CHAPTER 38 Bone Biology
GENERAL PRINCIPLES
Organization of bone
Macroscopically, the skeleton is composed of flat bones (e.g. bones of the skull) and long bones (e.g. tibia), based on development and growth occurring through an intramembraneous process or endochondral and intramembraneous processes, respectively.1 From a gross anatomical perspective, individual bones are made up of varying proportions of cortical (compact) and trabecular (cancellous) tissue, depending on location, and reflecting specific structural and functional differences. Compact bone is 80–90% mineralized tissue by volume, whereas trabecular bone is only 15–25% mineralized. Thus, trabecular bone has a much larger interface with soft tissue such as bone marrow, vascular, and connective tissue. In general terms, skeletal specialization is dictated by the relative abundance of cortical and trabecular tissue within specific bones; that is, cortical tissue accounts mainly for mechanical and protective functions while metabolic and cellular renewal functions are attributed mostly to trabecular tissue.
Cellular elements
Osteoclasts are giant multinucleated cells whose precursors are bone marrow monocytes/macrophages derived from hematopoietic stem cells and whose sole function is bone resorption.2 They are typically located in contact with a calcified bone surface within Howship’s lacunae. Their so-called zone of contact with bone is a ruffled membrane bounded circumferentially by an actin ring that serves to seal off the underlying bone as a discrete resorbing compartment. Specific attachment of osteoclasts to matrix components occurs through αvβ3, αvβ5, and α2β1 integrin receptors. The ruffled membrane contains both a proton ATPase and coupled chloride channel that acidifies the sealed-off bone resorbing compartment while maintaining electrical neutrality across the membrane. This is made possible by carbonic anhydrase within the cell and a HCO3−/Cl− exchanger on the basolateral membrane. RANK, the receptor for RANKL, and the macrophage-colony stimulating factor receptor are also located on the basolateral membrane, both being required for osteoclast differentiation.
Ossification in the spine
As in long bones, ossification in the spine occurs through an endochondral process.3 Mesenchymal cells give rise to prechondroblasts which then become progressively embedded within their cartilaginous matrix in lacunae. Now designated as chondrocytes, they continue to proliferate and then enlarge progressively to become hypertrophic, eventually undergoing apoptotic cell death. As cartilage cells die, and presumably under the influence of vascular endothelial growth factor (VEGF), the area is invaded by blood vessels. Osteoblasts move onto the cartilage surface and deposit bone in this area of primary spongiosa.
VERTEBRAL MORPHOGENESIS AND EMBRYOLOGICAL DEVELOPMENT
Cartilage models
Neural crest cells contribute to the branchial arch derivatives of the craniofacial skeleton, and the lateral plate mesoderm gives rise to the limbs. However, it is the paraxial mesoderm that gives rise to most of the axial skeleton, including the vertebral bodies, as well as the nonbranchial arch derivatives of the craniofacial skeleton. Mesenchymal cells condense to form regions with positional identity, which ultimately represent the future skeletal outline. The fate of mesenchymal cells within condensations dictates whether ossification occurs via an intramembranous or endochondral process. The former occurs as the result of osteoblast differentiation in the calvarium, maxilla, mandible, and subperiosteal bone-forming region of long bones. It is mediated by the transcription factors CBFA1/RUNX2 and osterix (OSX).4–7 Endochondral ossification requires differentiation of mesenchymal cells into chondrocytes, creating cartilage models of the remaining skeleton that serve as templates for bony replacement. Chondrocyte differentiation is influenced by members of the SOX-family of transcription factors.5–8
Cartilage models develop by interstitial and appositional growth until they take on the characteristic shapes and sizes of those intended bones. Patterning of the endochronal skeleton occurs even in the absence of eventual bone formation. Regulation of cartilage differentiation and growth in proper spatial sequence is essentially sufficient to complete a correctly shaped skeleton and this concept is supported by the phenotype of mice harboring two null alleles for the gene encoding CBFA1/RUNX2.5,6 Despite the lack of osteoblast differentiation in these mice, a nearly intact skeleton is formed with accurately placed and correctly shaped skeletal elements, albeit consisting totally of cartilage.
MESENCHYMAL CONDENSATION AND CHONDROCYTE DIFFERENTIATION
The regulation of events leading up to the formation of somites is thought to be tightly controlled by cell–cell interactions involving the Notch1 receptor and its ligands, which are also transmembrane molecules.9,10 Vertebral defects in mice can be seen as the result of somite condensation and patterning errors when the gene for Notch1 or one of its ligands is inactivated.8,11–13 Mutations in specific Notch1 ligands also occur in humans and are associated with the vertebral abnormalities found in the recessive form of spondylocostal dysostosis (semivertebrae) and Alagille syndrome (butterfly vertebrae).14–16
Disruption of sclerotome cell differentiation, and thus chondrocyte differentiation, also has profound effects on vertebral column formation. The notochord-secreted cytokine, Sonic hedgehog, is an important regulator of sclerotome cell differentiation. Its inactivation in mice leads to skeletal development without a vertebral column and posterior rib regions.17,18 Spinal deformities in mice and humans have also been reported to occur with mutations in PAX1, a transcription factor thought to be at least partly inducible by Sonic hedgehog.19,20
OSTEOBLAST DIFFERENTIATION AND BONE FORMATION
Regulatory pathways
Transcriptional control mechanisms regulate osteoblastogenesis. Examples of early transcriptional regulators include the homeodomain proteins (e.g. Msx-2, Dlx-2, Dlx-5, BAPX1), steroid receptors, as well as the helix-loop-helix (HLH) proteins Id, Twist, and Dermo. The HLH proteins play important roles in the proliferation of osteoprogenitor cells, but repress osteoblast differentiation and must be down-regulated before a mature bone cell phenotype can be expressed.21 The activating protein cFOS is also expressed in osteoprogenitor cells, resulting in osteosarcomas if overproduced, but is not detected in mature osteoblasts.22 A similar theme is true for some of the homeodomain transcription factors, where mesenchymal precursors abundantly express them, but mature bone-forming cells do not.23,24
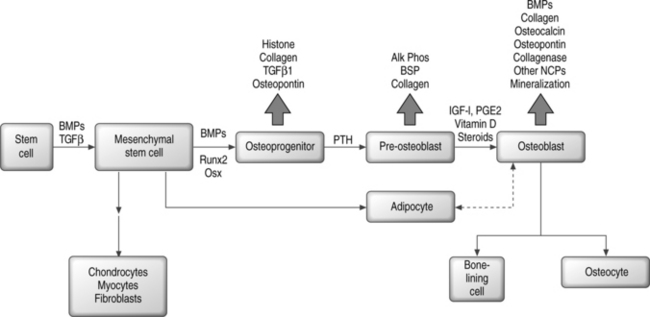
In contrast, the runt homology domain proteins (e.g. Runx2/Cbfa1) and the zinc finger protein Osterix and others activate genes expressed in the mature osteoblast, such as osteocalcin and osteopontin (Fig. 38.1).6,7,25–27 Null mouse mutants of Runx2 and Osterix result in inhibition of bone formation and perinatal mortality, demonstrating their requirement in osteogenesis.6,7 Runx2 also plays important roles in the maturation of chondrocytes, cartilage mineralization, and endothelial cell migration necessary for vascular invasion.28–30 Runx2 mediates its plethora of effects by interaction with diverse coregulatory proteins, including CBFβ1, Wnt pathway regulator LEF-1, and TGF-β/BMP responsive SMAD proteins, to name a few.31–38 Temporal expression of Runx2 followed by Osterix is thought to be essential for the later stages of osteoblast differentiation.7
Hormonal regulation of osteoblast differentiation includes steroid as well as polypeptide signaling molecules. Glucocorticoids enhance the differentiation of bone marrow stromal cells to osteoblasts in vitro, but in vivo have negative effects on bone that contribute to osteoporosis by inducing apoptotic cell death.39–41 Vitamin D3, sex steroids, adrenomedullin, and leptin all have osteogenic or anabolic effects on bone.42–50 Retinoic acid contributes to skeletal development in the embryo and also plays a role in postnatal bone formation.51–53 Among the polypeptide hormones, PTH and PTHrP have stimulatory effects on osteoprogenitor cells and regulatory effects on differentiation, both mediated by coupled G protein interactions with their receptors.54–61
Osteoprogenitor cells
Osteoprogenitor cells can arise from stem cells in a variety of tissues. There are as yet no unique identifying markers for the mesenchymal stem cell (MSC) that gives rise to bone, fat, cartilage, and muscle. Bone marrow stroma contains highly proliferative cells that will form single colonies or colony-forming units-fibroblasts (CFU-Fs) and are thought to contain mesenchymal stem cells that can be distinguished from early hematopoietic precursors.62–65 Stromal cells grown in vitro are heterogeneous with respect to the capacity for differentiation, and only a low percentage of all CFU-Fs have stem cell properties.62,63,66,67 Further, only a small fraction of CFU-Fs are bone-forming cells.68,69 Although a specific osteoprogenitor marker has not been identified, antibodies have been developed (e.g. STRO-1, SP-10, SH2, HOP-26) that recognize some subsets of precursor cells.70–77
Mesenchymal differentiation may proceed by a multistep hierarchical process with greater cell lineage restriction to terminal cell types at each subsequent step.78 This idea is supported by identification of bipotential adipocyte-osteoblast precursors, and has suggested that there may be an inverse relationship between adipocyte and osteoblast differentiation.79,80 A related observation is the transdifferentiation of bone to fat cells, or fat to bone cells.81–84 Depending on the local cellular environment, ‘committed’ MSC progeny may also dedifferentiate into another lineage.85 Thus, plasticity may be a common phenomenon; however, at least some reports suggest that cell fusion events may confound this paradigm.86–90 Figure 38.1 shows a simplified scheme for osteoblast differentiation.
MSCs similar to those found in bone marrow have been found in adult peripheral blood, fetal cord blood, fetal liver, tooth pulp, muscle satellite cells, and extramedullary adipose tissue and have osteogenic potential.91–100 Multipotent adult progenitor cells (MAPCs) derived from bone marrow can contribute to most somatic cell types, including skeletal tissue.101,102 Pericytes can also be induced toward the osteogenic lineage.103
Osteogenic growth factors
Bone morphogenetic proteins (BMPs), members of the transforming growth factor β (TGF-β) superfamily, are produced by skeletal and extraskeletal tissues. BMP-2, −4, and −6 are secreted by osteoblasts and play an autocrine role by enhancing the differentiated function of bone-forming cells.104 BMPs induce the differentiation of cells toward the osteoblast lineage, increase the terminally differentiated pool of osteoblasts, and induce endochondral ossification and cartilage formation in concert with Indian/Sonic hedgehog.105–107 BMPs interact with three distinct serine/threonine kinase receptors which, after dimerization, can signal through at least eight different Smads that become phosphorylated and/or heterodimerize to regulate transcription.108–110 Smads have activating and inhibitory functions, and also mediate signals initiated through TGF-β and activin receptors. BMPs and TGF-β signaling may also use Smad-independent pathways that rely on Ras or MAP kinase.111 Suppression of BMP activity occurs at several levels, including nonsignaling pseudoreceptors, inhibitory Smads, Smurf-mediated ubiquitination and degradation of signaling Smads, intracellular Smad binding proteins, and extracellular BMP antagonists such as noggin, chordin, follistatin, and twisted gastrulation.112
TGF-β1, -β2, and -β3 are polypeptides synthesized by bone cells and function as mitogens for preosteoblasts, stimulatory factors for collagen synthesis, and inducers of osteoclast apoptosis.113 The effect of BMPs on osteoblast differentiation is inhibited by TGF-β, and transgenic mice overproducing TGF-β are, in fact, osteopenic.114,115 Like BMPs, TGF-β signals through distinct serine-threonine kinase receptors that activate specific Smads to regulate transcription.110,111
Insulin-like growth factors (IGFs) are produced in a variety of tissues and have been characterized as IGF-I and IGF-II, both sharing similar properties but the former being more potent. IGFs116 stimulate proliferation of the osteoblastic lineage, prevent apoptosis of mature osteoblasts, and enhance net type I collagen abundance by stimulation of transcription and inhibition of collagen protein degradation. IGF-I knockout mice have decreased bone formation while those overexpressing IGF-I have enhanced bone formation.117,118 In humans, IGF-I has a generalized anabolic effect on musculoskeletal tissue with stimulation of bone remodeling.119 Parathyroid hormone (PTH) induces IGF-I synthesis in osteoblasts, and the anabolic effects of PTH on bone is diminished in the absence of IGF-I.117 Steroids decrease IGF-I expression and this may, in part, explain glucocorticoid-induced bone loss.120
β-catenin in association with specific transcription factors can regulate BMP and TGF-β signaling.121 The so-called Wnts prevent the degradation of β-catenin. Low-density lipoprotein receptor-related protein 5 (LPR5) is a coreceptor for Wnt that is required for optimal signaling with resultant accumulation of β-catenin.122 BMP-stimulated osteoblasts and stromal cells express LRP5 and knockout mice lacking LRP5 are osteopenic.123 In humans, inactivating mutations in LRP5 cause decreased bone mass, while other mutations in LRP5 cause a high bone mass phenotype.124,125
Fibroblast growth factor (FGF) 1 and 2 stimulate osteoblast proliferation and in vivo are important in the maintenance of bone mass. FGF null mice display low numbers of osteoblasts and decreased bone formation.126 Conversely, systemic FGF2 increases the preosteoblast pool that ultimately forms mature bone.127 FGF receptor signaling is essential for chondrogenesis and normal skeletal and limb development, with an activating mutation in FGF receptor-3 causing achondroplasia associated with dwarfism and early closure of cranial sutures.128 Platelet-derived growth factor (PDGF) has activities similar to those of FGF.129
Synthesis of bony matrix
In early bone formation, a chondroitin sulfate proteoglycan (versican) and the glycosaminoglycan hyaluronan are secreted and may delineate areas of future bone deposition.130 Versican is eventually replaced by two chondroitin sulfate proteoglycans, decorin and biglycan, each containing leucine-rich tandem repeat sequences. One possible function for these leucine-rich repeat proteins is their ability to bind and regulate TGF-β family members in the extracellular milieu.130 Biglycan-deficient transgenic mice have underdeveloped trabecular bone.131
The most abundant noncollagenous glycoprotein secreted by bone cells is osteonectin, with putative functions in osteoblast proliferation and matrix mineralization. Other glycoproteins mediate cell attachment and have been designated members of the SIBLING family (e.g. fibronectin, osteopontin, bone sialoprotein).132 All members contain the cell consensus sequence RGD (Arg-Gly-Asn) that binds to integrin cell surface molecules. Although most members of the SIBLING family are found in other tissues, bone sialoprotein is specific to mineralized tissue.133
The bone-specific gla-containing protein osteocalcin may function as an inhibitor of mineral deposition, as judged by osteocalcin-deficient mice who have increased bone mineral density.134 Similarly, mice deficient in the matrix gla protein (MGP), found in many connective tissues, develop extracellular sites of calcification.135
Bone mineral contains numerous impurities and is a carbonate-substituted hydroxyapatite with solubility properties that enable its constituent imperfect crystals to serve as a reservoir for calcium, phosphate, and magnesium ions.135 While the mineral content provides mechanical rigidity and load-bearing strength, the organic matrix provides elasticity, flexibility, and microstructural organization largely secondary to the presence of type I collagen.135
Extracellular matrix vesicles released from chondrocytes and osteoblasts accumulate calcium and phosphate ions, enzymes that degrade inhibitors of mineralization, and components of a nucleation core that induce apatite formation.133 It is unclear if there is an association of vesicle mineral with mineral in the collagen matrix or if the matrix vesicle directly provides a calcium and phosphate reservoir that supports mineralization initiated in collagen fibrils. In either case, the first stable crystal formed is followed by the addition of ions and ion clusters that allows for the expansion of crystal structure with addition of secondary nucleation sites.136 Removal of noncollagenous protein from matrix, particularly phosphoprotein, inhibits crystal nucleation and apatite formation.137 Alkaline phosphatase may contribute to enhanced mineralization by increasing the local phosphate concentration, removing phosphate-containing inhibitors of apatite growth, or modifying phosphoproteins to control their function as potential nucleators.133
Growth of mineral crystals is primarily dictated by the collagen matrix template. Noncollagenous proteins that bind crystals can also influence the extent and direction of mineral deposition.133 The presence of ions other than calcium and phosphate may influence bone apatite crystal growth, size, and solubility, and they include magnesium, strontium, cadmium, carbonate, and fluoride.133
MECHANISMS OF BONE RESORPTION
Osteoclast differentiation
Cells of the mononuclear/phagocytic lineage give rise to osteoclasts. Early precursors are committed to the myeloid series by the action of transcription factors PU-1 and MiTf. The lineage is further narrowed to monocytic cells by macrophage-colony stimulating factor (M-CSF).2,138 Proliferation and survival of monocytic cells, with concomitant RANK receptor expression, is mediated by M-CSF. The presence of RANKL is then required for commitment to the osteoclast differentiation program. Osteoclastogenesis depends on the association of M-CSF and RANKL, found on stromal cells and osteoblasts, with their receptors on monocytes/macrophage cells.2,138 Subsequent fusions of preosteoclasts result in the formation of giant multinucleated cells containing 4–20 nuclei and expressing characteristic markers of mature osteoclasts.
The osteoclast membrane has lost several macrophage markers and is lacking Fc and C3 receptors, but retains phagocytic non-specific esterases, lysozyme synthetic function, and CSF-1 receptors.2,138 Osteoclasts are distinguishable from macrophages by the abundance of RANK, vitronectin (integrin αvβ3) receptors, and calcitonin receptors. Osteoclasts undergo an estrogen-promoted apoptotic cell death after a cycle of resorption.2,138
Regulation of osteoclast activity
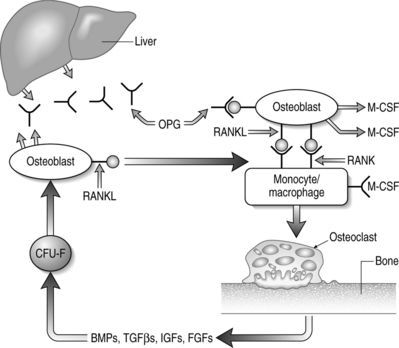
The key regulators of osteoclastic bone resorption are RANK ligand and its two confirmed receptors, RANK and osteoprotegerin (OPG). OPG is a member of the TNF receptor superfamily that lacks a transmembrane domain and, as such, is a secreted molecule. OPG regulates osteoclast activity by serving as a decoy receptor for RANKL.138–141 The only other known ligand for OPG is TRAIL which, like RANKL, is also a type II membrane-bound TNF homolog. The significance of OPG–TRAIL interaction is unknown. Deficiency of OPG in mice causes increased osteoclast formation, activity, and survival that results in destruction of growth plates, loss of trabeculae, decreased bone strength, and reduced bone mineral density (BMD).142,143 Parenteral administration of OPG causes a decrease in the number of active osteoclasts with a concomitant increase in BMD.144 In vitro, OPG abrogates osteoclast formation mediated by M-CSF and RANKL, in the absence of osteoblasts or stromal cells.139,140 OPG also inhibits osteoclastogenesis induced by a diverse group of osteotropic hormones.138–141 Finally, there is a direct effect of OPG on isolated mature osteoclasts due, in part, to limiting osteoclast survival.
RANKL expression is obligatory for osteoclastic resorption and physiologic bone remodeling (Fig. 38.2). Membrane-bound RANKL is more potent than soluble forms and mature osteoblasts express RANKL constitutively.145–147 Knockout mice lacking RANKL are devoid of osteoclasts and exhibit osteopetrosis with occlusion of the marrow cavity.148 The bone-resorbing activity and survival of mature osteoclasts is greatly increased by exposure to recombinant RANKL in the absence of stromal cells or osteoblasts.138–141
The signaling receptor for RANKL is RANK, a transmembrane protein that is sufficient to induce, upon RANKL binding, all downstream signals for osteoblast differentiation as well as later activation of mature osteoclasts. Mutations in the RANK gene that cause constitutive activation have been detected in familial expansile osteolysis, resulting in osteolytic lesions and osteopenia.149,150 Engineered mice lacking RANK have severe osteopetrosis secondary to the absence of osteoclasts.151,152
M-CSF or CSF-1 is required for normal osteoclast formation in the neonatal period and in rodent models of osteopetrosis there is impaired CSF-1 production. CSF-1 is secreted by stromal cells and cells of the osteoclast lineage express CSF-1 receptors. Interleukin (IL)-1α and β are potent stimulators of osteoclasts that work at all stages of formation and activation whose effects are mediated through RANKL.141,153 IL-1 has been implicated in conditions of increased bone turnover, including osteoporosis, bone loss seen in some malignancies, and in rheumatoid arthritis.154 Lymphotoxin and TNF-α are functionally related to IL-1 and their effects on bone are synergistic with IL-1.155
The list of other locally-acting factors is growing. Those that stimulate osteoclast formation and activity include IL-6, IL-15, IL-17, vitamin A, and TGF-α. Inhibitors of osteoclast formation and activity include interferon-γ (IFN-γ), TGF-β, IL-18, inorganic phosphate, and high extracellular calcium. In humans, exogenous TGF-β inhibits proliferation and differentiation of osteoclast precursors and enhances osteoclast apoptosis, presumably by increasing OPG expression in osteoblasts and bone marrow stromal cells. However, endogenous TGF-β may actually stimulate osteoclastogenesis.156,157 The role of other factors such as prostaglandins and leukotrienes is less clear.
The major systemic hormones that influence osteoclast activity are PTH, vitamin D3, calcitonin, glucocorticoids, and the sex hormones. PTH has dual effects on bone mediated through its regulation of Cbfa1, which is obligatory for bone formation and also influences RANKL expression on osteoblasts.158 Intermittent administration of PTH stimulates bone formation while continuous administration stimulates osteoclastic bone resorption. PTH-related protein has identical skeletal effects to those of PTH. The D vitamin 1,25 (OH)2D3 stimulates osteoclastic bone resorption by stimulating differentiation and fusion of osteoclast progenitors.159 These effects of 1,25 dihydroxyvitamin D are mediated indirectly by osteoblasts through RANKL signaling. Calcitonin, made by the parafollicular cells of the thyroid gland, is a potent inhibitor of osteoclastic bone resorption whose effects are transient owing to downregulation of its receptor.160 Glucocorticoids have inhibitory effects on formation of osteoclasts in vitro and on bone resorption in organ culture, but in vivo are associated with increased bone resorption. The latter effect may, in part, be due to glucocorticoid inhibition of calcium absorption from the gut and secondary hyperparathyroidism. For up to 10 years after the menopause, lack of estrogen is associated with enhanced osteoclastic bone resorption. Estrogens may have a direct effect on osteoclasts, but they may also suppress peripheral blood monocyte production of cytokines such as IL-1, TNF-α, and IL-6 that stimulate osteoclast formation.154,161–163
REMODELING IN THE SPINE
Coupling of bone formation and resorption
Remodeling of bone occurs in focal and discrete microscopic sites throughout the skeleton. Complete remodeling at each site takes 3–6 months, and is probably longer in cancellous than in cortical bone. Cancellous bone is more abundant in the vertebral column, comprising more than 66% of total bone in the lumbar spine. This is in contrast to the intertrochanteric portion of the femur, where bone is 50% cortical and 50% cancellous, and in the mid-radius where greater than 95% of bone is cortical bone. At sites of bone turnover an activation phase is followed by a resorption phase that is followed by a bone formation phase. Although anatomically different from each other, cortical and cancellous bone follow the same general remodeling sequence. However, turnover events in cancellous bone are probably influenced more by factors produced by adjacent bone marrow cells, whereas these events in cortical bone, distant from marrow-derived cytokines, are likely influenced more by systemic hormones such as PTH and 1,25 dihydroxyvitamin D3.164
In the initial step of the remodeling sequence, osteoclasts are activated in focal sites by mechanisms that are not well understood and the rate of bone turnover is dependent on the activation frequency of osteoclasts. Interactions between integrins on the osteoclast cell membrane and bony matrix proteins may initiate the activation phase. After a resorptive phase lasting up to 10 days, repair of resulting defects proceeds by attraction of osteoblasts and subsequent bone formation. The mechanisms by which bone formation is coupled to previous osteoclastic resorption are still unclear, but several possibilities can be supported by existing evidence.
Coupling may be mediated by local humoral factors acting on osteoblasts or osteoblast precursors (see Fig. 38.2). These mediators may serve as osteoblast-stimulating factors, such as IGF-I or TGB-β, that are released from the bone matrix with osteoclastic resorption.165–167 A factor that stimulates resorption may also stimulate osteoblasts, but at a much slower rate, so as to time the initiation of bone formation to occur after osteoclast activity has ceased.168 Coupling may instead involve transcription factors, such as Runx2/Cbfa1, that are both obligatory for bone formation but also play a role in the regulation of osteoclast formation.169 Alternatively, systemic factors such as leptin may play a role in the regulation of the bone formation component of the coupling process by presumptive hypothalamus-mediated β2-adrenergic stimulation of osteoblasts and their precursors.170 Lastly, coupling may be a more simple process whereby osteoblasts or their precursors in proximity to resorption sites repopulate bony defects after osteoclasts leave or die after their last resorptive cycle.
Regulation of the formation phase
The formation phase of bone remodeling occurs through a series of events beginning with osteoblast precursor chemotaxis and proliferation. These events are followed by osteoblast differentiation, mineralized bone formation, and cessation of osteoblast activity. Chemotaxis of osteoblast precursors is likely mediated by soluble factors produced locally as well as factors released from the dissolution of bone by osteoclastic resorption. Likely candidates include TGF-β, PDGF, and digested fragments of type I collagen and osteocalcin.171–176
Proliferation of osteoblast precursors is also likely to be stimulated by local factors released during the resorptive process including IGFs, FGFs, TGF-βs, and PDGF. Differentiation of osteoblastic precursors into mature bone-forming cells is likely to be induced by bone-derived growth factors such as IGF-I and BMP2. The role of TGF-β may thus be to trigger the process of bone formation by first attracting and then expanding the pool of osteogenic precursors, after which its disappearance or inactivation allows these precursors to differentiate into osteoblasts. The cessation of osteoblast activity may also be mediated by TGF-β, since TGF-β decreases osteoblast activity and it is expressed by osteoblasts as they become terminally differentiated.177 Osteoblast life span and factors that regulate it (see below under Osteoblast Senescence) may be critically important to terminating the formation phase of bone turnover, particularly with aging.
Bone remodeling and disease
In diseases which tend to increase osteoclast activation, such as primary hyperparathyroidism, hyperthyroidism, and Paget’s disease, there is also a roughly balanced increase in the amount of new bone formation, although new bone is not necessarily architecturally equivalent to bone formed under normal physiologic conditions. In diseases that cause osteolytic lesions of bone, namely multiple myeloma and certain malignant solid tumors, osteoblasts do not completely repair defects made by osteoclastic resorption. In the former case there appears to be a defect(s) in osteoblast differentiation while in the later case malignant cells produce local factors that stimulate osteoclast activity.178,179 Osteoblastic lesions occur at sites where no previous bone resorption has occurred, and can be found with cancers of the prostate or breast, or with administration of pharmacologic doses of fluoride.
With aging, there appears to be an uncoupling of bone resorption and formation that begins after about 35 years of age, depending on the skeletal site. There is a resulting decrease in trabecular wall thickness owing to the inability of osteoblasts to repair osteoclastic resorptive defects, presumably secondary to decreased osteoblast activity or a decrease in the number of osteoblasts available to initiate bone formation.180
THE AGING SPINE
Age-related changes in biomechanical properties
The strength of cancellous bone declines 8–10% per decade in comparison to cortical bone strength which declines only 2–5% per decade. This translates into a much higher risk of fracture in bones of the vertebral column. Inherent fragility of bone increases with age, allowing damage to accumulate more readily with repeated loading.181 Damaged bone is less able to sustain further deformation and less able to absorb energy with any impact, predisposing it to failure.182 Fracture risk in older adults is greater than predicted by loss of bone mass alone, suggesting that age-related changes in bone quality or architecture are important.
Ultramicroscopic tears in bone or fatigue damage results in fracture whenever damage occurs faster than the remodeling apparatus can repair it (slower bone turnover) or when remodeling is defective (uncoupling of bone formation to resorption).183 Bones vertically loaded, such as the vertebral bodies, depend on horizontal, cross-hatching trabeculae, for support. Loss of trabecular connectivity is thought to be a major contributor to the female preponderance of vertebral bone loss and susceptibility to vertebral fractures.
The decline in collagen content with age is associated with a reduction in bone strength and stiffness, two parameters generally used to define bone health. Fewer reducible collagen cross-links per unit of total collagen are associated with increase bone fragility in osteoporotic women.184,185 Periosteal apposition of bone with age serves as a compensatory mechanism for reduced bone volume in men, but not in women, at both the femoral neck and for the vertebral bodies.186–188
Bone loss: secondary hyperparathyroidism, menopause, and somatopause
The uncoupling of bone formation to resorption leads to remodeling imbalances that ultimately result in age-related bone loss. With aging, bone formation is affected by the reduction of osteoblast differentiation, activity, and life span that is potentiated by secondary hyperparathyroidism and, in women, by estrogen deprivation and increased osteoclast activity with menopause.189 Secondary hyperparathyroidism is caused by decreased intestinal and renal calcium resorption, precipitated by vitamin D deficiency in the housebound (lack of sunlight) and undernourished older adult, and by age-onset changes in renal function. Both processes are affected by estrogen deficiency. However, secondary hyperparathyroidism is ameliorated by a diet high in calcium.
Osteoblast senescence
Telomeres shorten with age in most human tissues, including bone, and because telomere shortening is a cause of cellular replicative senescence in cultured cells, including osteoblasts and MSCs,190–192 it is hypothesized that telomere shortening contributes to the aging of bone. Conversely, after forced ectopic expression of telomerase in human MSCs, proliferative capacity is extended in vitro and the capacity for bone formation is enhanced in vivo.190–192 Telomerase not only extends the life span of osteogenic precursors, but accelerates the osteogenic differentiation of MSCs.193 These observations provide strong evidence that telomerase status in MSCs is likely a critical component of bone formation.
Functional deficits in osteoblasts that occur with cellular senescence play a major role in the uncoupling of bone formation and resorption, resulting in a net loss of bone tissue.194 Thus, recruitment of osteoblast precursors (MSCs) and osteoblast differentiation become a critical component in maintaining the balance between these two opposing processes. If the number and activity of osteoblasts responsible for synthesizing new bone matrix are substantially reduced, then the diminished recruitment of osteogenic precursors to replace senescent osteoblasts may potentially explain many aspects of age-related bone loss. The osteogenic potential of murine MSCs have been reported to decline with increasing donor age.195 There are conflicting reports regarding the effects of age on human MSCs with the weight of evidence in favor of modest declines in several measures associated with osteogenic potential, particularly after the age of 40.196–203 However, there are at least two well-conceived studies that suggest no significant age-related differences.201,202 Thus, decrements in osteogenic stem cell potential with age may only partially account for changes in bone integrity associated with osteoporosis and poor fracture repair.
It has also been suggested that reductions in bone density seen with age may be the result of osteoblast replicative senescence, either directly (decreased proliferation and apoptosis) or secondary to other changes that accompany replicative senescence (e.g. failure to express an osteoblast phenotype).204 It is unclear if these alterations are solely a consequence of decreased osteoblast responsiveness to extracellular signals, but reduced expression of osteoblast markers such as alkaline phosphatase, osteocalcin, and type I collagen has been shown in response to various hormones and growth factors including 1,25 (OH)2 vitamin D3 [1,25 (OH)2 D3], insulin-like growth factor-I (IGF-I), parathyroid hormone (PTH), and prostaglandin E2 (PGE2).205–212 Many of these changes have been documented in primary osteoblast cultures from old donors as well as in osteoblasts aged in vitro by serial passage.
1 Marks SC, Odgren PR. 2nd edn.. Structure and development of the skeleton. Principles of bone biology, Vol. 1.. Academic Press, San Diego, 2002;3-15.
2 Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289(5484):1504-1508.
3 Baron R. General principles of bone biology. Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th edn., Washington, DC: The American Society for Bone and Mineral Research; 2003:1-8.
4 Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747-754.
5 Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765-771.
6 Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755-764.
7 Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17-29.
8 Akiyama H, Chaboissier MC, Martin JF, et al. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16(21):2813-2828.
9 Gossler A, Hrabe de Angelis M. Somitogenesis. Curr Top Dev Biol. 1998;38:225-287.
10 Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121(5):1533-1545.
11 Kusumi K, Sun ES, Kerrebrock AW, et al. The mouse pudgy mutation disrupts Delta homologue Dll3 and initiation of early somite boundaries. Nat Genet. 1998;19(3):274-278.
12 Wong PC, Zheng H, Chen H, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387(6630):288-292.
13 Saga Y, Hata N, Koseki H, et al. Mesp2: a novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev. 1997;11(14):1827-1839.
14 Bulman MP, Kusumi K, Frayling TM, et al. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;24(4):438-441.
15 Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235-242.
16 Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243-251.
17 Johnson RL, Laufer E, Riddle RD, et al. Ectopic expression of Sonic hedgehog alters dorsal-ventral patterning of somites. Cell. 1994;79(7):1165-1173.
18 Chiang C, Litingtung Y, Lee E, et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407-413.
19 Hol FA, Geurds MP, Chatkupt S, et al. PAX genes and human neural tube defects: an amino acid substitution in PAX1 in a patient with spina bifida. J Med Genet. 1996;33(8):655-660.
20 Wilm B, Dahl E, Peters H, et al. Targeted disruption of Pax1 defines its null phenotype and proves haploin sufficiency. Proc Natl Acad Sci USA. 1998;95(15):8692-8697.
21 Ogata T, Noda M. Expression of Id, a negative regulator of helix-loop-helix DNA binding proteins, is down-regulated at confluence and enhanced by dexamethasone in a mouse osteoblastic cell line, MC3T3E1. Biochem Biophys Res Commun. 1991;180(3):1194-1199.
22 Machwate M, Jullienne A, Moukhtar M, et al. Temporal variation of c-Fos proto-oncogene expression during osteoblast differentiation and osteogenesis in developing rat bone. J Cell Biochem. 1995;57(1):62-70.
23 Sumoy L, Wang CK, Lichtler AC, et al. Identification of a spatially specific enhancer element in the chicken Msx-2 gene that regulates its expression in the apical ectodermal ridge of the developing limb buds of transgenic mice. Dev Biol. 1995;170(1):230-242.
24 Orestes-Cardoso S, Nefussi JR, Lezot F, et al. Msx1 is a regulator of bone formation during development and postnatal growth: in vivo investigations in a transgenic mouse model. Connect Tissue Res. 2002;43(2–3):153-160.
25 Javed A, Barnes GL, Jasanya BO, et al. Runt homology domain transcription factors (Runx, Cbfa, and AML) mediate repression of the bone sialoprotein promoter: evidence for promoter context-dependent activity of Cbfa proteins. Mol Cell Biol. 2001;21(8):2891-2905.
26 Gutierrez S, Javed A, Tennant DK, et al. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J Biol Chem. 2002;277(2):1316-1323.
27 Shui C, Spelsberg TC, Riggs BL, et al. Changes in Runx2/Cbfa1 expression and activity during osteoblastic differentiation of human bone marrow stromal cells. J Bone Miner Res. 2003;18(2):213-221.
28 Stricker S, Fundele R, Vortkamp A, et al. Role of Runx genes in chondrocyte differentiation. Dev Biol. 2002;245(1):95-108.
29 Sun L, Vitolo M, Passaniti A. Runt-related gene 2 in endothelial cells: inducible expression and specific regulation of cell migration and invasion. Cancer Res. 2001;61(13):4994-5001.
30 Zelzer E, Glotzer DJ, Hartmann C, et al. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106(1–2):97-106.
31 Lian JB, Stein JL, Stein GS, et al. Runx2/Cbfa1 functions: diverse regulation of gene transcription by chromatin remodeling and co-regulatory protein interactions. Connect Tissue Res. 2003;44(Suppl 1):141-148.
32 Westendorf JJ, Hiebert SW. Mammalian runt-domain proteins and their roles in hematopoiesis, osteogenesis, and leukemia. J Cell Biochem. 1999;Suppl 32–33:51-58.
33 Sierra J, Villagra A, Paredes R, et al. Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol Cell Biol. 2003;23(9):3339-3351.
34 Miller J, Horner A, Stacy T, et al. The core-binding factor beta subunit is required for bone formation and hematopoietic maturation. Nat Genet. 2002;32(4):645-649.
35 Kahler RA, Westendorf JJ. Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. J Biol Chem. 2003;278(14):11937-11944.
36 Zaidi SK, Sullivan AJ, van Wijnen AJ, et al. Integration of Runx and Smad regulatory signals at transcriptionally active subnuclear sites. Proc Natl Acad Sci USA. 2002;99(12):8048-8053.
37 Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95(6):737-740.
38 Zhang YW, Yasui N, Ito K, et al. A RUNX2/PEBP2alpha A/CBFA1 mutation displaying impaired transactivation and Smad interaction in cleidocranial dysplasia. Proc Natl Acad Sci USA. 2000;97(19):10549-10554.
39 Kream BE, Lukert BE. Clinical and basic aspects of glucocorticoid action in bone. Principles of bone biology, 2nd edn., San Diego, CA: Academic Press; 2002:723-740.
40 Weinstein RS, Jilka RL, Parfitt AM, et al. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102(2):274-282.
41 Noble BS, Reeve J. Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol. 2000;159(1–2):7-13.
42 van Leeuwen JP, van Driel M, van den Bemd GJ, et al. Vitamin D control of osteoblast function and bone extracellular matrix mineralization. Crit Rev Eukaryot Gene Expr. 2001;11(1–3):199-226.
43 Cornish J, Callon KE, Coy DH, et al. Adrenomedullin is a potent stimulator of osteoblastic activity in vitro and in vivo. Am J Physiol. 1997;273(6 Pt 1):E1113-A1120.
44 Gordeladze JO, Drevon CA, Syversen U, et al. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem. 2002;85(4):825-836.
45 Reseland JE, Syversen U, Bakke I, et al. Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J Bone Miner Res. 2001;16(8):1426-1433.
46 Ducy P, Amling M, Takeda S, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100(2):197-207.
47 Takeda S, Elefteriou F, Levasseur R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305-317.
48 Thomas T, Gori F, Khosla S, et al. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140(4):1630-1638.
49 Burguera B, Hofbauer LC, Thomas T, et al. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142(8):3546-3553.
50 Pasco JA, Henry MJ, Kotowicz MA, et al. Serum leptin levels are associated with bone mass in nonobese women. J Clin Endocrinol Metab. 2001;86(5):1884-1887.
51 Meyer T, Kneissel M, Mariani J, et al. In vitro and in vivo evidence for orphan nuclear receptor RORalpha function in bone metabolism. Proc Natl Acad Sci USA. 2000;97(16):9197-9202.
52 Steinmayr M, Andre E, Conquet F, et al. Staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci USA. 1998;95(7):3960-3965.
53 Dussault I, Fawcett D, Matthyssen A, et al. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70(1–2):147-153.
54 Karaplis AC, Goltzman D. PTH and PTHrP effects on the skeleton. Rev Endocr Metab Disord. 2000;1(4):331-341.
55 Chung UI, Schipani E, McMahon AP, et al. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107(3):295-304.
56 Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8(3):277-289.
57 Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273(5275):663-666.
58 Schipani E, Lanske B, Hunzelman J, et al. Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA. 1997;94(25):13689-13694.
59 St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072-2086.
60 Schipani E, Langman CB, Parfitt AM, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. N Engl J Med. 1996;335(10):708-714.
61 Vortkamp A, Lee K, Lanske B, et al. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613-622.
62 Friedenstein AJ. Stromal mechanisms of bone marrow: cloning in vitro and retransplantation in vivo. Haematol Blood Transfus. 1980;25:19-29.
63 Friedenstein AJ. Osteogenic stem cells in the bone marrow. Bone and mineral research, 7th edn., New York, NY: Elsevier Science BV; 1990:243-270.
64 Bianco P, Riminucci M, Kuznetsov S, et al. Multipotential cells in the bone marrow stroma: regulation in the context of organ physiology. Crit Rev Eukaryot Gene Expr. 1999;9(2):159-173.
65 Owen ME. The marrow stromal cell system. Marrow stromal stem cells in culture. Cambridge, UK: Cambridge University Press, 1998;88-110.
66 Kuznetsov SA, Krebsbach PH, Satomura K, et al. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. 1997;12(9):1335-1347.
67 Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143-147.
68 Aubin JE. Osteoprogenitor cell frequency in rat bone marrow stromal populations: role for heterotypic cell–cell interactions in osteoblast differentiation. J Cell Biochem. 1999;72(3):396-410.
69 Wu X, Peters JM, Gonzalez FJ, et al. Frequency of stromal lineage colony forming units in bone marrow of peroxisome proliferator-activated receptor-alpha-null mice. Bone. 2000;26(1):21-26.
70 Simmons PJ, Torok-Storb B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood. 1991;78(1):55-62.
71 Gronthos S, Graves SE, Ohta S, et al. The STRO-1+ fraction of adult human bone marrow contains the osteogenic precursors. Blood. 1994;84(12):4164-4173.
72 Bruder SP, Horowitz MC, Mosca JD, et al. Monoclonal antibodies reactive with human osteogenic cell surface antigens. Bone. 1997;21(3):225-235.
73 Bruder SP, Ricalton NS, Boynton RE, et al. Mesenchymal stem cell surface antigen SB-10 corresponds to activated leukocyte cell adhesion molecule and is involved in osteogenic differentiation. J Bone Miner Res. 1998;13(4):655-663.
74 Bowen MA, Aruffo AA, Bajorath J. Cell surface receptors and their ligands: in vitro analysis of CD6–CD166 interactions. Proteins. 2000;40(3):420-428.
75 Barry FP, Boynton RE, Haynesworth S, et al. The monoclonal antibody SH-2, raised against human mesenchymal stem cells, recognizes an epitope on endoglin (CD105). Biochem Biophys Res Commun. 1999;265(1):134-139.
76 Zannettino AC, Harrison K, Joyner CJ, et al. Molecular cloning of the cell surface antigen identified by the osteoprogenitor-specific monoclonal antibody, HOP-26. J Cell Biochem. 2003;89(1):56-66.
77 Joyner CJ, Bennett A, Triffitt JT. Identification and enrichment of human osteoprogenitor cells by using differentiation stage-specific monoclonal antibodies. Bone. 1997;21(1):1-6.
78 Aubin JE. Bone stem cells. J Cell Biochem. 1998;30–31(Suppl):73-82.
79 Aubin JE, Heersche JNM. Vitamin D and osteoblasts. Vitamin D. San Diego, CA: Academic Press, 1997;313-328.
80 Nuttall ME, Gimble JM. Is there a therapeutic opportunity to either prevent or treat osteopenic disorders by inhibiting marrow adipogenesis? Bone. 2000;27(2):177-184.
81 Jeon MJ, Kim JA, Kwon SH, et al. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem. 2003;278(26):23270-23277.
82 Lecka-Czernik B, Gubrij I, Moerman EJ, et al. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J Cell Biochem. 1999;74(3):357-371.
83 Skillington J, Choy L, Derynck R. Bone morphogenetic protein and retinoic acid signaling cooperate to induce osteoblast differentiation of preadipocytes. J Cell Biol. 2002;159(1):135-146.
84 Nuttall ME, Patton AJ, Olivera DL, et al. Human trabecular bone cells are able to express both osteoblastic and adipocytic phenotype: implications for osteopenic disorders. J Bone Miner Res. 1998;13(3):371-382.
85 Park SR, Oreffo RO, Triffitt JT. Interconversion potential of cloned human marrow adipocytes in vitro. Bone. 1999;24(6):549-554.
86 Ying QL, Nichols J, Evans EP, et al. Changing potency by spontaneous fusion. Nature. 2002;416(6880):545-548.
87 Terada N, Hamazaki T, Oka M, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416(6880):542-545.
88 Wang X, Willenbring H, Akkari Y, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422(6934):897-901.
89 Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422(6934):901-904.
90 Wurmser AE, Gage FH. Stem cells: cell fusion causes confusion. Nature. 2002;416(6880):485-487.
91 Huss R, Lange C, Weissinger EM, et al. Evidence of peripheral blood-derived, plastic-adherent CD34 hematopoietic stem cell clones with mesenchymal stem cell characteristics. Stem Cells. 2000;18(4):252-260.
92 Kuznetsov SA, Mankani MH, Gronthos S, et al. Circulating skeletal stem cells. J Cell Biol. 2001;153(5):1133-1140.
93 Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. 2000;109(1):235-242.
94 Campagnoli C, Roberts IA, Kumar S, et al. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98(8):2396-2402.
95 Shi S, Gronthos S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Miner Res. 2003;18(4):696-704.
96 Miura M, Gronthos S, Zhao M, et al. SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci USA. 2003;100(10):5807-5812.
97 Asakura A, Komaki M, Rudnicki M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation. 2001;68(4–5):245-253.
98 Gronthos S, Franklin DM, Leddy HA, et al. Surface protein characterization of human adipose tissue-derived stromal cells. J Cell Physiol. 2001;189(1):54-63.
99 Zuk PA, Zhu M, Mizuno H, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7(2):211-228.
100 Huang JI, Beanes SR, Zhu M, et al. Rat extramedullary adipose tissue as a source of osteochondrogenic progenitor cells. Plast Reconstr Surg. 2002;109(3):1033-1041. discussion 1042–1043
101 Reyes M, Lund T, Lenvik T, et al. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood. 2001;98(9):2615-2625.
102 Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418(6893):41-49.
103 Doherty MJ, Ashton BA, Walsh S, et al. Vascular pericytes express osteogenic potential in vitro and in vivo. J Bone Miner Res. 1998;13(5):828-838.
104 Pereira RC, Rydziel S, Canalis E. Bone morphogenetic protein-4 regulates its own expression in cultured osteoblasts. J Cell Physiol. 2000;182(2):239-246.
105 Thies RS, Bauduy M, Ashton BA, et al. Recombinant human bone morphogenetic protein-2 induces osteoblastic differentiation in W-20–17 stromal cells. Endocrinology. 1992;130(3):1318-1324.
106 Hughes FJ, Collyer J, Stanfield M, et al. The effects of bone morphogenetic protein-2, -4, and -6 on differentiation of rat osteoblast cells in vitro. Endocrinology. 1995;136(6):2671-2677.
107 Zeng L, Kempf H, Murtaugh LC, et al. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16(15):1990-2005.
108 Kretzschmar M, Massague J. SMADs: mediators and regulators of TGF-beta signaling. Curr Opin Genet Dev. 1998;8(1):103-111.
109 Yamashita H, Ten Dijke P, Heldin CH, et al. Bone morphogenetic protein receptors. Bone. 1996;19(6):569-574.
110 Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95(6):737-740.
111 Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296(5573):1646-1647.
112 Canalis E, Economides AN, Gazzerro E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev. 2003;24(2):218-235.
113 Centrella M, McCarthy TL, Canalis E. Transforming growth factor-beta and remodeling of bone. J Bone Joint Surg Am. 1991;73(9):1418-1428.
114 Spinella-Jaegle S, Roman-Roman S, Faucheu C, et al. Opposite effects of bone morphogenetic protein-2 and transforming growth factor-beta1 on osteoblast differentiation. Bone. 2001;29(4):323-330.
115 Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132(1–2):195-210.
116 Hill PA, Tumber A, Meikle MC. Multiple extracellular signals promote osteoblast survival and apoptosis. Endocrinology. 1997;138(9):3849-3858.
117 Bikle DD, Sakata T, Leary C, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17(9):1570-1578.
118 Zhao G, Monier-Faugere MC, Langub MC, et al. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141(7):2674-2682.
119 Grinspoon S, Thomas L, Miller K, et al. Effects of recombinant human IGF-I and oral contraceptive administration on bone density in anorexia nervosa. J Clin Endocrinol Metab. 2002;87(6):2883-2891.
120 Canalis E, Giustina A. Glucocorticoid-induced osteoporosis: summary of a workshop. J Clin Endocrinol Metab. 2001;86(12):5681-5685.
121 Nishita M, Hashimoto MK, Ogata S, et al. Interaction between Wnt and TGF-beta signalling pathways during formation of Spemann’s organizer. Nature. 2000;403(6771):781-785.
122 Mao J, Wang J, Liu B, et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7(4):801-809.
123 Kato M, Patel MS, Levasseur R, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303-314.
124 Gong Y, Slee RB, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513-523.
125 Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513-1521.
126 Montero A, Okada Y, Tomita M, et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105(8):1085-1093.
127 Nakamura T, Hanada K, Tamura M, et al. Stimulation of endosteal bone formation by systemic injections of recombinant basic fibroblast growth factor in rats. Endocrinology. 1995;136(3):1276-1284.
128 Shiang R, Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335-342.
129 Hock JM, Canalis E. Platelet-derived growth factor enhances bone cell replication, but not differentiated function of osteoblasts. Endocrinology. 1994;134(3):1423-1428.
130 Robey PG. Bone proteoglycans and glycoproteins. Principles of bone biology. San Diego, CA: Academic Press, 2002;225-238.
131 Xu T, Bianco P, Fisher LW, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20(1):78-82.
132 Gorski JP. Is all bone the same? Distinctive distributions and properties of non-collagenous matrix proteins in lamellar vs. woven bone imply the existence of different underlying osteogenic mechanisms. Crit Rev Oral Biol Med. 1998;9(2):201-223.
133 Gokhale JA, Robey PG, Boskey AL. The biochemistry of bone. Osteoporosis, Vol. 1 . Academic Press, San Diego, CA, 2001;107-188.
134 Ducy P, Desbois C, Boyce B, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382(6590):448-452.
135 Glimcher MJ. The nature of the mineral phase in bone: biological and clinical implications. Metabolic bone disease and clinically related disorders, 3rd edn., San Diego, CA: Academic Press; 1998:23-51.
136 Eppell SJ, Tong W, Katz JL, et al. Shape and size of isolated bone mineralites measured using atomic force microscopy. J Orthop Res. 2001;19(6):1027-1034.
137 Termine JD, Belcourt AB, Conn KM, et al. Mineral and collagen-binding proteins of fetal calf bone. J Biol Chem. 1981;256(20):10403-10408.
138 Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337-342.
139 Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappa B ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001;79(5–6):243-253.
140 Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795-823.
141 Walsh MC, Choi Y. Biology of the TRANCE axis. Cytokine Growth Factor Rev. 2003;14(3–4):251-263.
142 Bucay N, Sarosi I, Dunstan CR, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12(9):1260-1268.
143 Mizuno A, Amizuka N, Irie K, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247(3):610-615.
144 Akatsu T, Murakami T, Ono K, et al. Osteoclastogenesis inhibitory factor exhibits hypocalcemic effects in normal mice and in hypercalcemic nude mice carrying tumors associated with humoral hypercalcemia of malignancy. Bone. 1998;23(6):495-498.
145 Khosla S, Arrighi HM, Melton LJ3rd, et al. Correlates of osteoprotegerin levels in women and men. Osteoporos Int. 2002;13(5):394-399.
146 Sakata M, Shiba H, Komatsuzawa H, et al. Expression of osteoprotegerin (osteoclastogenesis inhibitory factor) in cultures of human dental mesenchymal cells and epithelial cells. J Bone Miner Res. 1999;14(9):1486-1492.
147 Atkins GJ, Kostakis P, Pan B, et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18(6):1088-1098.
148 Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph node organogenesis. Nature. 1999;397(6717):315-323.
149 Hughes AE, Ralston SH, Marken J, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24(1):45-48.
150 Johnson-Pais TL, Singer FR, Bone HG, et al. Identification of a novel tandem duplication in exon 1 of the TNFRSF11A gene in two unrelated patients with familial expansile osteolysis. J Bone Miner Res. 2003;18(2):376-380.
151 Li J, Sarosi I, Yan XQ, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97(4):1566-1571.
152 Dougall WC, Glaccum M, Charrier K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13(18):2412-2424.
153 Boyce BF, Aufdemorte TB, Garrett IR, et al. Effects of interleukin-1 on bone turnover in normal mice. Endocrinology. 1989;125(3):1142-1150.
154 Pacifici R, Rifas L, McCracken R, et al. Ovarian steroid treatment blocks a postmenopausal increase in blood monocyte interleukin 1 release. Proc Natl Acad Sci USA. 1989;86(7):2398-2402.
155 Tashjian AHJr, Voelkel EF, Lazzaro M, et al. Tumor necrosis factor-alpha (cachectin) stimulates bone resorption in mouse calvaria via a prostaglandin-mediated mechanism. Endocrinology. 1987;120(5):2029-2036.
156 Chenu C, Pfeilschifter J, Mundy GR, et al. Transforming growth factor beta inhibits formation of osteoclast-like cells in long-term human marrow cultures. Proc Natl Acad Sci USA. 1988;85(15):5683-5687.
157 Kaneda T, Nojima T, Nakagawa M, et al. Endogenous production of TGF-beta is essential for osteoclastogenesis induced by a combination of receptor activator of NF-kappa B ligand and macrophage-colony-stimulating factor. J Immunol. 2000;165(8):4254-4263.
158 Krishnan V, Moore TL, Ma YL, et al. Parathyroid hormone bone anabolic action requires Cbfa1/Runx2-dependent signaling. Mol Endocrinol. 2003;17(3):423-435.
159 Roodman GD, Ibbotson KJ, MacDonald BR, et al. 1,25-Dihydroxyvitamin D3 causes formation of multinucleated cells with several osteoclast characteristics in cultures of primate marrow. Proc Natl Acad Sci USA. 1985;82(23):8213-8217.
160 Takahashi S, Goldring S, Katz M, et al. Downregulation of calcitonin receptor mRNA expression by calcitonin during human osteoclast-like cell differentiation. J Clin Invest. 1995;95(1):167-171.
161 Jilka RL, Hangoc G, Girasole G, et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257(5066):88-91.
162 Oursler MJ, Osdoby P, Pyfferoen J, et al. Avian osteoclasts as estrogen target cells. Proc Natl Acad Sci USA. 1991;88(15):6613-6617.
163 Girasole G, Jilka RL, Passeri G, et al. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest. 1992;89(3):883-891.
164 Mundy GR, Chen D, Oyajobi BO. Bone remodeling. Primer on the metabolic bone diseases and disorders of mineral metabolism, 5th edn., Washington, DC: American Society for Bone and Mineral Research; 2003:46-58.
165 Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption – a hypothesis. Calcif Tissue Int. 1981;33(4):349-351.
166 Howard GA, Bottemiller BL, Turner RT, et al. Parathyroid hormone stimulates bone formation and resorption in organ culture: evidence for a coupling mechanism. Proc Natl Acad Sci USA. 1981;78(5):3204-3208.
167 Locklin RM, Khosla S, Turner RT, et al. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89(1):180-190.
168 Manolagas SC, Jilka RL. Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N Engl J Med. 1995;332(5):305-311.
169 Enomoto H, Shiojiri S, Hoshi K, et al. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2–/– mice by RANKL transgene. J Biol Chem. 2003;278(26):23971-23977.
170 Takeda S, Elefteriou F, Levasseur R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305-317.
171 Mundy GR, Rodan SB, Majeska RJ, et al. Unidirectional migration of osteosarcoma cells with osteoblast characteristics in response to products of bone resorption. Calcif Tissue Int. 1982;34(6):542-546.
172 Mundy GR, Poser JW. Chemotactic activity of the gamma-carboxyglutamic acid containing protein in bone. Calcif Tissue Int. 1983;35(2):164-168.
173 Dallas SL, Rosser JL, Mundy GR, et al. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002;277(24):21352-21360.
174 Pfeilschifter J, Wolf O, Naumann A, et al. Chemotactic response of osteoblastlike cells to transforming growth factor beta. J Bone Miner Res. 1990;5(8):825-830.
175 Grotendorst GR, Seppa HE, Kleinman HK, et al. Attachment of smooth muscle cells to collagen and their migration toward platelet-derived growth factor. Proc Natl Acad Sci USA. 1981;78(6):3669-3672.
176 Seppa H, Grotendorst G, Seppa S, et al. Platelet-derived growth factor is chemotactic for fibroblasts. J Cell Biol. 1982;92(2):584-588.
177 Canalis E, Pash J, Varghese S. Skeletal growth factors. Crit Rev Eukaryot Gene Expr. 1993;3(3):155-166.
178 Valentin-Opran A, Charhon SA, Meunier PJ, et al. Quantitative histology of myeloma-induced bone changes. Br J Haematol. 1982;52(4):601-610.
179 Stewart AF, Vignery A, Silverglate A, et al. Quantitative bone histomorphometry in humoral hypercalcemia of malignancy: uncoupling of bone cell activity. J Clin Endocrinol Metab. 1982;55(2):219-227.
180 Darby AJ, Meunier PJ. Mean wall thickness and formation periods of trabecular bone packets in idiopathic osteoporosis. Calcif Tissue Int. 1981;33(3):199-204.
181 Courtney AC, Hayes WC, Gibson LJ. Age-related differences in post-yield damage in human cortical bone. Experiment and model. J Biomech. 1996;29(11):1463-1471.
182 McCalden RW, McGeough JA, Barker MB, et al. Age-related changes in the tensile properties of cortical bone. The relative importance of changes in porosity, mineralization, and microstructure. J Bone Joint Surg Am. 1993;75(8):1193-1205.
183 Seeman E. Pathogenesis of bone fragility in women and men. Lancet. 2002;359(9320):1841-1850.
184 Oxlund H, Mosekilde L, Ortoft G. Reduced concentration of collagen reducible cross links in human trabecular bone with respect to age and osteoporosis. Bone. 1996;19(5):479-484.
185 Bailey AJ, Wotton SF, Sims TJ, et al. Biochemical changes in the collagen of human osteoporotic bone matrix. Connect Tissue Res. 1993;29(2):119-132.
186 Martin RB, Atkinson PJ. Age and sex-related changes in the structure and strength of the human femoral shaft. J Biomech. 1977;10(4):223-231.
187 Beck TJ, Oreskovic TL, Stone KL, et al. Structural adaptation to changing skeletal load in the progression toward hip fragility: the study of osteoporotic fractures. J Bone Miner Res. 2001;16(6):1108-1119.
188 Duan Y, Turner CH, Kim BT, et al. Sexual dimorphism in vertebral fragility is more the result of gender differences in age-related bone gain than bone loss. J Bone Miner Res. 2001;16(12):2267-2275.
189 Chan GK, Duque G. Age-related bone loss: old bone, new facts. Gerontology. 2002;48(2):62-71.
190 Shi S, Gronthos S, Chen S, et al. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat Biotechnol. 2002;20:587-591.
191 Simonsen JL, Rosada C, Serakini N, et al. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol. 2002;20:592-596.
192 Yudoh K, Matsuno H, Nakazawa F, et al. Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shortening and replicative senescence in human osteoblasts. J Bone Miner Res. 2001;16:1453-1464.
193 Gronthos S, Chen S, Wang C-Y, et al. Telomerase accelerates osteogenesis of bone marrow stromal stem cells by upregulation of cbfa1, Osterix, and osteocalcin. J Bone Miner Res. 2003;18(4):716-722.
194 Chan GK, Duque G. Age-related bone loss: old bone, new facts. Gerontology. 2002;48:62-71.
195 Bergman RJ, Gazit D, Kahn AJ, et al. Age-related changes in osteogenic stem cells in mice. J Bone Miner Res. 1996;11(5):568-577.
196 Erdmann J, Kogler C, Diel I, et al. Age-associated changes in the stimulatory effect of transforming growth factor beta on human osteogenic colony formation. Mechanisms Aging Devel. 1999;110:73-85.
197 Long MW, Ashcraft EK, Normalle D, et al. Age-related phenotypic alterations in populations of purified human bone precursor cells. J Gerontol A Biol Sci Med Sci. 1999;54:B54-B62.
198 Mueller SM, Glowacki J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional sponges. J Cell Biochem. 2001;82:583-590.
199 Muschler GF, Nitto H, Boehm CA, et al. Age- and gender-related changes in the cellularity of human bone marrow and the prevalence of osteoblastic progenitors. J Orthop Res. 2001;19:117-125.
200 Nishida S, Endo N, Yamagiwa H, et al. Number of osteoprogenitor cells in human bone marrow markedly decreases after skeletal maturation. J Bone Miner Metab. 1999;17:171-177.
201 Oreffo RO, Bord S, Triffitt JT. Skeletal progenitor cells and ageing human populations. Clin Sci (Lond). 1998;94:549-555.
202 Stenderup K, Justesen J, Eriksen EF. Number and proliferative capacity of osteogenic stem cells are maintained during aging and in patients with osteoporosis. J Bone Miner Res. 2001;16:1120-1129.
203 D’Ippolito G, Schiller PC, Ricordi C, et al. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. 1999;14:1115-1122.
204 Kassem M, Ankersen L, Eriksen EF, et al. Demonstration of cellular aging and senescence in serially passaged long-term cultures of human trabecular osteoblasts. Osteoporos Int. 1997;7(6):514-524.
205 Kveiborg M, Flyvbjerg A, Rattan SI, et al. Changes in the insulin-like growth factor-system may contribute to in vitro age-related impaired osteoblast functions. Exp Gerontol. 2000;35(8):1061-1074.
206 Fujieda M, Kiriu M, Mizuochi S, et al. Formation of mineralized bone nodules by rat calvarial osteoblasts decreases with donor age due to a reduction in signaling through EP(1) subtype of prostaglandin E(2) receptor. J Cell Biochem. 1999;75(2):215-225.
207 Martinez ME, Medina S, Sanchez M, et al. Influence of skeletal site of origin and donor age on 1,25(OH)2D3-induced response of various osteoblastic markers in human osteoblastic cells. Bone. 1999;24(3):203-209.
208 Martinez ME, del Campo MT, Medina S, et al. Influence of skeletal site of origin and donor age on osteoblastic cell growth and differentiation. Calcif Tissue Int. 1999;64(4):280-286.
209 Long MW. Osteogenesis and bone-marrow-derived cells. Blood Cells Mol Dis. 2001;27(3):677-690.
210 Battmann A, Battmann A, Jundt G, et al. Endosteal human bone cells (EBC) show age-related activity in vitro. Exp Clin Endocrinol Diabetes. 1997;105(2):98-102.
211 Kveiborg M, Rattan SI, Clark BF, et al. Treatment with 1,25-dihydroxyvitamin D3 reduces impairment of human osteoblast functions during cellular aging in culture. J Cell Physiol. 2001;186(2):298-306.
212 Martinez P, Moreno I, De Miguel F, et al. Changes in osteocalcin response to 1,25-dihydroxyvitamin D(3) stimulation and basal vitamin D receptor expression in human osteoblastic cells according to donor age and skeletal origin. Bone. 2001;29(1):35-41.