72 Cancer Stem Cell Biology and Its Role in Radiotherapy
Radiotherapy is one of the most frequently employed and most effective forms of cancer treatment. Advances in medical physics are leading to an ever-increasing ability to deliver conformal radiotherapy, thus allowing dose escalation to tumors while minimizing toxicity to surrounding normal tissues. Radiation therapy can be employed alone or in combination with surgery and chemotherapy, and plays a critical role in achieving local control and cure for many patients. However, even with modern treatment approaches, local control cannot always be attained. In these cases, toxicity to normal structures in the vicinity of a tumor limit the total dose that can be delivered, and thus force radiation oncologists to limit treatment to doses that are insufficient to eliminate every clonogenic cancer cell. In such cases, combined chemoradiotherapy can be a potentially effective therapeutic strategy. Unfortunately, the generally relatively limited levels of tumor cell killing achievable with cytotoxic chemotherapeutics (compared with ionizing radiation)1–3 and the tendency to cause overlapping toxicities with radiation continue to pose significant restrictions on the applications of this approach. It would therefore be ideal to identify new molecular agents that can radiosensitize tumor cells and thus increase the probability of local control or even decrease the dose of radiation necessary to sterilize a given site. Recent insights into tumor biology, gained by applying the techniques and principles of normal stem cell biology to the study of cancers, suggest that in many tumors only a subset of cancer cells has the potential to divide indefinitely and to lead to tumor recurrence after treatment. It is this subset of cells, often called cancer stem cells, that needs to be targeted and radiosensitized for clinical outcomes to be further improved. The cancer stem cell hypothesis implies that identification of survival, self-renewal, and treatment resistance mechanisms in these cells may ultimately lead to improved outcomes for cancer patients.
Concepts and Principles of Normal Stem Cell Biology
The most-studied and best-understood stem cells are hematopoietic stem cells (HSCs), which serve as a model for understanding and studying all other normal-tissue stem cells. HSCs can give rise to the entire lymphohematopoietic system, including red blood cells, lymphocytes, granulocytes, monocytes, macrophages, platelets, and so on (Fig. 72-1).4 Radiation biologists played critical roles in the development of stem cell concepts and principles. James Till and Ernest McCulloch provided the first evidence for the existence of HSC in their efforts to study the ability of bone marrow transplantation to rescue lethally irradiated mice.5–7 They developed a method, called the spleen colony–forming assay, which allowed detection and quantification of stem cells based on the ability of transplanted bone marrow cells to form colonies in the spleens of previously irradiated recipients. Importantly, these colonies could be harvested and retransplanted into secondary recipients, where some gave rise to multilineage colonies, thus providing evidence for both self-renewal and differentiation potential. To this day, adaptation of the clonal in vivo assays pioneered by Till and McCulloch to other tissues and organs is the foundation of all stem cell analyses, because these assays best measure the core stem cells’ properties.
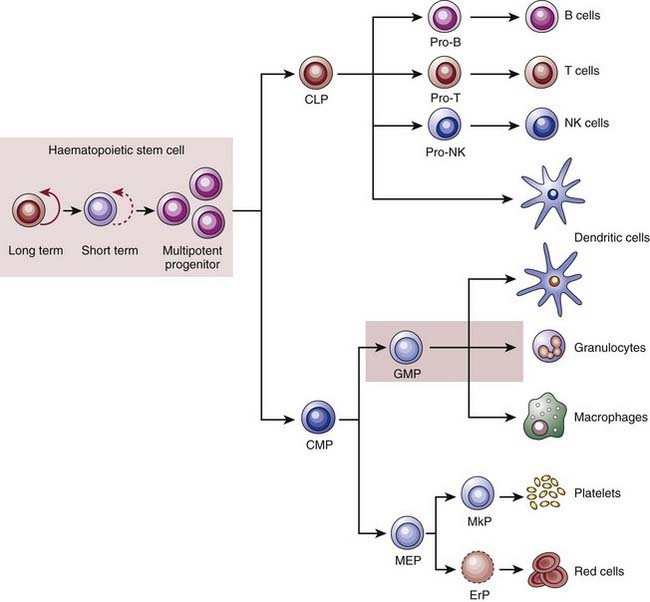
(From Reya T, Morrison SJ, Clarke MF, et al: Stem cells, cancer, and cancer stem cells, Nature 414(6859):105–111, 2001.)48
Extensive work since Till and McCulloch’s original observations has confirmed that HSCs display the three basic properties of stem cells: they self-renew, their numbers are under strict genetic control, and they give rise to a diverse collection of differentiated cells.8,9 The next critical advance in stem cell studies was the application of fluorescent-activated cell sorting (FACS) to isolate subpopulations of bone marrow cells with varied expression of specific surface proteins. This technique allows the prospective isolation of cells that can then be injected into irradiated animals to assay for repopulating activity. Using this approach, it has been determined that a small number of long-term self-renewing HSCs (which make up approximately 0.01% of mouse bone marrow cells) gives rise to short-term self renewing HSCs, which can only reconstitute an animal’s hematopoietic system for approximately 8 weeks (see Fig. 72-1). These then give rise to multipotent progenitor cells that cannot self-renew. The multipotent progenitors then further differentiate into one of several more restricted progenitor cells, including (1) the common myeloid progenitor, which ultimately gives rise to granulocytes, monocytes, erythrocytes, and platelets; (2) the common lymphoid progenitor, which gives rise to B and T cells. With the exception of the special case of memory B and T cells, HSCs are the only cells in the hierarchy that have the capacity to self-renew for extended periods under normal conditions.
A second normal-tissue stem cell that has been well characterized in recent years is the mammary stem cell. Mammary glands are made up of tubuloalveolar units consisting of a stratified epithelium made up of an inner layer of luminal epithelial cells and an outer layer of myoepithelial cells. As with HSCs, a critical step in the identification of mammary stem cells (also known as mammary repopulating units [MRUs]) was the development of an in vivo clonogenic assay. This assay consists of “clearing” mammary fat pads in 21-day-old recipient mice of endogenous epithelium and then injecting populations of dissociated mammary epithelial cells from donor animals. Approximately 6 weeks later, the injected fat pads can be harvested and analyzed for the presence of donor epithelial structures.10 Using such assays, it was initially shown that entire tubuloalveolar structures can develop from transplantation of short segments of existing mammary glands and that these result from clonal outgrowths.11–13 More recently, multiple investigators have applied FACS to prospectively isolate MRUs, just as had previously been done for HSCs. These experiments revealed that mammary glands contain a relatively small subpopulation of cells (~0.2%-0.5% of epithelial cells within a gland) that are able to generate entire epithelial outgrowths starting from a single cell.14,15 These cells give rise to differentiated luminal and myoepithelial cells and are able to do this for multiple generations of transplants, thus documenting self-renewal.
Stem Cells and Cancer
Comparisons have long been made between normal-tissue stem cells and cancer cells, and the idea that undifferentiated cells may be the origin of cancers dates back as far as the work of Rudolf Virchow.16 Clearly, at least some cancer cells must self-renew, although the tight control on this process apparent in normal stem cells appears to be relaxed or even absent in tumors. Furthermore, the concept of differentiation has long been a critical diagnostic and prognostic marker for human cancer. Thus, when pathologists “grade” tumors, they closely evaluate the degree to which a tumor represents the differentiation pattern of its tissue of origin. In many tumor types, this attribute predicts patient outcome. These observations are consistent with the idea that cancers may contain stem cells. Above all else, the concept of tumor heterogeneity suggests that cancers may contain a population of self-renewing cells that can give rise to differentiating progeny.
Competing Models of Tumor Heterogeneity
Two important clinical and experimental observations related to heterogeneity of cancer cells within tumors served as further impetus to the development of the hypothesis that tumors may contain malignant stem cells. Tumor heterogeneity refers to observations that although cancer formation is believed to be a clonal process beginning with a single transformed cell, not all malignant cells within a tumor are the same. Often, the variability within a tumor’s malignant cellular compartment is reminiscent of the normal tissue from which it is derived, and specifically of the various stages of differentiation that characterize these tissues. As such, cancer can be viewed as a caricature of the normal tissue from which it arose. For example, many moderately or well-differentiated squamous cell carcinomas form organized layers of tumor cells that are histologically similar to normal stratified squamous epithelia. Furthermore, interrogation of tumors with immunohistochemical techniques often reveals heterogeneity in the expression of differentiation markers relevant to the tissue of origin (Fig. 72-2). An extreme example of tumor heterogeneity can be found in teratocarcinomas, which can contain histologically diverse structures, including some resembling cartilage, muscle, bone, teeth, and hair. Conversely, many teratocarcinomas contain a small population of cells expressing immaturity markers such as placental alkaline phosphatase, beta–human chorionic gonadotropin, or α-fetoprotein, and elimination of these cells by chemotherapy is associated with positive clinical outcomes.17,18 Thus, many tumors contain evidence for partially intact differentiation mechanisms appropriate to their tissue of origin. This suggests that there may be a hierarchical relationship among tumor cells analogous to that seen in normal tissues.
(From Dalerba P, Dylla SJ, Park IK, et al: Phenotypic characterization of human colorectal cancer stem cells, Proc Natl Acad Sci U S A 104(24):10158–10163, 2007.)38
The second major observation regarding heterogeneity of tumor cells stems from data indicating that only a small subset of cells within most tumors has the capacity to form new tumors upon in vivo transplantation or colonies upon in vitro culture. In other words, tumor cells are heterogeneous in their capacity to proliferate indefinitely. For in vivo tumor transplantation, this is reflected in the large numbers of tumor cells that generally need to be transplanted to yield a tumor. For primary human and mouse tumors, this number often resides in the thousands or millions.19–21 Similarly, less than 1% of primary tumor cells cultured in vitro are generally able to give rise to colonies (i.e., low plating efficiencies) even under the most optimal culture conditions available.21–23
Two predominant models were developed to explain tumor heterogeneity (Fig. 72-3). The first, known as the stochastic model, is the classic view and argues that all malignant cells within a tumor have the potential to give rise to new tumors, but that this can only occur under ideal conditions and therefore is a relatively rare event. In this model, histologic and molecular heterogeneity are attributed to microenvironmental differences or to genomic instability24 of cancer cells. The second model, known as the cancer stem cell model, argues that, like the normal tissues from which they are derived, tumors contain a cellular hierarchy in which only a subpopulation of malignant stem cells can proliferate indefinitely and thus give rise to new tumors. Although the remaining cancer cells, often called nontumorigenic cancer cells, can proliferate, the number of divisions they can undergo is limited and they therefore can contribute to tumor bulk but cannot form new tumors. In the cancer stem cell model, the histologic and molecular heterogeneity seen in most tumors is largely explained by the partially intact differentiation programs that exist within tumors and that attempt to recapitulate the normal development of the tissue of origin. Importantly, the well-established notion of genomic instability within cancers fits within the framework of the cancer stem cell model, and suggests that tumors may contain subclones of cancer stem cells that give rise to genomically distinct nontumorigenic cells.
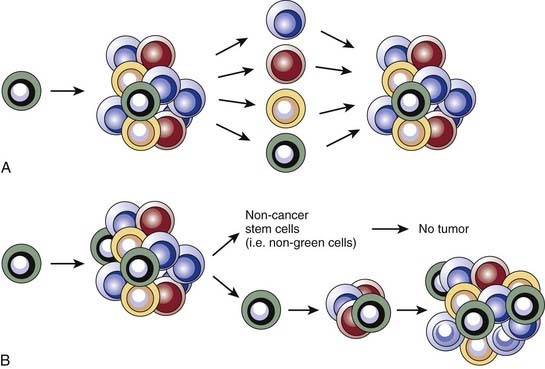
Cancer Stem Cell Nomenclature
As with any field of scientific inquiry, it is critical that the terminology used in the burgeoning cancer stem cell field is similar among various investigators. This has been a problem in recent years, as various investigators have used terms such as cancer stem cells, tumorigenic cells, or tumor-initiating cells to refer to ostensibly similar but not necessarily identical subpopulations of tumor cells. With this in mind, a recent American Association for Cancer Research workshop defined cancer stem cells as those cells within a tumor that possess the capacity to self-renew and to give rise to the heterogeneous lineages of cancer cells that compose the tumor.25 For the remainder of this chapter, we use the term cancer stem cell in accordance with this definition. It is important to point out that the definition hinges on the concept of self-renewal, which, as previously discussed, is not synonymous with cellular proliferation because it also reflects the differentiation and future mitotic potential of daughter cells.
Evidence Supporting the Cancer Stem Cell Model
If the cancer stem cell hypothesis is correct in stating that only a specific subset of tumor cells has the capacity to self-renew and give rise to all of the other cells within a tumor, then it follows that immature cancer cells should be able to drive tumor formation, whereas mature tumor cells should be unable to do so. Investigations of mouse teratocarcinoma and human leukemia provided some of the earliest evidence for the existence of cancer stem cells. Teratocarcinomas arise from germ cells and produce mature cells from all three embryonic layers upon transplantation. Illmensee and Mintz showed that when placed in a normal blastocyst, teratocarcinoma cells can differentiate into mature, nontumor tissues,26,27 thus supporting the existence of differentiation mechanisms within tumors akin to those of normal tissues.
Genetic studies of leukemia by Fialkow and colleagues provided the first strong evidence for the existence of cancer stem cells. They looked at the expression of isozymes of the X chromosome–linked enzyme glucose-6-phosphate dehydrogenase (G6PD) in mature blood cells of women with leukemia and found that some of these postmitotic cells were derived from the leukemic clone.28–30 In some cases, the leukemic clone gave rise to mature cells in multiple lineages (e.g., erythrocytes, granulocytes, and platelets). This definitively showed that immature cancer cells maintained the leukemia, whereas at least some of their progeny had lost the ability to proliferate.
Given that maturity markers can, at least in part, be used to distinguish cells that maintain a tumor from those that cannot, it should be possible to prospectively isolate cancer stem cells and test their clonogenic ability. Proof of this principle was first established for acute myelogenous leukemia (AML) by applying the techniques of normal stem cell biology first pioneered in the discovery of HSCs31,32 to the study of leukemias. The general approach, which has become standard in the cancer stem cell field, was to use flow cytometry to isolate phenotypically distinct subpopulations of leukemia cells based on the variable expression of surface proteins. The sorted subpopulations were then injected into immunocompromised nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice and the mice were monitored for engraftment of human leukemia. Using this approach it was determined that for the majority of AMLs, leukemic stem cells (LSCs) reside in the CD34+CD38− subpopulation, and that only these cells can lead to engraftment of human leukemia in NOD-SCID mice. The leukemic blasts, generally displaying the CD34−CD38+ immunophenotype, were unable to transplant the disease.33,34
Within a few years of this pioneering discovery, other investigators showed that cancer stem cells can also be prospectively isolated from solid tumors. This was first accomplished for human breast cancers. As for AML, NOD-SCID mice were used as xenotransplant recipients for flow cytometrically sorted subpopulations of tumor cells from primary human tumors. It was found that for all tumors analyzed, tumorigenic activity was highly enriched in cancer cells with the CD44+CD24−/low-lineage− immunophenotypes (Fig. 72-4A). Injection of as few as 100 of these cells led to tumor formation in mice whereas injection of tens of thousands of the remaining nontumorigenic cancer cells did not.35 Importantly, tumors arising in mice injected with CD44+CD24−/low-Lineage− cells contained the entire diversity of cancer cells found in the primary tumor, including cells with the cancer stem cell immunophenotype and a large population of cells with the nontumorigenic cell immunophenotype. Retransplantation of cells from first-generation xenografts led to similar results. These data indicate that human breast cancers contain a hierarchy of cancer cells.
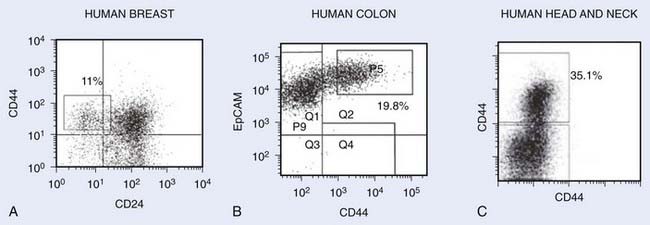
FIGURE 72-4 • Representative flow cytometry analysis of various tumors after dissociation into single-cell suspensions. Cells shown on the plots do not include dead cells and noncancer cells (i.e., lineage cells consisting of hematopoietic cells, endothelial cells, and fibroblasts). The numbers represent the percentage of cells with the markers of cancer stem cells. A, Human breast tumor with cancer stem cells exhibiting CD44+CD24−Lin− phenotype.35 B, Human colon tumor with cancer stem cells exhibiting EpCAM+CD44+Lin− phenotype.38 C, Head and neck tumor exhibiting CD44+Lin− phenotype.40
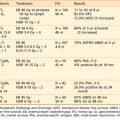
Since these initial studies, cancer stem cell–enriched populations have been isolated from a large number of human malignancies, including those arising in the brain,36 prostate,37 colon,38,39 head and neck,40 pancreas,41,42 and liver43 (Table 72-1). In most human epithelial tumor types, cancer stem cells express CD44, suggesting that commonalities may exist between cancer stem cells from different types of tumors (see Fig. 72-4A-C).
| Tumor | Markers of Tumorigenicity | Percentages of Cells in Patient Samples With Tumorigenic Markers |
|---|---|---|
| Colon tumors38 | ESA+/CD44+ |
Where indicated, the percentages are of viable (DAPI−, 7-AAD−, or propridium iodide−) cells that are lineage negative (Lin−). The markers used “enrich” for cancer stem cells, meaning that all cells staining for the markers are not necessarily tumor-forming cells. Percentages of cells staining for markers enriching for cancer stem cells vary between samples.
The Origin of Cancer Stem Cells
A common point of confusion surrounding the cancer stem cell hypothesis concerns the relationship between cancer stem cells and normal-tissue stem cells. There are two main models for the origin of cancer stem cells. The first posits that normal-tissue stem cells are the cell of origin, because their longevity allows mutations to accumulate and because they already possess the capacity to self-renew. The second model suggests that cancer stem cells can also arise from progenitor cells that have newly acquired the ability to self-renew (Fig. 72-5). Evidence for the second model came from the aforementioned genetic studies of Fialkow and colleagues,28–30 which suggested that leukemia stem cells could arise from either HSCs or progenitor cells. Using X-linked inactivation of G6PD in women with heterozygous alleles of this enzyme, they showed that in patients with chronic myeloid leukemia (CML) and some with AML, cells of both the myelocytic and lymphocytic lineages arose from the leukemic clone. However, in many patients with AML, the leukemic clone only contributed to the myelocytic lineage cells. They therefore favored a model in which the LSC could arise from either a transformed HSC that had lost the normal restrictions on expansion or from a progenitor cell that had acquired self-renewal capacity. However, the first model gained favor after the initial isolation of leukemia stem cells, because in many patients with AML, the phenotype of both the normal AML stem cell and normal HSC is CD34+CD38−. In this model, the lack of contribution to the lymphocytic lineages by the AML stem cell was thought to be due to differentiation blocks by the oncogenic mutations.
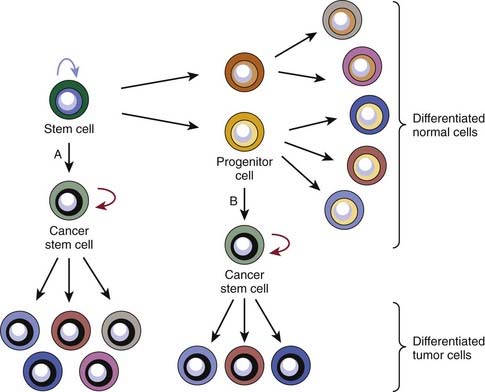
More recent work suggests that the second model is likely to be correct. The strongest evidence comes from investigation of the progression of chronic-phase CML to blast crisis. Jamieson and colleagues have shown that although human leukemia stem cells expressing a marker profile similar to HSCs are the self-renewing cells in chronic-phase CML, leukemic cells expressing a marker profile similar to that of more mature normal progenitor cells are the self-renewing cells in blast crisis CML.44 A better understanding of the hematopoietic cell developmental hierarchy also suggests that the cell of origin of the AML stem cells is a progenitor cell. Although both the HSC and the leukemia stem cells are CD34+/CD38−, in most patients there are clear phenotypic differences between the two cells. HSCs express Thy-1, whereas their malignant counterparts usually do not. Majeti and colleagues have found a normal hematopoietic progenitor cell compartment that is phenotypically indistinguishable from the leukemia stem cell.45 Although it is possible that oncogenic mutations alter the phenotype of the leukemia stem cell, the simplest explanation for these findings is that the cell of origin of the leukemia stem cell in many patients is an early progenitor cell.
For a cancer to arise from a progenitor cell, these cells must gain the ability to self-renew. Work by Akala and colleagues provides a molecular framework for progenitor cell renewal.46 The polycomb gene Bmi1 is critical for the maintenance of multiple types of adult stem cells. It functions via epigenetic repression of genes that induce differentiation, senescence, and cell death. Otherwise-normal early hematopoietic progenitor cells mutated for two loci functionally repressed by Bmi1, Trp53 and Cdkn2a (encoding Ink4a/p16 and Arf/p19), gained the ability to self-renew (triple-mutant cells). Because the self-renewing triple-mutant progenitor cells are more numerous and proliferate more rapidly than stem cells, they might be more likely to sustain the final oncogenic mutations that initiate a tumor. Mutations involving the Arf/Trp53 and Ink4a/RB1 pathways are very common in solid tumors. If these mutations also confer self-renewal capacity on early epithelial progenitor cells, then mutant progenitor cells that harbor mutations in the Trp53 and RB1 pathways could be a cell of origin for common solid tumors, such as those arising in the lung, colon, breast, and prostate.
Regardless of the cell of origin, there is growing evidence that in many tumors cancer stem cells often use many of the same pathways for self-renewal as do normal stem cells. If the cell of origin of a cancer is a normal stem cell, then these pathways were never shut down. If the cell of origin is a progenitor cell, certain stem cell pathways appear to be reactivated. For example, in a mouse model of AML, Meis1 expression is needed for self-renewal of normal stem cells as well as leukemia stem cells that arose from progenitor cells.47 These data argue for the importance of better understanding both normal and cancer stem cell self-renewal mechanisms in the hopes of developing therapies that could specifically interrupt self-renewal in cancer stem cells.
Cancer Stem Cells and Resistance to Chemotherapy
Many of the most commonly used systemic therapies for cancer induce cell death via the induction of deoxyribonucleic acid (DNA) damage. Normal stem cells have evolved multiple mechanisms to protect themselves from toxins and genotoxic stress. The observation that important regulatory pathways are often shared by normal stem cells and cancer stem cells led to the hypothesis that cancer stem cells might be relatively resistant to common therapeutic agents compared with their more differentiated nontumorigenic counterparts.48
Mounting evidence suggests that cancer stem cells are indeed relatively resistant to cytotoxic chemotherapy. A number of studies have shown that “cancer stem–like cells” found in some established cell lines and in long-term xenografts appear to be relatively resistant to cytotoxic chemotherapeutics.49–52 One recent study analyzed human CD44+/ESA+ colon cancer stem cells in early passage xenografts and found that these were enriched in xenografts that had been treated with cyclophosphamide.53 Enrichment was observed both at the level of immunophenotypes (i.e., percentage of cancer stem cells as assessed by flow cytometry) and in limiting dilution assays of unfractionated tumor cells, indicating both a phenotypic and functional enrichment of tumor forming cells. Although data from preclinical models such as these are highly suggestive of cancer stem cell chemoresistance, it is critical to study the same question in human cancer patients. Recently, Li and colleagues measured the frequency of cancer stem cells in tumors of women receiving neoadjuvant chemotherapy for locally advanced breast cancer.54 As had been predicted, they found that cancer stem cells accumulated after treatment with cytotoxic chemotherapy.
What are the mechanisms by which cancer stem cells resist chemotherapy-induced cell killing? Actively cycling cells are more sensitive to cytotoxic therapies than quiescent cells. Multiple investigators have shown that leukemia stem cells can be quiescent and resistant to chemotherapy.55 It is not yet clear whether this holds true for normal and malignant epithelial stem cells. Although at least some skin stem cells are quiescent,56 at least a portion of the normal intestinal stem cells divide daily.57 Also, cell cycle profiles of human breast cancer stem cells and nontumorigenic cells appear to be similar, arguing against large difference in cell cycle status between these populations.35 Another commonly cited mechanism for stem cell resistance to toxic agents is the expression of multiple drug-resistant transporters. These have long been known to be expressed by normal immature cells such as HSCs and hematopoietic progenitors58 and can lead to a characteristic appearance on flow cytometry known as side population, which reflects the ability to export fluorescent dyes. There is evidence that cancer stem-like cells in some established cell lines also display chemoresistance because of expression of MDR genes,59–61 although the importance of this mechanism in primary tumor stem cells remains to be elucidated. Finally, enzymes that detoxify certain toxins also tend to be expressed at high levels by stem cells. Members of the aldehyde dehydrogenase (ALDH) pathway protect cells from the alkylating agent cyclophosphamide. These enzymes are highly expressed by many types of normal stem cells and thus help to protect them from this drug. Many of the cancer stem cells in some, but not all, breast and colon tumors sometimes overexpress ALDH and are thus relatively resistant to cyclophosphamide.38,53,62 Ultimately, it is likely that cancer stem cells in many tumors are relatively chemoresistant because of a combination of molecular mechanisms.
Cancer Stem Cells and Resistance to Radiotherapy
The notion that cancer stem cells may be resistant to ionizing radiation began to be investigated shortly after the discovery of cancer stem cells in solid tumors. Unlike in the case of chemotherapy, there is less evidence indicating that normal-tissue stem cells are more resistant to ionizing radiation than their more differentiated progeny. However, work in both the hematopoietic and mammary systems suggests that more immature cells are relatively radioresistant compared with their more differentiated progeny.63,64 Thus, the obvious question arises whether cancer stem cells in solid tumors may also display this property.
In addition to the groundbreaking work of Till and McCullough on normal-tissue stem cells, radiobiologists were also among the earliest investigators studying cancer stem cells through the use of local control assays, which test the ability of tumors to regrow after treatment. By definition, cancer stem cells are the only cells that can lead to significant regrowth of tumors and thus local control assays test the ability of cancer stem cells to survive a given therapy. A significant body of data in the radiobiologic literature suggests that cancer stem cell content (as assessed by in vivo and in vitro clonogenic assays) and their intrinsic radiosensitivity correlate with radioresistance.21,65–67 Caveats of these studies include the fact that many used long-term cell lines or xenografts that may not faithfully recapitulate properties of primary tumors and that in vitro clonogenic activity does not necessarily reflect in vivo repopulating ability.15
Recently, investigators have begun to apply the modern tools of stem cell biology to the question of cancer stem cell radiosensitivity.64,68–74 Attention has particularly focused on models of breast and brain tumors, which were the first solid tumors for which cancer stem cell markers became available and for which clinical treatment plans often include radiotherapy. Most of these studies focused on cancer stem–like cells within established cell lines that had previously been selected for in vitro growth for many generations, thus again raising concerns that results may not necessarily reflect the behavior of cancer stem cells in primary tumors. However, taken in aggregate, these studies indicate that in many tumors, irradiation in vivo or in vitro leads to a relative enrichment of cancer stem cells as measured by flow cytometry. For example, Bao and colleagues examined the effects of irradiation on glioblastoma multiforme (GBM) xenografts and cells from primary tumor specimens.68 It had previously been shown that the CD133+ subpopulation is significantly enriched in cancer stem cells in at least a subset of GBMs.36 Bao et al.68 showed that CD133+ cells accumulated after irradiation both in vitro and in vivo (i.e., xenografts), increasing by a factor of approximately two- to threefold (Fig. 72-6). This degree of enrichment seemed to have biologic relevance, because tumors initiated from cell suspensions that were only slightly enriched for CD133+ cells grew much more rapidly. The authors also performed several molecular assays of radiation damage and showed that as measured by apoptosis and DNA damage, GBM cancer stem cells appeared relatively radioresistant compared with their nontumorigenic counterparts.
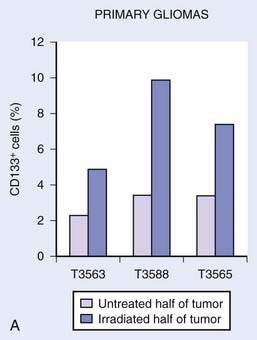
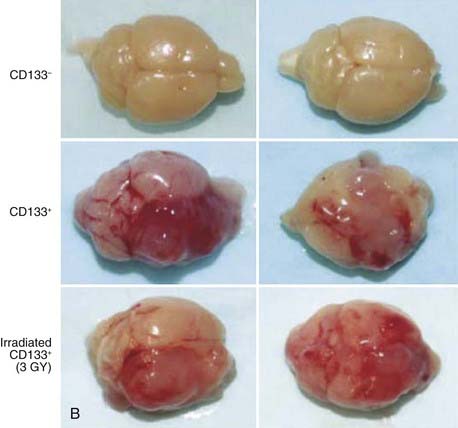
(From Bao S, et al: Glioma stem cells promote radioresistance by preferential activation of the DNA damage response, Nature 444:756–760, 2006.68)
The mechanisms leading to the observed cancer stem cell radioresistance have not been firmly established, but they are likely to be multifactorial. Pathways or mechanisms that have been implicated include DNA repair, resistance to apoptosis, resistance to oxidative stress, activation of Wnt/beta-catenin, the sonic hedgehog pathway, and Notch signaling.* It is also likely that cancer stem cells from different tumors, even within the same histologic subgroup, will be found to rely on overlapping but nonidentical resistance mechanisms. Because normal-tissue stem cells and their progenitors appear to differ in radiosensitivity and because cancer stem cells can originate either from normal-tissue stem cells or more differentiated cells, the cell of origin may play an important role in determining the radiosensitivity of cancer stem cells.
Acquired Treatment Resistance and Cancer Stem Cells
Although cancer stem cells in some tumors appear to use intrinsic stem cell pathways to protect themselves from cytotoxic therapies, this does not minimize the importance of classic models of drug and radiation resistance. The typical pattern of tumor response to cytotoxic therapy gives clues to mechanisms of resistance. For example, in tumors responsive to chemotherapy, one often observes an initial shrinkage followed by subsequent regrowth, even while still on drug therapy. Given the aforementioned observations of relative chemoresistance of cancer stem cells, this shrinkage is likely due in large part to the disproportionate death of nontumorigenic cancer cells. The fraction of cancer stem cells that were not killed can then regrow the tumor (Fig. 72-7).
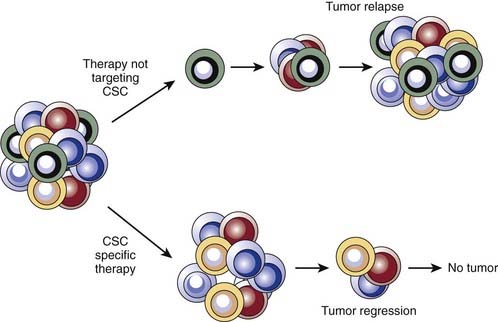
Even when tumors are initially responsive to radiotherapy and chemotherapy, many eventually fail to respond to therapy, particularly in the metastatic setting. This is often accompanied by the appearance of a clone of cancer cells that has upregulated the expression of specific resistance genes. A hybrid model can be envisioned in which intrinsic stem cell pathways partially protect the stem cells, which allows for the initial shrinkage but eventual regrowth. Over time, a cancer stem cell clone that has acquired mutations that make it resistant to the therapy is selected for, making the tumors completely resistant to the drug. Along these lines, preliminary evidence from colon cancer studies suggests that genomic instability within cancer stem cells can lead to new clones of cancer stem cells, which have a similar but nonidentical karyotype compared with the parental clone.75 These stem cells can give rise to differentiating progeny that also contain the new karyotype. An alternative model is that during therapy, ongoing mutations expand the cancer stem cell pool, because these cells are intrinsically resistant to therapy. A prediction of this approach is that the cancer stem cell frequency would increase after treatment and might remain elevated even after removal of the drug. Thus, the natural history of tumor response to chemotherapy or radiation in the clinic suggests that intrinsic stem cell resistance pathways and acquisition of new mutations may both contribute to resistance.
Location and Motility of Cancer Stem Cells
Given the geographically localized nature of radiation therapy, the location of cancer stem cells within tumors is a question of critical importance in radiation oncology. Although data addressing this question are still rather scant, a recent report by Prince et al.40 contains some intriguing information. Moderately and well-differentiated head and neck squamous cell carcinomas form structures reminiscent of normal mucosa, in which there is an apparent hierarchy of differentiation, with immature cells in the basal layer and more mature cells in higher layers. Using immunohistochemistry, Prince et al.40 analyzed the localization of cancer stem cells using markers such as CD44 and showed that these cells are found in the “basal” layer, adjacent to stromal cells. This suggests that, at least in head and neck tumors, cancer stem cells are found adjacent to stromal cords within tumors and at the stromal-tumor interface at the margins of tumors.
Indeed, one likely could have predicted this observation for a variety of reasons, including the aforementioned similarities between cancer stem cells and normal stem cells. Normal stem cells in many organs must be able to migrate to form fully functional tissues. For example, lactation is only possible because normal breast stem cells are able to migrate from a rudimentary duct into the surrounding mammary fat pad to generate a milk-producing network of ducts and glands. Neural crest stem cells migrate from the neural tube into the periphery to form the enteric nervous system, and central nervous system stem cells migrate from the hippocampus and subventricular zones to various regions of the brain. In the fetus, a genetic pathway called epithelial mesenchymal transition (EMT) regulates stem cell migrations and critical steps of morphogenesis.76,77 Gene expression profiling of human breast cancer stem cells revealed that these overexpress genes involved in cell migration and invasion.78 Elegant experiments by Mani and colleagues have shown that EMT genes are often active in both normal breast stem cells and their malignant counterparts, breast cancer stem cells, but not in more differentiated normal or malignant cells.79 Thus, expression of EMT genes in both normal and cancer stem cells likely promotes their invasiveness and tendency to be located near stromal elements.
The propensity of cancer stem cells to migrate affects the design of local therapies and local failure rates in many clinical situations. One example is GBM, in which tumor cells are often widely disseminated in the brain at the time of diagnosis, and standard radiation treatment fields cover volumes much larger than the contrast-enhancing portion of the tumors in an attempt to eliminate malignant cells that have migrated away from the primary site. Experiments consisting of injections of either normal human neural stem cells or human glioblastoma stem cells into the brains of immunodeficient mice provide graphic evidence of the potential effect of stem cell migration on local therapies. Using identical migration routes, both the human normal and malignant stem cells migrate to all regions of the brain. The cancer stem cells, but not their normal counterparts, also form tumors (Fig. 72-8).
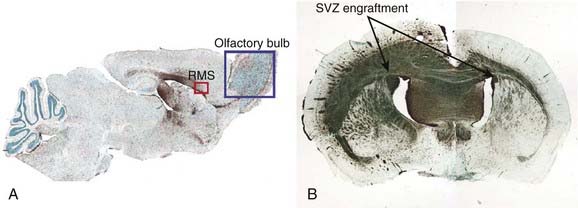
Therapeutic Targeting of Cancer Stem Cells
The best therapeutics would specifically target survival, drug resistance, or other critical pathways in cancer stem cells. Such agents would thus kill cancer stem cells while sparing normal stem cells. Preliminary studies by Jordan and colleagues suggest that the drug parthenolide, which inhibits the prosurvival transcription factor NF-κB and elevates intracellular reactive oxygen species, may be selectively toxic for human AML stem cells compared with normal HSCs.80 If cancer stem cells in a particular tumor arise from a progenitor cell rather than a normal stem cell, then differences in the two cell types could possibly exploited. For example, the aforementioned self renewing hematopoietic progenitor cells in mice mutant for Trp53, Arf, and Ink4a do not respond to growth factors used by HSCs to self-renew, although they do respond to growth factors used by progenitors. This suggests that antibodies or small molecules against the progenitor cells or their growth factors would spare HSCs while inhibiting the mutant progenitor cells. Similar approaches may work against cancer stem cells that arose from normal progenitors.
Even if a critical pathway is regulated similarly in both malignant and normal stem cells, selective delivery of the therapy to the cancer stem cells could still be used successfully in the clinic. Radiation, of course, is the paradigm for spatially selective delivery of toxic therapies. Recent advances in intensity-modulated radiation therapy and stereotactic radiosurgery have increased the doses that can be delivered to many types of tumor while still limiting normal-tissue toxicity to acceptable levels. Nonetheless, there still are patients who relapse locally after irradiation. Understanding the resistance pathways used by the cancer stem cells could point to new avenues for radiosensitization and thus improve the local control of tumors. One potential pathway was discovered by Bao and colleagues in the studies on GBM stem cells.68 They showed that GBM stem cells are relatively resistant to radiation and that a CHK1 inhibitor sensitized them to ionizing radiation. It is conceivable that such an inhibitor could be used to improve local control of brain tumors, although it will be critical to assess its effects on radiosensitization of normal brain cell types.
1 Krause M, Zips D, Thames HD, et al. Preclinical evaluation of molecular-targeted anticancer agents for radiotherapy. Radiother Oncol. 2006;80:112-122.
2 Steel GG. The search for therapeutic gain in the combination of radiotherapy and chemotherapy. Radiother Oncol. 1988;11:31-53.
3 Tannock IF. Treatment of cancer with radiation and drugs. J Clin Oncol. 1996;14:3156-3174.
4 Kondo M, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759-806.
5 McCulloch EA, Till JE. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat Res. 1960;13:115-125.
6 McCulloch EA, Till JE. Perspectives on the properties of stem cells. Nat Med. 2005;11:1026-1028.
7 Till JE, Mc CE. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res. 1961;14:213-222.
8 Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267-284.
9 Morrison SJ, et al. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J Immunol. 2002;168:635-642.
10 Stingl J, Raouf A, Eirew P, et al. Deciphering the mammary epithelial cell hierarchy. Cell Cycle. 2006;5:1519-1522.
11 Deome KB, Faulkin LJJr, Bern HA, et al. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959;19:515-520.
12 Kordon EC, Smith GH. An entire functional mammary gland may comprise the progeny from a single cell. Development. 1998;125:1921-1930.
13 Tsai YC, et al. Contiguous patches of normal human mammary epithelium derived from a single stem cell: implications for breast carcinogenesis. Cancer Res. 1996;56:402-404.
14 Shackleton M, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84-88.
15 Stingl J, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993-997.
16 Virchow R. Die Cellularpathologie in ihrer Begrundung auf physiologische und pathologische Gewebelehre. Berlin: August Hirschwald; 1858.
17 Donohue JP, et al. Persistent cancer in postchemotherapy retroperitoneal lymph-node dissection: outcome analysis. World J Urol. 1994;12:190-195.
18 Sarosdy MF, Einhorn LH, Donohue JP, et al. Correlation of clonogenic growth with histologic diagnosis after resection of residual tissue following chemotherapy for nonseminomatous testis cancer. Urology. 1989;34:396-398.
19 Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199:79-80.
20 Hewitt HB, Blake ER, Walder AS. A critique of the evidence for active host defence against cancer, based on personal studies of 27 murine tumours of spontaneous origin. Br J Cancer. 1976;33:241-259.
21 Hill RP, Milas L. The proportion of stem cells in murine tumors. Int J Radiat Oncol Biol Phys. 1989;16:513-518.
22 Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461-463.
23 West CM, Davidson SE, Hunter RD. Evaluation of surviving fraction at 2 Gy as a potential prognostic factor for the radiotherapy of carcinoma of the cervix. Int J Radiat Biol. 1989;56:761-765.
24 Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643-649.
25 Clarke MF, et al. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339-9344.
26 Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A. 1976;73:549-553.
27 Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72:3585-3589.
28 Fialkow PJ. Use of genetic markers to study cellular origin and development of tumors in human females. Adv Cancer Res. 1972;15:191-226.
29 Fialkow PJ, Faguet GB, Jacobson RJ, et al. Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood. 1981;58:916-919.
30 Fialkow PJ. Stem cell origin of human myeloid blood cell neoplasms. Verh Dtsch Ges Pathol. 1990;74:43-47.
31 Baum CM, Weissman IL, Tsukamoto AS, et al. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992;89:2804-2808.
32 Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58-62.
33 Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737.
34 Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-648.
35 Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-3988.
36 Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
37 Collins AT, Berry PA, Hyde C, et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-10951.
38 Dalerba P, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158-10163.
39 O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-110.
40 Prince ME, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973-978.
41 Li C, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030-1037.
42 Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313-323.
43 Yang ZF, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153-166.
44 Jamieson CH, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657-667.
45 Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635-645.
46 Akala OO, et al. Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a−/−p19Arf−/− multipotent progenitors. Nature. 2008;453:228-232.
47 Wong P, Iwasaki M, Somervaille TC, et al. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762-2774.
48 Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111.
49 Dangles-Marie V, et al. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 2007;67:398-407.
50 Ho MM, Ng AV, Lam S, et al. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827-4833.
51 Patrawala L, et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207-6219.
52 Todaro M, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389-402.
53 Dylla SJ, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE. 2008;3:e2428.
54 Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672-679.
55 Ishikawa F, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315-1321.
56 Kobielak K, Stokes N, de la Cruz J, et al. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci U S A. 2007;104:10063-10068.
57 Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-1007.
58 Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507-512.
59 Hirschmann-Jax C, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228-14233.
60 Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275-284.
61 Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872-877.
62 Ginestier C, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555-567.
63 Down JD, Boudewijn A, Van OS, et al. Variations in radiation sensitivity and repair among different hematopoietic stem cell subsets following fractionated irradiation. Blood. 1995;86:122-127.
64 Woodward WA, et al. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104:618-623.
65 Baumann M, Dubois W, Suit HD. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiat Res. 1990;123:325-330.
66 Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8:545-554.
67 Yaromina A, et al. Pre-treatment number of clonogenic cells and their radiosensitivity are major determinants of local tumour control after fractionated irradiation. Radiother Oncol. 2007;83:304-310.
68 Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-760.
69 Bar EE, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524-2533.
70 Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133− cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys. 2007;67:1-5.
71 Chiou SH, et al. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE. 2008;3:e2090.
72 Kang MK, et al. Potential identity of multi-potential cancer stem-like subpopulation after radiation of cultured brain glioma. BMC Neurosci. 2008;9:15.
73 Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777-1785.
74 Wang J, Guo LP, Chen LZ, et al. Identification of cancer stem cell-like side population cells in human nasopharyngeal carcinoma cell line. Cancer Res. 2007;67:3716-3724.
75 Odoux C, et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008;68:6932-6941.
76 Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19:294-308.
77 Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131-142.
78 Liu R, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217-226.
79 Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-715.
80 Guzman ML, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163-4169.