[level-membership-for-hematology-oncology-and-palliative-medicine-category]
34 Cancer of the Thyroid
Epidemiology
Primary thyroid cancer is the most common endocrine malignancy, with an estimated incidence of 37,340 cases in the United States per year.1 This incidence is increasing, and thyroid cancer is now the eighth most common malignancy in women and the second most common malignancy in women under 40 years of age.2 Most, but not all, of this increase is a consequence of the increased detection of smaller WDTC lesions.2 Autopsy studies suggest that there is a large number of people in the general population with small, subclinical WDTC lesions.3
With regards to gender, women represent 76% of newly diagnosed cases.1 With regards to race, the incidence per 100,000 Caucasian, Asian, Hispanic, and Black women, is 4.9, 4.1, 3.5, and 2.7, respectively.4 In the San Francisco Bay area, well-controlled epidemiological studies indicate that the rate of thyroid cancer is higher in women of Southeast Asian descent.5,6
Environmental and dietary factors may also impact the frequency and distribution of thyroid cancer subtypes in a population. Ionizing radiation,7 goitrogens,8 and familial syndromes,9 have all been associated with PTC. People exposed to ionizing radiation during the atomic exposures at Hiroshima and Nagasaki in 194510 and children living near the Chernobyl nuclear accident in 198611 have a higher incidence of PTC. Furthermore, external beam radiation treatments in children for conditions such as acne, ringworm, birthmarks, enlarged thymus glands, or tonsillitis are associated with a higher risk of PTC later in life.12 Iatrogenic radioiodine exposure also slightly increases the risk of developing thyroid cancer and benign thyroid disorders.13 The incidence of FTC is higher in iodine-deficient regions, while PTC rates are higher in iodine-replete regions.14 Familial PTC has been described in numerous families, although the gene or genes responsible for this syndrome has not been elucidated.15 With regards to prognosis, patients with familial PTC with more than two members with PTC have a worse prognosis than those with sporadic disease.16
ATC accounts for more than 50% of the thyroid cancer deaths in the United States, despite the fact that it represents less than 2% of all thyroid cancers diagnosed.17 A higher incidence of ATC is seen in areas with low socioeconomic status and a high incidence of endemic goiter.18 There is ample evidence that ATC originates through the dedifferentiation of WDTC cells; thus, incomplete or delayed treatment of WDTC increases the risk of anaplastic transformation.19
Molecular Basis of Thyroid Cancer
Although thyroid oncogenesis is not completely understood, some of the genetic changes that drive malignant transformation have been described for each histological subtype of thyroid cancer (Fig. 34-1). Thyroid oncogenesis entails the stepwise progression of molecular alterations that dysregulate cellular proliferation, apoptosis, and invasion.
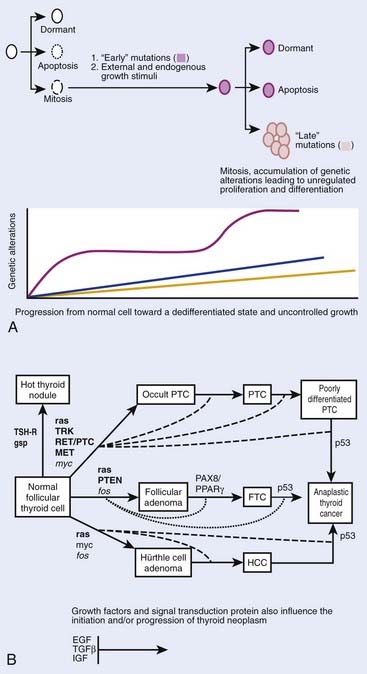
Most sporadic PTCs harbor an activating mutation of receptor tyrosine kinases or the RAS-RAF-MEK-ERK pathway.20 These mutations are generally nonoverlapping, suggesting that such activation plays a central role in thyroid oncogenesis.20 The B type RAF kinase (BRAF) is the most prominent RAF kinase in thyrocytes, and point mutations in BRAF are found in approximately 45% of sporadic PTC.21 The presence of BRAF mutations in PTC microcarcinomas suggests that it plays a role in the initiation of thyroid oncogenesis.22 The BRAF V600E point mutation is reportedly associated with aggressive tumor behavior. PTCs with this mutation have higher rates of lymph node metastases and local invasion, resulting in higher rates of recurrent and persistent disease after thyroidectomy.23 Lastly, intra- and interchromosomal inversions of the RET proto-oncogene (RET/PTC) can also be found in PTC, which results in constitutive activation.24 These RET/PTC rearrangements are seen in approximately 75% of PTCs associated with childhood exposure to ionizing radiation.25,26
Approximately 25% of all MTC cases are inherited in an autosomal dominant fashion in one of three familial syndromes.27 In MEN2A, MTC is associated with pheochromocytoma and hyperparathyroidism.28 In MEN 2B, MTC is associated with pheochromocytoma but not with hyperparathyroidism, and patients are characterized by a Marfanoid appearance and oral mucosal neuromas.28 Lastly, isolated familial MTC is marked by the autosomal dominant transmittance of MTC with no other associated endocrinopathy.28
The confirmation that MTC could be part of a familial syndrome prompted a considerable effort to uncover its molecular basis, which was later identified as a point mutation in the RET proto-oncogene.29 The RET proto-oncogene is a tyrosine kinase receptor located on chromosome 10. Interestingly, somatic mutations in the RET proto-oncogene also account for approximately 25% to 30% of sporadic cases of MTC.30 MTC tumor behavior and clinical outcomes are heterogeneous, and prior studies have demonstrated that tumor behavior is contingent upon genotype. For example, the somatic 918 RET mutation is associated with aggressive MTC behavior. These important molecular discoveries have shifted the focus of MTC to early identification and prophylactic surgery, which have improved patient outcomes.31,32
Although virtually all ATC have a p53-related defect, the mechanisms of anaplastic transformation remain incompletely understood.33,34 ATC is likely derived from the dedifferentiation of WDTC and studies suggest that activation of both RAS-RAF-MAPK and PI3K pathways may play a role in anaplastic transformation.35,36 One study found an increasing frequency of genetic alterations in RAS-RAF-MAPK and PI3K/Akt in ATC relative to DTC when these histological subtypes coexisted in the same specimen.37 In the same study, they found that the only overlapping mutations in matched WDTC and ATC were in BRAF and PIK3CA.37 Conversely, a different study showed that BRAF and p53 were both altered in ATC, but that they were not overlapping with RAS or RET/PTC mutations.38
Anatomic Considerations
The thyroid gland wraps around the anterolateral trachea with two shield-like lobes connected by an isthmus, giving an appearance that is vaguely similar to a butterfly (Fig. 34-2). The most useful surface anatomy landmark is the cricoid cartilage, which is found just cephalad to the thyroid isthmus. The cricoid cartilage is palpable caudal to the tracheal cartilage, or Adam’s apple. Alternatively, the cricoid cartilage can be found by identifying the sternal notch and palpating the trachea in a cephalad direction; the cricoid cartilage will usually be the first prominent cartilage palpated using this technique. The pyramidal lobe, which is the distal remnant of the thyroglossal duct, is a narrow piece of thyroid tissue that extends cranially from the isthmus. An unresected pyramidal lobe is occasionally the cause of remnant thyroid tissue detected on postoperative RAI scans. Aberrant thyroid tissue can be found in the midline, and very rarely, lateral or inferior to the main body of the thyroid gland within the central neck. More commonly, thyroid cells discovered from FNA samples lateral to the carotid sheath and thyroid gland represent metastatic WDTC.
The lymphatic drainage of the thyroid gland flows in all directions and is inconsistent. The anatomic classification scheme that defines the cervical lymphatic subdivisions was most recently updated by the Committee for Head and Neck Surgery and the American Academy of Otolaryngology-Head and Neck Surgery (Fig. 34-3).39 The central neck compartment, designated level VI, is located between the carotid sheaths. The lateral neck compartments, including the jugular chain (level II to IV) and posterior neck (level V), are generally secondary drainage basins after the central neck.
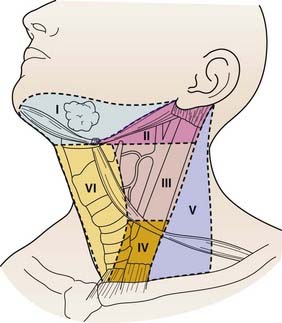
FIGURE 34-3 • Lymph node anatomic classification: the six levels of cervical lymph nodes in the neck.
(From Robbins KT, Clayman G, Levine PA, et al: Neck dissection classification update; Revisions proposed by the American Head and Neck Society and the American Academy of Otolaryngology—Head and Neck Surgery, Archives of Otolaryngol Head Neck Surg 128:751–758, 2002, p 752.)
The pattern of metastases in PTC typically begins with spread to ipsilateral central and then ipsilateral lateral lymph nodes, and then later to contralateral cervical lymph nodes.40 However, reports of “skip” metastasis to the lateral neck with normal central neck lymph nodes have been reported in about 20% of patients with PTC and MTC and may be more common in patients with primary tumors located in the superior thyroid poles. Perithyroidal, pretracheal lymph nodes, termed “Delphian” lymph nodes, when found to harbor metastatic disease, are associated with higher incidence of central and lateral compartment metastases.41 PTC can also spread to distant sites, including the lungs and bones in approximately 4% of newly diagnosed patients.42
Lymph node metastases are found in less than 6% of FTC in contrast to 6% to 30% of patients with HCC.43 Lung metastases from hematogenous spread are more common in FTC, HCC, and ATC. Liver metastases are relatively common in MTC, but are rare in thyroid cancers of follicular cell origin.
Physiology
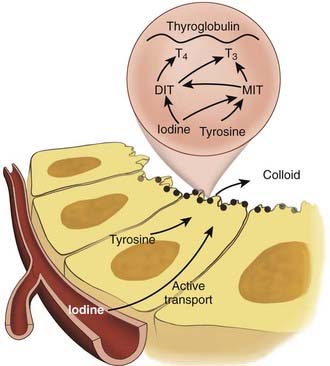
The principal endocrine function of the thyroid gland is the production and controlled release of thyroid hormone and calcitonin. Systemic thyroid hormone levels impact development, growth and metabolism. The two forms of thyroid hormone, triiodothyronine (T3) and thyroxine (T4) are synthesized within thyroid follicles. Thyroglobulin is a large glycoprotein synthesized by follicular cells, which is the source of tyrosyl residues needed for thyroid hormone synthesis. Both T3 and T4 are synthesized through the coupling of iodide with tyrosyl residues in a reaction involving the enzyme thyroperoxidase (Fig. 34-4) Newly synthesized thyroid hormone is bound to thyroglobulin protein and stored in colloid, which can later be released into the systemic circulation. Hydrolysis of thyroglobulin protein and release of thyroid hormone is regulated by TSH levels and the availability of iodide.
Thyroid hormone levels are tightly regulated in a negative-feedback manner within the hypothalamic-pituitary-thyroid axis (Fig. 34-5). Thyroid stimulating hormone (TSH) is the central element in this axis, which directly controls the activity and proliferation of follicular cells. TSH is synthesized and secreted by the anterior pituitary, a process that is regulated by thyrotropin-releasing hormone and blood thyroid hormone levels. Thyrotropin-releasing hormone is synthesized in the hypothalamus and is transported to the pituitary via the hypophyseal portal system, a process that is suppressed when systemic T3 levels are high.
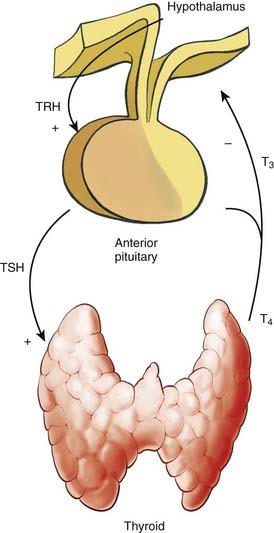
The TSH receptor is a G-protein-coupled receptor which upregulates thyroid hormone production and release via cAMP associated signaling. Because TSH can induce the proliferation of both normal thyrocytes and WDTC of follicular origin, suppression of TSH is an important feature of thyroid cancer treatment. Furthermore, TSH receptor mutations44 and a higher concentration of TSH receptors45 have been associated with autonomous follicular thyroid neoplasms.
Clinical Presentation and Diagnosis
In addition to a complete history and physical exam, patients that present with a neck mass suspicious for a thyroid neoplasm should undergo formal neck ultrasound by an experienced ultrasonographer to characterize the lesion, monitor the remainder of the thyroid gland for abnormalities, and to evaluate for abnormal cervical lymph nodes. FNA biopsy is a reliable diagnostic tool that should be performed on thyroid lesions with suspicious sonographic characteristics, such as internal microcalcifications. FNA has a sensitivity of 95% to 98% and specificity of 97% to 99%, when performed by an experienced cytopathologist.46 Unfortunately, FNA is not an effective for discriminating FTC from benign follicular adenomas, or HCC from Hürthle cell adenomas. Histologic architecture is required to determine if a follicular or Hürthle cell neoplasms are malignant; capsular or vascular invasion must be seen to diagnose carcinoma in these subtypes. The current standard of care in the evaluation of a follicular neoplasm requires diagnostic hemithyroidectomy with formal histological review to determine if the lesion has invasive features consistent with carcinoma. Molecular-based tools will likely eliminate the need to perform diagnostic surgery for such patients; several groups have recently used tumor gene expression to define molecular signatures that can discriminate benign from most malignant follicular tumors.47–49
Patients with sporadic MTC typically present with solitary thyroid nodules situated at the upper lateral regions of the thyroid gland, where the preponderance of C cells are located. On physical exam, MTC are described as hard, nodular tumors that are sometimes calcified. Patients with locally invasive MTC often complain of “aching” or painful thyroid tumors. Carcinoembryonic antigen (CEA) levels are almost always elevated in patients with MTC, which is the same serum protein marker utilized to screen colorectal cancer patients for recurrence after resection. Since MTC can secrete several proteins (e.g., somatostatin, vasoactive intestinal peptide, adrenocorticotropic hormone (ACTH), bombesin), patients with advanced disease and hepatic metastases can present with generalized symptoms, such as flushing, Cushing’s syndrome, and diarrhea is common.50
The diagnosis of MTC is reliably established by FNA, which reveals amyloid and may stain positive for calcitonin, CEA, and chromogranin A.51 In relatives of patients with familial MTC, small tumors and C-cell hyperplasia can be diagnosed by elevated basal or stimulated levels of calcitonin. High CEA levels appear to be a worse prognostic indicator than elevated blood calcitonin levels.
In comparison with sporadic MTC, hereditary MTC generally occurs in younger patients and can be seen in the context of the isolated familial MTC and MEN2 syndromes. A recent claim that former American president Abraham Lincoln may have had MEN2B seems unlikely,52 given that untreated patients with this syndrome typically die within the first 3 decades of life, with few reports of asymptomatic long-term survival.53 Although there are some RET proto-oncogene mutations that result in MTC in older patients, patients with MTC and MEN2B have the worst prognosis of all patients with MTC.
Patients with ATC typically present with a rapidly enlarging neck mass that is fixed to surrounding structures. ATC are generally large and cause local obstructive symptoms from their mass effect, including pressure in the neck, dysphagia, and dysphonia. Patients with thyroid lymphoma can also present in a similar fashion. In our own experience with ATC, the average tumor size was 7 cm and the majority of patients presented with symptomatic disease. Furthermore, invasive thyroid tumors can invade the recurrent laryngeal nerves and cause vocal cord dysfunction and regional pain. ATC can be readily confirmed by FNA biopsy with cytological review.54 Distant metastases are common at presentation and can be found in up to 43% of patients.55 Both distant and regional metastases essentially preclude curative therapy for patients with ATC.
Prognostic Models and Staging of Thyroid Cancer
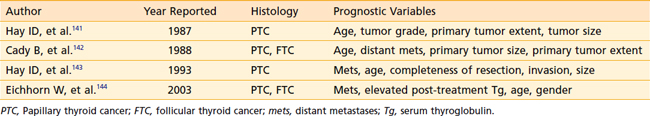
While the majority of patients with WDTC have excellent outcomes after surgical resection, some patients harbor aggressive disease. In an effort to select high-risk patients for adjuvant therapy, prognostic models utilize clinical and pathologic factors to predict survival (Table 34-1). Multiple large retrospective reviews have confirmed that age, tumor grade, distant metastases, tumor invasion, and tumor size are independent predictors of outcome after thyroidectomy.
The American Joint Committee on Cancer TNM system characterizes the anatomic status of the primary tumor, regional lymph nodes and distant metastases (Table 34-2). This system was updated in 2003, and is often applied to both WDTC and MTC. TNM staging can be used as a prognostic tool and also for research purposes to enable consistent stratification of patients.
Surgery for WDTC
Surgical resection is the primary treatment for patients with WDTC.56 Thyroidectomy is both safe and effective when performed by an experienced surgeon, with 10-year survival rates approximating 93% and 85% for PTC and FTC, respectively.17 Although most clinicians agree that total thyroidectomy is the primary treatment for patients with high-risk PTC, there is some controversy regarding the extent of surgery for patients with low-risk WDTC.57 Some clinicians argue that thyroid lobectomy is equivalent to total thyroidectomy, and avoids the potential morbidity of bilateral operation.58 We, and other experienced surgeons and endocrinologists, believe that total thyroidectomy should be employed for WDTCs larger than 1 cm, in the absence of advanced distant disease in patients with an acceptable performance status for neck surgery.57
There are several reasons for performing total thyroidectomy in all patients with WDTC, excluding occult micropapillary carcinomas. First, it improves the efficacy of adjuvant RAI therapy, particularly in the setting of distant metastases. In some instances, synchronous metastases outside of the neck, such as diffuse pulmonary metastases, may not be detected unless total thyroidectomy is performed. Furthermore, total thyroidectomy facilitates the use of serum thyroglobulin levels for postoperative screening. Also, PTCs are often multifocal, involving both thyroid lobes. Several studies have demonstrated superior outcomes for patients with WDTC greater than 1 cm when treated with total thyroidectomy rather than thyroid lobectomy.59–61
There are several potential complications related to thyroidectomy. The most dramatic complication is recurrent laryngeal nerve injury, which can result in temporary or permanent vocal cord dysfunction.62 Patients with recurrent or persistent thyroid cancer or with large invasive tumors are at the highest risk of recurrent laryngeal nerve injury. In the hands of an experienced thyroid surgeon, the incidence of permanent recurrent laryngeal nerve palsy should be less than 1 percent.63 The voice weakness and risk of aspiration from permanent recurrent laryngeal nerve palsy can be treated by vocal cord medialization, and the majority of patients that undergo this procedure can eventually generate a near-normal voice.64 Bilateral recurrent laryngeal nerve damage is a most serious complication and often requires tracheostomy placement to prevent respiratory distress and aspiration. This complication is exceedingly rare in the hands of an experienced thyroid surgeon. All reoperative cases in the central neck should have preoperative direct laryngoscopy to check for vocal cord dysfunction from prior recurrent laryngeal nerve injury or loss of nerve function due to the related tumor.
Hypoparathyroidism from parathyroid gland damage or devascularization is another uncommon but serious complication from thyroid surgery.65 Patients with permanent hypoparathyroidism can suffer from perioral numbness, extremity paresthesias and muscle cramping from chronic hypocalcemia. These patients typically require regular vitamin D and calcium supplementation to avoid these symptoms. Other complications from total thyroidectomy include postoperative neck hematoma, infection, and seroma.
Surgery for MTC
Similar to WDTC, surgical resection is the primary therapy and the only curative treatment modality for patients with MTC. Complete resection is possible in MTC, particularly when the lesion is discovered incidentally or when prophylactic thyroidectomy is performed for familial disease prior to age 6. The incidence of ipsilateral, central, and lateral lymph node metastases is approximately 60% in patients with palpable primary lesions.66 Because regional lymph node metastases are common, bilateral central neck and ipsilateral or bilateral lateral neck dissection are recommended for all patients with MTCs larger than 1.5 cm.67
RET proto-oncogene testing and prophylactic thyroidectomy has proven to be the most effective treatment for patients with C cell hyperplasia and occult MTC.31,68 Genetic testing for RET mutations are routinely performed for patients with presumed sporadic MTC, family members of those with documented RET mutations, and related conditions such as pheochromocytoma and Hirschsprung’s disease. The importance of genetic screening is underscored by the effectiveness of prophylactic total thyroidectomy for family members harboring RET proto-oncogene mutations.69 Some researchers suggest that the timing and extent of prophylactic surgery be individualized based on the specific RET mutation status, as genotype-phenotype correlations have shown that different RET mutations carry different risks for MTC aggressiveness, age of onset, and lymph node metastases.70,71 Prophylactic thyroidectomy offers the best outcomes for patients that harbor RET proto-oncogene mutations.72 Studies correlating genotype with clinical outcomes have demonstrated that particular mutations carry a much higher risk of MTC at younger ages than other RET mutations. These mutations have been risk stratified, and the highest risk mutations, such as those associated with MEN2B, require prophylactic thyroidectomy in children within the first year of life or at diagnosis. Some moderately high risk mutations, such as the most common 634 RET mutation, warrant total thyroidectomy prior to age 6.31,32 Attaining complete extirpation of all C cells prior to their inevitable malignant transformation offers the best chance of cure. The optimal extent of surgery may also be determined by biochemical data; bilateral central neck dissection is not necessary in RET positive children less than 6 years of age with normal thyroid ultrasound studies and normal blood calcitonin levels.
Complete resection should result in a drop in serum calcitonin to undetectable levels. Periodic calcitonin level checks can be used effectively to screen for disease recurrence. While C cells can potentially secrete other peptides, such as CEA, chromogranin A, somatostatin, and ACTH, only CEA is of additionally utility and is a useful adjunct to monitor for progressive metastatic disease. If MTC progresses and dedifferentiates, the cancer cells may lose their ability to secrete calcitonin but maintain their secretion of CEA.73
Five-year survival for patients with MTC after attempted curative resection is 95%. In contrast, 5-year survival is only about 40% in patients with advanced disease and those with MEN2B.74 Although advanced MTC with distant metastases is not curable by surgery, resection to achieve local control can decrease the risk of airway and esophageal obstruction and provide long-term palliation.
Surgery for ATC
Surgical intervention for ATC is controversial for both curative and palliative purposes. ATC is a relatively aggressive tumor that often invades locoregional structures, such as the recurrent laryngeal nerve, trachea, esophagus, and internal jugular vein. Locoregional invasion that fails to respect tissue margins will often preclude complete resection. In our experience, aggressive tumor debulking, when possible, followed by adjuvant therapy extends survival in patients with localized disease.75
Outcomes in ATC are uniformly poor and prior reports have focused on prolonging survival on the order of months, rather than years. The median survival of newly diagnosed ATC ranges, in most series, from 4 to 12 months.76 Surgical therapy for ATC can be categorized as either curative or palliative, both of which are controversial. The majority of patients with ATC present with locally invasive and distant disease that is not amenable to complete resection. In patients with ATC that is amenable to complete extirpation, tumor resection with combination chemoradiotherapy may be beneficial. Although no studies have demonstrated reliable curative surgical therapy, several have demonstrated significant improvements in short-term survival.77–80
Thyroid Stimulating Hormone Suppression
After total thyroidectomy with complete extirpation of both normal and neoplastic tissue, patients become hypothyroid. The hypothyroid state negatively impacts patient quality of life, with symptoms including depression, hair and skin changes, fatigue, and weight gain.81 Since the hypothalmo-pituitary axis remains intact, TSH levels increase dramatically if patients are not supplemented with postoperative thyroid hormone. Because TSH stimulates proliferation in residual thyroid cancer cells, thyroid hormone supplementation after total thyroidectomy is a logical adjuvant modality.
However, thyroid cell proliferation is influenced by many growth factors and involves multiple signal transduction pathways. Although thyroid hormone replacement is universally accepted as appropriate after total thyroidectomy, the extent of postoperative thyroid hormone replacement and the possibility that such therapy improves cancer outcomes through a TSH-independent action on tumor cells is controversial.82
Regardless, there is ample evidence to support routine TSH suppression after thyroidectomy and consensus guidelines recommend TSH suppression to <0.1 mIU/L for high-risk patients and 0.1 to 0.5 mIU/L for low-risk WDTC.83 Multiple studies have shown that patients treated with supraphysiologic doses of thyroid hormone after total thyroidectomy have longer disease-free survival.84,85,86,87 Furthermore, high risk patients benefit more from TSH suppression than low-risk patients88 and the degree of TSH suppression is independently predictive of longer disease-free survival.89
Although postoperative thyroxine supplementation is routine at most centers, such therapy should be monitored closely. Symptoms of iatrogenic hyperthyroidism include heat intolerance, anxiety, and palpitations, some of which may require beta blocker therapy to control symptoms.90 Complications such as bone demineralization and cardiac morbidity can occur, particularly in elderly patients.91
Radioactive Iodine Therapy
RAI can be used as an adjuvant modality to thyroidectomy and a primary therapy to treat distant WDTC metastases. Adjuvant RAI is usually performed 4 to 12 weeks after thyroidectomy to destroy remnant normal thyroid tissue, in addition to occult or known metastatic disease. Prior to RAI ablation, iodine restriction should be performed for at least 14 days and TSH levels should exceed 30 uIU/mL.92 The later can be achieved either by either thyroid hormone withdrawal or synthetic TSH injections. Consensus guidelines recommend adjuvant RAI ablation for all patients with PTC, except for those with stage I disease. It is also reasonable to omit adjuvant ablative RAI in patients with WDTC when they have negative diagnostic RAI scans and undetectable thyroglobulin levels (<1 ng/mL) under TSH stimulation.93 Postoperative RAI ablation reduces the recurrence rate after thyroidectomy60,94 and facilitates the use of thyroglobulin as a screening marker for postoperative recurrence.95 Although disease-free survival and cancer-specific survival is longer after complete ablation of remnant thyroid tissue, the dose of adjuvant RAI is controversial.96 A recent meta-analysis suggested that a higher dose of adjuvant RAI, (approximately 100 mCi), is superior to a lower dose, especially after near-total thyroidectomy.97
About 75% of WDTC maintain the ability to trap iodine. RAI offers a rational treatment strategy for patients with disseminated disease, such as lung and bone metastases, that maintains RAI avidity. Tumor susceptibility to RAI is dependent upon tumor size and location. Patients with microscopic lung metastases have better outcomes than those with macroscopic disease or bone metastases (Fig. 34-6).98 Furthermore, patients with isolated lung metastases have better outcomes than patients with either bone or multiple metastatic sites; in a recent study, the 5-year disease-specific survival for patients with only lung metastases was 50% versus 32% for those with bone metastases.99
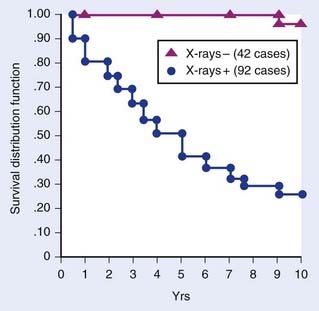
FIGURE 34-6 • Survival of patients with microscopic versus macroscopic well-differentiated thyroid cancer lung metastases.
(From Casara D, et al: Different features of pulmonary metastases in differentiated thyroid cancer: natural history and multivariate statistical analysis of prognostic variables, J Nucl Med 34:1626–1631, p 1629.)
Side effects from RAI are common, such as nausea, emesis, and parotitis, but are usually transient and well-tolerated. Sialadenitis rates can be decreased by proper hydration and low-dose steroids, as well as novel agents such as serotonin receptor antagonists and pilocarpine.100 Nausea can be treated medically with antiemetics. Oncologic complications such as an increased risk of leukemia101 and solid tumors102 have been reported after higher doses of RAI. Other reported side effects include pulmonary fibrosis in patients treated with extensive RAI for diffuse lung metastases.103
External Beam Radiation Therapy
External beam radiation therapy (EBRT) is an effective palliative treatment for some patients with unresectable disease and can be used as an adjuvant therapy for patients with residual tumor after inadequate or incomplete resection.104 The efficacy of adjuvant EBRT is controversial, as there are currently no prospective studies that have investigated this issue.105
Several studies have shown that EBRT is effective in patients with WDTC when residual disease is evident after surgical resection. In one retrospective review of 201 patients after incomplete resection of PTC, adjuvant EBRT decreased local recurrence.106 In a different retrospective series of 97 patients with residual WDTC, EBRT lowered the rate of local recurrence.107 One retrospective study of locally invasive WDTC showed that adjuvant EBRT provided additional benefit among patients who also received adjuvant RAI and TSH suppression after surgery.108 Furthermore, some studies showed that EBRT can benefit patients with microscopic residual disease.109
In contrast, several studies have shown no survival benefit to EBRT after resection of WDTC.90,110,111 Selection bias and inconsistent dosage and fields of treatment may have impacted these results.
EBRT is also a reasonable adjuvant treatment strategy for HCCs and other differentiated thyroid cancers that do not trap iodide and are therefore not amenable to radioactive iodine ablation.112 Additionally, some retrospective studies in patients with MTC showed that adjuvant EBRT resulted in improved local control,113–118 while others found that it was associated with worse outcomes.119 Although EBRT may not improve survival in all MTC subgroups, the association of EBRT with higher recurrence and death in some studies is likely secondary to selection bias. In our opinion, EBRT is an effective modality for patients with residual MTC after total thyroidectomy for local tumor control, with or without evidence of distal metastases.
Because no individual treatment modality is effective for ATC, aggressive combination therapy protocols that include EBRT are often advocated. Although some reports of multimodality regimens have demonstrated some success, no combination therapy has demonstrated consistent long-term survivorship. The two largest studies of ATC outcomes utilized the Surveillance, Epidemiology and End Results Program database to generate large study samples.55,120 In both studies, combined surgery and EBRT decreased cause-specific mortality in patients with regional and distant disease. Because of these reports and the lack of effective treatment alternatives, we advocate the use of EBRT as an adjuvant therapy for all patients with resected ATC.
EBRT also plays an important palliative role for the local control of ATC. The most common cause of death in ATC is asphyxiation secondary to local cervical disease. Since most ATCs are not amenable to curative resection and do not trap RAI, treatment options are generally limited to surgical debulking, systemic chemotherapy and EBRT. EBRT can reduce tumor bulk and relieve airway compression in patients with advanced ATC.121
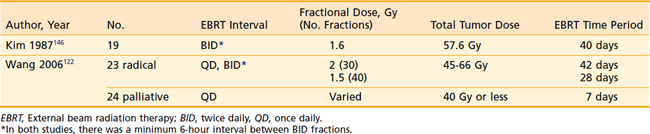
Despite the important role of EBRT in both curative and palliative therapy for ATC, there are few studies that detail the timing, mode of delivery, and dose of EBRT. Examples of EBRT protocols for ATC are shown in Table 34-3. Because of the rapid growth and proliferation of ATC, some support the use of hyperfractionated radiation therapy. One such group compared their EBRT results in patients with once-daily versus twice-daily hyperfractionated regimens.122 They found that EBRT was well-tolerated and that the patients receiving the twice-daily regimen trended towards longer survival. Among the patients treated with palliative intent, 65% had no local progression of disease, underlying the effectiveness of EBRT in preventing asphyxiation in the palliative setting.
Chemotherapy
Systemic chemotherapy treatments for thyroid cancer have been disappointing for all histologic variants. Generally speaking, chemotherapy is reserved for patients with thyroid cancer that is not amenable to curative intervention. In WDTC, chemotherapy is reserved for patients with progressive disease after they have failed surgical extirpation, RAI, and EBRT. In such patients, systemic chemotherapy has not improved outcomes in multiple studies; a recent report of doxorubicin showed a 5% partial response rate in patients with WDTC after 6 months.123
Similar to WDTC, several studies have had disappointing results using a variety of systemic chemotherapy agents for MTC.124,125 Alternative treatment strategies, such as radiolabeled antibodies specific for CEA, have been similarly disappointing.126 Because the well-characterized RET defect in MTC, several current efforts are directed toward utilizing receptor tyrosine kinase inhibitors.127 Clinical prospective trials are ongoing investigating a variety of tyrosine kinase inhibitors in the treatment of otherwise refractory, progressive MTC and WDTC, and the preliminary results are promising.128
Because results are generally poor after resection for patients with ATC, multimodality therapy including systemic chemotherapy is commonly employed. While chemotherapy may add little to long-term survival in this patient population, the grave nature of the disease warrants aggressive adjuvant therapy. Results from conventional chemotherapy treatment for ATC have been generally poor. Early studies of doxorubicin monotherapy demonstrated response rates approximating 22%, which was almost half that of the 42% response rate in MTC.129 Combination chemotherapy regimens that include paclitaxel,130 cisplatin,131 vincristine,132 bleomycin,133 and gemcitabine134 have demonstrated similarly disappointing results. Targeted therapeutics have demonstrated some efficacy in in-vitro and in-vivo studies.135–137 Other novel therapies that have demonstrated some promise in ATC either as single agents or in combination therapy include oncolytic virus therapy,138 histone deacetylase inhibitors,139 and combrestatins.140
1 American Cancer Society, Cancer Facts and Figures. www.cancer.org, 2008.
2 Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295:2164-2167.
3 Mortensen JD, Woolner LB, Bennett WA. Gross and microscopic findings in clinically normal thyroid glands. J Clin Endocrinol Metab. 1955;15:1270-1280.
4 Ries LAG, Melbert D, Krapcho M, et al, editors: SEER Cancer Statistics Review, 1975–2005, National Cancer Institute. Bethesda, MD http://seer.cancer.gov/csr/1975_2005/ based on November 2007 SEER data submission, posted to the SEER website, 2008.
5 Haselkorn T, Stewart SL, Horn-Ross PL. Why are thyroid cancer rates so high in Southeast Asian women living in the United States? The Bay Area thyroid cancer study. Cancer Epidem Biomark Prevent. 2003;12:144-150.
6 Iribarren C, Haselkorn T, Tekawa IS, Friedman GD. Cohort study of thyroid cancer in a San Francisco Bay area population. Int J Cancer. 2001;93:745-750.
7 De Groot LJ, Reilly M, Pinnameneni K, Refetoff S. Retrospective and prospective study of radiation-induced thyroid disease. Am J Med. 1983;74:852-862.
8 Paynter OE, Burin GJ, Jaeger RB, Gregorio CA. Goitrogens and thyroid follicular cell neoplasia: evidence for a threshold process. Regul Toxicol Pharmacol. 1988;8:102-119.
9 Sturgeon C, Clark OH. Familial nonmedullary thyroid cancer. Thyroid. 2005;15:588-593.
10 Sampson RJ, Key CR, Buncher CR, Iijima S. Thyroid carcinoma in Hiroshima and Nagasaki. I. prevalence of thyroid carcinoma at autopsy. JAMA. 1969;209:65-70.
11 Thomas GA, Bunnell H, Cook HA, et al. High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobyl thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J Clin Endocrinol Metab. 1999;84:4232-4238.
12 Kingston J. Thyroid cancer after neck irradiation during childhood. Lancet. 2005;365:1986-1987.
13 Franklyn JA, Maisonneuve P, Sheppard MC, Betteridge J, Boyle P. Mortality after the treatment of hyperthyroidism with radioactive iodine. N Engl J Med. 1998;338:712-718.
14 Williams ED, Doniach I, Bjarnason O, Michie W. Thyroid cancer in an iodine rich area, a histopathlogical study. Cancer. 1977;39:215-222.
15 Sippel RS, Caron NR, Clark OH. An evidence-based approach to familial nonmedullary thyroid cancer: screening, clinical management, and follow-up. World J Surg. 2007;31:924-933.
16 Triponez F, Wong M, Sturgeon C, et al. World J Surg. 2006;30:787-793.
17 Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A national cancer data base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985–1995. Cancer. 1998;83:2638-2648.
18 Bakiri F, Djemli FK, Mokrane LA, Djidel FK. The relative roles of endemic goiter and socioeconomic development status in the prognosis of thyroid carcinoma. Cancer. 1998;82:1146-1153.
19 DeMeter JG, De Jong SA, Lwrence AM, Paloyan E. Anaplastic thyroid carcinoma: risk factors and outcome. Surgery. 1991;110:956-963.
20 Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nature Rev. 2006;6:292-306.
21 Xing M. BRAF mutation in thyroid cancer. Endocrine Rel Cancer. 2005;12:245-262.
22 Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25-40.
23 Kebebew E, Weng J, Bauer J, et al. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann Surg. 2007;246:466-471.
24 Bongarzone I, Butt MG, Coronelli S, et al. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res. 1994;54:2979-2985.
25 Collins BJ, Chiappetta G, Schneider AB, et al. RET expression in papillary thyroid cancer from patients irradiated in childhood for benign conditions. J Clin Endocrinol Metab. 2002;87:3941-3946.
26 Nikiforov YE, Rowland JM, Bove K, et al. Distinct patterns of ret oncogene rearrangements in morphologic variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57:1690-1694.
27 Hoff AO, Hoff PM. Medullary thyroid carcinoma. Hematol Oncol Clin N Am. 2007;21:475-488.
28 Moley JF, Lairmore TC, Phay JE. Hereditary endocrinopathies. Curr Probl Surg. 1999;36:653-762.
29 Mathew CG, Chin KS, Easton DF, et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature. 1987;328:527-528.
30 Uchino S, Noguchi S, Yamashita Y, et al. Somatic mutations in RET exons 12 and 15 in sporadic medullary thyroid carcinomas: different spectrum of mutations in sporadic type from hereditary type. Jpn J Cancer Res. 1999;90:1231-1237.
31 Skinner MA, Moley JA, Dilley WG, Owzar K, Debenedetti MK, Wells SAJr. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med. 2005;353:1105-1113.
32 Machens A, Niccoli-Sire P, Hoegel J, et al. European Multiple Endocrine Neoplasia (EUROMEN) Study Group. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med. 2003;349:1517-1525.
33 Fagin JA, Matsuo K, Karmakar A, et al. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993;91:179-184.
34 Jossart GH, Epstein HD, Shaver JK, et al. J Clin Endocrinol Metab. 1996;81:3498-3504.
35 Nikiforova MN, Kimura ET, Gandhi M, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. 2003;88:5399-5404.
36 Miura D, Wada N, Chin K, et al. Anaplastic thyroid cancer: cytogenetic patterns by comparative genomic hybridization. Thyroid. 2003;13:283-290.
37 Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/Akt and Ras/Raf-Mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278-284.
38 Quiros RM, Ding HG, Gattuso P, Prinz RA, Xu X. Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations. Cancer. 2005;103:2261-2268.
39 Robbins KT, Clayman G, Levine P. Neck dissection classification update. Arch Otolaryngol Head Neck Surg. 2002;128:751.
40 Noguchi S, Noguchi A, Murakami N. Papillary carcinoma of the thyroid. I: Developing pattern of metastasis. Cancer. 1970;26:1053-1060.
41 Isaacs JD, Lundgren CI, Sidhu SB, Sywak MS, Edhouse PJ, Delbridge LW. The Delphian lymph node in thyroid cancer. Ann Surg. 2008;247:477-482.
42 Shoup M, Stojadinovic A, Nissan A, et al. Prognostic indicators of outcomes in patients with distant metastases from differentiated thyroid carcinoma. J Am Coll Surg. 2003;197:191-197.
43 D’Avanzo A, Treseler P, Ituarte PH, et al. Follicular thyroid carcinoma: histology and prognosis. Cancer. 2004;100:1123-1129.
44 Parma J, Duprez L, Van Sande J, et al. Somatic mutations in the thyrotropin receptor gene causing hyperfunctioning thyroid adenomas. Nature. 1993;365:649.
45 Duh QY, Siperstein AE, Miller RA, Smeds S, Clark OH. TSH binding correlates with TSH-stimulated thyroid adenylate cyclase activity in human thyroid tissues. Surgery. 1989;106:967-973.
46 Gharib H. Changing concepts in the diagnosis and management of thyroid nodules. Endocrinol Metab North Am. 1997;26:777-800.
47 Barden CB, Shister KW, Zhu B, et al. Classification of follicular thyroid tumors by molecular signature: results of gene profiling. Clin Cancer Res. 2003;9:1792-1800.
48 Kebebew E, Peng M, Reiff E, McMillan A. Diagnostic and extent of disease multigene assay for malignant thyroid neoplasms. Cancer. 2006;106:2592-2597.
49 Cerutti JM, Latini FRM, Nakabashi C, et al. Diagnosis of suspicious thyroid nodules using four protein biomarkers. Clin Cancer Res. 2006;12:3311-3317.
50 Kebebew E, Ituarte PH, Siperstein AE, Duh QY, Clark OH. Medullary thyroid carcinoma: clinical characteristics, treatment, prognostic factors, and a comparison of staging systems. Cancer. 2000;88:1139-1148.
51 Bugalho MJ, Santos JR, Sobrinho L. Preoperative diagnosis of medullary thyroid carcinoma: fine needle aspiration cytology as compared with serum calcitonin measurement. J Surg Oncol. 2005;91:56-60.
52 Sotos JG. The physical Lincoln: finding the genetic cause of Abraham Lincoln’s height, homeliness, pseudo-depression, and imminent cancer death. Mt Vernon, VA: Mt Vernon Book Systems; 2008.
53 Quayle FJ, Moley JF. Medullary thyroid carcinoma: including MEN2A and MEN2B syndromes. J Surg Oncol. 2005;89:122-129.
54 Us-Krasovec M, Golouh R, Auersperg M. Anaplastic carcinoma in fine needle aspirates. Acta Cytol. 1996;40:953-958.
55 Kebebew E, Greenspan FS, Clark OH, Woeber KA, McMillan A. Anaplastic thyroid carcinoma, treatment outcome and prognostic factors. Cancer. 2005;103:1330-1335.
56 Kebebew E, Clark OH. Differentiated thyroid cancer: “complete” rational approach. World J Surg. 2000;24:942-951.
57 Ruan DT, Clark OH. Is total thyroidectomy the procedure of choice for low-risk papillary thyroid cancer? Nat Clin Pract Endocrinol Metab. 2008;4:128-129.
58 Shah JP, Loree TR, Dharker D, Strong EW. Lobectomy versus total thyroidectomy for differentiated carcinoma of the thyroid: a matched-pair analysis. Am J Surg. 1993;166:331-335.
59 Mazzaferri EL, Jhiang SM. Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med. 1994;97:418-428.
60 Bilimoria KY, et al. Extent of surgery affects survival for papillary thyroid cancer. Ann Surg. 2007;246:375-384.
61 Hay ID, Grant CS, Bergstralh EJ, et al. Unilateral total lobectomy: is it sufficient surgical treatment for patients with AMES low-risk papillary thyroid carcinoma? Surgery. 1998;124:958-966.
62 Snyder SK, Lairmore TC, Hendricks JC, Roberts JW. Elucidating mechanisms of recurrent laryngeal nerve injury during thyroidectomy and parathyroidectomy. J Am Coll Surg. 2008;206:123-130.
63 Chiang FY, Wang LF, Huang YF, Lee KW, Kuo WR. Recurrent laryngeal nerve palsy after thyroidectomy with routine identification of the recurrent laryngeal nerve. Surgery. 2005;137:342-347.
64 Hartl DM, Travagli JP, Leboulleux S, Baudin E, Brasnu DF, Schlumberger M. Clinical review: Current concepts in the management of unilateral recurrent laryngeal nerve paralysis after thyroid surgery. J Clin Endocrinol Metab. 2005;90:3084-3088.
65 Asari R, Passler C, Kaczirek K, Scheuba C, Niederle B. Hypoparathyroidism after total thyroidectomy: a prospective study. Arch Surg. 2008;143:132-137.
66 Moley JF, DeBenedetti MK. Patterns of nodal metastases in palpable medullary thyroid carcinoma: recommendations for extent of node dissection. Ann Surg. 1999;229:880-887.
67 National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, Thyroid carcinoma V.2.2007. www.nccn.org.
68 Bergholm U, Bergstrom R, Ekbom A. Long-term follow-up of patients with medullary carcinoma of the thyroid. Cancer. 1997;79:132-138.
69 Pelizzo MR, Boschin IM, Bernante P, et al. Natural history, diagnosis, treatment and outcome of medullary thyroid cancer: 37 years experience on 157 patients. Eur J Surg Oncol. 2007;33:493-497.
70 Al-Rawi M, Wheeler MH. Medullary thyroid carcinoma: update and present management controversies. Ann R Coll Surg Engl. 2006;88:433-438.
71 Machens A, Gimm O, Hinze R, Hoppner W, Boehm BO, Dralle H. Genotype-phenotype correlations in hereditary medullary thyroid carcinoma: oncological features and biochemical properties. J Clin Endocrinol Metab. 2001;86:1104-1109.
72 Telander RL, Zimmerman D, van Heerden JA, et al. Results of early thyroidectomy for medullary thyroid carcinoma in children with multiple endocrine neoplasia type 2. J Pediatr Surg. 1986;21:1190-1194.
73 Saad MF, Fritsche HAJ, Samaan NA. Diagnostic and prognostic values of carcinoembryonic antigen in medullary carcinoma of the thyroid. J Clin Endocrinol Metab. 1984;58:889-894.
74 Ellenhorn JD, Shah JP, Brennan MF. Impact of therapeutic lymph node dissection for medullary thyroid carcinoma of the thyroid gland. Surgery. 1993;114:1078-1081.
75 Haigh PI, Ituarte PHG, Wu HS, et al. Completely resected anaplastic thyroid carcinoma combined with adjuvant chemotherapy and irradiation is associated with prolonged survival. Cancer. 2001;91:2335-2342.
76 Are C, Shaha AR. Anaplastic thyroid carcinoma: biology, pathogenesis, prognostic factors, and treatment approaches. Annals of Surgical Oncology. 2006;13:453-464.
78 Junor EJ, Paul J, Reed NS. Anaplastic thyroid carcinoma: 91 patients treated by surgery and radiotherapy. Eur J Surg Oncol. 1992;18:83-88.
79 Venkatesh YS, Ordonez NG, Schultz PN, Hickey RC, Goepfert H, Samaan NA. Anaplastic carcinoma of the thyroid: a clinicopathologic study of 121 cases. Cancer. 1990;66:321-330.
80 Kobayashi T, Asakawa H, Umeshita K, et al. Treatment of 37 patients with anaplastic carcinoma of the thyroid. Head Neck. 1996;18:36-41.
81 McMillan C, Bradley C, Razvi S, Weaver J. Evaluation of new measures of the impact of hypothyroidism on quality of life and symptoms: the ThyDQoL and ThySRQ. Value Health. 2008;11:285-294.
82 Brabant G. Thyrotropin suppressive therapy in thyroid carcinoma: what are the targets? J Clin Endocrinol Metab. 2008;93:1167-1169.
83 Cooper DS, Doherty GM, Haugen BR, et al. Management guidelines for patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association guidelines taskforce. Thyroid. 2006;16:109-142.
84 Landau D, Vini L, A’Hern R, Harmer C. Thyroid cancer in children: the Royal Marsden Hospital experience. Eur J Cancer. 2000;36:214-220.
85 Staunton MD, Greening WP. Treatment of thyroid cancer in 293 patients. Br J Surg. 1976;63:253-258.
86 Mazzaferri EL, Young RJ. Papillary thyroid carcinoma: a 10 year follow-up report of the impact of therapy in 576 patients. Am J Med. 1981;70:511-518.
87 Simpson WJ, Panzarella T, Carruthers JS, Gospodarowicz MK, Sutcliffe SB. Papillary and follicular thyroid cancer: impact of treatment in 1578 patients. Int J Radiat Oncol Biol Physiol. 1988;14:1063-1075.
88 Jonklaas J, Sarlis NJ, Litofsky D, et al. Outcomes of patients with differentiated thyroid carcinoma following initial therapy. Thyroid. 2006;16:1229-1242.
89 Pujol P, Daures JP, Nsakala N, Baldet L, Bringer J, Jaffiol C. Degree of thyrotropin suppression as a prognostic determinant in differentiated thyroid cancer. J Clin Endocrinol Metab. 1996;81:4318-4323.
90 Biondi B, Filetti S, Schlumberger M. Thyroid-hormone therapy and thyroid cancer: a reassessment. Nat Clin Pract Endocrinol Metab. 2005;1:32-40.
91 Toft AD. Thyroxine therapy. N Engl J Med. 1994;331:174.
92 Meier DA, Brill DR, Becker DV, et al. Procedure guideline for therapy of thyroid disease with 131Iodine. J Nucl Med. 2002;43:856-861.
93 Elaraj DM, Clark OH. Changing management in patients with papillary thyroid cancer. Cur Treat Options Oncol. 2007;8:305-313.
94 Wong JB, Kaplan MM, Meyer KB, Pauker SG. Ablative radioactive iodine therapy for apparently localized thyroid carcinoma. A decision analytic perspective. Endocrinol Metab Clin North Am. 1990;19:741-760.
95 Pagano L, Klain M, Pulcrano M, et al. Follow-up of differentiated thyroid carcinoma. Minerva Endocrinol. 2004;29:161-174.
96 Verberg FA, de Keizer B, Lips CJ, Zelissen PM, de Klerk JM. Prognostic significance of successful ablation with radioiodine of differentiated thyroid cancer patients. Eur J Endocrin. 2005;152:33-37.
97 Doi SAR, Woodhouse NJ, Thalib L, Onitilo A. Ablation of the thyroid remnant and I-131 dose in differentiated thyroid cancer: a meta-analysis revisited. Clin Med Res. 2007;5:87-90.
98 Casara D, Rubello D, Saladini G, et al. Different features of pulmonary metastases in differentiated thyroid cancer: natural history and multivariate statistical analysis of prognostic variables. J Nucl Med. 1993;34:1626-1631.
99 Haq M, Harmer C. Differentiated thyroid carcinoma with distant metastases at presentation: prognostic factors and outcome. Clin Endocrinol. 2005;63:87-93.
100 Silberstein EB. Reducing the incidence of 131I-induced sialadenitis: the role of pilocarpine. J Nucl Med. 2008;49:546-549.
101 Maxon HR, Smith HS. Radioiodine-131 in the diagnosis and treatment of metastatic well-differentiated thyroid cancer. Endocrinol Metab Clin North Am. 1990;19:685.
102 Edmonds CJ, Smith T. The long-term hazard of the treatment of thyroid cancer with radioiodine. Br J Radiol. 1986;59:45.
103 Lee J, Sogutlu G, Leard L, et al. Lung transplantation for pulmonary metastases and radiation-induced pulmonary fibrosis after radioactive iodine ablation of extensive lung metastases from papillary thyroid carcinoma. Thyroid. 2007;17:367-369.
104 Hyer SL, Dandekar P, Newbold K, Haq M, Wechalakar K, Hammer C. Thyroid cancer causing obstruction of the great veins in the neck. World J Surg Oncol. 2008;6:36.
105 Strasser JF, Raben A, Koprowski C. The role of radiation therapy in the management of thyroid cancer. Surg Oncol Clin N Am. 2008;17:219-232.
106 Simpson WJ, Panzarella T, Carruthers JS, et al. Papillary and follicular thyroid cancer: impact of treatment in 1578 patients. Int J Radiat Oncol Biol Phys. 1998;14:1063-1075.
107 Tubiana M, Haddad E, Schlumberger M, et al. External radiotherapy in thyroid cancers. Cancer. 1985;55:2062-2071.
108 Farahati J, Reinders C, Stuschke M, et al. Differentiated thyroid cancer: impact of adjuvant external radiotherapy with perithyroidal tumor infiltration (stage pT4). Cancer. 1996;77:172-180.
109 Tsang RW, Brierley JD, Simpson WJ, Panzarella T, Gospodarowicz MK, Sutcliffe SB. The effects of surgery, radioiodine, and external radiation therapy on the clinical outcome of patients with differentiated thyroid carcinoma. Cancer. 1998;82:375-388.
110 Benker G, Olbricht T, Reinwein D, et al. Survival rates in patients with differentiated thyroid carcinoma: influence of postoperative external radiotherapy. Cancer. 1990;65:1517-1520.
111 Chow SM, Law SC, Mendenhall WM, et al. Papillary thyroid carcinoma: prognostic factors and the role of radioiodine and external radiotherapy. Int J Radiat Oncol Biol Phys. 2002;52:784-795.
112 Vini L, Fisher C, A’Hern R, Harmer C. Hurthle cell cancer of the thyroid: the Royal Marsden experience. Thyroid. 1998;8:1228-1229.
113 Fife KM, Bower M, Harmer CL. Medullary thyroid cancer: the role of radiotherapy in local control. Eur J Surg Oncol. 1996;22:588-591.
114 Fersht N, Vini L, A’Hern R, et al. The role of radiotherapy in the management of elevated calcitonin after surgery for medullary thyroid cancer. Thyroid. 2001;11:1161-1168.
115 Steinfeld AD. The role of radiation therapy in medullary carcinoma of the thyroid. Radiology. 1977;123:745-746.
116 Rougier P, Parmentier C, Laplanche A, et al. Medullary thyroid carcinoma: prognostic factors and treatment. Int J Radiat Oncol Biol Phys. 1983;9:161-169.
117 Nguyen TD, Chassard JL, Lagarde P, et al. Results of postoperative radiation therapy in medullary carcinoma of the thyroid: a retrospective study by the French Federation of Cancer Institutes- the Radiotherapy Cooperative Group. Radiother Oncol. 1992;23:1-5.
118 Brierly J, Tsang R, Simpson WJ, et al. Medullary thyroid cancer: analyses of survival and prognostic factors and the role of radiation therapy in local control. Thyroid. 1996;6:305-310.
119 Samaan NA, Schultz PN, Hickey RC. Medullary thyroid carcinoma: prognosis of familial versus sporadic disease and the role of radiotherapy. J Clin Endocrinol Metab. 1988;67:801-805.
120 Chen J, Tward JD, Shrieve DC, et al. Surgery and radiotherapy improves survival in patients with anaplastic thyroid carcinoma. Am J Clin Oncol. 2008;31:460-464.
121 Nilsson O, Lindberg J, Zedenius J, et al. Anaplastic giant cell carcinoma of the thyroid gland: treatment and survival over a 25-year period. World J Surg. 1998;22:725-730.
122 Wang Y, Tsang R, Asa S, et al. Clinical outcome of anaplastic thyroid carcinoma treated with radiotherapy of once- and twice-daily fractionation regimens. Cancer. 2006;107:1786-1792.
123 Matuszczyk A, Petersenn S, Bockisch A, et al. Chemotherapy with doxorubicin in progressive medullary and thyroid carcinoma of the follicular epithelium. Horm Metab Res. 2008;40:210-213.
124 Wu LT, Averbuch SD, Ball DW, et al. Treatment of advanced thyroid carcinoma with a combination of cyclophosphamide, vincristine, and dacarbazine. Cancer. 1994;73:432-436.
125 Orlandi F, Caraci P, Berruti A, et al. Chemotherapy with dacarbazine and 5-flurorouracil in advanced medullary thyroid cancer. Ann Oncol. 1994;5:763-765.
126 Juweid ME, Hajjar G, Swayne LC, et al. Phase I/II trial of (131)I-MN014F(ab) 2 anti-carcinoembryonic antigen monoclonal antibody in the treatment of patients with metastatic medullary thyroid carcinoma. Cancer. 1999;85:1828-1842.
127 Frank-Raue K, Fabel M, Delorme S, Haberkorn U, Raue F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur J Endocrinol. 2007;157:215-220.
128 Sherman SI, Wirth LJ, Droz JP, et al. Motesanib Thyroid Cancer Study Group. N Engl J Med. 2008;359:31-42.
129 Ahuja S, Ernst H. Chemotherapy of thyroid carcinoma. J Endocrinol Invest. 1987;10:303-310.
130 Ain KB, Egorin MJ, DeSimone PA. Treatment of anaplastic thyroid carcinoma with paclitaxel: phase 2 trial using ninety-six hour transfusion. Collaborative Anaplastic Thryoid Cancer Health Intervention Trials (CATCHIT) Group. Thyroid. 2000;10:587-594.
131 Shimaoka K, Schoenffeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in advanced thyroid cancer. Cancer. 1985;5:2155-2160.
132 Sokal M, Harmer CL. Chemotherapy for anaplastic carcinoma of the thyroid. Clin Oncol. 1978;4:3-10.
133 De Besi P, Busnardo B, Toso S, et al. Combined chemotherapy with bleomycin, adriamycin, and platinum in advanced thyroid carcinoma. J Endocrinol Invest. 1991;14:475-480.
134 Voight W, Kegel T, Weiss M, Mueller T, Simon H, Schmoll HJ. Potential activity of paclitaxel, vinorelbine, and gemcitabine in anaplastic thyroid carcinoma. J Cancer Res Clin Oncol. 2005;131:585-590.
135 Schiff BA, McMurphy AB, Jasser SA, et al. Epidermal growth factor receptor (EGFR) is overexpressed in anaplastic thyroid cancer, and the EGFR inhibitor gefitinib inhibits the growth of anplastic thyroid cancer. Clin Cancer Res. 2004;10:8594-8602.
136 Mitsiades CS, McMillin D, Kotoula V, et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab. 2006;91:4013-4021.
137 Copland JA, Marlow LA, Kurakata S, et al. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits hman anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene. 2006;25:2304-2317.
138 Lin S-F, Gao SP, Price DL, et al. Synergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancer. Clin Cancer Res. 2008;14:1519-1528.
139 Catalano MG, Poli R, Pugliese M, Fortunati N, Boccuzzi G. Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocrin Rel Cancer. 2007;14:839-845.
140 Yeung S-C J, She M, Yang H, Pan J, Sun L, Chaplin D. Combination chemotherapy including combretastatin A4 phosphate and paclitaxel is effective against anaplastic thyroid cancer in a nude mouse xenograft model. J Clin Endocrinol Metab. 2007;92:2902-2909.
141 Hay ID, Grant CS, Taylor WF, et al. Ipsilateral lobectomy versus bilateral lobar resection in papillary thyroid carcinoma: A retrospective analysis of surgical outcomes using a novel prognostic scoring system. Surgery. 1987;102:1088-1095.
142 Cady B, Rossi R. An expanded view of risk-group definition in differentiated thyroid carcinoma. Surgery. 1988;104(6):947-953.
143 Hay ID, Bergstralh EJ, Goellner JR, et al. Predicting outcome in papillary thyroid carcinoma: development of a reliable prognostic scoring system in a cohort of 1779 patients surgically treated at one institution during 1940 through 1989. Surgery. 1993;114(6):1050-1057.
144 Eichhorn W, Tabler H, Lippold R, et al. Prognostic factors determining long-term survival in well-differentiated thyroid cancer: an analysis of four hundred eighty-four patients undergoing therapy and aftercare at the same institution. Thyroid. 2003;13(10):949-958.
145 Greene FL, Page DL, Fleming ID, et al, editors. AJCC Cancer Staging Manual, 6th ed, New York, NY: Springer, 2002.
146 Kim JH, Leeper RD. Treatment of locally advanced thyroid carcinoma with combination doxorubicin and radiation therapy. Cancer. 1987;60:2372-2375.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
34 Cancer of the Thyroid
Epidemiology
Primary thyroid cancer is the most common endocrine malignancy, with an estimated incidence of 37,340 cases in the United States per year.1 This incidence is increasing, and thyroid cancer is now the eighth most common malignancy in women and the second most common malignancy in women under 40 years of age.2 Most, but not all, of this increase is a consequence of the increased detection of smaller WDTC lesions.2 Autopsy studies suggest that there is a large number of people in the general population with small, subclinical WDTC lesions.3
With regards to gender, women represent 76% of newly diagnosed cases.1 With regards to race, the incidence per 100,000 Caucasian, Asian, Hispanic, and Black women, is 4.9, 4.1, 3.5, and 2.7, respectively.4 In the San Francisco Bay area, well-controlled epidemiological studies indicate that the rate of thyroid cancer is higher in women of Southeast Asian descent.5,6
Environmental and dietary factors may also impact the frequency and distribution of thyroid cancer subtypes in a population. Ionizing radiation,7 goitrogens,8 and familial syndromes,9 have all been associated with PTC. People exposed to ionizing radiation during the atomic exposures at Hiroshima and Nagasaki in 194510 and children living near the Chernobyl nuclear accident in 198611 have a higher incidence of PTC. Furthermore, external beam radiation treatments in children for conditions such as acne, ringworm, birthmarks, enlarged thymus glands, or tonsillitis are associated with a higher risk of PTC later in life.12 Iatrogenic radioiodine exposure also slightly increases the risk of developing thyroid cancer and benign thyroid disorders.13 The incidence of FTC is higher in iodine-deficient regions, while PTC rates are higher in iodine-replete regions.14 Familial PTC has been described in numerous families, although the gene or genes responsible for this syndrome has not been elucidated.15 With regards to prognosis, patients with familial PTC with more than two members with PTC have a worse prognosis than those with sporadic disease.16
ATC accounts for more than 50% of the thyroid cancer deaths in the United States, despite the fact that it represents less than 2% of all thyroid cancers diagnosed.17 A higher incidence of ATC is seen in areas with low socioeconomic status and a high incidence of endemic goiter.18 There is ample evidence that ATC originates through the dedifferentiation of WDTC cells; thus, incomplete or delayed treatment of WDTC increases the risk of anaplastic transformation.19
Molecular Basis of Thyroid Cancer
Although thyroid oncogenesis is not completely understood, some of the genetic changes that drive malignant transformation have been described for each histological subtype of thyroid cancer (Fig. 34-1). Thyroid oncogenesis entails the stepwise progression of molecular alterations that dysregulate cellular proliferation, apoptosis, and invasion.
Most sporadic PTCs harbor an activating mutation of receptor tyrosine kinases or the RAS-RAF-MEK-ERK pathway.20 These mutations are generally nonoverlapping, suggesting that such activation plays a central role in thyroid oncogenesis.20 The B type RAF kinase (BRAF) is the most prominent RAF kinase in thyrocytes, and point mutations in BRAF are found in approximately 45% of sporadic PTC.21 The presence of BRAF mutations in PTC microcarcinomas suggests that it plays a role in the initiation of thyroid oncogenesis.22 The BRAF V600E point mutation is reportedly associated with aggressive tumor behavior. PTCs with this mutation have higher rates of lymph node metastases and local invasion, resulting in higher rates of recurrent and persistent disease after thyroidectomy.23 Lastly, intra- and interchromosomal inversions of the RET proto-oncogene (RET/PTC) can also be found in PTC, which results in constitutive activation.24 These RET/PTC rearrangements are seen in approximately 75% of PTCs associated with childhood exposure to ionizing radiation.25,26
Approximately 25% of all MTC cases are inherited in an autosomal dominant fashion in one of three familial syndromes.27 In MEN2A, MTC is associated with pheochromocytoma and hyperparathyroidism.28 In MEN 2B, MTC is associated with pheochromocytoma but not with hyperparathyroidism, and patients are characterized by a Marfanoid appearance and oral mucosal neuromas.28 Lastly, isolated familial MTC is marked by the autosomal dominant transmittance of MTC with no other associated endocrinopathy.28
The confirmation that MTC could be part of a familial syndrome prompted a considerable effort to uncover its molecular basis, which was later identified as a point mutation in the RET proto-oncogene.29 The RET proto-oncogene is a tyrosine kinase receptor located on chromosome 10. Interestingly, somatic mutations in the RET proto-oncogene also account for approximately 25% to 30% of sporadic cases of MTC.30 MTC tumor behavior and clinical outcomes are heterogeneous, and prior studies have demonstrated that tumor behavior is contingent upon genotype. For example, the somatic 918 RET mutation is associated with aggressive MTC behavior. These important molecular discoveries have shifted the focus of MTC to early identification and prophylactic surgery, which have improved patient outcomes.31,32
Although virtually all ATC have a p53-related defect, the mechanisms of anaplastic transformation remain incompletely understood.33,34 ATC is likely derived from the dedifferentiation of WDTC and studies suggest that activation of both RAS-RAF-MAPK and PI3K pathways may play a role in anaplastic transformation.35,36 One study found an increasing frequency of genetic alterations in RAS-RAF-MAPK and PI3K/Akt in ATC relative to DTC when these histological subtypes coexisted in the same specimen.37 In the same study, they found that the only overlapping mutations in matched WDTC and ATC were in BRAF and PIK3CA.37 Conversely, a different study showed that BRAF and p53 were both altered in ATC, but that they were not overlapping with RAS or RET/PTC mutations.38
Anatomic Considerations
The thyroid gland wraps around the anterolateral trachea with two shield-like lobes connected by an isthmus, giving an appearance that is vaguely similar to a butterfly (Fig. 34-2). The most useful surface anatomy landmark is the cricoid cartilage, which is found just cephalad to the thyroid isthmus. The cricoid cartilage is palpable caudal to the tracheal cartilage, or Adam’s apple. Alternatively, the cricoid cartilage can be found by identifying the sternal notch and palpating the trachea in a cephalad direction; the cricoid cartilage will usually be the first prominent cartilage palpated using this technique. The pyramidal lobe, which is the distal remnant of the thyroglossal duct, is a narrow piece of thyroid tissue that extends cranially from the isthmus. An unresected pyramidal lobe is occasionally the cause of remnant thyroid tissue detected on postoperative RAI scans. Aberrant thyroid tissue can be found in the midline, and very rarely, lateral or inferior to the main body of the thyroid gland within the central neck. More commonly, thyroid cells discovered from FNA samples lateral to the carotid sheath and thyroid gland represent metastatic WDTC.
The lymphatic drainage of the thyroid gland flows in all directions and is inconsistent. The anatomic classification scheme that defines the cervical lymphatic subdivisions was most recently updated by the Committee for Head and Neck Surgery and the American Academy of Otolaryngology-Head and Neck Surgery (Fig. 34-3).39 The central neck compartment, designated level VI, is located between the carotid sheaths. The lateral neck compartments, including the jugular chain (level II to IV) and posterior neck (level V), are generally secondary drainage basins after the central neck.
FIGURE 34-3 • Lymph node anatomic classification: the six levels of cervical lymph nodes in the neck.
(From Robbins KT, Clayman G, Levine PA, et al: Neck dissection classification update; Revisions proposed by the American Head and Neck Society and the American Academy of Otolaryngology—Head and Neck Surgery, Archives of Otolaryngol Head Neck Surg 128:751–758, 2002, p 752.)
The pattern of metastases in PTC typically begins with spread to ipsilateral central and then ipsilateral lateral lymph nodes, and then later to contralateral cervical lymph nodes.40 However, reports of “skip” metastasis to the lateral neck with normal central neck lymph nodes have been reported in about 20% of patients with PTC and MTC and may be more common in patients with primary tumors located in the superior thyroid poles. Perithyroidal, pretracheal lymph nodes, termed “Delphian” lymph nodes, when found to harbor metastatic disease, are associated with higher incidence of central and lateral compartment metastases.41 PTC can also spread to distant sites, including the lungs and bones in approximately 4% of newly diagnosed patients.42
Lymph node metastases are found in less than 6% of FTC in contrast to 6% to 30% of patients with HCC.43 Lung metastases from hematogenous spread are more common in FTC, HCC, and ATC. Liver metastases are relatively common in MTC, but are rare in thyroid cancers of follicular cell origin.
Physiology
The principal endocrine function of the thyroid gland is the production and controlled release of thyroid hormone and calcitonin. Systemic thyroid hormone levels impact development, growth and metabolism. The two forms of thyroid hormone, triiodothyronine (T3) and thyroxine (T4) are synthesized within thyroid follicles. Thyroglobulin is a large glycoprotein synthesized by follicular cells, which is the source of tyrosyl residues needed for thyroid hormone synthesis. Both T3 and T4 are synthesized through the coupling of iodide with tyrosyl residues in a reaction involving the enzyme thyroperoxidase (Fig. 34-4) Newly synthesized thyroid hormone is bound to thyroglobulin protein and stored in colloid, which can later be released into the systemic circulation. Hydrolysis of thyroglobulin protein and release of thyroid hormone is regulated by TSH levels and the availability of iodide.
Thyroid hormone levels are tightly regulated in a negative-feedback manner within the hypothalamic-pituitary-thyroid axis (Fig. 34-5). Thyroid stimulating hormone (TSH) is the central element in this axis, which directly controls the activity and proliferation of follicular cells. TSH is synthesized and secreted by the anterior pituitary, a process that is regulated by thyrotropin-releasing hormone and blood thyroid hormone levels. Thyrotropin-releasing hormone is synthesized in the hypothalamus and is transported to the pituitary via the hypophyseal portal system, a process that is suppressed when systemic T3 levels are high.
The TSH receptor is a G-protein-coupled receptor which upregulates thyroid hormone production and release via cAMP associated signaling. Because TSH can induce the proliferation of both normal thyrocytes and WDTC of follicular origin, suppression of TSH is an important feature of thyroid cancer treatment. Furthermore, TSH receptor mutations44 and a higher concentration of TSH receptors45 have been associated with autonomous follicular thyroid neoplasms.
Clinical Presentation and Diagnosis
In addition to a complete history and physical exam, patients that present with a neck mass suspicious for a thyroid neoplasm should undergo formal neck ultrasound by an experienced ultrasonographer to characterize the lesion, monitor the remainder of the thyroid gland for abnormalities, and to evaluate for abnormal cervical lymph nodes. FNA biopsy is a reliable diagnostic tool that should be performed on thyroid lesions with suspicious sonographic characteristics, such as internal microcalcifications. FNA has a sensitivity of 95% to 98% and specificity of 97% to 99%, when performed by an experienced cytopathologist.46 Unfortunately, FNA is not an effective for discriminating FTC from benign follicular adenomas, or HCC from Hürthle cell adenomas. Histologic architecture is required to determine if a follicular or Hürthle cell neoplasms are malignant; capsular or vascular invasion must be seen to diagnose carcinoma in these subtypes. The current standard of care in the evaluation of a follicular neoplasm requires diagnostic hemithyroidectomy with formal histological review to determine if the lesion has invasive features consistent with carcinoma. Molecular-based tools will likely eliminate the need to perform diagnostic surgery for such patients; several groups have recently used tumor gene expression to define molecular signatures that can discriminate benign from most malignant follicular tumors.47–49
Patients with sporadic MTC typically present with solitary thyroid nodules situated at the upper lateral regions of the thyroid gland, where the preponderance of C cells are located. On physical exam, MTC are described as hard, nodular tumors that are sometimes calcified. Patients with locally invasive MTC often complain of “aching” or painful thyroid tumors. Carcinoembryonic antigen (CEA) levels are almost always elevated in patients with MTC, which is the same serum protein marker utilized to screen colorectal cancer patients for recurrence after resection. Since MTC can secrete several proteins (e.g., somatostatin, vasoactive intestinal peptide, adrenocorticotropic hormone (ACTH), bombesin), patients with advanced disease and hepatic metastases can present with generalized symptoms, such as flushing, Cushing’s syndrome, and diarrhea is common.50
The diagnosis of MTC is reliably established by FNA, which reveals amyloid and may stain positive for calcitonin, CEA, and chromogranin A.51 In relatives of patients with familial MTC, small tumors and C-cell hyperplasia can be diagnosed by elevated basal or stimulated levels of calcitonin. High CEA levels appear to be a worse prognostic indicator than elevated blood calcitonin levels.
In comparison with sporadic MTC, hereditary MTC generally occurs in younger patients and can be seen in the context of the isolated familial MTC and MEN2 syndromes. A recent claim that former American president Abraham Lincoln may have had MEN2B seems unlikely,52 given that untreated patients with this syndrome typically die within the first 3 decades of life, with few reports of asymptomatic long-term survival.53 Although there are some RET proto-oncogene mutations that result in MTC in older patients, patients with MTC and MEN2B have the worst prognosis of all patients with MTC.
Patients with ATC typically present with a rapidly enlarging neck mass that is fixed to surrounding structures. ATC are generally large and cause local obstructive symptoms from their mass effect, including pressure in the neck, dysphagia, and dysphonia. Patients with thyroid lymphoma can also present in a similar fashion. In our own experience with ATC, the average tumor size was 7 cm and the majority of patients presented with symptomatic disease. Furthermore, invasive thyroid tumors can invade the recurrent laryngeal nerves and cause vocal cord dysfunction and regional pain. ATC can be readily confirmed by FNA biopsy with cytological review.54 Distant metastases are common at presentation and can be found in up to 43% of patients.55 Both distant and regional metastases essentially preclude curative therapy for patients with ATC.
Prognostic Models and Staging of Thyroid Cancer
While the majority of patients with WDTC have excellent outcomes after surgical resection, some patients harbor aggressive disease. In an effort to select high-risk patients for adjuvant therapy, prognostic models utilize clinical and pathologic factors to predict survival (Table 34-1). Multiple large retrospective reviews have confirmed that age, tumor grade, distant metastases, tumor invasion, and tumor size are independent predictors of outcome after thyroidectomy.
The American Joint Committee on Cancer TNM system characterizes the anatomic status of the primary tumor, regional lymph nodes and distant metastases (Table 34-2). This system was updated in 2003, and is often applied to both WDTC and MTC. TNM staging can be used as a prognostic tool and also for research purposes to enable consistent stratification of patients.
Surgery for WDTC
Surgical resection is the primary treatment for patients with WDTC.56 Thyroidectomy is both safe and effective when performed by an experienced surgeon, with 10-year survival rates approximating 93% and 85% for PTC and FTC, respectively.17 Although most clinicians agree that total thyroidectomy is the primary treatment for patients with high-risk PTC, there is some controversy regarding the extent of surgery for patients with low-risk WDTC.57 Some clinicians argue that thyroid lobectomy is equivalent to total thyroidectomy, and avoids the potential morbidity of bilateral operation.58
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
