Chapter 15 Bone Modeling and Remodeling
Types of Bone
Based on general shape, bones can be classified into three groups: short, flat, and long or tubular. The femur, tibia, and phalanges are examples of long bones. Tubular or long bones have an expanded metaphysis and an epiphysis at either end of a thick cortical wall diaphysis.1 The shaft (diaphysis) is responsible for withstanding primarily torsional and bending stresses, whereas the metaphyseal portion, with its greater deformation under the same load, has become specialized in absorbing impact to protect the articular cartilage.1–3
Short bones, such as the vertebral bodies and tarsal and carpal bones, measure approximately the same length in all dimensions and are roughly cuboid in shape, with slight variations. They are all mainly composed of loose trabecular bone, like the metaphysis of the long bones. The main function of this bone aggregate is, again, absorbing the body’s weight. The carpals and tarsals have very thin cortices,1 whereas the vertebral bodies have a thin shell of compact trabecular bone with no true cortical structure.3
The iliac crests, the skull vault, and the vertebral laminae are examples of flat bones.
Long and short bones ossify using a previously formed cartilage model (endochondral ossification), whereas flat bones form from the condensation and mineralization of loose mesenchymal tissue (intramembranous ossification).3,4 A third type of ossification that occurs when osteoblasts line the periosteum of an existing bone surface and start secreting osteoid in layers, hence making the bone thicker, is termed appositional3–6
Immature bone is woven.1,3,4,7,8 It is found in the embryonic skeleton, fracture callus, and bone neoplasms. It is less organized, weaker, and more flexible, and has increased turnover compared with mature bone. Woven bone does not have the ability to remodel following the stress pattern.1,3,4
Mature bone is lamellar.1,3,4,7,8 Lamellar bone is stress oriented, stronger, and less flexible, and has slower turnover compared with woven bone.1,3,4 There are two different types of lamellar bone: cortical (compact) and cancellous (spongy or trabecular).1,3,4,8–12 Even though cortical and cancellous bone have the same structure and composition, their mechanical properties are very different because of their differences in density and distribution.9,13 Cancellous bone is 50% to 90% porous, whereas cortical bone has a porosity of approximately 10%. This difference in density makes cortical bone 10 times stronger in compression than the trabecular variant.8,14–16
Cortical bone, composed of tightly packed osteons, makes up 80% of the skeleton.1,4,10 Trabecular bone has a surface area per unit volume approximately 20 times that of cortical bone. Almost all of its cells lie between lamellae or on the surface of trabeculae, in close contact with the bone marrow, which makes them much more metabolically active than the cortical bone cells surrounded by bone matrix.9,17
Bone Formation
The formation and maintenance of the skeleton require that bone be produced constantly. Osteoblasts fabricate bone in response to many stimuli and under different conditions, including growth, physiologic remodeling, fracture healing, and heterotopic ossification.18–20 Several studies have also shown that new bone is formed in response to tumors and infections.21–24 It has been shown that osteoblasts have the ability to form bone during distraction osteogenesis,25–31 depositing new bone in the void initially filled by autologous or allogenic bone graft, demineralized bone matrix, or synthetic bone substitutes. In anterior cervical discectomy and fusion (ACDF) and plating, a 97.5% rate of fusion with new bone formation has been achieved with either autograft or allograft.32 In a recent study, Jensen et al. showed an 86% union rate after single- and multiple-level ACDF using patellar allograft and plating.33 In a study from Japan, Momma et al. reported complete bone remodeling on CT scan 6 to 12 months after the use of β-tricalcium phosphate to fill a partial vertebrectomy defect created for cervical decompression surgery.34
Vertebral Bone Formation
Because the vertebrae are short bones, they ossify through endochondral ossification.3–5 The process begins with the concentration of undifferentiated cells that transform into chondrocytes and secrete a hyaline or hyaline-like cartilaginous matrix.5,6,10,35,36 The chondrocytes enlarge and vascular buds invade the cartilage, bringing other progenitor cells that differentiate into osteoblasts that in turn start forming bone on the cartilaginous frame. Osteoclasts then reabsorb the ossified cartilage and immature bone. Osteoblasts finally fill this space with mature lamellar bone.3–5
Ossification Centers of the Vertebrae
By the sixth gestational week, centers of cartilage formation (chondrification) develop in each vertebra. Two chondrification centers develop in each half of the central vertebral body. A hemivertebra occurs when these centers fail to form in one side of the vertebral body. Centers of cartilage formation also develop in each half of the vertebral arches. Next, cartilaginous transverse and spinous processes develop from the primitive arches.37 It has been shown that bone morphogenetic protein 4 is required for the development of the cartilaginous spinous process.38
The primary ossification centers develop in utero. In the vertebra, three primary centers form around the eighth week of gestation. One is located in the center of the body and one in each vertebral arch. Bone forms on the pre-existing vertebral cartilage template.3–537 Primary ossification begins in the lower thoracic spine, then progresses in the cranial and caudal directions.39 The five secondary centers of ossification develop after birth: one at the tip of the spinous process, one at the tip of each transverse process, and one anular center at the ventral portion of the superior and inferior end plates. They start to ossify at approximately 15 to 16 years of age and fuse with the remaining osseous vertebra by the middle of the third decade of life.37,40
Bone Modeling and Remodeling
In general, modeling alludes to bone turnover that alters the shape of the bone, whereas remodeling is the turnover that recycles bone without changing its shape. Bone turnover approaches 100% during the first year of life.41 Most of the bone turnover during skeletal growth derives from modeling. After the completion of skeletal growth, bone turnover results primarily from remodeling. Bone modeling and remodeling are the end results of the activity of a vast array of cells that work in harmony to create bone while maintaining the body’s mineral homeostasis.3–510
Bone Modeling during Growth
During growth, coordinated osteal resorption and formation change the size and shape of bone.17 The physes grow and make the bone longer and narrower. The metaphysis also changes its shape, becoming narrower to match the rest of the bone. Appositional periosteal ossification increases the diaphysial diameter.5 At the same time, the cortices becomes thinner and the medullary canal larger owing to intensified bone resorption on the endosteal side.42,43
Physiologic Bone Remodeling after Growth
Throughout life, in situ removal and replacement of bone take place without changing bone form or density. Remodeling occurs on both the surface and the interior of the bone (internal remodeling). Both processes basically start with osteoclast activation. Internal remodeling commences with osteoclasts reabsorbing bone by cutting conical spaces through old osteonal systems.3–5,17,44 Spindle cells, osteoblasts, and blood vessels fill the conical spaces cut by the osteoclasts. Osteoblasts deposit successive lamellae of new osteoid matrix, which will later mineralize. It takes about 50 osteoblasts to fill the cone cut by 1 osteoclast. Internal remodeling is seen in cortical bone.
Surface remodeling occurs on trabecular (which comprises most of the vertebral body), endosteal, and periosteal bone and is very similar to internal remodeling, except that instead of cutting cones, osteoclasts run on the surface of the lamellae excavating a cavity, the so-called Howship lacuna. The rest of the process resembles internal remodeling. Physiologic remodeling serves to repair damaged bone matrix as well as to maintain mineral homeostasis.3–5
Bone Modeling and Remodeling and the Basic Multicellular Unit
Bone modeling and remodeling are performed by the basic multicellular unit (BMU), a temporary anatomic structure comprising osteoclasts and osteoblasts that replace older packets of bone with new bone tissue.44,45
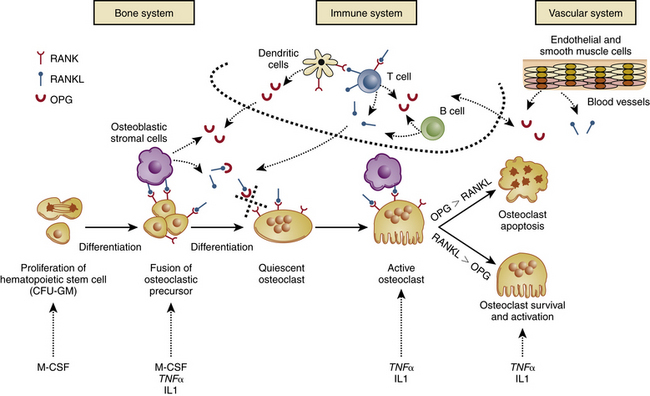
Osteoclasts are derived from hematopoietic stem cells. They exit the circulation close to the site to be remodeled.45 The mononuclear hematopoietic cell’s fusion into a polykaryon (immature osteoclast) requires the presence of macrophage colony-stimulating factor (M-CSF), a growth factor, and the receptor activator of nuclear factor κB ligand (RANKL), a tumor necrosis factor produced by osteoblasts.46,47 Further differentiation of the immature osteoclast occurs under the influence of RANKL and many other genes, including the activator protein-1 (AP-1) family member c-fos,48,49 microphthalmia-associated transcription factor (MITF),50,51 and nuclear factor of activated T cells, calcineurin dependent-1 (NFAT-c1).51,52
The receptor on the osteoclast for RANKL is called RANK.53 Concomitantly, another factor, also produced by stromal cells and osteoblasts, was found that inhibits the activity of RANKL; it was named osteoprotegerin (OPG).54 OPG is a soluble decoy receptor for RANKL, and its function is to reduce osteoclastogenesis by competitively occupying the stromal RANKL binding sites on osteoclast RANK receptors.55–57 The RANKL/OPG signaling axis provides a mechanism through which stromal cells control osteoblastic activity. Factors that exhibit a strong effect on resorption (e.g., parathyroid hormone, prostaglandins, interleukins, vitamin D, and corticosteroids) all signal to the osteoblast/stromal cell, which then appears to translate the message to the osteoclast through the RANKL/OPG axis.58 The only exception to this is the hormone calcitonin, which does not use the RANKL/OPG axis, instead acting directly on the osteoclast receptors.3,4 The mature osteoclast then engages in bone resorption by peripheral attachment to the bone matrix using the β3 integrin,59 which creates a microcompartment between the osteoclast’s ruffled basal border and the bone surface. Hydrogen ions are pumped into the compartment by the osteoclast to digest the mineral component. Next, protease is released to degrade the organic matrix60 (Fig. 15-1).
Osteoblasts are derived from mesenchymal stem cells from the bone marrow and periosteum.3–545 Expression of the transcription factors runt-related transcription factor-2 (Runx2), distal-less homeobox-5 (Dlx5), msh homeobox homologue-2 (Msx2),61–65 and osterix (Osx), as well as activation of several components of the Wnt signaling pathway,62,66–69 are required for osteoblastic differentiation (Fig. 15-2).
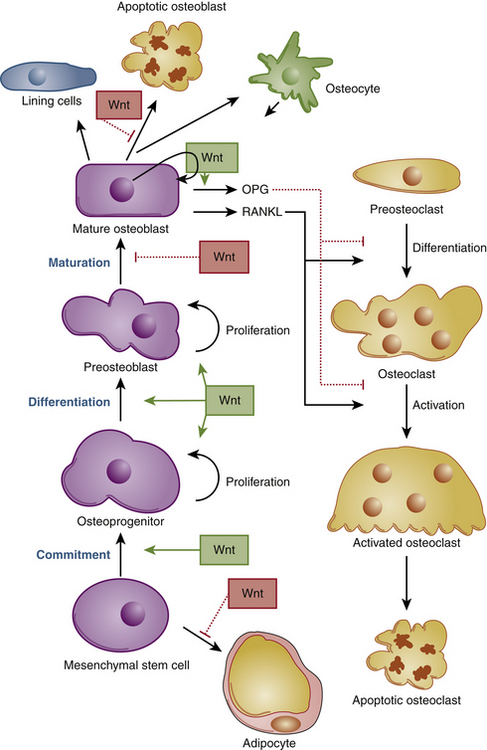
The mature osteoblast produces proteins like type I collagen, osteocalcin, and alkaline phosphatase, the latter a key enzyme in bone mineralization. Osteoblasts become entrapped in their own osteoid matrix and extrude long cytoplasmic processes to remain in contact with surrounding cells.70 They then start expressing a whole new set of genes to continue bone turnover and maintain mineral homeostasis. These cells are now considered osteocytes, the mature bone cell45 (Fig. 15-3).
Age-Related Bone Remodeling (Bone Loss)
Bone density changes drastically with age.3–5,41,45,71 Peak bone mass is reached approximately 10 years after cessation of skeletal growth. Subsequently, bone mass begins to decline and reaches approximately 50% of its peak value by the eighth or ninth decade of life.5 Men lose an average of 30% less bone mass than women in a lifetime. In women, extensive loss of bone density starts immediately after menopause and lasts for about 10 years. It is believed to be closely related to the decline in estrogen levels.41,72–74 Trabeculae decrease more in number than in thickness and the rate of endosteal resorption begins to exceed the amount of periosteal apposition. The bone, with fewer trabeculae and thinner cortices, becomes more fragile. Interestingly, Jaworski and Uhthoff have demonstrated that loss of bone mass due to disuse is caused by increased endosteal resorption in older dogs but mainly by slowing of periosteal apposition in younger dogs with growing skeletons.75,76
Modeling and Remodeling in Response to Mechanical Forces
For many years, the effect of mechanical forces on bone remodeling has intrigued investigators. In the 17th century, Galileo had already noted the correlation of bone size and body weight and activity.77 In the 19th century, Wolff made the landmark observation that bone structure and remodeling have a clear relationship with loading, and that this association can be expressed mathematically.3–5 The adaptive changes of bone in response to loading are therefore frequently referred to as following Wolff’s law. Several studies have shown that bone adapts to loading and that maintenance of adequate bone density requires cyclic loading.78–83 Goodship et al. have experimentally proved with animal studies that after resection of the ulna, the radius increases in size and compensates or nearly compensates for the loss.78 Excessive repetitive loading and vigorous exercise are also known to stimulate bone formation.79,80 Increased shaft circumference and bone density have been noted on the dominant humerus of tennis players.81,82 Although not precisely quantified, the absence of loading has a negative effect on bone mass. Uhthoff et al. have reported on loss of bone mass after immobilization/bed rest due to skeletal traction, and adjacent to rigid implants.76,83
Regaining bone mass after prolonged disuse may take several months, even in children. In some individuals, especially the elderly, it may never return to its previous level.5 Disuse results in suppressed periosteal apposition in growing bone and enhanced endocortical resorption in mature bone.75,76
Mechanotransduction
It is currently believed that the mechanical adaptation of bone is governed by the osteocytes, which respond to a loading-induced flow of interstitial fluid through the lacuno-canalicular network by producing signaling molecules70 (Fig. 15-4).
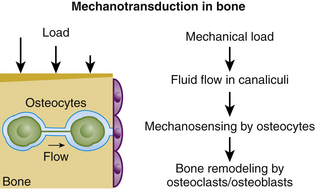
It has been shown that mechanical load induces fluid flow in the canalicular network.84 Weinbaum et al. suggested that this fluid flow is a physical mediator of mechanosensing by osteocytes in vivo.85 The osteocytes respond to mechanical stimuli with the production of signaling molecules that modulate the activities of osteoblasts and osteoclasts, thus converting mechanical stimuli into cellular signals that affect bone modeling and remodeling.86
Loading results in adaptive changes in bone, making it stronger. This adaptive response is regulated by the ability of resident bone cells to perceive and translate mechanical energy into a cascade of structural and biochemical changes within the cells, a process known as mechanotransduction44
Osteocytes probably do not respond directly to mechanical strain (deformation) of bone tissue, but respond indirectly to extracellular fluid flow caused by loading. When osteocytes and osteoblasts are subjected to these changes in fluid pressure, they release several bone-forming growth factors, including nitric oxide and prostaglandins.86 Certain prostaglandins, particularly PGE2, are anabolic with a demonstrated capacity to stimulate osteoblast activity and new bone formation.87 Nitric oxide, a strong inhibitor of bone resorption, works in part by suppressing the expression of RANKL and increasing the expression of OPG.88
Fluid flow along cell bodies produces drag force, fluid shear stress, and an electric potential. Each of these signals might activate bone cells, although cell culture experiments by Hung et al. and Reich et al. suggest that cells are less sensitive to electrical potentials than they are to fluid forces.89,90
There are several hormones that might amplify or transduce the effects of mechanical loading, including parathyroid hormone,91 estrogen,92 and insulin-like growth factors.93 Sawakami et al. suggest that an important event linking mechanical loading to bone formation is Wnt signaling through the LRP5 receptor pathway.94
Genetic Factors
Bone modeling and remodeling can be deeply affected by genetic imprint. Diseases like osteogenesis imperfecta, fibrodysplasia ossificans progressiva, and pycnodysostosis result from well-established genetic abnormalities.95–97 The same holds true for some types and grades of osteoporosis.98
Systemic Hormones
The hormones that most directly affect bone turnover and mineral hemostasis are parathyroid hormone (PTH) and calcitonin. Vitamin D also plays an important role. Modeling and remodeling of bone and mineral hemostasis are secondarily influenced by thyroxine (thyroid hormone), glucocorticoids, and estrogen.3–5
PTH is a single-chain polypeptide secreted by the chief cells of the parathyroid gland. It increases extracellular calcium levels by raising the renal tubular reabsorption of calcium and also by intensifying calcium release from bone due to increased bone resorption. PTH also stimulates the production of 1,25-dihydroxyvitamin D, which increases renal and gastrointestinal absorption of calcium, as well as its release from bone.3–5
Calcitonin is a polypeptide synthesized by the C cells of the thyroid gland. It lowers serum calcium levels by inhibiting osteoclastic bone resorption.3–5,99,100 Contrary to PTH, which acts on the osteoblasts to activate osteoclasts, calcitonin has a direct effect on osteoclast inhibition.3,4
The active form of vitamin D (1,25-dihydroxyvitamin D) is formed in the kidneys. Although its primary function is to increase the blood level of calcium by increasing calcium absorption in the gut and kidneys, it is also a powerful stimulator of bone resorption by osteoclasts.4,5,101,102
Thyroid hormone can stimulate resorption of bone by osteoclasts that can lead to loss of bone mass.103–106 Recent studies suggest that the bone loss seen in hyperthyroidism may be caused by the catabolic action of the elevated thyroid hormone itself or by the decreased anabolic action of thyroid-stimulating hormone.105,106
Glucocorticoids decrease bone mass not only by decreasing osteoid formation through osteoblast inhibition, they increase bone resorption by stimulating osteoclast activity.107 Studies have found that glucocorticoids cause an early and profound reduction in formation of bone through direct inhibition of osteoblasts.108,109 Glucocorticoids also increase bone resorption by stimulating production of OPG-L and inhibiting the production of OPG by osteoblasts, hence stimulating bone resorption by osteoclasts.110 Glucocorticoids have also been shown to stimulate the apoptosis of osteoblasts.111
Finally, estrogens exert a series of complex effects on bone, either directly by inhibiting bone resorption and total turnover or indirectly by acting on calcitonin, vitamin D, or parathyroid hormone.5 A recent study suggests that estrogen partially controls osteoblast and osteoclast function, is possibly involved in regulating mechanotransduction, and also interacts with the Wnt/β-catenin pathway.112
Exercise
Various authors have demonstrated that repetitive bone loading increases bone mass and decreased loading reduces it.75–83 Brighton et al. demonstrated that cyclical strain can stimulate bone cell function in culture.113,114 It is currently accepted that loading results in adaptive changes in bone, making it stronger. Bone’s adaptive response is regulated by the ability of resident bone cells to perceive and translate mechanical energy into a cascade of structural and biochemical changes within the cells, a process known as mechanotransduction44,45
Strain is defined as the deformation or change in dimension or shape caused by a load in any structure or structural material. Strain is expressed in microstrain units (millionths of a 100% strain), where 1000 microstrain units in compression would shorten a bone by 0.1% of its length.115 The amount of strain suffered by the bone during load application also influences the organization and density of newly formed bone. Minimal strain will cause the formation of a dense, well-organized bone. Moderate strain will result in formation of less dense, woven bone.116 A large amount of strain will lead to the formation of fibrous tissue. All of these strain effects are probably mediated through the mechanotransduction pathway.
Prostaglandins and Growth Factors
Cytokines play an important role in local control of normal bone turnover, as well as in neoplastic and inflammatory conditions. Cytokines can be divided into those that primarily form bone, those that primarily cause bone resorption, and those that do both. Interleukin (IL)-1 stimulates the resorption of bone by increasing the proliferation of osteoclast precursors and enhancing their activity.117–120 IL-1 stimulates osteoclasts through up-regulating activity of the RANK system.119,120 IL-1 has been also associated with the bone resorption of chronic inflammation and malignancy.5,120 Platelet-derived growth factor, IL-1, and IL-6 have been shown to be present at the implant-bone interface, contributing to the osteolysis that loosens joint implants.121–123 Debris from total-joint arthroplasties is associated with the secretion of proinflammatory cytokines such as IL-1β, tumor necrosis factor-α, IL-6, and IL-8. Activation of local (and systemic) inflammation results not only in decreased osteoblast function but in increased osteoclast activity.123 There is evidence that transforming growth factor beta (a member of the bone morphogenetic protein family) is released during bone resorption and that its presence further inhibits osteoclasts and stimulates osteoblastic activity.124,125
Certain prostaglandins, particularly PGE2, are anabolic and have a demonstrated capacity to stimulate osteoblast activity and new bone formation.87 As mentioned previously, nitric oxide is a strong inhibitor of bone resorption and works in part by suppressing the expression of RANKL and increasing the expression of OPG, which in turn leads to decreased recruitment of osteoclasts.88
Bone morphogenetic proteins (BMPs) are a group of growth factors originally defined by their ability to induce the formation of bone and cartilage. Seven proteins from this group were initially discovered. Of these, six (BMP2 through BMP7) belong to the transforming growth factor beta superfamily of proteins, whereas BMP1 is a metalloprotease involved in cartilage development. Since then, more BMPs have been discovered, making a total of approximately 20 today.126 Induction of bone formation by BMP is a sequential cascade. The key steps in this process are chemotaxis, mitosis, and differentiation, as shown in early studies by Reddi and Huggins.127 BMPs are known to stimulate osteoblasts and inhibit osteoclasts.124,125 Currently, only BMP2 is approved by the U.S. Food and Drug Administration for use in humans as a bone growth inducer.
Bono C., Parke W., Garfin S. Development of the spine. In: Herkowitz H.N., Garfin S.R., Eismont F.J., et al, editors. Rothman-Simeone the spine. ed 5. Philadelphia: Elsevier; 2006:3-15.
Buckwalter J.A., Glimcher M.J., Cooper R.R., et al. Bone biology. Part II: Formation, form, modeling, remodeling, and regulation of cell function. J Bone Joint Surg [Am]. 1995;77:1276-1289.
Klein-Nulend J., Bacabac R.G., Mullender M.G. Mechanobiology of bone tissue. Pathol Biol. 2005;53:576-580.
Miller J.D., McCreadie B.R., Alford A.I. Form and function of bone. In: Einhorn T.A., O’Keefe R.J., Buckwalter J.A., editors. Orthopaedic basic science. ed 3. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2007:129-159.
Robling A.G., Castillo A.B., Turner C.H. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng. 2006;8:455-498.
Theoleyre S., Wittrant Y., Kwan Tat S., et al. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15:457-475.
1. Buckwalter J.A., Glimcher M.J., Cooper R.R., et al. Bone biology. Part I: Structure, blood supply, cells, matrix and mineralization. J Bone Joint Surg [Am]. 1995;77:1256-1275.
2. Hoshino A., Wallace W.A. Impact-absorbing properties of the human knee. J Bone Joint Surg [Br]. 1987;69:807-811.
3. Miller J.D., McCreadie B.R., Alford A.I. Form and function of bone. In: Einhorn T.A., O’Keefe R.J., Buckwalter J.A., editors. Orthopaedic basic science. ed. Rosemont, IL: American Academy of Orthopaedic Surgeons; 2007:129-159.
4. Brinker R.M., O’Connor D.P. Basic sciences. In: Miller M.D., editor. Review of orthopaedics. ed 5. Philadelphia: Saunders Elsevier; 2008:1-132.
5. Buckwalter J.A., Glimcher M.J., Cooper R.R., et al. Bone biology. Part II: Formation, form, modeling, remodeling, and regulation of cell function. J Bone Joint Surg [Am]. 1995;77:1276-1289.
6. Arey L.B. The skeletal system. In Developmental anatomy: a textbook and laboratory manual of embryology, ed 7, Philadelphia: Saunders; 1965:396-425.
7. Buckwalter J.A. Musculoskeletal tissues and the musculoskeletal system. In: Weinstein S.L., Buckwalter J.A., editors. Turek’s orthopaedics: principles and their application. ed 5. Philadelphia: JB Lippincott; 1994:13-67.
8. Currey J.D. The mechanical adaptations of bones. Princeton, NJ: Princeton University Press; 1984.
9. Martin R.B., Burr D.B. Structure, function and adaptation of compact bone. New York: Raven Press; 1989.
10. Recker R.R. Embryology, anatomy, and microstructure of bone. In: Coe F.L., Favus M.J., editors. Disorders of bone and mineral metabolism. New York: Raven Press; 1992:219-240.
11. Revell P.A. Pathology of bone. New York: Springer; 1986. pp 1–34
12. Singh I. The architecture of cancellous bone. J Anat. 1978;127:305-310.
13. Currey J.D. Function and form of bone. Mow V.C., Ratcliffe A., Woo S.L.Y., editors. Biomechanics of diarthrodial joints. New York: Springer. 1990;vol. 2:3-30.
14. Cowin S.C. Properties of cortical hone and theory of bone remodeling. Mow V.C., Ratcliffe A., Woo S.L.Y., editors. Biomechanics of diarthrodial joints. New York: Springer. 1990;vol. 2:119-153.
15. Goldstein S.A., Hollister S.J., Kuhn J.L., et al. The mechanical and remodeling properties of trabecular bone. Mow V.C., Ratcliffe A., Woo S.L.Y., editors. Biomechanics of diarthrodial joints. New York: Springer. 1990;vol. 2:61-81.
16. Rubin C.T., McLeod K.J. Biologic modulation of mechanical influences in hone remodeling. Mow V.C., Ratcliffe A., Woo S.L.Y., editors. Biomechanics of diarthrodial joints. New York: Springer. 1990;vol. 2:97-118.
17. Sevitt S. Bone repair and fracture healing in man. New York: Churchill Livingstone; 1981. pp 1–24
18. Micheli A., Trapani S., Brizzi I., et al. Myositis ossificans circumscripta: a paediatric case and review of the literature. Eur J Pediatr. 2009;168:523-529.
19. Beiner J.M., Jokl P. Muscle contusion injury and myositis ossificans traumatica. Clin Orthop Relat Res. 2002;403:110-119.
20. Järvinen T.A., Järvinen T.L., Kääriäinen M., et al. Muscle injuries: biology and treatment. Am J Sports Med. 2005;33(5):745-764.
21. Aoki J., Yamamoto I., Hino M., et al. Reactive endosteal bone formation. Skeletal Radiol. 1987;16:545-551.
22. Daoud A., Saighi-Bouaouina A. Treatment of sequestra, pseudarthroses, and defects in the long bones of children who have chronic hematogenous osteomyelitis. J Bone Joint Surg [Am]. 1989;71:1448-1468.
23. Gebhardt M.C., Lippiello L., Bringhurst F.R., et al. Prostaglandin E2 synthesis by human primary and metastatic hone tumors in culture. Clin Orthop Relat Res. 1985;96:300-305.
24. Lerner U.H., Ohlin A. Tumor necrosis factors alpha and beta can stimulate bone resorption in cultured mouse calvariae by a prostaglandin-independent mechanism. J Bone Miner Res. 1993;8:147-155.
25. Ilizarov G.A. The tension-stress effect on the genesis and growth of tissues: Part I. The influence of stability of fixation and soft-tissue preservation. Clin Orthop Relat Res. 1989;238:249-281.
26. Ilizarov G.A. The tension-stress effect on the genesis and growth of tissues: Part II. The influence of the rate and frequency of distraction. Clin Orthop Relat Res. 1989;239:263-285.
27. Peltonen J.A., Kahn A.I., Lindberg L.A., et al. Bone formation after distraction osteotomy of the radius in sheep. Acta Orthop Scand. 1992;63:599-603.
28. Shearer J.R., Roach H.I., Parsons S.W. Histology of a lengthened human tibia. J Bone Joint Surg [Br]. 1992;74:39-44.
29. Delloye C., Delefortrie G., Coutelier L., et al. Bone regenerate formation in cortical bone during distraction lengthening: an experimental study. Clin Orthop Relat Res. 1990;250:34-42.
30. Lamm B.M. Percutaneous distraction osteogenesis for treatment of brachymetatarsia. J Foot Ankle Surg. 2010;49:197-204.
31. Kucukdeveci O., Sarisozen B., Atici T., et al. The effect of nicotine on distraction osteogenesis: an experimental study on rabbits. J Trauma. 2009;67:1376-1383.
32. Shen F.H., Matthews D.K., Yoon S.T., et al. Comparison of allograft to autograft in multilevel anterior cervical discectomy and fusion with rigid plate fixation. Spine J. 2003;3:451-459.
33. Jensen W.K., Moore T.A., Tribus C.B., et al. Use of patella allograft for anterior cervical diskectomy and fusion. J Spinal Disord Tech. 2009;226:392-398.
34. Momma F., Nakazawa T., Masaharu A. Repair and regeneration of vertebral body after antero-lateral partial vertebrectomy using β-tricalcium phosphate. Neurol Med Chir (Tokyo). 2008;48:337-342.
35. Caplan A.I. Cartilage begets bone versus endochondral myelopoiesis. Clin Orthop Relat Res. 1990;261:257-267.
36. Pechak D.G., Kujawa M.J., Caplan A.I. Morphological and histochemical events during first bone formation in embryonic chick limbs. Bone. 1986;7:441-458.
37. Bono C., Parke W., Garfin S. Development of the spine. In: Herkowitz H.N., Garfin S.R., Eismont F.J., et al, editors. Rothman-Simeone the spine. ed 5. Philadelphia: Elsevier; 2006:3-15.
38. Monsoro-Burq A.H., Duprez D., Watanabe Y., et al. The role of bone morphogenic proteins in vertebral development. Development. 1996;122:3607-3616.
39. Nolting D., Hansen B.F., Keeling J., et al. Prenatal development of the normal human vertebral corpora in different segments of the spine. Spine (Phila Pa 1976). 1998;23:2268-2271.
40. Noback C.R., Robertson C.C. Sequence of appearance of ossification centers in the human skeleton during the first five prenatal months. Am J Anat. 1951;89:1-28.
41. Avioli L.V., Lindsay R. The female osteoporotic syndrome(s). In: Avioli L.V., Krane S.M., editors. Metabolic bone disease and clinically related disorders. Philadelphia: WB Saunders; 1990:397-451.
42. Ruff C.B., Hayes W.C. Subperiosteal expansion and cortical remodeling of the human femur and tibia with aging. Science. 1982;217:945-948.
43. Keshawarz N.M., Recker R.R. Expansion of the medullary cavity at the expense of cortex in postmenopausal osteoporosis. Metab Bone Dis Relat Res. 1984;5:223-228.
44. Robling A.G., Turner C.H. Mechanical signaling for bone modeling and remodeling. Crit Rev Eukaryot Gene Expr. 2009;19:319-338.
45. Robling A.G., Castillo A.B., Turner C.H. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng. 2006;8:455-498.
46. Franzoso G., Carlson L., Xing L., et al. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997;11:3482-3496.
47. Takahashi N., Akatsu T., Udagawa N., et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123:2600-2602.
48. Wang Z.Q., Ovitt C., Grigoriadis A.E., et al. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360:741-745.
49. Partington G.A., Fuller K., Chambers T.J., et al. Mitf-PU.1 interactions with the tartrate-resistant acid phosphatase gene promoter during osteoclast differentiation. Bone. 2004;34:237-245.
50. So H., Rho J., Jeong D., et al. Microphthalmia transcription factor and PU.1 synergistically induce the leukocyte receptor osteoclast associated receptor gene expression. J Biol Chem. 2003;278:24209-24216.
51. Takayanagi H., Kim S., Koga T., et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901.
52. Matsuo K., Galson D.L., Zhao C., et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem. 2004;279:26475-26480.
53. Anderson D.M., Maraskovsky E., Billingsley W.L., et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175-179.
54. Yasuda H., Shima N., Nakagawa N., et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329-1337.
55. Simonet W.S., Lacey D.L., Dunstan C.R., et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309-319.
56. Yasuda H., Shima N., Nakagawa N., et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597-3602.
57. Theoleyre S., Wittrant Y., Kwan Tat S., et al. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15:457-475.
58. Martin T.J. Paracrine regulation of osteoclast formation and activity: milestones in discovery. J Musculoskel Neuron Interact. 2004;4:243-253.
59. McHugh K.P., Hodivala-Dilke K., Zheng M.H., et al. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433-440.
60. Vaananen K. Osteoclast function: biology and mechanisms. In: Bilezikian J.P., Raisz L.G., Rodan G.A., editors. Principles of bone biology. New York: Academic Press; 1996:3-13.
61. Bendall A.J., Abate-Shen C. Roles for Msx and Dlx homeoproteins in vertebrate development. Gene. 2000;247:17-31.
62. Ducy P., Zhang R., Geoffroy V., et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747-754.
63. Komori T., Yagi H., Nomura S., et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755-764.
64. Otto F., Thornell A.P., Crompton T., et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765-771.
65. Robledo R.F., Rajan L., Li X., et al. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002;16:1089-1101.
66. Glass D.A., Bialek P., Ahn J.D., et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751-764.
67. Hu H., Hilton M.J., Tu X., et al. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49-60.
68. Nakashima K., Zhou X., Kunkel G., et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17-29.
69. Baron R., Rawadi G., Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103-127.
70. Klein-Nulend J., Bacabac R.G., Mullender M.G. Mechanobiology of bone tissue. Pathol Biol. 2005;53:576-580.
71. Ott S.M. Bone density in adolescents [editorial]. N Engl J Med. 1991;325:1646-1647.
72. Riggs B.L., Wahner H.W., Melton L.J.III, et al. Rates of bone loss in the appendicular and axial skeletons of women: evidence of substantial vertebral bone loss before menopause. J Clin Invest. 1986;77:1487-1491.
73. Chilibeck P.D., Cornish S.M. Effect of estrogenic compounds (estrogen or phytoestrogens) combined with exercise on bone and muscle mass in older individuals. Appl Physiol Nutr Metab. 2008;33:200-212.
74. Richelson L.S., Wahner H.W., Melton L.J.III, et al. Relative contributions of aging and estrogen deficiency to postmenopausal bone loss. N Engl J Med. 1984;311:1273-1275.
75. Jaworski Z.F., Liskova-Kiar M., Uhthoff H.K. Effect of long-term immobilisation on the pattern of bone loss in older dogs. J Bone Joint Surg [Br]. 1980;62:104-110.
76. Uhthoff H.K., Jaworski Z.F. Bone loss in response to long-term immobilisation. J Bone Joint Surg [Br]. 1978;60:420-429.
77. Treharne R.W. Review of Wolff’s law and its proposed means of operation. Orthop Rev. 1981;10:35-47.
78. Goodship A.E., Lanyon L.E., McFie H. Functional adaptation of bone to increased stress: an experimental study. J Bone Joint Surg [Am]. 1979;61:539-546.
79. Leichter I., Simkin A., Margulies J.Y., et al. Gain in mass density of hone following strenuous physical activity. J Orthop Res. 1989;7:86-90.
80. Nilsson B.E., Westlin N.E. Bone density in athletes. Clin Orthop Relat Res. 1971;77:179-182.
81. Dalen N., Laftman P., Ohlsen H., et al. The effect of athletic activity on the bone mass in human diaphyseal bone. Orthopedics. 1985;8:1139-1141.
82. Jones H.H., Priest J.D., Hayes W.C., et al. Humeral hypertrophy in response to exercise. J Bone Joint Surg [Am]. 1977;59:204-208.
83. Uhthoff H.K., Boisvert D., Finnegan M. Cortical porosis under plates: reaction to unloading or to necrosis. J Bone Joint Surg [Am]. 1994;76:1507-1512.
84. Knothe-Tate M.L., Steck R., Forwood M.R., et al. In vivo demonstration of load-induced fluid flow in the rat tibia and its potential implications for processes associated with functional adaptation. J Exp Biol. 2000;203:2737-2745.
85. Weinbaum S., Cowin S.C., Zeng Y. A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J Biomech. 1994;27:339-360.
86. Burger E.H., Klein-Nulend J. Mechanotransduction in bone—role of the lacuno-canalicular network. FASEB J. 1999;13:S101-S112.
87. Jee W.S., Mori S., Li X.J., et al. Prostaglandin E2 enhances cortical bone mass and activates intracortical bone remodeling in intact and ovariectomized female rats. Bone. 1990;11:253-266.
88. Fan X., Roy E., Zhu L., et al. Nitric oxide regulates receptor activator of nuclear factor-κB ligand and osteoprotegerin expression in bone marrow stromal cells. Endocrinology. 2004;145:751-759.
89. Hung C.T., Allen F.D., Pollack S.R., et al. What is the role of the convective current density in the real-time calcium response of cultured bone cells to fluid flow? J Biomech. 1996;29:1403-1409.
90. Reich K.M., Gay C.V., Frangos J.A. Fluid shear stress as a mediator of osteoblast cyclic adenosine monophosphate production. J Cell Physiol. 1990;143:100-104.
91. Ma Y., Jee W.S., Yuan Z., et al. Parathyroid hormone and mechanical usage have a synergistic effect in rat tibial diaphyseal cortical bone. J Bone Miner Res. 1999;14:439-448.
92. Lee K., Jessop H., Suswillo R., et al. Endocrinology: bone adaptation requires estrogen receptor-alpha. Nature. 2003;424:389.
93. Gross T.S., Srinivasan S., Liu C.C., et al. Noninvasive loading of the murine tibia: an in vivo model for the study of mechanotransduction. J Bone Miner Res. 2002;17:493-501.
94. Sawakami K., Robling A.G., Pitner N.D., et al. Site-specific osteopenia and decreased mechanoreactivity in Lrp5-mutant mice. J Bone Miner Res. 2004;19(Suppl 1):38.
95. Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377-1385.
96. Shore E.M., Xu M., Feldman G.J., et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525-527.
97. Elmore S.M. Pycnodysostosis: a review. J Bone Joint Surg [Am]. 1967;49:153-162.
98. Shapiro J.R., Rowe D.W., Burn V. Familial osteoporosis pedigrees. J Bone Miner Res. 1987;2:344-349.
99. Friedman J., Raisz L.G. Thyrocalcitonin: inhibitor of bone resorption in tissue culture. Science. 1965;150:1465-1467.
100. Friedman J., Au W.Y., Raisz L.G. Responses of fetal rat bone to thyrocalcitonin in tissue culture. Endocrinology. 1968;82:149-156.
101. Holick M.F., Adams J.S. Vitamin D metabolism and biological function. In: Avioli L.V., Krane S.M., editors. Metabolic bone disease and clinically related disorders. 2nd ed. Philadelphia: WB Saunders; 1990:155-195.
102. Raisz L.G., Rodan G.A. Cellular basis for bone turnover. In: Avioli L.V., Krane S.M., editors. Metabolic bone disease and clinically related disorders. ed 2. Philadelphia: WB Saunders; 1990:1-41.
103. Mundy G.R., Raisz L.G. Thyrotoxicosis and calcium metabolism. Miner Electrolyte Metab. 1979;2:285-292.
104. Mundy G.R., Shapiro J.L., Bandelin J.G., et al. Direct stimulation of bone resorption by thyroid hormones. J Clin Invest. 1976;58:529-534.
105. Zaidi M., Davies T.F., Zallone A., et al. Thyroid-stimulating hormone, thyroid hormones, and bone loss. Curr Osteoporos Rep. 2009;7(2):47-52.
106. Galliford T.M., Murphy E., Williams A.J., et al. Effects of thyroid status on bone metabolism: a primary role for thyroid stimulating hormone or thyroid hormone? Minerva Endocrinol. 2005;30:237-246.
107. Hardy R., Cooper M.S. Bone loss in inflammatory disorders. J Endocrinol. 2009;201:309-320.
108. Pearce G., Tabensky D.A., Delmas P.D., et al. Corticosteroid-induced bone loss in men. J Clin Endocrinol Metab. 1988;83:801-806.
109. Cooper M.S., Blumsohn A., Goddard P.E., et al. 11β-Hydroxysteroid dehydrogenase type 1 activity predicts the effects of glucocorticoids on bone. J Clin Endocrinol Metab. 2003;88:3874-3877.
110. Hofbauer L.C., Gori F., Riggs B.L., et al. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140:4382-4389.
111. Eijken M., Koedam M., van Driel M., et al. The essential role of glucocorticoids for proper human osteoblast differentiation and matrix mineralization. Mol Cell Endocrinol. 2006;248:87-93.
112. Liedert A., Wagner L., Seefried L., et al. Estrogen receptor and Wnt signaling interact to regulate early gene expression in response to mechanical strain in osteoblastic cells. Biochem Biophys Res Commun. 2010;394:755-759.
113. Brighton C.T., Strafford B., Gross S.B., et al. The proliferative and synthetic response of isolated calvarial bone cells of rats to cyclic biaxial mechanical strain. J Bone Joint Surg [Am]. 1991;73:320-331.
114. Brighton C.T., Sennett B.J., Farmer J.C., et al. The inositol phosphate pathway as a mediator in the proliferative response of rat calvarial bone cells to cyclical biaxial mechanical strain. J Orthop Res. 1992;10:385-393.
115. Frost H.M. From Wolff’s law to the Utah paradigm: insights about bone physiology and its clinical applications. Anat Rec. 2001;262:398-417.
116. Turner C.H., Akhter M.P., Raab D.M., et al. A noninvasive in vivo model for studying strain adaptive bone modeling. Bone. 1991;12:73-79.
117. Russell R.G., Bunning R.A., Hughes D.E., et al. Humoral and local factors affecting bone formation and resorption. In: Stevenson J.C., editor. New techniques in metabolic bone disease. London: Butterworth; 1990:1-29.
118. Thomson B.M., Saklatvala J., Chambers T.J. Osteoblasts mediate interleukin I stimulation of bone resorption by rat osteoclasts. J Exp Med. 1986;164:104-112.
119. Trebec-Reynolds D.P., Voronov I., Heersche J.N., et al. IL-1alpha and IL-1beta have different effects on formation and activity of large osteoclasts. J Cell Biochem. 2010;109:975-982.
120. Trebec D.P., Chandra D., Gramoun A., et al. Increased expression of activating factors in large osteoclasts could explain their excessive activity in osteolytic diseases. J Cell Biochem. 2007;101:205-220.
121. Chiba J., Rubash H.E., Kim K.J., et al. The characterization of cytokines in the interface tissue obtained from failed cementless total hip arthroplasty with and without femoral osteolysis. Clin Orthop Relat Res. 1994;300:304-312.
122. Jiranek W.A., Machado M., Jasty M., et al. Production of cytokines around loosened cemented acetabular components: analysis with immunohistochemical techniques and in situ hybridization. J Bone Joint Surg [Am]. 1993;75:863-879.
123. Hallab N.J., Jacobs J.J. Biologic effects of implant debris. Bull NYU Hosp Jt Dis. 2009;67:182-188.
124. Centrella M., McCarthy T.L., Canalis E. Current concepts review: transforming growth factor-beta and remodeling of bone. J Bone Joint Surg [Am]. 1991;73:1418-1428.
125. Atfi A., Baron R. PTH battles TGF-beta in bone. Nat Cell Biol. 2010;12:205-207.
126. Chen D., Zhao M., Mundy G. Bone morphogenetic proteins. Growth Factors. 2004;22:233-241.
127. Reddi A.H., Huggins C. Biochemical sequences in the transformation of normal fibroblasts in adolescent rat. Proc Natl Acad Sci U S A. 1972;69:1601-1605.