[level-membership-for-opthalmology-category]
Chapter 27 Blood–Retinal Barrier, Immune Privilege, and Autoimmunity
Introduction
More than 100 years ago a fascinating discovery was made. Different tissue samples were placed into the anterior chamber of dogs or rabbits and were then observed for 4 months. Surprisingly, the induced intraocular inflammation was very limited. Most of the tissue samples were not rejected as they were known to occur from other parts of the body. At this time, the eye as one of the first sites of immune privilege was recognized.1
Today, we know that the eye is a unique and complex organ that has developed multiple mechanisms to protect itself against frivolous immunological attacks and inflammation in order to maintain its delicate structure and function. The blood–ocular barrier system is formed by the blood–aqueous barrier and the inner and outer blood–retinal barrier. It restrictively limits the passage of ions and proteins to maintain homeostasis and to keep tissue-specific antigens sequestrated. Furthermore, many active factors have been discovered in the eye that contribute to a downregulatory immune environment (DIE).2 The blood–ocular barrier and the DIE are mechanisms that depend on each other’s proper function. They are central to preserving the immune privilege of the eye. Immune privilege has served well to curb excessive inflammation and preserve function. However, this privilege may be lost under certain conditions and situations, resulting in retinal autoimmunity and ocular inflammation. A dysfunction of the inner blood–retinal barrier initiates diabetic retinopathy.3,4 A breakdown of the blood–retinal barrier can also be found in cystoid macular edema, while a loss of the DIE is essential to many forms of uveitis and in all probability to the development of age-related macular degeneration (AMD).5
For a long time, retinal autoimmunity has been perceived as pathogenic, and active suppression of retinal immunity was presumed necessary to maintain the health of the eye. However, studies have shown the presence of retinal autoantibodies in normal controls,6 and constitutive expression of proinflammatory ligands have been found in the normal retina in high concentration (B Kim, M Pillai, Z Li, unpublished data). Moreover, animal optic nerve injury studies revealed possible beneficial roles of retinal autoimmunity in controlling collateral damage to the retinal ganglion cells.7 Thus, retinal autoimmunity can be viewed as a “double-edged sword,” with both protective and destructive effects. To understand retinal autoimmunity, we first have to understand basic concepts in immunology and how some of these immunological components are at work to maintain the eye’s immune privilege.
Basic concepts of immunology
Adaptive immunity
In contrast, adaptive immunity, as the name implies, is antigen-specific, adaptive, transferable, and has other cardinal features like immunologic memory and tolerance. Adaptive immunity has evolved and developed novel molecules like the antibody and the T-cell receptor (TCR), which come in almost limitless varieties to recognize most molecules of biological interest, including both self and nonself. These molecules, developed in the absence of exogenous stimulation, have unique configurations that confer specificity in antigen recognition. The key cellular components of adaptive immunity are the T and B lymphocytes which possess unique receptors for recognizing billions of different antigenic epitopes.8 Each clone of lymphocyte expresses molecularly identical receptors on the surface; hence, in order to recognize billions of different epitopes, there are billions of unique lymphocytes. Upon maturation in the thymus (T cells) and the bone marrow (B cells), cells remain quiescent in G0 of the cell cycle until they encounter the complementary antigen of sufficient affinity to their receptors. Binding with the receptors in the presence of appropriate costimulation is the initiating event to trigger an immune response targeting elimination of the complementary molecule. If this molecule is pathogen-derived, activation will lead to the elimination of the pathogen, but if the molecule is self-derived then activation will potentially lead to autoimmunity, tissue injury, disease, and the destruction of host tissue. Activation of naive lymphocytes results in the emergence of clones of lymphocytes through proliferation, each with a unique receptor to recognize one epitope of the inciting antigen. The cytokine environment during activation of naive T cells is responsible for differentiation into three major types of effector T cells. Interferon (IFN)-γ leads to Th1 cell production, while interleukin (IL)-4 is responsible for Th2 production. Some Th1 and Th2 cell clones will become memory cells which are responsible for the enhanced immune response if the antigen is re-encountered. The rest of Th1 will secrete cytokines targeted at macrophages and other cells mediating cellular immunity. By contrast, Th2 will stimulate B cells into proliferation and antibody production, mediating humoral immunity. Th17 are a recently discovered family of T helper cells that are capable of producing IL-17. IL-1 and IL-23 seem to be needed for human Th17 differentiation.9 Th17 play an important role in combatting various bacterial and fungal species by producing IL-23. In contrast, large amounts of Th17 can be found in autoimmune diseases like uveitis, multiple sclerosis, and rheumatoid arthritis.10
Both immune and accessory cells, including those of the innate immunity system, are activated to eliminate pathogens. In this way, adaptive immunity serves to enhance immune protection through better-coordinated and more specific attacks using innate immunity mechanisms. Within the genome lies the ability to create antibodies and TCR for antigens that bind to self antigens with high affinity.11 Hence, the threat of autoimmunity is inborn and regulation is crucial. The ability to distinguish between foreign and self, and the ability to regulate autoimmunity, are critical for survival.
The immune response can be seen as analogous to the neural reflex arc, which has an afferent limb, a central process, and an efferent limb. In the immune afferent limb, antigens are captured, processed, transported, and finally presented to the lymphocytes. As the naive lymphocytes can only be activated in the organized lymph tissues in secondary lymphoid organs such as lymph nodes, spleen, tonsils, and Peyer’s patches, it needs specialized cells to execute the immune afferent limb. These cells are professional antigen-presenting cells (APCs). Dendritic cells and macrophages, both bone marrow-derived, have APC functions serving to capture antigens through phagocytosis and endocytosis, process the antigens, and present the processed antigen in conjunction with special major histocompatibility complex (MHC) molecules on their cell surface. In addition, the APCs provide the necessary costimulation needed for lymphocyte activation by upregulating an array of surface molecules (CD80, CD86, intercellular adhesion molecule-1, lymphocyte function-associated molecule-3, and CD40) that function as ligands for receptors expressed by the lymphocytes. They also secrete cytokines such as IL-12, IL-6, IL-10, and IL-1β serving similar costimulation functions. Costimulation is required as the second signal for full activation of the lymphocytes, independent of the first signal, which is the antigen and the lymphocyte receptor engagement.12 There are differences in antigen presentation to B and T lymphocytes. For B lymphocytes, the receptor, which is a surface-bound antibody, can engage a naive antigen directly while TCRs can only recognize peptide fragments presented on special surface molecules (MHC classes I and II). MHC class I, which is present in most cells, presents peptides derived from protein degradation in the cytoplasm (intracellular antigens such as viral and intracellular microbe products), while MHC class II molecules, which are present in APCs and lymphocytes, present peptides from phagocytic vesicles (extracellular antigens from the microenvironment).
Immune regulation
The immune system can be regulated at any part of the immune response loop, i.e., afferent limb, central process, and efferent limb. Regulation of the autoimmune response can be via any of the following mechanisms: central tolerance – clonal deletion and peripheral tolerance – clonal anergy, T-cell suppression, immune deviation, immunologic ignorance, and antigen sequestration. Since the genome has the capability to generate both self- and nonself-recognizing antibodies and TCRs, mechanisms must be in place to contain and prevent the activation of self-reactive T cells. During lymphocyte development, both T and B cells with receptors that recognize self molecules with high affinity are clonally deleted via apoptosis (central tolerance).13 However the process is not foolproof, as autoreactive T and B cells in the periphery exist even in normal individuals. Many tissue-specific antigens, like the eye-restricted molecules, may not be expressed in the thymus or cells may have escaped the selection process by the central tolerance mechanism in the thymus without encountering the specific self antigen. Hence, the potential for induction and expression of autoimmunity still exists, and mechanisms to contain these autoreactive immune cells are crucial to prevent autoimmunity. Evidence for peripheral tolerance can be found in all phases of the immune response, i.e., afferent limb, central process, and efferent limb.14,15 The afferent limb depends heavily on the functional properties of the APCs which offer many opportunities for modulation. First, the antigen can be sequestrated and prevented from contact with the APCs by physical barriers or by rendering APCs incapable of antigen capture in the microenvironment (sequestration). Second, the ability of the APC to degrade, process, and express the antigens can be inhibited. Third, the antigen-bearing APCs may be prevented from migrating to the lymphoid tissues. Lastly, the ability of the APCs to generate the costimulatory signal can be inhibited or different costimulatory signals may be produced (immune deviation).
Regulation of APC function can be seen as a form of immunologic ignorance where reactive autoimmune cells are present but never have the opportunity to encounter its antigen in a proper immunogenic form due to a deficiency in antigen presentation. In the central processing phase of the immune response, two signals are required for full activation of the lymphocyte. The absence of costimulation in the presence of antigen-specific stimulation renders the T cells unresponsive to further antigen stimulation, even in the presence of costimulatory signals (anergy).16 On the other hand, too much costimulation in the presence of high antigen dose may lead to profound cell activation and apoptosis. Both mechanisms will result in tolerance.
It is also known that APCs from certain immune-privileged sites such as the eye, and APCs in the gut processing orally ingested antigens, may cause some immune cells to develop into immunoregulatory cells like IL-10-secreting Tr1 cells or transforming growth factor-β (TGF-β) secreting Th3 cells. These cells have in common that they are adaptively regulatory. They receive a suppressive potential following specific antigen stimulation in a particular cytokine milieu.17
In the efferent limb, immune regulation can occur via targeting at the level of antigen-specific immune effector cells. One such mechanism would be through Fas ligand – Fas-induced apoptosis of lymphocytes.18,19 The immune-reactive lymphocytes express the CD95 receptor (Fas receptor). On encountering the Fas ligand, which is expressed in immuno-privileged eye tissues such as the cornea and retina, these immune cells undergo apoptosis. Other molecular mechanisms, such as TNF-α production by immune and accessory cells, such as Müller cells and retinal pigment epithelial (RPE) cells during antigen encounter, can also result in apoptosis, thereby deleting these immune-reactive cells. Similar to the central process, effector cells need costimulatory signals to be activated and the expression of inappropriate costimulatory signals such as IL-10 or TGF-β by APCs can lead to anergy or reprogramming of lymphocytes into immunoregulatory cells secreting immunosuppressive cytokines, thereby further suppressing autoimmunity.20,21
An important regulator of immune responses is the CD4+ CD25high forkhead box protein 3 (Foxp3)+ regulatory T cell (Treg). There are two groups of Treg in humans: the naturally occuring Treg and the induced Treg. The natural Treg is developmentally determined in the thymus as a distinct T-cell subpopulation that is specialized for suppressive function.17 It is distinguished from the induced Treg by a demethylated promoter region for Foxp3.22 Foxp3 seems to act as a repressor of transcription that regulates T-cell activation. A dysfunction of Foxp3 results in hyperactivation of T cells and severe autoimmune disease.23 Natural and induced Tregs maintain peripheral tolerance by modifying the functions of other T cells, both CD4+ and CD8+ populations, either directly through T-cell to T-cell interactions, or indirectly through APCs.24 Hence they are crucial for maintaining immune homeostasis and controlling autoimmunity in order to keep the immune system healthy.
Blood–ocular barrier
At the end of the 19th century, the German bacteriologist and immunologist Paul Ehrlich experimented with staining tissues in animal models. When he injected dyes into the bloodstream, almost all tissues were colored intensively. However, the brain remained unstained.25 A few years later, one of Ehrlich’s students, Edwin Goldmann, performed the experiment the other way around, by injecting dyes into the cerebrospinal fluid. This time, the brain became stained and the rest of the body remained dye-free. Goldmann correctly concluded that there was a restrictive barrier mechanism inhibiting the passage of solubles between the two compartments, the blood and the cerebrospinal fluid.26 In the same year, in 1913, Schnaudigel found a similar barrier for trypan blue in the retina of rabbits.27
Today, we know that the eye consists of several anatomical structures that limit flux between the blood and most parts of the eye itself.28 The blood–ocular barrier is represented by two main components: the blood–aqueous barrier and the blood–retinal barrier. The blood–aqueous barrier is formed by the nonpigmented layer of the ciliary body epithelium and by the endothelium of the iridal capillaries. The blood–retinal barrier is located at two levels. The inner barrier is the nonfenestrated capillaries of the retinal vessels, whereas the outer barrier is the tight junctions of the RPE, enabling a high degree of control of fluid permeability.29
Limiting the passage of ions enables the eye to maintain homeostasis in the aqueous humor and in the retina while the barrier also keeps small and large molecules like drugs from entering the eye. Specially hydrophilic substances show a low permeability across the blood–ocular barrier.30 Furthermore, the barrier has an essential immunologic function: the sequestration of tissue-specific antigens is crucial to preserve the immune privilege of the eye, and, therefore, to avoid intraocular inflammation. On the other hand, inflammation can cause breakdown of the blood–retinal barrier, leading to autoimmunity and inflammation, and allowing drugs and other molecules to penetrate into the eye. Considering its essential function it is not surprising that many of the most common eye diseases are directly associated with alterations of the blood–ocular barrier.
Blood–retinal barrier in diabetic retinopathy
The initial disorder causing both nonproliferative and proliferative diabetic retinopathy is chronic hyperglycemia leading to retinal hypoxia. Compensatory mechanisms like vasodilatation are a regulatory attempt to increase retinal blood flow. After exhaustion of autoregulation, a disruption of endothelial tight junctions in the retinal vasculature can occur.4 Furthermore, chronic hyperglycemia seems to be able to induce a hypoxia-mediated expression of growth factors and cytokines in the retinal endothelium, such as insulin-like growth factor-1, vascular endothelial growth factor-A, angiopoietin-2 and TNF-α, leading to leukocyte adhesion, endothelial cell injury, increased permeability of endothelial cells, and finally angiogenesis.31–33 The resulting plasma leakage from the inner blood–retinal barrier may cause diabetic macular edema and consecutive vision loss.
Blood–retinal barrier in cystoid macular edema
Cystoid macular edema is a major reason for vision loss in various ocular diseases. Different conditions such as trauma, inflammation, or vascular degeneration lead to an increased permeability of the blood–retinal barrier. The inner and outer plexiform layers of the retina act as diffusion barriers against continuous fluid distribution. Therefore, a breakdown of the inner blood–retinal barrier causes cysts mainly in the inner nuclear layer, whereas serum leakage from the outer blood–retinal barrier pools into cystic spaces in the Henle fiber layer.34
The eye as an immune-privileged site
The eye has been recognized as an immune-privileged site for more than 100 years. In the 1940s Medawar demonstrated this by showing prolonged, often indefinite, survival of organs or tissue grafts in the anterior chamber of the eye.35 It is now known that this immune privilege is a dynamic process in which immunoregulatory mechanisms combined with anatomical factors maintain the vitality of grafts in privileged sites and of privileged organs and tissues as grafts. Various tissues, including allogenic skin grafts, thyroid tissues, neuronal retinal tissue, and allogenic tumor cells, have been shown to survive in the anterior chamber of the eye for prolonged periods.36 However, this privilege is influenced by the immunogenic strength of the antigens expressed by the cells. Tumors expressing MHC-encoded alloantigens had only a transient extension of survival compared to those that expressed weaker transplantation antigens.37 Although most studies were done placing tissue in the anterior chamber of the eye, a number of studies demonstrated a similar immune privilege when allogenic tumor cells, foreign neuronal retinal tissue, and RPE were placed in the vitreous cavity and the subretinal space.38,39 Certain eye tissues are known to be immune-privileged, i.e., demonstrating an altered immune response and less rejection. Ocular tissues include the cornea, RPE, and probably the retina. When transplanted beneath the capsule of the kidney, a nonimmune-privileged site, cornea demonstrated extended survival compared with other ocular tissues like the conjunctiva.40 Both passive factors, such as the blood–retinal barrier, and active factors, such as the DIE, are essential to establish and maintain the ocular immune privilege (Table 27.1).
Table 27.1 Components contributing to ocular immune privilege
| Passive | |
| Blood–retinal barrier | Nonfenestrated vascular endothelium of the retinal vessels and tight junctions among the retinal pigment epithelium |
| Lack of lymphatic drainage | Absence of lymphatics in the retina – it is present in the choroid |
| Tissue fluid drainage | Tissue fluid drains via the hematogenous route |
| Reduced expression of MHC | Antigen presentation is reduced by reduction of MHC class I and II molecules and APCs. The APCs are altered by the microenvironment to promote immune regulation |
| Reduced APCs in the retina | |
| APCs with altered function | |
| Active | |
| Immunosuppressive | TGF-β, α-MSH, VIP, CGRP, MIF, IL-1 receptor antagonist and free cortisol microenvironment |
| Factors expressed on cell surface | CD95 ligand, CD59, CD55, CD46 |
MHC, major histocompatibility complex; APC, antigen-presenting cell; TGF, transforming growth factor; MSH, melanocyte-stimulating hormone; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; MIF, migration inhibitory factor; IL, interleukin; CD, nomenclature for surface proteins of human leukocytes.
Transportation of antigens
The blood–retinal barrier serves to keep tissue-specific antigens sequestrated. There is an absence of lymphatics in the retina, although there is evidence that antigen may still be able to be transported to the lymph nodes.41 Tissue drainage via the hematogenous route may alter the APC function.42 There is also a reduced expression of MHC class I and II molecules and an absence of bone marrow-derived cells that function within tissues as APCs in the normal retina.43
Downregulatory immune environment
The DIE is an evolutionary adaptation of the eye to protect itself against excessive inflammation. A small focus of inflammation in the eye has a far greater impact than a similar one in the kidney. The posterior segment seems to have several active factors that help to keep the local immunoenvironmental balance in a downregulatory state. Active factors include the downregulatory effect of Müller cells in cell-to-cell contact with lymphocytes.44 Furthermore, there are many other ocular cells that affect DIE (NK T cells, RPE, microglia, F4/80+ macrophages), as well as cell surface and soluble factors in the ocular microenvironment (e.g., CD95 ligand, CD55, CD59, CD46, IL-10, IL-11, TGF-β, α-melanocyte-stimulating hormone, vasoactive intestinal peptide, calcitonin gene-related peptide, macrophage migration-inhibitory factor, IL-1-receptor antagonist, and free cortisol). All of these factors contribute to active immunoregulation in an attempt to prevent any autoimmunity.2
Downregulatory immune environment in age-related macular degeneration
Several findings in patients with AMD (e.g., variants of CFH, HTRA-1, PLEKHA1 and Toll-like receptors, activated macrophages in the blood, autoantibodies to retinal components) allow it to be considered as an immune-mediated disease.5 Additionally, there is a masked randomized pilot study indicating that anti-inflammatory therapy was beneficial in treating wet AMD.45 Not all these associations may be directly related to AMD, but rather to the underlying DIE, weakening it and bringing it out of balance.46
Anterior-chamber-associated immune deviation
Anterior-chamber-associated immune deviation (ACAID) is the best-studied immune-privilege phenomenon in the eye.47 Although its name implies that it is an anterior-chamber phenomenon, there is much information to show that the same mechanisms are at work in the vitreous and subretinal space.39 Therefore understanding ACAID could further our knowledge of retinal autoimmunity. There are a large number of bone marrow-derived APCs in the iris, trabecular meshwork, and ciliary body. Studies have demonstrated that antigen captured in the anterior chamber by these APCs is processed in the spleen.48 These APCs reach the spleen via the hematogenous route, bypassing the afferent lymphatics and the lymph nodes. The result is a reduction of the antigen’s capacity to activate cell-mediated immune mechanisms. Functionally, these APCs are different. If transferred, these APCs induce ACAID rather than inducing conventional immune responses. The microenvironment of the eye also contains factors such as TGF-β2 that are able to reprogram the APCs from conventional sites, if exposed to the eye microenvironment, to secrete less IL-2, express less CD40 (a costimulatory signal for T-cell activation), and produce more mature TGF-β. These TGF-β2-treated APCs activate naive T cell to secrete IL-4 and trace amounts of IL-2, but not IFN-γ.49 The spleen is the critical lymphoid organ for the induction of ACAID. In the spleen, a unique population of CD4+ lymphocytes, which secrete IL-2 and TGF-β and develop a Th2 immune response upon antigen exposure, is activated. The B-cell response, occurring upon re-exposure to antigen with adjuvant, produces IgG1 isotype antibodies instead of IgG2a, IgG2b, and IgG3, consistent with a Th2 response.50 There appears to be a concerted effort to deviate the immune response from the usual Th1 pathway to a Th2 pathway, one that is seen in allergic responses. The role of CD95 and its ligand, Fas ligand, is still unclear. It is tempting to speculate that Fas ligand acts by causing active immune cells to apoptose upon engagement, thereby resulting in the elimination of the active immune cells.51 One would expect an upregulation of an inflammatory response if Fas ligand were absent in the eye. Contrary to the predictions, Fas ligand-deficient mice developed less severe inflammation during experimental autoimmune uveoretinitis (EAU) than wild types.52 Studies using Fas ligand-deficient mice have also revealed that ACAID is dependent on the presence of Fas ligands, as these mice are unable to induce ACAID.53 Hence, Fas–Fas ligand interaction may be more complex than just inducing apoptosis of activated immune cells, as initially presumed.
Although there are active soluble factors in the aqueous humor that can inhibit T-cell activation in vitro, decrease the ability of NK cells to lyse their target,54 and block complement activation,55,56 the eye is not left defenseless against pathogens. It still retains certain immune functions to deter pathogenic invasion. Antibodies in the aqueous are capable of neutralizing viruses and cytotoxic T cells (terminally differentiated) can bind and kill their target cells as elsewhere in the body.57
Retinal antigens and experimental autoimmune uveoretinitis
Several animal models for autoimmune posterior uveitis have been described (Table 27.2). EAU is one of the most studied of these models since its development in 1968 by Wacker and Lipton.58 (It is a good animal model for human ocular autoimmunity.) EAU is mostly self-limited and requires the use of adjuvant for disease induction. The EAU model has been instrumental in eliciting immune mechanisms, the identification of pathogenic epitopes of autoantigens in the eye in animals, and the evaluation of therapeutic strategies with clinical relevance.
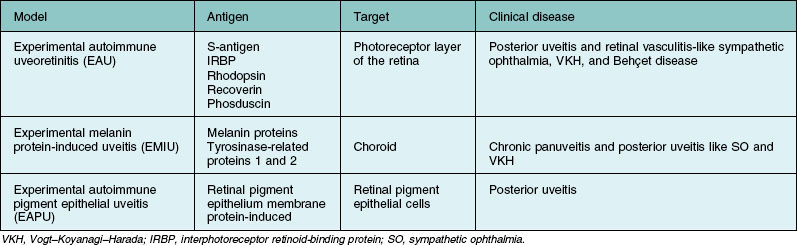
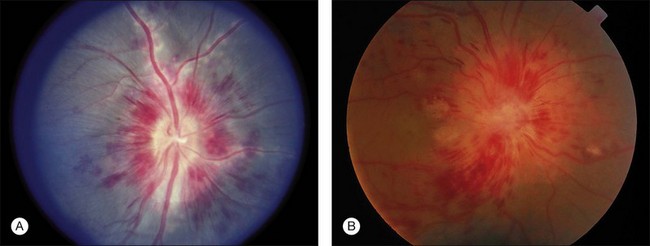
Several retinal antigens, including S-antigen (arrestin), interphotoreceptor retinoid-binding protein (IRBP), rhodopsin, recoverin, and phosducin, have been found to have uveitogenic properties. Immunization with these antigens or their fragments can induce ocular inflammation in susceptible strains of guinea pigs, rats, mice, and in rabbits and monkeys. The disease that is induced resembles various human uveitic conditions such as ocular sarcoidosis (Fig. 27.1), Vogt–Koyanagi–Harada disease, sympathetic ophthalmia, and Behçet disease (Fig. 27.2). Using advanced techniques, more novel eye autoantigens have been identified, such as uveal autoantigen with coiled coil domains and ankyrin repeats in patients with Vogt–Koyanagi–Harada disease,59 and an unknown retinal autoantigen targeting the connecting cilium region of photoreceptors in a patient with Waldenström’s macroglobulinemia, and retinal dysfunction similar to cancer-associated retinopathy.60 Much of our understanding of retinal autoantigens is derived from EAU models using S-antigen, IRBP (peptides derived from these proteins), and, to a lesser extent, rhodopsin, recoverin, and phosducin.
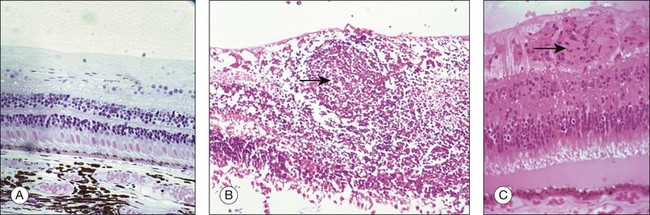
S-antigen
S-antigen, a 48-kDa protein also referred to as arrestin, is one of the antigens most commonly used to induce EAU.61 It was first identified in the soluble fraction of retinal extracts. As it is the first autoretinal antigen to be implicated in the pathogenesis of uveitis, the sequence and its role in phototransduction have been well characterized.62,63 It is a highly conserved protein found in the retinal photoreceptor cells and in the pinealocyte. The main function of arrestin is to block the interaction of rhodopsin with the G-protein transducin in the phototransduction cascade. Immunization of susceptible animals (such as Lewis rats but not mice) with S-antigen induces a predominantly CD4+ T-cell-mediated inflammatory response in the retina, uveal tract, and pineal gland. Six peptide fragments of S-antigen have been identified as uveitogenic. There are six regions of S-antigen identified as targets of the T-cell epitopes. One major uveitogenic peptide is M (sequence 303–320). Some of the fragments are species-specific in their uveitogenicity.64 Although the demonstration of uveitogenic properties of S-antigen has helped in the understanding of uveitis mechanisms, the relevance to human uveitis needs to be more fully explored. S-antigen has also been implicated in the pathogenesis of uveitis through molecular mimicry. It has been shown that several exogenous (baker’s yeast, Escherichia coli, hepatitis B virus, streptococcal M5 protein, Moloney murine sarcoma virus, and baboon endogenous virus) and endogenous (human leukocyte antigen (HLA) B-derived peptide, tropomyosin) antigens share sequence homology with peptide M of S-antigen.65 Some have been shown to be uveitogenic in the EAU models, and lymphocytes from animals immunized with M peptide cross-reacted and proliferated when stimulated with peptides derived from some of these exogenous or endogenous antigens. Antistreptococcal monoclonal antibodies were found to recognize several uveitogenic peptides of S-antigen, providing further evidence that immunological mimicry between self and exogenous antigens from an infectious agent may be a potential mechanism in the pathogenesis of uveitis in humans.66
Interphotoreceptor retinoid-binding protein
IRBP can be used to induce EAU in both mouse and rat.67 It is a major protein (1264 amino acid residues) of the interphotoreceptor matrix, functioning as a transporter of retinoids between the retina and RPE. Similar to S-antigen, it is also found in both the eye and the pineal gland, and induction of EAU with IRBP will lead to disease in both locations. Depending on the dose of antigen used and the species of animals, a spectrum of disease ranging from hyperacute to chronic relapsing disease could be induced. The inflammation is targeted at the photoreceptor layer, producing histopathology similar to human lesions seen in uveitis, such as retinal vasculitis, granuloma, focal serous detachments, loss of photoreceptors, and formation of sub-RPE infiltrates similar to Dalen–Fuchs nodules, seen in patients with Vogt–Koyanagi–Harada disease and sympathetic ophthalmia.68 The disease activity shows two peaks at the 5th and 10th weeks after immunization, simulating the relapsing course in human uveitis.69 The relatively long duration of disease activity in the murine IRBP EAU model makes it a good model for evaluation of therapeutic strategies in established disease.
Rhodopsin
Rhodopsin is the visual pigment of rod photoreceptor cells, belonging to the larger family of G-protein-coupled receptors.70 It is a membrane-bound protein comprising a polypeptide chain, the opsin, and a covalently bound chromophore, 11 cis-retinal. The function of rhodopsin is to capture photons and trigger the phototransduction cascade. The uveitis induced in susceptible animals such as guinea pigs, rabbits, rats, and monkeys is typically a retinitis with multifocal destruction of photoreceptor cells, showing dense mononuclear and polymorphonuclear cell infiltrates in the retina and anterior uvea in severe disease.71 There are three distinct immunopathogenic sites on the molecule: sequence 230–250 (extracellular loop V-VI) is the most uveitogenic, comparable to S-antigen and IRBP.72,73 The role of rhodopsin autoantigens in human diseases is still unclear. Studies in patients with normal-pressure glaucoma have demonstrated an elevated antirhodopsin antibody titer, indicating the possibility of its role in the optic neuropathy observed in these patients.74
Recoverin
Recoverin is a 23-kDa calcium-binding protein present in photoreceptor cells and functioning as a calcium sensor that regulates rhodopsin phosphorylation through inhibition of rhodopsin kinase. It is also found in bipolar cells, pinealocytes, and certain tumor cells. The function in these nonphotoreceptor cells is unknown as they do not contain rhodopsin kinase.75 Immunization of Lewis rats with high doses of recoverin produced EAU similar in severity, histology, and duration to other retinal autoantigens such as S-antigen.76 The major immunopathogenic epitope, sequence 62–81, has been characterized in Lewis rats. It has been identified as an autoantigen in paraneoplastic and degenerative diseases of the retina such as cancer-associated retinopathy.77 High levels of antirecoverin antibodies were detected in some patients with occult cancer and retinal dysfunction. More recently, these autoantibodies have been demonstrated to induce apoptosis of photoreceptors, supporting its pathogenic role in retinal autoimmunity.78
Phosducin
Phosducin is a 33-kDa protein, a cytosolic regulator of G-protein-mediated signaling, found in the retina and also in nonretinal tissues such as liver, lung, heart, and brain.79 Immunization with phosducin in Lewis rats produces mild to moderate EAU characterized by late onset, low-grade severity, and predominantly involving the posterior segment.80 Its role in human disease is unclear.
Retinal autoimmunity
Autoimmunity in human uveitis
More than four decades of work on EAU models using these retinal autoantigens have passed. However, it is still unclear whether there is an association between the occurrence of eye-specific autoantibodies and lymphocyte proliferation response and clinical disease, and exactly what their roles are in the pathogenesis of human uveitis and other potentially immune-mediated diseases like AMD. Lymphocytes reactive to retinal antigens have been demonstrated in normal individuals4 and those with retinal disease.81 In normal individuals, the presence of circulating autoreactive cells suggests that immunoregulatory mechanisms must exist to prevent retinal autoimmunity resulting in disease; in the latter it is tempting to hypothesize the possible roles of these cells in the induction of retinal autoimmunity. Both the humoral and cellular responses against S-antigen and IRBP have been investigated. T-lymphocyte responses to S-antigen, IRBP, and peptides derived from patients with uveitis were investigated in a few clinical studies. An increase in T-lymphocyte responses was reported in patients with Behçet disease during periods of active ocular inflammation.82 In another similar study, a large proportion of the patients had lymphocytes which responded to stimulation with S-antigen, IRBP, and several uveitogenic peptides such as S-antigen peptide M, but no difference was reported in the cellular response between Behçet disease patients with and without uveitis.83 Similar cellular responses to S-antigen, S-antigen-derived peptides (peptides M and G), and uveitogenic peptide derived from IRBP (R16) have been reported for patients with idiopathic uveitis.84
Lymphocyte proliferation responses to 20 peptides spanning the entire sequence of S-antigen were investigated in patients with uveitis in one study. Half of the patients with idiopathic uveitis responded to at least one peptide whereas there was no response to the peptides in the normal controls. The peptide fragments found to elicit more lymphocyte proliferation were 61–80, 81–100, 145–160, 161–180, 221–240, and 241–260.85 Other fragments of IRBP and S-antigen were studied in another similar study involving 82 patients with uveitis. Patients who responded to IRBP or S-antigen also responded to one or two of the fragments but to lesser magnitude. This suggests that these fragments are probably not the primary mediators of the disease.86 Isolation of S-antigen-specific T cells from the eyes of uveitis patients, which were expanded to antigen-specific T-cell lines, was reported.87 A study evaluating oral tolerance by feeding S-antigen to uveitis patients showed a good clinical response and the patients required less immunosuppression.88 Together, these studies provided further support for the possible pathogenic roles of retinal autoantigen in uveitis.
Although both, EAU in animals and uveitis in most humans can be a CD4+ Th1 or a Th17 effector response driving autoimmunity, there are differences in the T-cell specificities in antigen response.89 Patients with uveitis, due to epitope spread, can respond to more than one peptide, i.e., several epitopes are recognized in the same patient, while animals with EAU tend to respond only to the inciting antigen. There is no common pattern of lymphocyte response in clinical studies reported so far. Early clinical studies on the humoral response showed the presence of antiretinal autoantibodies, including anti-S-antigen antibodies in the sera of some uveitis patients90,91 (Fig. 27.3), but there was no good correlation between presence or levels of autoantibodies and clinical disease. Moreover, anti-IRBP antibody was reported to occur in the same frequency in the sera of patients with uveitis and normal controls. The only difference has been the stronger affinity of the antibodies in patients with uveitis.92
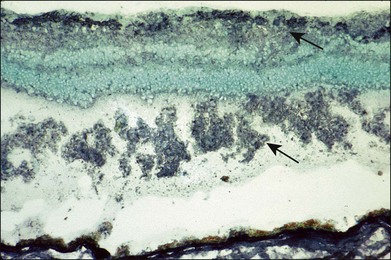
The relationship between humoral and cellular response to these autoantigens was studied in a group of patients with endogenous uveitis such as birdshot retinochoroidopathy, retinal vasculitis, and Behçet disease. The antibody titers to S-antigen and IRBP were found to be decreased during active inflammation periods and the lymphocyte responses to S-antigen were found to be most significant in the period preceding a relapse of ocular inflammation, supporting the pathogenic roles of these lymphocytes in retinal autoimmunity.93 The role of humoral and cellular responses to retinal autoantigens remains unclear and it is possible that different subsets of patients with uveitis, genetically determined, respond to different epitopes and antigens, resulting in the wide spectrum of clinical disease encountered.
Role of retinal autoimmunity in protection
Retinal autoimmunity may not always be pathogenic. Studies on central nervous system (CNS) injury and its reparative process have revealed a possible beneficial role of autoimmunity in limiting self-injury during insults to the CNS. Axonal injury in the CNS leads not only to the degeneration of the affected axon but also the neighboring axon through self-destructive compounds released by the degenerating axon into the microenvironment.94 Studies have revealed that autoreactive T cells directed against myelin antigens have beneficial effects on CNS myelinated axons after a mechanical crush injury95 and it has been proposed that this neuroprotective activity exhibited by autoimmune T cells may be a physiological process which the body develops to cope with stressful conditions.96 The observation seen in the CNS could be extrapolated to the retina. Supporting this are the findings of a study demonstrating that vaccination with peptides derived from IRBP resulted in protection of retinal ganglion cells from glutamate-induced death or death as a consequence of optic nerve injury.7 It appears that the immune system not only protects the body against invading pathogens but also protects it from toxic substances released by self tissues during trauma and stress. Therefore the autoreactive cells that induce neuroprotection and those that induce autoimmune disease may share the same specificity and phenotype, indicating their potential to be protective and destructive at the same time.97 The final outcome depends on how they are regulated. Correlation between the ability to manifest an autoimmune response with a beneficial outcome and the ability to resist the development of an autoimmune disease in animals has been reported.98 From these findings, it seems clear that the ability to protect the eye from inflammation and injury does not solely depend on mechanisms conferring immune privilege but rather a precise regulation of autoimmunity.
Role of retinal autoimmunity in infection
The role of infection in the pathogenesis of eye autoimmunity has long been suspected but there is no conclusive study to demonstrate it. The possible causative relationship between infections and ocular autoimmunity such as streptococcus, herpesvirus, and Behçet disease,99 Gram-negative bacteria, HLA B27-associated uveitis,100 herpesviruses, and serpiginous choroiditis101 has been reported. Infection as an exogenous trigger for inflammation in the eye is an enticing possibility as infections have been linked to the pathogenesis of many autoimmune diseases such as rheumatic fever,102 inflammatory bowel diseases,103 and sarcoidosis.104 Antiretinal antibodies in the serum of patients with toxoplasmosis retinochoroiditis,105 antiretinal and anti-RPE antibodies in murine coronavirus retinopathy and degeneration106 were reported. These reports provided support for a role of infection in retinal autoimmunity. Even in the current EAU models in animals, bacteria-derived products like mycobacteria in complete Freund’s adjuvant, lipopolysaccharides, and pertussis toxin are used to promote the development of autoimmune disease.107 An infection could possibly play a similar role, in sensitizing the immune system to eye-restricted antigens, which are “sequestrated” or expressed in low levels below the immune activation threshold. The mechanisms involved in the pathogenesis of uveitis may be similar to that of the adjuvant in EAU, including changes in the blood–tissue barrier to allow infiltration of inflammatory cells,108 promotion of Th1 response,109 stimulatory effects on the APCs,110 and enhancement of innate immune response.111 On the other hand, there is evidence that immunologic mimicry may play a role in pathogenesis as molecular homology exists between certain retinal autoantigens and peptides derived from microbes, such as the M peptide derived from S-antigen and the streptococcal cell wall peptide, the yeast’s histone peptide, and certain viral agents.66,112,113 Molecular mimicry has also been suspected of playing a role in conferring disease susceptibility in patients with certain HLA haplotypes, like HLA B27 for acute anterior uveitis,114 HLA B51 for Behçet’s disease,115 and HLA-A29 for birdshot retinochoroidopathy.116,117 Synthetic peptides derived from HLA-B molecules such as B27 and B51 with amino acid sequence homologous with S-antigen have been reported to induce EAU in animals.118 Lymphocytes from patients with uveitis, especially Behçet’s disease, respond to both peptides derived from HLA-B molecules and peptides derived from S-antigen, demonstrating possible immunologic mimicry.119 In addition, further studies also showed sequence homology between these peptides, rotavirus, and casein protein, a protein found in milk, and cross-reaction in cellular immunity between these peptides was found in animal studies.120 Together these studies bring forward the possibility of immunologic mimicry for exogenous factors like infection or foreign proteins breaking down tolerance and inducing autoimmunity.
The guardian that prevents pathologic autoimmunity is immune tolerance. Immune tolerance, usually maintained by mechanisms described earlier, could be lost or modulated. This was demonstrated in a study showing that heat shock protein (HSP)91 can modulate APC function, resulting in loss of tolerance in an animal model.121 HSPs, especially microbial, have been implicated in the pathogenesis of Behçet’s disease122 and demonstrated to induce inflammation in the eye in animal studies.123 Therefore, one may speculate that an infection and the subsequent release of microbial HSPs could be the initial triggering event, resulting in changes of APC function and a subsequent loss of tolerance, leading to autoimmunity. These notions remain as hypotheses for uveitis since it is a heterogeneous disease, and evidence of the initial triggering event is usually difficult to identify due to the protracted course and the delay between trigger and disease presentation. Furthermore, a disease could have multiple triggering mechanisms and different triggers may lead to the same disease, making clinical data difficult to interpret.
1 Van Dooremaal JC. Die Entwicklung der in fremden Grund versetzten lebenden Gewebe. Graefes Arch Ophthalmol. 1873;19:359–373.
2 Taylor AW. Ocular immunosuppressive microenvironment. Chem Immunol. 1999;73:72–89.
3 Cunha-Vaz J, Faria de Abreu JR, Campos AJ. Early breakdown of the blood–retinal barrier in diabetes. Br J Opthalmol. 1975;59:649–656.
4 Wallow IH, Engerman RL. Permeability and patency of retinal blood vessels in experimental diabetes. Invest Ophthalmol Vis Sci. 1977;16:447–461.
5 Nussenblatt RB, Liu B, Li Z. Age-related macular degeneration: an immunologically driven disease. Curr Opin Invest Drugs. 2009;10:434–442.
6 Yamamoto JH, Minami M, Inaba G, et al. Cellular autoimmunity to retinal specific antigens in patients with Behçet’s disease. Br J Ophthalmol. 1993;77:584–589.
7 Mizrahi T, Hauben E, Schwartz M. The tissue-specific self-pathogen is the protective self-antigen: the case of uveitis. J Immunol. 2002;169:5971–5977.
8 Davis MM. T cell receptor gene diversity and selection. Annu Rev Biochem. 1990;59:475–496.
9 Romagnani S, Maggi E, Liotta F, et al. Properties and origin of human Th17 cells. Mol Immunol. 2009;47:3–7.
10 Amadi-Obi A, Yu CR, Liu X, et al. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–718.
11 Croft M, Dubey C. Accessory molecule and co-stimulation requirements for CD4 T cell response. Crit Rev Immunol. 1997;17:89–118.
12 Schatz DG, Oettinger MA, Schlissel MS.V. D)J recombination. Annu Rev Immunol. 1992;10:359–383.
13 Sprent J. Central tolerance of T cells. Int Rev Immunol. 1995;13:95–105.
14 Arnold B, Schönrich G, Hämmerling GJ. Multiple levels of peripheral tolerance. Immunol Today. 1993;14:12–14.
15 Moudgil KD, Ametani A, Grewal IS, et al. Processing of self-proteins and its impact on shaping the T cell repertoire, autoimmunity and immune regulation. Int Rev Immunol. 1993;10:365–377.
16 Sinclair NR, Anderson CC. Co-stimulation and co-inhibition: equal partners in regulation. Scand J Immunol. 1996;43:597–603.
17 Sakaguchi S, Miyara M, Costantino CM, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500.
18 Van Parijs L, Abbas AK. Role of Fas-mediated cell death in the regulation of immune responses. Curr Opin Immunol. 1996;8:355–361.
19 Griffith TS, Ferguson TA. The role of FasL-induced apoptosis in immune privilege. Immunol Today. 1997;18:240–244.
20 De Vries JE. Immunosuppressive and antiinflammatory properties of interleukin 10. Ann Med. 1995;27:537–541.
21 Sosroseno W, Herminajeng E. The immunoregulatory roles of transforming growth factor beta. Br J Biomed Sci. 1995;52:142–148.
22 Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37:2378–2389.
23 Tommasini A, Ferrari S, Moratto D, et al. X-chromosome inactivation analysis in a female carrier of FOXP3 mutation. Clin Exp Immunol. 2002;130:127–130.
24 Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164.
25 Ehrlich P. Das Sauerstoffbedürfnis des Organismus, eine farbenanalytische Studie. Berlin: Hirschwald; 1885.
26 Goldmann EE. Vitalfärbung am Zentralnervensystem. Abh Preuss Akd Wiss Phys – Math Kl / 1. 1913:1–3.
27 Schnaudigel O. Die Vitalfarbung mit Trypanblau am Auge. Graefes Arch Ophthalmol. 1913;86:93–97.
28 Cunha-Vaz JG, Skakib M, Ashton N. Studies on the permeability of the blood–retinal barrier. I. On the existence, development, and site of a blood–retinal barrier. Br J Ophthalmol. 1966;50:441–453.
29 Runkle EA, Antonetti DA. The blood–retinal barrier: structure and functional significance. Methods Mol Biol. 2011;686:133–148.
30 Toda R, Kawazu K, Oyabu M, et al. Comparison of drug permeabilities across the blood–retinal barrier, blood–aqueous humor barrier, and blood–brain barrier. J Pharm Sci. 2011;100:3904–3911.
31 Haurigot V, Villacampa P, Ribera A, et al. Increased intraocular insulin-like growth factor-I triggers blood–retinal barrier breakdown. J Biol Chem. 2009;284:22961–22969.
32 Rangasamy S, Srinivasan R, Maestas J, et al. A potential role for angiopoietin 2 in the regulation of the blood–retinal barrier in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(6):3784–3791.
33 Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16(3):438–440.
34 Bringman A, Reichenbach A, Wiedemann P. Pathomechanisms of cystoid macular edema. Ophthalmic Res. 2004;36:241–249.
35 Medawar P. Immunity to homologous grafted skin. III. The date of skin homografts transplanted to the brain, to subcutaneous tissue and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69.
36 Niederkorn J, Streilein JW, Shadduck JA. Deviant immune responses to allogeneic tumors injected intracamerally and subcutaneously in mice. Invest Ophthalmol Vis Sci. 1980;20:355–363.
37 Niederkorn JY, Streilein JW. Immunogenetics basis for immunologic privilege in the anterior chamber of the eye. Immunogenetics. 1981;13:27–236.
38 Wenkel H, Streilein JW. Analysis of immune deviation elicited by antigens injected into the sub-retinal space. Invest Ophthalmol Vis Sci. 1998;39:1823–1834.
39 Jiang LQ, Jorquera M, Streilein JW. Subretinal space and vitreous cavity as immunologically privileged sites for retinal allografts. Invest Ophthalmol Vis Sci. 1993;34:3347–3354.
40 Streilein JW. Immune privilege and the cornea. In: Pleyes U, Hartmann C, Sterry W, editors. Proceedings of symposium on bullous oculo-muco-cutaneous disorders. Buren: Aeolus Press; 1997. p. 43–52.
41 Egan RM, Yorkey C, Black R, et al. Peptide-specific T cell clonal expansion in vivo following immunization in the eye, an immune-privileged site. J Immunol. 1996;157:2262–2271.
42 McMenamin PG, Forrester JV. Dendritic cells in the central nervous system and eye and their associated supporting tissues. In: Lotze MY, Thomson AW. Dendritic cells: biology and clinical applications. San Diego: Academic Press; 1999:205–254.
43 Zhang J, Wu GS, Ishimoto S, et al. Expression of major histocompatibility complex molecules in rodent retina: immunohistochemical study. Invest Ophthalmol Vis Sci. 1997;38:1848–1857.
44 Caspi RR, Roberge FG, Nussenblatt RB. Organ-resident, nonlymphoid cells suppress proliferation of autoimmune T-helper lymphocytes. Science. 1987;237:1029–1032.
45 Nussenblatt RB, Byrnes G, Sen HN, et al. A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina. 2010;30:1579–1587.
46 Nussenblatt RB, Ferris F, 3rd. Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol. 2007;144:618–626.
47 Stein-Streilein J, Streilein JW. Anterior chamber associated immune deviation (ACAID): regulation, biological relevance, and implications for therapy. Int Rev Immunol. 2002;21:123–152.
48 Kaplan HJ, Streilein JW. Do immunologically privileged sites require a functioning spleen? Nature. 1974;251:553–554.
49 Takeuchi M, Alard P, Streilein JW. TGF-beta promotes immune deviation by altering accessory signals of antigen-presenting cells. J Immunol. 1998;160:1589–1597.
50 Wilbanks GA, Streilein JW. Distinctive humoral immune responses following anterior chamber and intravenous administration of soluble antigen. Evidence for active suppression of IgG2-secreting B lymphocytes. Immunology. 1990;71:566–572.
51 Ferguson TA, Stuart PM, Herndon JM, et al. Apoptosis, tolerance, and regulatory T cells – old wine, new wineskins. Immunol Rev. 2003;193:111–123.
52 Wahlsten JL, Gitchell HL, Chan CC, et al. Fas and Fas ligand expressed on cells of the immune system, not on the target tissue, control induction of experimental autoimmune uveitis. J Immunol. 2000;165:5480–5486.
53 Kawashima H, Yamagami S, Tsuru T, et al. Anterior chamber inoculation of splenocytes without Fas/Fas-ligand interaction primes for a delayed-type hypersensitivity response rather than inducing anterior chamber-associated immune deviation. Eur J Immunol. 1997;27:2490–2494.
54 Apte RS, Niederkorn JY. Isolation and characterization of a unique natural killer cell inhibitory factor present in the anterior chamber of the eye. J Immunol. 1996;156:2667–2673.
55 Sohn JH, Kaplan HJ, Suk HJ, et al. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest Ophthalmol Vis Sci. 2000;41:3492–3502.
56 Sohn JH, Kaplan HJ, Suk HJ, et al. Complement regulatory activity of normal human intraocular fluid is mediated by MCP, DAF, and CD59. Invest Ophthalmol Vis Sci. 2000;41:4195–4202.
57 Koevary SB, Beaudry K. Effect of anterior chamber-associated immune deviation (ACAID) on rat islet allograft rejection. Ocul Immunol Inflamm. 2000;8:39–47.
58 Wacker WB, Lipton MM. Experimental allergic uveitis. II. Serologic and hypersensitive responses of the guinea pig following immunization with homologous retina. J Immunol. 1968;101:157–165.
59 Nishimura Y, Yamada K, Senju S, et al. Negi identification of a novel autoantigen UACA in patients with panuveitis. Biochem Biophys Res Commun. 2001;280:1169–1176.
60 Sen NH, Chan CC, Caruso RC, et al. Waldenström’s macroglobulinemia-associated retinopathy. Ophthalmology. 2004;111:535–539.
61 Singh VK, Nussenblatt RB, Donoso LA, et al. Identification of a uveitopathogenic and lymphocyte proliferation site in bovine S-antigen. Cell Immunol. 1988;115:413–419.
62 Hirose S, Singh VK, Donoso LA, et al. An 18-mer peptide derived from the retinal S antigen induces uveitis and pinealitis in primates. Clin Exp Immunol. 1989;77:106–111.
63 Merryman CF, Donoso LA, Zhang XM, et al. Characterization of a new, potent, immunopathogenic epitope in S-antigen that elicits T cells expressing V beta 8 and V alpha 2-like genes. J Immunol. 1991;146:75–80.
64 Gregerson DS, Fling SP, Obritsch WF, et al. A new perspective of S-antigen from immunochemical analysis. Curr Eye Res. 1990;9(Suppl.):145–153.
65 Shinohara T, Singh VK, Tsuda M, et al. S-antigen: from gene to autoimmune uveitis. Exp Eye Res. 1990;50:751–757.
66 Lerner MP, Donoso LA, Nordquist RE, et al. Immunological mimicry between retinal S-antigen and group A streptococcal M proteins. Autoimmunity. 1995;22:95–106.
67 Broekhuyse RM, Winkens HJ, Kuhlmann ED. Induction of experimental autoimmune uveoretinitis and pinealitis by IRBP. Comparison to uveoretinitis induced by S-antigen and opsin. Curr Eye Res. 1986;5:231–240.
68 Caspi RR, Roberge FG, Chan CC, et al. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol. 1988;140:1490–1495.
69 Chan CC, Caspi RR, Ni M, et al. Pathology of experimental autoimmune uveoretinitis in mice. J Autoimmun. 1990;3:247–255.
70 Hargrave PA, McDowell JH. Rhodopsin and phototransduction. Int Rev Cytol. 1992;137B:49–97.
71 Adamus G, Arendt A, Hargrave PA. Genetic control of antibody response to bovine rhodopsin in mice: epitope mapping of rhodopsin structure. J Neuroimmunol. 1991;34:89–97.
72 Adamus G, Schmied JL, Hargrave PA, et al. Induction of experimental autoimmune uveitis with rhodopsin synthetic peptides in Lewis rats. Curr Eye Res. 1992;11:657–667.
73 Adamus G, Ortega H, Witkowska D, et al. Recoverin: a potent uveitogen for the induction of photoreceptor degeneration in Lewis rats. Exp Eye Res. 1994;59:447–455.
74 Romano C, Barett DA, Li Z, et al. Antirhodopsin antibodies in sera from patients with normal-pressure glaucoma. Invest Ophthalmol Vis Sci. 1995;36:1968–1975.
75 Kawamura S, Cox JA, Nef P. Inhibition of rhodopsin phosphorylation by non-myristoylated recombinant recoverin. Biochem Biophys Res Commun. 1994;203:121–127.
76 Gery I, Chanaud NP, III., Anglade E. Recoverin is highly uveitogenic in Lewis rats. Invest Ophthalmol Vis Sci. 1994;35:3342–3345.
77 Adamus G, Guy J, Schmied JL, et al. Role of anti-recoverin autoantibodies in cancer-associated retinopathy. Invest Ophthalmol Vis Sci. 1993;34:2626–2633.
78 Adamus G. Autoantibody-induced apoptosis as a possible mechanism of autoimmune retinopathy. Autoimmun Rev. 2003;2:63–68.
79 Danner S, Lohse MJ. Phosducin is a ubiquitous G-protein regulator. Proc Natl Acad Sci U S A. 1996;93:10145–10150.
80 Dua HS, Lee RH, Loeely RN, et al. Induction of experimental autoimmune uveitis by the retinal photoreceptor cell protein, phosducin. Curr Eye Res. 1992;11(Suppl.):107–111.
81 Nussenblatt RB, Gery I, Ballintine EJ, et al. Cellular immune responsiveness of uveitis patients to retinal S-antigen. Am J Ophthalmol. 1980;89:173–179.
82 de Smet MD, Dayan M. Prospective determination of T-cell responses to S-antigen in Behçet’s disease patients and controls. Invest Ophthalmol Vis Sci. 2000;41:3480–3484.
83 Yamamoto JH, Minami M, Inaba G, et al. Cellular autoimmunity to retinal specific antigens in patients with Behçet’s disease. Br J Ophthalmol. 1993;77:584–589.
84 Rajasingh J, Singh VK, Singh V, et al. Cellular immune response to retinal S-antigen and interphotoreceptor retinoid binding protein fragments in idiopathic human uveitis. Ind J Med Res. 1996;103:222–226.
85 Rai G, Saxena S, Kumar H, et al. Human retinal S-antigen: T cell epitope mapping in posterior uveitis patients. Exp Mol Pathol. 2001;70:140–145.
86 de Smet MD, Yamamoto JH, Mochizuki M, et al. Cellular immune responses of patients with uveitis to retinal antigens and their fragments. Am J Ophthalmol. 1990;110:135–142.
87 Nussenblatt RB, Palestine AG, El-Saied M, et al. Long-term antigen specific and non-specific T-cell lines and clones in uveitis. Curr Eye Res. 1984;3:299–305.
88 Nussenblatt RB, Gery I, Weiner HL, et al. Treatment of uveitis by oral administration of retinal antigens: results of a phase I/II randomized masked trial. Am J Ophthalmol. 1997;123:583–592.
89 Luger D, Silver PB, Tang J, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810.
90 Chan CC, Palestine AG, Nussenblatt RB, et al. Anti-retinal auto-antibodies in Vogt–Koyanagi–Harada syndrome, Behçet’s disease, and sympathetic ophthalmia. Ophthalmology. 1985;92:1025–1028.
91 Doekes G, Luyendijk L, Gerritsen MJ, et al. Anti-retinal S-antigen antibodies in human sera: a comparison of reactivity in ELISA with human or bovine S-antigen. Int Ophthalmol. 1992;16:147–152.
92 Hoekzema R, Hwan SB, Rothova A, et al. Serum antibody response to human and bovine IRBP in uveitis. Curr Eye Res. 1990;9:1177–1183.
93 Jobin D, Thillaye B, de Kozak Y, et al. Severe retinochoroidopathy: variations of humoral and cellular immunity to S-antigen in a longitudinal study. Curr Eye Res. 1990;9(Suppl.):91–96.
94 Liu D, Yang R, Yan X, et al. Hydroxyl radicals generated in vivo kill neurons in the rat spinal cord: electrophysiological, histological, and neurochemical results. J Neurochem. 1994;62:37–44.
95 Hauben E, Nevo U, Yoles E, et al. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet. 2000;355:286–287.
96 Yoles E, Hauben E, Palgi O, et al. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci. 2001;21:3740–3748.
97 Kipnis J, Mizrahi T, Yoles E, et al. Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol. 2002;130:78–85.
98 Kipnis J, Yoles E, Schori H, et al. Neuronal survival after CNS insult is determined by a genetically encoded autoimmune response. J Neurosci. 2001;21:4564–4571.
99 Zouboulis CC, May T. Pathogenesis of Adamantiades–Behçet’s disease. Med Microbiol Immunol (Berl). 2003;192:149–155.
100 Sahly H, Podschun R, Kekow J, et al. Humoral immune response to Klebsiella capsular polysaccharides in HLA-B27-positive patients with acute anterior uveitis and ankylosing spondylitis. Autoimmunity. 1998;28:209–215.
101 Priya K, Madhavan HN, Reiser BJ, et al. Association of herpesviruses in the aqueous humor of patients with serpiginous choroiditis: a polymerase chain reaction-based study. Ocul Immunol Inflamm. 2002;10:253–261.
102 Guilherme L, Kalil J. Rheumatic fever: the T cell response leading to autoimmune aggression in the heart. Autoimmun Rev. 2002;1:261–266.
103 Cumming JH, Macfarlane GT, Macfarlane S. Intestinal bacteria and ulcerative colitis. Curr Issues Intest Microbiol. 2003;4:9–20.
104 Ishige I, Usui Y, Takemura T, et al. Quantitative PCR of mycobacterial and propionibacterial DNA in lymph nodes of Japanese patients with sarcoidosis. Lancet. 1999;354:120–123.
105 Whittle RM, Wallace GR, Whiston RA, et al. Human antiretinal antibodies in toxoplasma retinochoroiditis. Br J Ophthalmol. 1998;82:1017–1021.
106 Hooks JJ, Percopo C, Wang Y, et al. Retina and retinal pigment epithelial cell autoantibodies are produced during murine coronavirus retinopathy. J Immunol. 1993;151:3381–3389.
107 Sasamoto Y, Kawano YI, Bouligny R, et al. Immunomodulation of experimental autoimmune uveoretinitis by intravenous injection of uveitogenic peptides. Invest Ophthalmol Vis Sci. 1992;33:2641–2649.
108 Linthicum DS, Munoz JJ, Blaskett A. Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell Immunol. 1982;73:299–310.
109 Silver PB, Chan CC, Wiggert B, et al. The requirement for pertussis to induce EAU is strain-dependent: B10.RIII, but not B10.A mice, develop EAU and Th1 responses to IRBP without pertussis treatment. Invest Ophthalmol Vis Sci. 1999;40:2898–2905.
110 Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund’s adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol. 2002;169:117–125.
111 Agarwal RK, Sun SH, Su SB, et al. Pertussis toxin alters the innate and the adaptive immune responses in a pertussis-dependent model of autoimmunity. J Neuroimmunol. 2002;129:133–140.
112 Lerner MP, Donoso LA, Nordquist RE, et al. Immunological mimicry between retinal S-antigen and group A streptococcal M proteins. Autoimmunity. 1995;22:95–106.
113 Singh VK, Kalra HK, Yamaki K, et al. Molecular mimicry between a uveitopathogenic site of S-antigen and viral peptides: induction of experimental autoimmune uveitis in Lewis rats. J Immunol. 1990;144:1282–1287.
114 Suhler EB, Martin TM, Rosenbaum JT. HLA-B27-associated uveitis: overview and current perspectives. Curr Opin Ophthalmol. 2003;14:378–383.
115 Mizuki N, Inoko H, Mizuki N, et al. Human leukocyte antigen serologic and DNA typing of Behçet’s disease and its primary association with B51. Invest Ophthalmol Vis Sci. 1992;33:3332–3340.
116 LeHoang P, Ozdemir N, Benhamou A, et al. HLA-A29.2 subtype associated with birdshot retinochoroidopathy. Am J Ophthalmol. 1992;113:33–35.
117 Barnett LA, Fujinami RS. Molecular mimicry: a mechanism for autoimmune injury. FASEB J. 1992;6:840–844.
118 Wildner G, Thurau SR. Cross-reactivity between an HLA-B27-derived peptide and a retinal autoantigen peptide: a clue to major histocompatibility complex association with autoimmune disease. Eur J Immunol. 1994;24:2579–2585.
119 Wildner G, Diedrichs-Mohring M, Thurau SR. Induction of arthritis and uveitis in Lewis rats by antigenic mimicry of peptides from HLA-B27 and cytokeratin. Eur J Immunol. 2002;32:299–306.
120 Wildner G, Diedrichs-Mohring M. Autoimmune uveitis induced by molecular mimicry of peptides from rotavirus, bovine casein and retinal S-antigen. Eur J Immunol. 2003;33:2577–2587.
121 Millar DG, Garza KM, Odermatt B, et al. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat Med. 2003;9:1469–1476.
122 Zierhut M, Mizuki N, Ohno S, et al. Immunology and functional genomics of Behçet’s disease. Cell Mol Life Sci. 2003;60:1903–1922.
123 Lehner T, Bergmeier LA, Wang Y, et al. Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. Eur J Immunol. 2000;30:594–603.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 27 Blood–Retinal Barrier, Immune Privilege, and Autoimmunity
Introduction
More than 100 years ago a fascinating discovery was made. Different tissue samples were placed into the anterior chamber of dogs or rabbits and were then observed for 4 months. Surprisingly, the induced intraocular inflammation was very limited. Most of the tissue samples were not rejected as they were known to occur from other parts of the body. At this time, the eye as one of the first sites of immune privilege was recognized.1
Today, we know that the eye is a unique and complex organ that has developed multiple mechanisms to protect itself against frivolous immunological attacks and inflammation in order to maintain its delicate structure and function. The blood–ocular barrier system is formed by the blood–aqueous barrier and the inner and outer blood–retinal barrier. It restrictively limits the passage of ions and proteins to maintain homeostasis and to keep tissue-specific antigens sequestrated. Furthermore, many active factors have been discovered in the eye that contribute to a downregulatory immune environment (DIE).2 The blood–ocular barrier and the DIE are mechanisms that depend on each other’s proper function. They are central to preserving the immune privilege of the eye. Immune privilege has served well to curb excessive inflammation and preserve function. However, this privilege may be lost under certain conditions and situations, resulting in retinal autoimmunity and ocular inflammation. A dysfunction of the inner blood–retinal barrier initiates diabetic retinopathy.3,4 A breakdown of the blood–retinal barrier can also be found in cystoid macular edema, while a loss of the DIE is essential to many forms of uveitis and in all probability to the development of age-related macular degeneration (AMD).5
For a long time, retinal autoimmunity has been perceived as pathogenic, and active suppression of retinal immunity was presumed necessary to maintain the health of the eye. However, studies have shown the presence of retinal autoantibodies in normal controls,6 and constitutive expression of proinflammatory ligands have been found in the normal retina in high concentration (B Kim, M Pillai, Z Li, unpublished data). Moreover, animal optic nerve injury studies revealed possible beneficial roles of retinal autoimmunity in controlling collateral damage to the retinal ganglion cells.7 Thus, retinal autoimmunity can be viewed as a “double-edged sword,” with both protective and destructive effects. To understand retinal autoimmunity, we first have to understand basic concepts in immunology and how some of these immunological components are at work to maintain the eye’s immune privilege.
Basic concepts of immunology
Adaptive immunity
In contrast, adaptive immunity, as the name implies, is antigen-specific, adaptive, transferable, and has other cardinal features like immunologic memory and tolerance. Adaptive immunity has evolved and developed novel molecules like the antibody and the T-cell receptor (TCR), which come in almost limitless varieties to recognize most molecules of biological interest, including both self and nonself. These molecules, developed in the absence of exogenous stimulation, have unique configurations that confer specificity in antigen recognition. The key cellular components of adaptive immunity are the T and B lymphocytes which possess unique receptors for recognizing billions of different antigenic epitopes.8 Each clone of lymphocyte expresses molecularly identical receptors on the surface; hence, in order to recognize billions of different epitopes, there are billions of unique lymphocytes. Upon maturation in the thymus (T cells) and the bone marrow (B cells), cells remain quiescent in G0 of the cell cycle until they encounter the complementary antigen of sufficient affinity to their receptors. Binding with the receptors in the presence of appropriate costimulation is the initiating event to trigger an immune response targeting elimination of the complementary molecule. If this molecule is pathogen-derived, activation will lead to the elimination of the pathogen, but if the molecule is self-derived then activation will potentially lead to autoimmunity, tissue injury, disease, and the destruction of host tissue. Activation of naive lymphocytes results in the emergence of clones of lymphocytes through proliferation, each with a unique receptor to recognize one epitope of the inciting antigen. The cytokine environment during activation of naive T cells is responsible for differentiation into three major types of effector T cells. Interferon (IFN)-γ leads to Th1 cell production, while interleukin (IL)-4 is responsible for Th2 production. Some Th1 and Th2 cell clones will become memory cells which are responsible for the enhanced immune response if the antigen is re-encountered. The rest of Th1 will secrete cytokines targeted at macrophages and other cells mediating cellular immunity. By contrast, Th2 will stimulate B cells into proliferation and antibody production, mediating humoral immunity. Th17 are a recently discovered family of T helper cells that are capable of producing IL-17. IL-1 and IL-23 seem to be needed for human Th17 differentiation.9 Th17 play an important role in combatting various bacterial and fungal species by producing IL-23. In contrast, large amounts of Th17 can be found in autoimmune diseases like uveitis, multiple sclerosis, and rheumatoid arthritis.10
Both immune and accessory cells, including those of the innate immunity system, are activated to eliminate pathogens. In this way, adaptive immunity serves to enhance immune protection through better-coordinated and more specific attacks using innate immunity mechanisms. Within the genome lies the ability to create antibodies and TCR for antigens that bind to self antigens with high affinity.11 Hence, the threat of autoimmunity is inborn and regulation is crucial. The ability to distinguish between foreign and self, and the ability to regulate autoimmunity, are critical for survival.
The immune response can be seen as analogous to the neural reflex arc, which has an afferent limb, a central process, and an efferent limb. In the immune afferent limb, antigens are captured, processed, transported, and finally presented to the lymphocytes. As the naive lymphocytes can only be activated in the organized lymph tissues in secondary lymphoid organs such as lymph nodes, spleen, tonsils, and Peyer’s patches, it needs specialized cells to execute the immune afferent limb. These cells are professional antigen-presenting cells (APCs). Dendritic cells and macrophages, both bone marrow-derived, have APC functions serving to capture antigens through phagocytosis and endocytosis, process the antigens, and present the processed antigen in conjunction with special major histocompatibility complex (MHC) molecules on their cell surface. In addition, the APCs provide the necessary costimulation needed for lymphocyte activation by upregulating an array of surface molecules (CD80, CD86, intercellular adhesion molecule-1, lymphocyte function-associated molecule-3, and CD40) that function as ligands for receptors expressed by the lymphocytes. They also secrete cytokines such as IL-12, IL-6, IL-10, and IL-1β serving similar costimulation functions. Costimulation is required as the second signal for full activation of the lymphocytes, independent of the first signal, which is the antigen and the lymphocyte receptor engagement.12 There are differences in antigen presentation to B and T lymphocytes. For B lymphocytes, the receptor, which is a surface-bound antibody, can engage a naive antigen directly while TCRs can only recognize peptide fragments presented on special surface molecules (MHC classes I and II). MHC class I, which is present in most cells, presents peptides derived from protein degradation in the cytoplasm (intracellular antigens such as viral and intracellular microbe products), while MHC class II molecules, which are present in APCs and lymphocytes, present peptides from phagocytic vesicles (extracellular antigens from the microenvironment).
Immune regulation
The immune system can be regulated at any part of the immune response loop, i.e., afferent limb, central process, and efferent limb. Regulation of the autoimmune response can be via any of the following mechanisms: central tolerance – clonal deletion and peripheral tolerance – clonal anergy, T-cell suppression, immune deviation, immunologic ignorance, and antigen sequestration. Since the genome has the capability to generate both self- and nonself-recognizing antibodies and TCRs, mechanisms must be in place to contain and prevent the activation of self-reactive T cells. During lymphocyte development, both T and B cells with receptors that recognize self molecules with high affinity are clonally deleted via apoptosis (central tolerance).13 However the process is not foolproof, as autoreactive T and B cells in the periphery exist even in normal individuals. Many tissue-specific antigens, like the eye-restricted molecules, may not be expressed in the thymus or cells may have escaped the selection process by the central tolerance mechanism in the thymus without encountering the specific self antigen. Hence, the potential for induction and expression of autoimmunity still exists, and mechanisms to contain these autoreactive immune cells are crucial to prevent autoimmunity. Evidence for peripheral tolerance can be found in all phases of the immune response, i.e., afferent limb, central process, and efferent limb.14,15 The afferent limb depends heavily on the functional properties of the APCs which offer many opportunities for modulation. First, the antigen can be sequestrated and prevented from contact with the APCs by physical barriers or by rendering APCs incapable of antigen capture in the microenvironment (sequestration). Second, the ability of the APC to degrade, process, and express the antigens can be inhibited. Third, the antigen-bearing APCs may be prevented from migrating to the lymphoid tissues. Lastly, the ability of the APCs to generate the costimulatory signal can be inhibited or different costimulatory signals may be produced (immune deviation).
Regulation of APC function can be seen as a form of immunologic ignorance where reactive autoimmune cells are present but never have the opportunity to encounter its antigen in a proper immunogenic form due to a deficiency in antigen presentation. In the central processing phase of the immune response, two signals are required for full activation of the lymphocyte. The absence of costimulation in the presence of antigen-specific stimulation renders the T cells unresponsive to further antigen stimulation, even in the presence of costimulatory signals (anergy).16 On the other hand, too much costimulation in the presence of high antigen dose may lead to profound cell activation and apoptosis. Both mechanisms will result in tolerance.
It is also known that APCs from certain immune-privileged sites such as the eye, and APCs in the gut processing orally ingested antigens, may cause some immune cells to develop into immunoregulatory cells like IL-10-secreting Tr1 cells or transforming growth factor-β (TGF-β) secreting Th3 cells. These cells have in common that they are adaptively regulatory. They receive a suppressive potential following specific antigen stimulation in a particular cytokine milieu.17
In the efferent limb, immune regulation can occur via targeting at the level of antigen-specific immune effector cells. One such mechanism would be through Fas ligand – Fas-induced apoptosis of lymphocytes.18,19 The immune-reactive lymphocytes express the CD95 receptor (Fas receptor). On encountering the Fas ligand, which is expressed in immuno-privileged eye tissues such as the cornea and retina, these immune cells undergo apoptosis. Other molecular mechanisms, such as TNF-α production by immune and accessory cells, such as Müller cells and retinal pigment epithelial (RPE) cells during antigen encounter, can also result in apoptosis, thereby deleting these immune-reactive cells. Similar to the central process, effector cells need costimulatory signals to be activated and the expression of inappropriate costimulatory signals such as IL-10 or TGF-β by APCs can lead to anergy or reprogramming of lymphocytes into immunoregulatory cells secreting immunosuppressive cytokines, thereby further suppressing autoimmunity.20,21
An important regulator of immune responses is the CD4+ CD25high forkhead box protein 3 (Foxp3)+ regulatory T cell (Treg). There are two groups of Treg in humans: the naturally occuring Treg and the induced Treg. The natural Treg is developmentally determined in the thymus as a distinct T-cell subpopulation that is specialized for suppressive function.17 It is distinguished from the induced Treg by a demethylated promoter region for Foxp3.22 Foxp3 seems to act as a repressor of transcription that regulates T-cell activation. A dysfunction of Foxp3 results in hyperactivation of T cells and severe autoimmune disease.23 Natural and induced Tregs maintain peripheral tolerance by modifying the functions of other T cells, both CD4+ and CD8+ populations, either directly through T-cell to T-cell interactions, or indirectly through APCs.24 Hence they are crucial for maintaining immune homeostasis and controlling autoimmunity in order to keep the immune system healthy.
Blood–ocular barrier
At the end of the 19th century, the German bacteriologist and immunologist Paul Ehrlich experimented with staining tissues in animal models. When he injected dyes into the bloodstream, almost all tissues were colored intensively. However, the brain remained unstained.25 A few years later, one of Ehrlich’s students, Edwin Goldmann, performed the experiment the other way around, by injecting dyes into the cerebrospinal fluid. This time, the brain became stained and the rest of the body remained dye-free. Goldmann correctly concluded that there was a restrictive barrier mechanism inhibiting the passage of solubles between the two compartments, the blood and the cerebrospinal fluid.26 In the same year, in 1913, Schnaudigel found a similar barrier for trypan blue in the retina of rabbits.27
Today, we know that the eye consists of several anatomical structures that limit flux between the blood and most parts of the eye itself.28 The blood–ocular barrier is represented by two main components: the blood–aqueous barrier and the blood–retinal barrier. The blood–aqueous barrier is formed by the nonpigmented layer of the ciliary body epithelium and by the endothelium of the iridal capillaries. The blood–retinal barrier is located at two levels. The inner barrier is the nonfenestrated capillaries of the retinal vessels, whereas the outer barrier is the tight junctions of the RPE, enabling a high degree of control of fluid permeability.29
Limiting the passage of ions enables the eye to maintain homeostasis in the aqueous humor and in the retina while the barrier also keeps small and large molecules like drugs from entering the eye. Specially hydrophilic substances show a low permeability across the blood–ocular barrier.30 Furthermore, the barrier has an essential immunologic function: the sequestration of tissue-specific antigens is crucial to preserve the immune privilege of the eye, and, therefore, to avoid intraocular inflammation. On the other hand, inflammation can cause breakdown of the blood–retinal barrier, leading to autoimmunity and inflammation, and allowing drugs and other molecules to penetrate into the eye. Considering its essential function it is not surprising that many of the most common eye diseases are directly associated with alterations of the blood–ocular barrier.
Blood–retinal barrier in diabetic retinopathy
The initial disorder causing both nonproliferative and proliferative diabetic retinopathy is chronic hyperglycemia leading to retinal hypoxia. Compensatory mechanisms like vasodilatation are a regulatory attempt to increase retinal blood flow. After exhaustion of autoregulation, a disruption of endothelial tight junctions in the retinal vasculature can occur.4 Furthermore, chronic hyperglycemia seems to be able to induce a hypoxia-mediated expression of growth factors and cytokines in the retinal endothelium, such as insulin-like growth factor-1, vascular endothelial growth factor-A, angiopoietin-2 and TNF-α, leading to leukocyte adhesion, endothelial cell injury, increased permeability of endothelial cells, and finally angiogenesis.31–33 The resulting plasma leakage from the inner blood–retinal barrier may cause diabetic macular edema and consecutive vision loss.
Blood–retinal barrier in cystoid macular edema
Cystoid macular edema is a major reason for vision loss in various ocular diseases. Different conditions such as trauma, inflammation, or vascular degeneration lead to an increased permeability of the blood–retinal barrier. The inner and outer plexiform layers of the retina act as diffusion barriers against continuous fluid distribution. Therefore, a breakdown of the inner blood–retinal barrier causes cysts mainly in the inner nuclear layer, whereas serum leakage from the outer blood–retinal barrier pools into cystic spaces in the Henle fiber layer.34
The eye as an immune-privileged site
The eye has been recognized as an immune-privileged site for more than 100 years. In the 1940s Medawar demonstrated this by showing prolonged, often indefinite, survival of organs or tissue grafts in the anterior chamber of the eye.35 It is now known that this immune privilege is a dynamic process in which immunoregulatory mechanisms combined with anatomical factors maintain the vitality of grafts in privileged sites and of privileged organs and tissues as grafts. Various tissues, including allogenic skin grafts, thyroid tissues, neuronal retinal tissue, and allogenic tumor cells, have been shown to survive in the anterior chamber of the eye for prolonged periods.36 However, this privilege is influenced by the immunogenic strength of the antigens expressed by the cells. Tumors expressing MHC-encoded alloantigens had only a transient extension of survival compared to those that expressed weaker transplantation antigens.37 Although most studies were done placing tissue in the anterior chamber of the eye, a number of studies demonstrated a similar immune privilege when allogenic tumor cells, foreign neuronal retinal tissue, and RPE were placed in the vitreous cavity and the subretinal space.38,39 Certain eye tissues are known to be immune-privileged, i.e., demonstrating an altered immune response and less rejection. Ocular tissues include the cornea, RPE, and probably the retina. When transplanted beneath the capsule of the kidney, a nonimmune-privileged site, cornea demonstrated extended survival compared with other ocular tissues like the conjunctiva.40 Both passive factors, such as the blood–retinal barrier, and active factors, such as the DIE, are essential to establish and maintain the ocular immune privilege (Table 27.1).
Table 27.1 Components contributing to ocular immune privilege
| Passive | |
| Blood–retinal barrier | Nonfenestrated vascular endothelium of the retinal vessels and tight junctions among the retinal pigment epithelium |
| Lack of lymphatic drainage | Absence of lymphatics in the retina – it is present in the choroid |
| Tissue fluid drainage | Tissue fluid drains via the hematogenous route |
| Reduced expression of MHC | Antigen presentation is reduced by reduction of MHC class I and II molecules and APCs. The APCs are altered by the microenvironment to promote immune regulation |
| Reduced APCs in the retina | |
| APCs with altered function | |
| Active | |
| Immunosuppressive | TGF-β, α-MSH, VIP, CGRP, MIF, IL-1 receptor antagonist and free cortisol microenvironment |
| Factors expressed on cell surface | CD95 ligand, CD59, CD55, CD46 |
MHC, major histocompatibility complex; APC, antigen-presenting cell; TGF, transforming growth factor; MSH, melanocyte-stimulating hormone; VIP, vasoactive intestinal peptide; CGRP, calcitonin gene-related peptide; MIF, migration inhibitory factor; IL, interleukin; CD, nomenclature for surface proteins of human leukocytes.
Transportation of antigens
The blood–retinal barrier serves to keep tissue-specific antigens sequestrated. There is an absence of lymphatics in the retina, although there is evidence that antigen may still be able to be transported to the lymph nodes.41 Tissue drainage via the hematogenous route may alter the APC function.42 There is also a reduced expression of MHC class I and II molecules and an absence of bone marrow-derived cells that function within tissues as APCs in the normal retina.43
Downregulatory immune environment
The DIE is an evolutionary adaptation of the eye to protect itself against excessive inflammation. A small focus of inflammation in the eye has a far greater impact than a similar one in the kidney. The posterior segment seems to have several active factors that help to keep the local immunoenvironmental balance in a downregulatory state. Active factors include the downregulatory effect of Müller cells in cell-to-cell contact with lymphocytes.44 Furthermore, there are many other ocular cells that affect DIE (NK T cells, RPE, microglia, F4/80+
[/not-level-membership-for-opthalmology-category]
