Chapter 52G Cancer and the Nervous System
Paraneoplastic Disorders of the Nervous System
Paraneoplastic neurological syndromes (PNSs) are a heterogeneous group of disorders caused by cancers located outside the central nervous system. The pathophysiology of PNSs differs from that of metastases or other cancer complications such as metabolic and nutritional deficits, infections, coagulopathy, and side effects of cancer treatment. PNSs may affect any part of the nervous system (Box 52G.1), and their frequency varies according to the type of syndrome and type of cancer. The onset of symptoms of PNSs often begins prior to the diagnosis of systemic cancer; identification of specific antineuronal antibodies may facilitate the diagnosis. The onset of neurological symptoms is often acute or subacute and frequently associated with inflammatory changes in the cerebrospinal fluid (CSF). Once thought to be poorly responsive to treatment, it is now recognized that certain subgroups of PNSs are highly treatment responsive. For all PNSs, treatment of the tumor is the most effective step in controlling or at least stabilizing the neurological disorder (Graus et al., 2001).
Box 52G.1 Paraneoplastic Neurological Syndromes
Syndromes Affecting the Central Nervous System
Limbic and brainstem encephalitis
Motor neuron syndromes† (ALS, subacute motor neuronopathy, upper motor neuron dysfunction)
Syndromes Affecting the Peripheral Nervous System
Pathogenesis
Most PNSs are immune mediated (Darnell and Posner, 2003). It is believed that the expression of neuronal proteins by a tumor provokes an immune response that is misdirected against the nervous system. This hypothesis is supported by the detection in serum and CSF of antineuronal antibodies that react with antigens expressed by the tumor and the nervous system (Dalmau et al., 1999a) (Table 52G.1). Antibodies that target intracellular antigens (e.g., anti-Hu, Ri, Ma) are most commonly associated with PNSs of the central nervous system. The presence of these antibodies is highly predictive of the presence of a cancer. Antibodies that target cell-surface receptors or ion channels may occur in PNSs of the central (e.g., N-methyl-d-aspartate [NMDAR]) or peripheral (e.g., P/Q type voltage-gated calcium channels [VGCC]) nervous system and may be found in patients with or without cancer (Dalmau et al., 2008; Mason et al., 1997).
Table 52G.1 Paraneoplastic Antineuronal Antibodies, Associated Syndromes, and Cancers
| Antibody | Syndrome | Associated Cancers |
|---|---|---|
| Anti-Hu | Focal encephalitis, PEM, PCD, PSN, autonomic dysfunction | SCLC, other |
| Anti-Yo | PCD | Gynecological, breast |
| Anti-Ri | PCD, opsoclonus-myoclonus | Breast, gynecological, SCLC |
| Anti-Tr | PCD | Hodgkin lymphoma |
| Anti-CV2/CRMP5 | PEM, PCD, peripheral neuropathy chorea, uveitis | SCLC, other |
| Anti-Ma proteins* | Limbic, diencephalic, brainstem encephalitis, PCD | Germ cell tumors of testis, other solid tumors |
| Anti-NMDAR | Limbic encephalitis, seizures, psychiatric symptoms | Teratoma |
| Antiamphiphysin | Stiff man syndrome, PEM | Breast |
| Anti-VGCC† | LEMS, PCD | SCLC |
| Anti-AChR† | MG | Thymoma |
| Anti-LGI1 (previously attributed to VGKC)† | Limbic encephalitis | Thymoma, SCLC |
| Anti-CASPR2 (previously attributed to VGKC)† | Morvan syndrome PNH |
Thymoma |
| Anti-AMPAR | Limbic encephalitis, psychiatric symptoms | Lung, breast, thymus |
| Anti-GABA(B)R | Limbic encephalitis, seizures | SCLC, other neuroendocrine tumor of lung |
| Antirecoverin‡ | Retinopathy | SCLC |
| Antibipolar cells of the retina | Retinopathy | Melanoma |
AChR, Acetylcholine receptor; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CASPR, connectin-associated protein 2; GABA(B)R, γ-amino-butyric acid type B receptor; LEMS, Lambert-Eaton myasthenic syndrome; LGI1, leucine-rich glioma inactivated 1; MG, myasthenia gravis; NMDAR, N-methyl-d-aspartate receptor; PCD, paraneoplastic cerebellar degeneration; PEM, paraneoplastic encephalomyelitis; PNH, peripheral nerve hyperexcitability; PSN, paraneoplastic sensory neuronopathy; SCLC, small cell lung cancer; VGCC, voltage-gated calcium channels; VGKC, voltage-gated potassium channels.
* Patients with antibodies to Ma2 are usually men with testicular cancer. Patients with additional antibodies to other Ma proteins are men or women with a variety of solid tumors.
† These antibodies can occur with or without a cancer association.
‡ Other antibodies reported in a few or isolated cases include antibodies to tubby-like protein and the photoreceptor-specific nuclear receptor.
Data suggest that cytotoxic T-cell responses mediate disorders associated with antibodies to intracellular antigens. These data include the presence of prominent infiltrates of CD8+ and CD4+ T cells in the nervous system, as well as in vitro studies showing the cytotoxic effect of the lymphocytes on cells expressing the onconeuronal antigens (Albert et al., 1998; Benyahia et al., 1999; Tanaka et al., 1999). Because the T-cell and humoral immune responses appear directed against the same antigens, it is likely that PNSs result from the cooperation of both arms of the immune response.
In contrast, disorders associated with antibodies to cell-surface antigens are more likely antibody mediated. A pathogenic effect has been demonstrated for antibodies against the acetylcholine receptor (AChR) in myasthenia gravis (Drachman, 1994), P/Q type VGCC in Lambert-Eaton myasthenic syndrome (Fukuda et al., 2003), and ganglionic AChR in autonomic ganglionopathy (Vernino et al., 2000). Similarly, a direct effect of antibodies against the target autoantigen has been demonstrated using in vitro and in vivo models for antibodies to NMDA receptor (anti-NMDA receptor encephalitis) (Hughes et al., 2010) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (limbic encephalitis) (Lai et al., 2009).
The characterization of antibodies against intracellular, cell surface, or synaptic antigens has clinical relevance because in general, those related to antibodies to cell-surface or synaptic antigens are more treatment responsive (Tüzün and Dalmau, 2007).
General Diagnostic Approach
The specificity of paraneoplastic antineuronal antibodies for PNSs or some types of cancer makes them useful diagnostic tools (see Table 52G.2). In some 60% of patients with PNS, the neurological symptoms precede the tumor diagnosis, so in the right clinical context, the detection of a paraneoplastic antibody in the serum or CSF helps diagnose the PNS and focus the search for the neoplasm. Most paraneoplastic antineuronal antibodies are also detectable (usually at low titers) in the serum of some patients with cancer without PNSs. This is a consideration when detecting a paraneoplastic antineuronal antibody in the serum of a patient suspected to have PNSs. Consider other causes for the neurological dysfunction if the detected antibody is not usually associated with the neurological syndrome. Similarly, a second neoplasm should be suspected if the detected cancer is not the histological type typically found in association with the antibody (e.g., anti-Yo with lung cancer rather than breast or ovarian cancer). A search for another neoplasm is required if the tumor cells do not express the target antigen of the paraneoplastic antibody (Graus et al., 2001). Clinical experience suggests that finding high titers of paraneoplastic antibodies in the CSF is confirmatory evidence of PNSs of the central nervous system.
A set of diagnostic criteria has been developed that takes into consideration the type of syndrome, detection of a tumor, and presence or absence of paraneoplastic antibodies (Graus et al., 2004). The diagnosis of PNSs is relatively straightforward for patients who develop a well-defined syndrome typically associated with cancer. Patient age is important because symptoms often associated with paraneoplastic mechanisms in adults (e.g., subacute cerebellar dysfunction) are less typical in children. Conversely, the development of opsoclonus in children is often paraneoplastic but is less suggestive of a paraneoplastic cause in adults. In these settings, the detection of an antibody known to be associated with PNSs or cancer is practically confirmatory of paraneoplasia. If cancer is not present, assume the presence of an occult neoplasm unless proven otherwise. Body positron emission tomography (PET) scans may detect tumors that escape detection by other standard imaging methods (McKeon et al., 2010; Rees et al., 2001; Younes-Mhenni et al., 2004). Although almost any neoplasm can cause PNSs, the tumors most commonly involved are small-cell lung cancer (SCLC), cancers of the breast, ovary, thymoma, neuroblastoma, and plasma cell tumors. The development of PNSs frequently heralds tumor recurrence in patients with a history of cancer or those who have recently gone into tumor remission.
The diagnosis of PNSs is more difficult in patients who develop less characteristic symptoms (e.g., brainstem dysfunction, myelopathy), especially if no antibodies are found in the serum or CSF. Most PNSs have an acute or subacute onset compared with noninflammatory neurodegenerative disorders that are chronically progressive. If the patient is known to have cancer, the possibility of metastases and nonmetastatic neurological complications of cancer (side effects of treatment, metabolic encephalopathy, infection, or cerebrovascular disorders resulting from coagulopathy) should be considered before the diagnosis of PNSs. Neuroimaging, in particular magnetic resonance imaging (MRI), helps exclude some of these complications. The CSF profile in patients with PNSs of the central nervous system often suggests an inflammatory process: pleocytosis, increased protein concentration, intrathecal synthesis of immunoglobulin (Ig)G, and oligoclonal bands (Psimaras et al., 2010). If paraneoplasia remains a consideration, make every attempt to find the associated neoplasm.
Specific Paraneoplastic Syndromes and Their Treatment
Paraneoplastic Cerebellar Degeneration
Clinical Findings
Rapid onset of severe pancerebellar dysfunction characterizes paraneoplastic cerebellar degeneration (PCD). Clinical features include truncal and appendicular ataxia, dysarthria, and downbeat nystagmus. In adults, the subacute onset of PCD differentiates it from chronic degenerative diseases of the cerebellum. A subset of patients with SCLC develop PCD associated with Lambert-Eaton myasthenic syndrome (LEMS), often before the tumor is diagnosed (Mason et al., 1997). In some cases, one misses the diagnosis of LEMS unless it occurs prior to the onset of PCD.
Tumor Association
Small-cell lung cancer, cancer of the breast and ovary, and Hodgkin lymphoma are the tumors most commonly associated with PCD (Shams’ili et al., 2003).
Immune Responses
Anti-Yo is the most frequent and well-characterized antibody associated with PCD; it is usually associated with breast or gynecological tumors (Peterson et al., 1992). The presence of anti-Yo antibodies has been detected in a few male patients with PCD and cancer of the salivary gland, lung, and esophagus. Some patients with predominant truncal ataxia, opsoclonus, and other ocular movement abnormalities may harbor an antibody called anti-Ri. In such cases, the tumor is usually a breast carcinoma or, less frequently, a gynecological cancer, bladder cancer, or SCLC (Luque et al., 1991). These patients may also develop dementia, mixed peripheral neuropathy, axial rigidity, and myoclonus.
In patients with SCLC, the development of PCD may be the presenting symptom of a paraneoplastic encephalomyelitis (PEM). In such cases, other areas of the nervous system become involved and anti-Hu or anti-CV2/CRMP5 antibodies are usually present. Patients with symptoms restricted to cerebellar dysfunction and negative anti-Hu antibodies often harbor VGCC antibodies (Graus et al., 2002). Patients with PCD associated with Hodgkin disease develop anti-Tr antibodies (Graus et al., 1997). The neurological disorder may develop before or after the diagnosis of the lymphoma, sometimes heralding tumor recurrence.
Treatment
Several case reports describe patients with PCD who improved after treatment of the tumor, plasma exchange, intravenous immunoglobulin (IVIg), rituximab, or immunosuppression with cyclophosphamide or corticosteroids (Blaes et al., 1999; Shams’ili et al., 2006). However, most patients with PCD do not improve with any of these treatments (Vedeler et al., 2006).
Paraneoplastic Encephalomyelitis
Clinical Findings
Patients with PEM may develop clinical features of dysfunction at several different levels of the neuraxis (Dalmau et al., 1992; Graus et al., 2001; Sillevis et al., 2002). Many patients will develop a sensory neuronopathy (see Paraneoplastic Sensory Neuronopathy [PSN]) and cerebellar dysfunction—in particular, gait ataxia. Limbic and/or brainstem encephalopathy (see Limbic and Brainstem Encephalitis) are common and occur in up to one-third of patients with PEM. Lower motor neuron involvement secondary to myelitis occurs in approximately 20%; the presence of symptoms affecting other areas of the neuraxis helps rule out pure motor neuron disorders. Approximately one-fourth of patients with PEM develop autonomic nervous system dysfunction; symptoms include postural hypotension, gastroparesis and intestinal dysmotility, sweating abnormalities, neurogenic bladder, and impotence. Cardiac dysrhythmias and respiratory or autonomic failures are frequent causes of death. Paraneoplastic chorea and uveitis occur more frequently in a subset of patients with PEM and CV2/CRMP5 antibodies (Vernino et al., 2002).
Tumor Association
PEM, with or without PSN, can be associated with almost any tumor, but in most patients, the underlying tumor is lung carcinoma, particularly SCLC (Graus et al., 2001).
Immune Responses
Patients with PEM/PSN and SCLC often have anti-Hu and at a much lower frequency, anti-CV2/CRMP5 antibodies, or both. In these patients, neurological symptoms usually precede the cancer diagnosis. Anti-CV2/CRMP5 antibodies are also associated with thymoma and other cancers (Benyahia et al., 1999).
Limbic and Brainstem Encephalitis
Clinical Findings
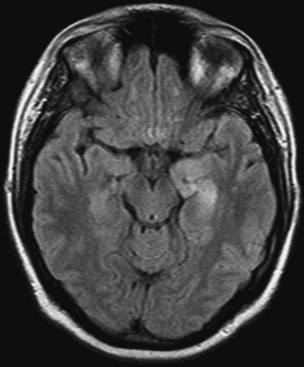
Limbic encephalitis is one of the few PNSs in which neuroimaging is often useful (Fig. 52G.1). Typical MRI findings include unilateral or bilateral mesial temporal lobe abnormalities, best seen on T2-weighted and fluid-attenuated inversion recovery (FLAIR) images (Ances et al., 2005; Gultekin et al., 2000). The temporal-limbic regions may be hypointense on T1-weighted sequences and may enhance with contrast.
Tumor Association
Limbic and brainstem encephalitis occur in both paraneoplastic and non-paraneoplastic settings. The cancers most commonly associated are SCLC, testicular germ-cell tumors, teratoma (usually of the ovary), thymoma, and Hodgkin lymphoma (Gultekin et al., 2000).
Immune Responses
The classification of immune responses in limbic and brainstem encephalitis is according to the location of the target antigen: intracellular or cell surface. The main intracellular antigens are Hu, Ma2, and less commonly, CV2/CRMP5. In patients with Hu antibodies, limbic encephalitis is a fragment of PEM, although the limbic encephalitis may predominate or be the initial presenting symptom (Dalmau et al., 1992). Patients often have signs of pontine dysfunction that progresses downward, with involvement of other areas of the neuraxis becoming more prominent with time. Most of these patients have SCLC.
Men younger than 45 years of age with symptoms of limbic, hypothalamic, and brainstem dysfunction are likely to have antibodies to Ma proteins and an underlying germ-cell tumor of the testis (Dalmau et al., 1999b; Hoffmann et al., 2008; Mathew et al., 2007). Ma antibodies are also encountered in older patients with similar neurological symptoms and other cancers (Dalmau et al., 2004). In contrast to patients with Hu antibodies, patients with Ma antibodies usually have a predominant mesencephalic syndrome with vertical gaze palsies (Saiz et al., 2009).
Antibodies to CV2/CRMP5 are seen in association with encephalomyelitis, sensorimotor neuropathy, cerebellar ataxia, chorea, uveitis and optic neuritis (Honnorat et al., 1997). Neurological findings rarely stay confined to the limbic and brainstem structures, and patients will often have frontostriatal and basal ganglia disturbances such as obsessive-compulsive behavior and cognitive deficits. CV2/CRMP5 antibodies can co-occur with anti-Hu or Zic antibodies, in which case patients often have multifocal deficits or PEM.
Patients with AMPA receptor antibodies usually present with acute limbic dysfunction or, less frequently, with prominent psychiatric symptoms (Graus et al., 2010; Lai et al., 2009). These disorders most commonly affect middle-aged women; about 70% have an underlying tumor in the lung, breast, or thymus.
Patients with antibodies to LGI1 develop memory disturbances, confusion, and seizures. Some patients develop hyponatremia or rapid eye movement (REM) sleep behavior disorders such as dream-enacting behavior and abnormal REM sleep patterns (Iranzo et al., 2006; Vincent et al., 2004). Most patients with limbic encephalitis and LGI1 antibodies do not have an underlying neoplasm. Only 20% of cases are paraneoplastic, and the commonly associated tumors are SCLC and thymoma (Pozo-Rosich et al., 2003). LGI1 is a secreted epilepsy-related protein that interacts with pre- and postsynaptic receptors (ADAM23 and ADAM22) (Lai et al., 2010). Mutations of LGI1 are associated with the syndrome of autosomal dominant lateral temporal lobe epilepsy (Gu et al., 2002; Kalachikov et al., 2002).
The encephalitis associated with GABA-B receptor antibodies is usually a limbic encephalitis and seizures. Approximately half of patients have an SCLC or a neuroendocrine tumor of the lung (Lancaster et al., 2010). These patients have a tendency to autoimmunity and frequently have additional antibodies to glutamic acid decarboxylase (GAD) and several non-neuronal proteins of unclear significance.
Limbic encephalitis with temporal lobe seizures occurs in association with GAD antibodies. The disorder is similar to other autoimmune limbic encephalitis; it is usually not paraneoplastic and is refractory to treatment (Blanc et al., 2009; Malter et al., 2010). Some patients have other more relevant antibodies against cell-surface antigens that may be causing the symptoms (Lancaster et al., 2010).
Treatment
Limbic encephalitis is the most likely PNS to improve with tumor treatment and immunomodulation with steroids and IVIg (Gultekin SH et al., 2000). The likelihood of improvement is higher if the disorder is associated with antibodies to cell-surface receptors or ion channels. Data suggest that in such cases, the antibodies are pathogenic; removal of the antigenic source (tumor) and antibodies with antibody-depleting treatments are often successful (Tüzün and Dalmau, 2007).
When the antigens are intracellular, response to treatment is often disappointing. An exception is the encephalitis associated with antibodies to Ma proteins. For these patients, treatment of the tumor (usually a testicular germ-cell neoplasm) and immunotherapy result in improvement in about one-third of cases (Dalmau et al., 2004).
Anti-N-Methyl-D-Aspartate Receptor Encephalitis
Clinical Findings
A characteristic clinical picture facilitates diagnosis of anti-NMDAR encephalitis. Patients experience a viral-like prodrome followed by the development of severe psychiatric symptoms, memory loss, seizures, decreased consciousness, dyskinesias (orofacial, limb, and trunk), and autonomic and breathing instability (Dalmau et al., 2008). Anti-NMDAR encephalitis is most common in young women and children but also occurs is men and older patients (Dalmau et al., 2008). The youngest reported patient was 20 months old (Wong-Kisiel et al., 2010). The differential diagnosis includes viral encephalitis, neuroleptic malignant syndrome, and (less frequently) hyperkinetic encephalitis lethargica (Dale et al., 2009; Gable et al., 2009).
Tumor Association
Over half of patients have an associated tumor, most commonly an ovarian teratoma that can be mistaken for a benign cyst. Patient age is a factor: about 50% of female patients older than 18 have unilateral or bilateral ovarian teratomas, but less than 9% of girls younger than 14 have a teratoma (Florance et al., 2009).
Immune Responses
The target antigen is the NR1 subunit of the NMDAR (Fig. 52G.2). Recent work shows that patients’ antibodies decrease the levels of NMDARs by a mechanism of cross-linking and internalization. These effects are demonstrable in cultures of hippocampal neurons, animal models, and patient autopsies. These data support a direct pathogenic effect of the antibodies (Hughes et al., 2010).

Treatment
Most patients with anti-NMDAR encephalitis respond to treatment regardless of the severity of the disorder. Patients with a teratoma who are treated early in the course often respond to removal of the tumor and corticosteroids, IVIg, or plasma exchange (Dalmau et al., 2008; Seki et al., 2008). Patients who are diagnosed at later stages of the disease or who do not have a tumor may need additional therapy with cyclophosphamide, rituximab, or both (Ishiura et al., 2008; Wong-Kisiel et al., 2010). In many cases, responses are slow, often requiring at least 2 or 3 months of hospitalization followed by physical and behavioral rehabilitation (Dalmau et al., 2008; Shimazaki et al., 2007). Identification and removal of the tumor is important because patients without tumor removal have prolonged recoveries and an increased risk of relapses (Iizuka et al., 2008).
Paraneoplastic Opsoclonus-Myoclonus
Clinical Findings
Opsoclonus-myoclonus consists of spontaneous, arrhythmic, large-amplitude conjugate saccades occurring in all directions of gaze that are associated with myoclonus of the head, trunk, or extremities. In children, paraneoplastic opsoclonus-myoclonus usually has an acute onset, with staggering and falling leading to a misdiagnosis of acute cerebellitis. These symptoms are followed by body jerks, drooling, refusal to walk or sit, ataxia, opsoclonus, hypotonia, irritability, and sleep disturbances (Russo et al., 1997; Tate et al., 2005). Although the ocular symptoms may resolve spontaneously, most children are left with behavioral abnormalities as well as language and psychomotor deficits. In adults, symptoms range from opsoclonus with mild truncal ataxia to a more severe syndrome characterized by opsoclonus, myoclonus, ataxia, and encephalopathy that can lead to stupor and death (Bataller et al., 2001). Spontaneous remissions are rare.
Tumor Association
In children, opsoclonus-myoclonus is usually a manifestation of neuroblastoma, although similar neurological symptoms may be associated with viral infections. The neurological symptoms precede the tumor diagnosis in 50% of patients. Children with neuroblastoma and opsoclonus have a better tumor prognosis than those without paraneoplastic symptoms (Tate et al., 2005). In adults, several underlying tumors have been reported, but the most common are SCLC and cancers of the breast and ovary.
Immune Responses
Many patients do not have paraneoplastic antibodies (Sabater et al., 2008). Patients with breast and gynecological cancers usually harbor anti-Ri antibodies (Luque et al., 1991). These patients often have additional brainstem and cerebellar dysfunction. Some adult patients, in particular those with SCLC and 5% of children with neuroblastoma, have anti-Hu antibodies in their sera. Some children with neuroblastoma have uncharacterized antibodies against postsynaptic or cell-surface antigens located on cerebellar granular cells (Blaes et al., 2008). These findings and the detection of CSF B-cell expansion and cellular immune activation support an immune-mediated pathogenesis of the PNS (Pranzatelli et al., 2004).
Treatment
Neuroblastoma-induced opsoclonus-myoclonus often responds to treatment of the tumor (chemotherapy) along with prednisone, adrenocorticotrophic hormone, IVIg, or rituximab (Bell et al., 2008; Tate et al., 2005). However, developmental and neurological sequelae are common (Russo et al., 1997). Sleep disturbance may respond to trazodone (Pranzatelli et al., 2005). Paraneoplastic opsoclonus-myoclonus in adults may partially respond to immunosuppression and IVIg. Patients whose tumors are treated promptly have a better neurological outcome than those whose tumors are not treated (Bataller et al., 2001). In the latter group, the disorder often progresses to severe encephalopathy and death.
Stiff Man Syndrome
Clinical Findings
Fluctuating rigidity of the axial musculature with superimposed spasms characterize stiff man syndrome. Muscle stiffness primarily affects the lower trunk and legs, but it can extend to the arms, shoulders, and neck. Emotional upset and auditory or somesthetic stimuli precipitate muscle spasms. Electrophysiological studies show continuous activity of motor units in the stiffened muscles that improve after treatment with diazepam. The rigidity disappears during sleep and after local or general anesthesia (Espay and Chen, 2006).
Tumor Association
The paraneoplastic form of stiff man syndrome is usually associated with breast cancer, although cases with SCLC and Hodgkin disease have been reported (Folli et al., 1993).
Immune Responses
The main autoantigen of the paraneoplastic form of the disorder is amphiphysin. Antibodies to amphiphysin may also occur in patients with other paraneoplastic syndromes such as encephalitis. In approximately 80% of patients with stiff man syndrome, the disorder develops as a non-paraneoplastic phenomenon in association with diabetes and polyendocrinopathy and often antibodies to GAD (Raju et al., 2005). A recent study reported antibodies to glycine receptors in a patient with progressive encephalomyelitis with rigidity and myoclonus (Hutchinson et al., 2008).
Treatment
Treatment of the tumor and the use of corticosteroids may improve paraneoplastic stiff man syndrome. IVIg is useful in patients with non-paraneoplastic stiff man syndrome (Dalakas et al., 2001;Vasconcelos and Dalakas, 2003). GABA-enhancing agents such as benzodiazepines, gabapentin, or baclofen, among others, provide symptomatic relief.
Paraneoplastic Sensory Neuronopathy
Clinical Findings
PSN is characterized by progressive sensory loss that may involve limbs, trunk, face, and sometimes sensorineural hearing loss. Painful dysesthesias are common. At onset, symptoms are usually asymmetrical and can be confused with radiculopathy or polyneuropathy. All modalities of sensation are eventually affected, and with progression, the sensory deficits result in ataxia, gait difficulty, and pseudoathetoid movements. PSN may develop alone or more commonly in association with PEM (Dalmau et al., 1992; Graus et al., 2001). Neurological dysfunction precedes the cancer diagnosis in two-thirds of patients. Typically, nerve conduction studies show small-amplitude or absent sensory nerve action potentials, although motor nerve and F-wave studies are usually normal. These findings are consistent with the pathological involvement of the dorsal root ganglia, but some patients also have electrophysiological evidence of axonal and demyelinating neuropathy (Camdessanche et al., 2002; Oh et al., 2005). In patients with no known cancer, suspect PSN when sensory symptoms develop subacutely and asymmetrically and involve the trunk and cranial nerves, particularly if the patient is a smoker. The CSF often shows an increased protein level with pleocytosis, oligoclonal bands, and intrathecal synthesis of IgG.
Immune Responses
The anti-Hu antibody is almost always detected in the serum of patients with PSN and SCLC but is rarely present in PSN associated with other tumors (Honnorat et al., 2009; Molinuevo et al., 1998). A few patients with PSN have been reported with antibodies to amphiphysin and CV2/CRMP5 (Antoine et al., 2001; Saiz et al., 1999).
Treatment
Studies of patients with SCLC and anti-Hu–associated PSN and PEM indicate that neurological symptoms in patients whose tumors completely responded to therapy were more likely to stabilize or improve compared to those with untreated tumors or tumors that did not respond to therapy (Graus et al., 2001; Sillevis et al., 2002). In some patients, prompt treatment with corticosteroids may partially improve sensory deficits. The effects of IVIg, cyclophosphamide, or rituximab are uncertain (Shams’ili et al., 2006; Vernino et al., 2004).
Vasculitis of the Nerve and Muscle
Clinical Findings
Vasculitis of the nerve and muscle usually occurs in older men. It can present as a painful symmetrical or asymmetrical subacute sensorimotor polyneuropathy or, less frequently, mononeuropathy multiplex (Oh, 1997). Electrophysiological findings are compatible with axonal degeneration involving motor and sensory nerves. Erythrocyte sedimentation rate and CSF protein concentration are elevated. Nerve and muscle histology shows intramural and perivascular inflammatory infiltrates composed of CD8+ T cells.
Subacute and Chronic Peripheral Neuropathies
Clinical Findings
A mild peripheral neuropathy is common in patients with cancer, especially in the advanced stages of the disease. The cause is multifactorial and includes metabolic and nutritional deficits and toxicity from chemotherapy. Paraneoplastic sensorimotor neuropathy may develop before or after the diagnosis of cancer. The onset may be subacute or acute, and the course is usually progressive. A relapsing and remitting course suggests chronic inflammatory demyelinating polyneuropathy (CIDP) (Antoine et al., 1999).
Peripheral Neuropathy Associated with Plasma Cell Dyscrasias and B-Cell Lymphoma
Several malignancies of plasma cells and lymphocytes are associated with neuropathy. Included are multiple myeloma, osteosclerotic myeloma, Waldenström macroglobulinemia, and B-cell lymphoma (Ropper and Gorson, 1998). A sensorimotor neuropathy similar to those seen in other advanced cancers may develop in patients with multiple myeloma. If amyloidosis complicates the myeloma, neuropathic symptoms often include autonomic dysfunction and lancinating and burning dysesthesias. In both cases, treatment of the myeloma does not affect the neurological symptoms.
Osteosclerotic myeloma is often associated with a symmetrical distal sensorimotor neuropathy with predominant motor symptoms that resemble CIDP. Some patients with osteosclerotic myeloma and neuropathy develop additional symptoms indicative of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes). A radiological survey demonstrates a solitary or reduced number of osteosclerotic lesions that tend to involve the axial skeleton (truncal and proximal long bones) and usually spare the skull. The neuropathy often improves with irradiation of the bone lesions or with prednisone alone or in combination with melphalan (Dispenzieri et al., 2003; Rotta and Bradley, 1997). Some patients with POEMS syndrome and a rapidly progressive neuropathy improved after peripheral blood stem cell transplant (Kuwabara et al., 2008).
Among patients with Waldenström macroglobulinemia, less than 10% develop a symmetrical demyelinating sensorimotor neuropathy, predominantly involving large sensory fibers, especially those for vibration sense. The IgM paraprotein sometimes reacts with myelin-associated glycoprotein or to gangliosides including GD1b and GM1 (Dimopoulos et al., 2000). The neuropathy may respond to treatment directed at Waldenström macroglobulinemia, plasma exchange, IVIg, rituximab, chlorambucil, cyclophosphamide, or fludarabine (Latov, 2000; Weide et al., 2000).
Castleman disease, or angiofollicular lymph node hyperplasia, is a rare disorder that overlaps with the POEMS syndrome. The multicentric presentation of this disorder is usually associated with plasma cell or mixed pathology variants. Systemic therapy is required, and prognosis is guarded. Castleman disease is frequently characterized by paraneoplastic neuropathies including a painful sensorimotor neuropathy, a chronic relapsing sensorimotor neuropathy, and a predominantly motor neuropathy (Vingerhoets et al., 1995). Additional symptoms that indicate POEMS syndrome are common. The incidence of Castleman disease is increased in patients with human immunodeficiency virus (HIV), usually in association with human herpesvirus 8. This viral association also occurs in 50% of patients without HIV. Specific serum antibodies are absent, and serum levels of interleukin 6 are often elevated (Dispenzieri and Gertz, 2005). Treatment of the neurological complications should be focused on the Castleman disease; improvements with plasma exchange or immunosuppression are reported (Fernandez-Torre et al., 1999; Vingerhoets et al., 1995).
An acute paraneoplastic polyradiculoneuropathy identical to Guillain-Barré syndrome (GBS) likely occurs with a higher frequency in patients with cancer (Vigliani et al., 2004). The neoplasm most commonly implicated is Hodgkin lymphoma, but other cancers including solid tumors are associated. In some patients, GBS may be the first manifestation of tumor recurrence. Treatment is the same as for the non-cancer related form and consists of plasma exchange and IVIg; neither is contraindicated in patients with cancer. Some evidence suggests that patients with cancer-associated GBS have a worse neurological outcome compared to patients with GBS alone (Vigliani et al., 2004).
Peripheral Nerve Hyperexcitability
Clinical Findings
The term peripheral nerve hyperexcitability (PNH) has been adopted to include a disorder previously described with several names (neuromyotonia, undulating myokymia, Isaacs syndrome) and the cramps-fasciculation syndrome (Hart et al., 2002). These terms characterize the spontaneous and continuous muscle fiber activity of peripheral nerve origin that follows voluntary muscle contraction. Symptoms associated with PNH include muscle twitches, cramps, weakness, increased sweating and salivation, pseudomyotonia, and sensory deficits. Approximately a quarter of patients with PNH have central nervous system abnormalities (Morvan syndrome) including changes in mood, irritability, insomnia, delusions, and hallucinations (Liguori et al., 2001). Electromyographic (EMG) studies show fibrillation, fasciculation, and myokymic discharges (doublet, triplet, or multiplet motor unit discharges) in most patients. Patients with the cramps-fasciculation syndrome do not have myokymic EMG discharges.
Tumor Association
About 25% of cases of PNH are paraneoplastic. The most commonly associated tumors are thymoma and lung cancers (Hart et al., 1997).
Immune Responses
Most patients with PNH do not have well-characterized immune responses. About 20% of patients have antibodies previously attributed to VGKC; these antibodies occur similarly in patients with Morvan syndrome and are directed against connectin-associated protein 2 (CASPR2) (Lai et al., 2010; Vincent, 2009). Patients with PNH and thymoma, with or without MG, may also harbor antibodies to AChRs (Hart et al., 2002).
Treatment
Tumor treatment or removal of the antibodies by plasma exchange may result in improvement (Newsom-Davis and Mills, 1993; van den Berg et al., 1999). Case reports exist of responses to IVIg, prednisolone with or without azathioprine, or methotrexate (Alessi et al., 2000; Nakatsuji et al., 2000). Some patients respond to symptomatic treatment with anticonvulsants that block sodium channels (carbamazepine, phenytoin).
Lambert-Eaton Myasthenic Syndrome
Clinical Findings
In patients with LEMS, neurological symptoms usually develop before the tumor diagnosis and gradually progress over weeks or months. Less often, the onset of symptoms is acute. Common features are fatigue, proximal muscle weakness, and paresthesias. More than half of patients have cholinergic dysautonomias that include dry mouth, erectile dysfunction, and blurry vision (O’Suilleabhain et al., 1998). Transitory cranial nerve dysfunction may produce diplopia, ptosis, or dysphagia. Neurological examination shows proximal weakness, occurring in the legs more than the arms, and absent or depressed tendon reflexes, which may potentiate after a brief muscle contraction. Strength may improve after brief exercise, but continued exercise increases weakness. The basis for diagnosis is electrophysiological studies. EMG shows small-amplitude compound muscle action potentials. Slow rates of repetitive nerve stimulation (2-5 Hz) produce a decremental response of greater than 10%. At fast rates of repetitive nerve stimulation (20 Hz or greater) or after maximal voluntary muscle contraction, facilitation occurs and an incremental response of at least 100% is seen.
LEMS can develop in association with other paraneoplastic syndromes such as PCD and PEM (Mason et al., 1997). Recurrence of LEMS after remission often heralds tumor recurrence.
Tumor Association
Approximately 60% of patients with LEMS have an underlying neoplasm, usually SCLC or, rarely, other tumors such as lymphoma. Non-paraneoplastic cases often have slower symptom presentation and are associated with other autoimmune conditions such as thyroiditis and insulin-dependent diabetes mellitus, among others (Wirtz et al., 2002). The presence of LEMS in a patient with SCLC correlates with improved tumor survival (Maddison et al., 1999).
Immune Responses
Patients with LEMS have serum antibodies against P/Q type VGCCs (Motomura et al., 1997). The antibodies interfere with the quantal release of acetylcholine at the presynaptic neuromuscular junction, resulting in failure of neuromuscular transmission. Antibodies to P/Q-type VGCCs also occur in a subset of patients with SCLC and paraneoplastic PCD (with or without LEMS). When LEMS develops in association with PEM, patients often have anti-Hu antibodies.
Treatment
Most patients with cancer improve neurologically with combined treatment of their cancer and therapy for LEMS. The latter includes medication to increase the release of acetylcholine and immunomodulation. The use of 3,4-diaminopyridine results in moderate to marked neurological improvement in 80% of patients (McEvoy et al., 1989). If 3,4-diaminopyridine is not available, a combination of pyridostigmine and guanidine may be beneficial (Oh et al., 1997). Plasma exchange and IVIg are useful for treating severe weakness; strength improves within days or weeks, but the benefits are transient (Bain PG et al., 1996). Long-term immunosuppression with prednisone or azathioprine should be considered if symptoms continue despite the use of 3,4-diaminopyridine.
Myasthenia Gravis
Immune Responses
Antibodies to AChR are found in 80% to 90% of patients with generalized MG and in 70% of those with ocular MG. A small percent of AChR-negative patients have antibodies to MusK. Thymoma-related MG almost invariably associates with AChR antibodies but not anti-MusK antibodies. Patients with thymoma often have additional antibodies against skeletal muscle proteins such as titin and others (Somnier and Engel, 2002).
Dermatomyositis
Clinical Findings
Dermatomyositis and polymyositis are immune-mediated inflammatory diseases of muscle. An association exists between cancer and dermatomyositis in adults, but an association with malignancy is less clear for polymyositis. Cohort studies report conflicting results (Antiochos et al., 2009; Chen et al., 2010). The symptoms of paraneoplastic dermatomyositis are the same as those in patients without cancer. Cutaneous changes include purplish discoloration of the eyelids (heliotrope rash) with edema and erythematous lesions over the knuckles. The presence of necrotic skin ulcerations and pruritus are suggested to indicate an underlying cancer (Mahe et al., 2003). The typical presentation is the subacute onset of proximal muscle weakness. Neck flexors, pharyngeal, and respiratory muscles are commonly involved and may lead to aspiration and hypoventilation. Tendon reflexes and sensation are normal. Serum creatine kinase concentrations are often elevated, although normal concentrations occur even in patients with profound muscle weakness. EMG shows increased spontaneous activity (fibrillations, positive sharp waves, and complex repetitive discharges) and short-duration, low-amplitude polyphasic units on voluntary activation. Muscle histology shows inflammatory infiltrates (CD4+ T cells predominate in dermatomyositis and CD8+ T cells in polymyositis) and muscle necrosis; the presence of perifascicular atrophy is characteristic of dermatomyositis.
Treatment
In some patients, muscle and dermatological symptoms improve coincidently with treatment of the tumor. No studies are available on the efficacy of immunosuppressants in cancer-associated dermatomyositis, but it seems reasonable to use strategies similar to those used in non-paraneoplastic dermatomyositis (steroids, azathioprine, IVIg) (Amato and Barohn, 1997; Dalakas et al., 1993).
Acute Necrotizing Myopathy
Clinical Findings
The characteristic feature of the rare disorder known as acute necrotizing myopathy is acute onset of painful proximal muscle weakness, which rapidly generalizes to involve respiratory and pharyngeal muscles. Serum creatine kinase concentrations are markedly elevated, and electrophysiological studies demonstrate myopathic findings. Muscle histology shows severe necrotic changes with minimal or no inflammatory infiltrates. The possible causes of an acute necrotizing myopathy in patients with cancer include chemotherapy and cytokine-induced rhabdomyolysis (Anderlini et al., 1995).
Paraneoplastic Visual Syndromes
Clinical Findings
Paraneoplastic involvement of the visual system may affect the retina and, less frequently, the uvea and optic nerves (Ko et al., 2008; Thirkill, 2005). Because paraneoplastic visual syndromes are rare, the more important considerations are metastatic infiltration of the optic nerves, toxic effects of chemotherapy or radiation therapy, and severe anemia. The symptoms of paraneoplastic retinopathy are photosensitivity, progressive loss of vision and color perception, central or ring scotomas, and night blindness. Electroretinogram (ERG) records attenuation of photopic and scotopic responses. Funduscopic examination is usually normal or may show nonspecific optic disc pallor and arteriolar narrowing. When one eye is affected, the other becomes symptomatic within days or weeks. Imaging studies and evaluation of the CSF are unrevealing.
Melanoma-associated retinopathy (MAR) affects patients with metastatic cutaneous melanoma (Boeck et al., 1997). Patients typically present with the acute onset of night blindness and shimmering, flickering, or pulsating photopsias. Symptoms often progress to complete visual loss. The ERG typically demonstrates reduction in the b-wave amplitude.
Immune Responses
Serum antibodies that specifically react with retinal proteins are detectable in some patients. The original use of the term cancer-associated retinopathy described patients with antibodies to recoverin, a retinal-specific calcium-binding protein. Antibodies to other retinal antigens such as tubby-like protein 1, enolase, and the photoreceptor-specific nuclear receptor are known. Anti-enolase antibodies are predominantly associated with central cone abnormalities and may also occur without cancer association (Adamus et al., 2004). Patients with MAR typically have antibodies that react with the bipolar cells of the retina. Anti-CV2/CRMP5 antibodies are reported in some patients with PEM, uveitis, and optic neuritis.
Adamus G., Ren G., Weleber R.G. Autoantibodies against retinal proteins in paraneoplastic and autoimmune retinopathy. BMC Ophthalmol. 2004;4:5.
Albert M.L., Darnell J.C., Bender A., et al. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med. 1998;4:1321-1324.
Alessi G., De Reuck J., De Bleecker J., et al. Successful immunoglobulin treatment in a patient with neuromyotonia. Clin Neurol Neurosurg. 2000;102:173-175.
Amato A.A., Barohn R.J. Idiopathic inflammatory myopathies. Neurol Clin. 1997;15:615-648.
Ances B.M., Vitaliani R., Taylor R.A., et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764-1777.
Anderlini P., Buzaid A.C., Legha S.S. Acute rhabdomyolysis after concurrent administration of interleukin-2, interferon-alfa, and chemotherapy for metastatic melanoma. Cancer. 1995;76:678-679.
Antiochos B.B., Brown L.A., Li Z., et al. Malignancy is associated with dermatomyositis but not polymyositis in Northern New England, USA. J Rheumatol. 2009;36:2704-2710.
Antoine J.C., Honnorat J., Camdessanche J.P., et al. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 2001;49:214-221.
Antoine J.C., Mosnier J.F., Absi L., et al. Carcinoma-associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry. 1999;67:7-14.
Bain P.G., Motomura M., Newsom-Davis J., et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47:678-683.
Bataller L., Graus F., Saiz A., et al. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain. 2001;124:437-443.
Bell J., Moran C., Blatt J. Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer. 2008;50:370-371.
Benyahia B., Liblau R., Merle-Béral H., et al. Cell-mediated auto-immunity in paraneoplastic neurologic syndromes with anti-Hu antibodies. Ann Neurol. 1999;45:162-167.
Blaes F., Pike M.G., Lang B. Autoantibodies in childhood opsoclonus-myoclonus syndrome. J Neuroimmunol. 2008;201-202:221-226.
Blaes F., Strittmatter M., Merkelbach S., et al. Intravenous immunoglobulins in the therapy of paraneoplastic neurological disorders. J Neurol. 1999;246:299-303.
Blanc F., Ruppert E., Kleitz C., et al. Acute limbic encephalitis and glutamic acid decarboxylase antibodies: a reality? J Neurol Sci. 2009;287:69-71.
Boeck K., Hofmann S., Klopfer M., et al. Melanoma-associated paraneoplastic retinopathy: case report and review of the literature. Br J Dermatol. 1997;137:457-460.
Camdessanche J.P., Antoine J.C., Honnorat J., et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain. 2002;125:166-175.
Chen Y.J., Wu C.Y., Huang Y.L., et al. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12:R70.
Dalakas M.C., Fujii M., Li M., et al. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345:1870-1876.
Dalakas M.C., Illa I., Dambrosia J.M., et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993-2000.
Dale R.C., Irani S.R., Brilot F., et al. N-methyl-d-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol. 2009;66:704-709.
Dalmau J., Gultekin H.S., Posner J.B. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9:275-284.
Dalmau J., Gultekin S.H., Voltz R., et al. Ma1, a novel neuronal and testis specific protein, is recognized by the serum of patients with paraneoplastic neurologic disorders. Brain. 1999;122:27-39.
Dalmau J., Gleichman A.J., Hughes E.G., et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091-1098.
Dalmau J., Graus F., Rosenblum M.K., et al. Anti-Hu-associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (Baltimore). 1992;71:59-72.
Dalmau J., Graus F., Villarejo A., et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127:1831-1844.
Darnell R.B., Posner J.B. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543-1554.
Dimopoulos M.A., Panayiotidis P., Moulopoulos L.A., et al. Waldenstrom’s macroglobulinemia: clinical features, complications, and management. J Clin Oncol. 2000;18:214-226.
Dispenzieri A., Gertz M.A. Treatment of Castleman’s disease. Curr Treat Options Oncol. 2005;6:255-266.
Dispenzieri A., Kyle R.A., Lacy M.Q., et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101:2496-2506.
Drachman D.B. Myasthenia gravis. N Engl J Med. 1994;330:1797-1810.
Espay A.J., Chen R. Rigidity and spasms from autoimmune encephalomyelopathies: stiff-person syndrome. Muscle Nerve. 2006;34:677-690.
Fernandez-Torre J.L., Polo J.M., Calleja J., et al. Castleman’s disease associated with chronic inflammatory demyelinating polyradiculoneuropathy: a clinical and electrophysiological follow-up study. Clin Neurophysiol. 1999;110:1133-1138.
Florance N.R., Davis R.L., Lam C., et al. Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66:11-18.
Folli F., Solimena M., Cofiell R., et al. Autoantibodies to a 128-kD synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med. 1993;328:546-551.
Fukuda T., Motomura M., Nakao Y., et al. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53:21-28.
Gable M.S., Gavali S., Radner A., et al. Anti-NMDA receptor encephalitis: report of ten cases and comparison with viral encephalitis. Eur J Clin Microbiol Infect Dis. 2009;28:1421-1429.
Graus F., Boronat A., Xifro X., et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology. 2010;74:857-859.
Graus F., Dalmau J., Valldeoriola F., et al. Immunological characterization of a neuronal antibody (anti-Tr) associated with paraneoplastic cerebellar degeneration and Hodgkin’s disease. J Neuroimmunol. 1997;74:55-61.
Graus F., Delattre J.Y., Antoine J.C., et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135-1140.
Graus F., Keime-Guibert F., Rene R., et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138-1148.
Graus F., Lang B., Pozo-Rosich P., et al. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59:764-766.
Gu W., Brodtkorb E., Steinlein O.K. LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol. 2002;52:364-367.
Gultekin S.H., Rosenfeld M.R., Voltz R., et al. Paraneoplastic limbic encephalitis: Neurological symptoms, immunological findings, and tumor association in 50 patients. Brain. 2000;123:1481-1494.
Hart I.K., Maddison P., Newsom-Davis J., et al. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. 2002;125:1887-1895.
Hart I.K., Waters C., Vincent A., et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol. 1997;41:238-246.
Hoffmann L.A., Jarius S., Pellkofer H.L., et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J Neurol Neurosurg Psychiatry. 2008;79:767-773.
Honnorat J., Aguera M., Guillon B., et al. Association of antineural autoantibodies in a patient with paraneoplastic cerebellar syndrome and small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1997;62:425-426.
Honnorat J., Cartalat-Carel S., Ricard D., et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2009;80:412-416.
Hughes E.G., Peng X., Gleichman A.J., et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866-5875.
Hutchinson M., Waters P., McHugh J., et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71:1291-1292.
Iizuka T., Sakai F., Ide T., et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology. 2008;70:504-511.
Iranzo A., Graus F., Clover L., et al. Rapid eye movement sleep behavior disorder and potassium channel antibody-associated limbic encephalitis. Ann Neurol. 2006;59:178-181.
Ishiura H., Matsuda S., Higashihara M., et al. Response of anti-NMDA receptor encephalitis without tumor to immunotherapy including rituximab. Neurology. 2008;71:1921-1923.
Kalachikov S., Evgrafov O., Ross B., et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335-341.
Keime-Guibert F., Graus F., Broet P., et al. Clinical outcome of patients with anti-Hu-associated encephalomyelitis after treatment of the tumor. Neurology. 1999;53:1719-1723.
Keltner J.L., Thirkill C.E., Yip P.T. Clinical and immunologic characteristics of melanoma-associated retinopathy syndrome: eleven new cases and a review of 51 previously published cases. J Neuroophthalmol. 2001;21:173-187.
Ko M.W., Dalmau J., Galetta S.L. Neuro-ophthalmologic manifestations of paraneoplastic syndromes. J Neuroophthalmol. 2008;28:58-68.
Kuwabara S., Misawa S., Kanai K., et al. Neurologic improvement after peripheral blood stem cell transplantation in POEMS syndrome. Neurology. 2008;71:1691-1695.
Lai M., Hughes E.G., Peng X., et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424-434.
Lai M., Huijbers M.G., Lancaster E., et al. The epilepsy related protein, Lgi1, is the antigen of limbic encephalitis previously attributed to potassium channels. Lancet Neurol. 2010. in press
Lancaster E., Lai M., Peng X., et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67-76.
Latov N. Prognosis of neuropathy with monoclonal gammopathy. Muscle Nerve. 2000;23:150-152.
Levin M.I., Mozaffar T., Al-Lozi M.T., et al. Paraneoplastic necrotizing myopathy: clinical and pathological features. Neurology. 1998;50:764-767.
Liguori R., Vincent A., Clover L., et al. Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001;124:2417-2426.
Luque F.A., Furneaux H.M., Ferziger R., et al. Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol. 1991;29:241-251.
Maddison P., Newsom-Davis J., Mills K.R., et al. Favourable prognosis in Lambert-Eaton myasthenic syndrome and small-cell lung carcinoma. Lancet. 1999;353:117-118.
Mahe E., Descamps V., Burnouf M., et al. A helpful clinical sign predictive of cancer in adult dermatomyositis: cutaneous necrosis. Arch Dermatol. 2003;139:539.
Malter M.P., Helmstaedter C., Urbach H., et al. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol. 2010;67:470-478.
Mason W.P., Graus F., Lang B., et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120:1279-1300.
Mathew R.M., Vandenberghe R., Garcia-Merino A., et al. Orchiectomy for suspected microscopic tumor in patients with anti-Ma2-associated encephalitis. Neurology. 2007;68:900-905.
McEvoy K.M., Windebank A.J., Daube J.R., et al. 3,4-Diaminopyridine in the treatment of the Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567-1571.
McKeon A., Apiwattanakul M., Lachance D.H., et al. Positron emission tomography-computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67:322-329.
Molinuevo J.L., Graus F., Rene R., et al. Utility of anti-Hu antibodies in the diagnosis of paraneoplastic sensory neuropathy. Ann Neurol. 1998;44:976-980.
Motomura M., Lang B., Johnston I., et al. Incidence of serum anti-P/O-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1997;147:35-42.
Nakatsuji Y., Kaido M., Sugai F., et al. Isaacs’ syndrome successfully treated by immunoadsorption plasmapheresis. Acta Neurol Scand. 2000;102:271-273.
Newsom-Davis J., Mills K.R. Immunological associations of acquired neuromyotonia (Isaac’s syndrome). Report of five cases and literature review. Brain. 1993;116:453-469.
O’Suilleabhain P., Low P.A., Lennon V.A. Autonomic dysfunction in the Lambert-Eaton myasthenic syndrome: serologic and clinical correlates. Neurology. 1998;50:88-93.
Oh S.J. Paraneoplastic vasculitis of the peripheral nervous system. Neurol Clin. 1997;15:849-863.
Oh S.J., Gurtekin Y., Dropcho E.J., et al. Anti-Hu antibody neuropathy: a clinical, electrophysiological, and pathological study. Clin Neurophysiol. 2005;116:28-34.
Oh S.J., Kim D.S., Head T.C., et al. Low-dose guanidine and pyridostigmine: Relatively safe and effective long-term symptomatic therapy in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1997;20:1146-1152.
Peterson K., Rosenblum M.K., Kotanides H., et al. Paraneoplastic cerebellar degeneration I: A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. 1992;42:1931-1937.
Pozo-Rosich P., Clover L., Saiz A., et al. Voltage-gated potassium channel antibodies in limbic encephalitis. Ann Neurol. 2003;54:530-533.
Pranzatelli M.R., Tate E.D., Dukart W.S., et al. Sleep disturbance and rage attacks in opsoclonus-myoclonus syndrome: response to trazodone. J Pediatr. 2005;147:372-378.
Pranzatelli M.R., Travelstead A.L., Tate E.D., et al. CSF B-cell expansion in opsoclonus-myoclonus syndrome: a biomarker of disease activity. Mov Disord. 2004;19:770-777.
Psimaras D., Carpentier A.F., Rossi C. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81:42-45.
Raju R., Foote J., Banga J.P., et al. Analysis of GAD65 autoantibodies in stiff-person syndrome patients. J Immunol. 2005;175:7755-7762.
Rees J.H., Hain S.F., Johnson M.R., et al. The role of [18F]fluoro-2-deoxyglucose-PET scanning in the diagnosis of paraneoplastic neurological disorders. Brain. 2001;124:2223-2231.
Ropper A.H., Gorson K.C. Neuropathies associated with paraproteinemia. N Engl J Med. 1998;338:1601-1607.
Rotta F.T., Bradley W.G. Marked improvement of severe polyneuropathy associated with multifocal osteosclerotic myeloma following surgery, radiation, and chemotherapy. Muscle Nerve. 1997;20:1035-1037.
Russo C., Cohn S.L., Petruzzi M.J., et al. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997;28:284-288.
Sabater L., Xifro X., Saiz A., et al. Analysis of antibodies to neuronal surface antigens in adult opsoclonus-myoclonus. J Neuroimmunol. 2008;196:188-191.
Saiz A., Bruna J., Stourac P., et al. Anti-Hu-associated brainstem encephalitis. J Neurol Neurosurg Psychiatry. 2009;80:404-407.
Saiz A., Dalmau J., Butler M.H., et al. Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1999;66:214-217.
Sampson J.B., Smith S.M., Smith A.G., et al. Paraneoplastic myopathy: response to intravenous immunoglobulin. Neuromuscul Disord. 2007;17:404-408.
Seki M., Suzuki S., Iizuka T., et al. Neurological response to early removal of ovarian teratoma in anti-NMDAR encephalitis. J Neurol Neurosurg Psychiatry. 2008;79:324-326.
Shams’ili S., de Beukelaar J., Gratama J.W., et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol. 2006;253:16-20.
Shams’ili S., Grefkens J., De Leeuw B., et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409-1418.
Shimazaki H., Ando Y., Nakano I., et al. Reversible limbic encephalitis with antibodies against the membranes of neurones of the hippocampus. J Neurol Neurosurg Psychiatry. 2007;78:324-325.
Sillevis S.P., Grefkens J., De Leeuw B., et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745-753.
Silvestre J., Santos L., Batalha V., et al. Paraneoplastic necrotizing myopathy in a woman with breast cancer: a case report. J Med Case Reports. 2009;3:95.
Skeie, G.O., Apostolski, S., Evoli, A., et al., 2010. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol Apr 12, E-pub ahead of print.
Somnier F.E., Engel P.J. The occurrence of anti-titin antibodies and thymomas: a population survey of MG 1970-1999. Neurology. 2002;59:92-98.
Tanaka K., Tanaka M., Inuzuka T., et al. Cytotoxic T lymphocyte-mediated cell death in paraneoplastic sensory neuronopathy with anti-Hu antibody. J Neurol Sci. 1999;163:159-162.
Tate E.D., Allison T.J., Pranzatelli M.R., et al. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J Pediatr Oncol Nurs. 2005;22:8-19.
Thirkill C.E. Cancer-induced, immune-mediated ocular degenerations. Ocul Immunol Inflamm. 2005;13:119-131.
Tüzün E., Dalmau J. Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13:261-271.
van den Berg J.S., Van Engelen B.G., Boerman R.H., et al. Acquired neuromyotonia: superiority of plasma exchange over high-dose intravenous human immunoglobulin. J Neurol. 1999;246:623-625.
Vasconcelos O.M., Dalakas M.C. Stiff-person syndrome. Curr Treat Options Neurol. 2003;5:79-90.
Vedeler C.A., Antoine J.C., Giometto B., et al. Management of paraneoplastic neurological syndromes: report of an EFNS Task Force. Eur J Neurol. 2006;13:682-690.
Vernino S., Low P.A., Fealey R.D., et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847-855.
Vernino S., O’Neill B.P., Marks R.S., et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro-oncol. 2004;6:55-62.
Vernino S., Tuite P., Adler C.H., et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002;51:625-630.
Vigliani M.C., Magistrello M., Polo P., et al. Risk of cancer in patients with Guillain-Barre syndrome (GBS). A population-based study. J Neurol. 2004;251:321-326.
Vincent A. Antibodies to contactin-associated protein 2 (CASPR2) in thymoma and Morvan’s syndrome. Ann Neurol. 2009;66:3.
Vincent A., Buckley C., Schott J.M., et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701-712.
Vingerhoets F., Kuntzer T., Delacretaz F., et al. Chronic relapsing neuropathy associated with Castleman’s disease (angiofollicular lymph node hyperplasia). Eur Neurol. 1995;35:336-340.
Weide R., Heymanns J., Koppler H. The polyneuropathy associated with Waldenstrom’s macroglobulinaemia can be treated effectively with chemotherapy and the anti-CD20 monoclonal antibody rituximab. Br J Haematology. 2000;109:838-841.
Wirtz P.W., Smallegange T.M., Wintzen A.R., et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
Wong-Kisiel L.C., Ji T., Renaud D.L., et al. Response to immunotherapy in a 20-month-old boy with anti-NMDA receptor encephalitis. Neurology. 2010;74:1550-1551.
Younes-Mhenni S., Janier M.F., Cinotti L., et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain. 2004;127:2331-2338.