[level-membership-for-obstetrics-gynecology-category]
CHAPTER 11 Biosynthesis of steroid hormones
Physiology
The classification of steroid hormones
All steroid hormones are ultimately derived from the 27 carbon (C) substrate cholesterol and so share the same cyclohexaphenanthrene ring structure (Figures 11.1 and 11.2). Steroids are classified into five families dependent upon the number of carbon atoms and the chemical groups present at key carbon residues (Table 11.1). Those steroid hormones with 21 carbon atoms are collectively termed ‘pregnenes’, and this category of steroid hormones can be subdivided into three steroid families: progestins (e.g. progesterone), glucocorticoids (e.g. cortisol) and mineralocorticoids (e.g. aldosterone). While the progestins are secreted predominantly from the ovary, the glucocorticoids and mineralocorticoids are collectively termed ‘corticosteroids’, reflecting their origin in the cortex of the adrenal gland. The major structural difference between the three pregnene families is that while progestins possess a methyl group (CH3) at position C21, both glucocorticoids and mineralocorticoids possess a C21 hydroxyl group (CH2OH) (see Table 11.1 and Figure 11.2). Progestins can be metabolized to generate 19 carbon steroids, termed ‘androgens’ [dehydroepiandrosterone (DHEA), androstenedione and testosterone], which are secreted from both the testis and the adrenal cortex. Within the ovary, androgens are usually metabolized to generate the oestrogens (e.g. oestradiol-17β) with their characteristic 18 carbon structure (see Table 11.1 and Figure 11.2).
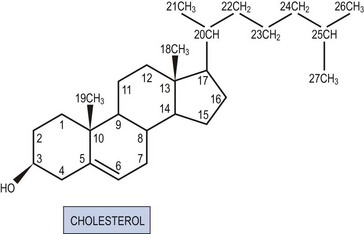
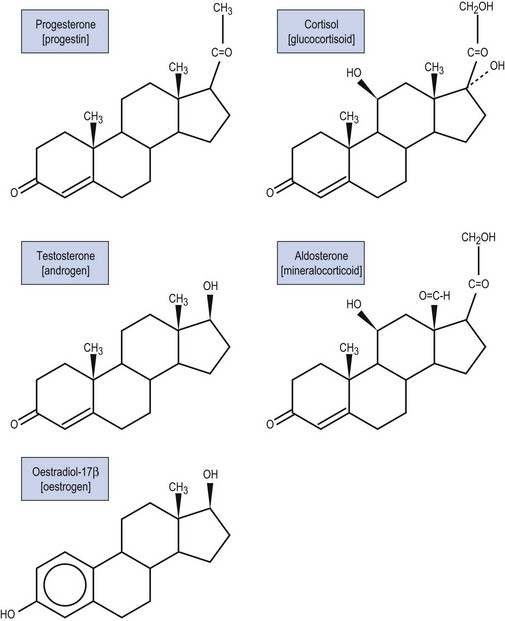
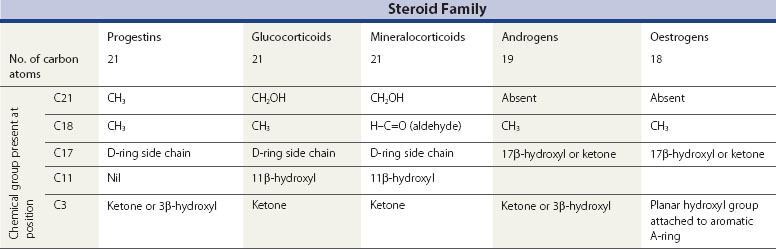
Within this classification scheme, there is a distinction to be made between Δ5 and Δ4 steroid hormones. Progestins and androgens of the Δ5 series are characterized by possessing a hydroxyl group at position C3 and a C C double bond between positions C5 and C6 in the steroid B-ring, as in cholesterol (Figure 11.1). In contrast, progestins and androgens of the Δ4 series possess a ketone (C
C double bond between positions C5 and C6 in the steroid B-ring, as in cholesterol (Figure 11.1). In contrast, progestins and androgens of the Δ4 series possess a ketone (C O) at position C3 and have their C
O) at position C3 and have their C C double bond between positions C4 and C5 in the A-ring of the steroid molecule (Figure 11.2). While this difference may seem a trivial biochemical detail, nothing could be further from the truth. The nature of the chemical group at position C3, together with the position of the C
C double bond between positions C4 and C5 in the A-ring of the steroid molecule (Figure 11.2). While this difference may seem a trivial biochemical detail, nothing could be further from the truth. The nature of the chemical group at position C3, together with the position of the C C double bond, profoundly alters the conformation of the steroid molecule and, in so doing, influences the ability of the hormone to activate intracellular receptors. Hence, Δ5 steroids, such as pregnenolone and DHEA, have low affinities for steroid receptors such that they can only exert limited biological actions. In contrast, the Δ4 steroids, such as progesterone and testosterone, are potent activators of steroid receptors and so act as the dominant physiological hormones.
C double bond, profoundly alters the conformation of the steroid molecule and, in so doing, influences the ability of the hormone to activate intracellular receptors. Hence, Δ5 steroids, such as pregnenolone and DHEA, have low affinities for steroid receptors such that they can only exert limited biological actions. In contrast, the Δ4 steroids, such as progesterone and testosterone, are potent activators of steroid receptors and so act as the dominant physiological hormones.
Origins and intracellular transport of cholesterol
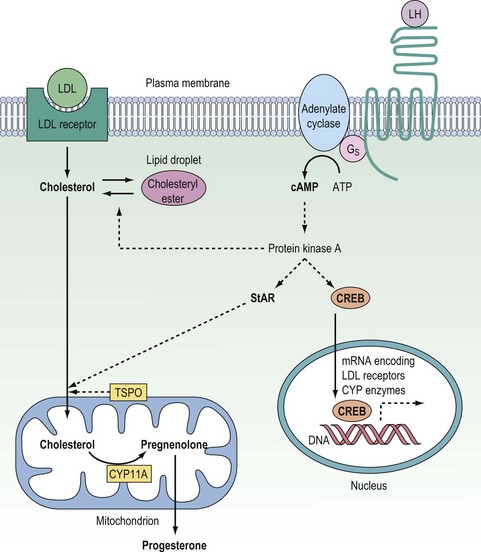
Once inside the cell, cholesterol must be transported across the cytosol and into the mitochondria where the first and rate-determining reaction of steroid synthesis occurs: the catabolism of cholesterol to form pregnenolone (see below). The transport of cholesterol across the two mitochondrial membranes and interceding aqueous intermembrane space is now recognized as the true rate-limiting step for steroidogenesis, and so with this realization, it is this process which has been the focus of research into steroid biosynthesis over the past two decades. Although the understanding of the molecular mechanisms by which cholesterol passes from the outer leaflet of the outer mitochondrial membrane (OMM) to the inner leaflet of the inner mitochondrial membrane (IMM) is still far from complete, significant advances have been made over the past decade. In the mid 1990s, the importance of the steroidogenic acute regulatory (StAR) protein was first indicated when Clark and Stocco showed StAR to be a short half-life protein which is rapidly upregulated in steroidogenic cells following stimulation either by trophic hormones (e.g. gonadotrophins) or by the common second messenger, cyclic adenosine 3′,5′-monophosphate (cAMP) (Stocco and Clark 1996). Analysis of the StAR peptide sequence suggested that this protein gets imported from the cytosol into the IMM, and it was postulated that the StAR protein might simply bind and transport cholesterol across the aqueous intermembrane space during the course of that import. However, subsequent site-directed mutagenesis studies revealed that the StAR protein could still drive cholesterol flux for steroidogenesis even when it was modified to prevent mitochondrial import, and it is now accepted that the StAR protein can act at the OMM to drive cholesterol passage from the outer leaflet of the OMM to the inner leaflet of the IMM without itself trafficking into the mitochondria (Miller 2007). This steroidogenic action of the StAR protein does, however, require: (i) that the StAR protein be phosphorylated on key serine residues by the cAMP-dependent protein kinase A (PKA); and (ii) that the phosphorylated form of the StAR protein interacts with the 18 kDa translocator protein (TSPO) expressed in the OMM (Figure 11.3) (Hauet et al 2005, Miller 2007). The TSPO protein, which was originally described as a peripheral benzodiazepine receptor (PBR), is very highly expressed in the OMM of steroidogenic cells. Recent studies have revealed that rather than reliance on a single protein to drive mitochondrial cholesterol uptake for steroidogenesis, this pivotal rate-limiting step involves a complex of proteins which includes the StAR and TSPO proteins in association with PBR-associated protein-7 (PAP7). By virtue of its structural interactions with both the TSPO protein and the PKAR1α regulatory subunit of PKA, PAP7 acts as a PKA-anchoring protein, localizing PKA to the OMM where it can phosphorylate the serine residues in the StAR protein to drive the StAR–TSPO interaction required to deliver cholesterol to the IMM for the first reaction in the steroidogenic pathway (Liu et al 2006, Miller 2007). While the StAR protein is not expressed in the placenta, this tissue does express a homologous protein, metastatic lymph node 64, which shares several functional and structural properties with the StAR protein, and is assumed to drive mitochondrial cholesterol uptake for placental steroidogenesis (Petrescu et al 2001).
Steroidogenic enzymes
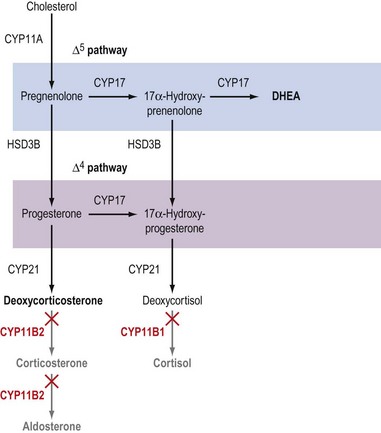
Although the biochemical pathway of steroid hormone synthesis, summarized in Figure 11.4, can look like a bewildering railway map, the whole pathway becomes more straightforward on recognizing that the eight major steroidogenic enzymes can all be classified into just two enzyme families (the cytochrome P450 enzymes and the hydroxysteroid dehydrogenases), each with definitive functions. The steroidogenic cytochrome P450 (CYP) enzymes are members of a larger superfamily of CYP oxidase enzymes. Each CYP enzyme catalyses a hydroxylation reaction by serving as the terminal electron acceptor in an electron transport pathway which transfers electrons from a pyridine nucleotide cofactor, nicotinamide adenine dinucleotide phosphate, via two sequential flavoproteins (ferrodoxin reductase and ferrodoxin) on to atomic oxygen (Miller 2008). This allows the atomic oxygen to be inserted, by the CYP enzyme, into a hydrocarbon (C․H) bond thus generating a polar hydroxyl group (C․O․H) which renders the substrate molecule more water soluble/hydrophilic. The non-steroidogenic CYP enzymes include those hepatic oxidase enzymes responsible for the inactivation and clearance of a wide range of drugs. Each of the CYP enzymes within the steroidogenic pathway has traditionally been referred to by a variety of names that reflect their ability to catalyse hydroxylation of a steroid substrate at a specific carbon position. However, following the completion of the human genome (HUGO) project, all steroidogenic CYP enzymes have been assigned new systematic names which relate to the CYP gene that encodes the enzyme protein (Table 11.2).
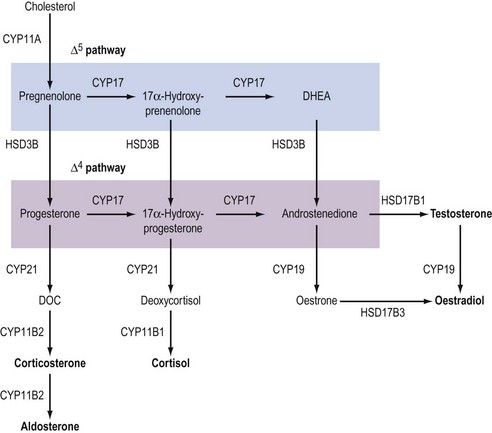
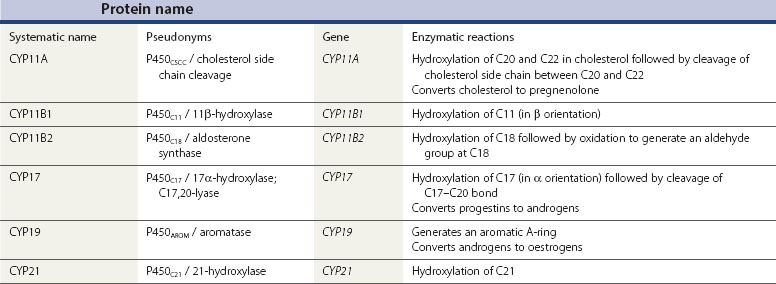
In addition to their characteristic hydroxylase activities, some steroidogenic CYP enzymes also catalyse lyase reactions. Specifically, the CYP11A (cytochrome P450 cholesterol side chain cleavage) and CYP17 (cytochrome P450 17α-hydroxylase/C17,20-lyase) enzymes can each catalyse cleavage of the C․C bond weakened by the hydroxylation reactions. CYP11A acts in the IMM to catalyse the rate-limiting reaction in steroid synthesis: the conversion of cholesterol to pregnenolone. This crucial CYP enzyme hydroxylates two adjacent carbons (C20 and C22) in the D-ring side chain of cholesterol, facilitating cleavage between C20 and C22 to leave the Δ5 21 carbon steroid, pregnenolone (Miller 2008). Similarly, CYP17 introduces a hydroxyl group at position C17 of either pregnenolone or progesterone, as a result of which the weakened C17–C20 bond breaks to generate either DHEA or androstenedione, respectively (Miller 2008) (see Figure 11.4). Other members of the CYP enzyme family simply catalyse introduction of oxygen to generate hydroxyl groups at specific carbon positions (see Table 11.2). The hydroxylations catalysed by CYP21 (21-hydroxylase) and CYP11B1 (11β-hydroxylase) are pivotal in the formation of corticosteroids by the adrenal cortex (Miller 2008). The hydroxyl group generated at position C18 by CYP11B2 (aldosterone synthase) undergoes rapid oxidation to form an aldehyde group (Curnow et al 1991), hence giving rise to the name ‘aldosterone’ (see Figure 11.2). Finally, CYP19 (aromatase) is the most complex member of the steroidogenic CYP enzyme family, catalysing a series of reactions that convert C19 androgens (androstenedione and testosterone) to their C18 oestrogen metabolites (oestrone and oestradiol, respectively). In this reaction sequence, the C19 methyl group is lost and the ketone at position C3 is reduced to a hydroxyl group (Miller 2008). This liberates electrons which are invested in the A-ring of the steroid to generate the aromatic phenol ring, the hallmark of oestrogens (see Figure 11.2) and a prerequisite for activation of the oestrogen receptor.
The second family of steroidogenic enzymes comprises the hydroxysteroid dehydrogenase (HSD) enzymes encoded by HSD genes (Penning 1997, Miller 2008). (As with the CYP enzymes, the HSD enzymes have all been renamed following the completion of the HUGO project, such that the systematic names for the 3βHSD and 17βHSD isoenzymes have changed to HSD3B and HSD17B, respectively.) All of the HSD enzymes catalyse the interconversion of a hydroxyl group with a ketone to influence the affinity of the steroid hormone for its intracellular receptors (Penning 1997). Each HSD enzyme is specified by a number and a Greek letter; the number denotes the position of the carbon atom at which the enzyme acts, and the Greek letter indicates the orientation of the OH group relative to the steroid molecule (where α and β indicate bonds below and above the plane of the molecule, respectively).
There are two HSD enzymes of significance in steroid hormone production. The two cloned HSD3B isoenzymes convert the 3β-hydroxyl group of weak Δ5 steroids to a ketone, and also catalyse movement of the C C double bond to generate the corresponding Δ4 steroid metabolite, thus increasing steroid potency (Penning 1997, Miller 2008). Hence, both the placental HSD3B1 enzyme and HSD3B2, expressed in the ovary, testis and adrenal gland, can convert weak Δ5 steroids, such as pregnenolone and DHEA, to the active Δ4 progestins and androgens, progesterone and androstenedione, respectively (see Figure 11.4). In contrast, the major HSD17B isoenzymes act at position C17 where they catalyse not oxidation but reduction, converting a ketone at C17 to a β-hydroxyl group. The significance of this reaction is attested to by the fact that androstenedione and oestrone (which have ketone groups at position C17) are relatively weak steroids, whereas their 17β-hydroxy-metabolites, testosterone and oestradiol-17β (see Figure 11.2), are the more potent androgen and oestrogen, respectively.
C double bond to generate the corresponding Δ4 steroid metabolite, thus increasing steroid potency (Penning 1997, Miller 2008). Hence, both the placental HSD3B1 enzyme and HSD3B2, expressed in the ovary, testis and adrenal gland, can convert weak Δ5 steroids, such as pregnenolone and DHEA, to the active Δ4 progestins and androgens, progesterone and androstenedione, respectively (see Figure 11.4). In contrast, the major HSD17B isoenzymes act at position C17 where they catalyse not oxidation but reduction, converting a ketone at C17 to a β-hydroxyl group. The significance of this reaction is attested to by the fact that androstenedione and oestrone (which have ketone groups at position C17) are relatively weak steroids, whereas their 17β-hydroxy-metabolites, testosterone and oestradiol-17β (see Figure 11.2), are the more potent androgen and oestrogen, respectively.
Regulation of Steroid Synthesis
In the ovary, testis and adrenal cortex, the synthesis of steroid hormones is regulated primarily by trophic hormones secreted from the anterior lobe of the pituitary gland, acting in conjunction with other endocrine, paracrine and autocrine modulators of steroidogenesis. The anterior pituitary hormones, being hydrophilic, have to act via cell surface receptors coupled to signal transduction pathways that increase the expression and activities of steroidogenic enzymes. While several second messengers are generated in response to trophic hormones, the steroidogenic responses to these proteins are mediated primarily through the generation of cAMP with activation of PKA. In the acute response to endocrine stimulation, activation of PKA phosphorylates: (i) cholesteryl ester hydrolase (increasing mobilization of cholesterol from intracellular lipid droplets); and (ii) the StAR protein (thus promoting mitochondrial uptake of cholesterol for steroid biosynthesis) (Niswender 2002, Miller 2008). In addition, activation of the cAMP–PKA system rapidly upregulates expression of the StAR protein, apparently via the cAMP-response element modulator (CREM) protein interacting with a non-classical cAMP response element half-site in the promoter region of the StAR gene (Sugawara et al 2006). In the chronic steroidogenic response, PKA activates the cAMP response element binding (CREB) protein, and this transcription factor upregulates the expression of apolipoprotein B100 [low-density lipoprotein (LDL)] receptors (Golos et al 1986) together with several of the steroidogenic CYP enzymes (Simpson et at 1990, Sewer et al 2007, Miller 2008) (see Figure 11.3).
Endocrine control of ovarian steroid synthesis
In the developing ovarian follicle, the synthesis of oestradiol is dependent upon both luteinizing hormone (LH) and follicle-stimulating hormone (FSH) acting in concert (Hillier et al 1994). Within the theca cells, LH stimulates the synthesis of androstenedione. Until the mid-1990s, it was assumed that this weak androgen was metabolized to testosterone prior to secretion from the theca. However, molecular studies demonstrated that human theca cells do not express the HSD17B3 isoenzyme required to convert androstenedione to testosterone (Zhang et al 1996). Instead, androstenedione passes from the theca cell layer across the membrana propria into the follicular interior. Within the granulosa cells, CYP19 converts androstenedione to oestrone which is subsequently metabolized to oestradiol by the HSD17B1 enzyme. Hence, the synthesis of oestradiol in the ovarian follicle requires LH to stimulate androgen output from the theca cells, while FSH stimulates the expression of CYP19 in the granulosa cells (Hillier et al 1994) (Figure 11.5).
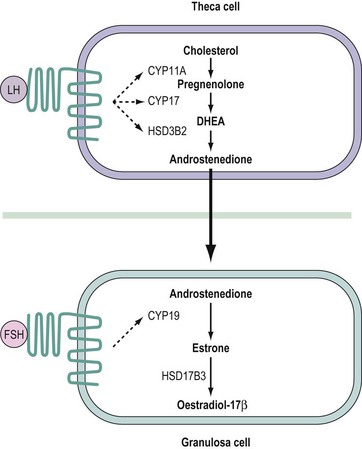
Figure 11.5 The two cell–two gonadotrophin model for follicular biosynthesis of oestradiol. See text for abbreviations.
Clinical experience from assisted conception cycles has called into question this ‘two cell–two gonadotrophin model’ for follicular oestradiol biosynthesis. Prior to the development of highly purified and recombinant preparations of FSH, it had been assumed that the in-vivo oestradiol response to human menopausal gonadotrophin reflected the fact that this gonadotrophin exhibited both LH and FSH activities. However, even after effective suppression of pituitary gonadotrophin secretion using a gonadotrophin-releasing hormone (GnRH) analogue, follicular growth and oestradiol output can be stimulated by highly purified or recombinant FSH without the need for administration of exogenous LH (Schoot et al 1994). Although this finding prompted some researchers to question the need for LH to support ovarian oestradiol production, it has been contested that the low levels of endogenous LH remaining in patients following administration of a GnRH agonist/antagonist may still be adequate to stimulate synthesis of androgen precursor in the theca cells of ovarian follicles.
In the periovulatory phase of the ovarian cycle, oestradiol induces the expression of LH receptors within the luteinizing granulosa cells (Kessel et al 1985, Farookhi and Desjardins 1986). Hence, in the CL, both the granulosa- and theca-derived luteal cells express LH receptors (Rao et al 1978, Yeko et al 1989). Moreover, the luteinizing granulosa cells cease to express FSH receptors and so become unresponsive to FSH. In the luteal phase of the ovarian cycle, the majority of progesterone is synthesized within and secreted from the granulosa-derived cells of the CL under the endocrine stimulus of LH, acting via the cAMP second messenger system (Devoto et al 2002). Potential conversion of progesterone to androstenedione within the granulosa-derived luteal cells is restricted by the negligible expression of CYP17 (Sasano et al 1989). Molecular investigations have revealed that in the CL of women and non-human primates, oestradiol biosynthesis continues to require cooperation between two morphologically distinct cell populations. In the theca-derived luteal cells, cholesterol is converted to androstenedione by the sequential actions of CYP11A, CYP17 and HSD3B2. Androstenedione then diffuses into the neighbouring granulosa-derived luteal cells where it serves as a substrate for CYP19 in the synthesis of oestradiol (via oestrone) (Fisch et al 1989). Whereas expression of CYP19 in the follicular granulosa cells is reliant on FSH, expression of the same enzyme in luteinized granulosa cells is dependent upon LH (Devoto et al 2002). Hence, the CL employs a two cell–one gonadotrophin system for oestradiol biosynthesis, as opposed to the two cell–two gonadotrophin system evident in the preovulatory follicle.
In the event of conception, the steroidogenic activity of the CL must be sustained until the luteo–placental shift, at which point placental steroidogenesis supersedes ovarian steroid production. The functional rescue of the CL, in a phenomenon referred to as the ‘maternal recognition of pregnancy’, relies on the fact that human chorionic gonadotrophin (hCG) can stimulate steroid synthesis in luteal cells via the same receptors as LH (Rao et al 1978, Jia et al 1991). Indeed, while hCG has a longer plasma half-life than LH, the glycoprotein structures that dictate receptor specificity are identical in LH and hCG, such that both hormones can bind the LH receptor with similar affinities (Jia et al 1991). In-vitro studies indicate that, on binding to the LH receptor in primate granulosa-derived luteal cells, hCG and LH are equipotent in increasing both intracellular cAMP levels and progesterone synthesis (Molskness et al 1991, Abayasekara et al 1993).
In addition to the gonadotrophins, other hormones, including growth hormone (GH) and insulin, have been implicated in the endocrine regulation of ovarian steroidogenesis, acting in synergy with the pituitary gonadotrophins (Franks 1998, Devoto et al 2002). Both GH and insulin activate receptor tyrosine kinases which phosphorylate different protein targets to those in the cAMP–PKA signal transduction cascade. This ability to operate through different cell signalling pathways may explain, at the cellular level, how insulin and GH can synergize with the gonadotrophins in stimulating ovarian steroidogenesis.
Glucocorticoids can also participate in the endocrine control of ovarian steroid biosynthesis. In general, these adrenal steroids antagonize the effects of gonadotrophins and suppress the activities of steroidogenic enzymes in ovarian cells, which may contribute to the dysregulation of ovarian steroid production in syndromes of adrenal hyperactivity (including chronic stress, anorexia nervosa and Cushing’s disease) (Michael and Cooke 1994).
Paracrine control of ovarian steroid synthesis
Within the ovary, a number of molecules exert paracrine/autocrine actions to modulate the steroidogenic response to the pituitary gonadotrophins. Recent interest has focused on such local actions of ovarian growth factors, some of which (e.g. vascular endothelial growth factor) influence ovarian activity (growth of ovarian follicles and development and maintenance of the CL) and steroid synthesis by mediating ovarian angiogenesis (Kaczmarek et al 2005). Others, such as the insulin-like growth factors (IGFs), act directly on follicular or luteal cells to synergize with LH or FSH in stimulating the activities of steroidogenic enzymes. For example, like insulin, IGFs I and II enhance the stimulation of theca cell androstenedione production by LH (Nahum et al 1995) and stimulate granulosa cell progesterone synthesis (Willis et al 1998). Members of the IGF family of growth factors provide the best example of how ovarian steroid production can be tightly controlled at the local level of the ovary. Not only are the paracrine/autocrine actions of IGFs dependent upon the local concentrations of each IGF and the appropriate plasma membrane receptors, but the effects of IGFs on ovarian steroidogenesis are further regulated by local concentrations of IGF-binding proteins (IGF-BP1 to IGF-BP5 inclusive) (Giudice et al 1995).
Inhibin and activin, which were originally identified as major secretory products of the ovary and testis, are members of the transforming growth factor-β superfamily of growth factors (De Kretser and Robertson 1989, Hillier and Miro 1993). Within the developing ovarian follicle, inhibin and activin appear to act in concert to regulate the stimulation of androstenedione production in the theca cells and of CYP19 expression in the granulosa cells (Hsueh et al 1987, Hillier and Miro 1993).
Research in the 1990s clearly demonstrated that immune cells, particularly macrophages and lymphocytes, actively participate in the paracrine control of ovarian steroid synthesis (Chryssikopoulos 1997). At this juncture, it is relevant to note that cytokines released within the ovary from ovarian leucocytes can also modulate the effects of LH and FSH on ovarian steroidogenesis. For example, interleukin-1β can inhibit LH-stimulated androgen synthesis in cultured theca cells (Hurwitz et al 1991).
Finally, prostaglandins synthesized either within the ovary or in the uterus (transported to the ovary by countercurrent exchange from the uterine vein to ovarian artery) can also affect ovarian steroidogenesis. In general, prostaglandin (PG) E2 stimulates steroid biosynthesis (apparently via the same cAMP–PKA pathway as LH), whereas PGF2α can act in ovarian cells to antagonize LH action and thus inhibit progesterone synthesis in the CL (Michael et al 1994, Olofsson and Leung 1994). Indeed, it has been speculated that the local synthesis of PGF2α within the CL may induce the functional regression of this transient ovarian gland at the end of the non-fertile menstrual cycle (Michael et al 1994, Olofsson and Leung 1994).
Endocrine control of adrenal steroid synthesis
In the same way as steroid biosynthesis by the ovary depends on LH and FSH from the gonadotroph cells of the anterior pituitary gland, the synthesis of corticosteroids by the cells of the adrenal cortex is dependent upon adrenocorticotrophic hormone (ACTH) secreted from the pituitary corticotroph cells. This hydrophilic protein hormone binds to cell surface receptors on adrenocortical cells to activate the cAMP–PKA signal transduction pathway (Schimmer 1995).Through increases in intracellular cAMP concentrations, ACTH stimulates cholesterol uptake (via LDL receptors), increases both the expression and activities of the CYP and HSD enzymes required to synthesize the adrenal steroid hormones (Simpson et al 1990, Sewer et al 2007), and crucially upregulates expression of the StAR protein which can then be activated by PKA-mediated phosphorylation (as in the ovary).
The adrenal cortex comprises three functional zones: the zona reticularis, zona fasciculata and zona glomerulosa. Cells in each adrenocortical zone express a different complement of steroidogenic enzymes and so synthesize different steroid products. Adrenal zona reticularis cells do not express HSD3B2 and so their major secreted product is DHEA: a 19 carbon, Δ5 steroid (i.e. weak androgen) which tends to be secreted as DHEA sulphate (Endoh et al 1996). The major product secreted from cells in the zona fasciculata of the adrenal cortex is the glucocorticoid cortisol which exerts negative feedback at the hypothalamus and anterior pituitary gland to control subsequent endocrine input from ACTH. Hence, if the biosynthesis of cortisol is compromised or if a patient is glucocorticoid resistant (commonly due to a loss-of-function mutation in the glucocorticoid receptor), pituitary ACTH secretion becomes elevated and the ACTH induces increased proliferation of the adrenocortical cells; a condition termed ‘congenital adrenal hyperplasia’ (CAH; see below). The third and final zone of the adrenal cortex, the zona glomerulosa, expresses the CYP11B2 enzyme required for the conversion of corticosterone to aldosterone (Muller 1987). Hence, the major function of zona glomerulosa cells is the synthesis and secretion of the mineralocorticoid required to control sodium–potassium exchange in the kidney and colon. While the synthesis of DHEA(S) and cortisol is under the endocrine control of ACTH, the synthesis of aldosterone is regulated in a multifactorial manner, with the most important stimulus to secretion being altered electrolyte and fluid status (Muller 1987). A fall in plasma volume triggers an increase in the release of prorenin from the glomeruli of individual nephrons in the kidney. In the general circulation, the precursor protein prorenin is converted to the active enzyme renin which in turn metabolizes the plasma protein angiotensinogen to angiotensin I. Within the lungs, angiotensin-converting enzyme converts angiotensin I to angiotension II and it is the latter peptide which acts at the zona glomerulosa to increase the synthesis of aldosterone. Acting via mineralocorticoid receptors in the distal segments of renal nephrons and in the colon, aldosterone stimulates the uptake of sodium ions in exchange for potassium ions. Since the net influx of sodium ions exceeds the efflux of potassium ions, water is reclaimed from the lumen of the nephron or colon, thus increasing plasma volume and hence blood pressure to close this homeostatic loop. As aldosterone increases the loss of potassium from the body, the synthesis of aldosterone in zona glomerulosa cells can also be stimulated directly by a rise in the plasma potassium concentration (Muller 1987).
Defects in Steroid Synthesis
Genital virilization/female pseuohermaphroditism
Virilization of the external female genitalia (see Chapter 13) usually reflects a pathological increase in androgen secretion from either the adrenal zona reticularis or the ovary. The severity of clinical presentation, ranging from clitoromegaly with labial hypertrophy to male external genitalia (usually with hypospadias), depends upon the source and age of onset of the elevation in the plasma androgen concentration (hyperandrogenaemia).
Congenital adrenal hyperplasia
Increased adrenal androgen output (adrenal hyperandrogenism) is a hallmark of the common forms of CAH and usually reflects loss-of-function mutations in CYP21 or CYP11B1 which prevent cells of the zona fasciculata from synthesizing cortisol (Figures 11.6 and 11.7). The consequent loss of negative feedback within the fetal hypothalamo–pituitary–adrenal axis causes an elevation in ACTH that stimulates adrenal hyperplasia with increased synthesis of DHEA in utero (White et al 1987). Although DHEA is only a weak androgen, this Δ5 steroid can be metabolized to androstenedione and testosterone in peripheral tissue, such as subcutaneous adipose tissue. Within the genital skin fibroblasts, the sequential actions of the HSD3B, HSD17B and 5α-reductase enzymes convert DHEA to 5α-dihydrotestosterone (DHT) which causes virilization of the external genitalia. Under the action of DHT, the genital tubercle differentiates into a glans, the urethral folds fuse to form a phallus and the labioscrotal swellings fuse to form an empty scrotum (see Chapter 13). In addition to causing virilization of the external genitalia, adrenal hyperandrogenism will also manifest as development of male secondary sexual characteristics in a woman (see Chapter 26). Of these, the most notable tend to be male-pattern hair loss, hirsutism and increased body odour, symptoms commonly observed in women with polycystic ovary syndrome (PCOS) (see below and Chapter 18).
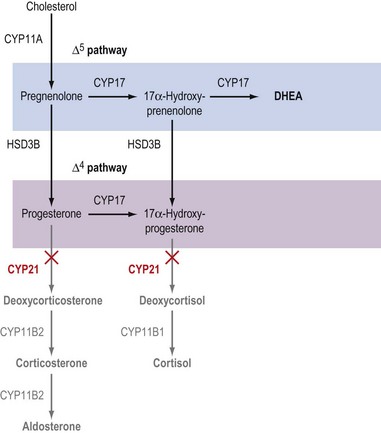
Although the effects of excessive DHEA production on the external genitalia and secondary sexual characteristics obviously affect a patient’s future sexual identity and fertility, the other endocrine sequelae of CAH are of greater medical importance. In patients with complete loss of CYP21 activity (see Figure 11.6), the total absence of corticosteroids renders the patient unable to maintain normal glucose homeostasis (due to the loss of glucocorticoids) and to regulate sodium–potassium exchange in the kidney and colon (due to the loss of aldosterone). Hence, complete loss of CYP21 activity causes ‘salt-wasting’ CAH, alternatively referred to as ‘simple virilizing’ CAH (White et al 1987). In contrast, ‘salt-sparing’ CAH can result from either a partial loss of CYP21 activity or from a complete loss of CYP11B1 activity (White et al 1987, Nimkarn and New 2008) (see Figure 11.7). In either event, the absence of glucocorticoids presents as hypoglycaemia with depleted hepatic glycogen stores. However, in salt-sparing CAH, deoxycorticosterone (DOC), a weak mineralocorticoid, is able to exert some control over renal and colonic ion balance. Indeed, because the synthesis of DOC is outside the control of the renin–angiotensin axis, this steroid tends to hyperstimulate sodium resorption and loss of potassium, and because water movement follows the flux of sodium ions, the elevation of DOC usually results in the hypervolaemic syndrome of ‘malignant hypertension’ in patients with salt-sparing CAH.
Ovarian hyperandrogenism
Essentially, there are three potential causes of ovarian hyperandrogenism:
As noted earlier in this chapter, insulin and IGFs can also stimulate thecal androgen production, significantly enhancing the steroidogenic response of theca cells to LH (Nahum et al 1995). By definition, PCOS is characterized by elevated plasma concentrations of insulin (hyperinsulinaemia) accompanied by clinical signs of hyperandrogenism (see Chapter 18). A number of studies have indicated that the hyperinsulinaemia reported in women with PCOS reflects resistance to the cellular actions of insulin in peripheral tissues (muscle and adipose tissue) (Dunaif et al 1989). However, the ability of insulin to stimulate ovarian steroidogenesis appears to be preserved in PCOS patients (Ben-Shlomo 2003), such that in PCOS, the elevated concentrations of insulin may act in concert with LH to stimulate excess ovarian androgen production.
Mutations in the CYP19 gene
A final mechanism for disturbing the delicate biochemical balance between the theca and granulosa cell compartments of the ovarian follicle is to decrease the capacity of the granulosa cells to metabolize thecal androstenedione substrate. The simplest mechanism to explain such a distortion in the steroidogenic pathway would be a complete loss-of-function mutation in the CYP19 gene that encodes the aromatase enzyme. Due to the requirements for oestradiol and oestriol in pregnancy, fetuses affected by complete loss of function in CYP19 are usually miscarried. However, a few patients with severely limited CYP19 activity have been reported. In such patients, failure of the granulosa cells to metabolize any androgen precursor results in profound ovarian hyperandrogenism with progressive clitoromegaly and accelerated linear growth (MacGillivray et al 1998). Interestingly, affected girls do not show the usual cessation of linear growth at puberty, despite having plasma testosterone concentrations far in excess of the reference range for normal adolescent boys. These patients have revealed the crucial role for the CYP19 enzyme in the fusion of the epiphyses in the long bones; in both sexes; closure of the epiphyseal plates requires testosterone to be aromatized within the bone to oestradiol, such that women with defective CYP19 activity require administration of exogenous oestrogen to halt their linear growth (MacGillivray et al 1998).
Although mutations in CYP19 are rare, there are other disease states in which the capacity of granulosa cells to aromatize androgens is limited to the extent that ovarian hyperandrogenism ensues. For example, loss-of-function mutations in the FSH receptor, which have been associated with hypergonadotrophic ovarian dysgenesis, can prevent FSH from upregulating CYP19 expression in the granulosa cells despite a normal plasma LH:FSH ratio (Tapanainen et al 1998). Alternatively, glucocorticoids can inhibit the expression and activity of CYP19 in cultured granulosa cells (Hsueh and Erickson 1978, Fitzpatrick and Richards 1991), an observation that explains, in part, the association between adrenal hyperactivity and dysregulation of ovarian steroid synthesis in conditions such as anorexia nervosa and other chronic stress syndromes.
KEY POINTS
Abayasekara DRE, Michael AE, Webley GE, Flint APF. Mode of action of prostaglandin F2α in human luteinized granulosa cells: role of protein kinase C. Molecular and Cellular Endocrinology. 1993;97:81-91.
Ben-Shlomo I. The polycystic ovary syndrome: what does insulin resistance have to do with it? Reproductive Biomedicine Online. 2003;6:36-42.
Chryssikopoulos A. The relationship between the immune and endocrine systems. Annals of the New York Academy of Sciences. 1997;816:83-93.
Curnow KM, Tusie-Luna M-T, Pascoe L, et al. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Molecular Endocrinology. 1991;5:1513-1522.
De Kretser DM, Robertson DM. The isolation and physiology of inhibin and related proteins. Biology of Reproduction. 1989;40:33-47.
Devoto L, Kohen P, Vega M, et al. Control of human luteal steroidogenesis. Molecular and Cellular Endocrinology. 2002;186:137-141.
Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165-1174.
Endoh A, Kristiansen SB, Casson PR, Buster JE, Hornsby PJ. The zona reticularis is the site of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3β-hydroxysteroid dehydrogenase. Journal of Clinical Endocrinology and Metabolism. 1996;81:3558-3565.
Farookhi R, Desjardins J. Luteinizing hormone receptor induction in dispersed granulosa cells requires estrogen. Molecular and Cellular Endocrinology. 1986;47:13-24.
Fisch B, Margara RA, Winston RML, Hillier SG. Cellular basis of luteal steroidogenesis in the human ovary. Journal of Endocrinology. 1989;122:303-311.
Fitzpatrick SL, Richards JS. Regulation of cytochrome P450 aromatase messenger ribonucleic acid and activity by steroids and gonadotropins in rat granulosa cells. Endocrinology. 1991;129:1452-1462.
Franks S. Growth hormone and ovarian function. Baillière’s Clinics in Endocrinology and Metabolism. 1998;12:331-340.
Giudice LC, van Dessel HJ, Cataldo NA, Chandrasekher YA, Yap O, Fauser BC. Circulating and ovarian IGF binding proteins: potential role in normo-ovulatory cycles and in polycystic ovarian syndrome. Progress in Growth Factor Research. 1995;6:397-408.
Golos TG, August AM, Strauss JFIII. Expression of low density lipoprotein receptor in cultured human granulosa cells: regulation by human chorionic gonadotropin, cyclic AMP and sterol. Journal of Lipid Research. 1986;27:1089-1096.
Hauet T, Yao ZX, Bose HS, et al. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into Leydig cell mitochondria. Molecular Endocrinology. 2005;19:540-554.
Hillier SG, Miro F. Inhibin, activin and follistatin. Potential roles in ovarian physiology. Annals of the New York Academy of Sciences. 1993;687:29-38.
Hillier SG, Whitelaw PF, Smyth CD. Follicular oestrogen synthesis: the ‘two-cell, two-gonadotrophin’ model revisited. Molecular and Cellular Endocrinology. 1994;100:51-54.
Hsueh AJW, Erickson GF. Glucocorticoid inhibition of FSH-induced estrogen production in cultured rat granulosa cells. Steroids. 1978;32:639-648.
Hsueh AJW, Dahl KD, Vaughan J, et al. Heterodimers and homodimers of inhibin subunits have different paracrine action in the modulation of luteinizing hormone-stimulated androgen biosynthesis. Proceedings of the National Academy of Sciences USA. 1987;84:5082-5086.
Hurwitz A, Payne DW, Packman JN, et al. Cytokine-mediated regulation of ovarian function: interleukin-1 inhibits gonadotropin-induced androgen biosynthesis. Endocrinology. 1991;129:1250-1256.
Jia X-C, Oikawa M, Bo M, et al. Expression of human luteinizing hormone (LH) receptor: interaction with LH and chorionic gonadotropin from human but not equine, rat and ovine species. Molecular Endocrinology. 1991;5:759-768.
Kaczmarek MM, Schams D, Ziecik A. Role of vascular endothelial growth factor in ovarian physiology — an overview. Reproductive Biology. 2005;5:111-136.
Kessel B, Liu YX, Jia XC, Hsueh AJW. Autocrine role for estrogens in the augmentation of luteinizing hormone receptor formation in cultured rat granulosa cells. Biology of Reproduction. 1985;32:1038-1050.
Liu J, Rone MB, Papadopoulos V. Protein–protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. Journal of Biological Chemistry. 2006;281:38879-38893.
MacGillivray MH, Morishima A, Conte F, Grumbach M, Smith EP. Pediatric endocrinology update: an overview. The essential roles of estrogens in pubertal growth, epiphyseal fusion and bone turnover: lessons from mutations in genes for aromatase and estrogen receptor. Hormone Research. 1998;49(Suppl 1):2-8.
Michael AE, Cooke BA. A working hypothesis for the regulation of steroidogenesis and germ cell development in the gonads by glucocorticoids and 11β-hydroxysteroid dehydrogenase (11βHSD). Molecular and Cellular Endocrinology. 1994;100:55-63.
Michael AE, Abayasekara DRE, Webley GE. Cellular mechanisms of luteolysis. Molecular and Cellular Endocrinology. 1994;99:R1-R9.
Miller WL. Mechanisms of StAR’s regulation of mitochondrial cholesterol transport. Molecular and Cellular Enodcrinology. 2007;265/266:46-50.
Miller WL. Steroidogenic enzymes. Endocrine Development. 2008;13:1-18.
Molskness TA, Zelinski-Wooten MB, Hild-Petito SA, Stouffer RL. Comparison of the steroidogeneic response of luteinized granulosa cells from rhesus monkeys to luteinizing hormone and chorionic gonadotropin. Biology of Reproduction. 1991;45:273-281.
Muller J. Regulation of aldosterone biosynthesis. In: Monographs on Endocrinology 29. Berlin: Springer-Verlag; 1987.
Nahum R, Thong KJ, Hillier SG. Metabolic regulation of androgen production by human thecal cells in vitro. Human Reproduction. 1995;10:75-81.
Nimkarn S, New MI. Steroid 11β-hydroxylase deficiency congenital adrenal hyperplasia. Trends in Endocrinology and Metabolism. 2008;19:96-99.
Niswender GD. Molecular control of luteal secretion of progesterone. Reproduction. 2002;123:333-339.
Olofsson J, Leung PCK. Auto/paracrine role of prostaglandins in corpus luteum function. Molecular and Cellular Endocrinology. 1994;100:87-91.
Penning TM. Molecular endocrinology of hydroxysteroid dehydrogenases. Endocrine Reviews. 1997;18:281-305.
Petrescu AD, Gallegos AM, Okamura Y, Strauss JF, Schroeder F. Steroidogenic acute regulatory protein binds cholesterol and modulates mitochondrial membrane sterol domain dynamics. Journal of Biological Chemistry. 2001;276:36970-36982.
Rao CV, Sanfilippo J, Carman FRJr. Gonadotropin receptors in human corpora lutea of term pregnancies. American Journal of Obstetrics and Gynecology. 1978;132:581-583.
Sasano H, Okanoto M, Mason JI, et al. Immunolocalization of aromatase, 17α-hydroxylase and side-chain-cleavage cytochromes P-450 in the human ovary. Journal of Reproduction and Fertility. 1989;85:163-169.
Schimmer BP. The 1994 Upjohn Award Lecture. Molecular and genetic approaches to the study of signal transduction in the adrenal cortex. Canadian Journal of Physiology and Pharmacology. 1995;73:1097-1107.
Schoot DC, Harlin J, Shoham Z, et al. Recombinant human follicle-stimulating hormone and ovarian response in gonadotrophin-deficient women. Human Reproduction. 1994;9:1237-1242.
Sewer MB, Dammer EB, Jagarlapudi S. Transcriptional regulation of adrenocortical steroidogenic gene expression. Drug Metabolism Reviews. 2007;39:371-388.
Simpson ER, Lund J, Ahlgren R, Waterman MR. Regulation by cyclic cAMP of the genes encoding steroidogenic enzymes: when the light finally shines. Molecular and Cellular Endocrinology. 1990;70:C25-C28.
Stocco DM, Clark BJ. Regulation of the acute production of steroids in steroidogenic cells. Endocrine Reviews. 1996;17:221-244.
Sugawara T, Sakuragi N, Minakami H. CREM confers cAMP responsiveness in human steroidogenic acute regulatory protein expression in NCI-H295R cells rather than SF-1/Ad4BP. Journal of Endocrinology. 2006;191:327-337.
Tapanainen JS, Vaskivuo T, Aittomaki K, Huhtaniemi IT. Inactivating FSH receptor mutations and gonadal dysfunction. Molecular and Cellular Endocrinology. 1998;145:129-135.
White PC, New MI, Dupont B. Congenital adrenal hyperplasia. New England Journal of Medicine. 1987;316:1519-1524.
Willis DS, Mason HD, Watson H, Franks S. Developmentally regulated responses of human granulosa cells to insulin-like growth factors (IGFs): IGF-1 and IGF-II action mediated via the type-1 IGF receptor. Journal of Clinical Endocrinology and Metabolism. 1998;83:1256-1259.
Yeko TR, Khan-Dawood FS, Dawood MY. Human corpus luteum: luteinizing hormone and chorionic gonadotropin receptors during the menstrual cycle. Journal of Clinical Endocrinology and Metabolism. 1989;68:529-534.
Zhang Y, Word RA, Fesmire S, Carr BR, Rainey WE. Human ovarian expression of 17β-hydroxysteroid dehydrogenase types 1, 2 and 3. Journal of Clinical Endocrinology and Metabolism. 1996;81:3594-3598.
[/level-membership-for-obstetrics-gynecology-category][not-level-membership-for-obstetrics-gynecology-category]
CHAPTER 11 Biosynthesis of steroid hormones
Physiology
The classification of steroid hormones
All steroid hormones are ultimately derived from the 27 carbon (C) substrate cholesterol and so share the same cyclohexaphenanthrene ring structure (Figures 11.1 and 11.2). Steroids are classified into five families dependent upon the number of carbon atoms and the chemical groups present at key carbon residues (Table 11.1). Those steroid hormones with 21 carbon atoms are collectively termed ‘pregnenes’, and this category of steroid hormones can be subdivided into three steroid families: progestins (e.g. progesterone), glucocorticoids (e.g. cortisol) and mineralocorticoids (e.g. aldosterone). While the progestins are secreted predominantly from the ovary, the glucocorticoids and mineralocorticoids are collectively termed ‘corticosteroids’, reflecting their origin in the cortex of the adrenal gland. The major structural difference between the three pregnene families is that while progestins possess a methyl group (CH3) at position C21, both glucocorticoids and mineralocorticoids possess a C21 hydroxyl group (CH2OH) (see Table 11.1 and Figure 11.2). Progestins can be metabolized to generate 19 carbon steroids, termed ‘androgens’ [dehydroepiandrosterone (DHEA), androstenedione and testosterone], which are secreted from both the testis and the adrenal cortex. Within the ovary, androgens are usually metabolized to generate the oestrogens (e.g. oestradiol-17β) with their characteristic 18 carbon structure (see Table 11.1 and Figure 11.2).
Within this classification scheme, there is a distinction to be made between Δ5 and Δ4 steroid hormones. Progestins and androgens of the Δ5 series are characterized by possessing a hydroxyl group at position C3 and a CC double bond between positions C5 and C6 in the steroid B-ring, as in cholesterol (Figure 11.1). In contrast, progestins and androgens of the Δ4 series possess a ketone (CO) at position C3 and have their CC double bond between positions C4 and C5 in the A-ring of the steroid molecule (Figure 11.2). While this difference may seem a trivial biochemical detail, nothing could be further from the truth. The nature of the chemical group at position C3, together with the position of the CC double bond, profoundly alters the conformation of the steroid molecule and, in so doing, influences the ability of the hormone to activate intracellular receptors. Hence, Δ5 steroids, such as pregnenolone and DHEA, have low affinities for steroid receptors such that they can only exert limited biological actions. In contrast, the Δ4 steroids, such as progesterone and testosterone, are potent activators of steroid receptors and so act as the dominant physiological hormones.
Origins and intracellular transport of cholesterol
Once inside the cell, cholesterol must be transported across the cytosol and into the mitochondria where the first and rate-determining reaction of steroid synthesis occurs: the catabolism of cholesterol to form pregnenolone (see below). The transport of cholesterol across the two mitochondrial membranes and interceding aqueous intermembrane space is now recognized as the true rate-limiting step for steroidogenesis, and so with this realization, it is this process which has been the focus of research into steroid biosynthesis over the past two decades. Although the understanding of the molecular mechanisms by which cholesterol passes from the outer leaflet of the outer mitochondrial membrane (OMM) to the inner leaflet of the inner mitochondrial membrane (IMM) is still far from complete, significant advances have been made over the past decade. In the mid 1990s, the importance of the steroidogenic acute regulatory (StAR) protein was first indicated when Clark and Stocco showed StAR to be a short half-life protein which is rapidly upregulated in steroidogenic cells following stimulation either by trophic hormones (e.g. gonadotrophins) or by the common second messenger, cyclic adenosine 3′,5′-monophosphate (cAMP) (Stocco and Clark 1996). Analysis of the StAR peptide sequence suggested that this protein gets imported from the cytosol into the IMM, and it was postulated that the StAR protein might simply bind and transport cholesterol across the aqueous intermembrane space during the course of that import. However, subsequent site-directed mutagenesis studies revealed that the StAR protein could still drive cholesterol flux for steroidogenesis even when it was modified to prevent mitochondrial import, and it is now accepted that the StAR protein can act at the OMM to drive cholesterol passage from the outer leaflet of the OMM to the inner leaflet of the IMM without itself trafficking into the mitochondria (Miller 2007). This steroidogenic action of the StAR protein does, however, require: (i) that the StAR protein be phosphorylated on key serine residues by the cAMP-dependent protein kinase A (PKA); and (ii) that the phosphorylated form of the StAR protein interacts with the 18 kDa translocator protein (TSPO) expressed in the OMM (Figure 11.3) (Hauet et al 2005, Miller 2007). The TSPO protein, which was originally described as a peripheral benzodiazepine receptor (PBR), is very highly expressed in the OMM of steroidogenic cells. Recent studies have revealed that rather than reliance on a single protein to drive mitochondrial cholesterol uptake for steroidogenesis, this pivotal rate-limiting step involves a complex of proteins which includes the StAR and TSPO proteins in association with PBR-associated protein-7 (PAP7). By virtue of its structural interactions with both the TSPO protein and the PKAR1α regulatory subunit of PKA, PAP7 acts as a PKA-anchoring protein, localizing PKA to the OMM where it can phosphorylate the serine residues in the StAR protein to drive the StAR–TSPO interaction required to deliver cholesterol to the IMM for the first reaction in the steroidogenic pathway (Liu et al 2006, Miller 2007). While the StAR protein is not expressed in the placenta, this tissue does express a homologous protein, metastatic lymph node 64, which shares several functional and structural properties with the StAR protein, and is assumed to drive mitochondrial cholesterol uptake for placental steroidogenesis (Petrescu et al 2001).
Steroidogenic enzymes
Although the biochemical pathway of steroid hormone synthesis, summarized in Figure 11.4, can look like a bewildering railway map, the whole pathway becomes more straightforward on recognizing that the eight major steroidogenic enzymes can all be classified into just two enzyme families (the cytochrome P450 enzymes and the hydroxysteroid dehydrogenases), each with definitive functions. The steroidogenic cytochrome P450 (CYP) enzymes are members of a larger superfamily of CYP oxidase enzymes. Each CYP enzyme catalyses a hydroxylation reaction by serving as the terminal electron acceptor in an electron transport pathway which transfers electrons from a pyridine nucleotide cofactor, nicotinamide adenine dinucleotide phosphate, via two sequential flavoproteins (ferrodoxin reductase and ferrodoxin) on to atomic oxygen (Miller 2008). This allows the atomic oxygen to be inserted, by the CYP enzyme, into a hydrocarbon (C․H) bond thus generating a polar hydroxyl group (C․O․H) which renders the substrate molecule more water soluble/hydrophilic. The non-steroidogenic CYP enzymes include those hepatic oxidase enzymes responsible for the inactivation and clearance of a wide range of drugs. Each of the CYP enzymes within the steroidogenic pathway has traditionally been referred to by a variety of names that reflect their ability to catalyse hydroxylation of a steroid substrate at a specific carbon position. However, following the completion of the human genome (HUGO) project, all steroidogenic CYP enzymes have been assigned new systematic names which relate to the CYP gene that encodes the enzyme protein (Table 11.2).
In addition to their characteristic hydroxylase activities, some steroidogenic CYP enzymes also catalyse lyase reactions. Specifically, the CYP11A (cytochrome P450 cholesterol side chain cleavage) and CYP17 (cytochrome P450 17α-hydroxylase/C17,20-lyase) enzymes can each catalyse cleavage of the C․C bond weakened by the hydroxylation reactions. CYP11A acts in the IMM to catalyse the rate-limiting reaction in steroid synthesis: the conversion of cholesterol to pregnenolone. This crucial CYP enzyme hydroxylates two adjacent carbons (C20 and C22) in the D-ring side chain of cholesterol, facilitating cleavage between C20 and C22 to leave the Δ5 21 carbon steroid, pregnenolone (Miller 2008). Similarly, CYP17 introduces a hydroxyl group at position C17 of either pregnenolone or progesterone, as a result of which the weakened C17–C20 bond breaks to generate either DHEA or androstenedione, respectively (Miller 2008) (see Figure 11.4). Other members of the CYP enzyme family simply catalyse introduction of oxygen to generate hydroxyl groups at specific carbon positions (see Table 11.2). The hydroxylations catalysed by CYP21 (21-hydroxylase) and CYP11B1 (11β-hydroxylase) are pivotal in the formation of corticosteroids by the adrenal cortex (Miller 2008). The hydroxyl group generated at position C18 by CYP11B2 (aldosterone synthase) undergoes rapid oxidation to form an aldehyde group (Curnow et al 1991), hence giving rise to the name ‘aldosterone’ (see Figure 11.2). Finally, CYP19 (aromatase) is the most complex member of the steroidogenic CYP enzyme family, catalysing a series of reactions that convert C19 androgens (androstenedione and testosterone) to their C18 oestrogen metabolites (oestrone and oestradiol, respectively). In this reaction sequence, the C19 methyl group is lost and the ketone at position C3 is reduced to a hydroxyl group (Miller 2008). This liberates electrons which are invested in the A-ring of the steroid to generate the aromatic phenol ring, the hallmark of oestrogens (see Figure 11.2) and a prerequisite for activation of the oestrogen receptor.
The second family of steroidogenic enzymes comprises the hydroxysteroid dehydrogenase (HSD) enzymes encoded by HSD genes (Penning 1997, Miller 2008). (As with the CYP enzymes, the HSD enzymes have all been renamed following the completion of the HUGO project, such that the systematic names for the 3βHSD and 17βHSD isoenzymes have changed to HSD3B and HSD17B, respectively.) All of the HSD enzymes catalyse the interconversion of a hydroxyl group with a ketone to influence the affinity of the steroid hormone for its intracellular receptors (Penning 1997). Each HSD enzyme is specified by a number and a Greek letter; the number denotes the position of the carbon atom at which the enzyme acts, and the Greek letter indicates the orientation of the OH group relative to the steroid molecule (where α and β indicate bonds below and above the plane of the molecule, respectively).
There are two HSD enzymes of significance in steroid hormone production. The two cloned HSD3B isoenzymes convert the 3β-hydroxyl group of weak Δ5 steroids to a ketone, and also catalyse movement of the CC double bond to generate the corresponding Δ4 steroid metabolite, thus increasing steroid potency (Penning 1997, Miller 2008). Hence, both the placental HSD3B1 enzyme and HSD3B2, expressed in the ovary, testis and adrenal gland, can convert weak Δ5 steroids, such as pregnenolone and DHEA, to the active Δ4 progestins and androgens, progesterone and androstenedione, respectively (see Figure 11.4). In contrast, the major HSD17B isoenzymes act at position C17 where they catalyse not oxidation but reduction, converting a ketone at C17 to a β-hydroxyl group. The significance of this reaction is attested to by the fact that androstenedione and oestrone (which have ketone groups at position C17) are relatively weak steroids, whereas their 17β-hydroxy-metabolites, testosterone and oestradiol-17β (see Figure 11.2), are the more potent androgen and oestrogen, respectively.
Regulation of Steroid Synthesis
In the ovary, testis and adrenal cortex, the synthesis of steroid hormones is regulated primarily by trophic hormones secreted from the anterior lobe of the pituitary gland, acting in conjunction with other endocrine, paracrine and autocrine modulators of steroidogenesis. The anterior pituitary hormones, being hydrophilic, have to act via cell surface receptors coupled to signal transduction pathways that increase the expression and activities of steroidogenic enzymes. While several second messengers are generated in response to trophic hormones, the steroidogenic responses to these proteins are mediated primarily through the generation of cAMP with activation of PKA. In the acute response to endocrine stimulation, activation of PKA phosphorylates: (i) cholesteryl ester hydrolase (increasing mobilization of cholesterol from intracellular lipid droplets); and (ii) the StAR protein (thus promoting mitochondrial uptake of cholesterol for steroid biosynthesis) (Niswender 2002, Miller 2008). In addition, activation of the cAMP–PKA system rapidly upregulates expression of the StAR protein, apparently via the cAMP-response element modulator (CREM) protein interacting with a non-classical cAMP response element half-site in the promoter region of the StAR gene (Sugawara et al 2006
[/not-level-membership-for-obstetrics-gynecology-category]
