[level-membership-for-pediatrics-category]
Chapter 694 Bone Structure, Growth, and Hormonal Regulation
Also see Chapters 48 and 564.
Rates of bone formation are coordinated with alterations in mineral metabolism in both the intestine and kidneys. Inadequate dietary intake or intestinal absorption of calcium causes a fall in serum levels of calcium and its ionized fraction. This serves as the signal for PTH synthesis and secretion, resulting in greater bone resorption to raise the serum calcium level, enhanced distal tubular reabsorption of calcium, and higher rates of synthesis by the kidneys of 1,25(OH]2D or calcitriol, the most active metabolite of vitamin D (Fig. 694-1). Calcium homeostasis thus is controlled by the intestine because the availability of 1,25(OH)2D ultimately determines the fraction of ingested calcium that is absorbed.
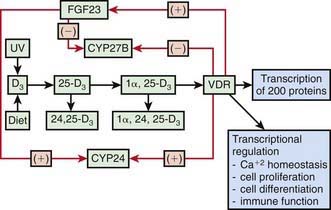
An understanding of the metabolism of vitamin D is necessary to appreciate metabolic bone disease and rickets. The skin contains 7-dehydrocholesterol, which is converted to vitamin D3 [25(OH)D3] by ultraviolet radiation; other inactive vitamin D sterols are also produced (Chapter 48). Vitamin D3 is then transported in the bloodstream to the liver by a vitamin D–binding protein (DBP); DBP binds all forms of vitamin D. The plasma concentration of free or nonbound vitamin D is much lower than the level of DBP-bound vitamin D metabolites.
Vitamin D also can enter the metabolic pathway by ingestion of dietary vitamin D2 (ergocalciferol) or vitamin D3 (cholecalciferol), both of which are absorbed from the intestine because of the action of bile salts. After absorption, ingested vitamin D is transported by chylomicrons to the liver, where, along with skin-derived vitamin D3, it is converted to 25-hydroxyvitamin D [25(OH)D] by the action of a hepatic microsomal enzyme requiring oxygen, NADPH, and magnesium to hydroxylate vitamin D at the 25th carbon atom. The 25(OH)D is next transported by DBP to the kidneys, where it undergoes further metabolism. 25(OH)D is the main circulating vitamin D metabolite in humans (Table 694-1). Because the synthesis of 25(OH)D is weakly regulated by feedback, its plasma level rises in summer and falls in winter. High vitamin D intake raises the plasma level of 25(OH)D to many times above normal, but the parent vitamin D compound itself is absorbed by adipose tissue.
Table 694-1 VITAMIN D METABOLIC VALUES IN PLASMA OF NORMAL HEALTHY SUBJECTS
| METABOLITE | PLASMA VALUE |
|---|---|
| Vitamin D2 | 1-2 ng/mL |
| Vitamin D3 | 1-2 ng/mL |
| 25(OH)D2 | 4-10 ng/mL |
| 25(OH)D3 | 26-70 ng/mL |
| TOTAL 25(OH)D | 30-80 ng/mL |
| 24,25(OH)2D | 1-4 ng/mL |
| 1,25(OH)2D | |
| Infancy | 70-100 pg/mL |
| Childhood | 30-50 pg/mL |
| Adolescence | 40-80 pg/mL |
| Adulthood | 20-35 pg/mL |
In the kidneys, 25(OH)D undergoes further hydroxylation, depending on the prevailing serum concentration of calcium, phosphate, and PTH. If the calcium or phosphate level is reduced or the PTH level is elevated, the enzyme 25(OH)D-1-hydroxylase is activated and 1,25(OH)2D is formed. This metabolite circulates at a level that is only 0.1% of the level of 25(OH)D (see Table 694-1) and acts on the intestine to increase the active transport of calcium and stimulate phosphate absorption. Because 1α-hydroxylase is a mitochondrial enzyme that is tightly feedback regulated, the synthesis of 1,25(OH)2D declines after serum calcium or phosphate values return to normal. Excessive 1,25(OH)2D is converted to an inactive metabolite. In the presence of normal or elevated serum calcium or phosphate concentrations, the renal 25(OH)D-24-hydroxylase is activated, producing 24,25-dihydroxyvitamin D [24,25(OH)2D], which is a pathway for the removal of excess vitamin D; serum levels of 24,25(OH)2D (1-5 ng/mL) increase after ingestion of large amounts of vitamin D (see Fig. 694-1). Although hypervitaminosis D and production of inactive metabolites can occur after oral dosing (Chapter 48), extensive skin exposure to sunlight does not usually produce toxic levels of 25(OH)D3, suggesting natural regulation of the production of this metabolite in cutaneous tissue.
Mineral deficiency prevents the normal process of bone mineral deposition. If mineral deficiency occurs at the growth plate, growth slows and bone age is retarded, a condition called rickets. Poor mineralization of trabecular bone resulting in a greater proportion of unmineralized osteoid is the condition of osteomalacia. Rickets is found only in growing children before fusion of the epiphyses, whereas osteomalacia is present at all ages. All patients with rickets have osteomalacia, but not all patients with osteomalacia have rickets. These conditions should not be confused with osteoporosis, a condition of equal loss of bone volume and mineral (Chapter 698).
Another class of proteins important in the regulation of mineral balance and vitamin D synthesis are the phosphatonins. Among these are fibroblast growth factor (FGF)-23, sFRP-4 and MEPE. Overexpression of FGF-23 results in hypophosphatemia, phosphaturia, reduced serum 1,25(OH)2D values, and rickets. Disorders of phosphate balance, including hyper- and hypophosphatemia, can relate to loss or gain of function of these phosphatonins (see Fig. 694-1).
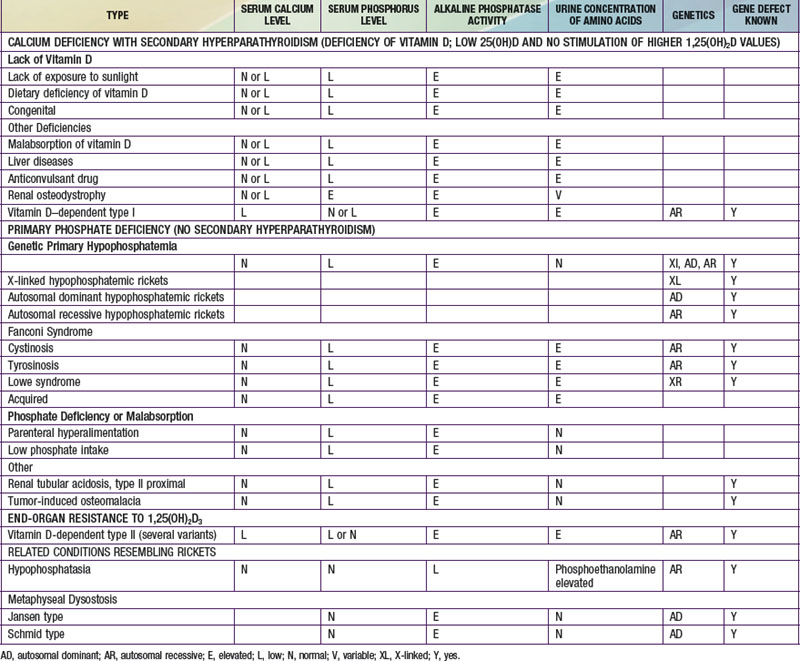
Rickets may be classified as calcium-deficient or phosphate-deficient rickets. Because both calcium and phosphate ions constitute bone mineral, the insufficiency of either type in the ECF that bathes the mineralizing surface of bone results in rickets and osteomalacia. The two types of rickets are distinguishable by their clinical manifestations (Table 694-2). Rickets can also occur in the face of mineral deficiency, despite adequate vitamin D stores. True dietary calcium deficiency rickets is found in some parts of Africa but rarely in North America or Europe. A form of phosphate-deficiency rickets can occur in infants given prolonged administration of phosphate-sequestering aluminum salts as a treatment for colic or gastroesophageal reflux. This results in the phosphate depletion syndrome.
Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology. 2009;24:17-25.
Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL-receptor related protein 5. N Engl J Med. 2002;346:1513-1521.
Carpenter TO. Disorders of mineral metabolism in childhood. In: Rosen CJ, Compston JE, Lian JB, editors. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society for Bone and Mineral Research; 2008:349-353.
Fewtrell MS. Bone densitometry in children assessed by dual x ray absorptiometry: uses and pitfalls. Arch Dis Child. 2003;88:795-798.
Gordon CM, Bachrach LK, Carpenter TO. Bone health in children and adolescents. Curr Prob Pediatr Adolesc Health Care. 2004;34:221-248.
Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656-1663.
Loud KJ, Gordon CM. Adolescent bone health. Arch Pediatr Adolesc Med. 2006;160:1026-1032.
Matlik L, Savaiano D, McCabe G, et al. Perceived milk intolerance is related to bone mineral content in 10- to 13-year-old female adolescents. Pediatrics. 2007;120:e669-e677.
Razzaque MS. FGF23-mediated regulation of systemic homeostasis: is Klotho an essential player? Am J Physiol. 2009;296:F470-F476.
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, et al. Multiple genetic loci for bone mineral density and fractures. N Engl J Med. 2008;358:2355-2365.
Van der Sluis IM, de Ridder MAJ, Boot AM, et al. Reference data for bone density and body composition measured with dual energy x-ray absorptiometry in white children and young adults. Arch Dis Child. 2002;87:341-347.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 694 Bone Structure, Growth, and Hormonal Regulation
Also see Chapters 48 and 564.
Rates of bone formation are coordinated with alterations in mineral metabolism in both the intestine and kidneys. Inadequate dietary intake or intestinal absorption of calcium causes a fall in serum levels of calcium and its ionized fraction. This serves as the signal for PTH synthesis and secretion, resulting in greater bone resorption to raise the serum calcium level, enhanced distal tubular reabsorption of calcium, and higher rates of synthesis by the kidneys of 1,25(OH]2D or calcitriol, the most active metabolite of vitamin D (Fig. 694-1). Calcium homeostasis thus is controlled by the intestine because the availability of 1,25(OH)2D ultimately determines the fraction of ingested calcium that is absorbed.
[/not-level-membership-for-pediatrics-category]
