Calcium Regulation, Calcium Homeostasis, and Genetic Disorders of Calcium Metabolism
Regulation of Calcium Homeostasis
Distribution and Metabolic Actions of Calcium
Mechanisms and Sites of Calcium Absorption
Parathyroid Hormone Gene Structure and Function
Parathyroid Adenoma 1 and PTH Genes
Multiple Endocrine Neoplasia 1 Gene
Multiple Endocrine Neoplasia 2 Gene (c-ret)
Hyperparathyroidism–Jaw Tumor Syndrome Gene
LRP5 and the WNT β-Catenin Pathway
Retinoblastoma-Interacting Zinc Finger Protein I Gene on Chromosome 1p
Nonsyndromic Familial Isolated Hyperparathyroidism
Hyperparathyroidism in Chronic Renal Failure
Disorders of the Calcium-Sensing Receptor
Familial Benign Hypercalcemia and Neonatal Severe Hyperparathyroidism
Parathyroid Hormone Gene Abnormalities
Glial Cells Missing B Abnormalities
X-linked Recessive Hypoparathyroidism
Pluriglandular Autoimmune Hypoparathyroidism
Hypoparathyroidism, Deafness, and Renal Anomalies Syndrome
Mitochondrial Disorders Associated With Hypoparathyroidism
Kenny-Caffey, Sanjad-Sakati, and Kirk-Richardson Syndromes
Calcium plays an important role in many physiologic pathways that include muscle contraction, the secretion of neurotransmitters and hormones, and the coagulation pathways, and it is an important component of the skeleton. Disturbances in extracellular calcium concentration may cause a variety of symptoms, the most common of which reflect abnormal neuromuscular activity. Hypercalcemia may lead to muscle weakness and areflexia, anorexia, constipation, vomiting, drowsiness, depression, confusion, other cognitive dysfunction, and coma. Hypercalcemia leads to hypercalciuria, which can result in medullary calcification, nephrocalcinosis, and renal failure. Hypocalcemia, conversely, may cause anxiety, seizures, muscle twitching, epilepsy, tetany, Chvostek’s and Trousseau’s signs, carpal or pedal spasm, stridor, bronchospasm, and intestinal cramps, as well as cataracts, skeletal malformations, and abnormal dentition. The control of body calcium involves a balance between the amounts that are absorbed from the gut, deposited into bone and cells, and excreted from the kidney (Fig. 5-1). This fine balance, involving three organs, is chiefly under the control of parathyroid hormone (PTH), which is synthesized and secreted by the parathyroid glands. Thus, hypocalcemia will lead to an increased secretion of PTH, whereas hypercalcemia will result in diminished PTH secretion. A number of clinical disorders characterized by derangements of calcium homeostasis are caused by abnormalities of the parathyroid glands themselves. Thus, PTH oversecretion due to parathyroid tumors, which affect 3/1000 of the population, is a major cause of hypercalcemia, which may be associated with kidney stones, osteoporosis, and peptic ulcers. PTH deficiency as part of a syndrome occurs in 1/4000 live births, isolated hypoparathyroidism resulting in hypocalcemia appears to occur less frequently. This chapter will review (1) the physiologic and biochemical mechanisms underlying extracellular calcium homeostasis and (2) the genetic basis for disorders of calcium metabolism.
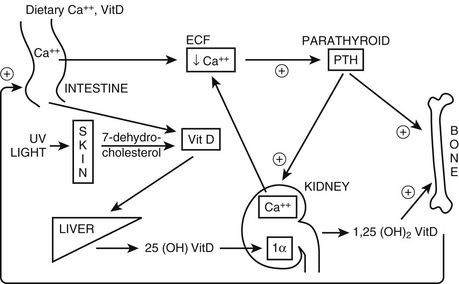
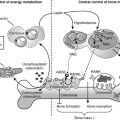
FIGURE 5-1 Regulation of extracellular fluid (ECF) calcium (Ca2+) by parathyroid hormone (PTH) action on kidney, bone, and intestine. A decrease in ECF Ca2+ is sensed by the calcium-sensing receptor, leading to an increase in PTH secretion, which predominantly acts directly on kidney and bone that possess the PTH/PTHrP receptor (PTHR1; see Fig. 5-3). The skeletal effects of PTH are to increase (+) osteoclastic bone reabsorption, but because osteoclasts do not express the PTHR1, this action is mediated via the osteoblasts, which do have this receptor and in response release cytokines and factors that activate osteoclasts. In the kidney, PTH stimulates (+) the 1α-hydroxylase (1α) to increase the conversion of 25-hydroxy vitamin D [25(OH)D] to the active metabolite 1,25-dihydroxyvitamin D [1,25(OH)2D]. In addition, PTH increases (+) reabsorption of Ca2+ from the renal distal tubule and inhibits reabsorption of phosphate from the proximal tubule, leading to hypercalcemia and hypophosphatemia. PTH also inhibits Na+/H+ antiporter activity and bicarbonate reabsorption, causing a mild hyperchloremic acidosis. Elevated 1,25(OH)2D acts on the intestine to increase (+) absorption of dietary calcium and phosphate; it is important to note that PTH does not appear to have a direct action on the gut. In response to hypocalcemia and the increase in PTH secretion, all of these direct and indirect actions of PTH on the kidney, bone, and intestine will help to increase ECF Ca2+, which in turn will act via the calcium-sensing receptor to decrease PTH secretion.
Regulation of Calcium Homeostasis
Distribution and Metabolic Actions of Calcium
The total body content of calcium in a normal adult is approximately 1000 g, of which nearly all exists within the crystal structure of bone mineral; less than 1% is soluble in extracellular and intracellular fluid.1,2 On average, bone mineral closely approximates the composition of hydroxyapatite [Ca10(PO4)6(OH)2], which means that 6 mmol of phosphate are released with every 10 mmol of calcium mobilized during bone resorption (or about 1 : 2 on a mg/mg basis).1,3
In blood, calcium is partly bound to proteins. Albumin accounts for about 70% of the protein-bound fraction.4 Another portion (about 6%) is associated with diffusible ion complexes.1 Thus normally, only half of total plasma calcium is freely ionized, but it is this fraction that is most important physiologically and subject to stringent endocrine regulation. The normal ranges of total and ionized serum calcium in the adult are 8.5 to 10.5 mg/dL and 1.17 to 1.33 mmol, respectively.5 The usual 2 : 1 ratio of total to ionized calcium may be disturbed by disorders such as metabolic acidosis (calcium binding to proteins is reduced at acid pH) or by changes in serum protein concentrations, as in starvation, cirrhosis, dehydration, or multiple myeloma. When precise knowledge of the ionized calcium concentration is clinically important, direct measurement with calcium-selective electrodes should be performed.
Serum calcium is higher during infancy/childhood and adolescence than in the adult6 but does not change at puberty or, in women, during the menstrual cycle. During pregnancy, total serum calcium and albumin decline progressively, but ionized calcium is minimally affected.7,8 Fetal calcium content rises dramatically during the third trimester (to about 30 g at term),9 and fetal serum total and ionized calcium concentrations both are higher than maternal levels, consistent with active placental transport of calcium.10 Despite daily losses of 200 to 300 mg/d of calcium in breast milk, lactating women maintain normal levels of ionized calcium in blood by increasing intestinal calcium absorption in response to augmented production of 1,25(OH)2D.11,12
Calcium entry into cells is strongly favored by a steep electrochemical gradient. Thus, the cell interior is electronegative, and cytosolic free calcium is in the range of 10−7 M, which is 10,000-fold lower than extracellular calcium concentrations. Calcium traverses the plasma membrane through various channels, including voltage-, receptor- and store-operated forms, the regulation of which is complex and tissue specific.13,14 Intracellularly, nearly all (99%) calcium is sequestered in pools within mitochondria, endoplasmic reticula, or sarcoplasmic reticula or is tightly bound to the inner surface of the plasma membrane.15 Sequestered calcium, especially that within the endoplasmic reticulum, may be released rapidly into the cytosol following activation of cell-surface receptors. In this way, it plays a critical role in signal transduction and in controlling calcium entry via store-operated channels. The extremely low cytosolic free calcium concentration is maintained by active calcium transport into intracellular pools or by extrusion out of the cell via high-affinity, low-capacity Ca2+/H+ adenosine triphosphatases (ATPases) and low-affinity, high-capacity Na+/Ca2+ exchangers driven by the transmembrane sodium gradient.16
With the discovery of a G protein–coupled calcium-sensing receptor (CaSR) expressed in parathyroid, renal epithelial, and other cells, it has become clear that calcium can act as an extracellular ligand to directly control cellular function.17 The principal actions of the CaSR with respect to calcium homeostasis include suppression of PTH secretion and, in the thick ascending loop of the renal tubule, inhibition of calcium, magnesium, and NaCl reabsorption. Extracellular calcium also is utilized directly for normal matrix mineralization in bone and cartilage and is required for activation of important circulating or extracellular enzymes and proteases. Intracellular calcium exerts a broad range of effects via interaction with key enzymes and effector molecules, including kinases, phosphatases, calmodulins, transcription factors, ion channels (including calcium channels), and troponins and other proteins involved in contraction, microtubule and microfilament assembly, and motility. The steep gradients between intracellular calcium and both extracellular and intracellularly sequestered calcium are crucial for normal neuromuscular activity and provide the potential required for rapid transients and waves of cytosolic free calcium that serve key second-messenger functions in both excitable and nonexcitable cells.14,18
Calcium Absorption
Intestinal calcium absorptive efficiency in humans ranges broadly between 20% and 70%, declines steadily with age, and is strongly influenced by previous calcium intake, the presence of other nutrients, pregnancy, lactation, overall calcium balance, and the availability of vitamin D.19–23 Fecal calcium includes the residual fraction of dietary calcium that is not absorbed, as well as a contribution from secreted calcium present in bile and other digestive juices (“endogenous fecal calcium”). Endogenous fecal calcium in humans normally amounts to 100 to 200 mg/d and is relatively unaffected by changes in dietary or serum calcium.24,25
Intestinal calcium absorption is adjusted physiologically in response to variations in calcium intake, as shown by radiocalcium kinetic and balance studies in normal subjects.26,27 Obligate renal and intestinal excretion of calcium is such that calcium balance cannot be maintained if dietary calcium consistently falls below 200 to 400 mg/d, even though the percentage of calcium absorbed may be very high (i.e., 70%). As calcium intake increases, overall calcium absorption (in mg/d) rises, but the fractional absorption of ingested calcium declines progressively such that within the physiologic range of calcium intake, total net calcium absorption tends to plateau at approximately 400 mg/d. Consequently, urinary calcium excretion tends also to plateau at higher intakes. Additional buffering of changes in dietary calcium results from control of renal tubular calcium reabsorption and skeletal calcium release, but regulation of intestinal absorptive efficiency is critical for calcium balance.26 The mechanisms of this inverse regulation of intestinal absorptive efficiency by changes in calcium availability are considered in the following discussion.
Mechanisms and Sites of Calcium Absorption
Calcium is absorbed throughout the intestine. In terms of rates of transport per unit length of mucosa, absorption is most efficient in the duodenum and proximal jejunum, which exhibit the highest levels of vitamin D–dependent calcium binding proteins and in which lower luminal pH (5 to 6) promotes dissociation of calcium from complexes with food constituents and other ions.28,29 On the other hand, longer residence times in the more distal small bowel segments may allow absorption of a larger proportion of total calcium intake in the distal jejunum and ileum.30,31 The ileum, for example, may become an important site of net calcium absorption during dietary restriction of calcium or when the residence time of luminal contents in more proximal bowel segments is reduced.30,32
Transcellular calcium absorption necessarily involves three steps: entry into the cell, diffusion across the cell, and extrusion from the cell (see Chapter 3). The first step, entry through the apical brush border, occurs via a member of the vanilloid (TRPV) superfamily of channels, namely TRPV6 (previously referred to as CaT1 or ECaC2).33,34 Another member of this family, TRPV5, appears to be dominantly involved in calcium reabsorption in the kidney (see later). The TRPV6 and TRPV5 genes are located on chromosome 7q33-35. Their structure includes six transmembrane regions, a short hydrophobic region between segments 5 and 6 that likely forms the calcium pore, and large intracellular domains at the N- and C-termini.33 The pore region is highly selective for calcium,35,36 and mutations in this region have been shown to abolish calcium permeability.37 Posttranslational modification and glycosylation of TRPV6 occurs, but the functional effects of these modifications are not well defined. Similarly, the mechanisms regulating the activity of these molecules are not well understood. TRPV6 and TRPV5 expression varies significantly between species and in humans, and both genes are expressed in a variety of tissues.38 Specifically, TRPV5 seems to be the major isoform expressed in the kidney, whereas TRPV6 is most highly expressed in the small intestine (restricted to the apical surface of epithelial cells).39 Given the important role of 1,25(OH)2D3 and other hormonal regulators in calcium absorption, the effects of these hormones on TRPV6 expression has recently been investigated. 1,25(OH)D3 clearly increases TRPV6 expression in vitro (see Chapter 3).40 Furthermore, mice with targeted inactivation of the vitamin D receptor demonstrate a 90% reduction in TRPV6 expression and a threefold reduction in intestinal calcium absorption.41 Additionally, calcium exposure appears to independently affect TRPV6 expression insofar as high calcium intake decreases TRPV6 expression, and low intake increases it.41,42 Finally, TRPV6 expression is decreased in mice with targeted inactivation of their estrogen receptor-α gene and is increased in mice receiving exogenous estradiol administration.43
After calcium has entered the cell, it must be transported through the cytoplasm and extruded from the cell against a steep gradient. The mechanism by which calcium moves through the cell involves the small cytosolic protein calbindin D9k. Calcium entering the cell via the apical TRPV6 channel becomes tightly associated with the calbindin, which buffers the relatively large mass of entering calcium and minimizes its impact upon cytosolic free-calcium concentrations. The calbindin/calcium complex then diffuses across the cytosol to the basolateral membrane. Free calcium then dissociates into the low-cytosolic calcium environment maintained immediately subjacent to the basolateral membrane by high-affinity membrane Ca2+-ATPases located there. Finally, these calcium ATPases actively extrude calcium out of the cell.29 The known cellular concentrations and kinetic properties of the calbindin D9k molecule would support observed rates of duodenal calcium transport at submicromolar concentrations of cytosolic free calcium.44 Indeed, the importance of this buffered diffusional process predicts that enterocyte calbindin D9k content is likely to be a major determinant, along with TRPV6 activity, of the overall rate of enterocyte calcium transport.
Renal Calcium Excretion
Regulation of renal calcium excretion is an important mechanism for homeostatic control of blood ionized calcium in the face of fluctuations in filtered load, as derived from intestinal calcium absorption and net bone resorption. When urinary calcium is viewed as a function of the amount of calcium actually absorbed by the gut (i.e., after regulation of intestinal absorptive efficiency has been factored out), the precision with which the kidney adjusts tubular calcium reabsorption to residual changes in filtered load becomes obvious. Ordinarily, the daily load of calcium filtered at the glomerulus (the product of the glomerular filtration rate45 and ultrafilterable calcium46) is approximately 10,000 mg/d in adult humans, which means that the extracellular calcium pool is completely filtered several times a day. Since urinary calcium excretion (and net intestinal calcium absorption) is approximately 200 mg/d, only 2% of filtered calcium is excreted normally. This high ratio of filtered to excreted calcium affords ample opportunity for finely tuned hormonal control of calcium excretion, even though it may be difficult to measure the small changes involved.
Sites and Mechanisms of Renal Calcium Reabsorption
Approximately 60% to 70% of tubular calcium reabsorption, like that of sodium, occurs in the proximal tubule.46 Proximal tubular calcium reabsorption occurs mainly via passive diffusion along paracellular pathways, down the ambient (lumen-positive) electrochemical gradient.47 Another 20% to 25% of the filtered calcium load is reabsorbed in Henle’s loop.46 This occurs mainly by paracellular diffusion in the cortical thick ascending limb (cTAL),48–50 although active transcellular transport may play a minor role as well.51,52 Calcium (and magnesium) reabsorption in this segment is severely impaired in patients with homozygous inactivating mutations in claudin 16 (CLDN16) (previously referred to as paracellin-1) or claudin 19 (CLDN19) protein, strongly expressed in tight junctions of the cTAL and presumably required for normal divalent cation conductance.53–56 Hypercalcemia suppresses renal tubular calcium reabsorption even in the absence of PTH,57 an effect likely due to direct activation of basolateral CaSRs in the cTAL (and possibly other nephron segments).58 In the cTAL, activated CaSRs lower apical K+ exit by inhibiting renal outer medullary K+ channels (ROMKs), thereby blunting Na-K-2Cl cotransporter (NKCC2) activity and reducing the transepithelial voltage gradient that drives paracellular cation movement.17,59–61 The critical importance of normal rates of Na-K-2Cl reabsorption for adequate calcium transport in the cTAL is highlighted clinically by the occurrence of dramatic hypercalciuria in patients with Bartter’s syndromes, which involve defects in NKCC2, ROMK, or other transporters or channels required for effective Na-K-2Cl reabsorption.62 An analogous Bartter’s-like phenotype may occur in some patients with activating CaSR mutations.63 CaSR activation in the cTAL also antagonizes PTH-stimulated increases in calcium reabsorption, possibly by impairing cyclic adenosine monophosphate (cAMP) generation61 (see later).
Only about 8% to 10% of filtered calcium is reabsorbed in more distal segments of the tubule, but calcium transport in the distal nephron is a key control point for hormonal regulation and involves predominantly transcellular reabsorption.64–66 Cells in the late distal tubule exhibit striking co-localization of several unique proteins that are critical for effective transcellular calcium reabsorption. These include an apical epithelial calcium channel, TRPV5 (formerly ECaC1 or CaT2 and highly homologous to TRPV6, discussed earlier), the cytosolic calcium-binding protein, calbindin D28k, basolateral plasma membrane calcium ATPase(s) (PMCAs), and basolateral NA+/Ca2+ exchanger 1 (NCX1).67–72 As in the intestinal epithelium, these cells admit calcium across their apical membranes via opening of TRPV5. Expression of this channel is up-regulated by PTH, thus enhancing calcium reabsorption (see later). In contrast, high intracellular calcium concentrations and increased calbindin D28k levels reduce calcium reabsorption, possibly via enhanced buffering of subapical Ca2+ ions. Calcium bound to calbindin D28k is ferried across the cell to the basolateral membrane, where it ultimately is extruded against a steep electrochemical gradient, predominantly via NCX1 (70% of Ca2+ flux),73,74 but also by PMCA. As expected, mice lacking TRPV575 or calbindin D28k76 were found to manifest severe hypercalciuria.
Additional calcium absorption may occur in more distal nephron segments, such as cortical and medullary collecting ducts.47 Proteins other than those described above may be involved in renal calcium reabsorption as well, including voltage-operated calcium channels47,77 and Cl− channels, as evidenced by the hypercalciuria seen in Dent’s disease, an X-linked disorder due to inactivating mutations in the Cl− channel ClC-5 that may be involved in endocytic vesicle function and protein trafficking.78,79
Regulation of Renal Calcium Reabsorption
Calcium excretion is affected by a variety of hormones, ions, nutrients, and drugs.80,81 Among these, PTH is the principal physiologic regulator of renal tubular calcium transport, as appreciated clinically by the markedly abnormal overall relation of serum to urinary calcium in hyper- and hypoparathyroidism.80,82,83 PTH acts to enhance tubular calcium reabsorption in the cTAL, distal convoluted tubules (DCT), and connecting tubules (CNT).50,65,84,85 It augments DCT calbindin D28k expression86 and basolateral N+/Ca2+ exchange,87–91 increases the affinity for calcium of the basolateral Ca2+-ATPase,92 and hyperpolarizes the DCT cell,93 which then activates the TRPV5 channel.94 In immortalized murine distal tubular cells, PTH causes insertion into the apical membrane of new dihydropyridine-sensitive membrane calcium channels,77 and it stimulates stretch-activated nonselective cation channels in rabbit CNT.95 These are distinct from TRPV5 channels and may represent other mechanisms of apical calcium entry. PTH increases the surface expression or activity of TRPV5 epithelial calcium channels. In the cTAL, PTH also may increase some form of active transcellular transport,51,52 and it may stimulate paracellular diffusional calcium transport by augmenting the transepithelial voltage gradient, at least in some segments and species.96,97
Studies in patients with inactivating vitamin D receptor mutations have demonstrated that 1,25(OH)2D3 is required for the calcium-reabsorptive response to PTH.98 It furthermore accelerates the increase in DCT cell calcium entry initiated by PTH,99 and it directly increases DCT calcium reabsorption, apparently by driving higher expression of TRPV5, calbindin D28k, and PMCA (see Chapter 3).74,100–103 Another hormone affecting calcium reabsorption is calcitonin. When given in large doses, calcitonin acutely reduces proximal tubular calcium reabsorption by a mechanism independent of PTH;104 however, an important role for calcitonin in the physiologic regulation of calcium reabsorption is thought to be unlikely. Hypercalciuria observed in states of excess growth hormone or cortisol seems likely to be secondary to an increased filtered load of calcium rather than to direct tubular actions of these hormones.105–110 Estrogen treatment of normal postmenopausal women lowers urinary calcium excretion by increasing tubular calcium reabsorption.111,112 In DCT of ovariectomized rats, 17β-estradiol acutely increases mRNA expression of all major components of the calcium transport pathway, including TRPV5, calbindin D28k, NCX1, and PMCA1b.113 Reported effects on tubular calcium reabsorption of insulin,114 glucagon,96 antidiuretic hormone,96 and angiotensin II115 are of uncertain physiologic or clinical significance.
Parathyroid Hormone Gene Structure and Function
The PTH gene is located on chromosome 11p15 and consists of 3 exons which are separated by 2 introns.116 Exon 1 of the PTH gene is 85 bp in length and is untranslated (Fig. 5-2), whereas exons 2 and 3 encode the 115-amino-acid pre-proPTH peptide (see Chapter 1 for detailed discussion). Exon 2 is 90 bp in length and encodes the initiation (ATG) codon, the prehormone sequence, and part of the prohormone sequence. Exon 3 is 612 bp in size and encodes the remainder of the prohormone sequence, the 84 amino acids comprising the mature PTH peptide and the 3′ untranslated region.117 The 5′ regulatory sequence of the human PTH gene contains a vitamin D response element 125 bp upstream of the transcription start site, which down-regulates PTH mRNA transcription in response to vitamin D receptor binding.118,119 PTH gene transcription (as well as PTH peptide secretion) is also dependent upon the extracellular calcium and phosphate concentration,120,121 although the presence of specific upstream “calcium or phosphate response element(s)” has not yet been demonstrated.122,123 The secretion of mature PTH from the parathyroid chief cell is regulated through a G protein–coupled calcium-sensing receptor, which is also expressed in renal tubules and several other tissues, albeit at lower abundance. PTH mRNA is first translated into a pre-proPTH peptide. The “pre” sequence consists of a 25-amino-acid signal peptide (leader sequence) which is responsible for directing the nascent peptide into the endoplasmic reticulum to be packaged for secretion from the cell.124 The “pro” sequence is 6 amino acids in length, and although its function is less well defined than that of the “pre” sequence, it is also essential for correct PTH processing and secretion.124 After the 84 amino acid–containing mature PTH peptide is secreted from the parathyroid cell, it is cleared from the circulation (with a short half-life of about 2 minutes) via nonsaturable hepatic and renal uptake (see Chapter 1).
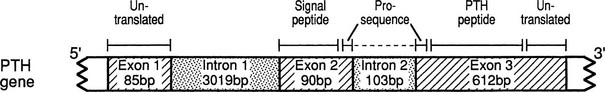
FIGURE 5-2 Schematic representation of the PTH gene, PTH mRNA, and PTH peptide. The PTH gene consists of 3 exons and 2 introns; the peptide is encoded by exons 2 and 3. The PTH peptide is synthesized as a precursor, which contains a presequence and a prosequence. The mature PTH peptide, which contains 84 amino acids, and larger carboxyl-terminal PTH fragments are secreted from the parathyroid cell. (From Parkinson D, Thakker R: A donor splice site mutation in the parathyroid hormone gene is associated with autosomal-recessive hypoparathyroidism, Nat Genet 1:149–153, 1992.)
PTH mediates its actions through a receptor it shares with PTH-related peptide (PTHrP also known as PTHrH, PTH-related hormone).125,126 This PTH/PTHrP receptor (Fig. 5-3) is a member of a subgroup of G protein–coupled receptors, and its gene is located on chromosome 3p21.3.127,128 The PTH/PTHrP receptor is highly expressed in kidney and bone, where it mediates the endocrine actions of PTH. However, during embryonic and postnatal development, the PTH/PTHrP receptor is most abundantly expressed in chondrocytes of the metaphyseal growth plate, where it mediates predominantly the autocrine/paracrine actions of PTHrP.129,130 Mutations involving the genes that encode PTH, the calcium-sensing receptor, the PTH/PTHrP receptor, and Gsα all affect the regulation of calcium homeostasis and can thus be associated with genetic disorders characterized by hypercalcemia or hypocalcemia (Table 5-1).
Table 5-1
Diseases of Calcium Homeostasis and Their Chromosomal Locations
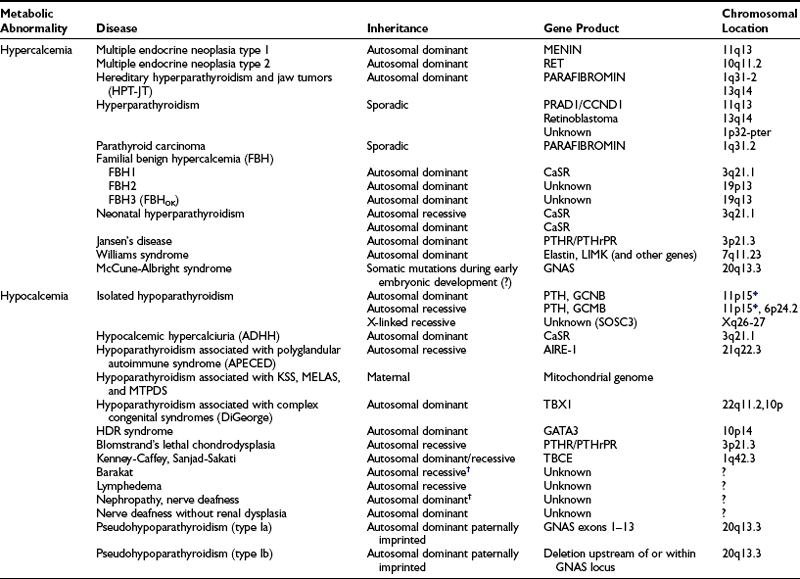
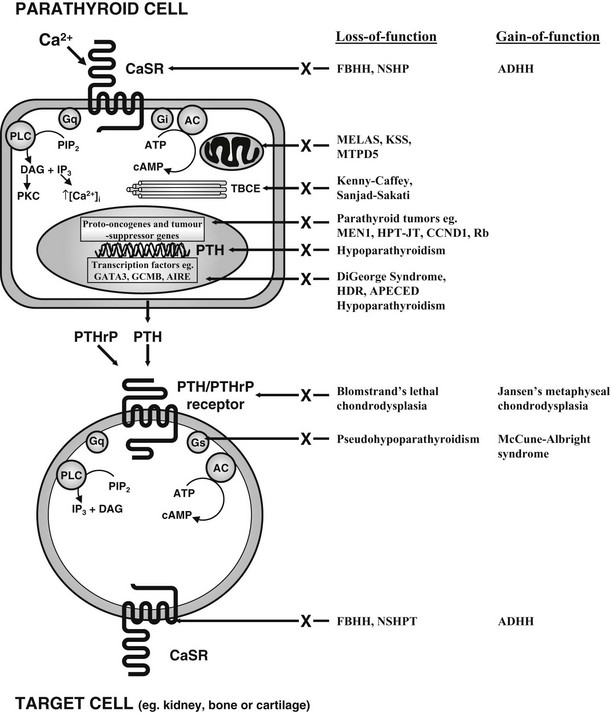
FIGURE 5-3 Schematic representation of some of the components involved in calcium homeostasis. Alterations in extracellular calcium are detected by the calcium-sensing receptor (CaSR), a 1078-amino-acid G protein–coupled receptor. The PTH/PTHrP receptor, which mediates the actions of PTH and PTHrP, is also a G protein–coupled receptor. Thus, Ca2+ and PTH and PTHrP involve G protein–coupled signaling pathways, and interaction with their specific receptors can lead to activation of Gs, Gi and Gq, respectively. Gs stimulates adenylcyclase (AC), which catalyses the formation of cAMP from ATP. Gi inhibits AC activity, and cAMP stimulates PKA, which phosphorylates cell-specific substrates. Activation of Gq stimulates PLC, which catalyses the hydrolysis of the phosphoinositide (PIP2) to inositol triphosphate (IP3), which increases intracellular calcium, and diacylglycerol (DAG), which activates PKC. These proximal signals modulate downstream pathways, resulting in specific physiologic effects. Abnormalities in several genes, which lead to mutations in proteins in these pathways, have been identified in specific disorders of calcium homeostasis (see Table 5-1).
Hypercalcemic Diseases
Similar to the findings in other tumor syndromes, the abnormal expression of an oncogene or the loss of a tumor-suppressor gene can result in abnormal proliferative activity of parathyroid cells. The molecular exploration of these genes has provided important novel insights into the pathogenesis of different forms of hyperparathyroidism. Oncogenes are genes whose abnormal expression may transform a normal cell into a tumor cell. The normal form of the gene is referred to as a proto-oncogene, and a single mutant allele may affect the phenotype of the cell; these genes may also be referred to as dominant oncogenes (Fig. 5-4A). The mutant versions (i.e., the oncogene), which are usually excessively or inappropriately active, may arise because of point mutations, gene amplifications, or chromosomal translocations. Tumor-suppressor genes, also referred to as recessive oncogenes or anti-oncogenes, normally inhibit cell proliferation, whereas their mutant versions in cancer cells have lost their normal function. In order to transform a normal cell into a tumor cell, both alleles of the tumor-suppressor gene must be inactivated. Inactivation arises by point mutations or, alternatively, by small or larger intragenic deletions that can involve substantial genomic portions or a whole chromosome. Larger deletions may be detected by cytogenetic methods, by Southern blot analysis, or by PCR-based analysis of polymorphic markers. Compared to genomic DNA from other cells (e.g., leukocytes), genomic DNA from the patient’s tumor cells typically lack certain chromosomal regions, and this finding is consequently referred to as loss of heterozygosity (LOH) (see Fig. 5-4B). Finding LOH, therefore, suggests an inactivating mutation or deletion in the other allele.
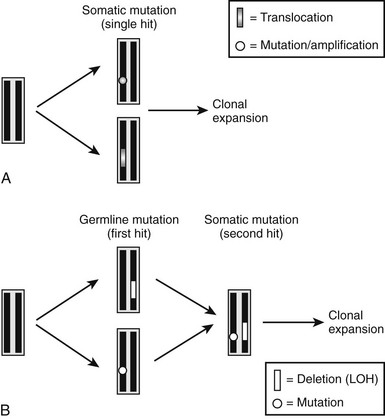
FIGURE 5-4 Schematic illustration of molecular defects that can lead to development of parathyroid tumors. A, Somatic mutation (point mutation or translocation) affecting a proto-oncogene (e.g., PRAD1 or RET) results in a growth advantage of single parathyroid cell and thus its clonal expansion. B, Inherited single point mutation or deletion affecting a tumor-suppressor gene (first hit) makes the parathyroid cell susceptible to a second, somatic “hit” (point mutation or deletion, i.e., LOH), which then leads to clonal expansion of a single cell.
Parathyroid Tumors
Parathyroid tumors may occur as an isolated and sporadic endocrinopathy or as part of inherited tumor syndromes,131 such as the multiple endocrine neoplasias (MEN) or hereditary hyperparathyroidism with jaw tumors (HPT-JT),132 or in response to chronic overstimulation, as in uremic hyperparathyroidism.133 Genetic analyses of kindreds with MEN1 and MEN2A and of tumor tissue from patients with single parathyroid adenomas have shown that some of the molecular mechanisms known to be involved in tumor genesis can also be responsible for the development of hyperparathyroidism.
Our current understanding indicates that sporadic parathyroid tumors are caused by single somatic mutations that lead to the activation or overexpression of proto-oncogenes such as parathyroid adenoma 1 (PRAD1) or RET (see Fig. 5-4A). Furthermore, different tumor-suppressor genes affecting the parathyroid glands are predicted to be located on several different chromosomes, and in a significant number of patients, LOH has been documented for one of these loci. For all these somatic mutations, a single point mutation or a deletion provides a growth advantage to a single parathyroid cell and its progeny, leading to their clonal expansion.
In hereditary forms of the disease, two distinct, sequentially occurring molecular defects are observed. The first “hit” (point mutation or deletion) is an inherited genetic defect, which affects only one allele that comprises a gene encoding an anti-oncogene (see Fig. 5-4B). Subsequently, a somatic mutation or deletion affecting the second allele occurs in a single parathyroid cell, and because of the resulting growth advantage, this mutation leads to its monoclonal expansion and thus the development of parathyroid tumors. Examples of this latter molecular mechanism in the development of hyperparathyroidism are the inactivation of tumor-suppressor genes such as the multiple endocrine neoplasia type 1 (MEN1) gene, the hyperparathyroidism–jaw tumor (HPT–JT) gene, and the retinoblastoma (Rb) gene (see Table 5-1).
Parathyroid Adenoma 1 and PTH Genes
Investigations of the PTH gene in sporadic parathyroid adenomas detected abnormally sized restriction fragment length polymorphisms (RFLPs) with a DNA probe for the 5′ part of the PTH gene in some adenomas,134 indicating disruption of the gene. Further studies of the tumor DNA demonstrated that the first exon of the PTH gene (see Fig. 5-2) was separated from the fragments containing the second and third exons, and that a rearrangement had occurred, juxtaposing the 5′ PTH regulatory elements with “new” non-PTH DNA.135 This rearrangement was not found in the DNA from the peripheral leukocytes of the patients, thereby indicating that it represented a somatic event and not an inherited germline mutation. Investigation of this rearranged DNA sequence localized it to chromosome 11q13, and detailed analysis revealed that it was highly conserved in different species and expressed in normal parathyroids and in parathyroid adenomas. The protein expressed as a result of this rearrangement, which was designated PRAD1, was demonstrated to encode a 295-amino-acid member of the cyclin-D family of cell-cycle regulatory proteins. Cyclins were initially characterized in the dividing cells of budding yeast, where they controlled the G1 to S transition of the cell cycle, and in marine mollusks, where they regulated the mitotic phase (M-phase) of the cell cycle.136 Cyclins have also been identified in man and have an important role in regulating many stages of cell-cycle progression. Thus PRAD1, which encoded a novel cyclin referred to as cyclin D1 (CCND1), is an important cell-cycle regulator, and overexpression of PRAD1 may be an important event in the development of at least 15% of sporadic parathyroid adenomas.137,138
Interestingly, more than 66% of the transgenic mice overexpressing PRAD1 under the control of a mammary tissue-specific promoter were found to develop breast carcinoma in adult life,139 and expression of this proto-oncogene under the control of the 5′ regulatory region of the PTH gene resulted in mild to moderate chronic hyperparathyroidism.137,138 Taken together, these findings in transgenic animals provide further evidence for the conclusion that PRAD1 can be involved in the development of a significant number of parathyroid adenomas.
In addition to the rearrangement of the PTH gene in some parathyroid adenomas, a nonsense mutation of the PTH gene, Arg83Stop, that occurred in association with LOH of the PTH locus has been reported in a parathyroid adenoma.140 The patient, who had presented with hypercalcemia and an undetectable serum PTH concentration, showed heterozygosity in the peripheral-blood leukocytes with wild-type and mutant alleles, but the parathyroid adenoma had a loss of the wild-type allele and retention of the mutant (Arg83Stop) allele, which predicts that the tumor secretes only a PTH peptide that is truncated after the 52nd amino acid. Following removal of the parathyroid adenoma, normocalcemia was restored.140 These findings demonstrate that PTH nonsense mutations, which result in truncated forms of PTH not recognizable by standard hormone assays, may be associated with parathyroid adenoma, and that endogenously produced N-terminal PTH fragments can be biologically active.140
Multiple Endocrine Neoplasia 1 Gene
MEN1 is characterized by the combined occurrence of tumors of the parathyroids, pancreatic islet cells, and anterior pituitary (Table 5-2).141,142 Parathyroid tumors occur in 95% of MEN1 patients, and the resulting hypercalcemia is the first manifestation of MEN1 in about 90% of patients. Pancreatic islet cell tumors occur in 40% of MEN1 patients; gastrinomas, leading to the Zollinger-Ellison syndrome, are the most common type and also the important cause of morbidity and mortality in MEN1 patients. Anterior pituitary tumors occur in 30% of MEN1 patients, with prolactinomas representing the most common type. Associated tumors, which may also occur in MEN1, include adrenal cortical tumors, carcinoid tumors, lipomas, and cutaneous angiofibromas and collagenomas.142,143 The gene causing MEN1 was localized to a less than 300-kb region on chromosome 11q13 by genetic mapping studies that investigated MEN1-associated tumors for LOH and by segregation studies in MEN1 families.144 The results of these studies, which were consistent with Knudson’s model for tumor development, indicated that the MEN1 gene represented a putative tumor-suppressor gene (see Fig. 5-4B). Characterization of genes from this region led to the identification of the MEN1 gene, which consists of 10 exons that encode a novel 610-amino-acid protein referred to as menin.145,146 Over 1100 germline MEN1 mutations have been identified, and the majority (>80%) are inactivating and are consistent with its role as a tumor-suppressor gene.147,148 These mutations are diverse in their types, and in approximate percentages, 25% are nonsense, 45% are frameshift deletions or insertions, 9% are splice-site mutations, 20% are missense mutations, and 1% are whole or partial gene deletions.144,147–149 In addition, the MEN1 mutations are scattered throughout the 1830-bp coding region of the gene with no evidence for clustering. Correlations between the MEN1 germline mutations and the clinical manifestations of the disorder appear to be absent.147–150 Tumors from MEN1 patients and non-MEN1 patients have been observed to harbor the germline mutation together with a somatic LOH involving chromosome 11q13, as expected from Knudson’s model and the proposed role of the MEN1 gene as a tumor suppressor.148,151–161 The role of the MEN1 gene in the etiology of familial isolated hyperparathyroidism (FIHP) has also been investigated, and germline MEN1 mutations have been reported in 29 families with FIHP.148,151,162–164 The sole occurrence of parathyroid tumors in these families is remarkable, and the mechanisms that determine the altered phenotypic expressions of these mutations remain to be elucidated.
Table 5-2
Multiple Endocrine Neoplasia Syndromes, Characteristic Tumors, and Associated Genetic Abnormalities*

*Autosomal-dominant inheritance of the multiple endocrine neoplasia (MEN) syndromes has been established.
The function of menin has been investigated by identifying its interactions with other proteins, and by under- or overexpression in in vitro studies. Menin has no homology to any known proteins or sequence motifs other than three nuclear localization signals (NLSs) in its C-terminal segment. Subcellular localization studies have shown that menin is predominantly a nuclear protein in nondividing cells, but in dividing cells, it is found in the cytoplasm. Menin has been shown to interact with a number of proteins that are involved in transcriptional regulation, genome stability, cell division, and proliferation.148
The functional role of menin as a tumor suppressor also has been investigated, and studies in human fibroblasts have revealed that menin acts as a repressor of telomerase activity via hTERT (a protein component of telomerase).165 Furthermore, overexpression of menin in the human endocrine pancreatic tumor cell line (BON1) resulted in an inhibition of cell growth166 that was accompanied by up-regulation of JunD expression but down-regulation of delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1, which is a negative regulator of c-Jun.166 These findings of growth suppression by menin were observed in other cell types. Thus, expression of menin in the RAS-transformed NIH3T3 cells partially suppressed the RAS-mediated tumor phenotype in vitro and in vivo.167 Overexpression of menin in CHO-IR cells also suppressed insulin-induced AP-1 transactivation, and this was accompanied by an inhibition of c-Fos induction at the transcriptional level.168 Furthermore, menin reexpression in Men1-deficient mouse Leydig tumor cell lines induced cell cycle arrest and apoptosis.169 In contrast, depletion of menin in human fibroblasts resulted in their immortalization.165 Thus, menin appears to have a large number of functions through interactions with proteins, and these mediate alterations in cell proliferation.
Multiple Endocrine Neoplasia 2 Gene (c-ret)
MEN2 describes the association (see Table 5-2) of medullary thyroid carcinoma (MTC), pheochromocytomas, and parathyroid tumors.141,144 Three clinical variants of MEN2 are recognized: MEN2a, MEN2b, and MTC-only (see Table 5-2). MEN2a is the most common variant, and the development of MTC is associated with pheochromocytomas (50% of patients), which may be bilateral, and parathyroid tumors (20% of patients). MEN2b, which represents 5% of all MEN2 cases, is characterized by the occurrence of MTC and pheochromocytoma in association with a Marfanoid habitus, mucosal neuromas, medullated corneal fibers, and intestinal autonomic ganglion dysfunction leading to multiple diverticula and megacolon. Parathyroid tumors do not usually occur in MEN2b. MTC-only is a variant in which medullary thyroid carcinoma is the sole manifestation of the syndrome. The gene causing all three MEN2 variants was mapped to chromosome 10cen-10q11.2, a region containing the c-ret proto-oncogene which encodes a tyrosine kinase receptor with cadherin-like and cysteine-rich extracellular domains, and a tyrosine kinase intracellular domain.170,171 Specific mutations of c-ret have been identified for each of the three MEN2 variants. Thus in 95% of patients, MEN2a is associated with mutations of the cysteine-rich extracellular domain, and mutations in codon 634 (Cys→Arg) account for 85% of MEN2A mutations. However, a search for c-ret mutations in sporadic non-MEN2a parathyroid adenomas revealed no codon 634 mutations.172,173 MTC-only is also associated with missense mutations in the cysteine-rich extracellular domain, and most mutations are in codon 618. However, MEN2b is associated with mutations in codon 918 (Met→Thr) of the intracellular tyrosine kinase domain in 95% of patients. Interestingly, the c-ret proto-oncogene is also involved in the etiology of papillary thyroid carcinomas and in Hirschsprung’s disease. Mutational analysis of c-ret to detect mutations in codons 609, 611, 618, 634, 768, and 804 in MEN2a and MTC-only, and codon 918 in MEN2b, has been used in the diagnosis and management of patients and families with these disorders.171,174
Hyperparathyroidism–Jaw Tumor Syndrome Gene
The HPT–JT syndrome is an autosomal-dominant disorder characterized by the development of parathyroid adenomas and carcinomas, and fibro-osseous jaw tumors.175,176 In addition, some patients may also develop uterine tumors and renal abnormalities, which include Wilms’ tumors, renal cysts, renal hamartomas, renal cortical adenomas, and papillary renal cell carcinomas.177 Other tumors, including pancreatic adenocarcinomas, testicular mixed germ cell tumors with a major seminoma component, and Hurthle cell thyroid adenomas have also been reported in some patients.132,177 It is important to note that the parathyroid tumors may occur in isolation and without any evidence of jaw tumors, and this may cause confusion with other hereditary hypercalcemic disorders such as MEN1, familial benign hypercalcemia (FBH) (which is also referred to as familial hypocalciuric hypercalcemia [FHH]), and FIHP.178 HPT–JT can be distinguished from FBH, because in FBH, serum calcium levels are elevated during the early neonatal or infantile period, whereas in HPT–JT, such elevations are uncommon in the first decade. In addition, HPT–JT patients, unlike FBH patients, will have associated hypercalciuria. The distinction between HPT–JT patients and MEN1 patients, who have only developed the usual first manifestation of hypercalcemia (>90% of patients), is more difficult and is likely to be influenced by operative and histologic findings and by the occurrence of other characteristic lesions in each disorder. It is important to note that HPT–JT patients will usually have single adenomas or a carcinoma, but MEN1 patients will often have multiglandular parathyroid disease. The distinction between FIHP and HPT–JT in the absence of jaw tumors is difficult but important; HPT–JT patients may be at a higher risk of developing parathyroid carcinomas.179–181 These distinctions may be helped by the identification of additional features, and a search for jaw tumors, renal, pancreatic, thyroid, and testicular abnormalities may help to identify HPT–JT patients. The jaw tumors in HPT–JT are different from the brown tumors observed in some patients with primary hyperparathyroidism and do not resolve after parathyroidectomy.178 Indeed, ossifying fibromas of the jaw are an important distinguishing feature of HPT–JT from FIHP, and the occurrence of these may occasionally precede the development of hypercalcemia in HPT–JT patients by several decades. The gene causing HPT–JT is located on chromosome 1q31.2 and consists of 17 exons that encode a ubiquitously expressed and evolutionarily conserved 531-amino-acid protein, designated parafibromin.132,182 This gene, CDC73, is also referred to as HRPT2 (i.e., hyperparathyroidism type 2). HRPT2 mutations associated with HPT–JT were found to be scattered throughout the 1593-bp coding region, with the majority (>80%) predicting a functional loss through premature truncation. A genotype-phenotype correlation was not apparent from these analyses.177,182,184 The observation of LOH involving the chromosome lq31.2 region in HPT–JT-associated tumors indicated that parafibromin may be acting as a tumor suppressor, consistent with Knudson’s two-hit hypothesis.177,178,182 This was supported by the observations of germline and somatic HRPT2 mutations in HPT–JT-associated tumors.177,182–185 Similar germline and somatic HRPT2 mutations have also been found in sporadic parathyroid carcinomas, and the frequency of such mutations is high, ranging from 67% to 100%183,186; however, the frequency of HRPT2 mutations in sporadic parathyroid adenomas is low at 0% to 4%, indicating that HRPT2 mutations likely confer an aggressive growth potential to the parathyroid cells.182–184,186,187 HRPT2 mutations and allelic imbalances have also been identified in sporadic renal tumors,188 and a loss or down-regulation of HRPT2 expression has been reported in both breast and gastric cancers.189,190 These studies indicate that HRPT2 and its encoded protein, parafibromin, play a critical role in inherited and sporadic parathyroid cancers as well as other nonhereditary solid tumors. The role of parafibromin, which is predominantly a nuclear protein with a monopartite NLS,191 was not readily apparent, inasmuch as it has no homologies to known proteins. However, the approximately 200 amino acids of the C-terminal domain shared over 25% sequence identity with the yeast Cdc73 protein which is a component of the yeast polymerase-associated factor 1 (PAF1) complex, a key transcriptional regulatory complex that interacts directly with RNA polymerase II.182,192,193 Studies of the PAF1 complex in yeast and Drosophila, as well as in mammalian cells, have revealed that parafibromin, as part of the PAF1 complex, is a mediator of the key transcriptional events of histone modification, chromatin remodeling, initiation and elongation, and the wnt/β-catenin signaling pathway.192–194 Studies of a mouse deleted for HRPTt2 have revealed that HRPT2 expression and the PARAFIBROMIN/PAF complex directly regulate genes (e.g., H19, IgF1, Igf3, Igfbp4, Hmga1, Hmga2, and Hmga3) that are involved in cell growth and apoptosis.195
LRP5 And The Wnt/β-Catenin Pathway
Aberrant Wnt/β-catenin signaling with an accumulation of β-catenin in the cytoplasm and nucleus is associated with several types of tumor development (e.g., adenomatous polyposis coli and colorectal cancer). Investigations of this pathway in parathyroid tumors have revealed that β-catenin accumulation occurs in parathyroid adenomas and in parathyroid tumors associated with chronic renal failure.196 In addition, a protein-stabilizing mutation, Ser37Ala, in exon 3 of β-catenin was detected in over 7% of parathyroid adenomas but not parathyroid tumors of chronic renal failure, from Swedish patients197,198 but not North American199 or Japanese200 patients. The Ser37Ala β-catenin mutations were homozygous in the parathyroid adenomas, which had a higher expression of β-catenin and the nonphosphorylated active form of β-catenin.198 In addition, MYC, which is a direct target of the Wnt/β-catenin signaling pathway in colorectal cancer cells and a critical mediator of the early stages of intestinal neoplasia, was also overexpressed, and the stable activity of endogenous β–catenin was found to be necessary for MYC and cyclin D1 expression.196 Stability of β-catenin is regulated by Wnt ligands, which bind to the cell-surface frizzled receptors and LRP5 and LRP6 co-receptors that alter phosphorylation of several intracellular second messengers and consequently accumulation of nonphosphorylated β-catenin. Investigation of the Wnt signaling pathway in parathyroid tumors revealed that over 85% of adenomas and 100% of tumors from chronic renal failure patients have a shorter LRP5 transcript, which contained an in-frame deletion of 142 amino acids (residues 666 to 809) that encompassed the third YWTD beta propeller domain between the second and third epidermal growth factor repeats.197 This internally truncated LRP5 receptor activated β-catenin signaling in parathyroid tumors by a mechanism that may involve an impaired inhibitory action of the WNT antagonist, DKK1.197 The parathyroid tumors expressing the internally truncated LRP5 receptor did not harbor the β-catenin stabilizing mutation, Ser37Ala, and those that had the stabilizing β-catenin mutation did not express the truncated LRP5 receptor.197 Thus, it seems that the presence of the stabilizing β-catenin mutation and the expression of the truncated LRP5 receptor are mutually exclusive. However, these studies demonstrate an important role for the WNT β-catenin signaling pathway in parathyroid tumorigenesis.
Rb Gene
The Rb gene, which is a tumor-suppressor gene201 located on chromosome 13q14, is involved in the pathogenesis of retinoblastomas and a variety of common sporadic human malignancies including ductal breast, small cell lung carcinoma, and bladder carcinomas. Allelic deletion of the Rb gene has been demonstrated in all parathyroid carcinomas and in 10% of parathyroid adenomas202,203 and was accompanied by abnormal staining patterns for the Rb protein in 50% of the parathyroid carcinomas but in none of the parathyroid adenomas.202 These results demonstrate an important role for the Rb gene in the development of parathyroid carcinomas and may be of help in the histologic distinction of parathyroid adenoma from carcinoma.202 The Rb protein may also be secondarily involved in parathyroid tumorigenesis by its interaction with the retinoblastoma-interacting zinc finger protein 1, RIZ1.204 The findings of extensive deletions of the long arm of chromosome 13 (including the Rb locus) in some parathyroid adenomas and carcinomas,203 and similar findings in pituitary carcinomas,205 suggest that other tumor-suppressor genes on chromosome 13q may also have a role in the development of such tumors.
Retinoblastoma-Interacting Zinc Finger Protein 1 Gene On Chromosome 1P
Loss of heterozygosity studies have revealed allelic loss of chromosome 1p32-pter in 40% of sporadic parathyroid adenomas.206 This region is estimated to be about 110 cM, equivalent to about 110 million base pairs (Mbp) of DNA, but additional studies narrowed the interval containing this putative tumor-suppressor gene(s) to an approximately 4-cM (i.e., about 4-Mbp) region.207 Investigations of one candidate gene, retinoblastoma-interacting zinc finger protein 1 (RIZ1), have revealed that over 25% of parathyroid tumors had LOH of the RIZ1 locus, and that over 35% of parathyroid tumors had hypermethylation of the RIZ1 promoter region.204 Moreover, the RIZ1 promoter hypermethylation was related to LOH in these tumors, indicating that these two events may represent the “two hits” (see Fig. 5-4B) required for tumor development in Knudson’s hypothesis for tumorigenesis.204
Nonsyndromic Familial Isolated Hyperparathyroidism
FIHP may represent an incomplete manifestation of a syndromic form such as MEN1, FHH, or HPT-JT.163,164,182,208 Twenty-nine MEN1, nine HRPT2 and five CaSR germline mutations have been reported to date in FIHP kindreds.164 However, it is important to note that the genetic etiology of nonsyndromic FIHP in the majority of families remains to be elucidated.209,210 Thus, studies of 32 kindreds with nonsyndromic FIHP for mutations of the MEN1, CaSR, and HRPT2 genes have revealed that only one family harbored a germline mutation, and this involved the HRPT2 gene that encodes parafibromin.209,210 However, studies of 10 other FIHP kindreds have indicated that another locus, referred to as HRPT3, on chromosome 2p13.3-p14, is likely to be involved in the etiology of nonsyndromic FIHP.211 Thus, the genes and their underlying abnormalities that lead to nonsyndromic FIHP remain to be identified.
Hyperparathyroidism In Chronic Renal Failure
Chronic renal failure is often associated with a form of secondary hyperparathyroidism that may subsequently result in the hypercalcemic state of “tertiary” hyperparathyroidism. The parathyroid proliferative response in this condition led to the proposal that the autonomous parathyroid tissue might have undergone hyperplastic change and therefore be polyclonal in origin. However, studies of X-chromosome inactivation in parathyroids from patients on hemodialysis with refractory hyperparathyroidism have revealed at least one monoclonal parathyroid tumor in over 60% of patients.133 In addition, LOH involving several loci on chromosome Xp11 was detected in one of these parathyroid tumors, thereby suggesting the involvement of a tumor-suppressor gene from this region in the pathogenesis of such tumors.133 Interestingly, none of the parathyroid tumors from these patients with chronic renal failure had LOH involving loci from chromosome 11q13. This unexpected finding of monoclonal parathyroid tumors in the majority of patients with “tertiary” hyperparathyroidism suggests that an increased turnover of parathyroid cells in secondary hyperparathyroidism may possibly render the parathyroid glands more susceptible to mitotic nondisjunction or other mechanisms of somatic deletions, which may involve loci other than those—MEN1 and PRAD1—located on chromosome 11q13. In addition, as noted above, parathyroid tumors from patients with chronic renal failure have been shown to accumulate β-catenin and to have a truncated form of the LRP5 receptor, which lacks 142 amino acids.196,197
Disorders of The Calcium-Sensing Receptor
Three hypercalcemic disorders due to mutations and/or reduced activity of CaSR have been reported,208,212–217 which are FBH, also referred to as FHH, neonatal severe hyperparathyroidism (NSHPT), and autoimmune hypocalciuric hypercalcemia (AHH) (Table 5-3).
Table 5-3
Diseases Associated With Abnormalities of the Extracellular Calcium-Sensing Receptor (CaSR)
| CaSR Abnormality and Disease | CaSR Genotype |
| Loss-of-function CaSR mutation | |
| Familial benign hypercalcemia | Heterozygous |
| Neonatal severe primary hyperparathyroidism | Heterozygous or homozygous (mutant) |
| Gain-of-function CaSR mutation | |
| Autosomal-dominant hypocalcemic hypercalciuria | Heterozygous |
| Bartter syndrome type V | Heterozygous |
| CaSR autoantibodies | |
| Autoimmune hypocalciuric hypercalcemia | Homozygous (normal) |
| Acquired hypoparathyroidism | Homozygous (normal) |
Familial Benign Hypercalcemia and Neonatal Severe Hyperparathyroidism
Mutational analyses of the human CaSR, which is a G protein–coupled receptor located on chromosome 3q21.1,218 have revealed different mutations that result in a loss-of-function of the CaSR in patients with FBH and NSHPT.212–217 Many of these mutations cluster around the aspartate- and glutamate-rich regions (codons 39-300) within the extracellular domain of the receptor, and this has been proposed to contain low-affinity calcium-binding sites, based on similarities to that of calsequestrin, in which the ligand-binding pockets also contain negatively charged amino acid residues.208,219 Approximately two-thirds of the FBH kindreds investigated have been found to have unique heterozygous mutations of the CaSR, and expression studies of these mutations have demonstrated a loss of CaSR function whereby there is an increase in the calcium ion–dependent set-point for PTH release from the parathyroid cell.212,217,220,221 NSHPT occurring in the offspring of consanguineous FBH families has been shown to be due to homozygous CaSR mutations.212,213,215,222,223 However, some patients with sporadic neonatal hyperparathyroidism have been reported to be associated with de novo heterozygous CaSR mutations,214 thereby suggesting that factors other than mutant gene dosage222—for example, the degree of set-point abnormality, the bony sensitivity to PTH, and the maternal extracellular calcium concentration—may also all play a role in the phenotypic expression of a CaSR mutation in the neonate. The remaining third of FBH families in whom a mutation within the coding region of the CaSR has not been demonstrated may either have an abnormality in the promoter of the gene or a mutation at one of the two other FBH loci that have been revealed by family linkage studies. One of these FBH loci is located on chromosome 19p and is referred to as FBH19p.224 Studies of another FBH kindred from Oklahoma that also suffered from progressive elevations in PTH, hypophosphatemia, and osteomalacia225,226 demonstrated that this variant, designated FBHOk, was linked to chromosome 19q13.227 These three FBH loci located on chromosomes 3q, 19p, and 19q have also been referred to as FBH (or FHH) types 1, 2, and 3, respectively.227
Autoimmune Hypocalciuric Hypercalcemia
Some patients who have the clinical features of FHH but not CaSR mutations may have AHH (see also Chapter 7). Four patients with AHH who all had other autoimmune manifestations have been reported228: three patients had antithyroid antibodies, and one had sprue with antigliadin and antiendomysial antibodies. These patients were shown to have circulating antibodies to the extracellular domain of the CaSR, and these antibodies stimulated PTH release from dispersed human parathyroid cells in vitro, probably by inhibiting the activation of the CaSR by extracellular calcium.228 Thus, AHH is a condition of extracellular calcium sensing that should be considered in FHH patients who do not have CaSR mutations.
Jansen’s Disease
Jansen’s disease (Figs. 5-5 and 5-6) is an autosomal-dominant disease that is characterized by short-limbed dwarfism caused by an abnormal regulation of chondrocyte proliferation and differentiation in the metaphyseal growth plate and associated, usually severe, hypercalcemia and hypophosphatemia despite normal or undetectable serum levels of PTH or PTHrP.229 These abnormalities are caused by mutations in the PTH/PTHrP receptor that lead to constitutive, PTH- and PTHrP-independent receptor activation.230–232 Three different heterozygous mutations of the PTH/PTHrP receptor have been identified in the severe form of Jansen’s disease, and these involve codon 223 (His→Arg), codon 410 (Thr→Pro), and codon 458 (Ile→Arg) (Fig. 5-7). Expression of the mutant receptors in COS-7 cells result in constitutive, ligand-independent accumulation of cAMP, while the basal accumulation of inositol phosphates is not measurably increased.230–232 Since the PTH/PTHrP receptor is most abundantly expressed in kidney and bone and in the metaphyseal growth plate, these findings provide a likely explanation for the abnormalities observed in mineral homeostasis and growth plate development associated with this disorder. This conclusion is supported further by observations in mice which express the human PTH/PTHrP receptor, with the His223Arg mutation under the control of the rat α1(II) promoter.233 This promoter targeted expression of the mutant receptor to the layer of proliferative chondrocytes, delayed their differentiation into hypertrophic cells, and led, at least in animals with multiple copies of the transgene, to a mild impairment in growth of long bones. These observations are consistent with the conclusion that expression of a constitutively active human PTH/PTHrP receptor in growth plate chondrocytes causes the characteristic metaphyseal changes in patients with Jansen’s disease.
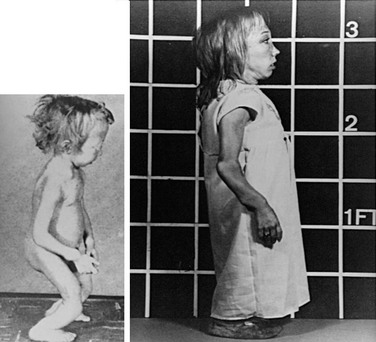
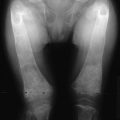
FIGURE 5-5 Patient with Jansen’s metaphyseal chondrodysplasia at ages 5 (left) and 22 years (right). (From Frame B, Poznanski AK: Conditions that may be confused with rickets. In DeLuca HF, Anast CS editors: Pediatric diseases related to calcium, New York, 1980, Elsevier, pp 269–289.341)
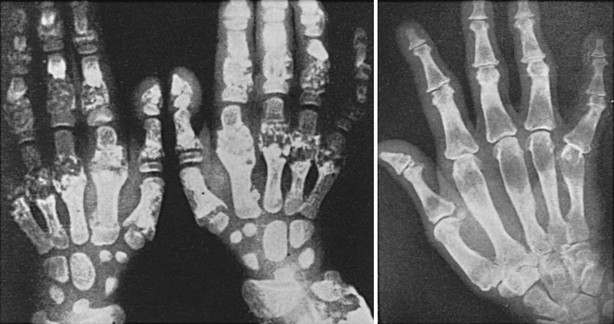
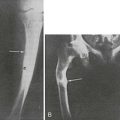
FIGURE 5-6 Hand radiographs at ages 10 (left) and 44 years (right) of the patient first described by Jansen. (From De Haas WHD, De Boer W, Griffioen F, et al: Metaphysial dysostosis: a late follow-up of the first reported case, J Bone Joint Surg Br 51:290–299, 1969.342)
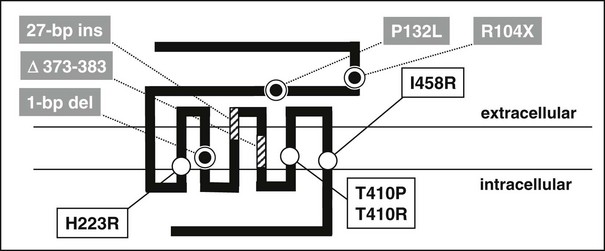
FIGURE 5-7 Schematic representation of the human PTH/PTHrP receptor. Approximate locations of heterozygous missense mutations that lead to constitutive receptor activation in patients with Jansen’s disease are indicated by open circles. Homozygous loss-of-function mutations identified in patients with Blomstrand’s disease are indicated by striped boxes or grey circles. The nucleotide exchange in exon M5 of the maternal PTH/PTHrP receptor allele introduces a novel splice acceptor site that leads to synthesis of an abnormal receptor protein which lacks portions of the fifth membrane-spanning domain (stippled box); for yet unknown reasons, the paternal allele is not expressed in this patient (see text for details).
Another novel heterozygous PTH/PTHrP receptor mutation, T410R, was identified in several members of a small kindred with an apparently mild form of Jansen’s disease.234 Affected individuals had, compared to patients with the previously identified activating PTH/PTHrP receptor mutations,230–232 less severe growth plate abnormalities, relatively normal stature, normal plasma calcium concentration, yet significant hypercalciuria and normal or suppressed plasma PTH levels. When tested in vitro, the PTH/PTHrP receptor with the T410R mutation showed less constitutive activity than that observed with the previously described T410P mutant.231,235 This less pronounced agonist-independent cAMP accumulation induced by the T410R mutation is consistent with the less severe skeletal and laboratory abnormalities observed in this milder form of Jansen’s disease.
Williams Syndrome
Williams syndrome is an autosomal-dominant disorder characterized by supravalvular aortic stenosis, elfin-like facies, psychomotor retardation, and infantile hypercalcemia. The underlying abnormality of calcium metabolism remains unknown, but abnormal 1,25(OH)2D3 metabolism or decreased calcitonin production have been implicated, although none have been consistently demonstrated. Studies have demonstrated hemizygosity at the elastin locus on chromosome 7q11.23 in over 90% of patients with the classical Williams phenotype,236–238 and only one patient had a cytogenetically identifiable deletion, thereby indicating that the syndrome is usually due to a microdeletion of 7q11.23.238 Interestingly, ablation of the elastin gene in mice results in vascular abnormalities similar to those observed in patients with Williams syndrome.239 However, the microdeletions that have been reported involve also another gene, designated LIM-kinase, that is expressed in the central nervous system.240 The calcitonin receptor gene, which is located on chromosome 7q21, is not involved in the deletions found in Williams syndrome and is therefore unlikely to be implicated in the hypercalcemia of such children.241 While deletion of the elastin and LIM-kinase genes can explain the respective cardiovascular and neurologic features of Williams syndrome, it seems likely that another, as yet uncharacterized, gene that is within this contiguously deleted region is involved in this disorder and could explain the abnormalities of calcium metabolism.
Hypocalcemic Disorders
Parathyroid Hormone Gene Abnormalities
DNA sequence analysis of the PTH gene (see Fig. 5-2) from one patient with autosomal-dominant isolated hypoparathyroidism has revealed a single base substitution (T→C) in exon 2,242 which resulted in the substitution of arginine (CGT) for cysteine (TGT) in the signal peptide. The presence of this charged amino acid in the midst of the hydrophobic core of the signal peptide impeded the processing of the mutant preproPTH, as demonstrated by in vitro studies. These revealed that the mutation impaired the interaction of the nascent protein with the translocation machinery and that cleavage of the mutant signal sequence by solubilized signal peptidase was ineffective.242,243 Studies using transfected HEK293 cells showed that the mutant PTH is trapped intracellularly, predominantly in the endoplasmic reticulum (ER), which is toxic for the cells and leads to apoptosis.244 In another family with autosomal-recessive hypoparathyroidism, a single base substitution (T→C) involving codon 23 of exon 2 was detected. This resulted in the substitution of proline (CCG) for the normal serine (TCG) in the signal peptide.245 This mutation alters the −3 position of the pre-pro-PTH protein cleavage site. Indeed, amino acid residues at the −3 and −1 positions of the signal peptidase recognition site have to conform to certain criteria for correct processing through the rough endoplasmic reticulum (RER), and one of these is an absence of proline in the region −3 and +1 of the site. Thus, the presence of a proline, which is a strong helix-breaking residue, at the −3 position is likely to disrupt cleavage of the mutant pre-pro-PTH that would be subsequently degraded in the RER, and PTH would not be available.245 Another abnormality of the PTH gene, involving a donor splice site at the exon 2/intron 2 boundary, has been identified in one family with autosomal-recessive isolated hypoparathyroidism.246 This mutation involved a single base transition (g→c) at position 1 of intron 2, and an assessment of the effects of this alteration in the invariant gt dinucleotide of the 5′ donor splice site consensus on mRNA processing revealed that the mutation resulted in exon skipping, in which exon 2 of the PTH gene was lost, and exon 1 was spliced to exon 3. The lack of exon 2 would lead to a loss of the initiation codon (ATG) and the signal peptide sequence (see Fig. 5-2), which are required for the commencement of PTH mRNA translation and the translocation of the PTH peptide, respectively.
Glial Cells Missing B Abnormalities
Glial cells missing B (GCMB), which is the human homolog of the Drosophila gene, Gcm, and of the mouse gcm2 gene, is expressed exclusively in the parathyroid glands, suggesting that it may be a specific regulator of parathyroid gland development.247 Mice that were homozygous (−/−) for deletion of gcm2 lacked parathyroid glands and developed the hypocalcemia and hyperphosphatemia observed in hypoparathyroidism.247 However, despite their lack of parathyroid glands, Gcm2-deficient (−/−) mice did not have undetectable serum PTH levels, but instead had levels indistinguishable from those of eucalcemic normal (+/+, wild-type) and heterozygous (+/−) mice. This endogenous level of PTH in the Gcm2-deficient (−/−) mice was too low to correct the hypocalcemia, but exogenous continuous PTH infusion could correct the hypocalcemia.247 Interestingly, there were no compensatory increases in PTHrP or 1,25(OH)2D3. These findings indicate that Gcm2 mice have a normal response (and not resistance) to PTH, and that the PTH in the serum of Gcm2-deficient mice was active. The auxiliary source of PTH was identified to be a cluster of PTH-expressing cells under the thymic capsule. These thymic PTH-producing cells also expressed the CaSR, and long-term treatment of the Gcm2-deficient mice with 1,25(OH)2D3 restored the serum calcium concentrations to normal and reduced the serum PTH levels, thereby indicating that the thymic production of PTH can be down-regulated.247 However, it appears that this thymic production of PTH cannot be up-regulated, because serum PTH levels are not high despite the hypocalcaemia in the Gcm2-deficient mice. This absence of up-regulation would be consistent with the very small size of the thymic PTH-producing cell cluster when compared to the size of normal parathyroid glands. The development of the thymic PTH-producing cells likely involves Gcm1, which is the other mouse homolog of Drosophila Gcm.248 Gcm1 expression, which could not be detected in parathyroid glands, colocalized with PTH expression in the thymus.247 The specific role of Gcm2 in the development of the parathyroids from the third pharyngeal pouch has been further investigated by studying the expression of the Hoxa3-Pax1/9-Eya1 transcription factor and Sonic hedgehog–bone morphogenetic protein 4 (Shh-Bmp4) signaling networks.249 These studies have revealed that Gcm2 (−/−) embryos that are 12 d.p.c. have a parathyroid-specific domain, but that this parathyroid domain undergoes coordinated programmed cell death (apoptosis) by 12.5 d.p.c. in the Gcm2-null mouse embryos.249 Moreover, the expression of the transcription factors Hoxa3, Pax1, Pax9, Eya1, Tbx1, and of Shh and Bmp4 was normal in the third pharyngeal pouch of these Gcm2-null mouse embryos. These findings indicate that the Hoxa3-Pax1/9-Eya transcription factor cascade, the transcription factor Tbx1, and the Shh-Bmp4 signaling network all act upstream of Gcm2.249 Moreover, these studies have revealed that Gcm2 has a role in promoting differentiation and survival of parathyroid cells in the developing embryo.249
Studies of patients with isolated hypoparathyroidism have shown that GCMB mutations are associated with autosomal-recessive and autosomal-dominant forms of the disease.250–252 A homozygous intragenic deletion of GCMB has been identified in a patient with autosomal-recessive hypoparathyroidism,250 while in another family, a homozygous missense mutation (Arg47Leu) of the DNA binding domain has been reported.251 Functional analysis, using electrophoretic mobility shift assays (EMSAs), of this Arg47Leu GCMB mutation revealed that it resulted in a loss of DNA binding to the GCM DNA binding site.251 More recently, heterozygous GCMB mutations, which consist of single nucleotide deletions (c1389deT and c1399delC) that introduce frameshifts and premature truncations, have been identified in two unrelated families with autosomal-dominant hypoparathyroidism (Fig. 5-8).252 By using a GCMB-associated luciferase reporter, both of these mutations were shown to inhibit the action of the wild-type transcription factor, thereby indicating that these GCMB mutants have dominant-negative properties.252
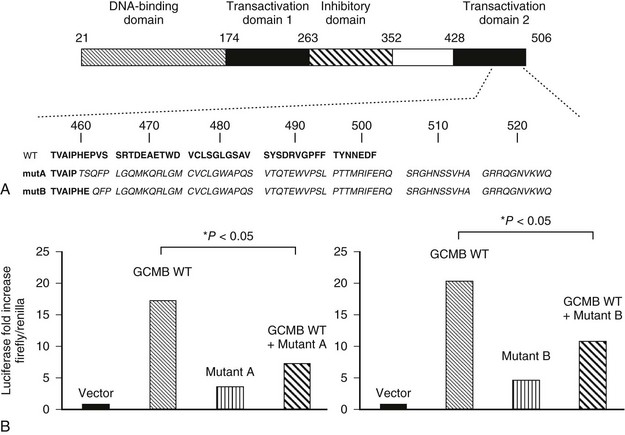
FIGURE 5-8 Heterozygous GCMB mutation can be a cause of autosomal-dominant isolated hypoparathyroidism. A, Domain structure of human GCMB and carboxyl-terminal amino acid sequence of wild-type human GCMB (WT) and of the mutants identified in families A (mutA) and B (mutB). Both single nucleotide deletions lead to a frameshift and translation of 65 novel amino acids after proline 464 and of 63 novel amino acids after glutamic acid 466, respectively. Novel amino acids are in italics. Numbering of amino acids above. B, Luciferase reporter assay using chicken fibroblast DF-1 cells. Cells were co-transfected with plasmids encoding wild-type human GCMB (GCMB WT), combined with either the c.1389delT mutant identified in family A (left), the c.1399delC mutant identified in family B (right), or empty vector; luciferase activity obtained with empty plasmid was defined as 1. (From Mannstadt M, Bertrand G, Muresan M, et al: Dominant-negative GCMB mutations cause an autosomal-dominant form of hypoparathyroidism, J Clin Endocrinol Metab 93:3568–3576, 2008.)
X-Linked Recessive Hypoparathyroidism
X-linked recessive hypoparathyroidism has been reported in two multigenerational kindreds from Missouri.253,254 In this disorder, only males are affected, and they suffer from infantile onset of epilepsy and hypocalcemia due to an isolated defect in parathyroid gland development.255 Relatedness of the two kindreds has been established by demonstrating an identical mitochondrial DNA sequence, inherited via the maternal lineage, in affected males from the two families.256 Studies utilizing X-linked polymorphic markers in these families localized the mutant gene to chromosome Xq26-q27,257 and a molecular deletion-insertion that involves chromosome 2p25 and Xq27 has been identified.258 This deletion-insertion is located approximately 67 kb downstream of SOX3, and hence it is likely to exert a position effect on SOX3 expression. Moreover, SOX3 was shown to be expressed in the developing parathyroids of mouse embryos, and this indicates a likely role for SOX3 in the embryonic development of the parathyroid glands.258 SOX3 belongs to a family of genes encoding high-mobility group (HMG) box transcription factors and is related to SRY, the sex-determining gene on the Y chromosome. The mouse homolog is expressed in the prestreak embryo and subsequently in the developing central nervous system (CNS) that includes the region of the ventral diencephalon, which induces development of the anterior pituitary and gives rise to the hypothalamus, olfactory placodes, and parathyroids.258–261 The location of the deletion-insertion approximately 67 kb downstream of SOX3 in X-linked recessive hypoparathyroid patients is likely to result in altered SOX3 expression, since SOX3 expression has been reported to be sensitive to position effects caused by X-chromosome abnormalities.262 Indeed, reporter construct studies of the mouse Sox3 gene have demonstrated the presence of both 5′ and 3′ regulatory elements,263 and thus it is possible that the deletion-insertion in the X-linked recessive hypoparathyroid patients may have a position effect on SOX3 expression and parathyroid development from the pharyngeal pouches. Indeed such position effects on SOX genes, which may be exerted over large distances, have been reported. For example, the very closely related Sox2 gene has been shown to have regulatory regions spread over a long distance, both 5′ and 3′ to the coding region,264 and disruption of sequences at some distance 3′ have been reported to lead to loss of expression in the developing inner ear and absence of sensory cells, whereas expression in other sites is unaffected.265 Similarly for the SRY gene, which probably originated from SOX3,266 both 5′ and 3′ deletions result in abnormalities of sexual development, and translocation breakpoints over 1 Mb upstream of the SOX9 gene have been reported to result in Campomelic dysplasia due to removal of elements that regulate SOX9 expression.262 The molecular deletion-insertion identified in X-linked recessive hypoparathyroidism may similarly cause position effects on SOX3 expression, and this points to a potential role for the SOX3 gene in the embryologic development of the parathyroid glands from the pharyngeal pouches.
Pluriglandular Autoimmune Hypoparathyroidism
Hypoparathyroidism may occur in association with candidiasis and autoimmune Addison’s disease, and the disorder has been referred to as either the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) syndrome or the autoimmune polyglandular syndrome type 1 (APS1).267 This disorder has a high incidence in Finland, and a genetic analysis of Finnish families indicated autosomal-recessive inheritance of the disorder.268 In addition, the disorder has been reported to have a high incidence among Iranian Jews, although the occurrence of candidiasis was less common in this population.269 Linkage studies of Finnish families mapped the APECED gene to chromosome 21q22.3.270 Further positional cloning approaches led to the isolation of a novel gene from chromosome 21q22.3. This gene, referred to as AIRE (autoimmune regulator), encodes a 545-amino-acid protein that contains motifs suggestive of a transcriptional factor and includes two zinc finger motifs, a proline-rich region, and three LXXLL motifs.271 Four AIRE1 mutations are commonly found in APECED families: (1) Arg257 Stop in Finnish, German, Swiss, British, and Northern Italian families; (2) Arg139 Stop in Sardinian families; (3) Tyr85Cys in Iranian Jewish families; and (4) a 13-bp deletion in exon 8 in British, Dutch, German, and Finnish families.271–275 AIRE1 has been shown to regulate the elimination of organ-specific T cells in the thymus, and thus APECED is likely to be caused by a failure of this specialized mechanism for deleting forbidden T cells and establishing immunologic tolerance.276 Patients with APS1 may also develop other autoimmune disorders in association with organ-specific autoantibodies, which are similar to those in patients with non-APS1 forms of the disease. Examples of such autoantibodies and related diseases are GAD6S autoantibodies in diabetes mellitus type 1A and 21-hydroxylase autoantibodies in Addison’s disease. Patients with APS1 may also develop autoantibodies that react with specific autoantigens that are not found in non-APS1 patients. Examples of this are autoantibodies to type 1 interferon omega (IFN-ω), which are present in all APS1 patients,277 and to NACHT leucine-rich-repeat protein 5 (NALP5), which is a parathyroid-specific autoantibody present in 49% of patients with APS1 associated hypoparathyroidism.278 NALP proteins are essential components of the inflammasome and activate the innate immune system in different inflammatory and autoimmune disorders such as vitiligo, which involves NALP1, and gout, which involves NALP3.279 The precise role of NALP5 in APS1-associated hypoparathyroidism remains to be elucidated.
Digeorge Syndrome
Patients with the DiGeorge syndrome (DGS) typically suffer from hypoparathyroidism; immunodeficiency; congenital heart defects; and deformities of the ear, nose, and mouth. The disorder arises from a congenital failure in the development of the derivatives of the third and fourth pharyngeal pouches, with resulting absence or hypoplasia of the parathyroids and thymus. Most cases of DGS are sporadic, but an autosomal-dominant inheritance of DGS has been observed. An association between the syndrome and an unbalanced translocation and deletions involving 22q11.2 have also been reported,280 and this is referred to as DGS type 1 (DGS1). In some patients, deletions of another locus on chromosome 10p have been observed in association with DGS,281 and this is referred to as DGS type 2 (DGS2). Mapping studies of the DGS1 deleted region on chromosome 22q11.2 have defined a 250-kb to 3000-kb critical region282,283 that contained approximately 30 genes. Studies of DGS1 patients have reported deletions of several of the genes (e.g., rnex40, nex2.2–nex3, UDF1L, and TBX1) from the critical region,280,284–286 and studies of transgenic mice deleted for such genes (e.g., Udf1l, Hira, and Tbx1) have revealed developmental abnormalities of the pharyngeal arches.287–289 However, point mutations in DGS1 patients have only been detected in the TBX1 gene,290 and TBX1 is now considered to be the gene causing DGS1.291 TBX1 is a DNA-binding transcriptional factor of the T-Box family and known to have an important role in vertebrate and invertebrate organogenesis and pattern formation. The TBX1 gene is deleted in roughly 96% of all DGS1 patients. Moreover, DNA sequence analysis of unrelated DGS1 patients who did not have deletions of chromosome 22q11.2 revealed the occurrence of three heterozygous point mutations.290 One of these mutations resulted in a frameshift with a premature truncation; the other two were missense mutations (Phe148Tyr and Gly310Ser). All of these patients had the complete pharyngeal phenotype but did not have mental retardation or learning difficulties. Interestingly, transgenic mice with deletion of Tbx1 have a phenotype that is similar to that of DGS1 patients.289 Thus, Tbx1-null mutant mice (−/−) had all the developmental anomalies of DGS1 (i.e., thymic and parathyroid hypoplasia; abnormal facial structures and cleft palate; skeletal defects; and cardiac outflow tract abnormalities), whereas Tbx1 haploinsufficiency in mutant mice (+/−) was associated only with defects of the fourth branchial pouch (i.e., cardiac outflow tract abnormalities). The basis of the phenotypic differences between DGS1 patients, who are heterozygous, and the transgenic (+/−) mice remain to be elucidated. It is plausible that Tbx1 dosage, together with the downstream genes that are regulated by Tbx1 could provide an explanation, but the roles of these putative genes in DGS1 remains to be elucidated.
Some patients may have late-onset DGS1, and these develop symptomatic hypocalcemia in childhood or during adolescence, with only subtle phenotypic abnormalities.292,293 These late-onset DGS1 patients have similar microdeletions in the 22q11 region. It is of interest to note that the age of diagnosis in the families of the three DGS1 patients with inactivating TBX1 mutations ranged from 7 to 46 years, which is in keeping with late-onset DGS1.290
Hypoparathyroidism, Deafness, and Renal Anomalies Syndrome
The combined inheritance of hypoparathyroidism, deafness, and renal dysplasia (HDR) as an autosomal-dominant trait was reported in one family in 1992.294 Patients had asymptomatic hypocalcemia, with undetectable or inappropriately normal serum concentrations of PTH and normal brisk increases in plasma cAMP in response to the infusion of PTH. The patients also had bilateral symmetrical sensorineural deafness involving all frequencies. The renal abnormalities consisted mainly of bilateral cysts that compressed the glomeruli and tubules and led to renal impairment in some patients. Cytogenetic abnormalities were not detected, and abnormalities of the PTH gene were excluded.294 However, cytogenetic abnormalities involving chromosome 10p14-10pter were identified in two unrelated patients with features that were consistent with HDR. These two patients suffered from hypoparathyroidism, deafness, and growth and mental retardation. One patient also had a solitary dysplastic kidney with vesico-ureteric reflux and a uterus bicornis unicollis, and the other patient, who had a complex reciprocal insertional translocation of chromosomes 10p and 8q, had cartilaginous exostoses.295 Neither of these patients had immunodeficiency or heart defects, which are key features of DGS2 (see earlier), and further studies defined two non-overlapping regions; thus, the DGS2 region was located on 10p13-14 and HDR on 10p14-10pter. Deletion mapping studies in two other HDR patients further defined a critical 200-kb region that contained GATA3,295 which belongs to a family of zinc finger transcription factors that are involved in vertebrae embryonic development. DNA sequence analysis in other HDR patients identified mutations that resulted in a haploinsufficiency and loss of GATA3 function.295–298 GATA3 has two zinc fingers; the C-terminal finger (ZnF2) binds DNA, and the N-terminal finger (2n-F1) stabilizes this DNA binding and interacts with other zinc finger proteins, such as the Friends of GATA (FOG).299 HDR-associated mutations involving GATA3 ZnF2 or the adjacent basic amino acids were found to result in a loss of DNA binding, whereas those involving ZnF1 either lead to a loss of interaction with FOG2 ZnFs or altered DNA binding affinity.297,298,300 These findings are consistent with the proposed three-dimensional model of GATA3 ZnF1, which has separate DNA-binding and protein-binding surfaces.297,298,301
Thus, the HDR-associated GATA3 mutations can be subdivided into two broad classes that depend upon (1) whether they disrupt ZnF1 or ZnF2 and (2) their subsequent effects on interactions with FOG2 and altered DNA binding, respectively. The majority (>75%) of these HDR-associated mutations are predicted to result in truncated forms of the GATA3 protein. Each proband and family will generally have its own unique mutation, and there appears to be no correlation with the underlying genetic defect and the phenotypic variation (e.g., presence or absence of renal dysplasia). Over 90% of patients with two or three of the major clinical features of the HDR syndrome—hypoparathyroidism, deafness, or renal abnormalities—have a GATA3 mutation.298 The remaining 10% of HDR of patients who do not have a GATA3 mutation of the coding region may harbor mutations in the regulatory sequences flanking the GATA3 gene, or else they may represent heterogeneity. The phenotypes of HDR patients with GATA3 mutations appear to be similar to those without GATA3 mutations.298
The HDR phenotype is consistent with the expression pattern of GATA3 during human and mouse embryogenesis in the developing kidney, otic vesicle, and parathyroids. However, GATA3 is also expressed in the developing central nervous system (CNS) and the hematopoietic organs in man and mice, and this suggests that GATA3 may have a more complex role. Indeed, studies of mice that are deleted for a Gata3 allele (+/−), or both Gata3 alleles (−/−) have revealed important roles for Gata3 in the development of the brain, spinal cord, peripheral auditory system, T-cells, fetal liver hematopoiesis, and urogenital system.302 Gata3+/− mice are viable, appear to be normal with a normal life span, and are fertile.302 However, Gata3+/− mice have hearing loss that is associated with cochlear abnormalities which consist of a significant progressive morphologic degeneration that starts with the outer hair cells at the apex and eventually involves all the inner hair cells, pillar cells, and nerve fibers.303,304 These studies have shown that hearing loss in Gata3 haploinsufficiency commences in the early postnatal period and is progressive through adulthood, and that it is peripheral in origin and is predominantly due to malfunctioning of the outer hair cells of the cochlea.303,304 Gata 3−/− mice are embryonically lethal, and these null embryos die between 11 to 12 dpc.302 Examination of these Gata3−/− embryos revealed a variety of abnormalities that included massive internal bleeding resulting in anemia, marked growth retardation, severe deformities of the brain and spinal cord, a hypopigmented retina, gross aberrations in fetal liver hematopoiesis, a total block of T-cell differentiation, and a retarded or missing lower jaw area.302,305 These Gata3−/− mice had an anatomically normal sympathetic nervous system, yet the sympathetic ganglia lacked tyrosine hydroxylase and dopamine β-hydroxylase, which are key enzymes that convert tyrosine to l-dihydroxyphenylalanine (l-dopa), and dopamine to noradrenaline, respectively, in the catecholamine synthesis pathway. Thus, the Gata3−/− mice lacked noradrenaline in the sympathetic neurons, and this was contributing to the early embryonic lethality.305 Feeding of catecholamine intermediates to the pregnant dams helped to partially rescue the Gata3−/− embryos to 12.5 to 16.5 dpc. These older, pharmacologically rescued Gata3−/− embryos showed abnormalities that could not be detected in the untreated mice.305 These late embryonic defects included thymic hypoplasia, a thin-walled ventricular septum, a poorly developed mandible, other developmental defects in structures derived from the cephalic neural crest cells, renal hypoplasia, a failure to form the metanephros, and an aberrant elongation of the nephric duct along the anteroposterior axis of the embryo.305,306 The defect of the nephric duct, which consisted of an abnormal morphogenesis and guidance in the developing kidney, was characterized by the loss of Ret expression that is an essential component of the glial-derived nerve factor (GDNF) signaling pathway involved in ureteric bud formation and nephric duct guidance.306 Thus, Gata3 has a role in the differentiation of multiple cell lineages during embryogenesis, as well as key regulation of nephric duct morphogenesis and guidance of the nephric duct in its caudal extension in the pro/mesonephric kidney.305,306
It is important to note that HDR patients with GATA3 haploinsufficiency do not have immune deficiency, and this suggests that the immune abnormalities observed in some patients with 10p deletions are most likely to be caused by other genes on 10p. Similarly, the facial dysmorphism and growth and developmental delay commonly seen in patients with larger 10p deletions were absent in the HDR patients with GATA3 mutations, further indicating that these features were likely due to other genes on 10p.295 These studies of HDR patients clearly indicate an important role for GATA3 in parathyroid development and in the etiology of hypoparathyroidism.
Mitochondrial Disorders Associated With Hypoparathyroidism
Hypoparathyroidism has been reported to occur in three disorders associated with mitochondrial dysfunction: the Kearns-Sayre syndrome (KSS), the MELAS syndrome, and a mitochondrial trifunctional protein deficiency syndrome (MTPDS). KSS is characterized by progressive external ophthalmoplegia and pigmentary retinopathy before the age of 20 years and is often associated with heart block or cardiomyopathy. The MELAS syndrome consists of a childhood onset of mitochondrial encephalopathy, lactic acidosis and stroke-like episodes. In addition, varying degrees of proximal myopathy can be seen in both conditions. Both the KSS and MELAS syndromes have been reported to occur with insulin-dependent diabetes mellitus and hypoparathyroidism.307,308 A point mutation in the mitochondrial gene tRNA leucine (UUR) has been reported in one patient with the MELAS syndrome who also suffered from hypoparathyroidism and diabetes mellitus.219 Large deletions, consisting of 6741 and 6903 bp and involving more than 38% of the mitochondrial genome, have been reported in other patients who suffered from KSS, hypoparathyroidism, and sensorineural deafness.309 Rearrangements and duplication of mitochondrial DNA have also been reported in KSS. Mitochondrial trifunctional protein deficiency (MTPDS) is a disorder of fatty acid oxidation that is associated with peripheral neuropathy, pigmentary retinopathy, and acute fatty liver degeneration in pregnant women who carry an affected fetus. Hypoparathyroidism has been observed in one patient with trifunctional protein deficiency.310 The role of these mitochondrial mutations in the etiology of hypoparathyroidism remains to be further elucidated.
Kenney-Caffey, Sanjad-Sakati, and Kirk-Richardson Syndromes
Hypoparathyroidism has been reported to occur in over 50% of patients with the Kenney-Caffey syndrome, which is associated with short stature, osteosclerosis and cortical thickening of the long bones, delayed closure of the anterior fontanel, basal ganglia calcification, nanophthalmos, and hyperopia.311 Parathyroid tissue could not be found in a detailed postmortem examination of one patient,312 and this suggests that hypoparathyroidism may be due to an embryologic defect of parathyroid development. In the Kirk-Richardson and Sanjad-Sakati syndromes, which are similar, hypoparathyroidism is associated with severe growth failure and dysmorphic features.313,314 This has been reported in patients of Middle Eastern origin.313,314 Consanguinity was noted in the majority of the families, indicating that this syndrome is inherited as an autosomal-recessive disorder. Homozygosity and linkage disequilibrium studies located this gene to chromosome 1q42-q43.315 Molecular genetic investigations have identified that mutations of the tubulin-specific chaperone (TBCE) are associated with the Kenney-Caffey and Sanjad-Sakati syndromes.316 TBCE encodes one of several chaperone proteins required for the proper folding of α-tubulin subunits and the formation of α-β tubulin heterodimers (see Fig. 5-3).316
Additional Familial Syndromes
Single familial syndromes in which hypoparathyroidism is a component have been reported (see Table 5-1). The inheritance of the disorder in some instances has been established, and molecular genetic analysis of the PTH gene has revealed no abnormalities. An association of hypoparathyroidism, renal insufficiency, and developmental delay has been reported in one Asian family in whom autosomal-recessive inheritance of the disorder was established; an analysis of the PTH gene in this family revealed no abnormalities.317 The occurrence of hypoparathyroidism, nerve deafness, and a steroid-resistant nephrosis leading to renal failure, which has been referred to as the Barakat syndrome,318 has been reported in four brothers from one family, and an association of hypoparathyroidism with congenital lymphedema, nephropathy, mitral valve prolapse, and brachytelephalangy has been observed in two brothers from another family.319 Molecular genetic studies have not been reported from these two families.
Calcium-Sensing Receptor Abnormalities
CaSR abnormalities are associated with three hypocalcemic disorders: autosomal-dominant hypocalcemic hypercalciuria (ADHH), Bartter syndrome type V (i.e., ADHH with a Bartter-like syndrome), and a form of autoimmune hypoparathyroidism due to CaSR autoantibodies (see Table 5-3).
Autosomal-Dominant Hypocalcemic Hypercalciuria
CaSR mutations that result in loss-of-function abnormalities are associated with familial benign (hypocalciuric) hypercalcemia.208,212–217 It was therefore postulated that gain-of-function mutations in CaSR lead to hypocalcemia with hypercalciuria, and the investigation of kindreds with autosomal-dominant forms of hypocalcemia have indeed identified such CaSR mutations.208,320–324 The hypocalcemic individuals generally had normal serum intact PTH concentrations and hypomagnesemia, and treatment with vitamin D or its active metabolites to correct the hypocalcemia resulted in marked hypercalciuria, nephrocalcinosis, nephrolithiasis, and renal impairment. The majority (>80%) of CaSR mutations that result in a functional gain are located within the extracellular domain,208,320–324 which is different from the findings in other disorders that are the result of activating mutations in G protein–coupled receptors.
Bartter Syndrome Type V
Bartter syndrome is a heterozygous group of autosomal-recessive disorders of electrolyte homeostasis characterized by hypokalemic alkalosis, renal salt wasting that may lead to hypotension, hyperreninemic hyperaldosteronism, increased urinary prostaglandin excretion, and hypercalciuria with nephrocalcinosis.325,326 Mutations of several ion transporters and channels have been associated with Bartter syndrome, and five types are now recognized.326 Type I is due to mutations involving the bumetanide-sensitive sodium-potassium-chloride co-transporter (NKCC2 or SLC12A2); type II is due to mutations of the outwardly rectifying renal potassium channel (ROMK); and type III is due to mutations of the voltage-gated chloride channel (CLC-Kb). Type IV is due to mutations of barttin, which is a beta subunit required for trafficking of CLC-Kb and CLC-Ka; this form is also associated with deafness, since barttin, CLC-Ka and CLC-Kb are also expressed in the marginal cells of the scala media of the inner ear that secrete potassium ion–rich endolymph. Type V is due to activating mutations of the CaSR. Patients with Bartter syndrome type V have the classic features of the syndrome: hypokalemic metabolic alkalosis, hyperreninemia, and hyperaldosteronism.63,327 In addition, they develop hypocalcemia, which may be symptomatic and lead to carpopedal spasm and an elevated fractional excretion of calcium that may be associated with nephrocalcinosis.63,327 Such patients have been reported to have heterozygous gain-of-function CaSR mutations, and in vitro functional expression of these mutations has revealed a more severe set-point abnormality for the receptor than that found in patients with ADHH.63,224,327 This suggests that the additional features occurring in Bartter syndrome type V but not in ADHH are due to severe gain-of-function mutations of the CaSR.326
Autoimmune Acquired Hypoparathyroidism
Twenty percent of patients who had acquired hypoparathyroidism in association with autoimmune hypothyroidism were found to have autoantibodies to the extracellular domain of the CaSR.228,328 The CaSR autoantibodies did not persist for long; 72% of patients who had acquired hypoparathyroidism for less than 5 years had detectable CaSR autoantibodies, whereas only 14% of patients with autoimmune hypoparathyroidism for more than 5 years had such autoantibodies.328 The majority of the patients who had CaSR autoantibodies were females, a finding that is similar to that found in other autoantibody-mediated diseases. Indeed, a few autoimmune hypoparathyroidism patients have also had features of autoimmune polyglandular syndrome type 1 (APS1). These findings establish that the CaSR is an autoantigen in autoimmune hypoparathyroidism.228,328
Blomstrand’s Disease
Blomstrand’s chondrodysplasia is an autosomal-recessive human disorder characterized by early lethality, dramatically advanced bone maturation, and accelerated chondrocyte differentiation.329 Affected infants are typically born to consanguineous healthy parents (only in one instance did unrelated healthy parents have two affected offspring),330–334 show pronounced hyperdensity of the entire skeleton (Fig. 5-9) and markedly advanced ossification, and particularly the long bones are extremely short and poorly modeled. PTH/PTHrP receptor mutations that impair its functional properties were identified as the most likely cause of Blomstrand’s disease (see Fig. 5-7). One of these defects is caused by a nucleotide exchange in exon M5 of the maternal PTH/PTHrP receptor allele, which introduces a novel splice acceptor site and thus leads to the synthesis of a receptor mutant that does not mediate, despite seemingly normal cell surface expression, the actions of PTH or PTHrP; the patient’s paternal PTH/PTHrP receptor allele is, for yet unknown reasons, only poorly expressed.335 In a second patient with Blomstrand’s disease, the product of a consanguineous marriage, a nucleotide exchange was identified that changes proline at position 132 to leucine.336,337 The resulting PTH/PTHrP receptor mutant showed, despite reasonable cell surface expression, severely impaired binding of radiolabeled PTH and PTHrP analogs, greatly reduced agonist-stimulated cAMP accumulation, and no measurable inositol phosphate response. Additional loss-of-function mutations of the PTH/PTHrP receptor have been identified in three unrelated patients with Blomstrand’s disease. Two of these mutations led to a frameshift and a truncated protein due either to a homozygous single nucleotide deletion in exon EL2 338 or a 27-bp insertion between exon M4 and EL2.339 The other defect was a nonsense mutation at residue 104 and thus resulted in a truncated receptor protein.339
FIGURE 5-9 Radiologic findings in a patient with Blomstrand’s disease. Note the markedly advanced ossification of all skeletal elements and the extremely short limbs, despite the comparatively normal size and shape of hands and feet. Furthermore, note that the clavicles are relatively long and abnormally shaped. (From Leroy JG, Keersmaeckers G, Coppens M, et al: Blomstrand lethal chondrodysplasia, Am J Med Genet 63:84–89, 1996.)
As in Jansen’s disease, the identification of mutant PTH/PTHrP receptors provided a plausible explanation for the severe abnormalities in endochondral bone formation in patients with Blomstrand’s disease. The disease is lethal, but it is likely that besides the striking skeletal defects, affected infants show abnormalities in other organs, including secondary hyperplasia of the parathyroid glands, presumably due to hypocalcemia. In addition, analysis of fetuses with Blomstrand’s disease have revealed abnormal breast development and tooth impaction, highlighting the involvement of the PTH/PTHrP receptor in the normal development of breast and tooth.340
References
1. Krane, SM. Calcium, phosphate and magnesium. In: Rasmussen H, ed. The International Encyclopedia of Pharmacology and Therapeutics. London: Pergamon Press; 1970:19.
2. Neer, R, Berman, M, Fisher, L, et al. Multicompartmental analysis of calcium kinetics in normal adult males. J Clin Invest. 1967;46:1364–1379.
3. Glimcher, MK, Krane, SM. Organization and structure of bone and the mechanism of calcification. In: Gould BS, Ramachandran GN, eds. Treatise on collagen, Part B. New York: Academic Press; 1968:68–241.
4. Carr, CW. Electrochemistry in biology and medicine. New York: John Wiley and Sons; 1955.
5. Bowers, GN, Brassard, C, Sena, SF. Measurement of ionized calcium in serum with ion-selective electrodes: a mature technology that can meet the daily service needs. Clin Chem. 1986;32:1437–1447.
6. Krabbe, S, Transbol, I, Christiansen, C. Bone mineral homeostasis, bone growth, and mineralization during years of pubertal growth: a unifying concept. Arch Dis Child. 1982;57:359–363.
7. Pitkin, RM, Reynolds, WA, Williams, GA, et al. Calcium metabolism in normal pregnancy: a longitudinal study. Am J Obstet. 1979;133:781–790.
8. Gertner, JM, Coustan, OR, Kliger, AS, et al. Pregnancy as a state of physiologic absorptive hypercalcemia. Am J Med. 1986;81:451–456.
9. Forbes, GB. Calcium accumulation by the human fetus. Pediatrics. 1976;57:976–977.
10. Pitkin, RM, Cruikshank, DP, Schauberger, CW, et al. Fetal calciotropic hormones and neonatal calcium homeostasis. Pediatrics. 1980;66:77–82.
11. Hillman, L, Sateesha, S, Haussler, M, et al. Control of mineral homeostasis during lactation: interrelationships of 25-hydroxyvitamin D, 24,25-dihydroxyvitamin D, 1,25-dihydroxyvitamin, parathyroid hormone, calcitonin, prolactin, and estradiol. Am J Obstet Gynecol. 1981;139:471–476.
12. Greer, FR, Tasang, RC, Searcy, JE, et al. Mineral homeostasis during lactation-relationship to serum 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D, parathyroid hormone, and calcitonin. Am J Clin Nutr. 1982;36:431–437.
13. Barritt, GJ. Receptor-activated Ca2+ inflow in animal cells: a variety of pathways tailored to meet different intracellular Ca2+ signalling requirements. Biochem J. 1999;337:153–169.
14. Berridge, MJ. Elementary and global aspects of calcium signaling. J Physiol. 1997;499:291–306.
15. Carafoli, E. Intracellular calcium homeostasis. Ann Rev Biochem. 1987;56:395–433.
16. Rasmussen, H, Barrett, PQ. Calcium messenger system: An integrated view. Physiol Rev. 1984;64:938–984.
17. Brown, E, Pollack, A, Hebert, S. The extracellular calcium-sensing receptor: its role in health and disease. Annu Rev Med. 1998;49:15–29.
18. Rasmussen, H. The calcium messenger system. N Engl J Med. 1986;314:1094–1101.
19. Heaney, RP, Gallagher, JC, Johnston, CC, et al. Calcium nutrition and bone health in the elderly. Am J Clin Nutr. 1982;36:986–1013.
20. Allen, LH. Calcium bioavailability and absorption: a review. Am J Clin Nutr. 1982;35:783–808.
21. Avioli, LV, McDonald, JE, Lee, SW. Influence of aging on the intestinal absorption of 47-Ca in women and its relation to 47-Ca absorption in postmenopausal osteoporosis. J Clin Invest. 1965;44:1960–1967.
22. Ireland, P, Fordtran, JS. Effect of dietary calcium and age on jejunal calcium absorption in humans studied by intestinal perfusion. J Clin Invest. 1973;52:2672–2681.
23. Gallagher, JC, Riggs, BL, Eisman, J, et al. Intestinal calcium absorption and serum vitamin D metabolites in normal subjects and osteoporotic patients. J Clin Invest. 1979;64:729–736.
24. Heaney, RP, Skillman, TG. Secretion and excretion of calcium by the human gastrointestinal tract. J Lab Clin Med. 1964;64:29–41.
25. Rose, GA, Reed, GW, Smith, AH. Isotopic method for measurement of calcium absorption from the gastrointestinal tract. Br Med J. 1965;1:690–692.
26. Phang, J, Berman, M, Finerman, G. Dietary perturbation of calcium metabolism in normal man: compartmental analysis. J Clin Invest. 1969;48:67–77.
27. Jung, A, Bartholdi, P, Mermillod, B. Critical analysis of methods of analyzing human calcium kinetics. J Theor Biol. 1978;73:131–157.
28. Fordtran, JS, Locklear, TW. Ionic constituents and osmolality of gastric and small-intestinal fluids after eating. Am J Dig Dis. 1966;11:503–521.
29. Bronner, F, Pansu, D, Stein, WD. An analysis of intestinal calcium transport across the rat intestine. Am J Physiol. 1986;250:G561–G569.
30. Marcus, CS, Lengemann, FW. Absorption of Ca45 and Sr85 from solid and liquid food at various levels of the alimentary tract of the rat. J Nutr. 1962;77:155–160.
31. Birge, J, Peck, WA, Berman, M, et al. Study of calcium absorption in man: a kinetic analysis and physiologic model. J Clin Invest. 1969;48:1705–1713.
32. Dano, P, Christiansen, C. Calcium absorption and bone mineral content following intestinal shunt operations for obesity: a comparison of three types of procedures. Scand J Gastroenterol. 1974;9:775–779.
33. Hoenderop, JG, van der Kemp, AW, Hartog, A, et al. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem. 1999;274:8375–8378.
34. Peng, JB, Chen, XZ, Berger, UV, et al. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J Biol Chem. 1999;274:22739–22746.
35. Vennekens, R, Prenen, J, Hoenderop, JG, et al. Pore properties and ionic block of the rabbit epithelial calcium channel expressed in HEK 293 cells. J Physiol. 2001;530:183–191.
36. Gunthorpe, MJ, Benham, CD, Randall, A, et al. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci. 2002;23:183–191.
37. Nilius, B, Vennekens, R, Prenen, J, et al. The single pore residue Asp542 determines Ca2+ permeation and Mg2+ block of the epithelial Ca2+ channel. J Biol Chem. 2001;276:1020–1025.
38. den Dekker, E, Hoenderop, JG, Nilius, B, et al. The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium. 2003;33:497–507.
39. Zhuang, L, Peng, JB, Tou, L, et al. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab Invest. 2002;82:1755–1764.
40. Wood, RJ, Tchack, L, Taparia, S. 1,25-Dihydroxyvitamin D3 increases the expression of the CaT1 epithelial calcium channel in the Caco-2 human intestinal cell line. BMC Physiol. 2001;1:11.
41. Van Cromphaut, SJ, Dewerchin, M, Hoenderop, JG, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A. 2001;98:13324–13329.
42. Song, Y, Kato, S, Fleet, JC. Vitamin D receptor (VDR) knockout mice reveal VDR-independent regulation of intestinal calcium absorption and ECaC2 and calbindin D9k mRNA. J Nutr. 2003;133:374–380.
43. Van Cromphaut, SJ, Rummens, K, Stockmans, I, et al. Intestinal calcium transporter genes are upregulated by estrogens and the reproductive cycle through vitamin D receptor-independent mechanisms. J Bone Miner Res. 2003;18:1725–1736.
44. Feher, JJ, Fullmer, CS, Wasserman, RH. Role of facilitated diffusion of calcium by calbindin in intestinal calcium absorption. Am J Physiol. 1992;262:C517–C526.
45. Ruchon, A, Tenenhouse, H, Marcinkiewicz, M, et al. Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. J Bone Miner Res. 2000;15:1440–1450.
46. Lassiter, WE, Gottschalk, CW, Mylle, M. Micropuncture study of renal tubular reabsorption on calcium in normal rodents. Am J Physiol. 1963;204:771–775.
47. Friedman, PA, Gesek, FA. Cellular calcium transport in renal epithelia: measurement, mechanisms, and regulation. Physiol Rev. 1995;75:429–471.
48. Bourdeau, JE, Burg, MB. Effects of PTH on calcium transport across the cortical thick ascending limb of Henle’s loop. Am J Physiol. 1979;238:F350.
49. Suki, WN, Rouse, D, Ng, RC, et al. Calcium transport in the thick ascending limb of Henle: heterogeneity of function in the medullary and cortical segments. J Clin Invest. 1980;66:1004–1009.
50. Shareghi, GR, Agus, ZS. Magnesium transport in the cortical thick ascending limb of Henle’s loop of the rabbit. J Clin Invest. 1982;69:759.
51. Bourdeau, JE, Burg, MB. Effect of PTH on calcium transport across the cortical thick ascending limb of Henle’s loop. Am J Physiol. 1980;239:F121–F126.
52. Friedman, PA. Basal and hormone-activated calcium absorption in mouse renal thick ascending limbs. Am J Physiol. 1988;254:F62–F70.
53. Simon, DB, Lu, Y, Choate, KA, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–106.
54. Weber, S, Hoffmann, K, Jeck, N, et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene. Eur J Hum Genet. 2000;8:414–422.
55. Blanchard, A, Jeunemaitre, X, Coudol, P, et al. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int. 2001;59:2206–2215.
56. Konrad, M, Schaller, A, Seelow, D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–957.
57. Quamme, GA. Effect of hypercalcemia on renal tubular handling of calcium and magnesium. Can J Physiol Pharmacol. 1982;60:1275–1280.
58. Riccardi, D, Hall, AE, Chattopadhyay, N, et al. Localization of the extracellular Ca2+/polyvalent cation-sensing protein in rat kidney. Am J Physiol. 1998;274:F611–F622.
59. Wang, WH, Lu, M, Hebert, SC. Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am J Physiol. 1996;271:C103–C111.
60. Wang, W, Lu, M, Balazy, M, et al. Phospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TAL. Am J Physiol. 1997;273:F421–F429.
61. Motoyama, HI, Friedman, PA. Calcium-sensing receptor regulation of PTH-dependent calcium absorption by mouse cortical ascending limbs. Am J Physiol Renal Physiol. 2002;283:F399–F406.
62. Barakat, AJ, Rennert, OM. Gitelman’s syndrome (familial hypokalemia-hypomagnesemia). J Nephrol. 2001;14:43–47.
63. Vargas-Poussou, R, Huang, C, Hulin, P, et al. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol. 2002;13:2259–2266.
64. Agus, ZS, Chiu, PJS, Goldberg, M. Regulation of urinary calcium excretion in the rat. Am J Physiol. 1977;232:F545.
65. Costanzo, LS, Windhager, EE. Effect of PTH, ADH and cAMP on distal tubular Ca and Na reabsorption. Am J Physiol. 1980;239:F478–F485.
66. Costanzo, LS. Comparison of calcium and sodium transport in early and late rat distal tubules: effect of amiloride. Am J Physiol. 1984;246:F937–F945.
67. Borke, JL, Caride, A, Verma, AK, et al. Plasma membrane calcium pump and 28-kDa calcium binding protein in cells of rat kidney distal tubules. Am J Physiol. 1989;257:F842–F849.
68. Ramachandran, C, Brunette, MG. The renal Na+/Ca++ exchange system is located exclusively in the distal tubule. Biochem J. 1989;257:259–264.
69. Reilly, RF, Ellison, DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80:277–313.
70. Hoenderop, JG, Hartog, A, Stuiver, M, et al. Localization of the epithelial Ca(2+) channel in rabbit kidney and intestine. J Am Soc Nephrol. 2000;11:1171–1178.
71. Kip, SN, Strehler, EE. Characterization of PMCA isoforms and their contribution to transcellular Ca2+ flux in MDCK cells. Am J Physiol Renal Physiol. 2003;284:F122–F132.
72. Loffing, J, Kaissling, B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am J Physiol Renal Physiol. 2003;284:F628–F643.
73. Shimizu, T, Yoshitomi, K, Nakamura, N, et al. Effects of PTH, calcitonin, and cAMP on calcium transport in rabbit distal nephron segments. Am J Physiol. 1990;259:F408–F414.
74. Bindels, RJ, Hartog, A, Timmermans, J, et al. Active Ca2+ transport in primary cultures of rabbit kidney CCD: stimulation by 1,25-dihydroxyvitamin D3 and PTH. Am J Physiol. 1991;261:F799–F807.
75. Hoenderop, JG, van Leeuwen, JP, van der Eerden, BC, et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest. 2003;112:1906–1914.
76. Lee, CT, Huynh, VM, Lai, LW, et al. Cyclosporine A-induced hypercalciuria in calbindin-D28k knockout and wild-type mice. Kidney Int. 2002;62:2055–2061.
77. Bacskai, BJ, Friedman, PA. Activation of latent Ca2+ channels in renal epithelial cells by parathyroid hormone. Nature. 1990;347:388–391.
78. Silva, IV, Cebotaru, V, Wang, H, et al. The ClC-5 knockout mouse model of Dent’s disease has renal hypercalciuria and increased bone turnover. J Bone Miner Res. 2003;18:615–623.
79. Christensen, EI, Devuyst, O, Dom, G, et al. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci U S A. 2003;100:8472–8477.
80. Epstein, FH. Calcium and the kidney. Am J Med. 1968;45:700–715.
81. Torikai, S, Wang, M-S, Klein, KL, et al. Adenylate cyclase and cell cyclic AMP of rat cortical thick ascending limb of Henle. Kidney Int. 1981;20:649–654.
82. Bijvoet, OLM, Kidney function in calcium and phosphate metabolism. Metabolic Bone Disease. Avioli, LV, Krane, SM, eds. Metabolic Bone Disease. New York: Academic Press; 1977;vol 1:49–140.
83. Peacock, M, Robertson, WG, Nordin, BEC. Relation between serum and urinary calcium with particular reference to parathyroid activity. Lancet. 1969;1:384–386.
84. Shareghi, GR, Stoner, LC. Calcium transport across segments of the rabbit distal nephron in vitro. Am J Physiol. 1978;235:F367–F375.
85. Suki, WN, Rouse, D. Hormonal regulation of calcium transport in thick ascending limb renal tubules. Am J Physiol. 1981;241:F171.
86. Hemmingsen, C. Regulation of renal calbindin-D28k. Pharmacol Toxicol. 2000;87:5–30.
87. Jayakumar, A, Cheung, L, Liang, CT, et al. Sodium gradient-dependent calcium uptake in renal basolateral membrane vesicles. J Biol Chem. 1984;259:10827–10833.
88. Bomsztyk, K, George, JP, Wright, FS. Effects of luminal fluid anions on calcium transport by proximal tubule. Am J Physiol. 1984;246:F600–F608.
89. Scoble, JE, Mills, S, Hruska, KA. Calcium transport in canine renal basolateral membrane vesicles, effect of parathyroid hormones. J Clin Invest. 1985;75:1096–1105.
90. Bouhtiauy, I, LaJeunesse, D, Brunette, MG. The mechanism of parathyroid hormone action on calcium reabsorption by the distal tubule. Endocrinology. 1991;128:251–258.
91. Shimizu, T, Nakamura, M, Yoshitomi, K, et al. Interaction of trichlormethiazide or amiloride with PTH in stimulating Ca2+ absorption in rabbit CNT. Am J Physiol. 1991;261:F36–F43.
92. Tsukamoto, Y, Saka, S, Saitoh, M. Parathyroid hormone stimulates ATP-dependent calcium pump activity by a different mode in proximal and distal tubules of the rat. Biochim Biophys Acta. 1992;1103:163–171.
93. Gesek, FA, Friedman, PA. On the mechanism of parathyroid hormone stimulation of calcium uptake by mouse distal convoluted tubule cells. J Clin Invest. 1992;90:749–758.
94. Hoenderop, JG, Nilius, B, Bindels, RJ. Molecular mechanism of active Ca2+ reabsorption in the distal nephron. Ann Rev Physiol. 2002;64:529–549.
95. Taniguchi, J, Takeda, M, Yoshitomi, K, et al. Pressure- and parathyroid-hormone-dependent Ca2+ transport in rabbit connecting tubule: role of the stretch-activated nonselective cation channel. J Membr Biol. 1994;140:123–132.
96. de Rouffignac, C, DiStefano, A, Wittner, M, et al. Consequences of differential effects of ADH and other peptide hormones on thick ascending limb of mammalian kidney. Am J Physiol. 1991;260:R1023–R1035.
97. Hebert, SC. Extracellular calcium-sensing receptor: implications for calcium and magnesium handling in the kidney. Kidney Int. 1996;50:2129–2139.
98. Even, L, Weisman, Y, Goldray, D, et al. Selective modulation by vitamin D of renal response to parathyroid hormone: a study in calcitriol-resistant rickets. J Clin Endocrinol Metab. 1996;81:2836–2840.
99. Friedman, PA, Gesek, FA. Calcium transport in renal epithelial cells. Am J Physiol. 1993;264:F181–F198.
100. Costanzo, LS, Sheehe, PR, Weiner, IM. Renal actions of vitamin D in D-deficient rats. Am J Physiol. 1974;226:1490–1495.
101. Christakos, S, Gabrielides, C, Rhoten, WB. Vitamin D-dependent calcium binding proteins: chemistry, distribution, functional considerations, and molecular biology. Endocr Rev. 1989;10:3–26.
102. Van Baal, J, Yu, A, Hartog, A, et al. Localization and regulation by vitamin D of calcium transport proteins in rabbit cortical collecting system. Am J Physiol. 1996;271:F985–F993.
103. Hoenderop, JG, Muller, D, Van Der Kemp, AW, et al. Calcitriol controls the epithelial calcium channel in kidney. J Am Soc Nephrol. 2001;12:1342–1349.
104. Singer, FR, Woodhouse, NJ, Parkinson, DK, et al. Some acute effects of administered porcine calcitonin in man. Clin Sci. 1969;37:181–190.
105. Chipman, JJ, Zerwekh, J, Nicar, M, et al. Effect of growth hormone administration: reciprocal changes in serum 1a,25-dihydroxyvitamin D and intestinal calcium absorption. J Clin Endocrinol Metab. 1980;51:321–324.
106. Gertner, JM, Tamborlane, WV, Hintz, RL, et al. The effects on mineral metabolism of overnight growth hormone infusion in growth hormone deficiency. J Clin Endocrinol Metab. 1981;53:818–822.
107. Wright, NM, Papadea, N, Wentz, B, et al. Increased serum 1,25-dihydroxyvitamin D after growth hormone administration is not parathyroid hormone-mediated. Calcif Tissue Int. 1997;61:101–103.
108. Hahn, TJ, Halstead, LR, Baran, DT. Effects of short-term glucocorticoid administration on intestinal calcium absorption and circulating vitamin D metabolite concentrations in man. J Clin Endocrinol Metab. 1981;52:111–114.
109. Lemann, J, Jr., Piering, WF, Lennon, EJ. Studies of the acute effects of aldosterone and cortisol on the interrelationship between renal sodium, calcium and magnesium excretion in normal man. Nephron. 1970;7:117–130.
110. Findley, JW, Adams, ND, Lemann, J, Jr., et al. Vitamin D metabolites and parathyroid hormone in Cushing’s syndrome: relationship to calcium and phosphorus homeostasis. J Clin Endocrinol Metab. 1982;54:1039–1044.
111. Gallagher, JC, Nordin, BEC. Treatment with oestrogens of primary hyperparathyroidism in post-menopausal women. Lancet. 1972;1:503–507.
112. McKane, W, Khosla, S, Burritt, M, et al. Mechanism of renal calcium conservation with estrogen replacement therapy in women in early menopause—a clinical research center study. J Clin Endocrinol Metab. 1995;80:3458–3464.
113. Van Abel, M, Hoenderop, JG, Dardenne, O, et al. 1,25-dihydroxyvitamin D(3)-independent stimulatory effect of estrogen on the expression of ECaC1 in the kidney. J Am Soc Nephrol. 2002;13:2102–2109.
114. DeFronzo, RA, Cooke, CR, Andres, R, et al. The effect of insulin on renal handling of sodium, potassium, calcium and phosphate in man. J Clin Invest. 1975;55:845–853.
115. Charbonneau, A, Leclerc, M, Brunette, MG. Effect of angiotensin II on calcium reabsorption by the luminal membranes of the nephron. Am J Physiol Endocrinol Metab. 2001;280:E928–36.
116. Naylor, SL, Sakaguchi, AU, Szoka, P, et al. Human parathyroid hormone gene (PTH) is on short arm of chromosome 11, Somat Cell. Gene. 1983;9:609–616.
117. Vasicek, T, McDevitt, BE, Freeman, MW, et al. Nucleotide sequence of genomic DNA encoding human parathyroid hormone. Proc Natl Acad Sci U S A. 1983;80:2127–2131.
118. Okazaki, T, Igarashi, T, Kronenberg, HM. 5′-Flanking region of the parathyroid hormone gene mediates negative regulation by 1,25(OH)2 vitamin D3. J Biol Chem. 1989;263:2203–2208.
119. Demay, MB, Kiernan, MS, DeLuca, HF, et al. Sequences in the human parathyroid hormone gene that bind the 1,25- dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1992;89:8097–8101.
120. Naveh-Many, T, Rahaminov, R, Livini, N, et al. Parathyroid cell proliferation in normal and chronic renal failure in rats, the effects of calcium, phosphate, and vitamin D. J Clin Invest. 1995;96:1786–1793.
121. Almaden, Y, Canalejo, A, Hernandez, A, et al. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res. 1996;11:970–976.
122. Russell, J, Lettieri, D, Sherwood, LM. Direct regulation by calcium of cytoplasmic messenger ribonucleic acid coding for pre-proparathyroid hormone in isolated bovine parathyroid cells. J Clin Invest. 1983;72:1851–1855.
123. Naveh-Many, T, Friedlaender, MM, Mayer, H, et al. Calcium regulates parathyroid hormone messenger ribonucleic acid (mRNA), but not calcitonin mRNA in vivo in the rat, Dominant role of 1,25-dihydroxyvitamin D. Endocrinology. 1989;125:275–280.
124. Kemper, B, Habener, JF, Mulligan, RC, et al. Preproparathyroid hormone: a direct translation product of parathyroid messenger RNA. Proc Natl Acad Sci U S A. 1974;71:3731–3735.
125. Jüppner, H, Abou-Samra, AB, Freeman, MW, et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–1026.
126. Abou-Samra, AB, Jüppner, H, Force, T, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol triphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992;89:2732–2736.
127. Gelbert, L, Schipani, E, Jüppner, H, et al. Chromosomal location of the parathyroid hormone/parathyroid hormone-related protein receptor gene to human chromosome 3p21.2–p24.2. J Clin Endocrinol Metab. 1994;79:1046–1048.
128. Pausova, Z, Bourdon, J, Clayton, D, et al. Cloning of a parathyroid hormone/parathyroid hormone-related peptide receptor (PTHR) cDNA from a rat osteosarcoma (UMR106) cell line: chromosomal assignment of the gene in the human, mouse, and rat genomes. Genomics. 1994;20:20–26.
129. Jüppner, H, Gardella, T, Brown, E, et al. Parathyroid hormone and parathyroid hormone-related peptide in the regulation of calcium homeostasis and bone development. In: DeGroot L, Jameson J, eds. Endocrinology. W.B. Saunders Company: Philadelphia, PA; 2000:969–998.
130. Kronenberg, H. Developmental regulation of the growth plate. Nature. 2003;423:332–336.
131. Thakker, RV. Molecular genetics of parathyroid disease. Curr Opin Endocrinol Diabetes Obes. 1996;3:521–528.
132. Szabo, J, Heath, B, Hill, VM, et al. Hereditary hyperparathyroidism-jaw tumor syndrome: the endocrine tumor gene HRPT2 maps to chromosome 1q21-q31. Am J Hum Genet. 1995;56:944–950.
133. Arnold, A, Brown, MF, Urena, P, et al. Monoclonality of parathyroid tumors in chronic renal failure and in primary parathyroid hyperplasia. J Clin Invest. 1995;95:2047–2053.
134. Arnold, A, Kim, HG, Gaz, RD, et al. Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest. 1989;83:2034–2040.
135. Motokura, T, Bloom, T, Kim, HG, et al. A BCL1-linked candidate oncogene which is rearranged in parathyroid tumors encodes a novel cyclin. Nature. 1991;350:512–515.
136. Nurse, P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508.
137. Hosokawa, Y, Yoshimoto, K, Bronson, R, et al. Chronic hyperparathyroidism in transgenic mice with parathyroid-targeted overexpression of cyclin D1/PRAD1. J Bone Miner Res. 1997;12(suppl 1):S110.
138. Imanishi, Y, Hosokawa, Y, Yoshimoto, K, et al. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J Clin Invest. 2001;107:1093–1102.
139. Wang, TC, Cardiff, RD, Zukerberg, L, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–671.
140. Au, AY, McDonald, K, Gill, A, et al. PTH mutation with primary hyperparathyroidism and undetectable intact PTH. N Engl J Med. 2008;359:1184–1186.
141. Thakker, RV. The molecular genetics of the multiple endocrine neoplasia syndromes. Clin Endocrinol (Oxf). 1993;38:1–14.
142. Trump, D, Farren, B, Wooding, C, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM. 1996;89:653–669.
143. Marx, S. Multiple endocrine neoplasia type 1. In: Vogelstein B, Kinzler K, eds. The genetic basis of human cancer. New York: McGraw Hill; 1998:489–506.
144. Thakker, RV. Multiple endocrine neoplasia–syndromes of the twentieth century. J Clin Endocrinol Metab. 1998;83:2617–2620.
145. Chandrasekharappa, SC, Guru, SC, Manickam, P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407.
146. Lemmens, I, Van de Ven, WJ, Kas, K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1, Hum Mol Genet. 1997;6:1177–1183.
147. Pannett, AA, Thakker, RV. Multiple endocrine neoplasia type 1. Endocr Relat Cancer. 1999;6:449–473.
148. Lemos, MC, Thakker, RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32.
149. Turner, JJ, Leotlela, PD, Pannett, AA, et al. Frequent occurrence of an intron 4 mutation in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 2002;87:2688–2693.
150. Bassett, JH, Forbes, SA, Pannett, AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet. 1998;62:232–244.
151. Teh, BT, Kytola, S, Farnebo, F, et al. Mutation analysis of the MEN1 gene in multiple endocrine neoplasia type 1, familial acromegaly and familial isolated hyperparathyroidism. J Clin Endocrinol Metab. 1998;83:2621–2626.
152. Heppner, C, Kester, MB, Agarwal, SK, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997;16:375–378.
153. Zhuang, Z, Vortmeyer, AO, Pack, S, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997;57:4682–4686.
154. Zhuang, Z, Ezzat, SZ, Vortmeyer, AO, et al. Mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res. 1997;57:5446–5451.
155. Prezant, TR, Levine, J, Melmed, S. Molecular characterization of the men1 tumor suppressor gene in sporadic pituitary tumors. J Clin Endocrinol Metab. 1998;83:1388–1391.
156. Debelenko, LV, Brambilla, E, Agarwal, SK, et al. Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum Mol Genet. 1997;6:2285–2290.
157. Vortmeyer, AO, Boni, R, Pak, E, et al. Multiple endocrine neoplasia 1 gene alterations in MEN1-associated and sporadic lipomas. J Natl Cancer Inst. 1998;90:398–399.
158. Farnebo, F, Teh, BT, Kytola, S, et al. Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab. 1998;83:2627–2630.
159. Carling, T, Correa, P, Hessman, O, et al. Parathyroid MEN1 gene mutations in relation to clinical characteristics of nonfamilial primary hyperparathyroidism. J Clin Endocrinol Metab. 1998;83:2960–2963.
160. Tanaka, C, Kimura, T, Yang, P, et al. Analysis of loss of heterozygosity on chromosome 11 and infrequent inactivation of the MEN1 gene in sporadic pituitary adenomas. J Clin Endocrinol Metab. 1998;83:2631–2634.
161. Pannett, AA, Thakker, RV. Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. J Clin Endocrinol Metab. 2001;86:4371–4374.
162. Teh, BT, Esapa, CT, Houlston, R, et al. A family with isolated hyperparathyroidism segregating a missense MEN1 mutation and showing loss of the wild-type alleles in the parathyroid tumors. Am J Hum Genet. 1998;63:1544–1549.
163. Pannett, AA, Kennedy, AM, Turner, JJ, et al. Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol (Oxf). 2003;58:639–646.
164. Hannan, FM, Nesbit, MA, Christie, PT, et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab. 2008;4:53–58.
165. Lin, SY, Elledge, SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–889.
166. Stalberg, P, Grimfjard, P, Santesson, M, et al. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J Clin Endocrinol Metab. 2004;89:2326–2337.
167. Kim, YS, Burns, AL, Goldsmith, PK, et al. Stable overexpression of MEN1 suppresses tumorigenicity of RAS. Oncogene. 1999;18:5936–5942.
168. Yumita, W, Ikeo, Y, Yamauchi, K, et al. Suppression of insulin-induced AP-1 transactivation by menin accompanies inhibition of c-Fos induction. Int J Cancer. 2003;103:738–744.
169. Hussein, N, Casse, H, Fontaniere, S, et al. Reconstituted expression of menin in Men1-deficient mouse Leydig tumour cells induces cell cycle arrest and apoptosis. Eur J Cancer. 2007;43:402–414.
170. Mulligan, LM, Kwok, JBJ, Healey, CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460.
171. Mulligan, LM, Ponder, BA. Genetic basis of endocrine disease: multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 1995;80:1989–1995.
172. Pausova, Z, Janicic, N, Konrad, EM, et al. Analysis of the RET proto-oncogene in sporadic parathyroid tumors. J Bone Mineral Research. 1994;9:S151.
173. Padberg, BC, Schroder, S, Jochum, W, et al. Absence of RET proto-oncogene point mutations in sporadic hyperplastic and neoplastic lesions of the parathyroid gland. Am J Pathol. 1995;147:1600–1607.
174. Heshmati, HM, Gharib, H, Khosla, S, et al. Genetic testing in medullary thyroid carcinoma syndromes: mutation types and clinical significance. Mayo Clin Proc. 1997;72:430–436.
175. Kennett, S, Pollick, H. Jaw lesions in familial hyperparathyroidism. Oral Surg Oral Med Oral Pathol. 1971;31:502–510.
176. Jackson, CE, Norum, RA, Boyd, SB, et al. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery. 1990;108:1006–1012.
177. Bradley, KJ, Hobbs, MR, Buley, ID, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005;257:18–26.
178. Cavaco, BM, Barros, L, Pannett, AA, et al. The hyperparathyroidism-jaw tumour syndrome in a Portuguese kindred. QJM. 2001;94:213–222.
179. Wassif, WS, Moniz, CF, Friedman, E, et al. Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab. 1993;77:1485–1489.
180. Williamson, C, Cavaco, BM, Jauch, A, et al. Mapping the gene causing hereditary primary hyperparathyroidism in a Portuguese kindred to chromosome 1q22-q31. J Bone Miner Res. 1999;14:230–239.
181. Weinstein, LS, Simonds, WF. HRPT2, a marker of parathyroid cancer. N Engl J Med. 2003;349:1691–1692.
182. Carpten, JD, Robbins, CM, Villablanca, A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680.
183. Shattuck, TM, Valimaki, S, Obara, T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729.
184. Howell, VM, Haven, CJ, Kahnoski, K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657–663.
185. Bradley, KJ, Cavaco, BM, Bowl, MR, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf). 2006;64:299–306.
186. Cetani, F, Pardi, E, Ambrogini, E, et al. Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (Oxf). 2006;64:146–152.
187. Krebs, LJ, Shattuck, TM, Arnold, A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2005;90:5015–5017.
188. Zhao, J, Yart, A, Frigerio, S, et al. Sporadic human renal tumors display frequent allelic imbalances and novel mutations of the HRPT2 gene. Oncogene. 2007;26:3440–3449.
189. Zheng, HC, Takahashi, H, Li, XH, et al. Downregulated parafibromin expression is a promising marker for pathogenesis, invasion, metastasis and prognosis of gastric carcinomas. Virchows Arch. 2008;452:147–155.
190. Selvarajan, S, Sii, LH, Lee, A, et al. Parafibromin expression in breast cancer: a novel marker for prognostication? J Clin Pathol. 2008;61:64–67.
191. Bradley, KJ, Bowl, MR, Williams, SE, et al. Parafibromin is a nuclear protein with a functional monopartite nuclear localization signal. Oncogene. 2007;26:1213–1221.
192. Rozenblatt-Rosen, O, Hughes, CM, Nannepaga, SJ, et al. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–620.
193. Yart, A, Gstaiger, M, Wirbelauer, C, et al. The HRPT2 tumor suppressor gene product parafibromin associates with human PAF1 and RNA polymerase II. Mol Cell Biol. 2005;25:5052–5060.
194. Mosimann, C, Hausmann, G, Basler, K. Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/Armadillo. Cell. 2006;125:327–341.
195. Wang, P, Bowl, MR, Bender, S, et al. Parafibromin, a component of the human PAF complex, regulates growth factors and is required for embryonic development and survival in adult mice. Mol Cell Biol. 2008;28:2930–2940.
196. Bjorklund, P, Akerstrom, G, Westin, G. Accumulation of nonphosphorylated beta-catenin and c-myc in primary and uremic secondary hyperparathyroid tumors. J Clin Endocrinol Metab. 2007;92:338–344.
197. Bjorklund, P, Akerstrom, G, Westin, G. An LRP5 receptor with internal deletion in hyperparathyroid tumors with implications for deregulated WNT/beta-catenin signaling. PLoS Med. 2007;4:e328.
198. Bjorklund, P, Lindberg, D, Akerstrom, G, et al. Stabilizing mutation of CTNNB1/beta-catenin and protein accumulation analyzed in a large series of parathyroid tumors of Swedish patients. Mol Cancer. 2008;7:53.
199. Costa-Guda, J, Arnold, A. Absence of stabilizing mutations of beta-catenin encoded by CTNNB1 exon 3 in a large series of sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2007;92:1564–1566.
200. Ikeda, S, Ishizaki, Y, Shimizu, Y, et al. Immunohistochemistry of cyclin D1 and beta-catenin, and mutational analysis of exon 3 of beta-catenin gene in parathyroid adenomas. Int J Oncol. 2002;20:463–466.
201. Weinberg, RA. Tumor suppressor genes. Science. 1991;254:1138–1146.
202. Cryns, VL, Thor, A, Xu, HJ, et al. Loss of the retinoblastoma tumor suppressor gene in parathyroid carcinoma. New Engl J Med. 1994;330:757–761.
203. Pearce, SH, Trump, D, Wooding, C, et al. Loss of heterozygosity studies at the retinoblastoma and breast cancer susceptibility (BRCA2) loci in pituitary, parathyroid, pancreatic and carcinoid tumours. Clin Endocrinol (Oxf). 1996;45:195–200.
204. Carling, T, Du, Y, Fang, W, et al. Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery. 2003;134:932–939.
205. Pei, L, Melmed, S, Scheithauer, B, et al. Frequent loss of heterozygosity at the retinoblastoma susceptibility gene (RB) locus in aggressive pituitary tumors: evidence for a chromosome 13 tumor suppressor gene other than RB. Cancer Res. 1995;55:1613–1616.
206. Cryns, VL, Yi, SM, Tahara, H, et al. Frequent loss of chromosomes arm 1p DNA in parathyroid adenomas. Genes Chromosomes & Cancer. 1995;13:9–17.
207. Williamson, C, Pannett, AA, Pang, JT, et al. Localisation of a gene causing endocrine neoplasia to a 4 cM region on chromosome 1p35-p36. J Med Genet. 1997;34:617–619.
208. Brown, EM, MacLeod, RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297.
209. Simonds, WF, James-Newton, LA, Agarwal, SK, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002;81:1–26.
210. Simonds, WF, Robbins, CM, Agarwal, SK, et al. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2004;89:96–102.
211. Warner, JV, Nyholt, DR, Busfield, F, et al. Familial isolated hyperparathyroidism is linked to a 1.7 Mb region on chromosome 2p13.3–14. J Med Genet. 2006;43:e12.
212. Pollak, MR, Brown, EM, WuChou, YH, et al. Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993;75:1297–1303.
213. Chou, YH, Pollak, MR, Brandi, ML, et al. Mutations in the human Ca(2+)-sensing-receptor gene that cause familial hypocalciuric hypercalcemia. Am J Hum Genet. 1995;56:1075–1079.
214. Pearce, S, Trump, D, Wooding, C, et al. Calcium-sensing receptor mutations in familial benign hypercalcaemia and neonatal hyperparathyroidism. J Clin Invest. 1995;96:2683–2692.
215. Janicic, N, Pausova, Z, Cole, DE, et al. Insertion of an Alu sequence in the Ca(2+)-sensing receptor gene in familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Am J Hum Genet. 1995;56:880–886.
216. Aida, K, Koishi, S, Inoue, M, et al. Familial hypocalciuric hypercalcemia associated with mutation in the human Ca(2+)-sensing receptor gene. J Clin Endocrinol Metab. 1995;80:2594–2598.
217. Heath, H, III., Odelberg, S, Jackson, CE, et al. Clustered inactivating mutations and benign polymorphisms of the calcium receptor gene in familial benign hypocalciuric hypercalcemia suggest receptor functional domains. J Clin Endocrinol Metab. 1996;81:1312–1317.
218. Janicic, N, Soliman, E, Pausova, Z, et al. Mapping of the calcium-sensing receptor gene (CASR) to human chromosome 3q13.3-21 by fluorescence in situ hybridization, and localization to rat chromosome 11 and mouse chromosome 16. Mamm Genome. 1995;6:798–801.
219. Morten, KJ, Cooper, JM, Brown, GK, et al. A new point mutation associated with mitochondrial encephalomyopathy. Hum Mol Genet. 1993;2:2081–2087.
220. Pearce, SH, Bai, M, Quinn, SJ, et al. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest. 1996;98:1860–1866.
221. Bai, M, Quinn, S, Trivedi, S, et al. Expression and characterization of inactivating and activating mutations in the human Ca2+-sensing receptor. J Biol Chem. 1996;271:19537–19545.
222. Pollak, MR, Chou, YH, Marx, SJ, et al. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism, Effects of mutant gene dosage on phenotype. J Clin Invest. 1994;93:1108–1112.
223. Bai, M, Pearce, SH, Kifor, O, et al. In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2+-sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest. 1997;99:88–96.
224. Heath, H, III., Jackson, CE, Otterrud, B, et al. Genetic linkage analysis in familial benign (hypocalciuric) hypercalcemia: evidence for locus heterogeneity. Am J Hum Genet. 1993;53:193–200.
225. McMurtry, CT, Schranck, FW, Walkenhorst, DA, et al. Significant developmental elevation in serum parathyroid hormone levels in a large kindred with familial benign (hypocalciuric) hypercalcemia. Am J Med. 1992;93:247–258.
226. Trump, D, Whyte, MP, Wooding, C, et al. Linkage studies in a kindred from Oklahoma, with familial benign (hypocalciuric) hypercalcaemia (FBH) and developmental elevations in serum parathyroid hormone levels, indicate a third locus for FBH. Hum Genet. 1995;96:183–187.
227. Lloyd, SE, Pannett, AA, Dixon, PH, et al. Localization of familial benign hypercalcemia, Oklahoma variant (FBHOk), to chromosome 19q13. Am J Hum Genet. 1999;64:189–195.
228. Kifor, O, Moore, FD, Jr., Delaney, M, et al. A syndrome of hypocalciuric hypercalcemia caused by autoantibodies directed at the calcium-sensing receptor. J Clin Endocrinol Metab. 2003;88:60–72.
229. Jüppner, H, Schipani, E, Silve, C. Jansen’s metaphyseal chondrodysplasia and Blomstrand’s lethal chondrodysplasia: two genetic disorders caused by PTH/PTHrP receptor mutations. In: Bilezikian J, Raisz L, Rodan G, eds. Principles of bone biology. San Diego, CA: Academic Press; 2002:1117–1135.
230. Schipani, E, Kruse, K, Jüppner, H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100.
231. Schipani, E, Langman, CB, Parfitt, AM, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. New Engl, J Med. 1996;335:708–714.
232. Schipani, E, Langman, CB, Hunzelman, J, et al. A novel PTH/PTHrP receptor mutation in Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 1999;84:3052–3057.
233. Schipani, E, Lanske, B, Hunzelman, J, et al. Targeted expression of constitutively active PTH/PTHrP receptors delays endochondral bone formation and rescues PTHrP-less mice. Proc Natl Acad Sci U S A. 1997;94:13689–13694.
234. Bastepe, M, Raas-Rothschild, A, Silver, J, et al. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating PTH/PTHrP receptor mutation. J Clin Endocrinol Metab. 2004;89:3595–3600.
235. Schipani, E, Jensen, GS, Pincus, J, et al. Constitutive activation of the cAMP signaling pathway by parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptors mutated at the two loci for Jansen’s metaphyseal chondrodysplasia. Mol Endocrinol. 1997;11:851–858.
236. Ewart, AK, Morris, CA, Atkinson, DL, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nature Genet. 1993;5:11–16.
237. Nickerson, E, Greenberg, F, Keating, MT, et al. Deletions of the elastin gene at 7q11.23 occur in approximately 90% of patients with Williams syndrome. Am J Hum Genet. 1995;56:1156–1161.
238. Lowery, MC, Morris, CA, Ewart, A, et al. Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: evaluation of 235 patients. Am J Hum Genet. 1995;57:49–53.
239. Li, D, Brooke, B, Davis, E, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280.
240. Tassabehji, M, Metcalfe, K, Fergusson, WD, et al. LIM-kinase deleted in Williams syndrome. Nat Genet. 1996;13:272–273.
241. Perez Jurado, LA, Li, X, Francke, U. The human calcitonin receptor gene (CALCR) at 7q21.3 is outside the deletion associated with the Williams syndrome. Cytogenet Cell Genet. 1995;70:246–249.
242. Arnold, A, Horst, SA, Gardella, TJ, et al. Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism. J Clin Invest. 1990;86:1084–1087.
243. Karaplis, AC, Lim, SK, Baba, H, et al. Inefficient membrane targeting, translocation, and proteolytic processing by signal peptidase of a mutant preproparathyroid hormone protein. J Biol Chem. 1995;270:1629–1635.
244. Datta, R, Waheed, A, Shah, GN, et al. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci U S A. 2007;104:19989–19994.
245. Sunthornthepvarakul, T, Churesigaew, S, Ngowngarmratana, S. A novel mutation of the signal peptide of the preproparathyroid hormone gene associated with autosomal-recessive familial isolated hypoparathyroidism. J Clin Endocrinol Metab. 1999;84:3792–3796.
246. Parkinson, D, Thakker, R. A donor splice site mutation in the parathyroid hormone gene is associated with autosomal-recessive hypoparathyroidism. Nature Genet. 1992;1:149–153.
247. Günther, T, Chen, ZF, Kim, J, et al. Genetic ablation of parathyroid glands reveals another source of parathyroid hormone. Nature. 2000;406:199–203.
248. Kim, J, Jones, B, Zock, C, et al. Isolation and characterization of mammalian homologs of the Drosophila gene glial cells missing. Proc Natl Acad Sci U S A. 1998;95:12364–12369.
249. Liu, Z, Yu, S, Manley, NR. Gcm2 is required for the differentiation and survival of parathyroid precursor cells in the parathyroid/thymus primordia. Dev Biol. 2007;305:333–346.
250. Ding, C, Buckingham, B, Levine, MA. Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J Clin Invest. 2001;108:1215–1220.
251. Baumber, L, Tufarelli, C, Patel, S, et al. Identification of a novel mutation disrupting the DNA binding activity of GCM2 in autosomal-recessive familial isolated hypoparathyroidism. J Med Genet. 2005;42:443–448.
252. Mannstadt, M, Bertrand, G, Muresan, M, et al. Dominant-negative GCMB mutations cause an autosomal dominant form of hypoparathyroidism. J Clin Endocrinol Metab. 2008;93:3568–3576.
253. Peden, V. True idiopathic hypoparathyroidism as a sex-linked recessive trait. Am J Human Genet. 1960;12:323–337.
254. Whyte, M, Weldon, V. Idiopathic hypoparathyroidism presenting with seizures during infancy: X-linked recessive inheritance in a large Missouri kindred. J Pediatr. 99, 1981. [628-611].
255. Whyte, M, Kim, G, Kosanovich, M. Absence of parathyroid tissue in sex-linked recessive hypoparathyroidism. J Paediatr. 1986;109:915.
256. Mumm, S, Whyte, MP, Thakker, RV, et al. mtDNA Analysis shows common ancestry in two kindreds with X-linked recessive hypoparathyroidism and reveals a heteroplasmic silent mutation. Am J Hum Genet. 1997;60:153–159.
257. Thakker, RV, Davies, KE, Whyte, MP, et al. Mapping the gene causing X-linked recessive idiopathic hypoparathyroidism to Xq26-Xq27 by linkage studies. J Clin Invest. 1990;86:40–45.
258. Bowl, M, Nesbit, M, Harding, B, et al. An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest. 2005;115:2822–2831.
259. Solomon, NM, Ross, SA, Morgan, T, et al. Array comparative genomic hybridisation analysis of boys with X-linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet. 2004;41:669–678.
260. Rizzoti, K, Brunelli, S, Carmignac, D, et al. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet. 2004;36:247–255.
261. Collignon, J, Sockanathan, S, Hacker, A, et al. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development. 1996;122:509–520.
262. Kleinjan, DA, van Heyningen, V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32.
263. Brunelli, S, Silva Casey, E, Bell, D, et al. Expression of Sox3 throughout the developing central nervous system is dependent on the combined action of discrete, evolutionarily conserved regulatory elements. Genesis. 2003;36:12–24.
264. Uchikawa, M, Ishida, Y, Takemoto, T, et al. Functional analysis of chicken Sox2 enhancers highlights an array of diverse regulatory elements that are conserved in mammals. Dev Cell. 2003;4:509–519.
265. Kiernan, AE, Pelling, AL, Leung, KK, et al. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035.
266. Stevanovic, M, Lovell-Badge, R, Collignon, J, et al. SOX3 is an X-linked gene related to SRY. Hum Mol Genet. 1993;2:2013–2018.
267. Ahonen, P, Myllarniemi, S, Sipila, I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis- ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836.
268. Ahonen, P. Autoimmune polyendocrinopathy–candidosis–ectodermal dystrophy (APECED): autosomal-recessive inheritance. Clin Genet. 1985;27:535–542.
269. Zlotogora, J, Shapiro, MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet. 1992;29:824–826.
270. Aaltonen, J, Bjorses, P, Sandkuijl, L, et al. An autosomal locus causing autoimmune disease: autoimmune polyglandular disease type I assigned to chromosome 21. Nat Genet. 1994;8:83–87.
271. Nagamine, K, Peterson, P, Scott, HS, et al. Positional cloning of the APECED gene. Nat Genet. 1997;17:393–398.
272. Pearce, SH, Cheetham, T, Imrie, H, et al. A common and recurrent 13-bp deletion in the autoimmune regulator gene in British kindreds with autoimmune polyendocrinopathy type 1. Am J Hum Genet. 1998;63:1675–1684.
273. Scott, HS, Heino, M, Peterson, P, et al. Common mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients of different origins. Mol Endocrinol. 1998;12:1112–1119.
274. Rosatelli, MC, Meloni, A, Devoto, M, et al. A common mutation in Sardinian autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy patients. Hum Genet. 1998;103:428–434.
275. Bjorses, P, Halonen, M, Palvimo, JJ, et al. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J Hum Genet. 2000;66:378–392.
276. Liston, A, Lesage, S, Wilson, J, et al. AIRE regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4:350–354.
277. Meager, A, Visvalingam, K, Peterson, P, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 2006;3:e289.
278. Alimohammadi, M, Bjorklund, P, Hallgren, A, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. 2008;358:1018–1028.
279. Eisenbarth, SC, Colegio, OR, O’Connor, W, et al. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126.
280. Scambler, PJ, Carey, AH, Wyse, RK, et al. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–206.
281. Monaco, G, Pignata, C, Rossi, E, et al. DiGeorge anomaly associated with 10p deletion. Am J Med Genet. 1991;39:215–216.
282. Gong, W, Emanuel, BS, Collins, J, et al. A transcription map of the DiGeorge and velo-cardio-facial syndrome minimal critical region on 22q11. Hum Mol Genet. 1996;5:789–800.
283. Scambler, PJ. The 22q11 deletion syndromes. Hum Mol Genet. 2000;9:2421–2426.
284. Augusseau, S, Jouk, S, Jalbert, P, et al. DiGeorge syndrome and 22q11 rearrangements. Hum Genet. 1986;74:206.
285. Budarf, ML, Collins, J, Gong, W, et al. Cloning a balanced translocation associated with DiGeorge syndrome and identification of a disrupted candidate gene. Nat Genet. 1995;10:269–278.
286. Yamagishi, H, Garg, V, Matsuoka, R, et al. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science. 1999;283:1158–1161.
287. Lindsay, EA, Botta, A, Jurecic, V, Carattini-Rivera, S, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401:379–383.
288. Magnaghi, P, Roberts, C, Lorain, S, et al. HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors, interacts with Pax3. Nat Genet. 1998;20:74–77.
289. Jerome, LA, Papaioannou, VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291.
290. Yagi, H, Furutani, Y, Hamada, H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373.
291. Baldini, A. DiGeorge’s syndrome: a gene at last. Lancet. 2003;362:1342–1343.
292. Scire, G, Dallapiccola, B, Iannetti, P, et al. Hypoparathyroidism as the major manifestation in two patients with 22q11 deletions. Am J Med Genet. 1994;52:478–482.
293. Sykes, K, Bachrach, L, Siegel-Bartelt, J, et al. Velocardiofacial syndrome presenting as hypocalcemia in early adolescence. Arch Pediatr Adolesc Med. 1997;151:745–747.
294. Bilous, R, Murty, G, Parkinson, D, et al. Autosomal dominant familial hypoparathyroidism, sensorineural deafness and renal dysplasia. N Engl J Med. 1992;327:1069–1084.
295. Van Esch, H, Groenen, P, Nesbit, MA, et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature. 2000;406:419–422.
296. Muroya, K, Hasegawa, T, Ito, Y, et al. GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. J Med Genet. 2001;38:374–380.
297. Nesbit, MA, Bowl, MR, Harding, B, et al. Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. J Biol Chem. 2004;279:22624–22634.
298. Ali, A, Christie, PT, Grigorieva, IV, et al. Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet. 2007;16:265–275.
299. Tsang, AP, Visvader, JE, Turner, CA, et al. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell. 1997;90:109–119.
300. Zahirieh, A, Nesbit, MA, Ali, A, et al. Functional analysis of a novel GATA3 mutation in a family with the hypoparathyroidism, deafness, and renal dysplasia syndrome. J Clin Endocrinol Metab. 2005;90:2445–2450.
301. Dai, YS, Markham, BE. p300 Functions as a coactivator of transcription factor GATA-4. J Biol Chem. 2001;276:37178–37185.
302. Pandolfi, PP, Roth, ME, Karis, A, et al. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995;11:40–44.
303. van der Wees, J, van Looij, MA, de Ruiter, MM, et al. Hearing loss following Gata3 haploinsufficiency is caused by cochlear disorder. Neurobiol Dis. 2004;16:169–178.
304. van Looij, MA, van der Burg, H, van der Giessen, RS, et al. GATA3 haploinsufficiency causes a rapid deterioration of distortion product otoacoustic emissions (DPOAEs) in mice. Neurobiol Dis. 2005;20:890–897.
305. Lim, KC, Lakshmanan, G, Crawford, SE, et al. Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nat Genet. 2000;25:209–212.
306. Grote, D, Souabni, A, Busslinger, M, et al. Pax 2/8-regulated Gata 3 expression is necessary for morphogenesis and guidance of the nephric duct in the developing kidney. Development. 2006;133:53–61.
307. Moraes, CT, DiMauro, S, Zeviani, M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293–1299.
308. Zupanc, ML, Moraes, CT, Shanske, S, et al. Deletion of mitochondrial DNA in patients with combined features of Kearns-Sayre and MELAS syndromes. Ann Neurol. 1991;29:680–683.
309. Isotani, H, Fukumoto, Y, Kawamura, H, et al. Hypoparathyroidism and insulin-dependent diabetes mellitus in a patient with Kearns-Sayre syndrome harbouring a mitochondrial DNA deletion. Clin Endocrinol (Oxf). 1996;45:637–641.
310. Dionisi-Vici, C, Garavaglia, B, Burlina, AB, et al. Hypoparathyroidism in mitochondrial trifunctional protein deficiency. J Pediatr. 1996;129:159–162.
311. Franceschini, P, Testa, A, Bogetti, G, et al. Kenny-Caffey syndrome in two sibs born to consanguineous parents: evidence for an autosomal recessive variant. Am J Med Genet. 1992;42:112–116.
312. Boynton, JR, Pheasant, TR, Johnson, BL, et al. Ocular findings in Kenny’s syndrome. Arch Ophthalmol. 1979;97:896–900.
313. Richardson, RJ, Kirk, JM. Short stature, mental retardation, and hypoparathyroidism: a new syndrome. Arch Dis Child. 1990;65:1113–1117.
314. Sanjad, SA, Sakati, NA, Abu-Osba, YK, et al. A new syndrome of congenital hypoparathyroidism, severe growth failure, and dysmorphic features. Arch Dis Child. 1991;66:193–196.
315. Parvari, R, Hershkovitz, E, Kanis, A, et al. Homozygosity and linkage-disequilibrium mapping of the syndrome of congenital hypoparathyroidism, growth and mental retardation, and dysmorphism to a 1-cM interval on chromosome 1q42–43. Am J Hum Genet. 1998;63:163–169.
316. Parvari, R, Hershkovitz, E, Grossman, N, et al. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal-recessive Kenny-Caffey syndrome. Nat Genet. 2002;32:448–452.
317. Parkinson, D, Shaw, N, Himsworth, R, et al. Parathyroid hormone gene analysis in autosomal hypoparathyroidism using an intragenic tetranucleotide (AAAT)n polymorphism. Hum Genet. 1993;91:281–284.
318. Barakat, AY, D’Albora, JB, Martin, MM, et al. Familial nephrosis, nerve deafness, and hypoparathyroidism. J Pediatr. 1977;91:61–64.
319. Dahlberg, PJ, Borer, WZ, Newcomer, KL, et al. Autosomal or X-linked recessive syndrome of congenital lymphedema, hypoparathyroidism, nephropathy, prolapsing mitral valve, and brachytelephalangy. Am J Med Genet. 1983;16:99–104.
320. Pollak, MR, Brown, EM, Estep, HL, et al. Autosomal dominant hypocalcaemia caused by a Ca2+-sensing receptor gene mutation. Nat Genet. 1994;8:303–307.
321. Finegold, DN, Armitage, MM, Galiani, M, et al. Preliminary localization of a gene for autosomal-dominant hypoparathyroidism to chromosome 3q13. Pediatr Res. 1994;36:414–417.
322. Pearce, SH, Williamson, C, Kifor, O, et al. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335:1115–1122.
323. Baron, J, Winer, K, Yanovski, J, et al. Mutations in the Ca(2+)-sensing receptor gene cause autosomal dominant and sporadic hypoparathyroidism. Hum Mol Genet. 1996;5:601–606.
324. Okazaki, R, Chikatsu, N, Nakatsu, M, et al. A novel activating mutation in calcium-sensing receptor gene associated with a family of autosomal dominant hypocalcemia. J Clin Endocrinol Metab. 1999;84:363–366.
325. Thakker, RV. Molecular pathology of renal chloride channels in Dent’s disease and Bartter’s syndrome. Exp Nephrol. 2000;8:351–360.
326. Hebert, SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527–532.
327. Watanabe, S, Fukumoto, S, Chang, H, et al. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet. 2002;360:692–694.
328. Li, Y, Song, YH, Rais, N, et al. Autoantibodies to the extracellular domain of the calcium sensing receptor in patients with acquired hypoparathyroidism. J Clin Invest. 1996;97:910–914.
329. Blomstrand, S, Claësson, I, Säve-Söderbergh, J. A case of lethal congenital dwarfism with accelerated skeletal maturation, Pediatr. Radiol. 1985;15:141–143.
330. Young, ID, Zuccollo, JM, Broderick, NJ. A lethal skeletal dysplasia with generalised sclerosis and advanced skeletal maturation: Blomstrand chondrodysplasia. J Med Genet. 1993;30:155–157.
331. Leroy, JG, Keersmaeckers, G, Coppens, M, et al. Blomstrand lethal chondrodysplasia. Am J Med Genet. 1996;63:84–89.
332. Loshkajian, A, Roume, J, Stanescu, V, et al. Familial Blomstrand chondrodysplasia with advanced skeletal maturation: further delineation. Am J Med Genet. 1997;71:283–288.
333. den Hollander, NS, van der Harten, HJ, Vermeij-Keers, C, et al. First-trimester diagnosis of Blomstrand lethal osteochondrodysplasia. Am J Med Genet. 1997;73:345–350.
334. Oostra, RJ, Baljet, B, Dijkstra, PF, et al. Congenital anomalies in the teratological collection of museum Vrolik in Amsterdam, The Netherlands. II: skeletal dysplasia. Am J Med Genet. 1998;77:116–134.
335. Jobert, AS, Zhang, P, Couvineau, A, et al. Absence of functional receptors parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34–40.
336. Zhang, P, Jobert, AS, Couvineau, A, et al. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab. 1998;83:3365–3368.
337. Karaplis, AC, Bin He, MT, Nguyen, A, et al. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. 1998;139:5255–5258.
338. Karperien, MC, van der Harten, HJ, van Schooten, R, et al. A frame-shift mutation in the type I parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand lethal osteochondrodysplasia. J Clin Endocrinol Metab. 1999;84:3713–3720.
339. Karperien, M, Sips, H, Harten, Hvd, et al. Novel mutations in the type I PTH/PTHrP receptor causing Blomstrand lethal osteochondrodysplasia (abstract). J Bone Mineral Res. 2001;16(Suppl 1):S549.
340. Wysolmerski, JJ, Cormier, S, Philbrick, W, et al. Absence of functional type 1 PTH/PTHrP receptors in humans is associated with abnormal breast development and tooth impactation. J Clin Endocrinol Metab. 2001;86:1788–1794.
341. Frame, B, Poznanski, AK. Conditions that may be confused with rickets. In: DeLuca HF, Anast CS, eds. Pediatric diseases related to calcium. New York: Elsevier; 1980:269–289.
342. De Haas, WHD, De Boer, W, Griffioen, F. Metaphysial dysostosis. A late follow-up of the first reported case. J Bone Joint Surg. 1969;51B:290–299.