[level-membership-for-cardiothoracic-surgery-category]
Chapter 30 Bleeding, Hemostasis, and Transfusion Medicine
The major components of the hemostatic system are the vasculature, platelets, and coagulation proteins. Deficiencies or abnormalities in any of these components can lead to coagulopathy and pathologic bleeding. Almost every imaginable quantitative and qualitative abnormality of the hemostatic system has been described following cardiac surgery, and managing postoperative bleeding remains one of the greatest challenges in caring for patients after cardiac surgery.
In this chapter, issues relating to bleeding and hemostasis in cardiac surgery patients are reviewed. Routine management of postoperative bleeding, including a protocol for blood component therapy, is outlined in Chapter 17.
PHYSIOLOGY AND PATHOPHYSIOLOGY
Normal Hemostasis
The vascular endothelium is a nonthrombogenic surface that secretes prostacyclin, tissue plasminogen activator, heparin sulfate, antithrombin III, protein C, and nitric oxide to inhibit platelet activation, and preserve vascular patency. However, if a blood vessel is cut or otherwise damaged, the subendothelial vascular basement membrane is exposed, and tissue factor and other molecular promoters are released, leading to platelet activation and thrombin generation which, in turn, leads to the formation of fibrin from fibrinogen and the creation of a stable fibrin-platelet clot.1,2
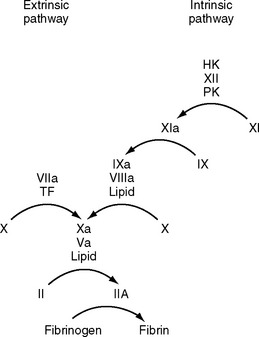
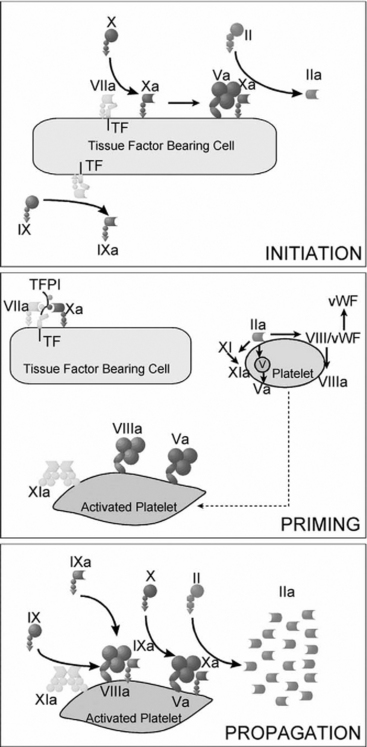
Coagulation is classically represented as two separate pathways that converge and form a final common pathway that leads to thrombin generation (Fig. 30-1). However, this model is derived from in vitro tests of coagulation and does not represent the in vivo state. It does not explain how deficiencies of some “intrinsic” pathway factors (factor XII, high molecular weight kininogen, prekallikrein) do not cause clinical bleeding, whereas deficiencies of other intrinsic factors (factors VIII and IX) do. Further, under normal circumstances hemostasis is initiated by the release of tissue factor by damaged endothelium (i.e., the “extrinsic” pathway), not by contact activation of factor XII (i.e., the intrinsic pathway). Although hemostasis is initiated by tissue factor, the regulation of thrombin generation at the site of endothelial injury is determined almost entirely by platelets. Platelet adhesion is initiated when receptors on platelet membranes bind to the subendothelial collagen by forming a bridge with von Willebrand factor (vWF). Once platelets adhere, they expose factors on their surface that provide a template for initiation of the coagulation cascade and the formation of the hemostatic plug. The cell-based model of coagulation shown in Figure 30-2 describes this central role for platelets in the generation of thrombin. Thrombin cleaves fibrinogen to fibrin monomer, which automatically polymerizes to long-chain fibrin fibers in a few seconds. Over the next few minutes, cross-linking between fibrin strands occurs, forming a stable fibrin mesh that traps platelets and forms a hemostatic plug.
Abnormal Bleeding Following Cardiac Surgery
Excess bleeding following cardiac surgery may reflect inadequate surgical hemostasis. Commonly, however, abnormal blood clotting due to coagulopathy, platelet dysfunction, and excess fibrinolysis contribute to or are the primary causes of postoperative bleeding. The factors that contribute to postoperative bleeding may be categorized as patient-related, intraoperative, and postoperative (Table 30-1).
Table 30-1 Risk Factors for Excessive Postoperative Bleeding in Patients Undergoing Cardiac Surgery
| Patient Variables |
| Advanced age (>70 years) |
| Preoperative anemia |
| Female gender |
| Increased body size |
| Cardiogenic shock, congestive heart failure, or poor left ventricular function |
| Renal insufficiency |
| Peripheral vascular disease |
| Insulin-dependent diabetes mellitus |
| Liver failure or hypoalbuminemia |
| Preoperative sepsis |
| Preoperative antithrombotic therapy |
| High intensity: abciximab, clopidogrel, direct thrombin inhibitors, low molecular weight heparin, fibrinolytic therapy |
| Low intensity: aspirin, dipyridamole, eptifibatide, tirofiban |
| Preoperative coagulopathy |
| Hereditary coagulopathy or platelet defect (e.g., von Willebrand disease, hemophilia A or B) |
| Acquired coagulopathy or platelet defect (e.g., nonspecific prolongation of bleeding time, chronic lymphocytic leukemia, lupus anticoagulant, polycythemia, myelodysplastic syndrome, idiopathic thrombocytopenic purpura, β-thalassemia) |
| Intraoperative and Postoperative Variables |
| Prolonged CPB time |
| Reoperation |
| Type of operation: aortic/complex >valve + CABG >valve >CABG |
| Use of internal mammary artery (either one or two) for CABG surgery |
| Reduced heparin dose |
| Increased protamine dose after CPB |
| Increased cell saver volume |
| Intraoperative autologous donation |
| Need for transfusion while on CPB |
| Low body temperature in ICU |
| Use of hydroxyethyl starch for volume expansion |
| Lack of transfusion algorithm with point-of-care testing |
CABG, coronary artery bypass graft; CPB, cardiopulmonary bypass; ICU, intensive care unit.
Modified from Ferrasis VA, Spiess BD: Perioperative blood transfusion and blood conservation in cardiac surgery: The Society of Thoracic Surgeons and The Society of Cardiovascular Anesthesiologists Practice Guidelines Series. Ann Thorac Surg [in press].
Patient Factors
Platelet Dysfunction and Thrombocytopenia.
Treatment with antiplatelet drugs (aspirin, nonsteroidal antiinflammatory drugs, clopidogrel, and glycoprotein IIb/IIIa inhibitors) is common in patients undergoing cardiac surgery (see Chapter 3). Acquired platelet dysfunction occurs with chronic liver disease and renal failure. A blood urea nitrogen concentration greater than 35 mmol/l (> 100 mg/dl) contributes to postoperative bleeding. Treatment of uremic coagulopathy involves dialysis, desmopressin, and platelets.
Causes of low platelet counts include: (1) heparin-induced thrombocytopenia (see subsequent material); (2) drugs (Table 30-2); (3) splenic sequestration, commonly due to splenomegaly secondary to portal hypertension or tumor infiltration; (4) reduced bone marrow activity (e.g., cytotoxic drugs, fibrosis, or tumor infiltration); (5) HIV/AIDS; and (6) idiopathic thrombocytopenic purpura, which is a condition characterized by immunologic destruction of platelets; it may be acute or chronic, mild or severe. The treatment of severe thrombocytopenia due to idiopathic thrombocytopenic purpura is usually corticosteroids. Patients with chronic cyanosis due to right-to-left intracardiac shunting have increased bleeding tendencies due to thrombocytopenia, platelet dysfunction, and reduced levels of some coagulation factors.
Table 30-2 Drugs Associated With Thrombocytopenia That May Be Used in Patients in the ICU
| Antimicrobials | Analgesics |
|---|---|
| Trimethoprim/sulfamethoxazole | Acetaminophen |
| NSAIDs | |
| Rifampin | H2-receptor antagonists |
| Amphotericin | Ranitidine |
| Vancomycin | Cimetidine |
| Piperacillin | CNS drugs |
| Ampicillin | Haloperidol |
| Cardiovascular drugs | Chlorpromazine |
| Thiazide diuretics | Diazepam |
| Amiodarone | Lithium |
| Quinidine | Carbamazapine |
| Amrinone | Glycoprotein IIb/IIIa antagonists |
| Captopril | |
| Digoxin | Abciximab |
| Nitroglycerine | Eptifibatide |
| Alprenolol | Tirofiban |
| Oxprenolol | |
| Diazoxide | |
| Methyldopa |
Modified from Rizvi MA, Shah SR, Raskob GE, et al: Drug-induced thrombocytopenia. Curr Opin Hematol 6:349-353, 1999. CNS, central nervous system; ICU, intensive care unit; NSAIDs, nonsteroidal antiinflammatory drugs.
Coagulopathy.
Acquired coagulopathy may occur as the result of treatment with heparins, warfarin or, less commonly, thrombin inhibitors (e.g., bivalirudin). Herbal supplements, notably garlic, gingko, and ginseng,3 can also cause coagulopathy. The most important nonpharmacologically acquired coagulopathy is that associated with chronic liver disease, which can lead to deficiency of all coagulation proteins (especially factors I, II, V, VII, IX, and X). Chronic liver disease also causes portal hypertension, which is associated with esophageal varices and splenomegaly (with platelet sequestration). Esophageal varices can rupture, either spontaneously or during instrumentation of the esophagus, causing catastrophic bleeding.
Vitamin K is a fat-soluble vitamin that is required for the hepatic synthesis of factors II, VII, IX, and X. Vitamin K deficiency is caused by poor dietary intake and malabsorption; it often coexists with chronic liver disease, particularly biliary cirrhosis. Coagulopathy may also occur due to sepsis (see subsequent material), most commonly in patients with acute endocarditis. Congenital defects of coagulation, such as hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency), are uncommon.
Intraoperative Factors
Intraoperative factors that contribute to postoperative bleeding include the effects of cardiopulmonary bypass (CPB), the type and duration of the surgical procedure, and the management of anticoagulation. The effects of CPB and surgery on hemostasis are complex.4 CPB and surgical trauma elicit a marked systemic inflammatory response characterized by leukocyte activation, release of inflammatory mediators, and activation of platelets and the coagulation and complement cascades (see Chapter 2). Activation of platelets and coagulation occurs through blood contact with the CPB circuit, through surgical trauma, and through the reinfusion of pericardial blood. Platelets are also activated by heparin and hypothermia. Hemodilution of platelets and coagulation proteins occurs because of the CPB prime solution and other intravenous fluids. Platelets are sequestered onto Dacron grafts. Fibrinolysis is stimulated by hypothermia, contact activation of thrombin and factor XII, and the release of tissue plasminogen activator from the vascular walls. These adverse effects are more pronounced when CPB is prolonged and deep hypothermia is utilized.
Various intraoperative strategies may be employed to reduce bleeding and allogenic (i.e., donor) blood transfusion. They include: (1) antifibrinolytics; (2) heparin-coated CPB circuits; (3) avoidance of deep hypothermia; (4) autologous (i.e., the patient’s) blood donation; (5) normovolemic hemodilution; (6) intraoperative blood recovery and reinfusion through a cell saver. Surgical strategies to reduce postoperative blood loss include meticulous surgical hemostasis and the use of topical agents such as surgical adhesives (e.g., BioGlue) and thrombin preparations (e.g., Thrombostat).
Postoperative Factors
Beyond the early postoperative period, pathologic bleeding is usually related to antithrombotic drugs. Acute gastrointestinal stress ulceration and hemorrhage is the most common nonsurgical cause of bleeding. Stress ulcer prophylaxis is discussed in Chapter 34. Thrombocytopenia is common in patients with critical illness. The causes include: (1) heparin-induced thrombocytopenia (see subsequent material); (2) drugs (see Table 30-2); (3) platelet destruction due to intraaortic balloon counterpulsation, renal replacement therapy, or mechanical cardiac support; (4) sepsis and disseminated intravascular coagulation (see subsequent material); (5) posttransfusion purpura (see subsequent material). Usually, platelet transfusion is required only to treat bleeding or before invasive procedures.
Disseminated Intravascular Coagulation
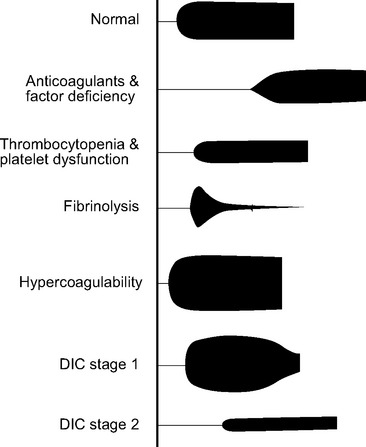
Disseminated intravascular coagulation (DIC) is a generalized disorder of the coagulation system involving the inappropriate activation of platelets, coagulation, and fibrinolysis, resulting in a consumptive coagulopathy and pathologic thrombosis. It is characterized by pathologic bleeding from surgical sites, intravenous cannula sites, and mucosal surfaces and, rarely, by clinically significant thrombosis. Following an initiating event, there are normally two stages of DIC. Stage 1 (early) involves excessive activation of platelets and coagulation proteins, resulting in generalized microvascular thrombosis and obstruction. This obstruction leads to red cell fragmentation and coagulation factor and platelet consumption, resulting in bleeding due to hemostatic factor and platelet depletion (stage 2, late). DIC has multiple causes, including trauma, hemolytic transfusion reaction, obstetric complications and, most important, sepsis. In addition, the consumptive coagulopathy that accompanies prolonged CPB or extracorporeal membrane oxygenation (ECMO) shares many of the hemostatic features of late DIC, namely, low platelet numbers, low fibrinogen, prolonged prothrombin time, prolonged aPTT, elevated D-dimer, and characteristic changes on the thromboelastogram (see Fig. 30-4).5 Management of DIC involves treating the underlying causes and controlling bleeding by means of blood component therapy. In the relatively rare circumstance of clinically important thrombosis, systemic heparinization is indicated.
Prothrombotic States
Important inherited prothrombotic conditions include factor V Leiden (the product of a specific mutation in the factor V gene that confers resistance to factor V cleavage by activated protein C); deficiencies of protein C and protein S (a cofactor for activated protein C); antithrombin III deficiency; and a mutant gene for prothrombin that causes increased levels of the normal protein. Antiphospholipid syndrome and elevated plasma homocysteine are two important acquired prothrombotic conditions. Antiphospholipid syndrome is caused by antibodies (including lupus anticoagulant and anticardiolipin antibodies) directed against cell membrane phospholipids. The term lupus anticoagulant is misleading because patients typically do not have systemic lupus erythematosus and, although the aPTT is elevated, patients have a prothrombotic tendency. Severe systemic illnesses are also associated with an increased risk for pathologic thrombosis. Cancer, major surgery (including cardiac and thoracic surgery), congestive heart failure, myocardial infarction, and stroke all provoke a hypercoagulable state and are risk factors for thromboembolism. High platelet counts and fibrinogen levels, as part of systemic inflammation, also contribute.
Patients with known or suspected hypercoagulability should receive prophylaxis against deep vein thrombosis (DVT) and pulmonary embolism (see Chapter 17) as soon as postoperative bleeding has settled. If an inherited or acquired hypercoagulable state (excluding pregnancy or systemic illness) is suspected, a thrombophilia screen should be obtained.
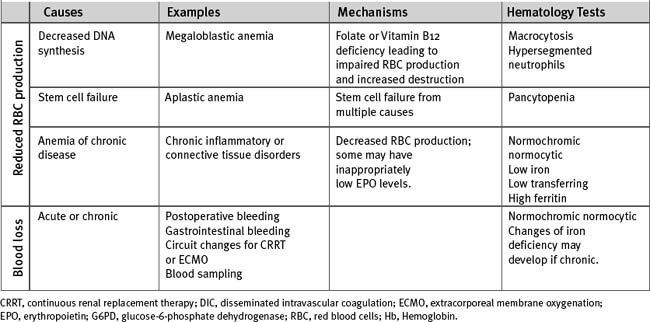
Anemia
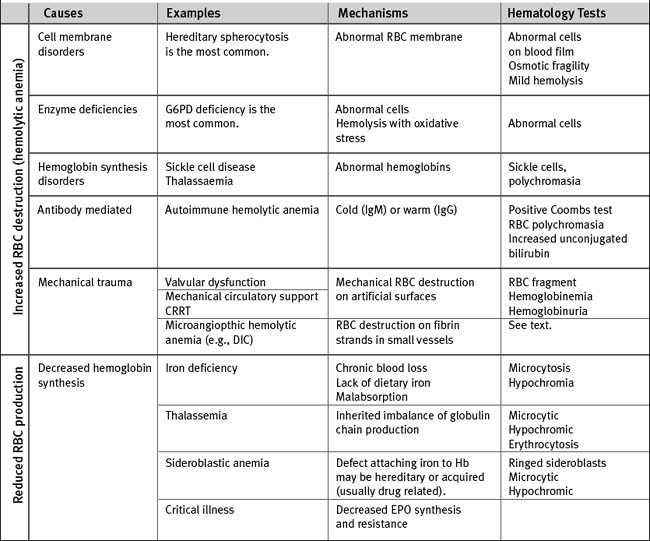
There are multiple causes of anemia in cardiac surgery patients (Table 30-3). Gastrointestinal hemorrhage is reasonably common, particularly in patients taking antithrombotic drugs. Patients may report an alteration in bowel habit and stool, but gastrointestinal blood loss is commonly asymptomatic. Occult retroperitoneal hematoma must be considered in those who have recently undergone angiography via femoral arterial puncture. Intravascular hemolysis is a potential cause in patients with regurgitant or stenotic heart valves. In patients with severe anemia who need urgent surgery, preoperative transfusion of packed red cells is indicated. For elective surgery and in those who belong to Jehovah’s Witnesses (for whom red cell transfusion may be unacceptable), preoperative treatment with iron supplements and erythropoietin is beneficial. In one study, a 14-day regimen of 300 mg per day of oral iron plus 5 doses of 500 U/kg erythropoietin was effective in increasing hemoglobin concentration by about 1.6 g/dl.6
TESTS OF HEMOSTASIS
Laboratory Versus Point-of-Care Testing
Traditionally, tests of hemostatic function (other than activated clotting time) have been performed in the hematology/coagulation laboratory. However, in an actively bleeding patient, the time required to label, transport, and report on the test causes a significant delay in obtaining the results. This has led to efforts to develop hemostatic tests that can be performed at or near the bedside (i.e., the point of care). Most of the hemostatic tests described here can be performed as point-of-care tests. By incorporating point-of-care testing into management algorithms, bleeding and transfusion requirements can be reduced.7,8
Activated Partial Thromboplastin Time
The aPTT monitors primarily the competency of the intrinsic coagulation pathway (i.e., high molecular weight kininogen, prekallikrein, and factors VIII, IX, X, XI, XII) and, to a lesser extent, the common pathway. The test involves adding a phospholipid tissue extract—which activates factor XII and calcium ions—to citrated plasma. The aPTT is used to monitor the effectiveness of anticoagulation with unfractionated heparin and direct thrombin inhibitors (see subsequent material). The normal range is 20 to 35 seconds but values vary slightly among laboratories. The therapeutic range for systemic heparinization is 50 to 80 seconds. Causes of prolonged aPTT include (1) deficiency of intrinsic pathway factors (e.g., due to consumptive coagulopathy or hemophilia); (2) unfractionated heparin; (3) the presence of acquired inhibitors (i.e., antibodies) of factors VIII and IX; and (4) the presence of lupus anticoagulant. Although typically associated with an elevated prothrombin time (PT), a mildly elevated aPTT may also be seen with warfarin overdose, liver disease, and vitamin K deficiency.
Fibrinogen
The normal range for the plasma fibrinogen concentration is 200 to 400 mg/dl. Levels increase as part of the acute-phase response and decrease with consumptive coagulopathy and advanced liver and renal disease. Several days following cardiac surgery, fibrinogen levels can increase to more than 500 mg/dl and remain elevated for up to a month.9
D-Dimer
D-dimer is a degradation product of cross-linked fibrin. Levels are normally low (<500 μg/l). Higher levels occur in any situation in which there is excess clotting followed by fibrinolysis; in particular, higher levels occur with venous thromboembolism (see Chapter 23), major hemorrhage, therapy, and DIC. Moderate elevations in the D-dimer are also seen in patients with severe systemic illness. Following cardiac surgery, the fibrinolytic D-dimer concentration is commonly greater than 1000 μg/l and remains elevated for as long as 60 days.9
Activated Clotting Time
The activated clotting time (ACT) is a bedside test that is used to assess the effectiveness of heparin anticoagulation. Whole blood is added to a tube or cartridge containing an activator (celite, kaolin, glass beads, or phospholipid tissue extract), which stimulates contact activation of the intrinsic coagulation pathway. The activated blood sample is then placed in a device that warms the blood and records the time it takes for clot to form. Devices from different manufacturers use different activators and different methodologies of assessing clot formation; thus, ACT values from different machines cannot be used interchangeably. The normal range for ACT depends on the device being used, but it is usually within the range of 70 to 150 seconds. Values above 400 seconds are generally used for CPB; values of 160 to 180 seconds are used for continuous renal replacement therapy (CRRT; see Chapter 33) and ECMO (see Chapter 22).
The ACT has the advantage of being able to be rapidly performed at the bedside. Most ACT devices have two channels, one of which can be used with a heparinase-containing cartridge. Heparinase is an enzyme that neutralizes heparin. An elevated ACT with a normal heparinase ACT indicates a heparin effect; if both ACTs are elevated, coagulopathy is suggested. The ACT has several limitations. First, as outlined earlier, values vary among devices and may not reliably correlate with plasma heparin concentrations.10 Second, values for ACT (more so than values for aPTT) are influenced by factor depletion, thrombocytopenia, platelet dysfunction, and antifibrinolytic therapy. Finally, as a point-of-care test, a quality assurance program must be maintained, but most ACT machines do not allow patient and user identification to be recorded.
Platelet Count and Platelet Function Tests
The normal value for the platelet count is 140,000 to 450,000/mm3. This parameter is easily measurable, but specific tests of platelet function are not widely available. Currently available point-of-care tests of platelet function, such as the PFA-100 and the HemoSTATUS analyzers, have proved disappointing as sole measures of hemostatic function in cardiac surgery patients.11–13 Important information about platelet function can be inferred from the thromboelastogram (see subsequent material).
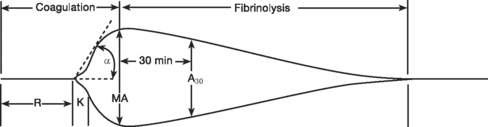
Thromboelastography
Standard thromboelastogram parameters are shown in Figure 30-3; typical abnormal patterns are shown in Figure 30-4. Point-of-care thromboelastogram-guided blood transfusion algorithms, either alone7,14 or in conjunction with other tests,8 have been shown to reduce transfusion requirements. As with ACT, point-of-care thromboelastogram analysis requires a quality assurance program.
TRANSFUSION MEDICINE
Blood Products
Red Blood Cells
Red blood cells are usually administered as packed cells from which much of the non-red-cell component has been removed and the blood has been resuspended in an anticoagulant storage solution, usually citrate-phosphate-dextrose-adenine. A unit of packed red cells has a volume of about 300 ml and a hematocrit of about 60%. Without continuing blood loss, transfusion of 4 to 5 ml/kg of packed red cells increases hemoglobin concentration by about 1 g/dl. Packed red cells are stored at 4°C and have a shelf life of 35 to 42 days. During storage, red cells metabolize dextrose into lactate, resulting in a fall in pH and an increase in lactate. Red cell fragility increases progressively, and temperature-sensitive failure of the red cells’ sodium-potassium pump occurs, resulting in an increase in the concentrations of free hemoglobin and extracellular potassium. There is also progressive loss of cellular 2,3-diphosphoglycerate (2,3-DPG), which is probably responsible for the short-lived (12 to 24 hours) leftward shift in patients’ oxygen-hemoglobin curves (and therefore the reduced tissue unloading of oxygen) that occurs with the transfusion of old blood. Despite these effects, no convincing data indicate an association between length of storage of red blood cells and adverse outcomes in cardiac surgery patients.15
Blood Groups and Compatibility Testing.
Red blood cell membranes contain many blood group antigens, the most important of which are those of the ABO and rhesus (Rh) blood groups. The ABO blood group is composed of two antigens, A and B, and their respective antibodies, anti-A and anti-B (Table 30-4). Antibodies naturally develop to nonself antigens during early childhood. Administration of red cells containing A or B antigen to a patient with a corresponding antibody results in a life-threatening hemolytic transfusion reaction. The compatibility of ABO groups with red cell and plasma products are shown in Table 30-5.
| Blood Group | Red Cells | Plasma |
|---|---|---|
| A | A antigen | Anti-B antibody |
| B | B antigen | Anti-A antibody |
| O | Neither A nor B antigens | Both anti-A and anti-B antibodies |
| AB | Both A and B antigens | Neither anti-A nor anti-B antibodies |
Table 30-5 Compatibility of ABO Groups for Red Cells and Plasma oxygen-hemoglobin
| Recipient ABO Blood Group | Compatible Donor ABO Groups |
|---|---|
| Red Cells | |
| Unknown | O |
| O | O only |
| A | A or O |
| B | B or O |
| AB | AB or B or O |
| Plasma Components | |
| Unknown | AB |
| O | O or A or B or AB |
| A | A or AB |
| B | B or AB |
| AB | AB (or A if AB is unavailable) |
Before blood is issued for transfusion, three processes usually occur: (1) ABO and Rh typing of the recipient’s blood; (2) antibody screening, in which recipient’s plasma and commercially supplied red cells containing antigens responsible for hemolytic transfusion reactions are combined; (3) cross-matching of the donor red blood cells and the recipient’s plasma. These processes usually take about 45 minutes. Before surgical procedures that are unlikely to require a blood transfusion a “type and screen” is performed, so if blood is subsequently required, only a cross-match is necessary. Most transfusion services issue type-specific blood, that is, group A blood to a group A recipient, Rh (+) blood to an Rh (+) recipient. Alternatively, cross-matched group O blood may be issued (see Table 30-5). If blood is required urgently, and a type and screen has been performed, type-specific, uncross-matched blood may be issued. In a true emergency, uncross-matched, group O Rh (−) blood may be used.
Transfusion Triggers.
The appropriate hemoglobin concentration at which to administer packed red cells is the subject of intense debate. Clinical reports indicate that untreated severe (<6 g/dl) anemia is associated with increased mortality rates, particularly in patients with cardiovascular disease.16,17 Against these are data indicating that in multiple large series, administering red blood cells has been shown to be an independent predictor of higher morbidity and mortality rates in cardiac surgery patients18–21 and in unselected patients in intensive care units (ICUs).22,23 However, these data are all nonrandomized, and it remains an open question as to whether the observed adverse outcomes relate to harmful effects of transfused blood or are due to inadequate adjustment for confounding variables. In a large randomized trial of ICU patients (from which cardiac surgery patients were specifically excluded), a restrictive transfusion policy targeting a hemoglobin concentration of 7 to 9 g/dl was not associated with a difference in survival rates when compared with maintaining the hemoglobin concentration above 10 g/dl.24 Subgroup analysis revealed improved survival rates for the restrictive group in younger patients (<55 years) and in those with less severe disease (Acute Physiology and Chronic Health Evaluation II score <20). No survival benefit was seen in the subgroup of patients with a primary or secondary diagnosis of cardiac disease.
Practice guidelines for transfusing red blood cells and other blood components in perioperative settings have been developed by the American Society of Anesthesiologists; published in 199625 and updated in 2006,26 they state that red cell transfusions are rarely indicated when the hemoglobin concentration is greater than 10 g/dl and are almost always indicated when it is less than 6 g/dl. The decision to transfuse red cells within this range should be based on evidence of organ ischemia, the rate and likelihood of bleeding, and risk factors for complications of inadequate oxygenation (such as low cardiorespiratory reserve and high oxygen consumption). Thus, transfusion at the higher end of this range is indicated in patients who are bleeding rapidly, have low arterial or venous oxygen saturation, or show evidence of organ (cardiac, renal, or brain) dysfunction. A transfusion trigger of 7 to 8 g/l has been suggested for patients older than 80 years, patients with severe coronary artery disease or heart failure, and patients who are febrile or hypermetabolic.27 A practical approach to red cell transfusion in cardiac surgery patients is presented in Table 17-4.
Fresh-frozen Plasma
Fresh-frozen plasma is derived from whole blood or plasma (obtained via apheresis) donations. Apheresis is a process whereby blood is drawn from a donor and split into its constituent parts, some of which are retained (usually the plasma and platelets); the remaining components (usually red blood cells) are returned to the patient. Plasma is rapidly frozen, stored at below −25°C, and rapidly thawed prior to use. Each unit of fresh-frozen plasma is derived from a single donor and has a volume of about 250 ml. It contains a range of coagulation factors but virtually no platelets. The fibrinogen concentration is 2 to 4 mg/ml. Once thawed, it should be kept at 4°C and should be administered within 24 hours.28 It may not be refrozen.
Because plasma contains ABO antibodies, ideally, fresh-frozen plasma should be of the same ABO type as the recipient. If ABO type-specific plasma is not available, plasma of a different ABO group may be used as long as it does not have high titers of anti-A or anti-B.28 Group O plasma, which contains anti-A and anti-B, should be given only to group O recipients (see Table 30-5). Fresh-frozen plasma of any Rh type may be given, and no anti-D prophylaxis is required if Rh (−) patients receive plasma from an Rh (+) donor because there are no red cells.28
Fresh-frozen plasma is used to treat bleeding associated with a raised PT (>1.5 times normal) or aPTT (>2 times normal).26 It may also be used in conjunction with prothrombin complex concentrate to treat warfarin excess when there is clinically significant bleeding or a high risk for it (Table 30-11). In an average-sized adult, one unit of fresh-frozen plasma increases coagulation factors by about 3%. For coagulopathic bleeding, a dose of 10 to 15 ml/kg (typically about 4 units) is an appropriate initial treatment.26 Fresh-frozen plasma is not indicated for intravascular volume expansion and is generally overused in cardiac surgery patients, in whom the main hemostatic problem is usually platelet dysfunction. Fresh-frozen plasma (1 to 2 units) is a source of antithrombin III in patients who display marked heparin resistance.
Table 30-11 Suggested Management of Elevated INR Due to Warfarin Therapy With or Without Bleeding
| Clinical Setting | Action |
|---|---|
| INR greater than therapeutic range but <5 | Lower or omit next dose of warfarin. |
| No bleeding | |
| INR 5 to 9 | Cease warfarin. |
| No bleeding | Consider possible causes. |
| If bleeding risk is high, give 2.5 to 5 mg vitamin K orally or 1 mg intravenously. | |
| Measure INR in 24 hr. | |
| When INR is in therapeutic range, resume warfarin at reduced dose. | |
| INR >9 | If bleeding risk is low, cease warfarin and give 2.5 to 5 mg vitamin K orally or 1 mg intravenously. |
| No bleeding | |
| Measure INR 6 to 12 hourly. | |
| Resume warfarin at reduced dose when INR <5. | |
| If bleeding risk is high, cease warfarin, give 1 mg vitamin K intravenously, and consider prothrombin complex concentrate 25 to 50 U/kg and fresh frozen plasma (150 to 300 ml). | |
| Measure INR 6 to 12 hourly. | |
| Resume warfarin at reduced dose when INR <5. | |
| Clinically significant bleeding when warfarin-induced coagulopathy is considered a contributing factor | Cease warfarin. |
| Give 5 to 10 mg vitamin K intravenously, as well as prothrombin complex 25 to 50 U/kg and fresh-frozen plasma (150 to 300 ml). | |
| Assess patient continuously until INR <5 and bleeding stops. | |
| or | |
| If prothrombin complex concentrate is not available: | |
| Cease warfarin. | |
| Give 5 to 10 mg vitamin K intravenously and fresh-frozen plasma 10 to 15 ml/kg. | |
| Assess patient continuously until INR <5 and bleeding stops. |
INR, International Normalized Ratio.
Modified from Baker RI, Coughlin PB, Gallus AS, et al: Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust 181:492-497, 2004. Note: Patients considered to be at high risk for bleeding are those with peptic ulcer disease, those receiving concomitant antiplatelet therapy, those who have undergone a major surgical procedure in the preceding 2 weeks, and those with throm-bocytopenia.
Cryoprecipitate
Cryoprecipitate contains the insoluble proteins that precipitate when frozen plasma is thawed in the cold at 1° to 6°C. When refrozen and stored at −30°C, cryoprecipitate has a shelf life of several months. The plasma used to produce cryoprecipitate may be obtained from multiple whole-blood donations or from a single donor by using apheresis. Cryoprecipitate contains fibrinogen, factor VIIIc, factor vWF, and factor XIII. The bag size, and therefore the factor content, varies among blood transfusion services. In the United States, a single unit of cryoprecipitate contains approximately 80 to 100 units of factor VIIIc and 150 to 250 mg of fibrinogen. The units are prepared from many single donors and then pooled. The fibrinogen concentration in cryoprecipitate is as much as 10 times that present in fresh-frozen plasma (typically 10 to 30 mg/ml, compared with 2 to 4 mg/ml for plasma). The principles of storage and blood group compatibility are the same as for fresh-frozen plasma.
Cryoprecipitate is used to treat bleeding associated with hypofibrinogenemia. It is usually indicated when the fibrinogen concentration is less than 80 to 100 mg/dl, and it may be indicated when the concentration is between 100 and 150 mg/dl and ongoing bleeding is anticipated.26
Platelets
Platelets are prepared from whole-blood donations or by means of platelet apheresis. Platelets obtained from 4 to 8 whole-blood donations (of the same ABO and Rh blood type) are randomly pooled and issued in a single multiunit bag. With apheresis, the equivalent of 4 to 6 pooled platelet units can be obtained from a single donor. Platelet concentrates contain small quantities of red blood cells and plasma. Unlike other blood products, platelets must be kept at room temperature (20° to 24°C)—and must be continuously agitated. They have a storage life of only 5 days and must be administered within a few hours of being pooled. Ideally, platelets should be of the same ABO and Rh group as the recipient. However, this is frequently not possible because of shortages of donor units, and ABO-incompatible transfusions may be necessary. Platelets from group O donors should be administered only to patients with blood groups A, B, and AB if they do not contain high titers of anti-A and anti-B (see Table 30-5).29 Whenever possible, Rh (−) platelets should be given only to Rh (−) recipients. If Rh (+) platelets are given to Rh (−) women of childbearing potential, anti-D immunoglobulin (passive immunization) should be given as soon as possible (within 72 hours at the outside)29 to prevent anti-D antibody formation (active immunization) by the recipient. If an Rh (−) woman with anti-D antibodies becomes pregnant with an Rh (+) fetus, maternal anti-D can cross the placenta and cause hemolytic disease in the newborn.
Thrombocytopenia and platelet dysfunction are common in cardiac surgery patients, and platelet concentrates are widely used to treat postoperative bleeding. In patients who are bleeding, platelet transfusion is usually indicated if the platelet count is less than 50,000/mm3 and is rarely indicated if the platelet count is greater than 100,000/mm3.26,29 However, if the thromboelastogram is suggestive of platelet dysfunction (maximum amplitude <45 mm), platelets are likely to be beneficial, even if the platelet count is greater than 100,000/mm3. One unit of pooled platelets is usually adequate for initial therapy. In patients who are not bleeding, platelet transfusion is not usually indicated. However, there are some exceptions to this recommendation, such as patients on ECMO (see Chapter 22) or thrombocytopenic patients who need invasive procedures. For most invasive procedures, a platelet count above 30,000 to 50,000 mm3 is adequate. For high-risk procedures, such as instrumenting the epidural or subarachnoid spaces, a platelet count above 100,000 mm3 is advisable.
As with packed red cells, platelet transfusion too has been linked to adverse outcomes, including prolonged hospital stay, stroke, and death, in a large nonrandomized series.30
Prothrombin Complex Concentrate
Prothrombin complex is a sterile, freeze-dried powder containing purified human coagulation factors II, IX, and X, with antithrombin III. It commonly contains some factor VII, but the amount may vary. In surgery patients, prothrombin complex is indicated only for the rapid reversal of warfarin (see Table 30-11). Prothrombin complex may have only low levels of factor VII, so it should be given with fresh-frozen plasma (see subsequent material). Each vial contains 500 IU of factors IX, II, and X. The dose for reversal of warfarin is 25 to 50 IU/kg.31 Prothrombin complex is not available in all countries.
Leukodepletion
Leukodepletion is the removal (usually of more than 99%) of white blood cells from blood components by centrifugation or filtration. It may be done at the time of administration in selected patients, or it may be performed before storage in all blood products issued by the blood transfusion service. Several countries—notably Canada, the United Kingdom, and New Zealand—have adopted universal leukodepletion for all blood products. Transmission by blood components of leukocyte-associated viruses (cytomegalovirus, Epstein-Barr virus, and herpes viruses) is prevented, and variant Creutzfeldt-Jakob disease is reduced by leukodepletion. Prevention of cytomegalovirus transmission is particularly important in transplant recipients who are cytomegalovirus antibody negative (see Chapter 13). Leukodepletion also reduces the incidence of some transfusion reactions and postoperative infections,32 and in some studies it has been associated with improved outcome in cardiac surgery patients.33–35 A metaanalysis of randomized trials did not establish a consistent benefit for long-term survival with leukodepletion, although a short-term benefit to mortality rates was seen in cardiac surgery patients.36 It is interesting to note that many of the trials that indicate an association between increased mortality rates and red cell transfusion have involved the use of nonleukodepleted blood.
Complications of Blood Component Therapy
Complications of blood transfusion may be classified as immune or nonimmune. Immune reactions may be hemolytic or nonhemolytic. The incidences of various complications are listed in Table 30-6.
| Type | Incidence |
|---|---|
| Transmission of Infection | |
| HIV | 1 in 1,400,000 to 2,400,000 |
| Hepatitis B | 1 in 58,000 to 149,000 |
| Hepatitis C | 1 in 872,000 to 1,700,000 |
| Bacterial | 1 in 2000 |
| Immunologic Reactions | |
| Febrile reactions | 1 in 100 |
| Anaphylactic reactions | 1 in 20,000 to 50,000 |
| ABO mismatch | |
| Hemolysis | 1 in 60,000 |
| Death | 1 in 600,000 |
| TRALI | 1 in 2000 |
| Posttransfusion purpura | Rare |
TRALI, transfusion-related acute lung injury.
Modified from Ferraris VA, Spiess BD: Perioperative blood transfusion and blood conservation in cardiac surgery: The Society of Thoracic Surgeons and The Society of Cardiovascular Anesthesiologists Practice Guidelines Series. Ann Thorac Surg [in press].
Immune-Mediated Nonhemolytic Reactions
Allergic and Anaphylactic Reactions.
Anaphylaxis is rare. It usually occurs in patients with IgA deficiency who have preformed IgE anti-IgA antibodies. IgA deficiency is relatively common (1 in 600 to 1 in 800 people), and if its presence is known, it is an indication to use washed red cells or IgA-free blood.
Transfusion-Related Acute Lung Injury.
Transfusion-related acute lung injury (TRALI)37–39 is a form of noncardiogenic pulmonary edema that develops within minutes to a few hours after a transfusion. Patients may complain of dyspnea and cough and show signs of respiratory distress. Marked—often profound—hypoxemia develops rapidly. The chest radiographic findings are usually striking, with bilateral diffuse alveolar infiltrates and sometimes almost complete whiteout of the lungs. In contrast to cardiogenic pulmonary edema, auscultation of the lungs is usually unremarkable (i.e., few crackles or wheezes). Fever and hypotension are common. The differential diagnosis includes anaphylaxis, circulatory overload, cardiac dysfunction, and acute lung injury due to other causes (aspiration, sepsis, etc.). The distinction between cardiogenic and noncardiogenic pulmonary edema is discussed in Chapter 27.
Nonimmune-Mediated Adverse Effects
Infection.
The risks for infectious complications are low in the developed world and are further reduced by leukodepletion (see earlier discussion). Since routine donor testing was instituted, the transmission of HIV, hepatitis B, and hepatitis C has become uncommon. Bacterial contamination can occur (particularly with platelets), causing severe reactions that are clinically similar to those in acute hemolytic transfusion reaction: fever, shock, and DIC. Yersinia species, a psychrophilic (cold-loving) microorganism, is responsible for most bacterial contaminations of red blood cells. Usual nosocomial microorganisms (see Chapter 35) are responsible for the contamination of platelets.
Metabolic Sequelae.
PHARMACOLOGY
Unfractionated Heparin
Heparin41 is the most widely used anticoagulant in cardiac surgery. It is an acidic polysaccharide that is isolated from either porcine intestine or bovine lung, where it is bound to histamine and stored in mast cell granules. The purification process results in a heterogeneous mixture of molecules that have molecular weights ranging from 5000 to 30,000 Daltons. Heparin acts indirectly as an anticoagulant by binding to antithrombin III, greatly enhancing the rate of formation of thrombin-antithrombin III complex. Antithrombin III is a natural anticoagulant that inhibits thrombin (factor IIa) along with factors Xa, IXa, VIIa, XIa, and XIIa. Heparin also binds to many other blood and endothelial proteins, including high molecular weight kininogen, vWF, fibronectin, lipoproteins, and platelet and endothelial receptors. Each of these potentially influences the ability of heparin to act as an anticoagulant and may, with antithrombin III levels, affect the patient’s dose response to heparin.
Pharmacokinetics and Dosing
Heparin anticoagulation is monitored on the basis of the aPTT or the ACT. The target values for these parameters vary according to the indication for anticoagulation; typical therapeutic ranges are listed under the earlier heading Tests of Hemostasis. For therapeutic anticoagulation in the ICU, doses in the range of 15 to 20 IU/kg/hr, preceded by a loading dose of 5000 IU, are typical. A weight-based dosing nomogram is shown in Table 30-7.
Table 30-7 Weight-Based Nomogram for Unfractionated Heparin Dosing
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Hirsh J, Warkentin TE, Shaughnessy SG, et al: Heparin and low molecular weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 119:64S-94S, 2001.
Heparin resistance—defined as the need for more than 35,000 IU/day to achieve therapeutic anticoagulation—is reasonably common in cardiac surgery patients. Causes include antithrombin III deficiency, increased heparin clearance, elevations in heparin-binding proteins, and increased levels of factor VIII, fibrinogen, and platelet factor 4. If heparin resistance is severe, anti-factor Xa levels should be measured to guide dosing, with the goal of a level of 0.35 to 0.7 IU/ml.41,42 Also, for marked heparin resistance, 1 to 2 units of fresh-frozen plasma, which contains antithrombin III, may be given.
Adverse Effects
The main adverse effect of heparin is bleeding. This risk is increased when heparin is combined with other antithrombotic agents. When used in the early postoperative period, unfractionated and low molecular weight heparins are associated with a two- to threefold increase in the need for surgical reexploration.43 The other main complication of heparin is heparin-induced thrombocytopenia (HIT).44,45
Heparin-Induced Thrombocytopenia.
Laboratory tests for HIT involve either immunoassays for platelet factor 4 antibodies or platelet activation assays (e.g., a platelet serotonin release assay or a heparin-induced platelet activation assay). Both types of test have a high negative predictive value, so negative tests virtually rule out the disease. However, there is a high rate of seroconversion in the absence of clinical disease; thus, the positive predictive value of antibody tests is only moderate. Positive predictive value is higher with activation tests. The diagnosis of HIT is established by a fall in the platelet count of more than 50% in association with a positive laboratory test. However, following CPB, the platelet count may drop acutely by 50% due to dilutional changes alone.
Options for anticoagulation in patients with HIT include a direct thrombin inhibitor (e.g., lepirudin, argatroban, bivalirudin) or a factor Xa inhibitor (e.g., danaparoid or fondaparinux) (Table 30-8). In the United States, only lepirudin and argatroban are approved for anticoagulation for HIT. Lepirudin is recombinant hirudin, which is a naturally occurring thrombin inhibitor produced by the salivary gland of the leech. Bivalirudin is approved for use during percutaneous coronary intervention and has been used off-label for patients with HIT requiring cardiac surgery. Danaparoid is not available in the United States but is widely used elsewhere. Fondaparinux has only recently been introduced in the United States, and experience with its use is limited.
Table 30-8 Nonheparin Anticoagulants for Use in Heparin-Induced Thrombocytopenia
| Drug | Dose | Comments |
|---|---|---|
| Direct Thrombin Inhibitors | ||
| Lepirudin | Infusion 0.15 mg/kg/hr (with or without bolus of 0.4 mg/kg) | Approved for HIT in United States |
| With normal renal function, effect rapidly | ||
| Dissipates (<1 hr) after discontinuation of infusion | ||
| With renal failure, reduce dose and avoid loading dose | ||
| Adjust dose to achieve 1.5 to 2 × baseline increase in aPTT | ||
| Minimal effect on INR | ||
| Argatroban | Infusion 2 μg/kg/min (no bolus) | Adjust dose to achieve 1.5 to 2 · baseline increase in aPTT |
| Can increase INR, which makes overlap with warfarin anticoagulation difficult | ||
| Bivalirudin | Infusion 0.15 to 0.2 mg/kg/hr (no bolus) | Adjust dose to achieve 1.5 to 2.5 × baseline increase in aPTT |
| Minimal effect on INR | ||
| Factor Xa Inhibitor | ||
| Danaparoid | Thrombosis prevention: | Not available in United States |
| 750 U SC 2 to 3 × daily (<90 kg) | Monitor anti-Xa levels: | |
| 1250 U SC 2 to 3 × daily (>90 kg) | For prevention: 0.2 U/ml on day 1 and 0.2 to 0.4 U/ml on day 5 | |
| Thrombosis treatment: | ||
| 2500 U IV bolus, then | Test level 6 hr after dose | |
| 400 U IV for 4 hr, then | For treatment: 0.5 to 0.7 U/ml 10 min after IV bolus and 0.5 to 0.8 during maintenance | |
| 200 U IV thereafter | ||
| Patients may be converted to SC dosing as for prevention after 5 to 10 days | ||
| Bolus dose adjustment: | ||
| <55 kg = 1250 U; >90 kg = 3750 U |
aPTT, activated partial thromboplastin time; HIT, heparin-induced thrombocytopenia; INR, international normalized ratio; IV, intravenous; SC, subcutaneous; U, units.
Note: These doses pertain to therapeutic anticoagulation for heparin-induced thrombocytopenia; different doses are required for other indications.
Modified from Warkentin TE, Greinacher A: Heparin-induced thrombocytopenia: recognition, treatment, and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 126:311S-337S, 2004.
Heparin Alternatives
Because of the short duration of action, the potential for resistance, and the side effects that occur with unfractionated heparin, alternative drugs for anticoagulation may be considered. For prophylaxis and treatment of thromboembolism, low molecular weight heparins (LMWHs) may be a good choice (see subsequent material). For patients with HIT, a direct thrombin inhibitor or factor Xa inhibitor is appropriate (see earlier material). For patients undergoing CRRT, regional citrate anticoagulation (i.e., anticoagulation of the dialysis circuit only) is a good alternative (see Chapter 33).
Fondaparinux is a pure antithrombin-III-dependent inhibitor of activated factor X. Unlike unfractionated heparin and LMWH, fondaparinux has no effect on thrombin and does not bind to platelets. It has a long duration of effect and is minimally antigenic. Fondaparinux has been investigated for the prophylaxis and treatment of venous thromboembolism and for acute coronary syndromes, and it appears to be at least as effective as LMWH.46 For the prevention of DVT, a once-daily dose of 2.5 mg subcutaneously is effective. As noted previously, fondaparinux may also be an acceptable anticoagulant for HIT. The drug is renally eliminated and should be avoided in patients with severe renal failure.
Low Molecular Weight Heparins
LMWHs41 are formed by the depolymerization of unfractionated heparin, which produces fragments with a mean molecular weight of approximately 5000 daltons. Compared with unfractionated heparin, LMWHs cause less inhibition of thrombin. Their anticoagulant effect is mainly through antithrombin III-dependent inhibition of activated factor X. This lack of antithrombin effect means that LMWHs do not elevate the aPTT. LMWHs have a long duration of action and are administered subcutaneously once or twice daily (Table 30-9). They are renally eliminated and their anticoagulant effect is prolonged with renal failure. The incidence of HIT is much lower with LMWH than with unfractionated heparin, but it can occur.
| Drug | High Dose (full anticoagulation) | Low Dose (prophylaxis) |
|---|---|---|
| Nadroparin | 86 anti-Xa U/kg sc twice daily | 38 anti Xa U/kg sc once or twice daily |
| Dalteparin | 120 anti-Xa U/kg sc twice daily | 60 anti-Xa U/kg sc twice daily |
| Enoxaparin | 1 mg/kg sc twice daily | 0.25 to 0.5 mg/kg daily |
sc, subcutaneous.
Laboratory monitoring is not routinely performed, but in patients with renal failure or obesity, anti-factor Xa levels should be checked initially. For therapeutic anticoagulation (as opposed to thromboembolism prophylaxis), an anti-factor Xa level of 0.6 to 1.0 U/ml 4 hours after dosing is an appropriate target.41
LMWHs are indicated primarily for the prophylaxis and treatment of venous thromboembolism and for the treatment of acute coronary syndromes. In most circumstances, the incidences of bleeding and thrombotic complications are comparable to those that occur with unfractionated heparin. However, administration of LWMH within 12 hours of cardiac surgery is associated with an increased incidence of postoperative bleeding compared with that found with unfractionated heparin.47 As noted both unfractionated heparin and LMWH are associated with increased bleeding and need for surgical reexploration when used in the early postoperative period.
Warfarin
Warfarin48,49 is a member of the family of drugs known as coumarins, which are vitamin K antagonists. Vitamin K is required for the posttranslational carboxylation of factors II, VII, IX, and X (and protein C and protein S). Without vitamin K, these coagulation factors are incapable of chelating calcium, which is required for binding coagulation factors to phospholipid membranes during the normal clotting process.
Pharmacokinetics and Dosing
Warfarin is rapidly absorbed from the gastrointestinal tract; peak plasma concentrations are reached 1 to 4 hours after ingestion. The plasma half-time is about 48 hours but, as explained earlier, the therapeutic effect is related to the half-time of the vitamin K-dependent coagulation proteins, not the drug itself. Warfarin is 97% protein-bound to albumin, and only the unbound form of the drug is pharmacologically active. Warfarin undergoes extensive hepatic metabolism by the cytochrome P450 (2C9) enzyme system and is involved in several clinically important drug interactions, which both potentiate and inhibit its anticoagulant effect.50 Drug interactions that are relevant to practice in the ICU are listed in Table 30-10. Of these, interaction with amiodarone is one of the most clinically important. Amiodarone displaces warfarin from its protein binding sites and inhibits the P450 (2C9) enzyme system, increasing the anticoagulation effect. The effect of warfarin is also potentiated by congestive heart failure, poor nutritional state, hyperthyroidism, and hepatic dysfunction. Thus, cardiac surgery patients are often very sensitive to warfarin, and the initial daily dose should not usually exceed 3 to 5 mg. In most circumstances, a target INR of 2 to 3 is appropriate (see Chapter 10). To achieve this, a maintenance dose of 4 to 6 mg/day is required for most patients.
Table 30-10 Drugs That Potentiate or Inhibit the Anticoagulant Effect of Warfarin
| Potentiation | Inhibition |
|---|---|
| Highly Probable | Highly Probable |
| Acetaminophen | Barbiturates |
| Alcohol (only if liver disease) | Carbamazepine |
| Amiodarone | Cholestyramine |
| Cimetidine | Griseofulvin |
| Ciprofloxacin | Nafcillin |
| Clofibrate | Ribavirin |
| Cotrimoxazole | Rifampin |
| Diltiazem | Sucralfate |
| Erythromycin | High vitamin K content enteral feeds |
| Fenofibrate | |
| Fluconazole | |
| Isoniazid | |
| Metronidazole | |
| Miconazole | |
| Ompeprazole | |
| Piroxicam | |
| Propafenone | |
| Propranalol | |
| Voriconazole | |
| Probable | Probable |
| Amoxicillin/clavulanic acid | Azathioprine |
| Celecoxcib | Bosentan |
| Clarithromycin | Candesartan |
| Fluvastatin | Dicloxacillin |
| Itraconazole | Multivitamin supplements containing vitamin K |
| Levofloxacin | |
| Phenytoin (biphasic with later inhibition) | |
| Quinidine | |
| Simvastatin | |
| Tamoxifen | |
| Tetracycline | |
| Tramadol | |
| Possible | Possible |
| Amoxicillin | Cyclosporin |
| Gemfibrozil | Telmisartan |
| Lovastatin | |
| Metolazone | |
| Norfloxacin | |
| Ofloxacin | |
| Rofecoxib | |
| Sulindac | |
| Ticlopidine |
Modified from Holbrook AM, Pereira JA, Labiris R, et al: Systematic overview of warfarin and its drug and food interactions. Arch Int Med 165: 1095-1106, 2005.
Adverse Effects
The main adverse effect of warfarin is bleeding, and the main determinant of this risk is the INR. The risk for a bleeding complication doubles as the INR increases from between 2.0 and 2.9 to between 3.0 and 4.4, and it increases eightfold as the INR increases to between 4.5 and 6.9.51 Bleeding complications are also higher in the elderly (>70 years) and during the first 3 months of treatment.51
Warfarin Reversal
Rapid reversal of warfarin31 anticoagulation is sometimes required because of bleeding, high INR, or planned surgery. Most procedures and surgeries can be safely performed with an INR of between 1.3 and 1.5. However, for high-risk interventions such as instrumenting the epidural space, the INR should be in the normal range (i.e., ≤1.2). Vitamin K reduces the INR within 6 to 12 hours, but high doses (5 to 10 mg) can make reanticoagulation difficult. If subsequent anticoagulation is required, only low doses (2 to 5 mg orally or 0.5 to 1 mg intravenously) should be used. For urgent reversal, fresh-frozen plasma, either alone or in combination with prothrombin complex, should be used. Guidelines for warfarin reversal are provided in Table 30-11.
Desmopressin
Desmopressin (1-desamino-8-D-arginine vasopressin; DDAVP) is a synthetic analog of vasopressin that mediates vasopressin type-2 receptor effects but without the usual vasopressor action. Desmopressin causes a twofold to twentyfold increase in the plasma concentration of factor VIII and stimulates vascular endothelium to release vWF. Desmopressin also causes the release of tissue plasminogen activator and prostacyclin from vascular endothelium. The vWF facilitates platelet adhesion to vascular subendothelium by functioning as a protein bridge between platelet receptors and the subendothelial vascular basement membrane. Desmopressin has been used to treat bleeding associated with platelet dysfunction and is specifically indicated for bleeding due to von Willebrand disease, uremia, and mild or moderate hemophilia A. However, most studies indicate that desmopressin has minimal efficacy in reversing bleeding abnormalities after cardiac surgery.52,53 Also, desmopressin is associated with an increased risk for perioperative myocardial infarction in cardiac surgery patients.52 If desmopressin is used, a dose of 0.3 μg/kg infused over 30 minutes is appropriate.
Antifibrinolytic Agents
Antifibrinolytic agents are widely used to reduce blood loss in patients undergoing cardiac surgery. They are used primarily for prophylaxis during the intraoperative period but may also be used in the ICU for treating documented fibrinolysis. Three antifibrinolytic drugs are in widespread use: the lysine analogs epsilon aminocaproic acid and tranexamic acid; and a serine protease inhibitor, aprotinin. In metaanalyses of randomized trials, these agents have been shown to reduce bleeding, allogenic blood transfusion, and the need for reoperation in patients undergoing cardiac surgery.52–55 Although there are fewer data on the efficacy of epsilon aminocaproic acid and tranexamic acid than on aprotinin, in head-to-head comparisons, no differences between the agents has been demonstrated.54
Lysine Analogs
The synthetic lysine analogs epsilon aminocaproic acid and tranexamic acid inhibit fibrinolysis by attaching to the lysine binding site on plasminogen, preventing its cleavage to form plasmin and therby reducing the breakdown of fibrin.56 Both agents have predominantly renal elimination, but dose reductions during cardiac surgery are not required because the duration of treatment is short. A tranexamic acid dose of 20 mg/kg may be used for treating fibrinolysis in the ICU. Side effects are minimal.
Aprotinin
Aprotinin is a broad-spectrum serine protease inhibitor that attenuates multiple aspects of the inflammatory responses to CPB, including fibrinolysis.56,57 It has a half-time of about 5 hours and is renally eliminated. Aprotinin may have a transient adverse effect on renal proximal tubular cells or may alter intrarenal blood flow through inhibition of renin and kallikrein activity, but clinically important postoperative renal dysfunction has not been demonstrated.58 Aprotinin is a bovine-derived protein and is therefore potentially antigenic. The risk for anaphylaxis is 2.7% in patients reexposed to aprotinin.59,60 Most cases of anaphylaxis occur when the reexposure interval is less than 6 months, and the risk-benefit ratio of early readministration must be considered in each individual.
Any drug or blood product that decreases bleeding and transfusion requirements has the potential to cause thrombotic complications. Studies of patients undergoing CABG surgery have generally not demonstrated differences in graft patency between patients treated with aprotinin and those not treated with aprotinin.61–63 In one randomized study, a slightly higher incidence of early vein graft occlusion was identified in aprotinin-treated patients (15.5% versus 10.9%, p = 0.03), but after adjusting for differences between groups, this difference was not found to be significant.64 A recent metaanalysis of randomized trials found no difference in the incidence of renal failure, myocardial infarction, or death in aprotinin-treated patients, but aprotinin was associated with a reduced risk for stroke.55 Against these findings are the results of a recent propensity-adjusted observational study, which suggest an increased risk of myocardial infarction, renal failure, stroke, and death with aprotinin.65 This paper has been subject to methodologic criticisms66,67 relating mainly to the risk of selection bias.
Aprotinin is widely used intraoperatively in patients at high risk of postoperative bleeding. In the ICU, aprotinin is indicated in patients who are bleeding and have documented fibrinolysis on a thromboelastogram; see Figure 30-4. This circumstance arises most commonly in the early period following prolonged CPB, but it may also arise in patients on ECMO. The appropriate dose of aprotinin for use in the ICU has not been established; however, a bolus dose of 1,000,000 to 2,000,000 kallikrein inhibitor units (KIU) followed by an infusion of 500,000 KIU per hour is reasonable. Because of the small risk for anaphylaxis, a test dose of 10,000 KIU should be given prior to the main dose. The thromboelastogram should be repeated after 1 to 2 hours.
Activated Factor VII
Recombinant activated factor VII is approved for treating bleeding episodes in patients with hemophilia who have inhibitors to factor VIII or factor IX. However, it is being increasingly used to treat life-threatening bleeding following cardiac and other surgery. Activated factor VII produces a prohemostatic effect by forming a complex with tissue factor. The tissue factor-factor VII complex activates factor X, leading to the generation of thrombin, which in turn leads to platelet activation and to fibrin formation (see Figs. 30-1 and 30-2).
Many clinical reports have described the off-label use of activated factor VII to treat life-threatening and refractory bleeding in cardiac surgery patients.68–71 Although 90 μg/kg is the initial dose for treating bleeding due to hemophilia, doses of 30 to 45 μg/kg have been reported to be effective in surgery patients.72 For activated factor VII to be effective, there must be adequate concentrations of coagulation factors and sufficient numbers of functioning platelets. Thus, conventional blood component therapy (platelets, fresh-frozen plasma, cryoprecipitate) should be administered to achieve relatively normal values for the platelet count, aPTT, and PT before administering activated factor VII. Other causes of bleeding, such as hypothermia and inadequate surgical hemostasis, should also be corrected. Additional studies are needed to further evaluate the dosing, safety, and efficacy of this product in cardiac surgery patients. Currently, there are insufficient data to indicate whether there is an increased risk for thrombotic complications in patients who receive this drug for postoperative bleeding.
AN APPROACH TO PATIENTS WITH SEVERE POSTOPERATIVE BLEEDING
Bleeding following cardiac surgery is common and, as outlined under the earlier heading Physiology and Pathophysiology, it has many causes. Treatment involves blood component therapy, hemostatic drugs, and timely surgical reexploration. Blood products and drugs should be administered according to a management algorithm incorporating laboratory or, ideally, point-of-care tests of hemostatic function.7,8 A transfusion algorithm is provided in Chapter 17.
Massive Transfusion Coagulopathy
Cardiac surgery and CPB can cause consumptive coagulopathy that contributes to postoperative bleeding.5,73 In addition, massive transfusion (>4 units of packed red cells/hr over several hours) can itself cause or exacerbate coagulopathy due to hypothermia, dilutional coagulopathy, platelet dysfunction, low fibrinogen levels, and fibrinolysis. Unless an intravenous-fluid warming device is used, hypothermia is common in massive transfusion. At a temperature of less than 35°C, the enzymatic activity of coagulation factors is slowed, the coagulation factor synthesis is reduced, fibrinolysis is increased, and platelet function is impaired. However, these effects may not be apparent on hemostatic tests, which are usually performed on blood samples warmed to 37°C. Resuscitation of blood loss with crystalloids, colloids, and packed red cells causes dilution of coagulation proteins and platelets. Platelet function may be further compromised by large-volume (>20 ml/kg) fluid resuscitation with hydroxyethyl starch solutions (see Chapter 32).
Patient Management
Anemia and Blood Loss
Transfusion triggers were discussed earlier. Irrespective of a patient’s hemoglobin concentration, transfusion is recommended when acute blood loss exceeds 1500 ml or 30% of a patient’s blood volume.25 In practice, this means targeting the upper limit of the range, which is 8 to 9 g/l. In patients who are bleeding rapidly, at least 4 units of cross-matched packed red cells should be kept in an ICU’s blood refrigerator at all times. Guidelines concerning the degree of blood loss that warrants surgical reexploration are provided in Chapter 17. Beyond the first hour, if there is more than 300 ml/hr of blood loss for 3 hours, surgical reexploration should be strongly considered. Persistent bleeding at this volume will lead to the complications of massive transfusion listed earlier and most likely expose the patient to unnecessary blood component therapy.
Hemostatic Dysfunction
Tests of hemostatic function (ACT, aPTT, PT, fibrinogen, complete blood count, thromboelastogram) should be obtained every 1 to 2 hours during major bleeding. Detection of a heparin effect with a heparinase ACT or thromboelastogram is important because rebound heparinization can occur following successful heparin reversal with protamine. Blood component therapy should be administered on the basis of hemostatic tests: platelet concentrates for low platelet levels (<100,000 to 150,000 mm3) and platelet dysfunction (thromboelastogram maximum amplitude <45 mm); fresh-frozen plasma for prolonged PT (>1.5 times normal) and aPTT (> 2 times normal); and cryoprecipitate for hypofibrinogenemia (<80 to 100 mg/dl).26 If there is documented fibrinolysis, an antifibriolytic should be administered. Platelet dysfunction is a key component of bleeding following cardiac surgery. Platelet transfusion may be ineffective with concomitant hypofibrinogenemia74; thus, co-administration of fresh-frozen plasma or cryoprecipitate should be considered.
Once appropriate blood components have been administered, tests of hemostatic function should be repeated immediately. With point-of-care hemostatic testing and an efficient blood transfusion service, a cycle of blood product administration followed by further testing can be repeated every 60 to 90 minutes. If laboratory tests of hemostatic function are used, the wait time for test results is commonly the limiting step in the cycle. If bleeding persists despite adequate protamine and blood component therapy, aprotinin (if indicated), surgical reexploration, and activated factor VII should be considered.
Hemodynamic Instability
Hemodynamic instability is common with massive transfusion, and its causation is multifactorial. Massive transfusion is associated with prolonged cardiac surgery, which is more common in patients with complex cardiac lesions who commonly have ventricular dysfunction. Coagulopathy due to prolonged CPB is also associated with marked systemic inflammation, which can cause pathologic vasodilatation and ventricular dysfunction (see Chapter 2). Resuscitation of major blood loss is associated with hypocalcemia, hypovolemia, and pericardial tamponade. Rhythm disturbances are common. Severe anemia can exacerbate myocardial ischemia. In addition to standard monitoring, pulmonary artery catheterization and transesophageal echocardiography should be available, the latter to rule out pericardial tamponade if it is suspected clinically. If patients are unstable, sedation and neuromuscular paralysis may be beneficial to prevent patient ventilator dysynchrony, which can exacerbate hypotension due to hypovolemia. Modest pharmacologic hypotension (e.g., mean arterial pressure 60 to 70 mmHg) may help to control excessive bleeding, particularly following complex aortic surgery, but it can lead to profound hypotension in the presence of hypovolemia.
Respiratory Insufficiency
Impaired gas exchange is common in patients undergoing massive transfusion. There may be pulmonary edema secondary to ventricular dysfunction and fluid overload (i.e., cardiogenic pulmonary edema) or acute lung injury secondary to systemic inflammation (i.e., noncardiogenic pulmonary edema). Occasionally, noncardiogenic pulmonary edema occurs due to TRALI. An underrecognized cause of impaired respiratory (and cardiac) function in patients who are bleeding is hemothorax. Any bleeding patient who experiences a significant deterioration in their respiratory status should undergo a chest radiograph to rule out this problem. A large postoperative hemothorax should be treated with surgical reexploration rather than thoracocentesis. If hemothorax is excluded, treatment of hypoxemia is primarily supportive and includes increasing the inspired oxygen concentration, pressure control ventilation, and increasing positive end-expiratory pressure (see Chapter 29). A positive end-expiratory pressure of 10 cm H2O is not effective in reducing mediastinal blood loss75,76 and, because it can contribute to hemodynamic instability, should not be used for this purpose.
Hypothermia and Metabolic Derangement
Hypothermia is more easily prevented than treated. If major blood loss is anticipated, a forced-air warming device should be applied and an intravenous fluid warmer used. A temperature of 36° to 36.5°C should be targeted. Shivering can exacerbate hemodynamic instability and hypoxemia; it should be treated with sedation and neuromuscular paralysis. Hyperkalemia occasionally occurs but does not usually require specific treatment. Metabolic acidosis is common and usually is related to cardiovascular dysfunction rather than to massive transfusion; treatment should be directed to supporting the circulation. Hypocalcemia requires treatment only if there is hemodynamic instability; modest hypocalcemia (0.8 to 1.0 mmol/l) does not contribute to coagulopathy. Hyperglycemia should be treated with intravenous insulin (see Chapter 36).
1 Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381-1389.
2 Hoffman M, Monroe DM3rd. A cell-based model of hemostasis. J Thromb Haemost. 2001;85:958-965.
3 Ang-Lee MK, Moss J, Yuan CS. Herbal medicines and perioperative care. JAMA. 2001;286:208-216.
4 Paparella D, Brister SJ, Buchanan MR. Coagulation disorders of cardiopulmonary bypass: a review. Intens Care Med. 2004;30:1873-1881.
5 Levy JH. Massive transfusion coagulopathy. Semin Hematol. 2006;43:S59-S63.
6 Sowade O, Messinger D, Franke W, et al. The estimation of efficacy of oral iron supplementation during treatment with epoetin beta (recombinant human erythropoietin) in patients undergoing cardiac surgery. Eur J Haematol. 1998;60:252-259.
7 Shore-Lesserson L, Manspeizer HE, DePerio M, et al. Thromboelastography-guided transfusion algorithm reduces transfusions in complex cardiac surgery. Anesthes Analg. 1999;88:312-319.
8 Avidan MS, Alcock EL, Da Fonseca J, et al. Comparison of structured use of routine laboratory tests or near-patient assessment with clinical judgment in the management of bleeding after cardiac surgery. Br J Anaesth. 2004;92:178-186.
9 Parolari A, Colli S, Mussoni L, et al. Coagulation and fibrinolytic markers in a two-month follow-up of coronary bypass surgery. J Thorac Cardiovasc Surg. 2003;125:336-343.
10 Despotis GJ, Summerfield AL, Joist JH, et al. Comparison of activated coagulation time and whole blood heparin measurements with laboratory plasma anti-Xa heparin concentration in patients having cardiac operations. J Thorac Cardiovasc Surg. 1994;108:1076-1082.
11 Forestier F, Coiffic A, Mouton C, et al. Platelet function point-of-care tests in post-bypass cardiac surgery: are they relevant ? Br J Anaesth. 2002;89:715-721.
12 Fattorutto M, Pradier O, Schmartz D, et al. Does the platelet function analyser (PFA-100) predict blood loss after cardiopulmonary bypass ? Br J Anaesth. 2003;90:692-693.
13 Ereth MH, Nuttall GA, Klindworth JT, et al. Does the platelet-activated clotting test (HemoSTATUS) predict blood loss and platelet dysfunction associated with cardiopulmonary bypass ? Anesth Analg. 1997;85:259-264.
14 Royston D, von Kier S. Reduced haemostatic factor transfusion using heparinase-modified thromboelastography during cardiopulmonary bypass. Br J Anaesth. 2001;86:575-578.
15 Vamvakas EC, Carven JH. Length of storage of transfused red cells and postoperative morbidity in patients undergoing coronary artery bypass graft surgery. Transfusion. 2000;40:101-109.
16 Carson JL, Duff A, Poses RM, et al. Effect of anaemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348:1055-1060.
17 Carson JL, Noveck H, Berlin JA, et al. Mortality and morbidity in patients with very low postoperative Hb levels who decline blood transfusion. Transfusion. 2002;42:812-818.
18 Kuduvalli M, Oo AY, Newall N, et al. Effect of peri-operative red blood cell transfusion on 30-day and 1-year mortality following coronary artery bypass surgery. Eur J Cardiothorac Surg. 2005;27:592-598.
19 Koch CG, Li L, Duncan AI, et al. Morbidity and mortality risk associated with red blood cell and blood-component transfusion in isolated coronary artery bypass grafting. Crit Care Med. 2006;34:1608-1616.
20 Koch CG, Li L, Duncan AI, et al. Transfusion in coronary artery bypass grafting is associated with reduced long-term survival. Ann Thorac Surg. 2006;81:1650-1657.
21 Engoren MC, Habib RH, Zacharias A, et al. Effect of blood transfusion on long-term survival after cardiac operation. Ann Thorac Surg. 2002;74:1180-1186.
22 Vincent JL, Baron JF, Reinhart K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288:1499-1507.
23 Corwin HL, Gettinger A, Pearl RG, et al. The CRIT Study: anemia and blood transfusion in the critically ill —current clinical practice in the United States. Crit Care Med. 2004;32:39-52.
24 Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340:409-417.
25 Practice guidelines for blood component therapy. a report by the American Society of Anesthesiologists Task Force on Blood Component Therapy. Anesthesiology. 1996;84:732-747.
26 Practice guidelines for perioperative blood transfusion and adjuvant therapies. An updated report by the American Society of Anesthesiologists Task Force on Perioperative Blood Transfusion and Adjuvant Therapies. Anesthesiology. 2006;105:198-208.
27 Madjdpour C, Spahn DR, Weiskopf RB. Anemia and perioperative red blood cell transfusion: a matter of tolerance. Crit Care Med. 2006;34:S102-S108.
28 O’Shaughnessy DF, Atterbury C, Bolton Maggs P, et al. Guidelines for the use of fresh-frozen plasma, cryoprecipitate and cryosupernatant. Br J Haematol. 2004;126:11-28.
29 British Committee for Standards in Hematology. Guidelines for the use of platelet transfusions. Br J Haematol. 2003;122:10-23.
30 Spiess BD, Royston D, Levy JH, et al. Platelet transfusions during coronary artery bypass graft surgery are associated with serious adverse outcomes. Transfusion. 2004;44:1143-1148.
31 Baker RI, Coughlin PB, Gallus AS, et al. Warfarin reversal: consensus guidelines, on behalf of the Australasian Society of Thrombosis and Haemostasis. Med J Aust. 2004;181:492-497.
32 Hebert PC, Fergusson D, Blajchman MA, et al. Clinical outcomes following institution of the Canadian universal leukoreduction program for red blood cell transfusions. JAMA. 2003;289:1941-1949.
33 Fung MK, Rao N, Rice J, et al. Leukoreduction in the setting of open heart surgery: a prospective cohort-controlled study. Transfusion. 2004;44:30-35.
34 Fung MK, Moore K, Ridenour M, et al. Clinical effects of reverting from leukoreduced to nonleukoreduced blood in cardiac surgery. Transfusion. 2006;46:386-391.
35 van de Watering LM, Hermans J, Houbiers JG, et al. Beneficial effects of leukocyte depletion of transfused blood on postoperative complications in patients undergoing cardiac surgery: a randomized clinical trial. Circulation. 1998;97:562-568.
36 Vamvakas EC. WBC-containing allogeneic blood transfusion and mortality: a meta-analysis of randomized controlled trials. Transfusion. 2003;43:963-973.
37 Moore SB. Transfusion-related acute lung injury (TRALI): clinical presentation, treatment, and prognosis. Crit Care Med. 2006;34:S114-S117.
38 Gajic O, Gropper MA, Hubmayr RD. Pulmonary edema after transfusion: how to differentiate transfusion-associated circulatory overload from transfusion-related acute lung injury. Crit Care Med. 2006;34:S109-S113.
39 Curtis BR, McFarland JG. Mechanisms of transfusion-related acute lung injury (TRALI): anti-leukocyte antibodies. Crit Care Med. 2006;34:S118-S123.
40 Walsh TS, McArdle F, McLellan SA, et al. Does the storage time of transfused red blood cells influence regional or global indexes of tissue oxygenation in anemic critically ill patients ? Crit Care Med. 2004;32:364-371.
41 Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest. 2001;119:64S-94S.
42 Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large daily doses of heparin. Arch Intern Med. 1994;154:49-56.
43 Jones HU, Muhlestein JB, Jones KW, et al. Early postoperative use of unfractionated heparin or enoxaparin is associated with increased surgical re-exploration for bleeding. Ann Thorac Surg. 2005;80:518-522.
44 Warkentin TE. Heparin-induced thrombocytopenia: pathogenesis and management. Br J Haematol. 2003;121:535-555.
45 Warkentin TE, Greinacher A. Heparin-induced thrombocytopenia: recognition, treatment, and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:311S-337S.
46 Davidson BL. Preparing for the new anticoagulants. J Thromb Thrombol. 2003;16:49-54.
47 Kincaid EH, Monroe ML, Saliba DL, et al. Effects of preoperative enoxaparin versus unfractionated heparin on bleeding indices in patients undergoing coronary artery bypass grafting. Ann Thorac Surg. 2003;76:124-128. discussion 128
48 Hirsh J, Dalen J, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119:8S-21S.
49 Ansell J, Hirsh J, Poller L, et al. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:204S-233S.
50 Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165:1095-1106.
51 Palareti G, Leali N, Coccheri S, et al. Bleeding complications of oral anticoagulant treatment: an inception-cohort, prospective collaborative study (ISCOAT). Italian Study on Complications of Oral Anticoagulant Therapy. Lancet. 1996;348:423-428.
52 Levi M, Cromheecke ME, de Jonge E, et al. Pharmacological strategies to decrease excessive blood loss in cardiac surgery: a meta-analysis of clinically relevant endpoints. Lancet. 1999;354:1940-1947.
53 Laupacis A, Fergusson D. Drugs to minimize perioperative blood loss in cardiac surgery: meta-analyses using perioperative blood transfusion as the outcome. The International Study of Peri-operative Transfusion (ISPOT) Investigators. Anesth Analg. 1997;85:1258-1267.
54 Henry DA, Moxey AJ, Carless PA, et al. Anti-fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Systematic Reviews. 2001:CD 001886.
55 Sedrakyan A, Treasure T, Elefteriades JA. Effect of aprotinin on clinical outcomes in coronary artery bypass graft surgery: a systematic review and meta-analysis of randomized clinical trials. J Thorac Cardiovasc Surg. 2004;128:442-448.
56 Levy JH. Pharmacologic preservation of the hemostatic system during cardiac surgery. Ann Thorac Surg. 2001;72:S1814-S1820.
57 Levy JH. Hemostatic agents. Transfusion. 2004;44:58S-62S.
58 Lemmer JHJr, Stanford W, Bonney SL, et al. Aprotinin for coronary artery bypass grafting: effect on postoperative renal function. Ann Thorac Surg. 1995;59:132-136.
59 Beierlein W, Scheule AM, Dietrich W, et al. Forty years of clinical aprotinin use: a review of 124 hypersensitivity reactions. Ann Thorac Surg. 2005;79:741-748.
60 Dietrich W. Incidence of hypersensitivity reactions. Ann Thorac Surg. 1998;65:S60-S64. discussion S74-S76
61 Bidstrup BP, Underwood SR, Sapsford RN, et al. Effect of aprotinin (Trasylol) on aorta-coronary bypass graft patency. J Thorac Cardiovasc Surg. 1993;105:147-152. discussion 153
62 Havel M, Grabenwoger F, Schneider J, et al. Aprotinin does not decrease early graft patency after coronary artery bypass grafting despite reducing postoperative bleeding and use of donated blood. J Thorac Cardiovasc Surg. 1994;107:807-810.
63 Lemmer JHJr, Stanford W, Bonney SL, et al. Aprotinin for coronary bypass operations: efficacy, safety, and influence on early saphenous vein graft patency: a multicenter, randomized, double-blind, placebo-controlled study. J Thorac Cardiovasc Surg. 1994;107:543-551. discussion 551-553
64 Alderman EL, Levy JH, Rich JB. Analyses of coronary graft patency after aprotinin use: results from the International Multicenter Aprotinin Graft Patency Experience (IMAGE) trial. J Thorac Cardiovasc Surg. 1998;116:716-730.
65 Mangano DT, Tudor IC, Dietzel C. The risk associated with aprotinin in cardiac surgery. N Engl J Med. 2006;354:353-365.
66 Levy JH, Ramsay JG, Guyton RA. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953-1957. author reply 1953-1957
67 Ferraris VA, Bridges CR, Anderson RP. Aprotinin in cardiac surgery. N Engl J Med. 2006;354:1953-1957. author reply 1953-1957
68 Halkos ME, Levy JH, Chen E, Reddy VS, et al. Early experience with activated recombinant factor VII for intractable hemorrhage after cardiovascular surgery. Ann Thorac Surg. 2005;79:1303-1306.
69 Steiner ME, Key NS, Levy JH. Activated recombinant factor VII in cardiac surgery. Curr Opin Anaesthesiol. 2005;18:89-92.
70 Tanaka KA, Waly AA, Cooper WA, et al. Treatment of excessive bleeding in Jehovah ’s Witness patients after cardiac surgery with recombinant factor VIIa (NovoSeven). Anesthesiology. 2003;98:1513-1515.
71 Karkouti K, Beattie WS, Wijeysundera DN, et al. Recombinant factor VIIa for intractable blood loss after cardiac surgery: a propensity score-matched case-control analysis. Transfusion. 2005;45:26-34.
72 Goodnough LT, Lublin DM, Zhang L, et al. Transfusion medicine service policies for recombinant factor VIIa administration. Transfusion. 2004;44:1325-1331.
73 Hardy JF, de Moerloose P, Samama M. Massive transfusion and coagulopathy: pathophysiology and implications for clinical management. Can J Anaesthesiol. 2004;51:293-310.
74 Murray DJ, Olson J, Strauss R, et al. Coagulation changes during packed red cell replacement of major blood loss. Anesthesiology. 1988;69:839-845.
75 Collier B, Kolff J, Devineni R, et al. Prophylactic positive end-expiratory pressure and reduction of postoperative blood loss in open-heart surgery. Ann Thorac Surg. 2002;74:1191-1194.
76 Zurick AM, Urzua J, Ghattas M, et al. Failure of positive end-expiratory pressure to decrease postoperative bleeding after cardiac surgery. Ann Thorac Surg. 1982;34:608-611.
77 Ferraris VA, Spiess BD: Perioperative blood transfusion and blood conservation in cardiac surgery: The Society of Thoracic Surgeons and The Society of Cardiovascular Anesthesiologists Practice Guidelines Series. Ann Thorac Surg (in press)
78 Rizvi MA, Shah SR, Raskob GE, et al. Drug-induced thrombocytopenia. Curr Opin Hematol. 1999;6:349-353.
[/level-membership-for-cardiothoracic-surgery-category][not-level-membership-for-cardiothoracic-surgery-category]
Chapter 30 Bleeding, Hemostasis, and Transfusion Medicine
The major components of the hemostatic system are the vasculature, platelets, and coagulation proteins. Deficiencies or abnormalities in any of these components can lead to coagulopathy and pathologic bleeding. Almost every imaginable quantitative and qualitative abnormality of the hemostatic system has been described following cardiac surgery, and managing postoperative bleeding remains one of the greatest challenges in caring for patients after cardiac surgery.
In this chapter, issues relating to bleeding and hemostasis in cardiac surgery patients are reviewed. Routine management of postoperative bleeding, including a protocol for blood component therapy, is outlined in Chapter 17.
PHYSIOLOGY AND PATHOPHYSIOLOGY
Normal Hemostasis
The vascular endothelium is a nonthrombogenic surface that secretes prostacyclin, tissue plasminogen activator, heparin sulfate, antithrombin III, protein C, and nitric oxide to inhibit platelet activation, and preserve vascular patency. However, if a blood vessel is cut or otherwise damaged, the subendothelial vascular basement membrane is exposed, and tissue factor and other molecular promoters are released, leading to platelet activation and thrombin generation which, in turn, leads to the formation of fibrin from fibrinogen and the creation of a stable fibrin-platelet clot.1,2
Coagulation is classically represented as two separate pathways that converge and form a final common pathway that leads to thrombin generation (Fig. 30-1). However, this model is derived from in vitro tests of coagulation and does not represent the in vivo state. It does not explain how deficiencies of some “intrinsic” pathway factors (factor XII, high molecular weight kininogen, prekallikrein) do not cause clinical bleeding, whereas deficiencies of other intrinsic factors (factors VIII and IX) do. Further, under normal circumstances hemostasis is initiated by the release of tissue factor by damaged endothelium (i.e., the “extrinsic” pathway), not by contact activation of factor XII (i.e., the intrinsic pathway). Although hemostasis is initiated by tissue factor, the regulation of thrombin generation at the site of endothelial injury is determined almost entirely by platelets. Platelet adhesion is initiated when receptors on platelet membranes bind to the subendothelial collagen by forming a bridge with von Willebrand factor (vWF). Once platelets adhere, they expose factors on their surface that provide a template for initiation of the coagulation cascade and the formation of the hemostatic plug. The cell-based model of coagulation shown in Figure 30-2 describes this central role for platelets in the generation of thrombin. Thrombin cleaves fibrinogen to fibrin monomer, which automatically polymerizes to long-chain fibrin fibers in a few seconds. Over the next few minutes, cross-linking between fibrin strands occurs, forming a stable fibrin mesh that traps platelets and forms a hemostatic plug.
Abnormal Bleeding Following Cardiac Surgery
Excess bleeding following cardiac surgery may reflect inadequate surgical hemostasis. Commonly, however, abnormal blood clotting due to coagulopathy, platelet dysfunction, and excess fibrinolysis contribute to or are the primary causes of postoperative bleeding. The factors that contribute to postoperative bleeding may be categorized as patient-related, intraoperative, and postoperative (Table 30-1).
Table 30-1 Risk Factors for Excessive Postoperative Bleeding in Patients Undergoing Cardiac Surgery
| Patient Variables |
| Advanced age (>70 years) |
| Preoperative anemia |
| Female gender |
| Increased body size |
| Cardiogenic shock, congestive heart failure, or poor left ventricular function |
| Renal insufficiency |
| Peripheral vascular disease |
| Insulin-dependent diabetes mellitus |
| Liver failure or hypoalbuminemia |
| Preoperative sepsis |
| Preoperative antithrombotic therapy |
| High intensity: abciximab, clopidogrel, direct thrombin inhibitors, low molecular weight heparin, fibrinolytic therapy |
| Low intensity: aspirin, dipyridamole, eptifibatide, tirofiban |
| Preoperative coagulopathy |
| Hereditary coagulopathy or platelet defect (e.g., von Willebrand disease, hemophilia A or B) |
| Acquired coagulopathy or platelet defect (e.g., nonspecific prolongation of bleeding time, chronic lymphocytic leukemia, lupus anticoagulant, polycythemia, myelodysplastic syndrome, idiopathic thrombocytopenic purpura, β-thalassemia) |
| Intraoperative and Postoperative Variables |
| Prolonged CPB time |
| Reoperation |
| Type of operation: aortic/complex >valve + CABG >valve >CABG |
| Use of internal mammary artery (either one or two) for CABG surgery |
| Reduced heparin dose |
| Increased protamine dose after CPB |
| Increased cell saver volume |
| Intraoperative autologous donation |
| Need for transfusion while on CPB |
| Low body temperature in ICU |
| Use of hydroxyethyl starch for volume expansion |
| Lack of transfusion algorithm with point-of-care testing |
CABG, coronary artery bypass graft; CPB, cardiopulmonary bypass; ICU, intensive care unit.
Modified from Ferrasis VA, Spiess BD: Perioperative blood transfusion and blood conservation in cardiac surgery: The Society of Thoracic Surgeons and The Society of Cardiovascular Anesthesiologists Practice Guidelines Series. Ann Thorac Surg [in press].
Patient Factors
Platelet Dysfunction and Thrombocytopenia.
Treatment with antiplatelet drugs (aspirin, nonsteroidal antiinflammatory drugs, clopidogrel, and glycoprotein IIb/IIIa inhibitors) is common in patients undergoing cardiac surgery (see Chapter 3). Acquired platelet dysfunction occurs with chronic liver disease and renal failure. A blood urea nitrogen concentration greater than 35 mmol/l (> 100 mg/dl) contributes to postoperative bleeding. Treatment of uremic coagulopathy involves dialysis, desmopressin, and platelets.
Causes of low platelet counts include: (1) heparin-induced thrombocytopenia (see subsequent material); (2) drugs (Table 30-2); (3) splenic sequestration, commonly due to splenomegaly secondary to portal hypertension or tumor infiltration; (4) reduced bone marrow activity (e.g., cytotoxic drugs, fibrosis, or tumor infiltration); (5) HIV/AIDS; and (6) idiopathic thrombocytopenic purpura, which is a condition characterized by immunologic destruction of platelets; it may be acute or chronic, mild or severe. The treatment of severe thrombocytopenia due to idiopathic thrombocytopenic purpura is usually corticosteroids. Patients with chronic cyanosis due to right-to-left intracardiac shunting have increased bleeding tendencies due to thrombocytopenia, platelet dysfunction, and reduced levels of some coagulation factors.
Table 30-2 Drugs Associated With Thrombocytopenia That May Be Used in Patients in the ICU
| Antimicrobials | Analgesics |
|---|---|
| Trimethoprim/sulfamethoxazole | Acetaminophen |
| NSAIDs | |
| Rifampin | H2-receptor antagonists |
| Amphotericin | Ranitidine |
| Vancomycin | Cimetidine |
| Piperacillin | CNS drugs |
| Ampicillin | Haloperidol |
| Cardiovascular drugs | Chlorpromazine |
| Thiazide diuretics | Diazepam |
| Amiodarone | Lithium |
| Quinidine | Carbamazapine |
| Amrinone | Glycoprotein IIb/IIIa antagonists |
| Captopril | |
| Digoxin | Abciximab |
| Nitroglycerine | Eptifibatide |
| Alprenolol | Tirofiban |
| Oxprenolol | |
| Diazoxide | |
| Methyldopa |
Modified from Rizvi MA, Shah SR, Raskob GE, et al: Drug-induced thrombocytopenia. Curr Opin Hematol 6:349-353, 1999. CNS, central nervous system; ICU, intensive care unit; NSAIDs, nonsteroidal antiinflammatory drugs.
Coagulopathy.
Acquired coagulopathy may occur as the result of treatment with heparins, warfarin or, less commonly, thrombin inhibitors (e.g., bivalirudin). Herbal supplements, notably garlic, gingko, and ginseng,3 can also cause coagulopathy. The most important nonpharmacologically acquired coagulopathy is that associated with chronic liver disease, which can lead to deficiency of all coagulation proteins (especially factors I, II, V, VII, IX, and X). Chronic liver disease also causes portal hypertension, which is associated with esophageal varices and splenomegaly (with platelet sequestration). Esophageal varices can rupture, either spontaneously or during instrumentation of the esophagus, causing catastrophic bleeding.
Vitamin K is a fat-soluble vitamin that is required for the hepatic synthesis of factors II, VII, IX, and X. Vitamin K deficiency is caused by poor dietary intake and malabsorption; it often coexists with chronic liver disease, particularly biliary cirrhosis. Coagulopathy may also occur due to sepsis (see subsequent material), most commonly in patients with acute endocarditis. Congenital defects of coagulation, such as hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency), are uncommon.
Intraoperative Factors
Intraoperative factors that contribute to postoperative bleeding include the effects of cardiopulmonary bypass (CPB), the type and duration of the surgical procedure, and the management of anticoagulation. The effects of CPB and surgery on hemostasis are complex.4 CPB and surgical trauma elicit a marked systemic inflammatory response characterized by leukocyte activation, release of inflammatory mediators, and activation of platelets and the coagulation and complement cascades (see Chapter 2). Activation of platelets and coagulation occurs through blood contact with the CPB circuit, through surgical trauma, and through the reinfusion of pericardial blood. Platelets are also activated by heparin and hypothermia. Hemodilution of platelets and coagulation proteins occurs because of the CPB prime solution and other intravenous fluids. Platelets are sequestered onto Dacron grafts. Fibrinolysis is stimulated by hypothermia, contact activation of thrombin and factor XII, and the release of tissue plasminogen activator from the vascular walls. These adverse effects are more pronounced when CPB is prolonged and deep hypothermia is utilized.
Various intraoperative strategies may be employed to reduce bleeding and allogenic (i.e., donor) blood transfusion. They include: (1) antifibrinolytics; (2) heparin-coated CPB circuits; (3) avoidance of deep hypothermia; (4) autologous (i.e., the patient’s) blood donation; (5) normovolemic hemodilution; (6) intraoperative blood recovery and reinfusion through a cell saver. Surgical strategies to reduce postoperative blood loss include meticulous surgical hemostasis and the use of topical agents such as surgical adhesives (e.g., BioGlue) and thrombin preparations (e.g., Thrombostat).
Postoperative Factors
Beyond the early postoperative period, pathologic bleeding is usually related to antithrombotic drugs. Acute gastrointestinal stress ulceration and hemorrhage is the most common nonsurgical cause of bleeding. Stress ulcer prophylaxis is discussed in Chapter 34. Thrombocytopenia is common in patients with critical illness. The causes include: (1) heparin-induced thrombocytopenia (see subsequent material); (2) drugs (see Table 30-2); (3) platelet destruction due to intraaortic balloon counterpulsation, renal replacement therapy, or mechanical cardiac support; (4) sepsis and disseminated intravascular coagulation (see subsequent material); (5) posttransfusion purpura (see subsequent material). Usually, platelet transfusion is required only to treat bleeding or before invasive procedures.
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a generalized disorder of the coagulation system involving the inappropriate activation of platelets, coagulation, and fibrinolysis, resulting in a consumptive coagulopathy and pathologic thrombosis. It is characterized by pathologic bleeding from surgical sites, intravenous cannula sites, and mucosal surfaces and, rarely, by clinically significant thrombosis. Following an initiating event, there are normally two stages of DIC. Stage 1 (early) involves excessive activation of platelets and coagulation proteins, resulting in generalized microvascular thrombosis and obstruction. This obstruction leads to red cell fragmentation and coagulation factor and platelet consumption, resulting in bleeding due to hemostatic factor and platelet depletion (stage 2, late). DIC has multiple causes, including trauma, hemolytic transfusion reaction, obstetric complications and, most important, sepsis. In addition, the consumptive coagulopathy that accompanies prolonged CPB or extracorporeal membrane oxygenation (ECMO) shares many of the hemostatic features of late DIC, namely, low platelet numbers, low fibrinogen, prolonged prothrombin time, prolonged aPTT, elevated D-dimer, and characteristic changes on the thromboelastogram (see Fig. 30-4).5 Management of DIC involves treating the underlying causes and controlling bleeding by means of blood component therapy. In the relatively rare circumstance of clinically important thrombosis, systemic heparinization is indicated.
Prothrombotic States
Important inherited prothrombotic conditions include factor V Leiden (the product of a specific mutation in the factor V gene that confers resistance to factor V cleavage by activated protein C); deficiencies of protein C and protein S (a cofactor for activated protein C); antithrombin III deficiency; and a mutant gene for prothrombin that causes increased levels of the normal protein. Antiphospholipid syndrome and elevated plasma homocysteine are two important acquired prothrombotic conditions. Antiphospholipid syndrome is caused by antibodies (including lupus anticoagulant and anticardiolipin antibodies) directed against cell membrane phospholipids. The term lupus anticoagulant is misleading because patients typically do not have systemic lupus erythematosus and, although the aPTT is elevated, patients have a prothrombotic tendency. Severe systemic illnesses are also associated with an increased risk for pathologic thrombosis. Cancer, major surgery (including cardiac and thoracic surgery), congestive heart failure, myocardial infarction, and stroke all provoke a hypercoagulable state and are risk factors for thromboembolism. High platelet counts and fibrinogen levels, as part of systemic inflammation, also contribute.
Patients with known or suspected hypercoagulability should receive prophylaxis against deep vein thrombosis (DVT) and pulmonary embolism (see Chapter 17) as soon as postoperative bleeding has settled. If an inherited or acquired hypercoagulable state (excluding pregnancy or systemic illness) is suspected, a thrombophilia screen should be obtained.
Anemia
There are multiple causes of anemia in cardiac surgery patients (Table 30-3). Gastrointestinal hemorrhage is reasonably common, particularly in patients taking antithrombotic drugs. Patients may report an alteration in bowel habit and stool, but gastrointestinal blood loss is commonly asymptomatic. Occult retroperitoneal hematoma must be considered in those who have recently undergone angiography via femoral arterial puncture. Intravascular hemolysis is a potential cause in patients with regurgitant or stenotic heart valves. In patients with severe anemia who need urgent surgery, preoperative transfusion of packed red cells is indicated. For elective surgery and in those who belong to Jehovah’s Witnesses (for whom red cell transfusion may be unacceptable), preoperative treatment with iron supplements and erythropoietin is beneficial. In one study, a 14-day regimen of 300 mg per day of oral iron plus 5 doses of 500 U/kg erythropoietin was effective in increasing hemoglobin concentration by about 1.6 g/dl.6
TESTS OF HEMOSTASIS
Laboratory Versus Point-of-Care Testing
Traditionally, tests of hemostatic function (other than activated clotting time) have been performed in the hematology/coagulation laboratory. However, in an actively bleeding patient, the time required to label, transport, and report on the test causes a significant delay in obtaining the results. This has led to efforts to develop hemostatic tests that can be performed at or near the bedside (i.e., the point of care). Most of the hemostatic tests described here can be performed as point-of-care tests. By incorporating point-of-care testing into management algorithms, bleeding and transfusion requirements can be reduced.7,8
Activated Partial Thromboplastin Time
The aPTT monitors primarily the competency of the intrinsic coagulation pathway (i.e., high molecular weight kininogen, prekallikrein, and factors VIII, IX, X, XI, XII) and, to a lesser extent, the common pathway. The test involves adding a phospholipid tissue extract—which activates factor XII and calcium ions—to citrated plasma. The aPTT is used to monitor the effectiveness of anticoagulation with unfractionated heparin and direct thrombin inhibitors (see subsequent material). The normal range is 20 to 35 seconds but values vary slightly among laboratories. The therapeutic range for systemic heparinization is 50 to 80 seconds. Causes of prolonged aPTT include (1) deficiency of intrinsic pathway factors (e.g., due to consumptive coagulopathy or hemophilia); (2) unfractionated heparin; (3) the presence of acquired inhibitors (i.e., antibodies) of factors VIII and IX; and (4) the presence of lupus anticoagulant. Although typically associated with an elevated prothrombin time (PT), a mildly elevated aPTT may also be seen with warfarin overdose, liver disease, and vitamin K deficiency.
Fibrinogen
The normal range for the plasma fibrinogen concentration is 200 to 400 mg/dl. Levels increase as part of the acute-phase response and decrease with consumptive coagulopathy and advanced liver and renal disease. Several days following cardiac surgery, fibrinogen levels can increase to more than 500 mg/dl and remain elevated for up to a month.9
D-Dimer
D-dimer is a degradation product of cross-linked fibrin. Levels are normally low (<500 μg/l). Higher levels occur in any situation in which there is excess clotting followed by fibrinolysis; in particular, higher levels occur with venous thromboembolism (see Chapter 23), major hemorrhage, therapy, and DIC. Moderate elevations in the D-dimer are also seen in patients with severe systemic illness. Following cardiac surgery, the fibrinolytic D-dimer concentration is commonly greater than 1000 μg/l and remains elevated for as long as 60 days.9
Activated Clotting Time
The activated clotting time (ACT) is a bedside test that is used to assess the effectiveness of heparin anticoagulation. Whole blood is added to a tube or cartridge containing an activator (celite, kaolin, glass beads, or phospholipid tissue extract), which stimulates contact activation of the intrinsic coagulation pathway. The activated blood sample is then placed in a device that warms the blood and records the time it takes for clot to form. Devices from different manufacturers use different activators and different methodologies of assessing clot formation; thus, ACT values from different machines cannot be used interchangeably. The normal range for ACT depends on the device being used, but it is usually within the range of 70 to 150 seconds. Values above 400 seconds are generally used for CPB; values of 160 to 180 seconds are used for continuous renal replacement therapy (CRRT; see Chapter 33) and ECMO (see Chapter 22).
The ACT has the advantage of being able to be rapidly performed at the bedside. Most ACT devices have two channels, one of which can be used with a heparinase-containing cartridge. Heparinase is an enzyme that neutralizes heparin. An elevated ACT with a normal heparinase ACT indicates a heparin effect; if both ACTs are elevated, coagulopathy is suggested. The ACT has several limitations. First, as outlined earlier, values vary among devices and may not reliably correlate with plasma heparin concentrations.10 Second, values for ACT (more so than values for aPTT) are influenced by factor depletion, thrombocytopenia, platelet dysfunction, and antifibrinolytic therapy. Finally, as a point-of-care test, a quality assurance program must be maintained, but most ACT machines do not allow patient and user identification to be recorded.
Platelet Count and Platelet Function Tests
The normal value for the platelet count is 140,000 to 450,000/mm3. This parameter is easily measurable, but specific tests of platelet function are not widely available. Currently available point-of-care tests of platelet function, such as the PFA-100 and the HemoSTATUS analyzers, have proved disappointing as sole measures of hemostatic function in cardiac surgery patients.11–13 Important information about platelet function can be inferred from the thromboelastogram (see subsequent material).
Thromboelastography
Standard thromboelastogram parameters are shown in Figure 30-3; typical abnormal patterns are shown in Figure 30-4. Point-of-care thromboelastogram-guided blood transfusion algorithms, either alone7,14 or in conjunction with other tests,8 have been shown to reduce transfusion requirements. As with ACT, point-of-care thromboelastogram analysis requires a quality assurance program.
TRANSFUSION MEDICINE
Blood Products
Red Blood Cells
Red blood cells are usually administered as packed cells from which much of the non-red-cell component has been removed and the blood has been resuspended in an anticoagulant storage solution, usually citrate-phosphate-dextrose-adenine. A unit of packed red cells has a volume of about 300 ml and a hematocrit of about 60%. Without continuing blood loss, transfusion of 4 to 5 ml/kg of packed red cells increases hemoglobin concentration by about 1 g/dl. Packed red cells are stored at 4°C and have a shelf life of 35 to 42 days. During storage, red cells metabolize dextrose into lactate, resulting in a fall in pH and an increase in lactate. Red cell fragility increases progressively, and temperature-sensitive failure of the red cells’ sodium-potassium pump occurs, resulting in an increase in the concentrations of free hemoglobin and extracellular potassium. There is also progressive loss of cellular 2,3-diphosphoglycerate (2,3-DPG), which is probably responsible for the short-lived (12 to 24 hours) leftward shift in patients’ oxygen-hemoglobin curves (and therefore the reduced tissue unloading of oxygen) that occurs with the transfusion of old blood. Despite these effects, no convincing data indicate an association between length of storage of red blood cells and adverse outcomes in cardiac surgery patients.15
Blood Groups and Compatibility Testing.
Red blood cell membranes contain many blood group antigens, the most important of which are those of the ABO and rhesus (Rh) blood groups. The ABO blood group is composed of two antigens, A and B, and their respective antibodies, anti-A and anti-B (Table 30-4). Antibodies naturally develop to nonself antigens during early childhood. Administration of red cells containing A or B antigen to a patient with a corresponding antibody results in a life-threatening hemolytic transfusion reaction. The compatibility of ABO groups with red cell and plasma products are shown in Table 30-5.
| Blood Group | Red Cells | Plasma |
|---|---|---|
| A | A antigen | Anti-B antibody |
| B | B antigen | Anti-A antibody |
| O | Neither A nor B antigens | Both anti-A and anti-B antibodies |
| AB | Both A and B antigens | Neither anti-A nor anti-B antibodies |
Table 30-5 Compatibility of ABO Groups for Red Cells and Plasma oxygen-hemoglobin
| Recipient ABO Blood Group | Compatible Donor ABO Groups |
|---|---|
| Red Cells | |
| Unknown | O |
| O | O only |
| A | A or O |
| B | B or O |
| AB | AB or B or O |
| Plasma Components | |
| Unknown | AB |
| O | O or A or B or AB |
| A | A or AB |
| B | B or AB |
| AB | AB (or A if AB is unavailable) |
Before blood is issued for transfusion, three processes usually occur: (1) ABO and Rh typing of the recipient’s blood; (2) antibody screening, in which recipient’s plasma and commercially supplied red cells containing antigens responsible for hemolytic transfusion reactions are combined; (3) cross-matching of the donor red blood cells and the recipient’s plasma. These processes usually take about 45 minutes. Before surgical procedures that are unlikely to require a blood transfusion a “type and screen” is performed, so if blood is subsequently required, only a cross-match is necessary. Most transfusion services issue type-specific blood, that is, group A blood to a group A recipient, Rh (+) blood to an Rh (+) recipient. Alternatively, cross-matched group O blood may be issued (see Table 30-5
[/not-level-membership-for-cardiothoracic-surgery-category]
