166 Calcium, Magnesium, and Phosphorus
 Key Points
Key Points• Regulation of calcium, magnesium, and phosphorus is interrelated, and abnormalities in one are highly correlated with other electrolyte abnormalities.
• Severely depressed mental status and precipitation of arrhythmias are the most dangerous consequences of severe abnormalities in calcium, magnesium, or phosphorus.
• Proper correction of electrolyte deficiencies requires knowledge of the various oral and intravenous electrolyte preparations available.
Calcium
Approximately 99% of total body calcium is located in bone as the calcium phosphate salt hydroxyapatite. Of the remaining total body calcium, 45% is bound to albumin; 10% is complexed with circulating ions such as bicarbonate, phosphate, citrate, or sulfate1; and the remaining 45% is found in the free, ionized form. The normal range for serum calcium is 8.5 to 10.5 mg/dL, with some variability among different laboratories. The normal range of ionized (unbound) calcium is 4.5 to 5.6 mg/dL, but this is often reported in the international units (SI units) of mmol/L, with the normal range being 1.1 to 1.4 mmol/L. This ionized fraction is responsible for the physiologic actions of calcium and is not dependent on albumin levels. The total serum calcium level can be corrected for the amount of serum albumin (see the “Facts and Formulas” box), but such correction can be unreliable, so an ionized calcium level should be obtained whenever true hypercalcemia or hypocalcemia is a concern.
 Facts and Formulas
Facts and Formulas
| Normal serum calcium level | 8.5-10.5 mg/dL (2.1-2.6 mmol/dL) |
| Normal ionized calcium level | 4.5-5.6 mg/dL (1.1-1.4 mmol/L) |
| Normal serum magnesium level | 1.8-2.5 mg/dL (0.74-0.94 mmol/L) |
| Normal serum phosphorus level | 2.5-4.5 mg/dL (0.81-1.45 mmol/L) |
| Total serum calcium level corrected for albumin: | For every 1 g/dL in albumin, serum calcium drops 0.8 mg/dL |
| Corrected calcium (mg/dL) | Measured calcium (mg/dL) + 0.8[4.4 − albumin (g/dL)] |
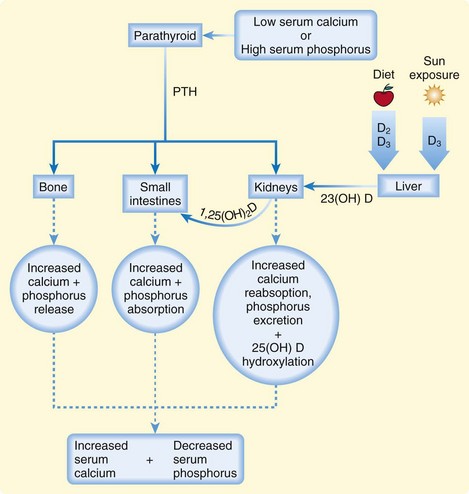
The plasma concentration of calcium is tightly maintained within the normal range by a feedback-regulated endocrine system that balances interactions among the small intestines, kidneys, bones, parathyroid glands, thyroid gland, and bloodstream. The key regulatory molecules in this system include calcium, phosphorus, parathyroid hormone (PTH), and 1,25-dihydroxyvitamin D (calcitriol)1,2 (Fig. 166.1).
Hypercalcemia
Epidemiology
The prevalence of hypercalcemia is approximately 0.5% to 3% in hospitalized adults.3 Hyperparathyroidism is the most common cause of hypercalcemia, and the incidence of primary hyperparathyroidism is approximately 21 cases per 100,000 person-years.4
The paraneoplastic syndrome hypercalcemia of malignancy is the second most common cause of hypercalcemia and occurs in approximately 10% to 30% of patients with cancer. Multiple myeloma and lung, breast, and prostate malignancies are most often associated with this disorder. It is typically seen in the end stages of disease and indicates a poor prognosis.5
Pathophysiology
Under normal conditions, excess calcium is excreted together with sodium in the proximal tubules of the kidneys. With hypercalcemia, dehydration caused by vomiting, poor oral intake, and osmotic diuresis results in reabsorption of sodium instead of excretion. This concurrent calcium reabsorption exacerbates the underlying hypercalcemia. PTH regulates the renal excretion of calcium. The excess production of PTH in primary hyperparathyroidism results in inappropriate calcium reabsorption. Causes of primary hyperparathyroidism include solitary adenomas (most common), ectopic adenomas in the mediastinum, diffuse hyperplasia of one or more parathyroid glands, and parathyroid carcinoma.6 These parathyroid abnormalities may be independent or a component of the multiple endocrine neoplasia syndromes (MEN 1 or 2a).
In the paraneoplastic syndrome hypercalcemia of malignancy, the majority of cases of hypercalcemia arise from tumor secretion of parathyroid hormone–related protein (PTHrP), a PTH homologue that acts on tissues like PTH does. Osteolytic bone metastases and ectopic tumor production of calcitriol and PTH cause the remaining cases of hypercalcemia of malignancy.5
Milk-alkali syndrome is the third most common cause of hypercalcemia severe enough to result in hospitalization.7 The clinical definition of milk-alkali syndrome is hypercalcemia, alkalosis, and renal failure in a patient ingesting excessive amounts of calcium and an alkali. Diagnosis is based on the patient history when other causes of hypercalcemia are excluded. Over-the-counter calcium carbonate supplements are commonly used for dyspepsia and prevention of osteoporosis and are currently the most frequent cause of milk-alkali syndrome. Historically, ingestion of milk and sodium bicarbonate for the treatment of peptic ulcer disease was the most common cause of milk-alkali syndrome, but this medication regimen went out of favor with the availability of H2 receptor antagonists and proton pump inhibitors. Serum PTH is low in these patients, indicative of no concurrent hyperparathyroidism.
Several medications rarely cause hypercalcemia. Thiazide diuretics, lithium, and the vitamin A derivatives all-trans-retinoic acid and cis-retinoic acid have been implicated. Some systemic illnesses also have the potential to cause hypercalcemia, including the granulomatous diseases sarcoidosis, leprosy, coccidiomycosis, histoplasmosis, and tuberculosis. The mechanism of hypercalcemia in these conditions is thought to be production of calcitriol by macrophages within granulomas.8 Additionally, rare inherited disorders such as familial hypocalciuric hypercalcemia cause hypercalcemia.9
Presenting Signs and Symptoms
Patients often become symptomatic from hypercalcemia at levels near 12 mg/dL, and nearly all patients with levels higher than 14 mg/dL will be symptomatic. Hypercalcemia affects a broad array of organ systems (Box 166.1).
A common renal manifestation of hypercalcemia is osmotic diuresis manifested as polyuria and excessive thirst. Nephrolithiasis and nephrocalcinosis are hallmarks of hypercalcemia. In patients with primary hyperparathyroidism, up to 20% have a history of symptomatic nephrolithiasis. Case series of patients with kidney stones have demonstrated a 2% to 8% incidence of primary hyperparathyroidism.10 It is thought that excessive calciuria combined with dehydration and decreased urine output leads to stone formation.
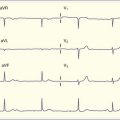
Cardiac manifestations of hypercalcemia are generally manifested as asymptomatic electrocardiographic (ECG) changes. Shortening of the QT interval (QTc <0.4 msec) is common, and ST elevations that may mimic acute myocardial infarction have been reported11,12 (Fig. 166.2). Symptomatic cardiac manifestations are rare and generally limited to bradydysrhythmias.
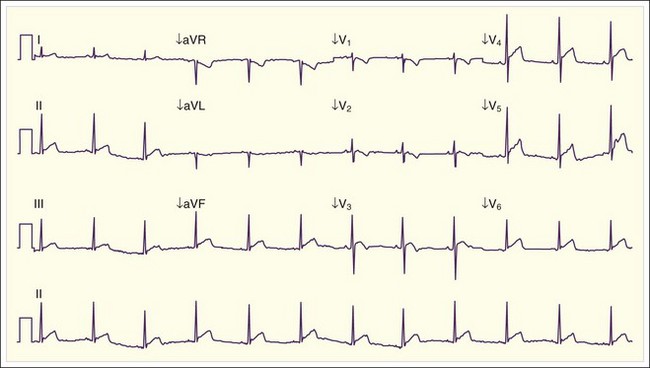
Fig. 166.2 Electrocardiographic manifestations of hypercalcemia.
Note the shortened QT interval, T-wave inversions, and ST-segment elevation.
(Courtesy Loren K. Rood, MD, Indiana University School of Medicine, Indianapolis.)
Musculoskeletal symptoms of hypercalcemia include muscle weakness, bone pain, and osteopenia.
Treatment
Initial therapy for hypercalcemia includes correction of dehydration and facilitation of renal excretion of calcium through volume reexpansion with normal saline at a rate of 200 to 500 mL/hr. Patients with severe hypercalcemia may require several liters of fluid resuscitation. For example, in a case series of patients with severe hyperparathyroid crisis requiring parathyroidectomy, a mean of 16 ± 6 L of isotonic fluid was administered over a period of several days before surgery.6
An additional therapy that has been studied most extensively in patients with hypercalcemia of malignancy is the use of bisphosphonates. These medications act on osteoclasts and limit release of calcium from bone. Their maximum calcium-lowering effects do not occur until several days after administration and can last for several weeks to months.13–15 Side effects of the bisphosphonates include hypophosphatemia, hypomagnesemia, osteonecrosis of the jaw, and postadministration acute phase reactions (fever, arthralgias, fatigue, malaise, myalgias). Table 166.1 summarizes the dosing regimens for available bisphosphonates.
Table 166.1 Bisphosphonates Available in the United States for the Treatment of Hypercalcemia of Malignancy

A treatment of hypercalcemia that is immediately effective in lowering serum calcium is calcitonin. Calcitonin inhibits urinary reabsorption of calcium and osteoclast maturation. The most commonly available form of this medication is salmon calcitonin administered at 4 to 8 U/kg subcutaneously every 8 to 12 hours. Lowering of the serum calcium level can occur as quickly as 2 hours after administration, but the effects are generally modest (lowering calcium by up to 3.8 mg/dL)16 and short-lived. Tachyphylaxis to this treatment occurs within 2 days. Side effects of salmon calcitonin include flushing, nausea, vomiting, and abdominal cramps.
Glucocorticoids inhibit conversion of 25-hydroxyvitamin D to calcitriol, which causes a decrease in intestinal absorption of calcium and an increase in renal calcium excretion. Efficacy in lowering serum calcium has been demonstrated only in the treatment of certain types of lymphoma that secrete calcitriol, vitamin D intoxication, and the granulomatous diseases.8 Additionally, administration of glucocorticoids may delay tachyphylaxis to calcitonin, so they are often used in conjunction with salmon calcitonin. A common regimen for the treatment of hypercalcemia is hydrocortisone, 200 to 300 mg/day intravenously for 3 to 5 days.
Follow-Up, Next Steps in Care, and Patient Education
Hospital admission is indicated for patients with symptomatic hypercalcemia, especially in the setting of altered mental status, dehydration, or acute renal failure. Definitive treatment of any underlying diseases causing hypercalcemia should be undertaken, as clinically appropriate. This may include parathyroidectomy for primary hyperparathyroidism or initiation of therapy for malignancy. Patients should be educated about the proper use of dietary supplements or antacids containing calcium and vitamin D (Table 166.2).
Hypocalcemia
Epidemiology
Hypocalcemia is defined as a serum calcium level lower than 8.5 mg/dL, although symptoms of hypocalcemia typically do not occur until serum calcium is below 7.0 to 7.5 mg/dL or ionized calcium is below 2.8 mg/dL (0.7 mmol/L).17 The incidence of hypocalcemia has not been well quantified.
Presenting Signs and Symptoms
The primary symptoms of hypocalcemia are manifestations of calcium’s critical role in the contraction and relaxation of skeletal and smooth muscle, as well as neurotransmission (Box 166.2). Neurologic symptoms often include both sensory and motor complaints. Sensory findings include perioral and extremity paresthesias. The most common motor abnormalities are neuromuscular irritability, including muscle cramps, hyperreflexia, carpal-pedal spasms, tetany, and seizures. Smooth muscle manifestations of hypocalcemia include bronchospasm, laryngospasm, biliary and small bowel cramping, dysphagia, bladder dysfunction, and painful menses or preterm labor from uterine contractions. Laryngospasm can be life-threatening.
Two well-recognized findings on physical examination that are pathognomonic for hypocalcemia are the Chvostek and Trousseau signs. The Chvostek sign is defined as facial muscle spasm elicited by tapping the facial nerve 1 to 2 cm anterior to the tragus of the ear. This sign is neither sensitive nor specific for hypocalcemia, with 25% of healthy people having a positive sign and only 71% of hypocalcemic patients having a positive sign.17 The Trousseau sign is more reliable, with only 1% to 4% of healthy people having a positive sign and 94% of hypocalcemic patients having a positive sign. The Trousseau sign is defined as carpal and digit spasm elicited by occluding the brachial artery with a sphygmomanometer inflated to 20 to 30 mm Hg above systolic blood pressure for 3 minutes (Fig. 166.3).
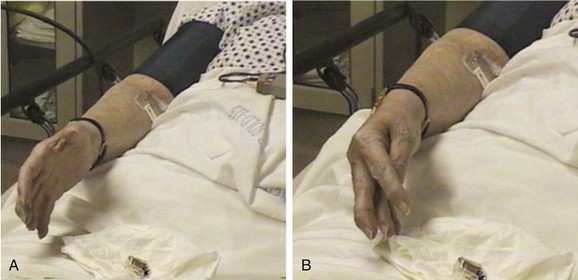
Fig. 166.3 Trousseau sign in a patient with hypocalcemia and hypomagnesemia.
(From Meininger ME, Kendler JS. Images in clinical medicine. Trousseau’s sign. N Engl J Med 2000;343:1855.)
In addition to neuromuscular and neuropsychiatric abnormalities, cardiac conduction abnormalities and dysrhythmias may occur. The most common cardiac manifestation is QT prolongation, but bradycardia, congestive heart failure, hypotension, and triggered ventricular dysrhythmias, including torsades de pointes, may occur (Fig. 166.4).
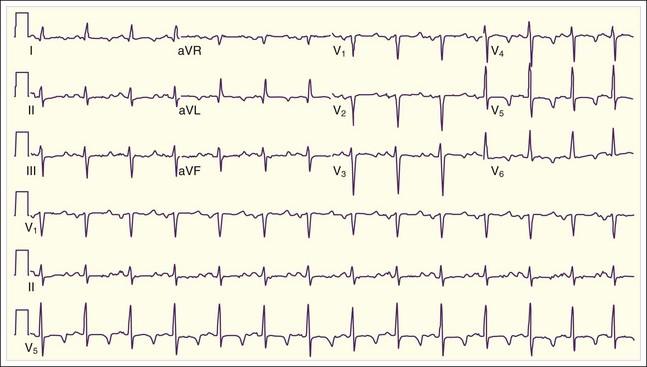
Treatment
The goals of therapy for hypocalcemia are to stop the uncomfortable symptoms of tetany and muscle spasm and prevent seizures, dangerous dysrhythmias, and laryngospasm. Severe, symptomatic hypocalcemia should be treated with a 10% intravenous solution of calcium gluconate or calcium chloride. The 10% solution of calcium chloride contains nearly three times the elemental calcium per milliliter as the 10% solution of calcium gluconate (Table 166.3). The dose of 10% calcium gluconate is 10 to 30 mL infused intravenously over a 10-minute period (provides 93 to 279 mg of elemental calcium). The dose of 10% calcium chloride is 10 mL infused intravenously over a 10-minute period (provides 270 mg of elemental calcium). The effects of a single bolus administration of intravenous calcium are temporary, so repeated boluses or a continuous infusion may be required in the setting of severe hypocalcemia or an ongoing process of calcium loss (pancreatitis or after parathyroidectomy). The rate of continuous infusion of calcium gluconate is 0.5 to 1.5 mg/kg/hr of elemental calcium, and it is generally supplied as 100 mL of 10% calcium gluconate in 900 mL of 5% dextrose in water (0.5 to 1.5 mL/kg/hr). Calcium chloride should be administered through a central vein. It is highly irritating to smaller peripheral veins and can cause phlebitis, and extravasation into subcutaneous tissues can result in significant local tissue necrosis.
Table 166.3 Comparison of Intravenous Solutions of Calcium for the Treatment of Hypocalcemia
| SOLUTION | ELEMENTAL CALCIUM (IN 10 mL) | DOSE |
|---|---|---|
| 10% Calcium chloride (1 g/10 mL) | 270 mg | 10 mL IV over 10-min period |
| 10% Calcium gluconate (1 g/10 mL) | 93 mg | 10-30 mL IV over 10-min period |
Adapted from Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bars Endocrinol Metab 2006;50:664–73.
Supplementation of calcium and vitamin D is the mainstay of therapy for chronic hypocalcemia. Virtually all forms of chronic hypocalcemia are associated with some degree of vitamin D deficiency. Vitamin D is available in a variety of forms. Selection of the appropriate form is based on the underlying cause of the vitamin D deficiency and the patient’s ability to appropriately hydroxylate the supplement in the liver and kidney (Table 166.4).
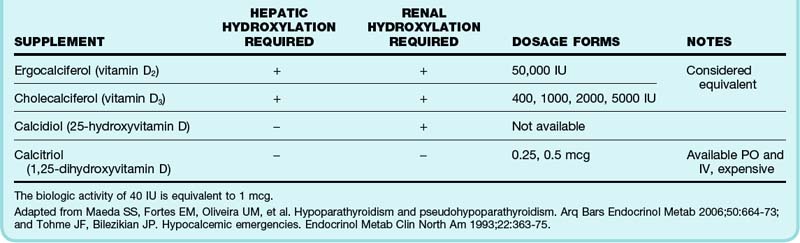
Oral calcium is available as a variety of salts, each with a different concentration of elemental calcium (Table 166.5). Calcium carbonate is generally preferred because it has the highest concentration of elemental calcium per tablet and thus fewer tablets are required to reach the appropriate dose of 1 to 1.5 g of elemental calcium daily.17 In the elderly, calcium citrate is preferred because it is more easily absorbed in the setting of low gastric acid.18
Table 166.5 Comparison of Calcium Salt Preparations
| SUPPLEMENT | ELEMENTAL CALCIUM CONTENT | AMOUNT OF SALT REQUIRED TO OBTAIN THE RECOMMENDED DAILY DOSE FOR ADULTS (1 g ELEMENTAL CALCIUM/DAY) |
|---|---|---|
| Calcium carbonate | 40% | 2.5 g |
| Calcium phosphate | 38% | 2.6 g |
| Calcium chloride | 27% | 3.7 g |
| Calcium citrate | 21% | 4.8 g |
| Calcium lactate | 13% | 7.7 g |
| Calcium gluconate | 9% | 11.1 g |
Adapted from Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bars Endocrinol Metab 2006;50:664–73.
Magnesium
Approximately 50% of total body magnesium is found in bone as a component of hydroxyapatite, similar to calcium. The remaining magnesium is located primarily intracellularly. Only 1% of total body magnesium is found in the extracellular space, with this amount further divided into protein-bound (30%) and ionized fractions.19
The balance between gastrointestinal absorption and renal excretion determines the serum magnesium level, with bone acting as a buffer to increase serum magnesium when levels are low. Neither gastrointestinal absorption nor renal excretion of magnesium is hormonally regulated. Circulating PTH regulates bone metabolism. Because serum magnesium levels influence release of PTH from the parathyroid glands, magnesium has an effect on serum calcium and phosphorus levels. The kidneys have the ability to reabsorb all but 0.5% of the filtered magnesium in the setting of hypomagnesemia and excrete up to 80% in the setting of hypermagnesemia.20 The normal serum range of magnesium is 1.8 to 2.5 mg/dL (0.74 to 0.94 mmol/L). No ionized magnesium laboratory test is available.
The recommended daily intake of elemental magnesium for adults is 320 mg for women and 420 mg for men. Dietary sources of magnesium include fish, nuts, cereals, and green vegetables. A variety of magnesium salt formulations are used therapeutically as laxatives and magnesium supplements (Table 166.6).
| SUPPLEMENT | ELEMENTAL MAGNESIUM CONTENT (%) | AMOUNT OF ELEMENTAL MAGNESIUM IN COMMON PREPARATIONS |
|---|---|---|
| Magnesium oxide | 61 | 242 mg in 400-mg tablet |
| Magnesium hydroxide (milk of magnesia) | 42 | 167 mg in 400 mg/5 mL oral suspension |
| Magnesium citrate | 16 | 48 mg in 290 mg/5 mL oral solution |
| Magnesium gluconate | 5 | 27 mg in 500-mg tablet |
| Magnesium chloride | 12 | 64 mg in 535-mg tablet |
| Magnesium sulfate solution | 10 | 500 mg in 10 mL of a 50% solution (5 mg MgSO4/10 mL) |
| Magnesium sulfate Epson salts |
10 | 98 mg in 1 g of salts |
| Magnesium lactate | 12 | 10 mg in 84-mg tablet |
| Magnesium aspartate | 10 | 122 mg in 1230-mg tablet |
Adapted from Guerrera MP, Volpe SL, Mao JJ. Therapeutic uses of magnesium. Am Fam Physician 2009;80:157–62.
Magnesium acts as a cofactor for an extensive number of intracellular enzymatic reactions and is necessary for protein and DNA synthesis, as well as for adenosine triphosphate (ATP) function and glucose metabolism.21 It is also critical for neuromuscular conduction and muscle contraction.
Hypomagnesemia
Epidemiology
Hypomagnesemia is defined as a serum magnesium level lower than 1.8 mg/dL, but most patients are not symptomatic until lower levels are reached. Two older studies demonstrated an incidence of hypomagnesemia of approximately 10% in the general hospitalized population and up to 65% in the intensive care unit patient population.22,23 It is unclear whether this incidence has changed over time. The incidence of hypomagnesemia in patients seen in the emergency department has not been determined.
Pathophysiology
Magnesium is absorbed over the entire length of the gastrointestinal tract. Absorption is passive in the small bowel. Active transcellular absorption occurs in the colon. The direction of passive flow of magnesium can change to favor magnesium excretion into the bowel lumen in patients with conditions such as excessive diarrhea, vomiting, persistent nasogastric suction, or bulimia.19,24 Failure to properly absorb dietary magnesium because of malabsorption syndromes such as short gut syndrome, gastric bypass, steatorrhea, celiac disease, and radiation-induced enteritis can result in chronic hypomagnesemia.
Presenting Signs and Symptoms
Hypomagnesemia is highly correlated with abnormalities in other electrolytes. Hypokalemia is common because magnesium is necessary for proper function of the sodium-potassium adenosine triphosphatase (Na+,K+-ATPase) pump, which maintains an appropriate transcellular potassium gradient. Hypomagnesemia impairs secretion of PTH, causes renal and bone resistance to PTH, and causes intestinal resistance to vitamin D, thereby resulting in hypocalcemia.19
Magnesium has been demonstrated to be helpful in the treatment of dangerous arrhythmias, especially torsades de pointes and atrial fibrillation, thus suggesting that hypomagnesemia plays a role in the development of these arrhythmias. ECG changes associated with hypomagnesemia include prolongation of the PR and QTc intervals and the development of U waves (Fig. 166.5).
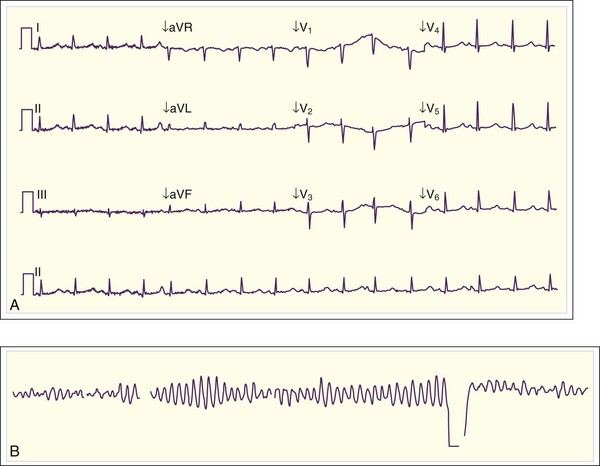
Hypermagnesemia
Epidemiology
Hypermagnesemia is defined as a serum magnesium level higher than 2.5 mg/dL. Hypermagnesemia is rare because of efficient renal excretion of excessive magnesium. In 1990 a published series of more than 1000 hospitalized adult patients demonstrated a 5.7% prevalence of hypermagnesemia in this population.25 There are multiple case reports in the emergency medicine literature of hypermagnesemia caused by iatrogenic and accidental overdoses.26
Pathophysiology
In the setting of iatrogenic or accidental overdose of magnesium, renal excretion of magnesium can be overwhelmed. The imbalance of absorption over excretion is worsened in the setting of acquired or chronic renal insufficiency, especially in patients with a glomerular filtration rate lower than 30 mL/min. Symptomatic hypermagnesemia is seen most often in the setting of iatrogenic administration of intravenous magnesium or after an overdose of magnesium-containing supplements or enemas. Concomitant use of anticholinergic or opiate medications that slow gastrointestinal motility can increase the risk for toxicity from magnesium-containing medications.27
Presenting Signs and Symptoms
The symptoms of hypermagnesemia progress in a dose-related fashion (Table 166.7). Early signs of hypermagnesemia include headache, nausea, vomiting, flushing, and hypotension as a result of peripheral vasodilation. Reflexes diminish and are eventually lost. Mental status worsens from somnolence to coma. Muscle weakness can progress to include the muscles of respiration and result in ventilatory failure and the need for endotracheal intubation and mechanical ventilation. At higher magnesium levels, cardiac complications begin to develop, with progression from bradycardia to atrioventricular block, intraventricular conduction block, complete heart block, or asystole.
| MAGNESIUM LEVEL | SYMPTOMS |
|---|---|
| 5-8 mg/dL | Nausea, vomiting, flushing, hyporeflexia |
| 9-12 mg/dL | Somnolence, areflexia, hypotension, bradycardia, prolongation of the QRS, PR, and QT intervals |
| >15 mg/dL | Respiratory depression, complete heart block, paralysis, coma |
| >20 mg/dL | Asystole, death |
Adapted from Birrer RB, Shallash AH, Totten V. Hypermagnesemia-induced fatality following Epson salt gargles. J Emerg Med 2002;22:185–8.
Phosphorus
Phosphorus exists as organic and inorganic forms—it is the inorganic forms that are measured in the laboratory determination of serum phosphorus. Within the range of typical body pH, inorganic phosphorus exists as a balance between the phosphate anions H2PO4− and HPO4−2. At neutral pH the ratio of H2PO4− to HPO4−2 is 1 : 4. The normal serum range of phosphorus is 2.5 to 4.5 mg/dL (0.81 to 1.45 mmol/L).28
The balance between gastrointestinal absorption, bone anabolism and catabolism, and renal excretion determines serum phosphorus levels. Passive absorption of phosphorus from the diet occurs in the small intestines. Vitamin D–dependent active absorption also occurs and is responsible for approximately 30% of phosphorus absorption.29 Foods rich in phosphorus include animal proteins, milk, eggs and multiple food preservatives.30
Excretion of phosphorus occurs in the kidneys. Serum phosphorus is freely filtered by the glomeruli, with 80% to 90% being reabsorbed though an Na/PO4 cotransporter in the proximal tubules. PTH enhances the excretion of phosphorus by inhibiting this transporter. When PTH is released from the parathyroid gland, it acts on bone to release calcium and phosphorus. It also stimulates the kidney to increase production of 1,25-dihydroxyvitamin D, which results in increased gastrointestinal absorption of calcium and phosphorus. Both these mechanisms result in efficient increases in serum calcium, but the increase in serum phosphorus is modest. When combined with the increased excretion of phosphorus by the kidneys in response to PTH, the net effect of a rise in PTH is a decrease in serum phosphorus and an increase in serum calcium (see Fig. 166.1).
Hypophosphatemia
Epidemiology
Hypophosphatemia is rare in the general population, occurs in only 2% to 3% of hospitalized patients,31,32 but is much more common in the setting of critical illness, in which the incidence of phosphorus levels lower than 2.5 mg/dL ranges from 24% to 100%, depending on the intensive care unit population studied.33
Pathophysiology
There are three predominant mechanisms of hypophosphatemia.33 Acute hypophosphatemia occurs in the setting of forces that drive phosphate excessively from the extracellular space into the intracellular space. Respiratory alkalosis and treatment of diabetic ketoacidosis with insulin are two common examples of this form of hypophosphatemia. Refeeding syndrome, in which severely malnourished patients are fed a diet high in carbohydrate, is a rare cause of hypophosphatemia but may be encountered in the treatment of patients with severe anorexia nervosa.
Treatment
Patients with severe phosphorus deficiency (serum phosphorus <1 mg/dL) should receive intravenous replacement in the form of either sodium phosphate or potassium phosphate. Potassium phosphate is preferred in patients with concomitant hypokalemia. Multiple studies have examined the safety of various doses and rates of administration of intravenous phosphorus supplementation. Doses up to 45 mmol and rates of 20 mmol/hr have been demonstrated to be safe when administered through a central venous catheter.31,33 The typical dose recommended for adults is 0.08 to 0.16 mmol/kg (2.5 to 5 mg/kg) administered over a period of 2 to 6 hours.34 Intravenous administration of phosphate-containing supplements can precipitate hypocalcemia, as well as hyperkalemia when potassium phosphate is used.
Mild hypophosphatemia (serum phosphorus level of 1 to 2 mg/dL) may be treated with oral phosphate-containing supplements at a dose of up to 1000 to 2000 mg daily, divided three or four times per day. A combination sodium phosphate and potassium phosphate tablet is available (K-Phos Neutral) that contains 250 mg of phosphorus, 298 mg (13 mEq) of sodium, and 45 mg of potassium (1.1 mEq). Cow’s milk is a rich dietary source of phosphorus that contains approximately 1 mg of phosphorus per 1 mL of milk.35
Hyperphosphatemia
Epidemiology
Hyperphosphatemia is defined as a serum phosphorus level higher than 4.5 mg/dL, with severe hyperphosphatemia being defined as serum levels higher than 14 mg/dL. The most common cause of hyperphosphatemia is chronic renal failure requiring hemodialysis. Nearly 90% of these patients are managed with phosphorus-binding therapy and diets low in phosphorus to limit hyperphosphatemia.36
Pathophysiology
In patients with chronic renal failure, gastrointestinal absorption of phosphorus continues, but renal excretion ceases. Hemodialysis removes approximately 800 to 1000 mg of phosphorus per session,30 which is less than the typical dietary intake of phosphorus—a mainstay of treatment of hyperphosphatemia is limiting dietary intake and absorption of phosphorus. Excessive phosphorus combines with calcium to form salts that are deposited in the soft tissues of the body. Deposition in the soft tissues of the heart, kidneys, and vasculature has the potential to cause abnormalities in cardiac electrical conduction and cardiac muscle relaxation and contraction, as well as renal disease, coronary artery disease, and peripheral vascular disease. Some evidence indicates that calcium phosphate salt deposition increases mortality in patients undergoing hemodialysis.36
Presenting Signs and Symptoms
The symptoms of hyperphosphatemia are generally the consequence of associated renal failure and hypocalcemia and include altered mental status, seizures, dysrhythmias, prolongation of the QTc interval, muscle weakness, and neuromuscular excitability (see Box 166.2).
Treatment
Phosphate-binding salts are frequently used in the treatment of hyperphosphatemia in patients with renal failure.36 The cheapest and best-tolerated phosphate binders are calcium carbonate and calcium acetate. These medications may increase deposition of calcium phosphate salt in soft tissues, but considerable debate exists about the cost-effectiveness and side effect profile of alternative phosphate binders. Gastrointestinal side effects of nausea and diarrhea are common. Magnesium hydroxide, magnesium carbonate, sevelamer hydrochloride, sevelamer carbonate, and lanthanum are alternative phosphate-binding salts used for the treatment of hyperphosphatemia in patients with renal failure.
Amanzadeh J, Reily RF, Jr. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract Nephrol. 2006;2:136–148.
Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35:215–237. v–vi
Pellitteri PK. Evaluation of hypercalcemia in relation to hyperparathyroidism. Otolaryngol Clin North Am. 2010;43:389–397.
Phitayakorn R, McHenry CR. Hyperparathyroid crisis: use of bisphosphonates as a bridge to parathyroidectomy. J Am Coll Surg. 2008;206:1106–1115.
Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362:1312–1324.
1 Kleeman CR, Massry SG, Coburn JW. The clinical physiology of calcium homeostasis, parathyroid hormone, and calcitonin. I. Calif Med. 1971;114(3):16–43.
2 Kleeman CR, Massry SG, Coburn JW. The clinical physiology of calcium homeostasis, parathyroid hormone, and calcitonin. II. Calif Med. 1971;114(4):19–30.
3 Frolich A. Prevalence of hypercalcemia in normal and hospitalized populations. Dan Med Bull. 1998;45:436–439.
4 Wermers RA, Khosla S, Atkinson EJ, et al. Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993–2001: an update on the changing epidemiology of the disease. J Bone Miner Res. 2006;21:171–177.
5 McMahan J, Linneman T. A case of resistant hypercalcemia of malignancy with a proposed treatment algorithm. Ann Pharmacother. 2009;43:1532–1538.
6 Phitayakorn R, McHenry CR. Hyperparathyroid crisis: use of bisphosphonates as a bridge to parathyroidectomy. J Am Coll Surg. 2008;206:1106–1115.
7 Ulett K, Wells B, Centor R. Hypercalcemia and acute renal failure in milk-alkali syndrome: a case report. J Hosp Med. 2010;5:E18–E20.
8 Ariyan CE, Sosa JA. Assessment and management of patients with abnormal calcium. Crit Care Med. 2004;32:S146–S154.
9 Pellitteri PK. Evaluation of hypercalcemia in relation to hyperparathyroidism. Otolaryngol Clin North Am. 2010;43:389–397.
10 Berger AD, Wu W, Eisner BH, et al. Patients with primary hyperparathyroidism—why do some form stones? J Urol. 2009;181:2141–2145.
11 Donovan J, Jackson M. Hypercalcemia mimicking STEMI on electrocardiography. Case Report Med. 2010;2010:563–572.
12 Turhan S, Kilickap M, Kilinc S. ST segment elevation mimicking acute myocardial infarction in hypercalcemia. Heart. 2005;91:999.
13 Mundy GR, Wilkinson R, Heath DA. Comparative study of available medical therapy for hypercalcemia of malignancy. Am J Med. 1983;74:421–432.
14 Ralston SH, Gardner MD, Dryburg FJ. Comparison of aminohydroxypropylidene diphosphonate, mithramycin, and corticosteroids/calcitonin in treatment of cancer-associated hypercalcemia. Lancet. 1985;2:907–910.
15 Ralston SH, Gallacher SJ, Dryburg FJ. Treatment of severe hypercalcemia with mithracmycin and aminohydroxypropylidene bisphosphonate. Lancet. 1988;2:277.
16 Heath DA. The emergency management of disorders of calcium and magnesium. Clin Endocrinol Metab. 1980;9:487–502.
17 Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bars Endocrinol Metab. 2006;50:664–673.
18 Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am. 1993;22:363–375.
19 Rude RK. Magnesium metabolism and deficiency. Endocrinol Metab Clin North Am. 1993;22:377–395.
20 Nadieri ASA, Reilly RF, Jr. Hereditary etiologies of hypomagnesemia. Nat Clin Pract Nephrol. 2008;4:80–89.
21 Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35:215–237. v–vi
22 Wong ET, Rude RK, Singer FR, et al. A high prevalence of hypomagnesemia and hypermagnesemia in hospitalized patients. Am J Clin Pathol. 1983;79:348–352.
23 Ryzen E, Wagers PW, Singer FR, et al. Magnesium deficiency in a medical ICU population. Crit Care Med. 1985;13:19–21.
24 Alexander RT, Hoenderop JG, Bindels RJ. Molecular determinant of magnesium homeostasis: insights from human disease. J Am Soc Nephrol. 2008;19:1451–1458.
25 Whang R, Ryder KW. Frequency of hypomagnesemia and hypermagnesemia. Requested vs. routine. JAMA. 1990;263:3063–3064.
26 Vissers RJ, Purssell R. Iatrogenic magnesium overdose: two case reports. J Emerg Med. 1996;14:187–191.
27 Birrer RB, Shallash AH, Totten V. Hypermagnesemia-induced fatality following Epson salt gargles. J Emerg Med. 2002;22:185–188.
28 Hodgson SF, Hurley DL. Acquired hypophosphatemia. Endocrinol Metab Clin North Am. 1993;22:363–375.
29 Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104.
30 Uribarri J. Phosphorus homeostasis in normal health and in chronic kidney disease patients with special emphasis on dietary phosphorus intake. Semin Dial. 2007;20:295–301.
31 Brunelli SM, Goldfarb S. Hypophosphatemia: clinical consequences and management. J Am Soc Nephrol. 2007;18:1999–2003.
32 Shiber JR, Mattu A. Serum phosphate abnormalities in the emergency department. J Emerg Med. 2002;23:395–400.
33 Geerse DA, Bindels AJ, Kuiper MA, et al. Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care. 2010;14:R147.
34 Amanzadeh J, Reily RF, Jr. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract Nephrol. 2006;2:136–148.
35 Gaasbeek A, Meinder E. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101.
36 Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362:1312–1324.