[level-membership-for-neurology-category]
Chapter 19 Brainstem Syndromes
Ocular Motor Syndromes
Combined Vertical Gaze Ophthalmoplegia
Combined vertical gaze ophthalmoplegia is defined as paresis of both upward and downward gaze. Vertical gaze ophthalmoplegia is an example of a brainstem syndrome in which the objective physical findings dictate the diagnostic approach to the problem. Symptoms of vertical gaze ophthalmoplegia, when present, are relatively nonspecific and usually occur in patients who have difficulty looking down, as required in reading, eating from a table, and walking down a flight of stairs. In addition, the patient’s report of symptoms may be unobtainable because of mental status changes caused by dysfunction of the reticular formation that lies adjacent to the vertical gaze generator in the rostral midbrain (see Chapter 35).
The neurological examination discloses associated signs of the disorders listed in the differential diagnosis (see Box 19.1) (Graff-Redford et al., 1985). Coma may be associated with reticular system involvement. Long-tract signs and loss of pupillary reflexes are commonly associated. The syndrome of combined vertical gaze ophthalmoplegia is diagnosed when the ocular findings occur in isolation from long-tract signs.
Box 19.1 Differential Diagnosis for Combined Vertical Gaze Ophthalmoplegia
Corticobasal ganglionic degeneration
Thalamic and mesencephalic tumors
AVM, Arteriovenous malformation; MS, multiple sclerosis; PSP, progressive supranuclear palsy.
With combined vertical gaze ophthalmoplegia, vertical saccades and pursuit are lost. This gaze limitation may be overcome by the oculocephalic (doll’s head or doll’s eye) maneuver, which tests the vestibulo-ocular reflex (VOR) (see Chapter 35). It is demonstrated by having the patient focus on an object and rotating the patient’s head; a conjugate eye movement in the opposite direction is the expected response with this maneuver. The Bell phenomenon (reflex movement of the eyes up and out in response to forced eye closure) often is absent. Skew deviation (vertical malalignment of the eyes) may occur. Absence of convergence and loss of pupillary reactions to light are common.
The location of the lesion of combined vertical gaze ophthalmoplegia is the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) for loss of vertical pursuit and saccades (Leigh and Zee, 2006). Box 19.1 lists the disorders involving the rostral mesodiencephalic region (for the differential diagnosis) that cause combined vertical gaze ophthalmoplegia (see Chapter 35). The most common causes of isolated combined vertical gaze ophthalmoplegia are stroke and progressive supranuclear palsy (PSP). In cortical-basal ganglionic degeneration, ocular motility findings are similar to those in PSP but are less severe. Whereas the supranuclear vertical gaze ophthalmoplegia may be prominent early in the course of PSP, obvious vertical and horizontal gaze restriction usually is a late finding in cortical-basal ganglionic degeneration (Rottach et al., 1996).
The diagnostic formulation varies with the age of the patient. Isolated combined vertical gaze ophthalmoplegia usually is due to infarction of the rostral dorsal midbrain. When onset is gradual instead of abrupt or if the patient is young, other disorders should be considered (see Box 19.1). In the elderly, PSP (see Chapter 66) is likely if the onset is gradual. PSP can be mimicked by the treatable Whipple disease (Averbuch-Heller et al., 1999). For Whipple disease, the movement disorder, oculomasticatory myorhythmia, is pathognomonic. Laboratory investigations used to evaluate combined vertical gaze ophthalmoplegia include computed tomography (CT) scan or, preferably, magnetic resonance imaging (MRI). Care should be taken not to overlook lesions inferior to the floor of the third ventricle. Lumbar puncture (LP), syphilis serology, erythrocyte sedimentation rate, and an antinuclear antibody test complete the evaluation when the cause is not obvious. Small-bowel biopsy should be considered if Whipple disease is a possible diagnosis. A polymerase chain reaction (PCR) assay of small-bowel biopsy specimen, cerebrospinal fluid (CSF), or other tissues for the 16S ribosomal ribonucleic acid (RNA) gene of Tropheryma whippelii appears to have both sensitivity and specificity for the diagnosis of Whipple disease (Lee, 2002).
Upgaze Paresis (Dorsal Midbrain or Parinaud Syndrome)
The location of the lesion causing the upgaze paresis of the dorsal midbrain syndrome is the posterior commissure and its interstitial nucleus (Leigh and Zee, 2006). The presence of the full syndrome implies a lesion of the dorsal midbrain (including the posterior commissure), a bilateral lesion of the pretectal region, or a large unilateral tegmental lesion.
The differential diagnosis is presented in Box 19.2. Other than the mild upgaze limitation that occurs with age, the most common cause of loss of upgaze is a tumor of the pineal region. The next most common causes are stroke and trauma. The upgaze palsy portion of the syndrome can be mimicked by any of several conditions: double elevator palsy, PSP, orbital causes such as thyroid ophthalmopathy and the bilateral Brown superior oblique tendon sheath syndrome, pseudo–dorsal midbrain syndrome secondary to myasthenia gravis (MG) or Guillain-Barré syndrome, and congenital upgaze limitation. Forced ductions (see Chapter 16) may be performed by grasping anesthetized sclera with forceps and moving the globe through its range of motion. The presence of restriction of movement with forced ductions implies a lesion within the orbit, as distinct from a midbrain lesion.
Internuclear Ophthalmoplegia
The MLF carries information for vertical pursuit and the vertical VOR. Consequently, other associated findings with MLF lesions are abnormal vertical smooth pursuit and impaired reflex vertical eye movements (doll’s eye maneuver, Bell phenomenon). Voluntary vertical eye movements (pursuit and saccades) are unaffected. Gaze-evoked vertical nystagmus (usually on upgaze) and skew deviation may be present with the higher eye usually present on the side of the lesion. Skew deviation is a pure vertical ocular deviation that is not due to a cranial nerve palsy, orbital lesion, or strabismus but is caused by disturbed supranuclear input to the third and fourth cranial nerve nuclei. It is thought to be due to unilateral damage to the otolith-ocular pathways or the pathways mediating the VOR (Zwergal et al., 2008).
Internuclear ophthalmoplegia, discussed further in Chapter 35, may occur as a false localizing sign. Brainstem compression due to subdural hematoma with transtentorial herniation and cerebellar masses may cause INO. Myasthenia gravis and Guillain-Barré syndrome also may simulate INO.
The diagnostic considerations are many and varied. Examination can differentiate a lesion of the MLF from a partial third cranial nerve palsy, MG, strabismus, or thyroid ophthalmopathy. The common causes of INO are stroke (including vertebral artery dissection) in older age groups and MS in the young. Keane’s series (Keane, 2005) from a large inner-city hospital reveals that approximately one third of INO cases are due to stroke, one third to MS, and one third to other causes. The less common causes include trauma, herniation, infections, tumor, vasculitis, and surgical procedures.
Horizontal Gaze Paresis
Lesions of the sixth cranial nerve nucleus cause horizontal gaze paresis with inability of the oculocephalic maneuver to overcome the gaze limitation. Although an associated ipsilateral peripheral facial palsy is usually associated from involvement of the fascicle of the seventh cranial nerve coursing over the sixth cranial nerve nucleus, cases of isolated horizontal gaze paresis caused by sixth nerve nuclear lesions have been reported (Miller et al., 2002). With bilateral lesions, loss or limitation of horizontal saccades and (usually) pursuit in both directions is characteristic. Gaze-paretic nystagmus may be present. In the acute phase, transient vertical gaze paresis and vertical nystagmus or upgaze paresis can occur. In the chronic phase, vertical eye movements are full, although nystagmus may be noted on upgaze.
The location of the lesion for horizontal gaze paresis is the frontopontine tract, mesencephalic reticular formation, PPRF, and sixth cranial nerve nucleus. The explanation of gaze palsy occurring with a nuclear lesion is given later in the chapter (see Syndromes Involving Ocular Motor Nuclei).
Global Paralysis of Gaze
The location of the lesion is the frontopontine tract for saccades, and the parieto-occipitopontine tract for pursuit, where they converge at the subthalamic and upper midbrain level (Thurtell and Halmagyi, 2008). The differential diagnosis for total ophthalmoplegia is given in Box 19.3. The common causes for this presentation are diseases outside the CNS, such as Guillain-Barré syndrome, MG, and chronic progressive external ophthalmoplegia (CPEO); for intraaxial lesions, considerations include stroke, Wernicke encephalopathy, and PSP.
Box 19.3 Differential Diagnosis for Total Ophthalmoplegia
The diagnostic formulation usually is concerned with extraaxial (cranial nerve, neuromuscular junction, or muscle) pathology, because isolated complete ophthalmoplegia is rarely caused by a brainstem lesion. Myasthenia gravis (sometimes in combination with thyroid ophthalmopathy), bilateral cavernous sinus metastases (Ebert et al., 2009), and Guillain-Barré syndrome are more likely possibilities if the onset is subacute. If the presentation is long-standing, slowly progressive, and accompanied by eyelid ptosis, the CPEO syndromes, such as Kearns-Sayre syndrome, should be considered. In these extraaxial disorders, oculocephalic reflexes do not overcome the gaze limitations. PSP is a diagnostic possibility in the elderly, whereas Wernicke encephalopathy should be considered in alcoholics and nutritionally deprived patients. Whipple disease also can cause this rare clinical presentation.
Syndromes Involving Ocular Motor Nuclei
Sixth Cranial Nerve Nucleus
Patients with isolated horizontal gaze paresis usually are asymptomatic. If they do have symptoms, they complain of difficulty looking to one side. On examination, conjugate horizontal gaze paresis is present that is not overcome by an oculocephalic maneuver or caloric stimulation. This occurs because the fibers mediating this response, the VOR, synapse in the sixth cranial nerve nucleus. A peripheral seventh cranial nerve palsy invariably accompanies a lesion of the sixth cranial nerve nucleus. Considerations in the differential diagnosis include stroke (Miller et al., 2002), Wernicke encephalopathy, MS, and a tumor of the pontomedullary junction.
Other Brainstem and Associated Syndromes
Diencephalic Syndrome (Russell Syndrome)
The common symptoms of diencephalic syndrome are emaciation with increased appetite, euphoria, vomiting, and excessive sweating (Fleischman et al., 2005). Patients also may have an alert appearance with motor hyperactivity. Most cases occur in children younger than 3 years.
Thalamic Syndrome
The usual location of the lesion for this type of pain is the ventroposterolateral nucleus of the thalamus. In addition to the thalamus, thalamic-type pain can occur with lesions of the parietal lobe, medial lemniscus, and dorsolateral medulla (MacGowan et al., 1997). The differential diagnosis is between stroke and tumor. The diagnostic formulation depends on the rate of onset of symptoms, associated signs, and findings on neuroimaging studies. The apoplectic onset of symptoms implicates cerebrovascular disease. Gradual onset with progressive worsening of symptoms and signs is characteristic of brain tumor. Neuroimaging studies should confirm the clinical impression. The imaging modality of choice is MRI.
Tectal Deafness
On examination, deafness that usually spares pure tones is confirmed. Pure word deafness with lesions of the inferior colliculi has been reported (Vitte et al., 2002). Other brainstem signs, including the dorsal midbrain syndrome, often are associated. The location of the lesion is the inferior colliculi; the most common causes are trauma, stroke, or a tumor of the brainstem, cerebellum, or pineal region. The diagnostic formulation for hearing loss caused by lesions rostral to the cochlear nuclei is the presence of hearing loss characterized by sparing of pure tone, with marked deterioration when background noise distortion or competing messages are added. In addition, signs of damage to adjacent nervous system structures are present. Neuroimaging studies may confirm the diagnosis.
The pertinent laboratory investigations include MRI and an audiogram. Tests that reveal CNS auditory loss are distorted speech audiometry, dichotic auditory testing, and auditory brainstem evoked responses, although findings on the last test may be normal (Vitte et al., 2002).
Foramen Magnum Syndrome
Foramen magnum syndrome is characterized by upper motor neuron–type weakness and sensory loss in any modality below the head. Detecting this syndrome is important because it often is caused by benign tumors such as meningiomas or fibromas, which may be removed completely when detected early in their course. Its only manifestations may be those of a high spinal cord syndrome (see Chapter 24).
Syringobulbia
Syringobulbia is a disorder of the lower brainstem caused by progressive enlargement of a fluid-filled cavity that involves the medulla and almost invariably the spinal cord (syringomyelia). The symptoms and signs are primarily those of a disorder of the central spinal cord region (see Syringomyelia in Chapter 73).
Brainstem Ischemic Stroke Syndromes
Vertebrobasilar ischemic lesions often have a rostrocaudal or patchy localization (Fig. 19.1), rather than the simplified transverse localization that usually is schematized (Kubik and Adams, 1946). In addition, all of the clinical features may not be explainable in anatomical terms; that is, clinicopathological correlation may not be precise.
Ischemic stroke syndromes are outlined next. These syndromes occur in isolation, as presented here, and in combination. The combinations can be medial, with lateral or often rostrocaudal extension. Several classic stroke syndromes, such as Wallenberg later medullary syndrome and Weber cerebral peduncle syndrome, exist. However, an investigation that looked prospectively at 304 cases of brainstem stroke found 1 of 24 named syndromes in only 20. About 20% of cases showed different unnamed crossed brainstem syndromes (Marx and Thomke, 2009).
Thalamic Stroke Syndromes
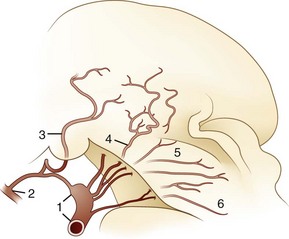
The blood supply of the thalamus is from the posterior cerebral, posterior communicating, basilar communicating (Fig. 19.2), and anterior and posterior choroidal arteries. Thalamic stroke syndromes are listed in Table 19.1. Fig. 19.3 illustrates the five arterial territories of the thalamus.
| ANTEROLATERAL |
| Common Symptoms |
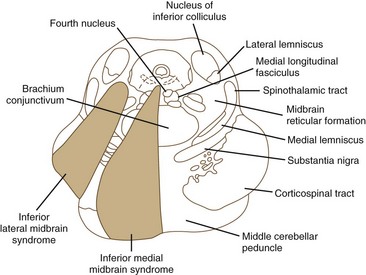
Midbrain Stroke Syndromes
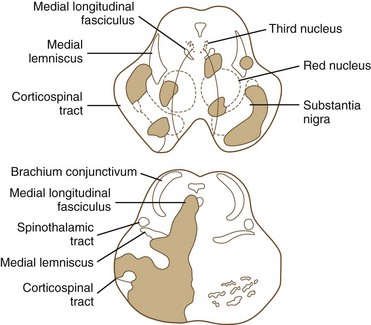
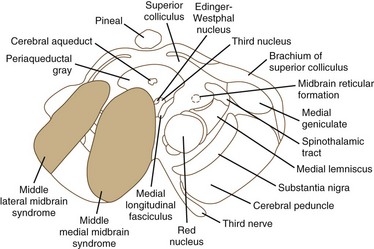
Ischemia of the midbrain is characterized by long-tract signs combined with involvement of the third and fourth cranial nerves (Bogousslavsky et al., 1994). Supratentorial (anterior circulation) stroke syndromes may manifest with midbrain signs when rostrocaudal deterioration occurs, causing transtentorial herniation. Classifications of the blood supply to the brainstem are numerous, and this variability is nowhere more apparent than in the midbrain.
The medial midbrain syndromes are characterized by an ipsilateral third cranial nerve palsy associated with a contralateral hemiparesis. Loss of the discriminative sensations (proprioception, vibration, and stereognosis) with involvement of the medial lemniscus may occur. The lateral syndromes are composed of contralateral loss of pain and temperature sensation and ipsilateral Horner syndrome and loss of facial sensation. Ataxia may occur on either side. Ischemic stroke syndromes of the mesencephalon are outlined in Table 19.2, and Figs. 19.4 and 19.5 show the territories involved with occlusive and ischemic stroke syndromes in this area. Eponymic designations are given later in the text in Table 74.1. Mossuto-Agatiello (2006) identified a caudal paramedian midbrain syndrome with a distinctive clinical picture: bilateral cerebellar (truncal and gait) ataxia, eye movement disorders (nuclear third nerve palsy, INO), and palatal myoclonus.
| MIDDLE MEDIAN MIDBRAIN SYNDROME |
| Common Symptoms |
Pontine Stroke Syndromes
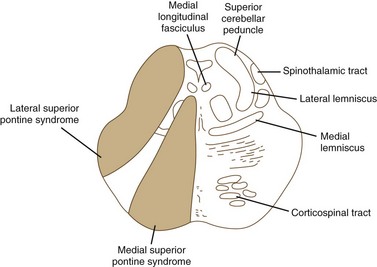
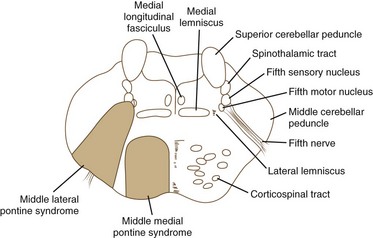
The pons is supplied by numerous penetrating branches of the basilar artery. These arteries have little collateral supply; consequently, lacunar syndromes (see Chapter 51A) can occur (Table 19.3). These syndromes (Figs. 19.6, 19.7, and 19.8) may be clinically indistinguishable from lacunar syndromes due to lesions of the internal capsule. More extensive paramedian syndromes are accompanied by characteristic pontine findings. The findings may fluctuate (pontine warning syndrome) and in this situation may be treated by intravenous venous tissue plasminogen activator (Saposnik et al., 2008). Contralateral hemiparesis, ipsilateral ataxia, INO, and conjugate horizontal gaze paresis characterize the medial syndromes.
| SUPERIOR MEDIAL PONTINE SYNDROME (see Fig. 19.6) |
| Common Symptoms |
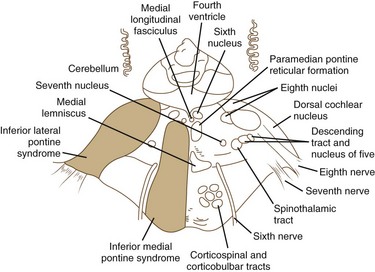
Medullary Stroke Syndromes
Medial medullary ischemia (Fig. 19.9) can cause crossed hypoglossal hemiparesis syndrome (Table 19.4). In addition, patients may have loss of discriminative-type sensation (position sense, graphesthesia, and stereognosis) when there is associated medial lemniscus involvement. Kumral and associates (2002) have described four patterns of medial medullary stroke: (1) the most frequent, classic crossed hypoglossal hemiparesis syndrome, (2) sensorimotor stroke without lingual palsy, (3) pure hemiparesis, and (4) bilateral medial medullary stroke. Ocular motor findings often are prominent (Kim et al., 2005), with ocular contrapulsion, ocular tilt reaction, ipsilesional nystagmus (gaze-evoked, upbeating, and hemi-seesaw).
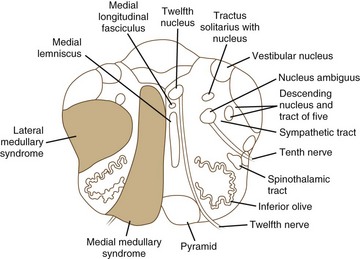
| MEDIAL MEDULLARY SYNDROME (see Fig. 19.9) |
| Common Symptoms |
Lateral medullary syndrome (Wallenberg syndrome; see Fig. 19.9) is one of the most dramatic clinical presentations in neurology (see Table 19.4). Long-tract signs (i.e., contralateral loss of pain and temperature sensation over half of the body, ipsilateral ataxia, ipsilateral axial lateropulsion [Eggers et al., 2009], Horner syndrome) are accompanied by involvement of the nuclei and fasciculi of cranial nerves V, VIII, IX, and X. Nystagmus often is present. The critical sign that distinguishes this from a lateral pontine syndrome is involvement of the nucleus ambiguus or its fasciculus and consequent weakness of the ipsilateral palate and vocal cord. It is increasingly found in the setting of vertebral artery dissection. (A more detailed discussion of stroke is presented in Chapter 51A.)
Averbuch-Heller L., Paulson G.W., Daroff R.B., et al. Whipple’s disease mimicking progressive supranuclear palsy: The diagnostic value of eye movement recording. J Neurol Neurosurg Psychiatry. 1999;66:532-535.
Bogousslavsky J., Meader P., Regli F., et al. Pure midbrain infarction: clinical syndromes, MRI, and etiologic patterns. Neurology. 1994;44:2032-2040.
Ebert S., Pilgram S.M., Bahr M., et al. Bilateral ophthalmoplegia due to symmetric cavernous sinus metastasis from gastric adenocarcinoma. J Neurol Sci. 2009;279:106-108.
Eggers C., Fink G.R., Moller-Hartmann W., et al. Correlation of anatomy and function in medulla oblongata infarction. Eur J Neurol. 2009;16:201-204.
Fleischman A., Brue C., Poussaint T.Y., et al. Diencephalic syndrome: a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics. 2005;115:742-748.
Graff-Redford N.R., Damasio H., Yamada T. Nonhaemorrhagic thalamic infarction: clinical, neuropsychological and electrophysiological findings in four anatomical groups defined by computerized tomography. Brain. 1985;108:485-516.
Keane J. Internuclear ophthalmoplegia; unusual causes in 114 of 410 patients. Arch Neurol. 2005;62:714-717.
Kim J.S., Choi K.D., Oh S.Y., et al. Medial medullary infarction: abnormal ocular motor findings. Neurology. 2005;65:1294-1298.
Kubik C.S., Adams R.D. Occlusion of the basilar artery: a clinical and pathological study. Brain. 1946;69:73-121.
Kumral E., Afsar N., Kirbas D., et al. Spectrum of medial medullary infarction: Clinical and magnetic resonance imaging findings. J Neurol. 2002;249:85-93.
Lee A.G. Whipple disease with supranuclear ophthalmoplegia diagnosed by polymerase chain reaction of cerebrospinal fluid. J Neuroophthalmol. 2002;22:18-21.
Leigh R.J., Zee D.S. The Neurology of Eye Movements, 4th ed. New York: Oxford University Press; 2006.
MacGowan D.G., Janal M.N., Clark W.C., et al. Central poststroke pain and Wallenberg’s lateral medullary infarction. Frequency, character, and determinants in 63 patients. Neurology. 1997;49:120-125.
Marx A.E., Thomke F. Classical crossed brain stem syndromes: myth or reality? J Neurol. 2009;256:898-903.
Miller N.R., Biousse V., Hwang T., et al. Isolated acquired unilateral horizontal gaze paresis from a putative lesion of the abducens nucleus. J Neuroophthalmol. 2002;22:204-207.
Mossuto-Agatiello L. Caudal paramedian midbrain syndrome. Neurology. 2006;66:1668-1671.
Rottach K.G., Riley D.E., DiScenna A.O., et al. Dynamic properties of horizontal and vertical eye movements in parkinsonian syndromes. Ann Neurol. 1996;39:368-377.
Saposnik G., Noel de Tilly L., Caplan L.R. Pontine warning syndrome. Arch Neurol. 2008;65:1375-1377.
Thurtell M.J., Halmagyi G.M. Complete ophthalmoplegia an unusual sign of bilateral paramedian midbrain-thalamic infarction. Stroke. 2008;39:1355-1357.
Vitte E., Tankere F., Bernat I., et al. Midbrain deafness with normal brainstem auditory evoked potentials. Neurology. 2002;58:970-973.
Zwergal A., Cnyrim C., Arbusow V., et al. Unilateral INO is associated with ocular tilt reaction in pontomesencephalic lesions: INO plus. Neurology. 2008;71:590-593.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 19 Brainstem Syndromes
Ocular Motor Syndromes
Combined Vertical Gaze Ophthalmoplegia
Combined vertical gaze ophthalmoplegia is defined as paresis of both upward and downward gaze. Vertical gaze ophthalmoplegia is an example of a brainstem syndrome in which the objective physical findings dictate the diagnostic approach to the problem. Symptoms of vertical gaze ophthalmoplegia, when present, are relatively nonspecific and usually occur in patients who have difficulty looking down, as required in reading, eating from a table, and walking down a flight of stairs. In addition, the patient’s report of symptoms may be unobtainable because of mental status changes caused by dysfunction of the reticular formation that lies adjacent to the vertical gaze generator in the rostral midbrain (see Chapter 35).
The neurological examination discloses associated signs of the disorders listed in the differential diagnosis (see Box 19.1) (Graff-Redford et al., 1985). Coma may be associated with reticular system involvement. Long-tract signs and loss of pupillary reflexes are commonly associated. The syndrome of combined vertical gaze ophthalmoplegia is diagnosed when the ocular findings occur in isolation from long-tract signs.
Box 19.1 Differential Diagnosis for Combined Vertical Gaze Ophthalmoplegia
Corticobasal ganglionic degeneration
Thalamic and mesencephalic tumors
AVM, Arteriovenous malformation; MS, multiple sclerosis; PSP, progressive supranuclear palsy.
With combined vertical gaze ophthalmoplegia, vertical saccades and pursuit are lost. This gaze limitation may be overcome by the oculocephalic (doll’s head or doll’s eye) maneuver, which tests the vestibulo-ocular reflex (VOR) (see Chapter 35). It is demonstrated by having the patient focus on an object and rotating the patient’s head; a conjugate eye movement in the opposite direction is the expected response with this maneuver. The Bell phenomenon (reflex movement of the eyes up and out in response to forced eye closure) often is absent. Skew deviation (vertical malalignment of the eyes) may occur. Absence of convergence and loss of pupillary reactions to light are common.
The location of the lesion of combined vertical gaze ophthalmoplegia is the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) for loss of vertical pursuit and saccades (Leigh and Zee, 2006). Box 19.1 lists the disorders involving the rostral mesodiencephalic region (for the differential diagnosis) that cause combined vertical gaze ophthalmoplegia (see Chapter 35). The most common causes of isolated combined vertical gaze ophthalmoplegia are stroke and progressive supranuclear palsy (PSP). In cortical-basal ganglionic degeneration, ocular motility findings are similar to those in PSP but are less severe. Whereas the supranuclear vertical gaze ophthalmoplegia may be prominent early in the course of PSP, obvious vertical and horizontal gaze restriction usually is a late finding in cortical-basal ganglionic degeneration (Rottach et al., 1996).
The diagnostic formulation varies with the age of the patient. Isolated combined vertical gaze ophthalmoplegia usually is due to infarction of the rostral dorsal midbrain. When onset is gradual instead of abrupt or if the patient is young, other disorders should be considered (see Box 19.1). In the elderly, PSP (see Chapter 66) is likely if the onset is gradual. PSP can be mimicked by the treatable Whipple disease (Averbuch-Heller et al., 1999). For Whipple disease, the movement disorder, oculomasticatory myorhythmia, is pathognomonic. Laboratory investigations used to evaluate combined vertical gaze ophthalmoplegia include computed tomography (CT) scan or, preferably, magnetic resonance imaging (MRI). Care should be taken not to overlook lesions inferior to the floor of the third ventricle. Lumbar puncture (LP), syphilis serology, erythrocyte sedimentation rate, and an antinuclear antibody test complete the evaluation when the cause is not obvious. Small-bowel biopsy should be considered if Whipple disease is a possible diagnosis. A polymerase chain reaction (PCR) assay of small-bowel biopsy specimen, cerebrospinal fluid (CSF), or other tissues for the 16S ribosomal ribonucleic acid (RNA) gene of Tropheryma whippelii appears to have both sensitivity and specificity for the diagnosis of Whipple disease (Lee, 2002).
Upgaze Paresis (Dorsal Midbrain or Parinaud Syndrome)
The location of the lesion causing the upgaze paresis of the dorsal midbrain syndrome is the posterior commissure and its interstitial nucleus (Leigh and Zee, 2006). The presence of the full syndrome implies a lesion of the dorsal midbrain (including the posterior commissure), a bilateral lesion of the pretectal region, or a large unilateral tegmental lesion.
The differential diagnosis is presented in Box 19.2. Other than the mild upgaze limitation that occurs with age, the most common cause of loss of upgaze is a tumor of the pineal region. The next most common causes are stroke and trauma. The upgaze palsy portion of the syndrome can be mimicked by any of several conditions: double elevator palsy, PSP, orbital causes such as thyroid ophthalmopathy and the bilateral Brown superior oblique tendon sheath syndrome, pseudo–dorsal midbrain syndrome secondary to myasthenia gravis (MG) or Guillain-Barré syndrome, and congenital upgaze limitation. Forced ductions (see Chapter 16) may be performed by grasping anesthetized sclera with forceps and moving the globe through its range of motion. The presence of restriction of movement with forced ductions implies a lesion within the orbit, as distinct from a midbrain lesion.
Internuclear Ophthalmoplegia
The MLF carries information for vertical pursuit and the vertical VOR. Consequently, other associated findings with MLF lesions are abnormal vertical smooth pursuit and impaired reflex vertical eye movements (doll’s eye maneuver, Bell phenomenon). Voluntary vertical eye movements (pursuit and saccades) are unaffected. Gaze-evoked vertical nystagmus (usually on upgaze) and skew deviation may be present with the higher eye usually present on the side of the lesion. Skew deviation is a pure vertical ocular deviation that is not due to a cranial nerve palsy, orbital lesion, or strabismus but is caused by disturbed supranuclear input to the third and fourth cranial nerve nuclei. It is thought to be due to unilateral damage to the otolith-ocular pathways or the pathways mediating the VOR (Zwergal et al., 2008).
Internuclear ophthalmoplegia, discussed further in Chapter 35, may occur as a false localizing sign. Brainstem compression due to subdural hematoma with transtentorial herniation and cerebellar masses may cause INO. Myasthenia gravis and Guillain-Barré syndrome also may simulate INO.
The diagnostic considerations are many and varied. Examination can differentiate a lesion of the MLF from a partial third cranial nerve palsy, MG, strabismus, or thyroid ophthalmopathy. The common causes of INO are stroke (including vertebral artery dissection) in older age groups and MS in the young. Keane’s series (Keane, 2005) from a large inner-city hospital reveals that approximately one third of INO cases are due to stroke, one third to MS, and one third to other causes. The less common causes include trauma, herniation, infections, tumor, vasculitis, and surgical procedures.
Horizontal Gaze Paresis
Lesions of the sixth cranial nerve nucleus cause horizontal gaze paresis with inability of the oculocephalic maneuver to overcome the gaze limitation. Although an associated ipsilateral peripheral facial palsy is usually associated from involvement of the fascicle of the seventh cranial nerve coursing over the sixth cranial nerve nucleus, cases of isolated horizontal gaze paresis caused by sixth nerve nuclear lesions have been reported (Miller et al., 2002). With bilateral lesions, loss or limitation of horizontal saccades and (usually) pursuit in both directions is characteristic. Gaze-paretic nystagmus may be present. In the acute phase, transient vertical gaze paresis and vertical nystagmus or upgaze paresis can occur. In the chronic phase, vertical eye movements are full, although nystagmus may be noted on upgaze.
The location of the lesion for horizontal gaze paresis is the frontopontine tract, mesencephalic reticular formation, PPRF, and sixth cranial nerve nucleus. The explanation of gaze palsy occurring with a nuclear lesion is given later in the chapter (see Syndromes Involving Ocular Motor Nuclei).
Global Paralysis of Gaze
The location of the lesion is the frontopontine tract for saccades, and the parieto-occipitopontine tract for pursuit, where they converge at the subthalamic and upper midbrain level (Thurtell and Halmagyi, 2008). The differential diagnosis for total ophthalmoplegia is given in Box 19.3. The common causes for this presentation are diseases outside the CNS, such as Guillain-Barré syndrome, MG, and chronic progressive external ophthalmoplegia (CPEO); for intraaxial lesions, considerations include stroke, Wernicke encephalopathy, and PSP.
Box 19.3 Differential Diagnosis for Total Ophthalmoplegia
The diagnostic formulation usually is concerned with extraaxial (cranial nerve, neuromuscular junction, or muscle) pathology, because isolated complete ophthalmoplegia is rarely caused by a brainstem lesion. Myasthenia gravis (sometimes in combination with thyroid ophthalmopathy), bilateral cavernous sinus metastases (Ebert et al., 2009), and Guillain-Barré syndrome are more likely possibilities if the onset is subacute. If the presentation is long-standing, slowly progressive, and accompanied by eyelid ptosis, the CPEO syndromes, such as Kearns-Sayre syndrome, should be considered. In these extraaxial disorders, oculocephalic reflexes do not overcome the gaze limitations. PSP is a diagnostic possibility in the elderly, whereas Wernicke encephalopathy should be considered in alcoholics and nutritionally deprived patients. Whipple disease also can cause this rare clinical presentation.
