Chapter 52B Cancer and the Nervous System
Pathology and Molecular Genetics
General Principles of Nervous System Tumor Biology
Nervous system tumors, like other human neoplasms, are clonal proliferations that develop secondary to changes in key growth regulatory genes. Such genes fall into several different classes: growth-promoting oncogenes that abnormally activate in tumors, growth-checking tumor suppressor genes that inactivate in tumors, cell death genes that are impaired in tumors, and deoxyribonucleic acid (DNA) repair genes that deregulate in tumors. In combination, these genetic changes result in powerful growth advantages that enable cells to proliferate, evolve, and disseminate (Louis, 2006).
Some of the particular genes affected in human nervous system tumors are newly discovered and are discussed with the respective tumor type. Interestingly, most of the identified genes are deregulated in many other types of human cancers (a notable exception being IDH1), and the manner in which such deregulation specifically affects nervous system cells remains unclear. Certain nervous system cells are probably more susceptible to neoplastic transformation than others. For example, the far greater frequency of neoplastic transformation of gliomas compared to neuronal tumors is attributable to the greater neoplastic vulnerability of glial cells. Because oncogenic transformation requires cell division, postmitotic cells such as neurons should not be susceptible to tumorigenic events. However, it remains possible that nervous system tumors arise when oncogenic changes occur in precursor cells rather than mature glial cells or neurons. Such precursor neural stem cells reside in the brain into adult life (Vescovi et al., 2006). In this sense, the greater frequency of gliomas may relate to the particular paths of differentiation that are followed after specific tumorigenic genetic changes (i.e., transformed neuroepithelial progenitors may have to undergo specific molecular events to become tumors, with these same events essentially restricting cells to glial differentiation).
It is unknown why nervous system cells rather than other cells in the body transform in these instances because the etiologies of brain tumors remain unknown. To date, radiation and hereditary predisposition are the only clearly implicated factors in the genesis of nervous system tumors. See Chapter 52A for a discussion of ionizing radiation as a risk factor for specific types of brain tumors. Hereditary predisposition to brain tumors is rare and confined to specific syndromes (e.g., neurofibromatosis type 1 [NF1], neurofibromatosis type 2 [NF2], tuberous sclerosis, von Hippel-Lindau disease, Li-Fraumeni syndrome, Turcot syndrome, and Gorlin syndrome). These conditions highlight the critical roles played by specific genes in brain tumorigenesis.
History of Nervous System Tumor Classification Schemes
The foundation of the first comprehensive classification of nervous system tumors, formulated by Percival Bailey and Harvey Cushing in 1926, presumed parallels between embryological and neoplastic cells. In large part, this histogenetic “cell of origin” model still forms the basis for current nomenclature; renewed interest in the role of developmental pathways in tumorigenesis has emerged in recent years. In 1949, as a means of enhancing the clinical utility of tumor classification, Kernohan contributed a tumor grading system whose purpose was assessing patient prognosis. Russell and Rubinstein modified and updated the Bailey and Cushing system during the 1960s, 70s, and 80s. Further updates were incorporated into the World Health Organization (WHO) classification, first completed in 1979 and then revised in 1993 and 2007 (Louis et al., 2007). The WHO classification system has become the system most widely used by neuropathologists today.
The WHO classification currently lists more than 100 types of nervous system tumors and their variants (Louis et al., 2007). This level of complexity may seem daunting at first, but consideration of key clinical and imaging characteristics typically narrows the differential diagnosis to only a few common possibilities (Table 52B.1). For example, the differential diagnosis varies substantially for supratentorial versus infratentorial, pediatric versus adult, and enhancing versus nonenhancing tumors.
Table 52B.1 Common Central Nervous System Tumor Diagnoses by Location, Age, and Imaging Characteristics
| Location | Child/Young Adult | Older Adult |
|---|---|---|
| Cerebral/supratentorial |
AT/RT, Atypical teratoid/rhabdoid tumor; DNT, dysembryoplastic neuroepithelial tumor; E, enhancing; MEN, mural enhancing nodule; MPNST, malignant peripheral nerve sheath tumor; NE, nonenhancing; NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2; PNET, primitive neuroectodermal tumor, SEGA, subependymal giant cell astrocytoma.
General Histopathological Features and Techniques
General Histopathological Features
Palisading
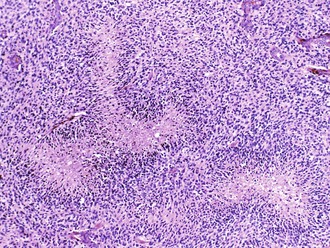
Palisading, or lining up of tumor cells, is characteristic of schwannomas, in which collections of palisaded nuclei are termed Verocay bodies. The term describes a collection of neoplastic cells arranged in parallel arrays, or palisades. In glioblastomas, palisading (also known as pseudopalisades) occurs around necrosis, with serpiginous zones of necrosis lined by hypercellular viable tumor nuclei (Fig. 52B.1).
Rosettes
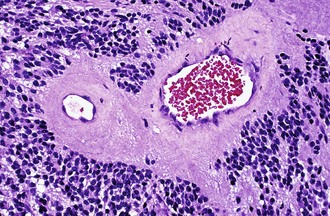
Characteristic of perivascular rosettes is a peripheral ring of tumor nuclei surrounding a central blood vessel with a nuclear-free eosinophilic zone in between (Fig. 52B.2). The nuclear-free zone derives from tapering cellular processes that radiate from the tumor cells to the vessel. Perivascular rosettes are most typical of ependymomas, and glial fibrillary acidic protein (GFAP) immunostains highlight the perivascular processes. Occasionally, similar axon-bearing structures appear in neuronal tumors as well (e.g., neurocytomas, pineocytomas, various forms of PNET).
Microvascular Proliferation
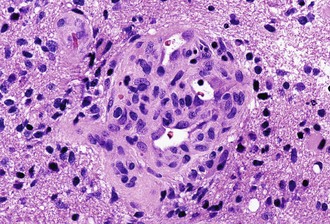
Microvascular (endothelial and pericytic/smooth muscle cell) proliferation represents an important and interesting response to tumor-associated and hypoxia-associated angiogenesis factors. It occurs in either benign or malignant CNS neoplasms, although it is more common in the malignant. In pilocytic astrocytoma, the new vessels may take on a delicate glomeruloid or “wicker-works” configuration, usually with a single-layered flat endothelial lining. In malignant tumors such as glioblastoma, these vessels are also glomeruloid but additionally display multilayering, with enlarged or “hyperplastic” endothelial cells (Fig. 52B.3). In either case, these neovascular structures lack the tight junctions normally contributing to the blood-brain barrier. Therefore, they typically leak intravascular contrast material, thus accounting for the tumor-associated enhancement detected radiologically.
Frozen Sections and Touch Imprints/Smears
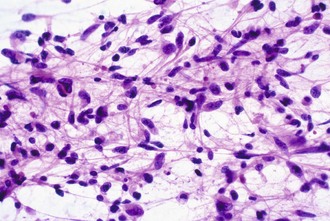
Intraoperative frozen sections are more difficult to interpret than fixed tissue sections, but their use has the advantage of speed, requiring only a few minutes to prepare. This technique is requested for several reasons, including simple curiosity and a desire to provide rapid feedback to patients and their families. However, the most important reasons for performing this technique are to ensure that the pathologist obtains a representative sample for permanent sections and to provide information that may alter the surgical procedure. For example, a neurosurgeon may opt to stop at a limited biopsy for a suspected lymphoma, aggressively resect for an ependymoma, or send additional material to the microbiology laboratory for an abscess. Because the process of tissue freezing produces significant cytological artifacts, another popular technique has been the preparation of touch imprints, or smears, from fresh tissue. Although the underlying architecture is lost, the tissue requirements are minimal, and the cytological preservation is excellent (Fig. 52B.4). This may be critical for diagnoses based primarily on nuclear cytology, such as reactive gliosis versus low-grade diffuse glioma. It is also important to realize that the frozen section technique results in substantial tissue loss and morphological artifacts in the thawed residual specimen. Therefore, it is best to omit its use in limited biopsies for which the surgical procedure will not be altered. In other words, it is imperative to save at least some optimally fixed nonfrozen tissue for final diagnosis on sections from the fixed embedded material.
Molecular Diagnostics
Few molecular diagnostic tests have become routinely used or standard of care in neuro-oncology. The most notable and most widely used is chromosome 1p and 19q testing as a prognostic/management tool for patients with oligodendroglial tumors. FISH has the advantage of simplicity, morphological preservation, minimal tissue and purity requirements, and a lack of necessity for microdissection or matching blood/nonneoplastic tissue. However, it requires some experience for accurate interpretation, especially in cases with aneuploid populations of tumor cells. It also utilizes large probes (100–300 kb) and is insensitive to very small deletions. In selected cases, FISH has been used to evaluate epidermal growth factor receptor (EGFR) amplification to distinguish small cell glioblastoma from anaplastic oligodendroglioma. It is likely that many more applications of molecular diagnostics will become incorporated into diagnostic neuropathology laboratories in the near future, but it is also important to note that specific immunohistochemical assays may “substitute” for molecular diagnostic approaches in certain situations. For example, immunohistochemical absence of the product of the INI1 gene on chromosome 22q is now widely accepted as a sensitive and specific marker for atypical teratoid/rhabdoid tumors (AT/RTs) (Judkins et al., 2004). Similarly, because IDH1 is a frequently mutated gene in lower-grade gliomas and secondary glioblastoma (Yan et al., 2009), antibodies to the most common mutant form of the IDH1 protein have demonstrated clinical utility in the pathological assessment of infiltrating glioma (Camelo-Piragua et al., 2009).
Primary Neuroepithelial Tumors
Diffuse Astrocytoma (WHO Grade II)
Microscopically, a slight to moderate increase in cellularity occurs, and the primary distinction from reactive astrocytosis rests on the finding of cytological atypia. Diffuse astrocytoma cells can take on various morphological appearances, including fibrillary, gemistocytic, and protoplasmic types. Fibrillary morphology is most common and consists of irregular elongated hyperchromatic nuclei, either appearing “naked” in an otherwise fibrillary background or displaying discernible cytoplasmic processes (see Fig. 52B.4). GFAP immunostains highlight the latter, although this is neither absolutely sensitive nor specific, because (1) some astrocytomas harbor minimal quantities of GFAP-positive cytoplasm, (2) interpretation may be difficult owing to staining of nonneoplastic astrocytic elements or high background in general, and (3) other gliomas display GFAP immunoreactivity. By definition, grade II astrocytomas lack mitotic activity, microvascular proliferation, and necrosis. Ancillary staining for MIB-1 (Ki-67) generally reveals a low proliferative index as well.
Recently, a large proportion of low-grade gliomas (including 90% of grade II astrocytomas) were found to have mutations in IDH1 and IDH2, even though the mutation was initially described in 12% of (largely secondary) glioblastomas (Parsons et al., 2008). IDH1 encodes isocitrate dehydrogenase, a component of the citric acid cycle. The role of IDH1 in glioma genesis remains uncertain, although recent work suggests that this loss of function mutation may cause accumulation of substrate, triggering HIF signaling pathways (Zhao et al., 2009). Others have proposed that tumorigenesis may be a consequence of 2-hydroxyglutarate formation (Dang et al., 2009). IDH1 mutations in low-grade glioma are tightly correlated with earlier age at diagnosis and tumor grade, appear to confer prognostic benefit beyond tumor grade, and are frequently associated with O6-methylguanine-DNA methyltransferase (MGMT) methylation (Sanson et al., 2009).
Chromosome 17p losses and mutations of TP53 are common molecular genetic events in diffuse astrocytomas, seen in approximately half of cases (Louis, 2006). Those with gene mutation usually display strong nuclear immunostaining for p53 protein, although the association is imperfect, since the protein is stabilizable by other mechanisms, and many immunopositive cases lack TP53 mutations. Those cases that evolve to glioblastoma (secondary glioblastoma) typically retain evidence of TP53 mutation and may even show additional TP53 gene mutations or clonal expansion of the tumor cells with mutation. On the other hand, de novo, or primary, glioblastomas rarely display such mutations. Other common alterations involve the overexpression of platelet-derived growth factor receptor alpha (PDGFα) in astrocytomas of all grades and losses of chromosome 22.
Anaplastic Astrocytoma (WHO Grade III)
Histologically, anaplastic astrocytomas are more cellular than grade II astrocytomas and display a greater degree of proliferation. The presence of mitotic figures primarily defines these tumors. One astrocytoma variant that commonly presents at the anaplastic or grade III level is the gemistocytic (“stuffed cell”) astrocytoma. Characterized by strongly GFAP-positive cells with eccentric bellies of eosinophilic cytoplasm, this tumor type has a high incidence of progression to glioblastoma. Interestingly, the small-cell astrocytoma elements in the background primarily constitute the proliferating element. Another feature that may help distinguish anaplastic astrocytomas from grade II astrocytomas is the generally higher Ki-67 or MIB-1 proliferative index on immunostains (Giannini et al., 1999a).
Anaplastic astrocytomas share the high frequency of TP53, IDH1, and IDH2 mutations seen in grade II astrocytomas. Inactivation of the retinoblastoma protein (Rb) cell-cycle regulatory pathway is also common and may be primarily responsible for the increased proliferation observed histologically. Homozygous deletion of the CDKN2A/p16 gene on 9p is the most common mechanism for disabling this pathway, although deletions of the RB gene and CDK4 amplifications are alternative aberrations (Louis, 2006). Amplifications of the EGFR gene and losses of PTEN on 10q occur less frequently in anaplastic astrocytomas than in glioblastomas and are virtually absent in tumors containing an IDH1 mutation (Sanson et al., 2009). When present, EGFR amplification may suggest either a worse prognosis or the possibility of undersampling/undergrading in a glioblastoma (Smith et al., 2001).
Glioblastoma (WHO Grade IV)
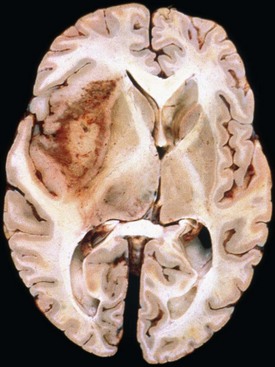
Glioblastoma most commonly occurs in the deep white matter, basal ganglia, or thalamus and is rarely found in the cerebellum or spinal cord. The gross and microscopic appearance is heterogeneous. Replacing the affected portion of the brain is a single mass that grossly may appear deceptively well circumscribed but microscopically infiltrates widely, often spreading to the opposite hemisphere via the corpus callosum (butterfly lesion). Multifocal tumors may occur and in most cases likely represent separate regions of malignant transformation within a widely disseminated lower-grade astrocytoma such as gliomatosis cerebri. In advanced stages, the tumor may extend into the meninges or the ventricle. Seeding of the neuraxis as multiple implants on the brain or ventricular surfaces is an atypical growth pattern, and extracranial metastases are extremely rare. The cut surface has a variegated appearance (Fig. 52B.5) characterized by central yellow or white zones of necrosis and hemorrhage surrounded by a hyperemic ring (endothelial hyperplasia) and “edematous” brain with variable mixtures of vasogenic edema, gliosis, and tumor infiltrates. Microscopically characterizing glioblastomas are all the features of anaplastic astrocytoma plus endothelial hyperplasia or necrosis. The endothelial hyperplasia is thickened or glomeruloid vessels with multilayering (see Fig. 52B.3), representing a form of tumor-induced angiogenesis, a potential target for novel therapies. The necrosis is often associated with a characteristic serpiginous distribution and associated nuclear pseudopalisading (see Fig. 52B.1). Several histological variants exist, including giant cell glioblastoma, small cell glioblastoma, and gliosarcoma. Characteristic of the latter is a sarcomatous element, currently felt to arise most likely from mesenchymal metaplasia within a glioblastoma. No significant clinical differences are identifiable in these variants when compared with conventional glioblastoma, although gliosarcomas more frequently occur in the temporal lobes.
Glioblastomas are among the best genetically characterized CNS tumors and often harbor numerous cytogenetic and molecular genetic aberrations (Louis, 2006). Mutations of TP53, IDH1, and IDH2 are less common than in diffuse astrocytomas or anaplastic astrocytomas and occur more often in secondary glioblastomas. Arising in younger patients after malignant transformation of a lower-grade precursor, TP53 mutations occur at greater frequency in giant cell glioblastomas (Louis et al., 2001). In contrast, 30% to 40% of glioblastomas harbor EGFR amplifications, often with an associated constitutively activating mutation, and these occur mainly in the primary, or de novo, glioblastoma (Louis et al., 2001). Defining the latter is the lack of a precursor lesion, typically in an older patient with rapid clinical onset. EGFR amplifications are also particularly common (≈70%) in the small-cell variant. These tumors are composed of small round cells with minimal cytoplasm and a remarkably brisk mitotic index. Owing to the rounded, uniform nuclei, a misdiagnosis of anaplastic oligodendrogliomas may occur, although small cell glioblastoma share the demographic features and poor prognosis of glioblastomas in general and do not harbor the 1p and 19q deletions seen in oligodendrogliomas. Alterations of the p16/CDK4/RB pathway are nearly universal in glioblastoma, and many cases harbor chromosome 10 losses (mostly primary glioblastomas). The PTEN gene located on chromosome 10q23 mutates in a subset of these and may have prognostic significance in certain patient populations. Additional tumor suppressor genes on both the long and short arms of chromosome 10 are suspected in the remaining group of PTEN wild-type tumors with demonstrable LOH. The list of additional glioblastoma-associated alterations is growing rapidly. Recent large-scale sequencing studies have shown that glioblastomas have somatic mutations of multiple genes (Parsons et al., 2008; TCGA Network, 2008). The studies have confirmed our knowledge of the incidence of mutated and/or amplified genes such as EGFR, PTEN, RB1, and PIK3CA and showed higher prevalence for mutations in TP53, NF1, ERBB2, and PIK3R1 (TCGA Network, 2008) as well as new candidate genes involved in tumorigenesis, such as IDH1 (Parsons et al., 2008). Nevertheless, the focus of intensive research is on mechanisms of invasion, cellular migration, proliferation, angiogenesis, apoptosis deregulation, and therapeutic resistance (Louis, 2006).
Early evidence suggests that molecular analysis of glioblastomas may have prognostic and predictive relevance. For example, methylation of the MGMT gene promoter, leading to down-regulation of MGMT expression, appears prognostic of improved survival in patients with glioblastoma treated with the alkylating agent, temozolomide (Hegi et al., 2005). However, emerging data suggest that prognostic benefit associated with MGMT methylation in glioblastoma may be unrelated to the mode of therapy administered (Rivera et al., 2010). Furthermore, resistance to alkylating chemotherapy in glioblastoma is associated with therapy-related inactivation of MSH6, a DNA mismatch repair enzyme, allowing increased mutagenesis and the emergence of a hypermutated phenotype (Cahill et al., 2007; Yip et al., 2009).
Molecular approaches to global-expression profiling potentially provide powerful means to distinguish glioblastomas from other malignant gliomas that follow distinct clinical courses (Nutt et al., 2003). Moreover, gene-expression profiling of glioblastoma has progressed from augmenting information provided by histological examination to characterization of molecular subtypes based on different gene expression patterns in each group (Sulman et al., 2009). For example, The Cancer Genome Atlas (TCGA) Research Network published a molecular classification of glioblastoma based on expression of signature genes (Verhaak et al., 2010). Of the four subgroups identified (named based on prior work and genes expressed as proneural, neural, mesenchymal, and classic subtypes), the proneural group showed distinctive clinical characteristics including overrepresentation among younger patients, mutations in IDH1, and prior or concomitant areas with low-grade histology—indicating that these are secondary glioblastomas (Verhaak et al., 2010). There also appears to be a CpG island methylator phenotype (CIMP) of glioma belonging to the same proneural molecular subgroup that is common among low-grade gliomas, with a high proportion showing mutations in IDH1 (Noushmehr et al., 2010). While perhaps allowing more coherent selection of potential targets for molecular therapy and validating the role of surrogate markers such as IDH1 for identifying prognostically significant subgroups of glioma, molecular profiling remains largely investigative to date.
Circumscribed (“Favorable Prognosis”) Astrocytomas
Pilocytic Astrocytoma (WHO Grade I)
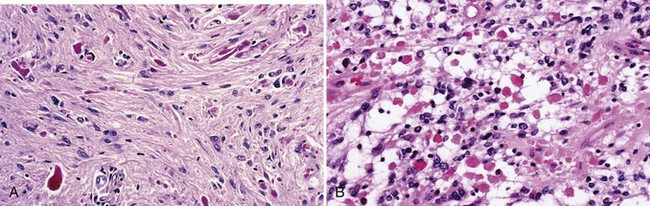
The distinctive histological feature of pilocytic astrocytoma is a biphasic pattern with compact pilocytic areas interspersed with microcystic, spongy, or loose areas (Fig. 52B.6). Dense portions contain piloid (hairlike) or bipolar astrocytes with long spindle-shaped processes. Rosenthal fibers are common. These are masses of intracellular astrocytic filaments, fusiform or corkscrew shaped, with a hyaline appearance (see Fig. 52B.6, A). In addition, mulberry-shaped eosinophilic granular bodies also occur in most cases (see Fig. 52B.6, B). Although not entirely specific, both Rosenthal fibers and eosinophilic granular bodies are generally signs of an indolent process and represent important diagnostic clues that distinguish pilocytic from diffuse astrocytomas. NF1 gene inactivation occurs in NF1-associated pilocytic astrocytomas but not in sporadic examples. A KIAA1549:BRAF fusion gene at 7q34 characterizes the majority of pilocytic astrocytomas (Jones et al., 2008). The fusion protein is a constitutively active mutant BRAF isoform with activity levels similar to or higher than wild-type BRAF, with demonstrable transforming potential in transfected cells.
Pleomorphic Xanthoastrocytoma (WHO Grade II or III)
PXA is a rare and distinctive form of astrocytoma associated with a favorable prognosis and often misdiagnosed in the past as glioblastoma. The average age at diagnosis is 26 years, and a history of seizures often precedes diagnosis (Giannini et al., 1999b). PXA usually involves the cerebral cortex and overlying meninges, and the preferred site is the temporal lobe. The histological features are hypercellularity with many atypical and pleomorphic tumor astrocytes. Bizarre giant cells are present, but mitoses are unusual. Probably the most helpful pathological finding is that of eosinophilic granular bodies, since these do not occur in glioblastomas. Despite its name, xanthomatous cells (lipidized astrocytes) with foamy lipid-filled cytoplasm only occur in approximately one-fourth of cases. PXA has a relatively favorable prognosis (WHO grade II), with postoperative survival averaging 81% at 5 years and 70% at 10 years. However, it is estimated that 15% to 20% of these tumors undergo malignant transformation (WHO grade III) and are associated with an aggressive clinical course. Few studies document consistent genomic derangements in PXA, but the relatively frequent loss (50%) of chromosome 9, especially at 9p21, suggests a role for CDKN2A/CDKN2B deletion in these tumors (Weber et al., 2007).
Subependymal Giant Cell Astrocytoma (WHO Grade I)
Some tumor cells are clearly of astrocytic origin, and GFAP fills the cytoplasm. Other tumor cells resemble neurons having prominent nucleoli, and many have intermediate features with astrocytoma-like cytoplasm and neuronal-like nuclei. Positive immunohistochemical staining for neuronal markers further suggests neuronal differentiation. Tumor cells may stain with both neuronal and glial markers or with neither, explaining the preference of some neuropathologists for the term subependymal giant cell tumor rather than SEGA. Because hamartomas of many organs characterize tuberous sclerosis, giant cell astrocytomas may also be hamartomas, rather than neoplasms. This is consistent with the benign behavior of these tumors. These tumors are related to alterations in the TSC1 (hamartin) and TSC2 (tuberin) genes and thus involve aberrant signaling in this growth-regulatory pathway. Subsequent to the finding that rapamycin could induce regression in these tumors (Franz et al., 2006), studies have identified novel proteins in these tumors that are likely mTOR (mammalian target of rapamycin) effectors (Tyburczy et al., 2010).
Oligodendroglioma (WHO Grade II or III)
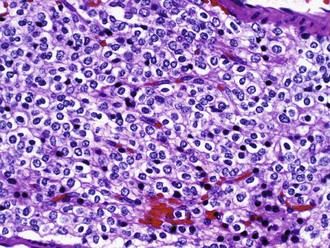
The classic and uniformly accepted oligodendroglial features include uniformly round nuclei, bland chromatin, clear perinuclear haloes imparting a “fried egg” appearance, and a rich branching capillary network reminiscent of “chicken wire” (Fig. 52B.7). Less specific findings include cortical involvement, microcalcifications, mucin-rich microcystic spaces, and perineuronal satellitosis. Although helpful, the so-called fried-egg appearance is a formalin fixation artifact that is neither necessary for diagnosis nor encountered in frozen sections or rapidly fixed specimens. The morphological spectrum includes two strongly GFAP-positive cells: minigemistocytes (or microgemistocytes) and gliofibrillary oligodendrocytes. The former are gemistocyte-like cells with small bellies of eosinophilic cytoplasm, round bland nuclei resembling those of classic oligodendroglioma nuclei, and no cytoplasmic processes. The latter are histologically identical to classic oligodendroglioma cells but exhibit a thin perinuclear rim of GFAP immunoreactivity. These two cell types (1) occur commonly in otherwise classic-appearing oligodendrogliomas, (2) are reminiscent of normal GFAP-positive oligodendroglial precursor cells, and (3) do not impact negatively on prognosis (although the presence of numerous microgemistocytes should prompt a careful search for anaplastic features). Hypercellularity, numerous mitoses, and microvascular proliferation defines anaplastic oligodendroglioma (grade III). Some oligodendrogliomas have regions that are histologically similar to diffuse astrocytoma, which can cause diagnostic difficulty because currently no specific oligodendroglioma markers exist.
The genetic characterization of oligodendrogliomas has provided some of our most important clues thus far in the diagnosis and treatment of diffuse gliomas. A characteristic loss of chromosomal arms 1p and 19q occurs in 50% to 80% of cases. More importantly, this genetic signature has been associated with both prolonged survival and a favorable response to chemotherapy or radiation therapy in both anaplastic (WHO grade III) (Cairncross et al., 1998, 2006; Van den Bent et al., 2006) and low-grade (grade II) oligodendrogliomas (Kaloshi et al., 2007). Based on these findings, ancillary testing for 1p and 19q status is a common request by both clinicians and patients; many centers now perform genetic testing routinely (Abrey et al., 2007). The most commonly utilized techniques include FISH and LOH, each with advantages and disadvantages (Perry et al., 2003; Yip et al., 2008). A subset of 1p/19q co-deleted tumors display polysomies or evidence of polyploidy. Patients with these tumors have shorter progression-free survival, although the overall survival is not affected (Snuderl et al., 2009). IDH1 (and IDH2) mutations occur very at high frequency in oligodendroglioma (84%) and anaplastic oligodendroglioma (94%) (Yan et al., 2009). Additional progression-associated alterations, such as those commonly seen in astrocytomas, sometimes occur in anaplastic oligodendrogliomas as well and are typically associated with worse prognosis.
Oligoastrocytoma (WHO Grade II or III)
Genetic studies of the biphasic variant, in which microdissection is feasible (Maintz et al., 1997), have identified the same alterations in both components, consistent with a monoclonal process rather than a collision tumor (i.e., coexistence of two neoplastic clones). Therefore, most biphasic oligoastrocytomas have genetically resembled either pure oligodendroglioma or astrocytoma, with only rare cases showing mixed patterns. One study suggested that WHO grade II and grade III oligoastrocytomas with an oligodendroglioma genotype (i.e., 1p and 19q loss) follow clinical courses similar to those of more typical oligodendrogliomas, whereas those with an astrocytic genotype (i.e., 17p loss) are more akin to astrocytomas in their behavior (Eoli et al., 2006).
Ependymoma (WHO Grade II or III)
Ependymomas are usually well-circumscribed masses that tend to compress rather than infiltrate the adjacent parenchyma. As such, some cases may be surgically curable, and the extent of resection constitutes a much more important prognostic variable in ependymomas than in diffuse gliomas. Cystic tumors more likely occur in the cerebrum. Ependymomas in contact with CSF pathways may seed the subarachnoid space and generate drop metastases in approximately 5% of cases, a condition associated with a poor prognosis. The characteristic features of ependymomas are sheets of cells interrupted by perivascular rosettes, nuclear free zones surrounding a central blood vessel (see Fig. 52B.2). True ependymal rosettes (i.e., containing a central lumen) and canals (i.e., slitlike structures resembling small ventricles) are even more specific but only occur in approximately 10% of cases.
Ependymomas are often aneuploid with complex, albeit nonspecific, alterations. Chromosome 22q deletions are among the most common, with associated NF2 mutations primarily restricted to spinal ependymomas, a fact that fits with the spinal location of most ependymomas in NF2 patients (Singh et al., 2002). Other 22q tumor suppressors are likely, and the existence of many additional ependymoma-associated genes is expected. Ependymomas may arise from radial glial cells (Taylor et al., 2005). Further molecular analysis of these tumors suggests that the gene-expression profile of ependymomas may differ according to site in the CNS, suggesting these are different entities at a molecular level (Taylor et al., 2005). In childhood tumors, 1q gain appears to be a frequent event and is possibly associated with aggressive behavior (Zacharoulis and Moreno, 2009). Gains at 9qter, specifically 9q33-44, appear associated with progression and up-regulation of the Notch pathway (Puget et al., 2009).
Choroid Plexus Tumors
Choroid Plexus Papilloma (WHO Grade I)
Choroid plexus papillomas have a pink or red, highly vascular, polypoid or cauliflower appearance, often with chalky calcifications (Fig. 52B.8). Large tumors in the third or fourth ventricle may occlude or even distend the ventricle, causing noncommunicating hydrocephalus. Histologically, CPP resemble normal choroid plexus in terms of a well-formed papillary structure with true fibrovascular cores and, in most cases, a single-layered epithelial covering. However, the lining of papillomas lacks the cobblestone-like appearance of normal choroid plexus and tends to form instead a uniform layer of tall cuboidal to columnar cells without intervening spaces. Calcifications and clear intracytoplasmic vacuoles are common. The mitotic index is low.
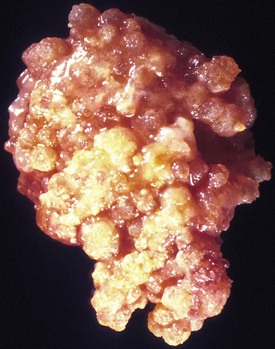
The majority of CPP are well-differentiated neoplasms (WHO grade I). The 2007 WHO classification also includes atypical CPP, which are distinguished from CPP by increased mitotic activity (more than 2 mitotic figures per 10 high-power fields). Other atypical histological features may be present but are not required for diagnosis (Louis et al., 2007).
Choroid Plexus Carcinoma (WHO Grade III)
Histologically, choroid plexus carcinoma resembles high-grade papillary adenocarcinoma but does not secrete mucin, an important distinction from the majority of metastatic adenocarcinomas. Some cases have a small-cell appearance reminiscent of PNET or rhabdoid cells similar to those of AT/RT. The genetics of these high-grade malignancies remains unknown, although the possible involvement of the INI-1/SMARCB1 gene on 22q11.2 has been raised. However, it remains possible that reported tumors with such genetic changes are really AT/RT. Choroid plexus carcinomas are observed in Li-Fraumeni syndrome kindreds (Wolburg and Paulus, 2010). In addition, sporadic choroid plexus carcinomas lacking a germline TP53 mutation may have a more favorable prognosis (Tabori et al., 2010).
Neuronal/Glioneuronal Tumors
Ganglioglioma/Gangliocytoma (WHO Grade I or II)
Portions of the tumor resemble a low-grade astrocytoma, either pilocytic or fibrillary in nature. Unlike native entrapped neurons within an infiltrative glioma, some of the tumor ganglion cells have a dysmorphic appearance, as evidenced by their lack of polarity, clustering, cytoplasmic vacuolation, increased nuclear pleomorphism, or multinucleation (Fig. 52B.9). Binucleate or multinucleate neurons are particularly helpful for diagnostic purposes when present. Otherwise, the most useful features to distinguish this tumor from diffuse gliomas include relative circumscription and eosinophilic granular bodies. Perivascular lymphocytic cuffing, microcystic spaces, and fibrosis with collagen deposition are other common findings. Rosenthal fibers are common, particularly at the edges of the lesion. Those without an obvious astrocytic component are sometimes referred to as gangliocytoma, although it is not yet clear that this distinction has any clinical relevance. The term ganglion cell tumor is less specific and incorporates both entities. Rare cases demonstrate signs of anaplasia (grade III), most often in the glial component, but the grading criteria and their predictive value have yet to be firmly established. GFAP activity is abundant in the astrocytic component, whereas the ganglion cell component expresses most markers of mature neurons such as synaptophysin, chromogranin, neurofilament, and NeuN. The molecular basis of these tumors remains poorly understood, with systematic investigation of altered gene expression hampered by their cellular complexity.
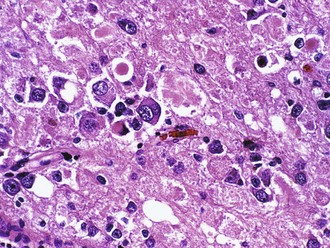
Dysembryoplastic Neuroepithelial Tumor (WHO Grade I)
Histologically, there is a wide range of appearances, although the most characteristic features include patterned (i.e., with ribbons or arcades), mucin-rich, cortical nodules and “floating neurons,” the latter consisting of ganglion cells that appear to float within a lacune-like mucin-filled space. The cytology of the tumor cells varies, although most resemble oligodendroglioma cells. In fact, on a small biopsy, it may be virtually impossible to distinguish these two entities, but the presence of diffuse cortical invasion on larger samples argues against a diagnosis of DNT. Little is known about the genetic basis of this tumor, although DNTs do not carry the 1p and 19q deletions typical of oligodendroglioma (Fuller and Perry, 2005).
Other Glioneuronal Tumors
Cerebellar liponeurocytoma was included as a grade I neoplasm in the 2000 edition of the WHO classification, but it is now assigned grade II (Louis et al., 2007). A rare neoplasm of adults, they are mainly located in the cerebellar hemispheres. The tumor is composed of neurocytic cells that become lipidized in areas. Although initially regarded as having little proliferative potential and possibly curable by resection, longer follow-up periods have shown a higher local recurrence rate without acquiring histological features of aggressive behavior.
Papillary glioneuronal tumor is possibly a ganglioglioma variant. These tumors are generally located in the cerebral hemispheres, particularly the temporal lobe, and have a characteristic pseudopapillary appearance conferred by single-layered cuboidal glial cells ensheathing fibrovascular cores with interpapillary aggregates of neurocytic and/or ganglioid cells. They correspond to WHO grade I (Louis et al., 2007).
Rosette-forming glioneuronal tumor of the fourth ventricle is a very rare entity found in the posterior fossa, usually presenting with obstructive hydrocephalus. It is a biphasic tumor with a predominant glial component, often resembling pilocytic astrocytoma, and a neurocytic component comprising uniform cells forming neurocytic rosettes or perivascular pseudorosettes (Preusser et al., 2003). They correspond to WHO grade I (Louis et al., 2007).
Central Neurocytoma (WHO Grade II)
Central neurocytomas are slow-growing tumors located in the lateral or third ventricle near the foramen of Monro, frequently involving the septum pellucidum (Schild et al., 1997). Age at diagnosis is usually in the second or third decade. They are usually sharply demarcated, sometimes lobulated masses that fill the ventricular space without significant infiltration of the surrounding brain.
The main histological feature is a proliferation of uniformly round tumor cells that mimic oligodendroglioma. Unlike oligodendrogliomas, these tumors often display neurocytic rosettes, exaggerated or irregular Homer Wright–like rosettes with central axon-rich neuropil. These rosettes are indistinguishable from the pineocytic rosettes encountered in pineocytoma, another tumor of small mature neurons, with similar cytological features to those of central neurocytomas. Ultrastructurally, their cytoplasm contains microtubules, synapses, and neurosecretory granules, belying their neuronal nature. Further, central neurocytomas are generally immunoreactive for the neuronal markers, synaptophysin and NeuN. Central neurocytomas with elevated proliferative indices (e.g., >2%) and vascular hyperplasia tend to have a higher rate of recurrence and are sometimes referred to as atypical neurocytomas, although grading criteria have not been firmly established. Rare examples of extraventricular neurocytomas and liponeurocytomas (with fat metaplasia) occur in the cerebral hemispheres, cerebellum, and spinal cord, but it is yet to be determined whether these represent the same family of tumors. Extraventricular neurocytoma is now recognized as a similarly behaving counterpart to central neurocytoma (Louis et al., 2007). The WHO also recognizes the cerebellar liponeurocytoma as a distinct entity (see later discussion). The genetic alterations of neurocytoma are largely unknown; as expected, central neurocytomas do not harbor the 1p and 19q deletions seen in oligodendrogliomas (Fuller and Perry, 2005), although rare extraventricular neurocytomas have been noted with 1p and 19q loss (Mrak et al., 2004). Further studies suggest this co-deletion may occur in as many as a quarter of the tumors and is associated with atypical histological features (Rodriguez et al., 2009). The relationship of these tumors to oligodendrogliomas with 1p/19q deletions remains to be clarified.
Embryonal Tumors/Primitive Neuroectodermal Tumors
The precise definition of primitive neuroectodermal tumor has long been debated, but essentially it refers to an extracerebellar “small blue cell tumor” that otherwise resembles medulloblastoma and in most cases shows evidence for primarily neuronal differentiation, albeit immature. Glial, mesenchymal, or melanotic elements may occur as well. Sometimes referred to as cerebellar PNET, medulloblastoma therefore represents the prototype and most common member of the PNET family. Adding further confusion to the nomenclature is the existence of a peripheral nervous system PNET (pPNET), which is felt to represent a completely different small blue cell tumor type with homologous genotypic and phenotypic features to those of Ewing sarcoma—t(11;22)(q24;q12) with EWS-FLI1 or variant fusion products. Other than medulloblastomas, the CNS variants are relatively uncommon and may include CNS PNET (cPNET), pineoblastoma, central neuroblastoma, ependymoblastoma, and medulloepithelioma. Despite the morphological and immunohistochemical overlap among these entities and the convenience of using the umbrella term PNET for all of them, this is likely overly simplified. We know, for example, that cPNETs have a significantly worse prognosis than medulloblastomas and seem to differ from them genetically (Reddy et al., 2000). Therefore, the overall heading of CNS primitive neuroectodermal tumors may be preferable to discuss this group of tumors and has been utilized in the 2007 WHO classification together with medulloblastoma and AT/RT to form the group of embryonal tumors.
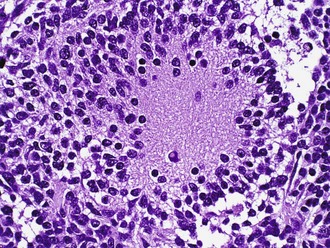
Medulloblastoma (WHO Grade IV)
Medulloblastomas consist of small immature cells with hyperchromatic round to carrot-shaped nuclei with minimal cytoplasm, as well as numerous mitoses and apoptotic bodies. These tumors typically display limited degrees of neuronal maturation, with neuropil formation, synaptophysin immunoreactivity, and occasional Homer Wright (neuroblastic) rosettes (Fig. 52B.10). Sometimes the terms cerebellar neuroblastomas or ganglioneuroblastomas apply to tumors with extensive rosette formation or neuronal maturation, respectively. The 2007 WHO Classification recognizes several medulloblastoma variants, some of which are clinically useful. These include the anaplastic/large-cell variant which shows widespread cellular anaplasia and has an aggressive clinical course with a high frequency of metastasis. The desmoplastic/nodular variant is recognized by the presence of pale nodular areas representing foci of neuronal differentiation surrounded by proliferating cells with hyperchromatic nuclei, which elaborate intercellular reticulin. When strictly applied, these histological features are associated with a better prognosis than “classic” medulloblastoma. Some evidence exists that the nodular or desmoplastic variant is encountered most frequently in adults, in the lateral cerebellar hemispheres, and in patients with Gorlin nevoid basal cell carcinoma syndrome (NBCCS) where there is loss of the tumor suppressor gene, PTCH, on chromosome 9q (see later discussion). The extensively nodular variant occurs in infants and features marked expansion of the pale areas described in desmoplastic medulloblastoma with elaboration of neuropil-like tissue. It is associated with much better survival than classic medulloblastoma (Eberhart et al., 2002).
Familial forms of medulloblastoma have provided important clues regarding the inherited and sporadic forms of the tumor (Ellison, 2002). These include the hedgehog/patched signaling pathway implicated from studies of NBCCS and the APC/Wnt pathway associated with a form of Turcot syndrome (polyposis coli and brain tumors). Unfortunately, medulloblastoma appears to be a heterogeneous genetic tumor, with no single alteration accounting for the majority of cases. Cytogenetically, a characteristic isochromosome 17q occurs in one-third to one-half of cases, but mutations in the TP53 gene are rare. Both MYCC and MYCN gene amplifications have been associated with particularly aggressive medulloblastomas and encountered more commonly in the anaplastic/large-cell variant, whereas 6q loss or monosomy has been associated with Wnt pathway activation and a favorable prognosis (Northcott et al., 2010). Expression of the neurotrophin receptor, TrkC, is associated with a significantly better prognosis. Interestingly, genomic screening with expression profile microarrays that characterize thousands of genes simultaneously may be a useful method for predicting biological behavior (Pomeroy et al., 2002). Further work has suggested the presence of clinically relevant subgroups with anomalies and activation of different molecular pathways that may, as for glioblastoma, guide therapy in the future (Kool et al., 2008). Therefore, the routine diagnostic workup of medulloblastoma may eventually involve several histopathological and genetic techniques for further stratification and customized therapy.
Atypical Teratoid/Rhabdoid Tumor (WHO Grade IV)
AT/RT is an embryonal CNS neoplasm often misdiagnosed as medulloblastoma or PNET because of the prominence of small blue cells in many cases (Packer et al., 2002). The name of this tumor derives from the fact that it may resemble either epithelial tumors (teratoid) or the malignant rhabdoid tumor seen in the kidney, soft tissue, and other organ sites throughout the body. Mostly restricted to infants, AT/RT is one of the most aggressive human tumors. Average survival times are in the range of 6 to 8 months, and these tumors typically do not respond to conventional medulloblastoma-associated therapies. These dramatic biological differences make it critical to distinguish AT/RT from medulloblastoma; it is probable that part of the poor prognosis reported in medulloblastoma patients younger than age 3 years stems from prior inclusion of misdiagnosed AT/RTs.
Genetically, the majority of AT/RTs harbor monosomy 22 or 22q deletions coupled with mutations in the INI1/hSNF5 tumor suppressor gene, which leads to loss of expression of the INI1 gene product. These alterations provide specific molecular markers for the diagnosis of AT/RT, and monoclonal antibodies have now found widespread implementation in diagnosis (Judkins et al., 2004). Germline mutations also occur in familial or disseminated forms of this disease.
Meningeal/Extraaxial Tumors
Meningioma (WHO Grade I)
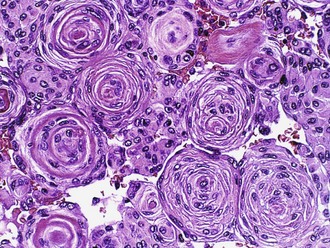
Meningiomas are histologically heterogeneous; the most recent WHO classification includes 13 morphological types (Louis et al., 2007). Four rare variants are considered more aggressive by definition: clear cell (grade II), chordoid (grade II), papillary (grade III), and rhabdoid (grade III). The other nine subtypes are considered benign unless they fulfill additional criteria for atypical (grade II) or anaplastic (grade IIII) meningioma. The majority of meningiomas have two basic histological patterns: meningothelial or fibroblastic, with the transitional variant having features of both. Meningothelial tumors are composed of arachnoidal epithelioid cells arranged in lobules, often with prominent whorls and psammoma bodies (Fig. 52B.11), which represent laminated calcifications of degenerated meningothelial whorls. Fibroblastic meningiomas are distinguished by their spindled appearance, fascicular or storiform architecture, and abundant collagen deposition. The most helpful immunohistochemical marker is EMA, which is detectable at least focally in the vast majority of meningiomas.
More than half of meningiomas are associated with losses of chromosome 22 or portions thereof. The tumor suppressor gene primarily involved in most of these cases is NF2, a finding that correlates well with the fact that meningiomas are the second most common tumor type in NF2 patients. A second gene with a high degree of homology, DAL1, or protein 4.1B on 18p11.3, has been implicated as well (Gutmann et al., 2000). Losses of both NF2 and protein 4.1B are common in meningiomas of all grades, suggesting that these alterations are early genetic events. Progesterone receptors are present in more than 50% of cases. Their significance is unclear, other than the fact that meningiomas can enlarge dramatically with pregnancy and regress after delivery.
Atypical Meningioma (WHO Grade II)
In most pathology series, the mitotic or proliferative index is the most powerful predictor of outcome. Based on a large Mayo Clinic series, the presence of at least 4 mitoses/10 HPF, even focally, qualifies for the diagnosis of atypical meningioma (Perry et al., 1999). With fewer mitoses, the presence of at least three of five additional parameters (sheeting architecture, hypercellularity, macronucleoli, small cell formation, and necrosis) also suffices. The issue of brain invasion has been debatable; although once considered the ultimate manifestation of malignancy, recent studies suggest that in the absence of frank anaplasia, these tumors have similar recurrence and mortality rates as those of atypical meningioma. Similar to mitotic counts, MIB-1 (Ki-67) labeling indices may be helpful for predicting the risk of recurrence, particularly in borderline atypical cases. Meningioma grade is also inversely proportional to progesterone receptor expression, so that in general, fewer atypical meningiomas are progesterone receptor immunoreactive than their benign counterparts.
A growing number of cytogenetic alterations have been associated with malignant progression of meningiomas, though the genes are unidentified. Most common in atypical meningiomas are deletions of 1p, 6q, 10, and 14q (Weber et al., 1997). Recently, aCGH analysis has found 59% of tumors with 1q gain, and that 1q gain is associated with shorter progression-free survival (Gabeau-Lacet et al., 2009).
Anaplastic Meningioma (WHO Grade III)
With the omission of brain invasion as a criterion for anaplastic meningioma, these tumors have become quite rare, accounting for no more than 1% to 2% of all cases. Many of these tumors start as benign or atypical meningiomas and progress over time, although de novo presentations also occur. As a group, anaplastic meningiomas are highly aggressive, rapidly growing, and highly infiltrative, with a median survival of less than 2 years (Perry et al., 1999). Nevertheless, extent of resection remains important, and long-term survival is still possible in a subset of patients. Histologically, anaplastic meningiomas are defined by the presence of excessive mitotic activity (>20/10 HPF) and/or frank anaplasia with a carcinoma-like or sarcoma-like appearance. These tumors are highly cellular, with extensive sheeting, necrosis, and nuclear atypia. Often, lower-grade elements, more easily recognizable as meningioma, occur. For those lacking this feature, immunohistochemistry or electron microscopy is often necessary to exclude hemangiopericytoma or other tumors such as dural-based sarcoma, metastatic carcinoma, or melanoma.
Hemangiopericytoma and Solitary Fibrous Tumor (WHO Grade II or III)
Hemangiopericytoma (HPC), once considered an angioblastic variant of meningioma, is now generally accepted to be a highly vascular dural-based sarcoma, analogous to those encountered in soft-tissue sites and of uncertain histogenesis. Solitary fibrous tumors (SFT) were initially characterized as primary mesenchymal neoplasms of the pleura but have in recent years been described in many extrapleural sites including the meninges. Although still debated, it has been suggested that HPC and SFT may represent ends of the same biological spectrum. It is clear, however, that tumors fulfilling criteria for HPC typically have high rates of local recurrence (60% to 80%) and systemic metastasis (25%). In contrast, SFTs of the meninges generally behave in a more indolent fashion, one study demonstrating excellent local control through surgical resection alone (Tihan et al., 2003). SFTs with malignant morphological features and aggressive behavior are rarely described. Both tumors occur at all ages, peaking in the fourth to sixth decades. Unlike meningiomas, there is no female predilection, and there is no association with NF2 or any of the known meningioma-associated genetic alterations.
Both SFT and HPC are EMA negative but positive for CD34, which is characteristically described as diffusely strong in SFT (Perry et al., 1997). Bcl2 is described as strongly staining cells in SFT, but HPC frequently also shows positive signal.
Nerve Sheath Tumors
Schwannoma (Neurilemoma) (WHO Grade I)
The vast majority of both sporadic and familial schwannomas are associated with loss of expression for the NF2 protein product, merlin or schwannomin (Stemmer-Rachamimov et al., 1997). NF2 gene deletions and LOH are common, though other mechanisms may also be involved. INI1 itself may be altered in familial schwannomatosis (Hulsebos et al., 2007). INI1 immunohistochemistry shows a mosaic pattern of staining in tumors from familial and sporadic schwannomatosis patients, as well as in schwannomas from NF2 patients, contrasting with strong uniform nuclear signal in sporadic tumors. These findings suggest a role for INI1 in multiple schwannoma syndromes (Patil et al., 2008).
Neurofibroma (WHO Grade I)
Studies suggest that the Schwann cell is the neoplastic component within neurofibromas, with remaining cell types likely representing reactive or entrapped elements (Perry et al., 2001). Deletions of the NF1 gene and losses of its protein product, neurofibromin, are detectable in a subset of both familial and sporadic neurofibromas.
Miscellaneous Tumors
Central Nervous System Lymphoma
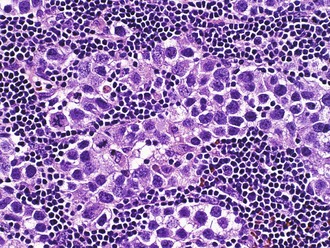
Of non-HIV-associated PCNSL, 90% are diffuse large B-cell type, with the remaining 10% being poorly characterized low-grade lymphomas, Burkitt lymphomas, or T-cell lymphomas. These tumors show a distinctive angiocentric pattern in which malignant cells surround and invade blood vessels in concentric layers (Fig. 52B.12). Reactive T cells are often numerous as well. Cases from immunosuppressed patients are characteristically necrotizing and EBV immunoreactive, whereas those from immunocompetent patients are not. Biopsies from patients treated preoperatively with corticosteroids are a common source of frustration and diagnostic difficulty for pathologists because the tumor cells often die, leaving behind a process that resembles either an inflammatory or demyelinating disorder. In such cases, an accurate diagnosis may await the time of recurrence. In addition, because sometimes the cellularity is low in comparison to lymph node biopsies and the typically mixed infiltrate, wherein reactive T cells may outnumber tumor cells, flow cytometry is often less sensitive for establishing the diagnosis than routine histology and may waste a considerable amount of limited tissue. Immunohistochemical expression of the germinal-center B-cell subgroup surrogate marker, BCL-6, occurs at a lower frequency in primary CNS lymphoma compared to nodal lymphoma and may be a favorable prognostic marker (Lin et al., 2006). Gene expression profiling of primary CNS lymphoma specimens have demonstrated that these tumors are distinguished from their nodal lymphoma counterparts by high expression of regulators of the unfolded protein response signaling pathway—the oncogenes c-Myc and Pim-1—and by regulators of apoptosis (Rubenstein et al., 2006).
Germ Cell Tumors
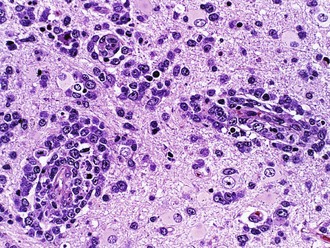
Histologically, germinomas are identical to testicular seminoma and ovarian dysgerminoma, with two distinct cell populations (Fig. 52B.13). The neoplastic element resembles primordial germ cells with abundant glycogen-rich clear cytoplasm and both PLAP and c-kit immunoreactivity. Immunoreactivity for the transcription factor, Oct4, is a useful diagnostic marker for the neoplastic cells. The stroma is often rich in reactive lymphocytes, primarily T cells. Sarcoid-like granulomas are also quite common and may obscure the diagnosis in biopsies where the reactive response overshadows the tumor cells. Embryonal carcinoma resembles a poorly differentiated carcinoma. Endodermal sinus tumor (yolk sac tumor) forms loose papillary epithelial structures known as Schiller-Duvall bodies. Combinations of mononucleated cytotrophoblasts and multinucleated syncytiotrophoblasts characterize choriocarcinoma. The tumor is highly vascular and particularly prone to hemorrhage. Teratomas differentiate into elements from all three germ layers. These tumors contain mature components such as teeth, hair, muscle, cartilage, and bronchial wall. These components arrange in a haphazard nonfunctional manner but are benign. Immature teratoma contains the same elements but has a fetal rather than mature appearance. Foci of immature brain with neural tubelike structures are particularly common.
Craniopharyngioma (WHO Grade I)
The advancing margins of the craniopharyngioma may appear deceptively sharp, but microscopic finger-like extensions into surrounding tissue are common. Both solid and cystic areas are intermingled. The cysts can become large and typically fill with a dark, viscous, cholesterol-rich fluid likened to motor oil. Irregularly shaped calcium deposits, varying in size from grains of sand to fine gravel, occur in approximately 75% of cases. The microscopic appearance is comparable to that of adamantinoma of the tibia and ameloblastoma of the jaw. Therefore, sometimes the classic craniopharyngioma is named adamantinomatous craniopharyngioma. The tumor demonstrates benign-appearing epithelium with central cobweb-like loosening (stellate reticulum) and peripheral palisading. Squamoid foci may occur, and the pattern of keratinization with ghostlike nests of keratinocytes is called wet keratin. Wet keratin differs from the dry, flaky keratin of epidermoid and dermoid cysts and is unique to craniopharyngioma. Therefore, it is diagnostic on a biopsy, even without the presence of viable epithelium. A rare variant is the papillary craniopharyngioma, which often presents in the third ventricle of adults. A characteristic feature is a true nonkeratinizing squamous lining over fibrovascular cores. Goblet cells are another feature. It is uncertain whether or not this variant has a better prognosis. Aberrant nuclear accumulation of β-catenin occurs in 94% of adamantinomatous craniopharyngiomas. In 77% of tumors, the dysregulation of the Wnt signaling pathway causing this phenomenon is due to genetic mutations which appear to target the GSK3β phosphorylation site of the β-catenin gene necessary for protein degradation (Buslei et al., 2005). Mutations in other components of the pathway such as APC were not found. This finding may help discriminate adamantinomatous from papillary craniopharyngiomas, which do not show nuclear translocation of β-catenin. It may also help distinguish adamantinomatous craniopharyngioma from other sellar tumors in the event of a small biopsy where diagnostic histological features are not readily apparent.
Metastatic Tumors
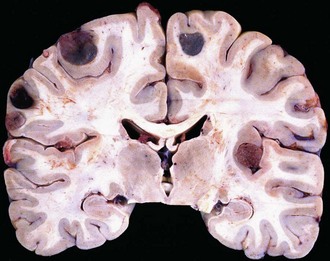
The typical metastasis is round, sharply demarcated, with central necrosis or hemorrhage (Fig. 52B.14). Spontaneous bleeding is characteristic of choriocarcinoma, melanoma, or renal carcinoma metastases. However, metastatic lung carcinoma is so much more common that it accounts for most hemorrhagic cases. In fact, lung carcinoma represents by far the most common primary tumor that metastasizes to the brain. Most other primary tumors metastasize to the lung before they gain access to the brain. The histological appearance is variable and recapitulates the morphology of the primary tumor. Most metastatic lesions are carcinomas or melanomas rather than sarcomas or lymphomas.
Abrey L.E., Louis D.N., Paleologos N., et al. Survey of practice patterns for anaplastic oligodendroglioma. Neuro Oncol. 2007;9:314-318.
Buslei R., Nolde M., Hofmann B., et al. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumors originating from the sellar region. Acta Neuropathol. 2005;109:589-597.
Cahill D.P., Levine K.K., Betensky R.A., et al. Loss of the mismatch repair protein MSH6 in human glioblastoma is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038-2045.
Cairncross J.G., Ueki K., Zlatescu M.C., et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendroglioma. J Natl Cancer Inst. 1998;90:1473-1479.
Cairncross G., Berkey B., Shaw E., for the Intergroup Radiation Therapy Oncology Group Trial 9402. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma. J Clin Oncol. 2006;24:2707-2714.
Camelo-Piragua S., Jansen M., Ganguly A., et al. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2009;119:509-511.
Dang L., White D.W., Gross S., et al. Cancer associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-744.
Eberhart C.G., Kepner J.L., Goldthwaite P.T., et al. Histopathologic grading of medulloblastomas. Cancer. 2002;94:552-560.
Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol. 2002;28:257-282.
Eoli M., Bissola L., Bruzzone M.G. Reclassification of oligoastrocytomas by loss of heterozygosity studies. Int J Cancer. 2006;119:84-90.
Franz D.N., Leonard J., Tudor C., et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-498.
Fuller C.E., Perry A. Molecular diagnostics in CNS tumors. Adv Anat Pathol. 2005;12:180-194.
Gabeau-Lacet D., Engler D., Gupta S., et al. Genomic profiling of atypical meningiomas associates gain of 1q with poor clinical outcome. J Neuropathol Exp Neurol. 2009;68:1155-1165.
Giannini C., Scheithauer B.W., Burger P.C., et al. Cellular proliferation in pilocytic and diffuse astrocytomas. J Neuropathol Exp Neurol. 1999;58:46-53.
Giannini C., Scheithauer B.W., Burger P.C., et al. Pleomorphic xanthoastrocytoma. What do we really know about it? Cancer. 1999;85:2033-2045.
Gutmann D.H., Donahoe J., Perry A., et al. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningioma. Hum Mol Genet. 2000;9:1495-1500.
Hegi M.E., Diserens A.C., Gorlia T., et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003.
Hulsebos T.J., Plomp A.S., Wolterman R.A., et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Pathol. 2007;80:805-810.
Jones D.T., Kocialkowski S., Liu L., et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673-8677.
Judkins A.R., Mauger J., Ht A., et al. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol. 2004;28:644-650.
Kaloshi G., Benouaich-Amiel A., Diakite F., et al. Temozolomide for low-grade gliomas: predictive impact of 1p/19q loss on response and outcome. Neurology. 2007;68:1831-1836.
Kool M., Koster J., Bunt J., et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088.
Lin C.H., Kuo K.T., Chuamg S.S., et al. Comparison of the expression and prognostic significance of differentiation markers between diffuse large B-cell lymphoma of central nervous system origin and peripheral nodal origin. Clin Cancer Res. 2006;12:1152-1156.
Louis D.N., Holland E.C., Cairncross J.G. Glioma classification: a molecular reappraisal. Am J Pathol. 2001;159:779-786.
Louis D.N., Ohgaki H., Wiestler O.D., et al. World Health Organization Classification of Tumours of the Central Nervous System. Louis D.N., Ohgaki H., Wiestler D.D.. Lyon, France: IARC Press. 2007.
Louis D.N. Molecular pathology of malignant glioma. Ann Rev Pathol Mech Dis. 2006;1:97-117.
Maintz D., Fiedler K., Koopmann J., et al. Molecular genetic evidence for subtypes of oligoastrocytomas. J Neuropathol Exp Neurol. 1997;56:1098-1104.
Mrak R.E., Yasargil M.G., Mohapatra G., et al. Atypical extraventricular neurocytoma with oligodendroglioma-like spread and an unusual pattern of chromosome 1p and 19q loss. Human Pathol. 2004;35:1156-1159.
Northcott P.A., Rutka J.T., Taylor M.D. Genomics of medulloblastoma: from Giemsa-banding to next-generation sequencing in 20 years. Neurosurg Focus. 2010;28:E6-E12.
Noushmehr H., Weisenberger D.J., Diefes K., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510-522.
Nutt C., Nutt C.L., Mani D.R., et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63:1602-1607.
Packer R.J., Biegel J.A., Blaney S., et al. Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol. 2002;24:337-342.
Parsons D.W., Jones S., Zhang X., et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-1812.
Patil S., Perry A., Maccollin M., et al. Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol. 2008;18:517-519.
Perry A. Pathology of low-grade gliomas: an update of emerging concepts. Neuro-Oncol. 2003;5:168-178.
Perry A., Roth K.A., Banerjee R., et al. NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and MPNSTs. Am J Pathol. 2001;159:57-61.
Perry A., Scheithauer B.W., Stafford S.L., et al. “Malignancy” in meningiomas: a clinicopathologic study of 116 patients. Cancer. 1999;85:2046-2056.
Perry A., Scheithauer B.W., Nacimento A. The immunophenotypic spectrum of meningeal hemangiopericytoma: a comparison with fibrous meningioma and solitary fibrous tumour of the meninges. Am J Surg Pathol. 1997;21:1354-1360.
Pomeroy S.L., Tamayo P., Gaasenbeek M., et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436-442.
Preusser M., Dietrich W., Czech T., et al. Rosette-forming glioneuronal tumor of the fourth ventricle. Acta Neuropathol. 2003;106:506-508.
Puget S., Grill J., Valent A., et al. Candidate genes on chromosome 9q33-34 involved in the progression of childhood ependymomas. J Clin Oncol. 2009;27:1884-1892.
Reddy A.T., Janss A.J., Philips P.C., et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189-2193.
Rodriguez F.J., Mota R.A., Scheithauer B.W., et al. Interphase cytogenics for 1p19q and t(1;19)(q10;p10) may distinguish prognostically relevant subgroups in extraventricular neurocytoma. Brain Pathol. 2009;19:623-629.
Rivera A.L., Pelloski C.E., Gilbert M.R., et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 2010;12:116-121.
Rubenstein J.L., Fridlyand J., Shen A., et al. Gene expression and anisotropism in primary CNS lymphoma. Blood. 2006;107:3716-3723.
Sanson M., Marie Y., Paris S., et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150-4154.
Schild S.E., Scheithauer B.W., Haddock M.G., et al. Central neurocytomas. Cancer. 1997;79:790-795.
Singh P.K., Gutmann D.H., Fuller C.E., et al. Differential involvement of protein 4.1 family members, DAL-1 and NF2 in intracranial and intraspinal ependymomas. Mod Pathol. 2002;15:526-531.
Smith J.S., Tachibana I., Passe S.M. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst. 2001;93:1246-1256.
Snuderl M., Eichler A.F., Ligon K.L., et al. Polysomy for chromosomes 1 and 19 predicts earlier recurrence in anaplastic oligodendrogliomas with concurrent 1p/19q loss. Clin Cancer Res. 2009;15:6430-6437.
Stemmer-Rachamimov A.O., Xu L., Gonzalez-Agosti C., et al. Universal absence of merlin, but not other ERM family members, in schwannomas. Am J Pathol. 1997;152:1649-1654.
Sulman E.P., Guerrero M., Aldape K. Beyond grade: molecular pathology of malignant gliomas. Semin Radiat Oncol. 2009;19:142-149.
Tabori U., Shilien A., Baskin B., et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28:1995-2001.
Taylor M.D., Poppleton H., Fuller C., et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323-335.
TCGA Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-1068.
Tihan T., Viglione M., Rosenblum M., et al. Solitary fibrous tumors in the central nervous system: a clinicopathological review of 18 cases and comparison to meningeal hemangiopericytomas. Arch Pathol Lab Med. 2003;127:432-439.
Tyburczy M.E., Kotulska K., Pokarowski P., et al. Novel proteins regulated by mTOR in subependymal giant cell astrocytoma of patients with tuberous sclerosis complex and new therapeutic implications. Am J Pathol. 2010;176:1878-1890.
Van den Bent M.J., Carpentier A.F., Brandes A.A., et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organization for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715-2722.
Verhaak R.G., Hoadley K.A., Purdom E., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell. 2010;6:98-110.
Vescovi A.L., Galli R., Reynolds B.A. Brain tumour stem cells. Natl Rev Cancer. 2006;6:425-436.
Weber R.G., Bostrom J., Wolter M., et al. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci U S A. 1997;94:14719-14724.
Weber R.G., Hoischen A., Ehrler M., et al. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytoma. Oncogene. 2007;26:1088-1097.
Wolburg H., Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75-88.
Yan H., Parsons D.W., Jin G., et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-773.
Yip S., Iafrate A.J., Louis D.N. Molecular diagnostic testing in malignant gliomas: a practical update on predictive markers. J Neuropathol Exp Neurol. 2008;67:1-15.
Yip S., Miao J., Cahill D.P., et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15:4622-4629.
Zacharoulis S., Moreno L. Ependymoma: an update. J Child Neurol. 2009;24:1431-1438.
Zhao S., Lin Y., Xu L., et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1 alpha. Science. 2009;324:261-265.