acid composition of adipose tissue and possibly the composition of structural lipids during periods of rapid weight gain in the first year of life. This may affect the functional ability of tissues and structures.
Summary
Growth “is an accretion of materials brought together in a synergism involving anabolism and catabolism.”82 Growth involves increases in cell size and in the complexity of cells, tissues, and organs. Alterations in growth during the perinatal period arise from maternal, fetal, or placental factors that alter the availability, accretion, or use of substrates or nutrients. These events are influenced by metabolic processes involved in carbohydrate, protein, and lipid metabolism. When this occurs the fetus or neonate may be unable to adapt to environmental stress and is at increased risk for morbidity and mortality.82 Implications for clinical practice are summarized in Table 16-6.
Table 16-6
Know the usual changes in carbohydrate, protein, and fat metabolism during the fetal and neonatal periods (pp. 573-581).
Monitor neonates for alterations in metabolic processes (pp. 578-582).
Monitor newborn glucose status during transition and in the early neonatal period (pp. 578-580 and Table 16-4).
Initiate early enteral feeding as appropriate (pp. 578-580 and Table 16-4).
Monitor neonates for signs of excessive and inadequate intake of carbohydrates, protein, and fat (pp. 578-582).
Recognize infants at risk for hypoglycemia (pp. 582-584 and Table 16-5).
Know the clinical signs of hypoglycemia (p. 582).
Assess and monitor infants at risk for neonatal hypoglycemia (p. 584 and Table 16-5).
Recognize infants at risk for hyperglycemia (p. 584).
Assess and monitor infants at risk for hyperglycemia (p. 584).
Monitor infants at risk for hyperglycemia for alterations in fluid and electrolyte balance (p. 584).
Recognize and monitor for problems for which the infant of a diabetic mother is at increased risk (pp. 572, 583-584 and Figure 16-11).
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
Carbohydrate, fat, and protein metabolism
Metabolism comes from the Greek word meaning “to change” and is the totality of chemical reactions within a living organism. Four principles that underlie and guide metabolic functions in humans are “(1) plasma glucose must be maintained within normal limits; (2) an optimal source of glycogen must be maintained as an emergency fuel; (3) an optimal supply of protein must be maintained for use in enzymatic mechanisms of metabolism as well as muscular mobility; excess protein is converted to fat and the nitrogen released is excreted in urine; and (4) protein must be conserved when it is scarce and stored fat used in time of caloric need.”47,p.2
Maternal physiologic adaptations
Metabolic adaptations during pregnancy are directed toward (1) ensuring satisfactory growth and development of the fetus; (2) providing the fetus with adequate stores of energy and substrates needed for transition to extrauterine life; (3) meeting maternal needs to cope with the increased physiologic demands of pregnancy; and (4) providing energy and substrate stores for the demands of pregnancy, labor, and lactation.6 The first two demands compete with the third and fourth demands. As a result, alterations in maternal metabolic processes can significantly affect maternal and fetal health status.
Pregnancy involves a “coordinated series of physiologic adjustments which act in concert to preserve maternal homeostasis while at the same time providing for fetal growth and development.”47 Pregnancy is primarily an anabolic state in which food intake and appetite are increased, activity is decreased, approximately 3.5 kg of fat is deposited, energy reserves of approximately 30,000 kcal (125,550 J) are established, and 900 g of new protein is synthesized by the mother, fetus, and placenta. The overall energy cost of reproduction is estimated at 75,000 to 85,000 kcal (313,875 to 355,725 J).6,80,82 Anabolic aspects are most prominent during the first two trimesters when, due to enhanced lipogenesis, accumulation of maternal fat and increased blood volume lead to maternal weight gain (Figure 16-1).19,78 Protein and glycogen synthesis increase in muscle with increased glycogenolysis (breakdown of glycogen to form glucose) and decreased glycolysis in the liver.78 Insulin increases in response to glucose with a normal or slight increase in peripheral insulin sensitivity and serum glucose levels. This results in uptake of nutrients and maternal fat accumulation. During the third trimester, the woman’s metabolic status becomes more catabolic as stored fat is used (see Figure 16-1). Gluconeogenesis (formation of glucose from amino acids and glycerol) in the liver decreases and intestinal dietary fat absorption increases.78 Lipolytic activity within adipose tissue is enhanced with increases in plasma free fatty acids and glycerol. Maternal ketone production is increased. Counterinsulin hormones increase, leading to insulin resistance. During this phase, maternal weight gain is primarily due to the growing fetus and placenta; the fetus gains 90% of its growth in the last half of pregnancy.12,19,57,80,82
Antepartum period
Pregnancy is associated with major changes in metabolic processes and endocrine function. Pregnancy has been characterized as a metabolic “tug of war” between the competing needs of the mother and the fetus.47 The fetus and placenta influence maternal metabolic alterations in that these tissues become an additional site for metabolism of maternal hormones as well as new sites for hormonal biosynthesis. Many of the metabolic changes during pregnancy are aimed at providing substances (especially glucose, lipids, and amino acids) for the growth and development of the fetus. As the fetus and placenta grow, maternal fuel economy is altered to support this growth.38,109
Basal metabolic rate
The basal metabolic rate (BMR) increases during pregnancy (Figure 16-2). The rate of change varies with maternal prepregnant nutritional status and fetal growth. Significant variations are seen among individual women with up to an eightfold increase reported. Similar variations are reported for fat accretion.17,79,91 If a woman has low energy reserves at conception, there is less of an increase in the BMR and energy is conserved.
The total energy required for pregnancy can be divided into three parts: (1) obligatory energy needed for the fetus, placenta, uterus, and breasts (which is the smallest part); (2) energy for maternal fat storage; and (3) energy maintenance of these new tissues.79 If a woman has lower energy stores, less of the maternal energy intake is needed to maintain new tissues and energy is conserved for maternal basic needs and the fetal-placental unit.79,80,107,109 For example, in undernourished women, fetal weight accounts for 60% of the pregnancy weight gain, versus 25% in a well-nourished woman.109 This energy-sparing response may allow the woman to sustain the pregnancy but is often at the expense of fetal growth, with decreased birth weight and risk of fetal growth restriction.109 Prentice and Goldberg suggest that leptin might monitor a woman’s prepregnancy energy stores and adjust or coordinate maternal metabolic resources.109 Women with large-for-gestational-age (LGA) infants tend to have increased BMR with less maternal energy storage.79,111
The pregnant woman meets the energy demands of pregnancy by increasing her intake, decreasing her activity, or limiting fat storage.80,107 King and colleagues propose three examples of how women in different situations might alter their energy to sustain pregnancy.79 First, an underweight impoverished woman with limited food, poor fat stores, and need for physical work cannot increase food intake or limit physical activity during pregnancy. Her body responds by decreasing basal energy expenditures so that pregnant energy needs are similar to prepregnant needs. Second, a normal weight woman in a developed country with fat stores before pregnancy and adequate nutrition during pregnancy increases fat stores in pregnancy and increases her BMR slightly. Finally, an overweight woman in a developed country increases her BMR by 20% or more, perhaps to reduce additional fat deposition.79
Carbohydrate metabolism
Basal endogenous hepatic glucose production remains sensitive to insulin and increases up to 30% by the third trimester to meet fetal and placental needs.12,16,72,98 Endogenous glucose production increases with gestational age, paralleling fetal and maternal needs.72 Maternal glucose levels are generally 10% to 20% lower than in nonpregnant women. In addition, during the overnight fasting period, maternal plasma glucose values fall to levels 15 to 20 mg/dL (0.8 to 1.1 mmol/L) lower than in nonpregnant women.88 This decrease in glucose leads to lower insulin levels during the postabsorptive state (between meals and overnight) and a tendency toward hypoglycemia and ketosis. The decreased glucose level in the postabsorptive state is due to increased plasma volume in early pregnancy and later to placental transport of glucose to the fetus, which increases during gestation as fetal glucose needs increase.98,101
During the first two trimesters, the pregnant woman is in an anabolic state. Insulin secretion increases with increased peripheral glucose use without increases in insulin resistance.47 As pregnancy progresses, peripheral glucose use by the mother decreases because of increasing insulin resistance. This reduces maternal glucose utilization and makes glucose more readily available to the fetus. The mother compensates by using fat stores to meet her energy needs with breakdown of glycerol to glucose.57 Insulin resistance in the latter part of pregnancy is believed to be a result of a decrease in sensitivity of cell receptors that results from the insulin antagonism effects of hPL, progesterone, and cortisol.16,47 The insulin antagonism is partially modulated by pancreatic β-cell hyperplasia and hypertrophy with increased insulin availability after a meal.16 Pregnancy is also characterized by greater oscillations in insulin and glucagon levels.47 A reduction in the extraction of insulin by the liver may contribute to the peripheral hyperinsulinemia. Variations in hepatic insulin binding may lead to alterations in the ratio of insulin to glucagon.6
Progesterone augments insulin secretion, decreases peripheral insulin effectiveness, and increases insulin levels after a meal. Estrogen increases the level of plasma cortisol (an insulin antagonist), stimulates β-cell hyperplasia (and thus insulin production), and enhances peripheral glucose utilization. Increased levels of both bound and free cortisol decrease hepatic glycogen stores and increase hepatic glucose production. These changes further increase glucose availability for the fetus.88,98
hPL levels correlate with fetal and placental weight and are higher in multiple pregnancies.16 hPL increases synthesis and availability of lipids. Lipids can be used by the mother as an alternative fuel, enhancing availability and transfer of glucose and amino acids to the fetus. A mild form of the metabolic changes seen during pregnancy can be induced by giving hPL to nonpregnant women. However, there is no consistent relationship between hPL levels and insulin requirements in the pregnant diabetic woman.
Protein metabolism
Decreased serum amino acid and serum protein levels are found in pregnancy.6,42,72 This decrease is related to increased placental uptake, increased insulin levels, hepatic diversion of amino acids for gluconeogenesis, and transfer of amino acids to the fetus.6,42 The fetus uses some of these amino acids for glucose formation. Maternal plasma levels of glucogenic amino acids (e.g., those that can be converted into glucose), such as alanine, threonine, glutamate, and serine, are reduced due to placental transfer of these amino acids.31,72 Maternal plasma alanine levels, in particular, are lower because alanine is a key precursor for glucose formation (via gluconeogenesis) by the fetal liver.72
Alterations in protein metabolism during pregnancy have a biphasic pattern.72 During the first half of gestation, maternal protein storage increases, with a net retention of 1.3 g/day of nitrogen.80 Most of this is transported to the fetus, but some is retained in maternal tissues. During the second half, maternal protein use is more economic, with decreased urinary nitrogen excretion, thus conserving protein.86 These changes may be mediated by decreased activity of hepatic enzymes involved in amino acid deamination and urea synthesis.
Lipid metabolism
Pregnancy results in marked alterations in lipid metabolism with a markedly different lipoprotein profile, with marked increases in triglycerides, as well as increases in phospholipids and cholesterol. Basal oxidation of fatty acids increases 70% in pregnancy leading to a relative hyperlipidemia.27 Synthesis of very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL), which are contained in triglycerides, and LDL and HDL, which are found in cholesterol, also increase.59
Lipid metabolism in pregnancy is characterized by two phases and is analogous to the patterns of change in carbohydrate and protein metabolism.19,57 During the first two trimesters, triglyceride synthesis and fat storage increase. VLDL increases threefold in the second and third trimesters. LDL decreases slightly initially, followed by a progressive rise.100 HDL increases progressively to 24 weeks, then decreases to 32 weeks and stabilizes for the remainder of pregnancy.100 Triglycerides increase 40% by 18 weeks and 250% by term.87 Phospholipids and cholesterol levels also increase; the triglyceride-to-cholesterol level remains stable since cholesterol also increases.59,58 By late pregnancy, cholesterol levels are 50% higher than prior to pregnancy, regardless of maternal dietary intake.42 These changes enhance the availability of substrates for the fetus.57
Maternal fat storage is most prominent from 10 to 30 weeks, before the peak of fetal energy demands.79 Promotion of lipogenesis and suppression of lipolysis during this phase are mediated by progressive increases in insulin responsiveness and enhanced by progesterone, cortisol, leptin, and prolactin.6,16,30,42,59 Estrogens decrease lipoprotein lipase activity.18 During this period the pregnant woman experiences a physiologic ketosis with a twofold to threefold increase in baseline ketone body production, with an acute increase after fasting, suggesting enhanced fat utilization.116,130
Lipid metabolism in late pregnancy is illustrated in Figure 16-3. The third trimester is characterized by both lipogenesis and lipolysis, with increased breakdown of fat deposits.19,32,57,58 These changes are mediated by increased adipose tissue lipolytic activity and decreased lipoprotein lipase (LPL) activity (due to estrogens).19,58 LPL, which is present in the capillary endothelium of extrahepatic tissues, hydrolyzes circulating triglycerides, including VLDL, producing free fatty acids and glycerol.58 Adipose tissue is broken down into free fatty acids and glycerol, which circulate to the liver and are converted to active forms (acyl-CoA and glycerol-3-phosphate) and re-esterified into triglycerides (circulate as VLDL).19,58 Glycerol can also be used for glucose synthesis (gluconeogenesis). The increased lipolysis is due to the rise of hPL levels with its antiinsulinogenic and lipolytic effects, as well as the effects of cortisol, glucagon, and prolactin, resulting in increased lipolytic activity in adipose tissue.16,19,57,58,78 Enhanced ketogenesis in the liver is a consequence of increased oxidation of free fatty acids for energy and release of ketone bodies.58 The fat mobilization is associated with increased glucose and amino acid uptake by the fetus. Thus fats are used by the mother as an alternative energy substrate, allowing the mother to conserve glucose for the fetus and her central nervous system (CNS) during the second half of gestation.6,16,59,58 Fetal glucose and ketone uptake increases. Ketones are used by the fetus for oxidative metabolism, lipogenesis, triglyceride production, and as substrate for brain lipid synthesis.58,78,116,130
The changes in lipid metabolism during pregnancy are reflected in changes in maternal serum free fatty acid concentrations as well as plasma triglyceride, cholesterol, and phospholipid levels. These changes may be exaggerated in obese women.27 The minimal change in free fatty acids during early pregnancy is probably due to increased fat storage and augmented fat utilization. As maternal fat stores are mobilized, serum levels of triglycerides, free fatty acids, glycerol, and triglyceride-rich lipoproteins (VLDL) increase to peak near term.19,58,59 Because elevations in free fatty acids are due to catabolism of stored triglycerides (into free fatty acid and glycerol), changes in free fatty acids are mirrored by changes in glycerol.6 Hypertriglyceridemia during the third trimester is primarily due to increases in VLDLs and decreased peripheral clearance.16,19,58,59 The increase is due to reduced VLDL clearance secondary to decreased activity of LPL in the liver and adipose tissue, enhanced activity of cholesterol ester transferase, increased gastrointestinal absorption of lipids, and increased hepatic triglyceride production.16,32,58,59 The decrease in lipoprotein lipase is due to increases in estrogens in late pregnancy and the increasing insulin resistance.58,78 Near term, lipoprotein lipase activity increases in the mammary glands. This enhances availability of triglycerides for milk production.57
Changes in lipid metabolism are accompanied by functional and morphologic changes in the adipocytes. Hypertrophy of these cells accommodates the increased fat storage during the first two thirds of pregnancy. In the last trimester, maximal glucose transport, glucose oxidation, and lipogenesis within the adipocytes decrease.42 The number of insulin receptors on the adipocytes increases in the first part of pregnancy and returns to prepregnant levels by term.6,42 Because responsiveness of adipose tissue to insulin is not diminished as much as that of other tissues, these changes in the adipocytes facilitate fat storage.42 After a meal, maternal fat stores are replenished by increased glucose uptake, incorporation of glucose into glycerol, and esterification of fatty acids by adipocytes.6
Insulin
Insulin levels and responsiveness of tissues to insulin change dramatically during pregnancy, leading to peripheral insulin resistance. Peripheral insulin resistance is “the decreased ability of insulin to affect glucose uptake, primarily in skeletal muscle and to a lesser degree in adipose tissue.”98 Actions of insulin are summarized in Table 16-1.40 Insulin production and sensitivity during pregnancy differ in early compared to later pregnancy.16 During early pregnancy (up to 12 to 14 weeks), insulin responses are enhanced, sending glucose to the embryo and young fetus. The pregnant woman has a normal glucose tolerance, basal glucose production, and peripheral muscle sensitivity to insulin.16,110 Adipose tissue is more sensitive to insulin during this period, resulting in lipogenesis and fat storage.110
Table 16-1
Compiled from Guyton, A.C., & Hall, J.E. (2010). Textbook of medical physiology (12th ed.). Philadelphia: Saunders Elsevier; Widmaier, E., Raff, H., & Strang, K.T. (2005). Vander’s Human physiology: The mechanism of body function (10th ed.). New York: McGraw-Hill.
In later pregnancy (from 20 weeks to term), insulin sensitivity decreases and insulin secretion and resistance increase with decreased glucose uptake by muscle and adipose tissue.16,131 Maternal insulin levels increase 2.5- to threefold by the third trimester.47,78,98 These changes are accompanied by maternal pancreatic hypertrophy and hyperplasia.110 Increased insulin secretion ensures adequate maternal protein synthesis in the face of increasing resistance of peripheral tissues to the effects of insulin. With increasing gestation, mean insulin sensitivity decreases up to 50% to 70%.78 Tissue resistance is most prominent in liver, adipose, and muscle cells, and is further altered in women with preeclampsia.119 The increasing insulin resistance promotes nutrient flux from the mother to the fetus and promotion of adipose tissue accumulation.78 Insulin resistance is often exaggerated in overweight and obese women.27
Insulin receptor binding does not change significantly during pregnancy, however, the postreceptor insulin signaling cascade, and thus handling of glucose by cells, is altered in skeletal muscle and adipose tissue.110 During pregnancy insulin receptor substrate-1 (IRS-1) is down-regulated, affecting insulin uptake and use by cells.98 The postreceptor handling of glucose during pregnancy is altered by decreased IRS-1 expression (especially in skeletal muscle), altered expression of tyrosine kinase activity, and decreased expression of GLUT-4 glucose transporter protein in adipose tissue, which normally promotes glucose uptake.98,110
Insulin resistance is mediated by the increasing levels of placental hormones, especially estrogens, progesterone, and hPL, as well as prolactin and cortisol, and is minimally affected by changes in blood glucose levels.98 Other factors that play an important role in the insulin resistance of pregnancy are tumor necrosis factor-α and other cytokines, circulating free fatty acids, leptin, and placental growth hormone, which stimulates insulin-like growth factor-I (IGF-I).15,78,197,124 In late pregnancy, although basal insulin levels are elevated, maternal blood glucose values are similar to prepregnant levels.6 Increased insulin secretion after a meal (in response to the higher blood glucose) offsets the contrainsulin effects of the placental hormones and facilitates movement of nutrients to the fetus. The insulin resistance of late pregnancy enhances maternal fat breakdown and increased gluconeogenesis and ketogenesis in the postabsorptive state, the state in which changes in insulin response are most apparent.16,47,58 If the pregnant woman is not able to elevate her insulin secretion to overcome the increasing pregnancy-induced insulin resistance, maternal and fetal hyperglycemia will result and metabolic abnormalities such as gestational diabetes may develop or existing metabolic problems such as diabetes will be aggravated.
Alterations in insulin production and responsiveness are critical in integrating changes in carbohydrate and fat metabolism throughout the course of pregnancy. Baird summarizes these interactions as follows: During early pregnancy, increased insulin in response to glucose, minimal changes in insulin sensitivity, and increased number of insulin receptors on the adipocytes result in normal or slightly enhanced carbohydrate tolerance. The increased hepatic synthesis and secretion of triglycerides during this period, along with a normal or slightly elevated removal of triglycerides from the circulation, lead to a net storage of fat. During late pregnancy, the elevated plasma insulin, decrease in numbers of adipocyte insulin receptors to prepregnant levels, and increasing insulin resistance result in reduced assimilation of glucose and triglycerides by maternal tissues, greater transfer of these substances to the fetus, and increased lipolysis. The net result is a decrease in maternal blood glucose, increased glucose turnover, and greater maternal reliance on lipid catabolism for energy.6 Changes in lipid, carbohydrate, and protein metabolism are summarized in Table 16-2.
Table 16-2
Maternal Metabolic Processes during Pregnancy: Relationship between Hormonal and Metabolic Changes
| HORMONAL CHANGE | EFFECT | METABOLIC CHANGE |
| Increased hPL | ||
| Increased prolactin | Insulin resistance | |
| Increased bound and free cortisol | Ensures glucose and amino acids to fetus | |
| Increased estrogen, progesterone, and insulin during early pregnancy | Anabolic fat storage during early pregnancy | |
| Increased hPL in late pregnancy | Lipolysis | Catabolic fat mobilization in late pregnancy |
Adapted from Moore, T.R. (2004). Diabetes and pregnancy. In R.K. Creasy, R. Resnik, & J.D. Iams (Eds.), Maternal-fetal medicine: Principles and practice (5th ed.). Philadelphia: Saunders. hPL, human placental lactogen.
Absorptive versus postabsorptive states
In addition to phasic changes in metabolic processes in early versus late pregnancy, metabolism of amino acids, carbohydrates, and fats also varies on a daily basis depending on whether the mother is in the absorptive (fed) or postabsorptive state (Figures 16-4 and16-5). As a result of these changes, pregnancy has been characterized as a time of both “accelerated starvation” and “facilitated anabolism.”96
Absorptive state.
During the absorptive (fed) state, ingested nutrients (amino acid, glucose, triglyceride) are entering the blood from the gastrointestinal tract (see Chapter 12) and must be oxidized for energy, used for protein synthesis, or stored. The average meal takes 4 to 6 hours for complete absorption. In this state anabolism exceeds catabolism and glucose is the major energy source. Small amounts of amino acid and fat are converted into energy or used to resynthesize body proteins or for structural fat. Most of the amino acid and fat and any extra carbohydrate are transformed into adipose tissue; carbohydrate is also stored as glycogen.
Insulin has an anabolic and anticatabolic role during this state.78 Insulin secretion increases and plasma insulin levels rise. Insulin promotes glucose uptake by the hepatocytes and peripheral tissues, inhibits glycogen breakdown, and inhibits lipolysis in adipose tissue. Glucose is converted to glycogen for storage in the liver, cardiac muscle, and skeletal muscle. Muscle amino acid uptake is enhanced and proteolysis inhibited.78
After a meal the pregnant woman has higher glucose, insulin, and triglyceride levels and suppression of glycogen compared to nonpregnant women. Thus the absorptive state during pregnancy (see Figure 16-4, A) is characterized by relative hyperinsulinemia (related to decreased insulin sensitivity), hyperglycemia (due to failure of liver glucose uptake), insulin resistance (especially in skeletal muscle), hypertriglyceridemia, and lipogenesis (more glucose is converted to triglyceride for storage).6,16,78,119,129 Pregnancy has been called a state of facilitated anabolism to describe these metabolic alterations that conserve energy, especially during early pregnancy.94 These changes increase glucose availability for transport to the fetus; increase availability of an alternate energy source (triglycerides) for maternal needs; and provide fewer stimuli for maternal gluconeogenesis, glycogenolysis, and ketogenesis.42,47
Maternal blood glucose levels may rise transiently to 130 to 140 mg/dL (7.2 to 7.8 mmol/L) (see Figure 16-5). Gluconeogenesis and circulating free fatty acids are decreased. Under the influence of placental hormones, resistance of the liver and peripheral tissues to insulin is increased by as much as 60% to 70%.119,129 The hyperinsulinemic response is most marked during the third trimester because of hypertrophy and hyperplasia of islet β cells. These cells become more responsive to alterations in blood glucose and amino acid levels. The increased insulin levels after eating overcome the insulin resistance to allow glucose uptake by muscles for storage as glycogen.119 Even with increased production of insulin, however, overall glucose levels are maintained, although at a relatively lower level than in nonpregnant women because of the counterbalancing effects of estrogen, progesterone, and hPL.
Postabsorptive and fasting state.
In the postabsorptive state (i.e., when nutrients are not entering the blood from the intestines, beginning 4 to 6 hours after a meal) and fasting state (12 or more hours after the last meal), energy must be supplied by body stores. Insulin levels are low. Fat and protein synthesis are decreased and catabolism exceeds anabolism. Gluconeogenesis (production of new glucose from lactate, amino acids [especially alanine], and glycerol) and catabolism of fat are the main sources of energy.78 Plasma glucose levels are maintained during the postabsorptive state by use of these alternate sources of glucose and glucose-sparing or fat-utilization reactions.16,42 The CNS continues to use glucose, while other organs and tissues become glucose sparing, depending on fat as the primary energy source.
During the fasting state (e.g., overnight between the evening meal and breakfast), plasma glucose levels decline. The magnitude of the decline is greater in pregnant women than in nonpregnant women, due to the continuous transfer of glucose to the fetus, and is associated with a more rapid conversion to fat metabolism. Fatty acids are liberated by breakdown of triglycerides. Lipolysis yields glycerol (converted to glucose by the liver) and free fatty acids, which are catabolized to ketone bodies (oxidized for energy). Under homeostatic conditions ketones do not accumulate in the body to produce ketoacidosis because excess ketones not needed for energy are rapidly cleared by the kidneys.78
This response is an exaggeration of the changes normally seen during the overnight fast in nonpregnant women. These changes would normally raise blood glucose levels, but in the pregnant woman the fasting glucose level tends to be lower because of the limited availability of substrate for gluconeogenesis. For example, as early as 15 weeks’ gestation, maternal glucose levels after a 12- to 14-hour overnight fast are 15 to 20 mg (0.8 to 1.1 mmol/L) lower than levels in nonpregnant women. The decrease in glucose during the overnight “fasting” period is especially prominent during the second and third trimesters.6,78
The exaggerated maternal responses during the fasting state are influenced by (1) continuous placental uptake of glucose and amino acids from the maternal circulation; (2) decreased peripheral utilization of glucose as plasma concentrations of ketones and free fatty acids increase; (3) decreased renal absorption of glucose; and (4) decreased hepatic glucose production. This response, seen primarily in late pregnancy, is characterized by lower fasting glucose and amino acid levels, increased blood glucose levels after eating, and increased plasma free fatty acids, triglycerides, ketones, and insulin secretion in response to glucose.12,42,78 Thus the postabsorptive fasting state in pregnancy is characterized by a relative hypoglycemia (due to the fetal siphon, increased renal losses, and decreased liver production), hyperketonuria (ketones used as an alternative energy source), hypoaminoacidemia (due to placental transfer for use in fetal glucose production), and hypoinsulinemia (see Figures 16-4, B, and 16-5).12,24,57,72,78,98 Hypoalaninemia also develops because maternal protein stores can provide only limited substrate, which is insufficient to meet both maternal and fetal amino acid needs.47 Increased insulin during the postabsorptive state enhances uptake of glucose into the maternal skeletal muscle and adipose tissue. In lean women, this leads to suppression of hepatic glucose production in late pregnancy. Less suppression is seen in obese women, therefore the alteration in insulin sensitivity is greater in obese than nonobese women.98
Levels of lipoprotein lipase are increased, enhancing triglyceride breakdown with release of free fatty acids and glycerol (which is broken down into glucose) and production of ketone bodies to provide energy when plasma glucose supply is low.16 Glycerol is a source for hepatic gluconeogenesis. The elevated free fatty acids prevent glucose uptake and oxidation by maternal cells, thus preserving glucose for the maternal CNS and the fetus.12 In the nonpregnant woman this switch to fat oxidation occurs after 14 to 18 hours of fasting; during pregnancy the switch occurs after 2 to 3 hours and has been termed accelerated starvation.12,19,42,96
Metabolic changes characteristic of this state are primarily due to hPL, which promotes lipolysis to increase free fatty acid levels and opposes insulin action, thus increasing glucose availability to the fetus. Other factors influencing this response include increased glucose utilization by the fetus (“the fetal siphon”) and mother, along with an increase in the volume of distribution for glucose (i.e., hemodilution).78 During the second half of pregnancy, effectiveness of insulin in translocating glucose into cells is reduced.78 Because insulin is the ultimate arbitrator of both the absorptive and postabsorptive states, alterations in insulin secretion alter substrate availability to the mother and fetus.47 The insulin antagonism in pregnancy is progressive, paralleling the growth of the fetoplacental unit, and disappears immediately after delivery. Placental hormones and other substances are major factors in producing this insulin antagonism.
Drainage of glucose and amino acids by the fetus may lead to increased maternal appetite and a feeling of faintness sometimes experienced in pregnancy. Pregnant women may experience more rapid development of ketosis and fasting hypoglycemia after food deprivation. With greater maternal reliance on fat utilization during pregnancy, production of ketone bodies and risks of abnormalities such as acidosis are increased. Dieting and caloric restriction during pregnancy should be considered potentially dangerous to both the mother and fetus.37
Effects of placental hormones and other substances
The phasic changes in carbohydrate, lipid, and protein metabolism during pregnancy are due to the interplay of placental hormones, especially estrogen, progesterone, hPL, and leptin.75,98 During the first half of pregnancy, metabolism is affected primarily by estrogens and progesterone. In late pregnancy the influences of increasing concentrations of hPL and leptin become more prominent. Maternal metabolic changes are also influenced by prolactin and cortisol.12
Estrogen stimulates islet β-cell hyperplasia and insulin secretion, enhances glucose utilization in peripheral tissues, and increases plasma cortisol, an insulin antagonist. As a result, particularly in the first half of gestation, estrogen decreases fasting glucose levels, improves glucose tolerance, and increases glycogen storage.78 Progesterone augments insulin secretion, increases fasting plasma insulin concentrations, and diminishes peripheral insulin effectiveness. Cortisol mediates these changes by inhibiting glucose uptake and oxidation, increasing liver glucose production, and possibly augmenting glucagon secretion.12 Cortisol increases to 2.5 times normal levels by late pregnancy.12,131 Prolactin increases fivefold to 10-fold and stimulates insulin production and, at least in animal models, increases the number of β-cell receptors.131
Human placental lactogen (hPL), also called human chorionic somatomammotropin (hCS), is a polypeptide hormone produced by the syncytiotrophoblast (see Chapter 3). hPL is the most potent insulin antagonist of the placental hormones. This hormone is secreted primarily into the maternal circulation although some is also secreted into fetal circulation after around 6 weeks.43 Levels of hPL increase markedly after 20 weeks. Because effects of hPL are similar to those of growth hormone, it has been called the “growth hormone” of the second half of pregnancy.12 hPL action increases availability of maternal glucose and amino acids to the fetus. Other effects of hPL include diminished tissue response to insulin; increased β-cell mass; lipolysis, which increases plasma free fatty acids; enhanced nitrogen retention; decreased urinary potassium excretion; and increased calcium excretion. The major effect is sparing of maternal carbohydrate (glucose) by providing alternative energy sources such as free fatty acids for the mother.16
Several adipokines, including leptin, adiponectin, and retinal binding protein 4 (RBP4), are also important for ensuring adequate substrate for fetal growth.58 Leptin, a protein product of the obese (ob) gene, was identified in 1994. It is produced and secreted by adipose tissue and other tissues, including the placenta during pregnancy. Leptin is involved in regulating appetite and food intake and enhancing energy expenditure, and may also be important in signaling readiness for sexual maturation at puberty.128 Leptin receptors are found in the hypothalamus, placenta, muscle, liver, lymphoid tissue, uterus, chorion, amnion, pancreas, ovary, and adipose tissue.78,124 Placental leptin is secreted into both maternal and fetal circulations.86
Leptin plays a role in maturation and regulation of reproduction and “may serve as a detector of long-term metabolic fuel availability signaling the presence of significant maternal fat stores to initiate reproduction.”16 Leptin modulates glucose metabolism, insulin sensitivity, and adipose tissue lipolysis during pregnancy, enhancing breakdown of maternal fat stores in the second half of pregnancy and availability of glucose and lipids for the fetus.58 Other roles of leptin in pregnancy may be to mediate changes in appetite, thermogenesis, and lipid metabolism.16,49 Leptin may also modulate fetal growth. Concentrations of leptin increase from 6 to 8 weeks’ gestation, rising further in the second and (especially) third trimesters.81 The increase is probably primarily due to increased placental leptin production. Levels in early pregnancy correlate with maternal weight and body mass index; this correlation is not found in later pregnancy and may reflect a form of leptin resistance (similar to that seen with obesity).16,81
Adiponectin is a protein, secreted by adipose tissue, that is involved in modulating glucose and lipid metabolism and enhancing insulin sensitivity.58 Changes in leptin and adiponectin have been reported in complicated pregnancies. For example, both leptin and adiponectin are increased in women with severe preeclampsia.7,84,86 Adiponectin is decreased in women with gestational diabetes mellitus, whereas tumor necrosis factor-α (an inflammatory marker) and cord blood leptin are increased.55,58,90,112 Low levels of adiponectin in women with gestational diabetes may play a role in the increased insulin resistance seen in these women.58 Low leptin levels are associated with spontaneous abortion.83 RBP4 may also have a role in glucose metabolism and insulin sensitivity. RBP4 increases with increasing gestational age (probably due to placental production) and is increased in pregnancies complicated by gestational diabetes.58
Intrapartum period
The processes of parturition are dependent on an available supply of glucose and triglycerides as energy sources. In addition, essential fatty acids are important as precursors of prostaglandins (arachidonic acid, from which prostaglandins are derived, is a derivative of essential fatty acid), which are critical to the onset of labor. These relationships are described in Chapter 4.
During labor and delivery, maternal glucose consumption increases markedly to produce the energy required by the uterus and skeletal muscles.47 As a result, maternal insulin requirements fall. Oxytocin may augment or supplant insulin during this period. In animals, oxytocin has been demonstrated to act similarly to insulin; that is, oxytocin stimulates glucose oxidation, lipogenesis, glycogen synthesis, and protein formation.47
Postpartum period
With removal of the placenta, concentrations of placental hormones such as hPL, estrogens, and progesterone fall rapidly within hours after delivery (see Chapter 5). The postpartum woman is in a state of relative hypopituitarism with blunted production of gonadotropins and growth hormone.47 This hypopituitarism may result from the feedback effects of elevated hPL and prolactin levels during pregnancy on the pituitary gland. hPL is similar to growth hormone, and its disappearance with removal of the placenta leaves the woman without its contrainsulin effects during a period of relative deficiency of growth hormone.47 Plasma leptin levels decrease by 24 hours after delivery.83
Fasting plasma glucose levels fall within a few days, then increase reaching late pregnancy fasting levels by 5 days.42 It is unclear when these levels return to prepregnant values.42 The insulin resistance of late pregnancy is reversed soon after delivery.98
Plasma free fatty acids fall to prepregnant levels by 3 days; triglycerides by 2 weeks.18,42 During the first week the decrease in triglycerides coincides with the fall in hPL. This fall is more rapid in women who are breastfeeding.18,58 In these women, free fatty acids rise to late pregnancy levels by 6 weeks, followed by a decrease to prepregnant levels by 3 to 6 months.6 The increasing levels of fatty acids from 1 to 6 weeks postpartum may reflect maternal use of other nutrients for milk production.6 Cholesterol levels slowly decrease to prepregnant levels over the first few weeks postpartum.42 Maternal plasma amino acid levels return to prepregnant values of approximately 4.3 mg/dL (0.043 g/L) (versus pregnant values of 3.5 mg/dL [0.35 g/L]) by several days after birth.1,35
Clinical implications for the pregnant woman and her fetus
The metabolic adaptations of pregnancy safeguard against variations in maternal caloric intake, changes in activity, increased metabolic efficiency, and changes in the metabolism of carbohydrates, fats, and proteins.6 These metabolic changes occur in a phasic pattern—probably programmed by placental hormones—that spreads the energy costs and protein requirements of pregnancy over the entire 9 months of gestation. In early pregnancy, energy is conserved (facilitated anabolism), followed by later redirection of energy (glucose) to the fetus (“accelerated starvation”), whereas throughout pregnancy the mother uses protein more economically to provide adequate amino acids for development of the fetal brain and other organs.6,47 Pregnancy has also been characterized as a diabetogenic state. This state is reflected in the elevated blood glucose levels in association with increasing insulin resistance. This state is described in this section along with the basis for alterations in the glucose tolerance test (GTT) and the effects of the normal metabolic changes of pregnancy on the diabetic woman and her fetus.
The diabetogenic effects of pregnancy are reflected by alterations in the GTT, with higher glucose values after a meal reflecting an acquired resistance to insulin. The alterations in carbohydrate metabolism are most evident during late pregnancy in the absorptive state (see Figure 16-4, A). When the woman is in this state and glucose is being added to the plasma from the gut, her blood glucose levels do not drop as rapidly as usual, even in the face of higher circulating insulin levels. This response results from decreased maternal sensitivity to insulin due to the action of hormones such as hPL, progesterone, and cortisol. Secretion of these hormones increases during the second half of pregnancy; therefore diabetogenic effects are most prominent during this period. Insulin resistance is somewhat compensated for by increased plasma insulin concentrations.78
Effects of metabolic changes on glucose tolerance tests
Two methods commonly used to evaluate glucose tolerance in pregnancy are (1) a one-step 75-g, 2-hour test (recommended by the World Health Organization); and (2) a two-step test with a 1-hour 50-g glucose challenge (screening test) followed by a 100-g, 3-hour oral glucose tolerance test (OGTT) if challenge levels are either 135 mg/dL (7.5 mmol/L) or greater (more women meet criteria for the 3-hour test, but a greater proportion of women with gestational diabetes mellitus [GDM] are identified) or 140 mg/dL (7.8 mmol/L).78,98 The one-step testing has a lower detection rate for GDM.97 The two-step test has been used most commonly in the United States, whereas the one-step test is used most commonly in other countries.95,98 The Fifth International Workshop Conference on Gestational Diabees Mellitus recommended the two-step test.95 However, following the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) recommended use of a 75-g glucose load followed by fasting, 12-, and 2-hour plasma glucose concentrations.64
During pregnancy, the initial fasting blood glucose value is lower than in nonpregnant individuals, because of decreased glucose utilization and increased fat utilization by the mother (making increased glucose available to the fetus) and the subsequent effects of the fetal siphon. Blood glucose levels tend to remain high after ingestion of carbohydrates for a longer period of time secondary to insulin antagonism and decreased insulin sensitivity. Normally the magnitude of the increase in blood glucose after a carbohydrate feeding is a reflection of failure in glucose uptake by the liver. During pregnancy the increased glucose response in the face of increased endogenous insulin confirms the relative insensitivity and resistance of the liver (as well as peripheral tissues such as muscle and adipose tissue) to insulin.78,98
Maternal-fetal relationships
Growth and development of the fetus is dependent upon the availability of a constant supply of glucose, amino acids, and lipids from the mother for energy, protein synthesis, and production of new tissues. The fetus must also develop adequate stores of these substances to meet the demands of the intrapartum period and transition to extrauterine life. Placental transfer of selected nutrients and hormones is summarized in Figure 16-6. Fetal requirements for substrates involved in carbohydrate, fat, and protein metabolism are discussed in the next section.
Alterations in maternal metabolic processes or in placental transfer of essential nutrients that increase the availability of specific substrates are usually an advantage to the fetus but can be a disadvantage in altered maternal metabolic states. For example, because fetal energy requirements are met almost exclusively by glucose, the metabolic changes in pregnancy increase availability of glucose in maternal plasma for placental transfer by reducing the efficiency of maternal glucose storage. If the usual metabolic changes of pregnancy or placental function are altered, however, variations in fetal growth—such as occur in the infant of a diabetic mother or the an infant with fetal growth restriction—may develop.122 Maternal glucose infusions during the intrapartum period can lead to a fetal hyperglycemia that stimulates insulin and inhibits glucagon secretion. This may delay gluconeogenesis after birth and increase the risk of neonatal hypoglycemia.
The placenta is a highly metabolic organ with its own substrate needs. Placental metabolic activities include glycolysis, gluconeogenesis, glycogenesis, oxidation, protein synthesis, amino acid interconversion, triglyceride synthesis, and alterations in the length of fatty acid chains (see Chapter 3). The placenta can modify nutrient uptake to meet fetal growth demands.115 The degree of glucose uptake by the placenta is similar to that of the brain. Cholesterol from the mother is essential for placental synthesis of estrogens and progesterone. Fatty acids are needed by the placenta for oxidation and membrane formation.16 Leptin may have a role in coordinating placental metabolism.81
Glucose is the nutrient that crosses the placenta in highest concentrations. The increased maternal reliance on fats as an alternative energy source, especially during the maternal postabsorptive fasting state, conserves maternal glucose for transfer to the fetus. The fetal-placental unit uses approximately 50% of the total maternal glucose needed for pregnancy.57 Glucose crosses via facilitated transport by insulin dependent glucose transporters.81 Amino acids are transferred via active transport because levels are higher in the fetus than in the mother. Amino acids are removed from maternal circulation and concentrated in the placental intercellular matrix. As fetal amino acid levels fall, these placental stores are transferred to the fetus. Mechanisms for amino acid transfer include direct transfer from mother to fetus without modification in the placenta, metabolism of maternally derived amino acids by the placenta to produce other amino acids (which are then transferred to the fetus), and production of new amino acids by the placenta for fetal transfer.57,61
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
