Chapter 52D Cancer and the Nervous System
Management of Primary Nervous System Tumors in Adults
Established Treatment Strategies
Radiation Therapy
Chemotherapy
Standard Cytotoxic Chemotherapy
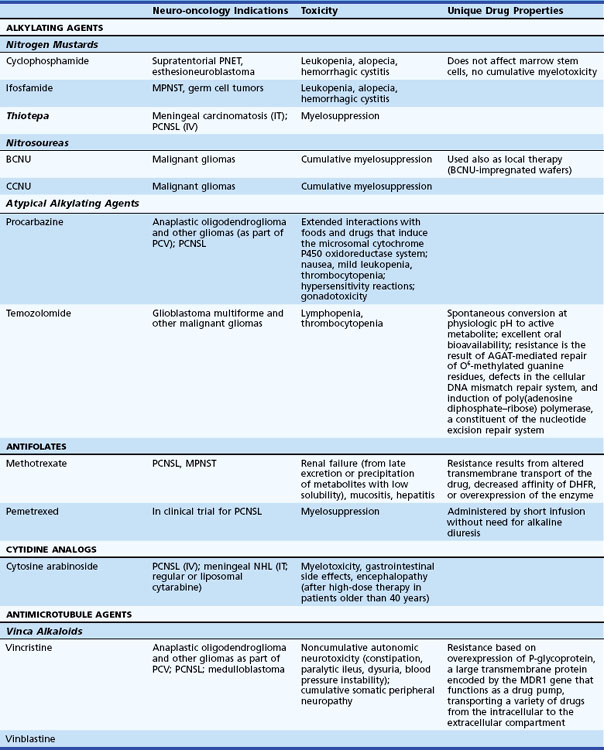
Chemotherapy is provided to most patients with malignant brain tumors. Less commonly treated are nonresected low-grade but symptomatic tumors prior to or following radiation therapy. Chemotherapy is becoming increasingly important for patients with brain lymphoma or anaplastic oligodendroglial tumors (Table 52D.1).
Table 52D.1 Cytotoxic Chemotherapeutic Agents, Applications in Neuro-oncology, Associated Toxicities, and Unique Properties
Myeloablative doses of chemotherapy followed by autologous peripheral-blood stem cell transplantation have failed to produce higher response rates in malignant gliomas when compared with conventional adjuvant chemotherapy (Finlay et al., 1996). Moreover, this approach is associated with significant treatment-related morbidity and mortality and thus has not found widespread use. Results of high-dose chemotherapy with peripheral-blood stem cell rescue in patients with chemosensitive brain tumors like anaplastic oligodendroglioma or primary central nervous system lymphoma (PCNSL) are more promising (Abrey et al., 2006).
Delivery Strategies
The BBB is the major anatomical obstacle for chemotherapy of primary brain tumors. It is composed of the endothelial cell layer of cerebral capillaries sealed by intercellular tight junctions, the vascular basal membrane, and astrocytic foot processes. Few studies have measured brain concentrations of systemically administered agents, but delivery strategies developed to circumvent the barrier include the following: (1) intrathecal administration of methotrexate, thiotepa, or cytosine-arabinoside for leptomeningeal metastases, (2) intracarotid infusion of hypertonic solutions (25% mannitol or 15% glycerol) to produce reversible opening of the BBB, and (3) biodegradable polymers impregnated with BCNU. Intracarotid infusion of hypertonic solutions, selectively used in specialized centers, produces 1 to 2 hours of barrier lysis during which hydrophilic chemotherapeutic agents such as methotrexate or cyclophosphamide are provided. The technology obligates general anesthesia and serial angiographic procedures and is associated with toxicity, including seizures and transient encephalopathy. Biodegradable polymers impregnated with BCNU increase local drug concentration without notable systemic toxicity. Dime-sized wafers of p-carboxyphenoxy (polybis) propane and sebacic acid release the chemotherapeutic agent over 7 to 10 days into tumor surrounding the resection site. The polymer-based delivery strategy is associated with median survival improvements of 2 months in patients with malignant glioma (Westphal et al., 2003). Complications include infection, wound healing impairment, brain necrosis, and cerebrospinal fluid (CSF) leak.
New Treatment Strategies
Several methods are under investigation to reduce resistance to alkylating agents. O6-benzylguanine is a potent inhibitor of AGAT that has been co-administered with alkylating agents (Quinn et al., 2005). Inhibitors of poly(adenosine diphosphate–ribose) polymerase (PARP), cell signaling enzymes implicated in cellular responses to DNA injury provoked by genotoxic stress, potentiate the effect of various chemotherapeutic agents, including alkylating compounds and inhibitors of topoisomerase 1.
Various compounds interfering with pathways regulating cell growth have been developed for numerous cancer types. Cell growth control can be attacked at different levels: growth factors, growth factor receptors, intracellular signal transducers, nuclear transcription factors, and cell-cycle control proteins. Various strategies are available to interfere with proteins or the transcription/translation of their encoding genes at each level. Modified peptides or peptidomimetics such as imatinib are molecules designed to bind to the active sites of proteins, such as the tyrosine kinase domain of growth factor receptors. Imatinib, a synthetic inhibitor of the tyrosine kinase receptors, abl and c-kit, has been of value in the therapy of chronic myelogenous leukemia and gastrointestinal stromal tumors. This has inspired the use of similar agents to target analogous brain tumor pathways. Antisense oligonucleotides injected into tumors hybridize with transcripts of growth control genes and inhibit their translation. Ribozymes degrade transcripts with high specificity. Monoclonal antibodies directly target growth-control proteins. Gene therapy may restore the function of mutated cell-cycle control proteins. A summary of new treatment strategies can be found in Box 52D.1.
Box 52D.1 Targeted Therapies for Intracranial Neoplasm*
Small-Molecule Inhibitors of Intracellular Signal Transduction Pathways
Epidermal growth factor receptor (EGFR) is an attractive target because it is commonly overexpressed or mutated. However, monotherapy with agents targeting this receptor (gefitinib, erlotinib, tyrphostin) has been disappointing (Rich et al., 2004). Molecular predictors of the rare responses have been identified (Mellinghoff et al., 2005). Inhibition of platelet-derived growth factor receptor (PDGFR) with imatinib has also been unsuccessful. This has led to the development of small-molecule inhibitors with a broader spectrum or “dual” inhibitors. AEE788 and vatalanib interfere with both EGFR and vascular endothelial growth factor receptor (VEGFR) signal transduction. Lapatinib inhibits EGFR and ErbB2, two members of the ErbB family of transmembrane tyrosine kinase receptors. These compounds are currently undergoing clinical evaluation, as are combination regimens using GFR and downstream signal transduction inhibitors (e.g., erlotinib, sirolimus).
Histone deacetylase (HDAC) inhibitors interfere with transcription. HDAC induces hyperacetylation of histones, resulting in chromatin relaxation and transcriptional activation. The anticancer properties of various HDACs have been recognized and are likely the complex result of activation of differentiation programs, cell-cycle inhibition, and induction of apoptosis in cancer cells (Johnstone, 2002). Compounds such as phenylacetate, phenylbutyrate, or valproic acid display HDAC-inhibiting properties but are unlikely to play a role as brain cancer therapeutics. Suberoylanilide hydroxamic acid (SAHA) and the fungal tetrapeptide, depsipeptide, are currently undergoing clinical evaluation in malignant gliomas.
Inhibition of angiogenesis or cell invasion represents another promising approach to brain tumor therapy. Gliomas larger than a few millimeters stimulate new blood vessel formation. This induction is affected by promoters including VEGF (hypoxia-inducible endothelial cell mitogen, vascular permeability factor), basic fibroblast growth factor (bFGF), platelet-derived growth factor, EGF, transforming growth factor (TGF), and tenascin. Endogenous inhibitors of angiogenesis include angiostatin, endostatin, thrombospondin, and heparin. Kinase insert domain receptor (KDR) and FMS-related tyrosine kinase 1 are receptors for VEGF. Bevacizumab, a humanized monoclonal antibody with murine complementarity-determining regions binding VEGF, is now approved for use in patients with relapsed glioblastoma. Several small-molecule inhibitors of VEGFR are at various stages of development. Sunitinib is already in clinical use for advanced renal cell cancer. Early experience with cediranib in glioblastoma has been promising, and a phase III study in patients with newly diagnosed disease was completed recently (Batchelor et al., 2007). Other potential therapies targeted to endothelial cells include thalidomide, interleukin (IL)-12, cyclooxygenase II inhibitors, and cilengitide, a cyclic pentapeptide inducing apoptosis of growing endothelial cells through inhibition of their αVβ3 integrin interaction with the matrix proteins, vitronectin and tenascin.
Gene therapy of brain tumors encompasses a wide spectrum of various strategies. A comprehensive review of these approaches goes beyond the scope of this chapter, and the interested reader is referred to excellent review articles (Lam and Breakefield, 2001). Viral vectors create localized inflammation while expressing transgenes that activate cytokines and chemotherapies. Transfection efficiency depends on the agent and the mode of introduction. Cells are killed not only by transfection but by the cellular reaction that damages adjacent tumor cells: the “bystander effect.” The delivery of a therapeutic gene can be enhanced by improving the vector or delivering it through infusional clysis. Ligands or antibodies targeted at receptors expressed on tumor cells (EGFR, transferrin receptor, integrin receptor) can be incorporated into the capsid of adenoviral vectors. Tumor-specific expression systems make use of the human telomerase reverse transcriptase (hTERT) promoter; hTERT is the catalytic subunit of the telomerase ribonucleoprotein and is expressed in glioma cells but not in normal glia cells. Tumor selectivity can also be accomplished by using replication-conditional viral vectors, retroviruses, or placement of genes essential to virus replication under the control of promoters that are selectively active in gliomas (e.g., nestin promoter). Currently used vector systems are either replication-defective or replication-conditional and include recombinant HSV, Ad, retrovirus, and hybrid vectors. Gene therapy delivery involves stereotactic injection into the tumor or intraoperative insertion into the wall of the resection cavity, convection-enhanced delivery, or intraarterial or intraventricular application. Nonviral strategies have made use of naked DNA, polycationic polymers, and liposomes.
Therapies based on immune-mediated strategies aim to increase immune responses to the tumor. Whether primary brain tumors suppress immune reaction, are poorly recognized by the immune system, or are protected by the immunosuppressive effects of concurrent glucocorticoid administration is uncertain. Tumor vaccination makes use of immunogenic peptides, attenuated autologous tumor cells, or dendritic cells loaded with tumor antigens. These tumor antigens create an immune reaction enhanced by irradiation, transfection with cytokine genes, or transfection with major histocompatibility complex (MHC) class II genes. The antigen can be presented in subcutaneous tissues, after which cytotoxic T cells infiltrate the site of injection as well as the brain. A “one-fits-all” immunization strategy targeting a somatic mutant of EGFR (EGFRvIII) has been tested in small phase II studies, with promising results (Sampson et al., 2009).
There are various mechanisms by which gliomas evade recognition by the immune system. Potential mediators include inhibitory cytokines (TGF-β, prostaglandin E, IL-10) and defective cytokine receptors on tumor-infiltrating T lymphocytes. Strategies rendering glioblastoma cells immunogenic have included transfection with antisense TGF-β or decorin, a TGF-β-binding and TGF-β-inhibiting proteoglycan. Cytokines can be linked to bacterial toxins as genetically engineered fusion proteins that enter the tumor cell via binding to selectively expressed receptors. A phase III clinical trial of cintredekin besudotox (IL-13 linked to Pseudomonas exotoxin) administered intracerebrally through convection-enhanced delivery failed to demonstrate a survival benefit (Debinski et al., 1998).
Oncolytic viruses are modified viruses that preferentially replicate in and destroy cancer cells. For example, ONYX-015 is a replication-competent E1B-attenuated adenovirus. E1B is a viral protein that binds and inactivates p53, a prerequisite for the virus’s ability to replicate in its host cell. Ad lacking E1B can only replicate in TP53-deficient cells. Loss of TP53 function is an early event in the pathogenesis of gliomas and thus renders them susceptible to lytic infection with this attenuated virus (Chiocca et al., 2004). Clinical trials have proven safe, but delivery systems have thus far proven insufficient.
New delivery strategies are designed to circumvent the BBB to treat malignant gliomas. Intraoperative injection of resection margins with various therapeutic agents (viral vectors, oncolytic viruses) does not depend on BBB permeability but is highly inefficient. Tissue penetration can be improved using convection-enhanced delivery. This technique requires intraoperative or stereotactic placement of infusion catheters in the wall of the resection cavity. Using microinfusion pumps, therapeutic agents are provided postoperatively over up to 96 hours. Exposure may be even further enhanced by packaging the therapeutic compound (cytotoxic chemotherapy agent, vector, antibody, etc.) into microspheres from which it is released over a modifiable period of time (Saltzman and Olbricht, 2002). Neuroprogenitor cells may deliver vectors or therapeutic genes to tumors. Animal experiments have shown that systemically administered neural stem cells home to brain tumors. Progenitor cells have been found within experimental brain tumors following injection into the contralateral cerebral hemisphere—an observation that may indicate the stem cells’ ability to track down migratory brain tumor cells (Aboody et al., 2000).
Management of Specific Brain Tumors
Neuroepithelial Tumors
Astrocytic Tumors
Noninfiltrative Tumors
Pilocytic Astrocytoma
Comprising 85% of infratentorial astrocytomas, most pilocytic astrocytomas are benign tumors located in the cerebellum and occur in the first and second decade (Burkhard et al., 2003). The remainder grow in the hypothalamus, the walls of the third ventricle, the optic pathway, and the brainstem. The tumor likely emerges from true astrocytes or subependymal precursors. Pilocytic astrocytoma, usually of the optic nerve, is the most common central nervous system (CNS) tumor associated with neurofibromatosis type 1. The tumor diagnosis is heralded by symptomatic obstructive hydrocephalus, headache, or hypothalamic-pituitary dysfunction. Posterior fossa signs include neck stiffness, head tilt, and incoordination. The masses enhance with gadolinium and appear in proximity to the ventricle or subarachnoid space. Cysts, focal hemorrhage, and calcification are described. Much of the tumor contains benign features: bipolar (piloid) cells with Rosenthal fibers in addition to microcysts surrounded by protoplasmic astrocytes and eosinophilic granular bodies. Three-quarters of patients receive surgical resection. For the unresectable case exhibiting progressive growth or symptoms refractory to treatment, involved field radiation therapy is given with a margin of 0.5 cm. Chemotherapy can be used prior to irradiation or for tumor progression. The 25-year survival rate is between 50% and 94% following surgical resection. Malignant transformation of pilocytic astrocytoma is highly unusual.
Pleomorphic Xanthoastrocytoma
Pleomorphic xanthoastrocytoma (PXA) is a rare superficial cortical glioma (Fig. 52D.1, A). Two-thirds of cases are diagnosed before age 25. Seizures anticipate diagnosis by 3 years. The masses are located in the temporal lobes and extend into the leptomeninges and Virchow-Robin spaces. PXAs are well demarcated from surrounding tissue and may contain cysts. The solid portion of the tumor usually enhances markedly on MRI with gadolinium. On unenhanced T1-weighted images, it is hypo- to isointense. The pleomorphic cells vary in size and shape and include single and multinucleated giant cells. The large cells accumulate lipids, hence the term xanthoastrocytoma. There is some uncertainty as to the true aggressive potential of the masses; mitoses and necrosis are seen, and malignant forms have been identified. Cells of origin are probably subpial astrocytes. Complete resection is feasible in most patients, and radiation or chemotherapy is provided for aggressive or recurrent tumors. Survival is better than 80% after 5 years and 70% after 10 years, with well-resected lesions faring best (Giannini et al., 1999). Anaplastic transformation occurs in 15% to 20% of patients, for whom the natural history may be indistinguishable from glioblastoma.
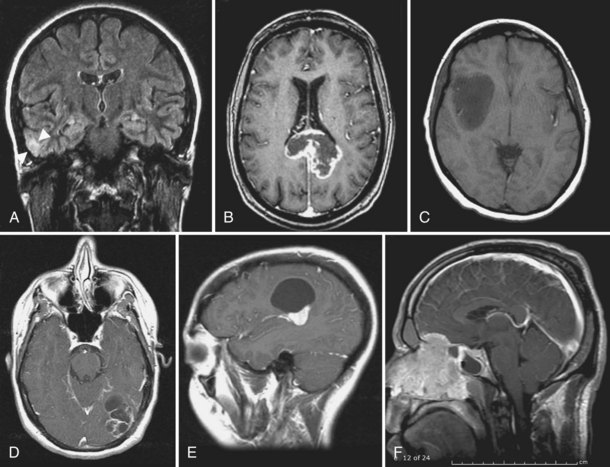
Subependymal Giant Cell Astrocytoma
Subependymal giant cell astrocytoma (SEGA) occurs in patients with tuberous sclerosis and may be unique to that disease. Shepherd, in a detailed search of the Mayo Clinic tissue registry, found no SEGA patient without tuberous sclerosis, although SEGA is only reported in 6.1% of patients afflicted with this phakomatosis (Shepherd et al., 1991). Hydrocephalus emerges before the third decade as tumors in the wall of the lateral ventricles or in the intraventricular foramen clog CSF outflow. Epilepsy of longstanding duration may change pattern. The well-circumscribed MRI masses rarely have hemorrhage or calcification but may show enhancement. Tumor cells stain with glial (GFAP) and neuronal (synaptophysin and neuron-specific enolase) markers and may be mixed with large cells which resemble gemistocytic astrocytes, elongated tumor cells, and giant multinucleated pyramidal cells. Surgery is curative and required when CSF flow is obstructed. Rapamycin at standard immunosuppressive doses can induce regression of SEGA (Franz et al., 2006).
Neuroepithelial Tumors of Unknown Origin: Chordoid Glioma of the Third Ventricle
Chordoid gliomas occur in the third ventricle of adult women (Brat et al., 1998). These tumors arise in the anterior third-ventricular surface and commonly are diagnosed when CSF flow is obstructed or visual field deficits or signs of pituitary dysfunction arise. They commonly fill the third ventricle and obstruct the lateral ventricles. On MRI, chordoid glioma is characterized by homogenous enhancement. Microscopically, tumors are composed of GFAP and vimentin-positive cells in a mucinous matrix. Some reports suggest that chordoid gliomas of the third ventricle arise from the lamina terminalis and perhaps from tanycytes in this region. Surgical resection is the treatment of choice, but complete removal is often impossible owing to the location of the tumor and adherence to vital structures. This procedure has been aided by endoscopic techniques or intraoperative MRI scanning. Adjuvant radiotherapy is used for tumors that progress after surgery. The excellent prognosis reflects the extent of surgical resection.
Infiltrative Low-Grade Astrocytomas
A tumor of young adulthood, symptoms of infiltrative low-grade astrocytoma appear either during the first or between the third and fourth decade. Representing fewer than 5% of brain tumors, there is a slight male predominance. Seizures herald the appearance of a lesion, and a careful history often discloses symptoms of many years or decades. Commonly afflicted are the frontal and temporal lobe of the cerebral hemispheres, where MRI studies reveal poorly demarcated hypointense T1- and hyperintense T2-weighted lesions that do not enhance after gadolinium administration. Surgically resected material contains one of three main subtypes: fibrillary, protoplasmic, and gemistocytic astrocytoma (in order of frequency). The cells appear cytologically benign but infiltrate surrounding brain tissue. The gemistocytic subtype is prone to anaplastic progression, usually after 4 to 5 years. The goal of surgery is tumor removal to the largest possible extent without risking the loss of neurological function. Surgery establishes a diagnosis. Symptoms are alleviated, including those associated with mass effect, hydrocephalus, hemorrhage, cyst formation, or seizure activity. Surgery reduces the cell pool at risk of malignant degeneration and removes potentially more aggressive foci within radiographically benign appearing tumors. Asymptomatic nonresectable diffuse astrocytomas are followed with quarterly and then 6-monthly MRI scans. After surgery, radiation or chemotherapy does not prolong the recurrence-free interval (7 years) or influence the expected survival (median 12 years) (Karim et al., 1996; Van den Bent et al., 2005). These treatments are reserved for patients with progressive symptoms or tumor expansion, uncontrolled seizures, or steroid dependence (Karim et al., 2002; Knisely et al., 1997). The radiation field includes the area of radiographically identifiable tumor (fluid-attenuated inversion recovery [FLAIR] or T2 margin) and an additional margin of 1 to 2 cm. Temozolomide has become the most widely used chemotherapeutic agent. Off-label use in low-grade neoplasms follows treatment protocols established for malignant gliomas. Favorable outcomes follow gross total resection of tumors in young patients. Unfavorable prognostic factors for survival are age older than 40 years, a tumor diameter exceeding 6 cm, tumor crossing the midline, and presence of neurological deficit before surgery (Piepmeier et al., 1996; Pignatti et al., 2002). Most patients suffer anaplastic tumor transformation.
High-Grade Astrocytomas
Anaplastic Astrocytoma
Anaplastic astrocytomas account for 5% of all primary brain tumors. Clinical presentation is similar to low-grade infiltrative gliomas, although neurological syndromes tend to evolve faster. MRI typically demonstrates heterogeneously enhancing lesions. Nonenhancing lesions are rarely anaplastic, but the older the patient, the less reliable this feature is (Barker et al., 1997). Diffusion-weighted imaging can identify areas of increased cellularity within low-grade neoplasms and thus may be useful in identifying foci of early anaplastic transformation. After tumor resection, adjuvant therapy is required. A recent study suggests that either irradiation or chemotherapy can be used at initial diagnosis, with similar outcome when the other modality is applied for disease progression (Wick et al., 2009). With this approach, median time to treatment failure is 42 months. Prognostically favorable are methylation of the methylguanine-methyltransferase (MGMT) promoter and somatic mutations in the isocitrate dehydrogenase gene (IDH1). Carmustine-impregnated wafers are available for patients with newly diagnosed and relapsed disease (Brem et al., 1995; Westphal et al., 2003). Based on a population-based registry, relative survival rate at 2 years is 42% and at 5 years, 27% (CBTRUS, 2010).
Glioblastoma Multiforme
Glioblastoma multiforme (GBM) is the most common neuroepithelial tumor (50%) and the second most common primary nervous system tumor. Approximately 10,000 cases are diagnosed each year. Common clinical presentations are headache, seizures, or rapid progression of a focal neurological deficit. The deep-seated infiltrative masses can also cause cognitive difficulties or personality change. MRI identifies a single rim-enhancing lesion with central necrosis surrounded by vasogenic edema (see Fig. 52D.1, B). “Multifocal” disease is characterized by the occurrence of more than one enhancing lesion—a misnomer, as GBM by definition is a diffuse or multifocal disease. FLAIR sequences reveal areas of nonenhancing brain infiltration. Prior to evolution of the typical rim-enhancing appearance, diffusion-weighted images are useful in the diagnosis of these tumors. Gross total resection is recommended whenever feasible to reduce mass effect, improve quality of life, and reduce the residual malignant cell pool to be targeted by adjuvant therapies and extend survival. Biopsy for histological confirmation is offered to older adult patients with lesions in proximity to sensitive cortical areas or deep-seated locations. Radiation therapy doubles the median survival of patients with GBM (Walker et al., 1980). The treatment consists of fractionated external beam irradiation to a total dose of 55 to 60 Gy. Standard therapy combines irradiation with concomitant and adjuvant temozolomide. Given continuously at low dose (75 mg/m2/day) during the 6 weeks of radiotherapy then followed by six 4-week cycles (150-200 mg/m2 on days 1-5), median overall survival is increased from 12.1 to 14.6 months, with acceptable toxicity (Stupp et al., 2005). The efficacy of temozolomide is particularly pronounced in patients whose tumors lack expression of MGMT (Hegi et al., 2005). Biodegradable polymers impregnated with carmustine are available for intracavitary application after tumor removal (Brem et al., 1995). At first tumor recurrence, patients are treated with bevacizumab or enrolled in a clinical trial. Bevacizumab was approved for this indication based on phase II trial data demonstrating a response rate of 28% to 35% and a median duration of response of about 4 months (Friedman et al., 2009; Kreisl et al., 2009). Bevacizumab is frequently combined with irinotecan or other cytotoxic agents, although evidence for superior benefit of this approach remains to be shown. Patients who have failed standard therapies and are ineligible for a clinical trial are provided nitrosoureas; platinum compounds (cisplatin, carboplatin); combination therapy with ifosfamide, carboplatin, and etoposide; and CPT-11, among others. Small-molecule inhibitors of EGFR and its signal transduction pathways may benefit a molecularly defined subset of patients (expression of the somatic mutant, EGFRvIII, and the tumor suppressor gene product, PTEN) (Mellinghoff et al., 2005). Patients with small, radiographically distinct areas of residual or recurrent tumor can be considered for stereotactic treatment, but this treatment fails for the same reason other localized therapies cannot contain these tumors—their diffusely infiltrative nature. The adverse effects of this aggressive approach include seizures, transient focal neurological deficits, and radiation-induced necrosis developing within 3 to 18 months (Shrieve et al., 1999). The prognosis of patients reflects age at diagnosis, performance status prior to treatment, MGMT promoter methylation status, and somatic IDH1 mutation. Median survival of glioblastoma remains less than 1 year in population-based statistics (CBTRUS, 2010). Concomitant radiochemotherapy results in a probability of survival of 20% at 2 years and 10% at 5 years (Stupp et al., 2009).
Oligodendroglial Tumors
Oligodendroglioma and Oligoastrocytoma
Oligodendroglial tumors account for 4% of newly diagnosed neoplasms. The clinical presentation is similar to that of other infiltrative glial tumors. Gross total resection prolongs survival (see Fig. 52D.1, C). Debulking is needed for symptoms of mass effect and hemorrhage. Asymptomatic patients are followed without immediate therapy after surgery. Benefit is achieved with adjuvant radiation therapy and chemotherapy at progression (Karim et al., 2002). Oligodendrogliomas respond to temozolomide or PCV (Hoang-Xuan et al., 2004; Stege et al., 2005). Chemotherapy is commonly administered to low-grade oligodendrogliomas that cannot be resected, are symptomatic, progress radiographically, or contain more aggressive features (contrast enhancement with gadolinium, MR spectroscopy with choline/creatine ratio higher than 3 : 1, elevated MR perfusion, and increased proliferative activity). Objective response is correlated with loss of heterozygosity on chromosome 1p. The median survival time of patients with oligodendrogliomas exceeds 10 years. Key prognostic variables are extent of resection and the provision of adjuvant therapy.
Anaplastic Oligodendroglioma and Anaplastic Oligoastrocytoma
Anaplastic oligodendroglial tumors are outnumbered by their low-grade equivalents (2% of all newly diagnosed neoplasms). A multimodality approach is always required. Resection or, if not possible, biopsy are followed by radiation or chemotherapy. The exquisite chemosensitivity of these tumors was discovered at the end of the 1980s. Three-quarters of patients respond to PCV (Cairncross et al., 1994), a finding since confirmed by others (Van den Bent et al., 1998). Temozolomide therapy is equally effective (Wick et al., 2009). Radiation therapy is given in 30 fractions to a cumulative dose of 55 to 60 Gy. A prospective trial demonstrated equivalence of outcome irrespective of which adjuvant treatment modality was used at initial diagnosis (Wick et al., 2009). Sequential use of radiation and chemotherapy at initial diagnosis may extend progression-free survival but not overall survival, suggesting that one adjuvant treatment strategy can be withheld until disease relapse (Cairncross et al., 2006; Van den Bent et al., 2006). Alternative treatment strategies include BCNU, melphalan, or high-dose myeloablative chemotherapy followed by peripheral stem cell rescue. However, the latter approach is still considered experimental in this disease setting (Abrey et al., 2003). Tumors with chromosome 1p loss or the combined loss of 1p and 19q are more likely to respond to PCV and live longer (Cairncross et al., 1998).
Ependymal Tumors
Ependymoma and Anaplastic Ependymoma
Some 6% of newly diagnosed neuroepithelial neoplasms (2% of all nervous system tumors) are of ependymal differentiation. Ependymomas occur along the neuraxis, usually in proximity to the ventricles or subarachnoid space. Intracranial ependymoma is more common in children (infratentorial more frequent than supratentorial), spinal ependymoma in adults. Ependymoma is the most common intramedullary spinal cord tumor in adults and is typically in proximity to the conus medullaris. In this location, most are of the myxopapillary subtype. Only a small fraction of ependymomas are high grade (see Fig. 52D.1, D). Grading of these tumors remains difficult and morphology based, resulting in inconsistent correlation with outcome. Based on their location, symptoms related to obstruction of CSF pathways and myelopathies are common in patients with ependymal tumors. The vast majority of tumors avidly enhance after gadolinium administration. Complete surgical removal is indicated and may cure the patient with a low-grade ependymoma. Resection of myxopapillary tumors is technically difficult because the masses adhere to nerve roots and have indistinct surgical margins that separate them from normal tissue of the base of the spinal cord. Radiation therapy is beneficial to patients with residual symptomatic tumor after operation, at recurrence, or with aggressive histology (Chang et al., 2002; Schild et al., 1998). Whether or not patients after gross total resection benefit from adjuvant radiotherapy remains a matter of debate. Low-grade tumors, especially those located within the spinal cord, require only involved field radiation. Conformal radiotherapy for supratentorial tumors results in good disease control and excellent neurocognitive outcome (Merchant et al., 2004). Craniospinal irradiation is only needed if leptomeningeal spread occurs. The benefits of prophylactic craniospinal irradiation for high-grade ependymoma are unclear. Local failure tends to herald leptomeningeal spread. Chemotherapy for ependymoma includes carboplatin, PCV, combination chemotherapy with cisplatin, lomustine, and vincristine, or alternating cycles of cisplatin/etoposide and cyclophosphamide/vincristine (Chamberlain, 2002). Chemotherapy is indicated when local therapies have failed.
Subependymoma
Subependymomas are benign tumors in proximity to the ventricles (Ragel et al., 2006). Tumors are commonly detected incidentally as calcified masses, and therapy is only indicated when symptoms ensue. Enhancement on MRI with gadolinium is variable. The fourth ventricle is the most common location, followed by the septum pellucidum and the lateral ventricles. The tumor is well demarcated, and surgical removal is curative. Subtotally resected tumors should be followed with serial MRI studies. Symptomatic residual tumors and those that show progressive growth should be irradiated.
Choroid Plexus Tumors
Choroid Plexus Papilloma and Carcinoma
Choroid plexus papillomas are tumors of childhood. In adults they account for only 0.2% of all intracranial neoplasms. The tumor is located in the fourth ventricle, the cerebellopontine angle, or the lateral ventricles. Patients most commonly present with deficits of the eighth cranial nerve, signs of cerebellar dysfunction, or overproduction of CSF. The masses enhance homogeneously after gadolinium administration. Total resection is accomplished in fewer than half of cases. Frequently the tumor is attached to lower cranial nerves, potentially increasing morbidity of surgical intervention. No role exists for adjuvant treatment modalities at diagnosis, because long-term survival follows even subtotal resection. At progression or recurrence, treatment with conventional external beam radiation or SRS is of benefit. Obstructive hydrocephalus, a frequent complication of choroid plexus papilloma, is relieved by tumor removal, and shunt procedures are only indicated in a few cases (Tacconi et al., 1996). Leptomeningeal seeding is a rare complication of low-grade neoplasms.
Neuronal and Mixed Neuronal-Glial Tumors
Ganglioglioma and Gangliocytoma
Seizures are the most common manifestations of ganglioma and gangliocytoma. These tumors contain two cell populations: gangliocytes and mature glial elements derived from precursor cells that can differentiate into both elements (see Fig. 52D.1, E). Enhancement pattern on MRI is variable. The slow growth of these tumors is reflected by intratumoral calcification. Gross total resection results in survival ranging from 7 to 17 years. Thus, adjuvant irradiation is only provided to tumors that are incompletely resected or those with anaplastic progression. These have a worse prognosis, with an overall survival of 3 years or less (Hakim et al., 1997). Chemotherapy regimens for the rare anaplastic tumors are identical to the ones used for high-grade gliomas.
Central Neurocytoma
Central neurocytoma, a neuronal tumor, occurs in young adults in proximity to the supratentorial ventricular system. Accounting for less than 1% of all primary brain tumors, the tumor arises from the fornix, septum pellucidum, or the walls of the lateral ventricles. Symptoms resulting from CSF flow obstruction commonly precede its discovery. Enhancement on postgadolinium MRI is variable. Intratumoral hemorrhage is rarely observed but can give rise to acute obstructive hydrocephalus. A benign growth, surgical removal is performed by operations through the corpus callosum or cortex, with attendant memory and cognitive problems which are transient. Fewer than 50% of patients experience a cure; for the remainder, there is bleeding or adherence of tumor to adjacent structures. However, long-term tumor control can be achieved with partial resection. Instances of anaplasia or dissemination along CSF pathways are rare. Thus radiation therapy is recommended for tumors with a high proliferative index or growth. For tumors smaller than 3 cm, SRS may be beneficial. Adjuvant chemotherapy benefits a minority of patients treated with a combination of cisplatin, etoposide, and cyclophosphamide. The 5-year survival rate exceeds 80% (Brandes et al., 2000; Schild et al., 1997).
Pineal Parenchymal Tumors
Tumors of pineal origin include pineocytoma, pineal parenchymal tumors of intermediate grade, and pineoblastoma. The more malignant tumors are seen in younger patients; more than 90% of patients with pineoblastoma are younger than age 23, whereas in patients older than age 40, a third of masses are tumors of low or intermediate grade (Chang et al., 1995). Fewer than 150 cases are diagnosed each year. Compression of tectal structures result in upgaze inhibition, dissociation of papillary response, and retractory nystagmus (Parinaud syndrome) or syndromes related to CSF flow obstruction. There is uncertainty regarding the role of surgery. CT- or MR-based stereotactic biopsies are performed in many centers. Tertiary neurosurgical hospitals advocate complete tumor resection. Radiation therapy to the pineal region and the craniospinal axis is provided to patients with high-grade neoplasms, residual tumor after surgery, and subarachnoid dissemination. Chemotherapy is used in the setting of pineoblastoma or parenchymal tumors that are disseminated at onset or at recurrence. Commonly, pre-irradiation chemotherapy reduces tumor size. As is the case with other neuroblastic tumors, platinum-based compounds are used, usually in combination with etoposide alkylating agents (CCNU, PCZ, cyclophosphamide) or vincristine. Long-term responders are reported, but estimates of survival are based on literature reviews or small retrospective institutional series.
Peripheral Neuroblastic Tumors
Esthesioneuroblastoma
Esthesioneuroblastomas arise from the neuroepithelium of the upper nasal cavity and are usually discovered when the nasal passage is obstructed. From there, the tumor invades the neurocranium through the cribriform plate, compresses the frontal lobes of the brain, and may infiltrate the brain parenchyma or subarachnoid spaces (see Fig. 52D.1, F). A standardized therapeutic approach has not been established for this exceedingly rare tumor. A grading (Hyams) and staging system (Kadish) have been established that correlate with outcome. Frequently, gross total resection via a craniofacial approach is followed by adjuvant external beam radiation therapy unless the tumor is excised completely or is low grade (Eich et al., 2001). Because gratifying responses are seen with chemotherapy, patients with disseminated or recurrent disease and those with high-grade tumors are treated with combinations such as cisplatin-etoposide, cyclophosphamide-vincristine-doxorubicin, or alternating cycles of cisplatin-etoposide and cyclophosphamide-vincristine (McElroy et al., 1998). Advocates of radiation and chemotherapy prior to surgery cite decreased tumor burden and facilitation of gross total removal with decreased morbidity as benefits of this approach. Others recommend chemotherapy with cisplatin and etoposide after biopsy, followed by proton-photon irradiation and postradiation chemotherapy (Fitzek et al., 2002). Patients with low-grade tumors have progression-free survival exceeding 10 years, in comparison with less than 3 years for those with more aggressive tumors. Late local recurrences or metastases to cervical lymph nodes are common. When the tumor invades through the dura into the subarachnoid space, intrathecal methotrexate, cytosine-arabinoside, or thiotepa may be used. Radiation is administered to symptomatic or nodular leptomeningeal recurrences.
Embryonal Tumors
Medulloblastoma
Medulloblastomas in adults are rare neoplasms (<1% of all primary brain tumors). The majority of patients present before 40 years of age with cerebellar syndromes, headache, or other signs of increased intracranial pressure. Unlike childhood tumors, these are prone to the cerebellar hemispheres, less commonly in contiguity with the fourth ventricle. As a result, dissemination into the CSF is seen in less than a third of cases. In general, the natural history is less aggressive. Extraneural metastases have been reported in fewer than 150 patients, and then most frequently to bone (Chan et al., 2000). Multidisciplinary management is critical for the successful treatment of these tumors. Resection, craniospinal radiation, and adjuvant chemotherapy results in 5-year survival rates of 65% (Brandes et al., 2003). Obstructive hydrocephalus is a frequent complication of medulloblastoma, requiring at least transient external ventricular drainage. Third ventriculostomy or ventriculoperitoneal shunting is provided to symptomatic patients with occlusion of the fourth ventricle persisting after resection. Resection is followed by evaluation of CSF and lumbar spine for tumor spread, and craniospinal irradiation (posterior fossa, 54 Gy; whole-brain, 40 Gy; and spine, 36 Gy) (Abacioglu et al., 2002). Chemotherapy is considered a standard component of medulloblastoma management in children and likely also benefits adults, especially high-risk patients with significant residual tumor, brainstem infiltration, or leptomeningeal metastasis. Commonly used drugs either prior to or following irradiation include cisplatin or carboplatin, cyclophosphamide, nitrosoureas, and etoposide. The Pediatric Oncology Group pioneered the pre-irradiation use of alternating cycles of cisplatin (90 mg/m2 on day 1, or 20 mg/m2 on days 1-5) and etoposide (100 mg/m2 on days 1-5) with cyclophosphamide (between 450 and 1580 mg/m2 on days 1 and 2) and vincristine (1.5 mg/m2 day 1) (Duffner et al., 1993). The Packer protocol, likewise designed for pediatric medulloblastoma, consists of weekly vincristine (1.5 mg/m2) during craniospinal radiation therapy and for 6 weeks thereafter, followed by 8 cycles (1 cycle every 6-8 weeks) of cisplatin (75 mg/m2) and CCNU (50 mg/m2) on day 1 and vincristine (1.5 mg/m2) given 3 times per cycle (Packer et al., 1994). The latter protocol appears to be considerably more toxic in adults than in children, and dose reductions of all drugs are required in the vast majority of cases (Greenberg et al., 2001). Rational administration of these drugs in adults provides for their use before irradiation because craniospinal irradiation limits bone marrow reserve, and the incidence of leukoencephalopathic complications is higher when chemotherapy follows radiation. In children, benefit from pre-radiation chemotherapy could only be demonstrated in the youngest patients (<3 years). Radiation therapy should certainly not be delayed by extended periods of myelosuppression from chemotherapy. Whether to offer prophylactic chemotherapy along with reduced-dose craniospinal irradiation to low-risk patients is uncertain. Ultimately this issue will be addressed by formal study.
Treatment of tumor recurrence after multimodality therapy is difficult due to the patients’ limited bone marrow reserve. Re-resection or SRS may be offered to selected patients. High-dose chemotherapy followed by stem cell rescue may benefit young patients. New insights into the molecular pathogenesis of medulloblastoma will hopefully soon result in the implementation of new therapeutic agents such as inhibitors of the sonic hedgehog signaling pathway (Rudin et al., 2009). Preliminary data on temozolomide in relapsed medulloblastoma have been published in the pediatric literature.
Tumors of Cranial and Peripheral Nerves
Schwannoma and Neurofibroma
Schwannomas and neurofibromas are benign tumors of the peripheral nerve sheath (PNSTs) and account for roughly 9% of tumors diagnosed each year. Nerve sheath tumors, while commonly found along large nerve trunks, have been described in almost any location in the body. Schwannomas have a predilection for head, neck, and flexor surfaces of the extremities. Intracranial schwannomas commonly arise from the sensory branches of cranial nerves such as the acoustic or trigeminal nerves. Naturally, the clinical spectrum varies widely, and symptoms arise from loss of function of the affected nerve or mass effect on adjacent structures. PNSTs display homogenous contrast enhancement. A rim-enhancing appearance reflects degenerative changes and should not be confused with a malignant neoplasm. Surgical removal is the treatment of choice for PNST and is curative. Schwannomas can be resected, with resolution of symptoms and preservation of function in 90% of cases (Kim et al., 2005; Tiel and Kline, 2004). Spinal PNSTs can adhere to spinal cord or critical extradural structures such as the vertebral artery, rendering complete removal difficult. Transsection of the dorsal rootlets rarely results in paresthesias; segmental loss of motor function results from sacrificing the anterior portion of nerve roots at the level of the upper and lower extremities (C5-T1, L3-S1) but is frequently only transient, suggesting that nerves afflicted by a nerve sheath tumor are not functional (Jinnai and Koyama, 2005).
Small and stable asymptomatic acoustic schwannomas are followed with serial MRI scans on an annual basis. Symptomatic tumors are treated with microsurgical resection or stereotactic radiosurgery. Surgical risks include postoperative CSF leak, facial and trigeminal neuropathy, and deafness. These risks are directly related to tumor size (Kaylie et al., 2001). The use of proton radiosurgery or fractionated stereotactic radiation therapy achieves tumor “control” without disappearance in over 90% of patients, with hearing complications in approximately 20% (Harsh et al., 2002). The risk of hearing loss has been decreased by limiting the dose to 12 Gy (prescribed to the 50% isodose line).
Malignant Peripheral Nerve Sheath Tumor
Treatment failure in half the patients is local within 3 years, but late local recurrences at 25 years have been described; median survival exceeds 2 to 5 years. Tumor size, extent of surgery, and age at diagnosis are predictors of survival (Baehring et al., 2003).
Meningeal Tumors
Meningioma
Meningiomas are the most common intracranial tumors (over one-third of newly diagnosed nervous system tumors; Fig. 52D.2, A). Clinical manifestation and management depend on location, coexistent morbidities, and the patient’s age and health (Chamberlain, 2001). Incidentally found asymptomatic meningiomas lacking mass effect or compression of a venous sinus can be followed conservatively with serial MRI evaluations. Many meningiomas will not grow over years, sparing older adult patients the need for craniotomy (Black et al., 1998). However, when seizures occur, tumors grow, or focal signs emerge, surgical resection can be curative, especially in meningiomas overlying the hemispheres. Technically more challenging are tumors invading dural venous sinuses, tumors arising from the dura overlying the medial portions of the sphenoid bone or other parts of the skull base, meningioma en plaque, posterior fossa meningiomas, and the rare intraventricular meningiomas. Complete surgical removal may not be possible in these situations and may be complicated by infection, CSF leakage, cerebral venous thrombosis, or cranial neuropathies. Less aggressive surgery in association with stereotactic radiation reduces treatment morbidity and may improve progression-free survival (Villavicencio et al., 2001). At recurrence or tumor progression, conventional external beam irradiation or SRS are the major treatment options.
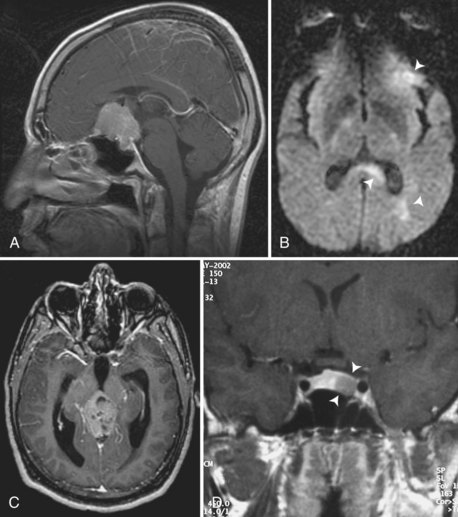
Hemangiopericytoma
Intracranial hemangiopericytoma is a rare meningeal neoplasm with a high rate of local recurrence and predisposition to metastases to bone and liver. Leptomeningeal spread has been described, as have recurrences beyond 5 years of diagnosis. Gross total resection emphasizes removal of surrounding normal dura or brain. Radiation therapy (fractionated to 48-60 Gy) reduces the local recurrence rate and prolongs the progression-free survival (Guthrie et al., 1989). Unfortunately, as with meningioma and schwannoma, tumor shrinkage cannot be expected until years after therapy.
At tumor recurrence, options include surgical resection, conventional external beam radiation, and SRS. Chemotherapy may benefit the patient with systemic metastases or local therapy-refractory disease (Galanis et al., 1998). Most protocols use doxorubicin in combination with cyclophosphamide, ifosfamide, cisplatin, or dacarbazine. Partial responses or stable disease for several months have been observed with these regimens. Anecdotal reports of successful therapy with interferon alfa-2A and small-molecule inhibitors of VEGFR are available. Up to 90% of tumors recur locally or systemically within 9 years of initial manifestation. Extraneural recurrence is predictive of worsened prognosis. Median survival after first recurrence is between 4 and 5 years, but with aggressive management, long-term survival is possible.
Neuraxis Tumors Derived from the Hematopoietic System
Primary Central Nervous System Lymphoma
Non–Acquired Immunodeficiency Syndrome–Related Disease
Approximately 1400 cases of PCNSL are diagnosed each year in the United States. The “chameleon” of modern clinical medicine, the disease presents with a wide variety of neurological syndromes. Common presentations are psychomotor decline and focal neurological symptoms. Diencephalic involvement results in disorders of sodium homeostasis, appetite control, and behavior. Neoplastic infiltrates have a predilection for ependymal or pial surfaces. Masses are homogeneously enhancing on postgadolinium MRI. Tumor cellularity results in their hyperdense appearance on unenhanced CT scans and restriction of water diffusion. Methotrexate-based chemotherapy given in high doses (HDMTX, above 3.5 g/m2) followed by leucovorin rescue has been shown to be the single most effective treatment for PCNSL. Its use, either alone or in combination before radiation therapy, has resulted in response rates of 70% to 95% and survival durations in excess of 3 years (Baehring and Hochberg, 2006). Methotrexate monotherapy is provided in 10- to 14-day intervals until complete radiographic remission is achieved, then followed by monthly consolidation treatments for 1 year (Batchelor et al., 2003). HDMTX seems to be well tolerated by the largely elderly patient population. The treatment produces no alopecia, minimal to modest myelotoxicity, and is compatible with normal cognitive function. Successfully used combination regimens consist of HDMTX, procarbazine, vincristine, cytarabine (Abrey et al., 2000), HDMTX, lomustine, procarbazine, methylprednisolone, and intrathecal chemotherapy with methotrexate and cytarabine (Hoang-Xuan et al., 2003), or HDMTX with cyclophosphamide, ifosfamide, vincristine/vindesine, dexamethasone, cytarabine, and intrathecal chemotherapy (Pels et al., 2003). Intraarterial application of methotrexate preceded by mannitol-induced BBB disruption in combination with intravenous cyclophosphamide and etoposide is effective but requires monthly triple-vessel angiography and is associated with a high frequency of procedure-related, albeit reversible, complications (Doolittle et al., 2000). The administration of intrathecal or intraocular chemotherapy is of uncertain benefit. High-dose intravenous methotrexate results in cytotoxic drug levels within the CSF and in the aqueous and vitreous humor of the eyes. However, high tumor burden within CSF may be associated with methotrexate resistance, and thus intrathecal chemotherapy may postpone the need for craniospinal irradiation.
Methotrexate monotherapy or combination chemotherapy has also been used in combination with external beam irradiation (DeAngelis et al., 2002). There seems to be a trend toward prolonged time to progression at the expense of earlier neurocognitive decline and without an increase in overall survival if radiation is used as part of the initial treatment protocol. Thus, WBRT is now commonly deferred until the time of recurrence, especially in patients older than age 60. Above doses of 35 to 45 Gy, escalation provides no survival benefit (Nelson, et al., 1992). Failures of radiation therapy occur within months. Similarly, surgical resection provides no survival benefit, and thus only biopsy is performed to establish the diagnosis.
In spite of major progress, most patients relapse, and systemic dissemination is encountered in up to 8%. At the time of recurrence, we and others have successfully used methotrexate even in prior recipients of this drug (Plotkin et al., 2004). Anecdotal responses at the time of relapse have also been reported with the use of topotecan, rituximab (a chimeric antibody targeting CD20), temozolomide, PCV, or cytosine-arabinoside/etoposide, as well as intensive chemotherapy supported by autologous or allogeneic peripheral blood stem cell transplantation. WBRT is an option for patients who either fail or are not candidates for salvage chemotherapy.
PCNSL is a highly aggressive tumor (see Fig. 52D.2, B); left untreated, most patients succumb within 6 months. Unfavorable prognostic factors include older age (>60 years), low performance status, multiple brain lesions, evidence of leptomeningeal dissemination, lack of radiographic complete response to treatment, and elevated serum lactate dehydrogenase level (Ferreri et al., 2003). While in small series of patients treated at referral centers, median survival of 3 to 5 years is reported, population-based registries document a survival rate of 47% at 1 year.
Germ Cell Tumors
Germinoma
Germinomas are infiltrative tumors with a tendency to subependymal and leptomeningeal spread (see Fig. 52D.2, C). Traditionally, treatment included surgical resection or biopsy followed by craniospinal irradiation and a boost to the tumor bed. It has been recognized that in patients with localized disease, long-term survival can be achieved by extended focal irradiation including the third and lateral ventricles, the sella, and the pineal region (Matsutani et al., 1997). Thus, craniospinal irradiation is now increasingly reserved for patients with cytopathological or radiographic evidence of leptomeningeal spread. Intracranial germinomas share with their extracranial counterparts a sensitivity to platinum-based chemotherapy, but relapses are common unless combined with radiotherapy (Allen et al., 1987; Balmaceda et al., 1996). The most commonly used protocols include cisplatin, etoposide, bleomycin, or cyclophosphamide. Long-term survivors of radiation are at risk of neuroendocrine morbidities reflecting dysfunction of the hypothalamic-pituitary axis. Pre-irradiation chemotherapy is given to high-risk patients to achieve complete responses and allows for significant reduction of radiation dose (Matsutani et al., 1998; Sawamura et al., 1998).
Nongerminomatous Germ Cell Tumors
Gross total resection prolongs survival of patients with nongerminomatous germ cell tumors but is achieved in less than half the cases. Each of these tumors is approached differently (Schild et al., 1996). Mature or immature teratomas are operated on and then receive involved field radiation therapy with good control. If the teratoma has undergone transformation to malignancy, there is an increased risk of leptomeningeal metastases. This argues strongly for prophylactic whole-brain or craniospinal irradiation. Nonteratomatous, nongerminomatous germ cell tumors are provided adjuvant radiation therapy (above 50 Gy) and platinum-based polychemotherapy. The radiation therapy fields include the craniospinal axis, followed by a “boost” to the tumor bed. These approaches are associated with complications of anterior pituitary dysfunction and intellectual decline—changes that have spurred the use of localized lower doses or hyperfractionated radiation in the setting of polychemotherapy (carboplatin, etoposide; ifosfamide, cisplatin, etoposide; cisplatin, etoposide, cyclophosphamide, bleomycin/carboplatin, etoposide, and bleomycin [Kellie et al., 2004]; carboplatin, cyclophosphamide, etoposide). Prognosis for mature teratomas is excellent. On the other side, only 10% to 15% of patients with embryonal carcinoma, yolk sac tumor (endodermal sinus tumor), or choriocarcinoma survive 3 years after diagnosis. Mixed germ cell tumors and immature teratomas fall between these two extremes. High-dose chemotherapy followed by autologous stem cell transplantation is an option for salvage therapy (Modak et al., 2004).
Tumors of the Sellar Region
Craniopharyngioma
Craniopharyngioma is a tumor derived from Rathke pouch epithelium, which may be present both within the sella turcica and other skull-base locations. Only a few hundred cases are diagnosed each year in adults. Masses may infiltrate the hypothalamus. Patients present with headache, visual field disturbances, or neuroendocrine dysfunction. Microsurgical resection via a subfrontal, perioral, or transsphenoidal approach is the primary treatment in symptomatic cases (Van Effenterre and Boch, 2002). External beam radiation therapy may extend progression-free survival after incomplete resection, although long-term survival can be accomplished with surgery alone (Yasargil et al., 1990). Stereotactic radiation techniques have been successfully used. Intracavitary irradiation (32P or 90Y) (Hasegawa et al., 2004) or instillation of bleomycin are available for solitary cystic craniopharyngioma or cystic components of a mixed tumor following stereotactic aspiration of the cyst contents.
Pituitary Adenoma
Pituitary adenoma is the third most common intracranial neoplasm (12.7% of newly diagnosed tumors). Clinical manifestation depends upon tumor size, hormone secretion by tumor cells, and compression of the normal gland and adjacent structures. Surgical resection is the treatment of choice for pituitary adenomas larger than 1 cm in diameter (see Fig. 52D.2, D) or those with compression of the optic chiasm, erosion of bone, or extension into the walls of the sella. The transsphenoidal approach (Hardy procedure), preferable to the transcranial route (most often right perioral craniotomy), achieves gross total resection in one-third of patients. Improvement in microsurgical technique and imaging has reduced mortality to below 2%.
Radiation therapy is provided as primary treatment to older adult patients, those who are not surgical candidates, and following partial resection (Sasaki et al., 2000). Tumor shrinkage is seen only years after treatment. Adverse effects are infrequent and include necrosis of the adjacent portions of the temporal lobe, hearing loss, optic neuropathy, and radiation-induced sarcomas. Multiple field techniques and radiosurgery have reduced the incidence of these complications. The majority of patients treated with surgery and radiation require replacement of pituitary gland–dependent hormones. Local control rates are higher than 80% for patients with nonsecreting pituitary adenomas. Prognosis for secreting adenomas is slightly worse.
Dopamine agonists such as bromocriptine, cabergoline, quinagolide, and pergolide are effective in micro- as well as macroprolactinomas and lead to reduction in tumor size, improvement of symptoms, and normalization of prolactin levels (Vance, 2003).
Abacioglu U., Uzel O., Sengoz M., et al. Medulloblastoma in adults: treatment results and prognostic factors. Int J Radiat Oncol Biol Phys. 2002;54(3):855-860.
Aboody K.S., Brown A., Rainov N.G., et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97(23):12846-12851.
Abrey L.E., Childs B.H., Paleologos N., et al. High-dose chemotherapy with stem cell rescue as initial therapy for anaplastic oligodendroglioma: long-term follow-up. Neuro Oncol. 2006;8(2):183-188.
Abrey L.E., Childs B.H., Paleologos N., et al. High-dose chemotherapy with stem cell rescue as initial therapy for anaplastic oligodendroglioma. J Neurooncol. 2003;65(2):127-134.
Abrey L.E., Yahalom J., DeAngelis L.M. Treatment for primary CNS lymphoma: the next step. J Clin Oncol. 2000;18(17):3144-3150.
Allen J.C., Kim J.H., Packer R.J. Neoadjuvant chemotherapy for newly diagnosed germ-cell tumors of the central nervous system. J Neurosurg. 1987;67(1):65-70.
Baehring J.M., Betensky R.A., Batchelor T.T. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology. 2003;61(5):696-698.
Baehring J.M., Hochberg F.H. Primary lymphoma of the nervous system. Cancer J. 2006;12(1):1-13.
Balmaceda C., Heller G., Rosenblum M., et al. Chemotherapy without irradiation–a novel approach for newly diagnosed CNS germ cell tumors: results of an international cooperative trial. The First International Central Nervous System Germ Cell Tumor Study. J Clin Oncol. 1996;14(11):2908-2915.
Barker F.G., Chang S.M., Huhn S.L., et al. Age and the risk of anaplasia in magnetic resonance-nonenhancing supratentorial cerebral tumors. Cancer. 1997;80(5):936-941.
Batchelor T., Carson K., O’Neill A., et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07. J Clin Oncol. 2003;21(6):1044-1049.
Batchelor T.T., Sorensen A.G., di Tomaso E., et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83-95.
Black P., Kathiresan S., Chung W. Meningioma surgery in the elderly: a case-control study assessing morbidity and mortality. Acta Neurochir (Wien). 1998;140(10):1013-1016.
Brandes A.A., Amista P., Gardiman M., et al. Chemotherapy in patients with recurrent and progressive central neurocytoma. Cancer. 2000;88(1):169-174.
Brandes A.A., Ermani M., Amista P., et al. The treatment of adults with medulloblastoma: a prospective study. Int J Radiat Oncol Biol Phys. 2003;57(3):755-761.
Brat D.J., Scheithauer B.W., Staugaitis S.M., et al. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. 1998;57(3):283-290.
Brem H., Piantadosi S., Burger P.C., et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-Brain Tumor Treatment Group. Lancet. 1995;345(8956):1008-1012.
Burkhard C., Di Patre P.L., Schuler D., et al. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003;98(6):1170-1174.
Cairncross G., Berkey B., Shaw E., et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24(18):2707-2714.
Cairncross G., Macdonald D., Ludwin S., et al. Chemotherapy for anaplastic oligodendroglioma. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1994;12(10):2013-2021.
Cairncross J.G., Ueki K., Zlatescu M.C., et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
CBTRUS, 2010 CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004-2006. CBTRUS, Central Brain Tumor Registry of the United States 2010.
Chamberlain M.C. Meningiomas. Curr Treat Options Neurol. 2001;3(1):67-76.
Chamberlain M.C. Salvage chemotherapy for recurrent spinal cord ependymomna. Cancer. 2002;95(5):997-1002.
Chan A.W., Tarbell N.J., Black P.M., et al. Adult medulloblastoma: prognostic factors and patterns of relapse. Neurosurgery. 2000;47(3):623-631.
Chang S.M., Lillis-Hearne P.K., Larson D.A., et al. Pineoblastoma in adults. Neurosurgery. 1995;37(3):383-390.
Chang U.K., Choe W.J., Chung S.K., et al. Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol. 2002;57(2):133-139.
Chiocca E.A., Abbed K.M., Tatter S., et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958-966.
DeAngelis L.M., Seiferheld W., Schold S.C., et al. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93-10. J Clin Oncol. 2002;20(24):4643-4648.
Debinski W., Gibo D.M., Obiri N.I., et al. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat Biotechnol. 1998;16(5):449-453.
Doolittle N.D., Miner M.E., Hall W.A., et al. Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors. Cancer. 2000;88(3):637-647.
Duffner P.K., Horowitz M.E., Krischer J.P., et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328(24):1725-1731.
Eich H.T., Staar S., Micke O., et al. Radiotherapy of esthesioneuroblastoma. Int J Radiat Oncol Biol Phys. 2001;49(1):155-160.
Ferreri A.J., Blay J.Y., Reni M., et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21(2):266-272.
Finlay J.L., Goldman S., Wong M.C., et al. Pilot study of high-dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children’s Cancer Group. J Clin Oncol. 1996;14(9):2495-2503.
Fitzek M.M., Thornton A.F., Varvares M., et al. Neuroendocrine tumors of the sinonasal tract. Results of a prospective study incorporating chemotherapy, surgery, and combined proton-photon radiotherapy. Cancer. 2002;94(10):2623-2634.
Franz D.N., Leonard J., Tudor C., et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59(3):490-498.
Friedman H.S., Prados M.D., Wen P.Y., et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733-4740.
Galanis E., Buckner J.C., Scheithauer B.W., et al. Management of recurrent meningeal hemangiopericytoma. Cancer. 1998;82(10):1915-1920.
Giannini C., Scheithauer B.W., Burger P.C., et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85(9):2033-2045.
Greenberg H.S., Chamberlain M.C., Glantz M.J., et al. Adult medulloblastoma: multiagent chemotherapy. Neuro Oncol. 2001;3(1):29-34.
Guthrie B.L., Ebersold M.J., Scheithauer B.W., et al. Meningeal hemangiopericytoma: histopathological features, treatment, and long-term follow-up of 44 cases. Neurosurgery. 1989;25(4):514-522.
Hakim R., Loeffler J.S., Anthony D.C., et al. Gangliogliomas in adults. Cancer. 1997;79(1):127-131.
Harsh G.R., Thornton A.F., Chapman P.H., et al. Proton beam stereotactic radiosurgery of vestibular schwannomas. Int J Radiat Oncol Biol Phys. 2002;54(1):35-44.
Hasegawa T., Kondziolka D., Hadjipanayis C.G., et al. Management of cystic craniopharyngiomas with phosphorus-32 intracavitary irradiation. Neurosurgery. 2004;54(4):813-820.
Hegi M.E., Diserens A.C., Gorlia T., et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
Hoang-Xuan K., Capelle L., Kujas M., et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions. J Clin Oncol. 2004;22(15):3133-3138.
Hoang-Xuan K., Taillandier L., Chinot O., et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21(14):2726-2731.
Jinnai T., Koyama T. Clinical characteristics of spinal nerve sheath tumors: analysis of 149 cases. Neurosurgery. 2005;56(3):510-515.
Johnstone R.W. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287-299.
Karim A.B., Afra D., Cornu P., et al. Randomized trial on the efficacy of radiotherapy for cerebral low-grade glioma in the adult: European Organization for Research and Treatment of Cancer Study 22845 with the Medical Research Council study BRO4: an interim analysis. Int J Radiat Oncol Biol Phys. 2002;52(2):316-324.
Karim A.B., Maat B., Hatlevoll R., et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36(3):549-556.
Kaylie D.M., Gilbert E., Horgan M.A., et al. Acoustic neuroma surgery outcomes. Otol Neurotol. 2001;22(5):686-689.
Kellie S.J., Boyce H., Dunkel I.J., et al. Primary chemotherapy for intracranial nongerminomatous germ cell tumors: results of the second international CNS germ cell study group protocol. J Clin Oncol. 2004;22(5):846-853.
Kim D.H., Murovic J.A., Tiel R.L., et al. A series of 397 peripheral neural sheath tumors: 30-year experience at Louisiana State University Health Sciences Center. J Neurosurg. 2005;102(2):246-255.
Knisely J.P., Haffty B.G., Christopher S.R. Early vs. delayed radiotherapy in a small cohort of patients with supratentorial low grade glioma. J Neurooncol. 1997;34(1):23-29.
Kreisl T.N., Kim L., Moore K., et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27(5):740-745.
Lam P.Y., Breakefield X.O. Potential of gene therapy for brain tumors. Hum Mol Genet. 2001;10(7):777-787.
Matsutani M., Sano K., Takakura K., et al. Combined treatment with chemotherapy and radiation therapy for intracranial germ cell tumors. Childs Nerv Syst. 1998;14(1-2):59-62.
Matsutani M., Sano K., Takakura K., et al. Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg. 1997;86(3):446-455.
McElroy E.A.Jr., Buckner J.C., Lewis J.E. Chemotherapy for advanced esthesioneuroblastoma: the Mayo Clinic experience. Neurosurgery. 1998;42(5):1023-1027.
Mellinghoff I.K., Wang M.Y., Vivanco I., et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012-2024.
Merchant T.E., Mulhern R.K., Krasin M.J., et al. Preliminary results from a phase II trial of conformal radiation therapy and evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol. 2004;22(15):3156-3162.
Modak S., Gardner S., Dunkel I.J., et al. Thiotepa-based high-dose chemotherapy with autologous stem-cell rescue in patients with recurrent or progressive CNS germ cell tumors. J Clin Oncol. 2004;22(10):1934-1943.
Nelson D.F., Martz K.L., Bonner H., et al. Non-Hodgkin’s lymphoma of the brain: can high dose, large volume radiation therapy improve survival? Report on a prospective trial by the Radiation Therapy Oncology Group (RTOG): RTOG 8315. Int J Radiat Oncol Biol Phys. 1992;23(1):9-17.
Packer R.J., Sutton L.N., Elterman R., et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81(5):690-698.
Pels H., Schmidt-Wolf I.G., Glasmacher A., et al. Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol. 2003;21(24):4489-4495.
Piepmeier J., Christopher S., Spencer D., et al. Variations in the natural history and survival of patients with supratentorial low-grade astrocytomas. Neurosurgery. 1996;38(5):872-878.
Pignatti F., van den B.M., Curran D., et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol. 2002;20(8):2076-2084.
Plotkin S.R., Betensky R.A., Hochberg F.H., et al. Treatment of relapsed central nervous system lymphoma with high-dose methotrexate. Clin Cancer Res. 2004;10(17):5643-5646.
Quinn J.A., Desjardins A., Weingart J., et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23(28):7178-7187.
Ragel B.T., Osborn A.G., Whang K., et al. Subependymomas: an analysis of clinical and imaging features. Neurosurgery. 2006;58(5):881-890.
Rich J.N., Reardon D.A., Peery T., et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22(1):133-142.
Rudin C.M., Hann C.L., Laterra J., et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173-1178.
Saltzman W.M., Olbricht W.L. Building drug delivery into tissue engineering. Nat Rev Drug Discov. 2002;1(3):177-186.
Sampson J.H., Archer G.E., Mitchell D.A., et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773-2779.
Sasaki R., Murakami M., Okamoto Y., et al. The efficacy of conventional radiation therapy in the management of pituitary adenoma. Int J Radiat Oncol Biol Phys. 2000;47(5):1337-1345.
Sawamura Y., Ikeda J., Shirato H., et al. Germ cell tumours of the central nervous system: treatment consideration based on 111 cases and their long-term clinical outcomes. Eur J Cancer. 1998;34(1):104-110.
Schild S.E., Haddock M.G., Scheithauer B.W., et al. Nongerminomatous germ cell tumors of the brain. Int J Radiat Oncol Biol Phys. 1996;36(3):557-563.
Schild S.E., Nisi K., Scheithauer B.W., et al. The results of radiotherapy for ependymomas: the Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42(5):953-958.
Schild S.E., Scheithauer B.W., Haddock M.G., et al. Central neurocytomas. Cancer. 1997;79(4):790-795.
Shepherd C.W., Scheithauer B.W., Gomez M.R., et al. Subependymal giant cell astrocytoma: a clinical, pathological, and flow cytometric study. Neurosurgery. 1991;28(6):864-868.
Shrieve D.C., Alexander E.III, Black P.M., et al. Treatment of patients with primary glioblastoma multiforme with standard postoperative radiotherapy and radiosurgical boost: prognostic factors and long-term outcome. J Neurosurg. 1999;90(1):72-77.
Stege E.M., Kros J.M., de Bruin H.G., et al. Successful treatment of low-grade oligodendroglial tumors with a chemotherapy regimen of procarbazine, lomustine, and vincristine. Cancer. 2005;103(4):802-809.
Stupp R., Hegi M.E., Mason W.P., et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
Stupp R., Mason W.P., Van den Bent M.J., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-996.
Tacconi L., Delfini R., Cantore G. Choroid plexus papillomas: consideration of a surgical series of 33 cases. Acta Neurochir (Wien ). 1996;138(7):802-810.
Tiel R., Kline D. Peripheral nerve tumors: surgical principles, approaches, and techniques. Neurosurg Clin N Am. 2004;15(2):167-175. vi
Van den Bent M.J., Afra D., De Witte O., et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366(9490):985-990.
Van den Bent M.J., Carpentier A.F., Brandes A.A., et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24(18):2715-2722.
Van den Bent M.J., Kros J.M., Heimans J.J., et al. Response rate and prognostic factors of recurrent oligodendroglioma treated with procarbazine, CCNU, and vincristine chemotherapy. Dutch Neuro-oncology Group. Neurology. 1998;51(4):1140-1145.
Van Effenterre R., Boch A.L. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97(1):3-11.
Vance M.L. Medical treatment of functional pituitary tumors. Neurosurg Clin N Am. 2003;14(1):81-87.
Villavicencio A.T., Black P.M., Shrieve D.C., et al. LINAC radiosurgery for skull base meningiomas. Acta Neurochir (Wien). 2001;143(11):1141-1152.
Walker M.D., Green S.B., Byar D.P., et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303(23):1323-1329.
Westphal M., Hilt D.C., Bortey E., et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5(2):79-88.
Wick W., Hartmann C., Engel C., Stoffels M., et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27(35):5874-5880.
Yasargil M.G., Curcic M., Kis M., et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73(1):3-11.